Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 94/03469 2141 ~~ 7 9 PCT/US93/07307
1
NEW DERIVATIVES OF :L~1EUR.AMINIC ACID
BACKGROUND OF 'THE INVENTION
Field of the Invention
The present invention rE~lates to new derivatives of
neuraminic acid, especially carboxylic amides of the
following formula,
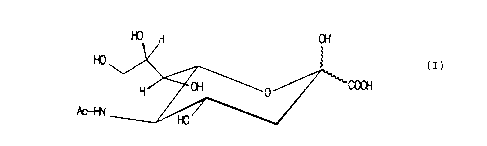
N OH
W ~ ~ ~ (I>
H/ ~~_ WO
Ar+N
where Ac represents an acyl residue of a carboxylic acid
of the aliphatic, araliphatic, aromatic, alicyclic or
heterocyclic series, comprie;ing the carboxylic amides,
their 2-hydrocarbyl-glycosides, and their peracylated
derivatives at the hydroxy groups of both these series
of amides.
These compounds have interesting pharmacological
properties, especially a protective effect against the
neurotoxicity induced by excitatory amino acids of the
of glutamic acid type, and can therefore be used in
therapies of the central :.~rvous syste,.,, such as those
following cerebral degener,ations or lesions, e.g.,
ischemia, hypoxia, epilepsy, trauma or compressions,
metabolic dysfunctions, aging, toxic-infective and
WO 94/03469
PCT/US93/07307
2
chronic neurodegenerative diseases, like Alzheimer's,
Parkinson's, and Huntington's diseases.
The carboxylic amides and their derivatives of
formula I according the present invention are new.
Description of Related Art
In the literature, there is a description of the
non-substituted amide of N-acetyl-neuraminic acid,
prepared as an intermediate in the synthesis of
tetrazolyl-2-decarboxy-N-acetyl-neuraminic acid (see
Ann. 1986, 2104-11).
In an article published in Hoppe Seyler's
Physiol. Chemie, 1983, 364 (109) 1411-17, there is a
description of the amides obtainable through the
reaction of the benzylketoside of N-acetyl-neuraminic
acid with L-glycine, L-glutamic acid, and
L-phenylalanine, followed by the elimination of the
benzyl group through catalytic hydrogenation; no
pharmacological action is described for these
derivatives.
SUMMARY OF THE INVENTION
In addition to providing new derivatives of
neuraminic acid, the present invention also provides
pharmacological preparations containing the aforesaid
derivatives for therapeutic use.
A third object of the present invention concerns
the therapeutic use of these preparations.
A final object of the present invention concerns
procedures for the production of these new derivatives.
Further scope of the applicability of the present
invention will become apparent from the detailed
description provided below. However, it should be
understood that the detailed description and specific
examples, while indicating preferred embodiments of the
invention, are given by way of illustration only, since
various changes and modifications within the spirit and
w~ 3 ~ l 41679
scope of the invention will become apparent to those
skilled in the art from this detailed description.
DETAILED DESCRIPTION OF THE INVENTION
The following detailed description of the
invention is provided to aid those skilled in the art
in practicing the present invention. Even so, the
following detailed description should not be construed
to unduly limit the present invention, as modifications
and variations in the embodiments herein discussed may
be made by those of 'ordinary skill in the art without
departing from the spirit or scope of the present
inventive discovery.
The amides and their derivatives according to
the present invention can derive from both possible
anomeric forms in position 2 of neuraminic acid, and
therefore all the new compounds can be of a type at
that position. The steric configuration of the other
carbon atoms of the neuraminic residue is the same as
that of the natural acid.
The acyl group Ac on the nitrogen of the
neuraminic acid residue in the aforesaid formula has at
least 4 and not more than 24 carbon atoms, and derives
from non-substituted or substituted acids, preferably
from 1 to 3 functions selected from the group
consisting of halogen atoms; free, esterified, or
etherified hydroxylic or mercapto groups; free or
esterified carboxylic or sulfonic groups, or such
groups transformed into amides; and free hydrocarbylic
groups or hydrocarbylic groups substituted aminic
groups.
These acids can be interrupted by -SO-, -S02-,
or phenylene groups in the carbon atom chain of the
hydrocarbylic residue. The halogen atoms are
preferentially fluorine, bromine, or chlorine.
Esterified hydroxylic or mercapto groups can derive
f rom
w 2141679
WO 94/03469 I'Cf/US93/07307
4
one of the acids mentioned regarding the Ac group, but
they preferentially derive from aliphatic or aromatic
acids with not more than 8 carbon atoms. Moreover, they
can derive from inorganic acids such as, for example,
sulfuric or phosphoric acid, or especially from their
partial esters with mono- or polyvalent aliphatic
alcohols, eventually with hydroxylic groups or aminic
functions substituting in the hydrocarbylic residues.
Finally, they can derive from hydrocarbylsulfonic acids .
Etherified hydroxy or mercapto groups,' or
esterified carboxylic or sulfonic groups, preferentially
derive from alcohols of the aliphatic series having no
more than 8 carbon atoms, or from the araliphatic series
with only one benzene ring and an alkylene of 1 or 2
carbon atoms. The hydrocarbylic groups which can
substitute the aminic groups preferentially derive from
these alcohols; the amino groups can also be in the form
of quaternary ammonium salts, e.g., tetraalkyl groups,
e.g., tetrabutylammonium.
Ac groups containing functionally modified hydroxy,
mercapto, or amino groups can also be present in the
form of hydrocarbylic residues of the Ac acyl group,
interrupted in the carbon atom chain by the heteroatoms
-O-, -S-, or -NH-, and, in the particular case of esters
of hydroxy or mercapto groups with partially esterified
sulfuric or phosphoric acid,, by groups of the type
O
-O-S-~-~ -O-p ~ , or -O_p~0
\OH \O _
and in those of esters with hydrocarbylsulfonic acids
(e.g., p-toluenesulfonic or methanesulfonic acid), by
-O-S02.
The hydrocarbylic reside Ac, as already stated,
can be blocked by sulfoxide or sulfdriyl residues. In
amides, converted carboxylic or sulfonic groups
preferentially derive from lower aliphatic amines with
WO 94/03469 ~ ~~ 416 7 9 , p~/US93/07307
not more than 4 carbon atom:, or from araliphatic amines
with only one benzene ring and one or two carbon atoms
in alkenyl residue.
The acids from which the Ac groups of the aliphatic
5 series derive can be saturated or unsaturated, and in
this case, they preferentially have only one double
bond, and can have linear or branched chains. Of
particular interest are the following acids: butyric,
valeric, particularly normal valeric and isovaleric,
trimethylacetic (pivalic acid), caproic, isocaproic,
enantic, caprylic, pelargonic, capric, undecilic,
di-tert-butyl-acetic, 2-propyl-valeric (valproic acid),
lauric, tridecilic, myristic, pentadecilic, palmitic,
margaric, stearic, arachic, behenic, and lignoceric.
Among substituted aliphatic acids, levulinic acid
must be mentioned; among dicarboxylic acids, succinic
acid; and among natural .amino acids, e.g., valine,
leucine, phenylalanine, tryptophan, aminobutyric acid,
methionine, lysine, aspartic acid, glutamic acid,
proline, hydroxyproline; among the acids substituted
with halogens, mono- and dichloroacetic acid,
trichlorobutyric acid, and dibromobutyric acid.
The Ac group of formula I can also derive from
natural or synthetic peptides preferentially having not
more than 12 amino acid:, selected from naturally
occurring amino acids, e.g., those aforesaid.
Of particular interest according to the present
invention are those in which Ac is an acyl residue
belonging to peptides of the thymus gland. Acids, from
which derive an Ac group of araliphatic nature, are
e.g., phenylacetic, cinnamic, phenylpropionic or atropic
acid. Among aromatic acids, benzoic acid and its
methylatPd homologues, salicylic acid, anthranilic acid,
trimethoxybenzoic acid, pht:halic or terephthalic acid,
o,o'-dicarbonic acid, chlorobenzoic acid, vanillic acid,
and veriatric or piperonilic acid must be mentioned.
WO 94/03469 ~ ~ ~ ~ ~ ~ PCT/US93/073~'1
6
Among Ac acyl groups belonging to acids of the
alicyclic series, there must be mentioned cyclohexane-
and cyclopentane-carbonic acids, hexahydrophthalic,
hexahydroisophhtalic and hexahydroterephthalic acids,
camphoric and apocamphoric acid, and, among acids with
a higher carbon atom content, prostaglandins and
steroidic acids such as, for example, cholanic or cholic
acid.
If Ac represents an acyl group belonging to an acid
of the heterocyclic series, this can be one of the
following acids: nicotinic or isonicotinic, cinconninic,
lysergic, isolysergic, dihydrolysergic,
2-bromo-lysergic, 2-bromo-dihydrolysergic,
1-methyl-lysergic, 1-methyl-dihydro-lysergic,
1-methyl-2-bromo-lysergic or teophyllinacetic.
The carboxylamido functions according to the
present invention can derive from ammonia (and in this
case it is the non-substituted amide, -CONH2), or from
primary or secondary aliphatic, aromatic, araliphatic,
alicyclic or heterocyclic amines, which can also be
substituted in the hydrocarbylic residue by one to three
functions selected from the group consisting of free,
esterified, or etherified hydroxylic or mercapto groups,
halogens, free, esterified, or amide-modified carboxylic
or sulfonic groups, and free or hydrocarbyl-substituted
amino groups, wherein the hydrocarbyl group is blocked
with an -SO- or-S02- group. These amines have no more
than 24 carbon atoms.
The functions which can eventually substitute the
carbon atom chain of the amide or the amine are
preferentially those mentioned for the Ac group of
formula I. Aliphatic amines can have an open, saturated,
unsaturated, linear, branched or cyclic chain. Of
particular interest are alkyl- and dialkylamines having
from 1 to 12 carbon atoms, such as, for example,
methylamine, ethylamine, propylamine, hexylamine,
diethylamine, dimethylamine, diisopropylamine,
WO 94/03469 2 1 . ~ ~ 9 PCT/US93/07307
dihexylamine and alkylenylamines having from 3 to 6
cyclic carbon atoms, wherein the rings are substituted
or non-substituted, preferentially between one and three
C1-14 alkyl groups, e.c~., methyl groups, like
pyrrolidine, piperidine, and azepine.
The hydrocarbylic chains can also be blocked with
heteroatoms such as, for e:xample, -O-, -S-, or -NH-
groups, or they can be substituted, as already
mentioned, with different: functions, particularly
alcoholic, amino, mercapto, carboxylic, and sulfonic
functions, or by their functionally modified forms, such
as esters, ethers, or alkylated derivatives. Of
particular interest are the following: aliphatic
diamines, like ethylenediamine, trimethylenediamine,
tetramethylenediamine, penta- and hexamethylenediamine,
piperazine and its N-alkyl o:r C-alkyl derivatives having
a C1-4 alkyl; aminoalcohols like aminoethanol or
aminopropanol; aminomercaptanes like mercaptoethylamine;
aliphatic aminoacids like all those mentioned for the Ac
group of formula I; and aminosulfonic acids like
taurine. Moreover, morphol.ine and thiomorpholine and
their alkylated derivatives such as, for example, those
which are N- or C-methylated, are also useful.
The amide groups eventually can derive from
peptides, such as those mentioned for The Ac group.
Of particular interest also are some derivatives of
amines having a large number of carbon atoms of an
aliphatic nature, and which .are related to phospholipids
or sphingolipids or similar derivatives.
According to the present invention, the carboxylic
group of the N-acyl-neuraminic acids can contain
saturated or unsaturated aminic groups having between 14
and 24 carbon atoms, or bases present in the following
lipids: phosphatidylethano7_amine, phosphatidylserine,
sphingosine, dihydrosphingosine, psychosine,
dihydropsychosine, phosphorylcholine-sphingosine,
WO 94/03469 '~, ~ ~ ~ ~~ ~ PCT/US93/073m
s
phosphorylcholine-dihydrosphingosine, and
phytosphingosine.
The carboxylamides of the present invention can
also derive from aromatic or araliphatic amines, but
preferentially from those having only one aromatic ring,
which can be substituted with 1 to 3 functional groups
selected from the group consisting of halogens,
hydroxylic or methoxylic groups, carboxylic or sulfonic
groups, or C1-4 lower aliphatic hydrocarbylic groups
such as, for example, aniline, ~anthranilic acid,
1-amino-4-sulfonic acid, and benzylamide. In the
aforesaid aliphatic amines, in one or more positions of
the hydrocarbylic chain, a phenyl group can be present
to block the carbon atom chain.
Amines which can be used for the conversion into
amides according to the present invention include, for
example, amines of pyrimidines such as cyanmethine,
i.e., 2,4-dimethyl-6-amino-pyrimidine, purine
derivatives such as adenine, 4-aminouracil, and guanine,
and alkaloids such as ephedrine, tyramine, and
adrenalin.
The 2-hydrocarbyl-glycosides of the aforesaid
amides of neuraminic acids of formula I derive from
alcohols of the aliphatic, cycloaliphatic, aromatic,
araliphatic or heterocyclic series, particularly from
alcohols of the aliphatic series having not more than 12
carbon atoms, or from the araliphatic series having
preferentially only one benzene ring, eventually
substituted with 1 - 3 lower C1-4 alkyl groups, for
example methyl groups, and not more than 4 carbon atoms
in the aliphatic chain, or from alcohols of the
alicyclic or aliphatic-alicyclic series having only one
cycloaliphatic ring and not more than 14 carbon atoms,
or from the heterocyclic series having not more than 12,
and especially 6 carbon atoms, and only one heterocyclic
ring containing 1 or 2 heteroatoms selected from the
group consisting of -NH-, -O- and -S-. These alcohols
°
~ WO 94/03469 PCT/US93/07307
2;141679
~3
can also be substituted, particularly with functions
selected from the group consisting of hydroxy, amino,
and alkoxy groups having not more than 4 carbon atoms,
and carboxylic and carbalkoxy groups having not more
than 4 carbon atoms in the alkyl residues.
The aforesaid alcohols can be mono- and polyvalent,
particularly bivalent. Among alcohols of the aliphatic
series, of particular interest are lower alcohols having
not more than 6 carbon atoms such as, for example,
methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, and
tert-butyl alcohol, and ethyleneglycol and
propyleneglycol, among divalent alcohols. Among
alcohols of the araliphat:ic series, of particular
interest are those having only one benzene residue, like
benzyl and phenetyl alcohol; among alcohols of the
alicyclic series, preferred are those having only one
cycloaliphatic ring, like cyclohexylic alcohol. Among
alcohols of the alicyclic series having more rings,
steroid alcohols such as, for example, those of the
pregnane group, like cortic:osteroids, and among these
methylprednisolone, must be mentioned. Among alcohols of
the heterocyclic series there must be mentioned
tetrahydrofuranol, tetrahyd:ropyranol, furfuryl alcohol
and pyridylcarbinol.
In peracylated derivatives of the amides and their
2-hydrocarbyl-glycosides, the hydroxy groups in position
2,4,7,8 and 9 are acylated 'with acids belonging to the
aliphatic, aromatic, araliphatic, alicyclic and
heterocyclic series. Peracylated derivatives derive
preferentially from acids of the aliphatic series having
not more than 10 carbon atoms, like formic, acetic, and
butyric acid and their isomers; valeric acids, like
normal valeric, or pivalic acid; and capronic or capric
acid. These acids can also be substituted, and the
peracylated derivatives c~~n therefore derive from
hydroxyacids like lactic a<:id, from amino acids like
glycine, or from dibasic acids like succinic, malonic or
y-WO 94/03469 ~ ~ 416 7 9 PCT/US93/07307
1. 0
malefic acid. Among aromatic acids, there must be
mentioned those with only one benzene ring, particularly
benzoic acid and its derivatives with methyl, hydroxy,
amino or carboxylic groups such as, for example,
5- p-aminobenzoic, salicylic and phthalic acid.
The new compounds according to present invention
can eventually be transforme=d into their acidic addition
salts or into metallic salts with organic bases, if the
corresponding basic or acidic functions are present.
These salts can also be used for the therapeutic
purposes described infra. With respect to this
equivalence between salts and amides in free form, it is
obvious that what will be de=scribed for the compounds in
free form, especially their pharmaceutical and medical
applications, is also.true for the corresponding salts,
provided that these salts are therapeutically
acceptable, and therefore they also form an object of
the present invention. These salts can also be used for
the purification of the amides, and in this case also,
therapeutically non-acceptable bases and acids can be
used, such as salts of picric and picrolonic acid.
Compounds of the Present Invention
Defined compounds according to the present
invention include the amide, methylamide, ethylamide,
dimethylamide, diethylamide, propylamide, glycine amide,
L-serine amide, aminobutyric amide, L-cysteine amide,
taurine amide, the amide of cysteic acid, homocysteic
acid, N-palmitoyl-neuraminic acid, N-stearoyl-neuraminic
acid, N-acetyl-neuraminic acid, N-propionyl-neuraminic
acid, N-pivaloyl-neuraminic: acid, N-valeroyl-neuraminic
acid, N-caproyl-neuraminic acid, N-lauroyl-neuraminic
acid, N-succinyl-neuraminic acid, phenylacetyl-
neuraminic acid, benzoyl-neuraminic acid, trimethoxy-
benzoyl-neuraminic acid, phthaloyl-neuraminic acid,
chlorobenzoyl-neuraminic acid, vanilloyl-neuraminic
~~ JO 94/U3469 1416 7 9 . , I'CI'/US93/U73U7
11
acid, cyclopenthane- and cycl.ohexane-carbonyl-neuraminic
acid, N-nicotinyl-neuraminic acid, N-isonicotinyl-
neuraminic acid, lisergyl-neuraminic acid, 2-bromo-
lisergyl-neuraminic acid, 1.-methyl-lisergyl-neuraminic
acid, theophillineacetyl-neuraminic acid, and their
2-glycosides derived from one of the following alcohols:
methyl alcohol, ethyl alcohol, propyl alcohol, isopropyl
alcohol, butyl alcohol, i;sobutyl alcohol, tertbutyl
alcohol, ethyleneglycol, propyleneglycol, benzyl
alcohol, methylprednisolone, tetrah~rdrofuranol,
tetrahydropyranol, fu:rfuryl alcohol, and
pyridylcarbinol.
Other compounds of the: present invention include
the amide, methylamide, ethylamide, dimethylamide,
diethylamide, propylamide, glycine amide, L-serine
amide, aminobutyric amide, L-cysteine amide, taurine
amide of N-acylneuraminic <~cids having an acyl group
deriving from one of the following acids: aminobutyric,
methionine, lysine, aspart is acid, glutamic acid,
proline, tryptophan, ~or from an acyl residue deriving
from a peptide present ~i.n the thymus, and their
2-glycosides deriving from one of the following
alcohols : methyl alcohol , etlhyl alcohol , propyl alcohol ,
isopropyl alcohol, butyl <~lcohol, isobutyl alcohol,
tertbutyl alcohol, ethyleneglycol, propyleneglycol,
benzyl alcohol, methyl-predr~isolone, tetrahydrofuranol,
tetrahydropyranol, fu:rfuryl alcohol, and
pyridylcarbinol.
Another group of interesting compounds according to
the present invention is foamed by the.amides deriving
from pyrrolidine, piperidine, azepine, ethylenediamine,
trimethylenediamine, hexamethylenediamine, piperazine or
N-methyl or N-ethyl-piperazine, aminoethanol,
aminopropanol, mercapto~ethylamine, morpholine,
tiomorpholine, or peptides like those present in the
thymus, and from phosphatidylethanolamine,
phosphatidylserine, sp:hingosine, psychosine,
':.
WO 94/03469 ~ PC1"/US93/07307
2141679
12
dihydropsychosine, sphingosylphosphorylcholine,
dihydrosphingosylphosphoryl~~holine, or from the
phytosphingosine of one of the following
N-acyl-neuraminic acids: N-palmitoyl-neuraminic acid,
N-stearoyl-neuraminic acid, N-acetyl-neuraminic acid,
N-propionyl-neuraminic acid, N-pivaloyl-neuraminic acid,
N-valeroyl-neuraminic acid, I~&-caproyl-neuraminic acid,
N-lauroyl-neuraminic acid, N,--'succinyl-neuraminic acid,
phenylacetyl-neuraminic acid, phthaloyl-neuraminic acid,
chlorobenzoyl-neuraminic acid, N-nicotinyl-neuraminic
acid, N-isonicotinyl-neuraminic acid, lisergyl-
neuraminic acid, 2-bromo-lisergyl-neuraminic acid,
1-methyl-lisergyl-neuraminic acid, theophillineacetyl-
neuraminic acid, and their 2.-glycosides derived from one
of the following alcoho7.s: methyl alcohol, ethyl
alcohol, propyl alcohol, isopropyl alcohol, butyl
alcohol, isobutyl alcohol, tertbutyl alcohol,
ethyleneglycol, propylene:glycol, benzyl alcohol,
methylprednisolone,tetrhydrofuranol,tetrahydropyranol,
furfuryl alcohol, and pyrid.ylcarbinol.
Other derivatives- are: those peracylated at the
hydroxy groups of N-aryl-neuraminic acid, and
particularly peracetates, perpropionates, perbutyrates,
pervalerianates, perpiva7.ates, persuccinates and
perbenzoates.
Synthesis of the compounds of the present invention
The present invention also comprises processes for
the preparation of the new amides of N-acyl-neuraminic
acids, their 2-hydrocarbylc~lycosides, and their salts.
These processes are a:Lready known, and consist of
the stepwise introduction of the amine function and
eventually of the 2-g:lycosidic group into an
N-acyl-neuraminic acid, and eventually of acyl groups
-into the hydroxy groups and the final formation of their
salts.
WO 94/03469 2 ~ 4 16 7 9 PCT/US93/07307
13
In a preferred embodirnent, the carboxylic group of
an N-acyl-neuraminic acid, in which the acyl group is
desired in the final compound, or its
2-hydrocarbyl-glycosidic dE~rivatives, is transformed to
an amide group and, if desired, the 2-hydrocarbyl group
can be eliminated, again forming the hydroxy group; if
desired, the obtained compound is converted into a
peracylated derivative at the hydroxy functions.
The carboxy group of the neuraminic acid can be
converted into the amide, and eventually the 2-hydroxy
group in its hydrocarbyl derivatives in both sequences
and acylate the free amino group with the desired acid
and, if desired, peracylatE~ the free hydroxy groups, or
perform this peracylation in every step of the
processes, e.g., at the beginning.
The conversion of the: carboxylic group of N-acyl
derivatives of neuraminic acid or of their
2-hydrocarbyl-glycosides to the corresponding amide can
be performed directly starting from the acid, or from
its metal or organic base soalt, or indirectly preparing
first the ester. of the acid, an anhydride, or a
halogenide of the acid, and then converting these
compounds into the amide.
A preferred method ~~onsists of activating the
carboxylic group and then reacting the intermediate with
the desired amine, utilizing methods known in peptide
chemistry, avoiding methods utilizing acidic or basic
conditions. If metal salts of the acid, like sodium, are
used, it is convenient to treat the salt with an ion
exchange resin of the Dowe~s~ type or a similar resin. As
an example, it is possible to use the condensation
method in presence of carbodiimides, e.g.,
dicyclohexylcarbodiimide,benzylisopropylcarbodiimide or
benzylethylcarbodiimide, in the presence of
1-hydroxybenzo-triazol, or the condensation in the
presence of N,N'carbonyl-di.imidazol. Starting from the
aforesaid acidic derivatives, like esters or
Trademark
WO 94/03469 ~ (~ PCT/US93/07307
:L 4
halogenides, e.g., bromides or chlorides, the
transformation into the amide is carried out by direct
treatment with the desired amine at relatively low
temperature, e.g., room temperature or -5 °C to 10 °C,
or higher temperatures, e.g., between 30 and 120 °C.
Ketones, aromatic hydrocarbides, dimethylformamide,
dimethylsulfoxide, dioxane or tetrahydrofuran can be
used as solvents. The starting esters can be aliphatic
esters, e.g., ethyl or methyl esters, or aromatic
esters, e.g., phenols.
The 2-O-hydrocarbyl derivatives of the starting
compounds or of the compounds already possessing the
amine function are prepared according to the conditions
known for the acetylation of aldehydes or ketones, or
for the preparation of glycosides. The 2-hydrocarbyl
groups of the glycosides can be transformed at the
hydroxy group at every step by hydrolysis with acids
under mild conditions.
If acylation of the amine group of neuraminic acid
is performed at the end of the procedure, e.g., after
amide or glycoside for;nation, known acylation methods
are used, e.g., treatment of the compound with acid
halogenides or anhydrides, eventually in the presence of
inorganic or organic bases, like pyridine or collidine.
This acylation can be performed contemporaneously with
the acylation of hydroxy groups.
The transformation of the final compounds into
their salts, as well as the interconversion of the
salts, is performed in a known manner, as for example
when intermediate salts are prepared for their
purification.
The aforesaid procedure according to the present
invention also comprises all variations in which the
procedure is stopped at evE=ry step, or in which the
starting compound is an intermediate, or in which the
starting compounds are prepared in situ.
WO 94/03469 2 : s "~ PCT/US93/07307
The synthesis of the compounds of the present
invention is illustrated by the following examples.
Examx~le 1
Butilamide of N-acetylneuraminic acid
5 3.23, g (10 ~ mM) of N-acetylneuraminic acid
methylester, prepared according to Kuhn et al., Chem
Ber. 99,611 (1966), were solubilized in 50 ml of
anhydrous methyl alcohol; 3.66 g (50 mM) of 2-butylamine
were added. The mixture was stirred for 5 hours at 40
10 °C. The solution was evaporated under vacuum and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/water, 110:40:6. The fractions containing the
butilamide of N-acetyl-neura~minic acid were gathered and
15 evaporated under vacuum. T'he residue was crystallized
from 50 ml of n-propyl alcohol. Yield: 85%.
Rf - 0.25, chloro:form/methyl alcohol/water,
110:40:6.
.Examt~le 2
Q-2-O-ethylalycoside of butilamide of N-acetylneuraminic
acid
3.65 g (10 mM) of t:he (3-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester, prepared according
to Kuhn et al., Chem Ber. 99, 611 (1966), were
solubilized in 80 ml of anhydrous methyl alcohol; 3.66
g (50 mM) of 2-butylamine were added. The mixture was
stirred for 5 hours at 40 °C. The solution was
evaporated under vacuum and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/water, 80:20:2. The
fractions containing the ~3-2-O-ethylglycoside of the
butilamide of N-acetyl-neuraminic acid were gathered and
evaporated under vacuum. The residue was crystallized
from 50 ml of n-propyl alcohol and 100 ml of ethyl
ether. Yield: 70%.
WO 94/03469 2 1 4 1 6 7 9
PCT/US93/07307
16
Rf = 0.37, chloroform/methyl alcohol/water,
110:40:6;
0.19, chloroform/methyl alcohol/2.5N NH40H,
80:20:2.
Example 3
a-2-O-ethvlctlvcoside of the benzylamide of
N-acetylneuraminic acid
3.65 g (10 mM) of the ~3-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester, prepared according
to Kuhn et al., Chem Ber. 99, 611 (1966), were
solubilized in 50 ml of anhydrous methyl alcohol; 5.36
g (50 mM) of benzylamine were added. The mixture was
stirred for 5 hours at 40 °C. The solution was
evaporated under vacuum and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/water, 80:20:2. The
fractions containing the ~i-2-O-ethylglycoside of the
benzylamide of N-acetyl-neuraminic acid were gathered
and evaporated under vacuum. The residue was
crystallized from 50 ml of isopropyl alcohol. Yield:
65%.
Rf = 0.50, chloroform/methyl alcohol/water,
110:40:6;
0.16, chloroform/methyl alcohol/2.5N NH40H,
80:20:2.
Example 4
a-2-O-ethvlalycoside of the dimethylaminopropylamide of
N-acetylneuraminic acid
3.65 g (10 mM) of the (3-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester were solubilized in
50 ml of anhydrous methyl alcoh~7.; 10.2 a (lOn mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
WO 94/03469 _ 214 I ~~ ~ 9 PCT/US93/07307
1'7
chloride/methyl alcohol/2.5N NH40H, 55:45:10. The
fractions containing the ~i-2-O-ethylglycoside of
dimethylaminopropylamide of N-acetyl-neuraminic acid
were gathered and evaporateol under vacuum. The residue
was dissolved in 50 ml of water. Yield: 75%.
Rf = 0.19, chloroform,/methyl alcohol/2.5N NH40H,
40:60:15.
Exam~~le 5
Q-2-O-ethvlctlvcoside of the dimethylaminopropylamide of
N-acetylneuraminic acid (mal.eic acid salt)
3.65 g (10 mM) of tine /3-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester were solubilized in
50 ml of anhydrous methyl alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue: was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/2.5N NH40H, 55:45:10. The
fractions containing the ~i-2-O-ethylglycoside of the
dimethylaminopropylamide of: N-acetyl-neuraminic acid
were gathered and evaporated under vacuum. The residue
was dissolved in 50 ml of wager, a stoichiometric amount
of malefic acid was added, and the material was
lyophilized. Yield: 75%.
Rf = 0.19, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
Exammle 6
a-2-O-ethylalycoside of the dimethylamide of
N-acetvlneuraminic acid
3.65 g (10 mM) of t:he ~i-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester were solubilized in
50 ml of anhydrous methyl alcohol; 4.5 g (100 mM) of
dimethylamine were added. The mixture was stirred
overnight at 25 °C. The solution was evaporated under
vacuum and the residue was purified by silica gel
WO 94/03469 ~ ~ ~ ~ ~ ~ (~
PCT/US93/07307
18
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 80:20:2. The fractions
containing the,-2-O-ethylglycoside of the dimethylamide
of N-acetyl-neuraminic acid were gathered and evaporated
under vacuum. The residue was crystallized from 30 ml
of methanol and 150 ml of ethylrether. Yield: 80%.
Rf = 0.38, chloroform/methyl alcohol/water,
110:40:6.
Example 7
a-2-O-ethvlctlvcoside of the dimethylaminopropylamide of
N-palmitoylneuraminic acid
5.62 g (10 mM) of the ~3-2-O-ethylglycoside of
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methyl alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 110:40:6. The fractions
containing the /3--2-O-ethylglycoside of the
dimethylaminopropylamide of N-palmitoyl-neuraminic acid
were gathered and evaporated under vacuum. The residue
was dissolved in 60 ml of water and lyophilized. Yield:
70 %.
Rf = 0.12, chloroform/methyl alcohol/2.5N NH40H,
80:20:2.
Example 8
-2-O-ethvlctlycoside of the dimethvlaminopropvlamide of
N-palmitoylneuraminic acid (malefic acid salt)
5.62 g (10 mM) of the ~i-2-O-ethylglycoside of
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methyl alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
WO 94/03469 PCT/US93/07307
2.141 ~'~9
....
19
chromatography, using as solventt a mixture of methylene
chloride/methyl alcohol/wat:er, 110:40:6. The fractions
containing the ~i-2-O-ethylglycoside of the
dimethylaminopropylamide of N-palmitoyl-neuraminic acid
were gathered and evaporated under vacuum. The residue
was dissolved in 50 ml of water, a stoichiometric amount
of malefic acid was added, and the material was
lyophilized. Yield: 70%.
Rf = 0.12, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
Example 9
a-2-O-ethvlglvcoside of the dimethylaminopropylamide of
N-nalmitoylneuraminic acid
5.48 g (10 mM) of t:he a-2-O-ethylglycoside of
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methy7_ alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 110:40:6. The fractions
containing the a-2-O-ethylglycoside of the
dimethylaminopropylamide of N-palmitoyl-neuraminic acid
were gathered and evaporated under vacuum. The residue
was dissolved in 60 ml of water and lyophilized. Yield:
70%.
Rf = 0.40, chloroform/methyl alcohol/0.3% CaCl2,
60:40:9.
Examp:L a 10
a-2-O-ethylalycoside of the dimethvlaminopropvlamide of
N-palmitoylneuraminic acid (male~~ acid salt?
5.48 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoylneuraminic acid Ethyl ester were solubilized
in 50 ml of anhydrous methyl alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
WO 94/03469 PCT/US93/07307
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 110:40:6. The fractions
5 containing the a-2-O-ethylglycoside of the
dimethylaminopropylamide of N.=~palmitoyl-neuraminic acid
were gathered and evaporated'under vacuum. The residue
was dissolved in 50 ml of water, a stoichiometric amount
of malefic acid was added, and the material was
10 lyophilized. Yield: 70%.
Rf = 0.40, chloroform/methyl alcohol/0.3% CaCl2,
60:40:9.
Example 11
Q-2-O-ethylglvcoside of the dimethylamide of
15 N-palmitoylneuraminic acid
5.56 g (10 mM) of the ~i-2-O-ethylglycoside of
N-palmitoyl-neuraminic acid, sodium salt, were dissolved
in 50 ml of pyridine, and 2.3 g (20 mM) of pyridinium
chloride and 4.12 g (20 mM) of N,N'dicyclohexyl-
20 carbodiimide were added. The mixture was stirred for 2
hours at 25 °C. 4.5 g (100 mM) of dimethylamine were
added and the reaction was conducted overnight at 25 °C.
The solution was evaporated under vacuum and the residue
was purified by silica gel chromatography, using as
solvent a mixture of methylene chloride/methyl
alcohol/water, 80:10:1. The fractions containing the
~i-2-O-ethylglycoside of the dimethylamide of
N-palmitoyl-neuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 50 ml of
acetone and precipitated in 20 volumes of hexane. Yield:
90%.
Rf = 0.69, chloroform/methyl alcohol/water,
110:40:6.
WO 94/03469
1 ~~.16 7 9 p~/US93/07307
2 :L
Example 12
a-2-O-ethylcrlycoside of the dimethylamide of
N-palmitoylneuraminic acid
5.48 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methyl alcohol; 4.5 g (100 mM) of
dimethylamine were added. The mixture was stirred
overnight at 25 °C. The so7Lution was evaporated under
vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 80:10:1. The fractions
containing the a-2-O-ethylglycoside of the dimethylamide
of N-palmitoyl-neuraminic acid were gathered and
evaporated under vacuum. The residue was dissolved in
50 ml of acetone and precipitated in 20 volumes of
hexane. Yield: 90%.
Rf = 0.69, chloroform/methyl alcohol/water,
110:40:6.
Examnl e: 13
a-2-O-ethylctl~rcoside ~ of the butyl amide of
N-acetvlneuraminic acid
3.65 g (10 mM) of the a-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester were solubilized in
50 ml of anhydrous methyl alcohol; 3.66 g (50 mM) of
butylamine were added. 'The mixture was stirred
overnight at 25 °C. The solution was evaporated under
vacuum and the residue way; purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 80:20:2. The fractions
containing the a-2-O-ethylglycoside of the butylamide of
N-acetyl-neuraminic acid were gathered and evaporated
under vacuum. The residue was crystallized from 50 ml
of methanol and 300 ml of ethyl ether. Yield: 75%.
WO 94/03469 PCT/US93/07zn;
~14~~79
22
Rf - 0.55, chloroform/methyl alcohol/water,
110:40:6;
Rf - 0.53, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
Example .14
a-2-O-ethylQlycoside of the dimethylaminopropvlamide of
N-acetylneuraminic acid
3.65 g (10 mM) of the a-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester were solubilized in
50 ml of anhydrous methyl alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/2.5N NH40H, 55:45:10. The
fractions containing the a-2-O-ethylglycoside of the
dimethylaminopropylamide of N-acetyl-neuraminic acid
were gathered and evaporated under vacuum. The residue
was dissolved in 50 ml of water and lyophilized. Yield:
700.
Rf = 0.21, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
Example 15
a-2-O-ethvlglycoside of thedimethvlaminopropvlamide of
N-acetylneuraminic acid (malefic acid salt)
3.65 g (10 mM) of the a-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester were solubilized in
50 ml of anhydrous methyl alcohol; 10.2 g (100 mM) of
dimethylaminopropylamine were added. The mixture was
stirred overnight at 25 °C. The solution was
evaporated under vacuum and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/2.5N NH40H, 55:45:10.
The fractions containing the a-2-O-ethylglycoside of the
dimethylaminopropylamide of N-acetyl-neuraminic acid
23
21416.79
were gathered and evaporated under vacuum. The residue
was dissolved in 50 ml of water, a stoichiometric amount
of malefic acid was added, and the material was
lyophilized. Yield: 70$.
Rf - 0.21, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
Example 16
a-2-O-ethylalycoside of N-ace~tvlneuraminic acid amide with
L-alanine-D-iso9~lutamine
3.65 g (10 mM) of the a-2-O-ethylglycoside of N-
acetylneuraminic acid ethyl ester were solubilized in
40 ml of water, and 10 ml (10 mM) of NaOH were added. The
solution was maintained at 25°C for 30 minutes,
neutralized with 1N HCl, and eluted with water from a
column containing 30 ml of Dowex*50x8 resin, pyridinium
form. The eluate was lyophilized and the residue was
solubilized in 100 ml of anhydrous pyridine. 1.15 g
(10 mM) of N-hydroxysuccinimide and 4.13 g (20 mM) of
N,N'-dicyclohexylcarbodiimide~ were added at -10°C. After
15 minutes, the temperature was raised to 25°C, and the
mixture was stirred for 5 hours. 5.14 g (15 mM) of L-
alanine-D-isoglutamine benzyl ester hydro-chloride
(prepared according to Le Francier (Bull. Soc. Chim.
Biol., Vol. 49, No. 10 (196;7)) and Kusumoto (Bulletin of
the Chemical Society of Japan, Vol. 49(2), pp 533-539
(1976)) were added at 25°C. The mixture was stirred
overnight and then evaporated under vacuum. The obtained
residue was dissolved in 200 ml of a mixture of n-
butanol/water/acetic acide, 4:1:1, and hydrogenated in an
H2 current in the presence of BaS04-supported palladium.
After filtration, the solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of: methylene chloride/methyl
alcohol/water, 60:35:8. The fractions containing the a-2-
0-ethylglycoside of N-acetyl-neuraminic acid amide with L-
alanine-D-iso-glutamine were gathered and evaporated under
vacuum. The residue was
*Trademark
r
WO 94/03469 2 1416 7 9 PCT/US93/07307
24
dissolved in 200 ml of wager and lyophilized. Yield:
600.
Rf - 0.56, chloroform~/methyl alcohol/2.5N NH40H,
60:35:8;
Rf = 0.18, chloroform/methyl alcohol/0.3% CaCl2,
60:40:9.
Exam~> 1 a 17
Peracetylated a-2-O-ethylglycoside of N-palmitovl-
neuraminic acid amide with L-alanine-D-isoglutamine
5.56 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoylneuraminic acid, sodium salt, were
solubilized in 50 ml of anhydrous N, N' -dimethylformamide
at 25 °C; 3.06 g (11 mM) of p-bromophenacyl bromide were
added and the solution was stirred overnight. 18 ml of
anhydrous pyridine and 10.:? g of acetic anhydride were
added and stirring was cond'.ucted for 24 hours at 35 °C.
The solution was evaporated under vacuum and the residue
was dissolved with 100 ml of water and extracted three
times with 200 ml of methy~lene chloride . The organic
phases were washed twice with 50 ml of water and then
gathered, anhydrified with anhydrous sodium sulfate, and
evaporated under vacuum. The obtained residue was
solubilized in 50 ml of anhydrous N,N'-dimethylformamide
at 25 °C; 2.64 g (20 mM) of sodium thiophenate were
added and the mixture was stirred for 4 hours. The
solution was evaporated under high vacuum. The residue
was extracted three times with 200 ml of ethyl acetate,
washed with 100 ml of cold 1N HC1 and twice with 50 ml
of water. The organic phases were anhydrified with
anhydrous sodium sulfate, gathered, and evaporated under
vacuum. The residue was di:~solved in 30 ml of water and
eluted with water from a column containing 30 ml of
Dowex 50x8 resin, pyridinium form. The eluate was
lyophilized and the residue was solubilized in 100 ml of
anhydrous pyridine. 1.15 g (10 mM) of
N-hydroxysuccinimide and 4.13 g (2o mM) of
Trademark
- ~ WO 94/03469 ~ ~41 G 7 9 pCT/U593/07307
c. 5
N,N'-dicyclohexylcarbodiimide were added at -10 °C.
After 15 minutes the temperature was raised to 25 °C and
the mixture was stirred for 5 hours. 5.14 g (15 mM) of
L-alanine-D-isoglutamine benzyl ester hydrochloride
(prepared according to Le Francier and Kusumoto) were
added at 25 °C. The mixture was stirred overnight and
then evaporated under vacuum. The obtained residue was
dissolved in 200 ml. of a mixture of
N-butanol/water/acetic acid, 4:1:1, and hydrogenated in
an H2 current in the presence of BaS04-supported
palladium. After filtration, the solution was
evaporated, and the residue: was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 60:35:8. The fractions
containing the a-2-O-ethylglycoside of N-acetyl-
neuraminic acid amide with L-alanine-D-isoglutamine were
gathered and evaporated under vacuum. The residue was
dissolved in 200 ml of water and lyophilized. Yield:
55%.
Rf = 0.54, chloroform/methanol, 90:10.
Examp:l a 18
2-O-ethvlalvcoside of N-~~cetvlneuraminic acid amide
with L-alanine-D-isog~lutamine
3.59 g (10 mM) of the ~i-2-O-ethylglycoside of
N-acetylneuraminic acid, sodium salt, prepared according
to Eschenfelder and Brossmer, Hoppe Seyler's Z. Physiol.
Chem. 360, 1253 (1979), were solubilized in 50 ml of
anhydrous N,N'-dimethylformamide at 25 °C; 3.06 g (11
mM) of p-bromophenacyl bromide were added and the
solution was stirred overnight. 18 ml of anhydrous
pyridine and 10.2 g of acetic anhydride were added and
stirring was canducted for 24 hours at 35 °C. The
solution was evaporated under vacuum and the residue was
dissolved with 100 ml of water and extracted three times
with 200 ml of methylene chloride. The organic phases
were washed twice with 50 ml of water and then gathered,
~~ JVO 94/03469 2 ~ 416 7 9 ~'~/US93/07307
:26
anhydrified with anhydrous sodium sulfate, and
evaporated under vacuum. The obtained residue was
solubilized in 50 ml of anhydrous N, N' -dimethylformamide
at 25 °C; 2.64 g (20 mM) of sodium thiophenate were
added and the mixture was stirred for 4 hours. The
solution was evaporated under high vacuum. The residue
was extracted three times with 200 ml of ethyl acetate,
washed with 100 ml of cold 1N HC1 and twice with 50 ml
of water. The organic phases were anhydrified~ with
anhydrous sodium sulfate, gathered, and evaporated under
vacuum. The residue was dissolved in 30 ml of water and
eluted with water from a column containing 30 ml of
Dowex* 50x8 resin, pyridinium form. The eluate was
lyophilized and the residue was solubilized in 100 ml of
anhydrous pyridine. 1.15 g (10 mM) of
N-hydroxysuccinimide and. 4.13 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added at -10 °C.
After 15 minutes, the temperature was raised to 25 °C
and the mixture was stirred for 5 hours. 5.14 g (15 mM)
of L-alanine-D-isoglutamine benzyl ester hydrochloride
(prepared according to Le Francier and Kusumoto) were
added at 25 °C. The mixture was stirred overnight and
then evaporated under vacuum. The obtained residue was
dissolved in 60 ml of anhydrous methanol at 25 °C. 100
mg of potassium terbutylat:e were added, and the mixture
was stirred for 30 minutes . 5 ml of anhydrous Dowex*
50x8 resin, H+ form, were added. The solution was
filtered and evaporated under vacuum, the residue was
dissolved in 30 ml of water, and 10 ml of 1N NaOH were
added. The solution was stirred for 15 minutes at 25 °C
and eluted with water from a column containing 30 ml of
Dowex*50x8 resin, H+ form. The eluate was lyophilized,
and purified by silica gel chromatography, using as
solvent a mixture of methylene chloride/methyl
alcohol/water, 60:40:9. The fractions containing the
(3-2-O-ethylglycoside of N-acetyl-neuraminic acid amide
with ~ L-alanine-D-isoglutamine were gathered and
Trademark
WO 94/03469 2 ~ 416 7 9 PCT/US93/07307
27
evaporated under vacuum. The residue was dissolved in
50 ml of water and lyophilized. Yield: 600.
Rf = 0.12, chloroform/methyl alcohol/2.5N NH40H,
60:35:8;
0.10, chloroform/methyl alcohol/0.3o CaCl2,
60:40:9.
Example 19
I3-2-O-ethylcxlycoside of N-palmitovlneuraminic acid amide
with L-alanine-D-isoctlutam~ine
5.56 g (10 mM) of the ~i-2-O-ethylglycoside of
N-palmitoylneuraminic acid, sodium salt, were
solubilized in 50 ml of anhydrous N,N'-dimethylformamide
at 25 °C; 3.06 g (11 mM) of p-bromophenacyl bromide were
added and the solution was stirred overnight. 18 ml of
anhydrous pyridine and 10.2 g of acetic anhydride were
added and stirring was conducted for 24 hours at 35 °C.
The solution was evaporated. under vacuum and the residue
was dissolved with 100 ml of water and extracted three
times with 200 ml of methylene chloride. The organic
phases were washed twice with 50 ml of water and then
gathered, anhydrified with anhydrous sodium sulfate and
evaporated under vacuum. The obtained residue was
solubilized in 50 ml of anh~rdrous N, N' -dimethylformamide
at 25 °C;.2.64 g (20 mM) of sodium thiophenate were
added and the mixture was stirred for 4 hours. The
solution was evaporated under high vacuum. The residue
was extracted three times with 200 ml of ethyl acetate,
washed with 100 ml of cold 1N HC1 and twice with 50 ml
of water. The organic phases were anhydrified with
anhydrous sodium sulfate, gathered, and evaporated under
vacuum. The residue was dissolved in 30 ml of water and
eluted with water from a column containing 30 ml of
Dowex* 50x8 resin, pyridinium form. The eluate was
lyophilized and the residue was solubilized in 100 ml of
anhydrous pyridine. 1.15 g (10 mM) of
N-hydroxysuccinimide and 4.13 g (20 mM) of
Trademark
WO 94/U34G9 2 ~ ~ ~ 6 7 9 I'Cf/US93/U7307
28
N,N'-dicyclohexylcarbodiimide were added at -10 °C.
After 15 minutes, the temperature was raised to 25 °C
and the mixture was stirred for 5 hours. 5.14 g (15 mM)
of L-alanine-D-isoglutamine benzyl ester hydrochloride
(prepared according' to Le Francier and Kusumoto) were
added at 25 °C. The mixture was stirred overnight and
then evaporated under vacuum. The obtained residue was
dissolved in 60 ml of anhydrous methanol at 25 °C, 100
mg of potassium terbutylate were added, and the mixture
was stirred for 30 minutes. 5 ml of anhydrous Dower
50x8 resin, H+ form, wera added. The solution was
filtered and evaporated under vacuum. The residue was
dissolved in 30 ml of water, and 10 ml of 1N NaOH were
added. The solution was stirred for 15 minutes at 25 °C
and eluted with water from a column containing 30 ml of
Dowex 50x8 resin, H+ form. The eluate was lyophilized,
and purified by silica gel chromatography, using as
solvent a mixture of methylene chloride/methyl
alcohol/water, 60:40:9. 'The fractions containing the
(3-2-O-ethylglycoside~ of N-palmitoyl-neuraminic acid
amide with L-alanine-D-isoglutamine were gathered and
evaporated under vacuum. The residue was dissolved in
50 ml of a mixture of water/dioxane, 4:1, and
lyophilized. Yield: 60%.
Rf = 0.12, chloroform/methyl alcohol/2.5N NH40H,
110:40:6.
0.57, chloroform/methyl alcohol/0.3o CaCl2,
60:40:9.
Example 20
a-2-O-ethylg~coside of N~-acetvlneuraminic acid amide
with arainine
3.65 g (10 mM) of the a-2-O-ethylglycoside of
N-acetylneuraminic acid ethyl ester, prepared according
to van der Vlengel et al., Carbohydr. Res. 102, 121
(1982), were solubilized in 40 ml of water and 10 ml (10
mM) of NaOH were added. The solution was maintained at
*Trademark
'VO 94/03469 21416 l 9 PCT/US93/07307
29
25 °C for 30 minutes, neutralized with 1N HCl, and
eluted with water from a column containing 30 ml of
Dowex* 50x8 resin, pyrid_Lnium form. The eluate was
lyophilized and the residue was solubilized in 100 ml of
anhydrous pyridine. 1.15 g (10 mM) of
N-hydroxysuccinimide and 4.13 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added at -10 °C.
After 15 minutes, the temperature was raised to 25 °C
and the mixture was stirred for 5 hours. 4.64 g (15 mM)
of N-nitro-L-arginine ben.zyl ester (prepared according
to Bonnaud, Bull Chim. Farcn. 121, 1982) were added at 25
°C. The mixture was stirred overnight and then
evaporated under vacuum. The obtained residue was
dissolved in 200 ml of a mixture of
n-butanol/water/acetic acid, 4:1:1, and hydrogenated in
an H2 current in the presence of BaS04-supported
palladium. After filtration, the solution was
evaporated, and the residue was purified by silica gel
chromatography, using as aolvent a mixture of methylene
chloride/methyl alcohol/water, 60:40:9. The fractions
containing the a-2-O-ethylglycoside of
N-acetyl-neuraminic acid. amide with arginine were
gathered and evaporated Lender vacuum. The residue was
dissolved in 50 ml of water and lyophilized. Yield: 50%.
Rf = 0.13, chlorofo~rm/methyl alcohol/0.3% CaCl2,
60:40:9.
Example 21
-2-O-ethylqlycoside of N-acetvlneuraminic acid amide
with arainine
3.59 g (10 mM) of the ~i-2-O-ethylglycoside of
N-acetylneuraminic acid, sodium salt, were solubilized
in 50 ml of anhydrous N.N'-dimethylformamide at 25 °C;
3.06 g (11 mM) of p-bromophenacyl bromide were added and
the solution was stirred overnight. 18 ml of anhydrous
pyridine and 10.2 g of acetic anhydride were added and
stirring was conducted iEor 24 hours at 35 °C. The
Trademark
r~WO94/03469 ~~ 6~~ ~ PCT/US93/07307
solution was evaporated under vacuum and the residue was
dissolved with 100 ml of water and extracted three times
with 200 ml of methylene chloride. The organic phases
were washed twice with 50 ml of water and then gathered,
5 anhydrified with anhydr~ous~ .. sodium sulfate, and
evaporated under vacuum. ,fihe obtained residue was
solubilized in 50 ml of anh~idrous N, N' -dimethylformamide
at 25 °C; 2.64 g (20 mM) of sodium thiophenate were
added and the mixture wa;~ stirred for 4 hours. The
10 solution was evaporated under high vacuum. The residue
was extracted three times with 200 ml of ethyl acetate,
washed with 100 ml of cold 1N HC1 and twice with 50 ml
of water. The organic phases were anhydrified with
anhydrous sodium sulfate, gathered, and evaporated under
15 vacuum. The residue was solubilized in 100 ml of
anhydrous pyridine; 7..15 g (10 mM) of
N-hydroxysuccinimide, 4.13 g (20 mM) of
N,N'-dicyclohexylcarbodiimide and 11.6 g (10 mM) of
pyridinium chloride were added at -10 °C. After 15
20 minutes, the temperature was raised to 25 °C and the
mixture was stirred for 5 hours. 4.64 g (15 mM) of
N-nitro-L-arginine benzyl ester hydrochloride (prepared
according to Bonnaud, Bull. Chim. Farm. 121, 1982) were
added at 25 °C. The mixtux-e was stirred overnight and
25 then evaporated under vacuum. The obtained residue was
dissolved in 60 ml of anhydrous methanol at 25 °C. 100
mg of potassium terbutylate were added, and the mixture
was stirred for 30 minutes. 5 ml of anhydrous Dowex 50x8
resin, H+ form, were added. The solution was filtered
30 and evaporated under vacuum, and the residue was
dissolved in 100 ml of a mixture of
N-butanol/water/acetic acid, 4:1:1, and hydrogenated in
an H2 current in the presence of BaSO~-supported
palladium. After filtration, the solution was
evaporated, and the residuE~ was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/wat=er, 110:40:6. The fractions
VO 94/03469 1 ~I 1 ~ 7 PCf/US93/07307
:31
containing the /3-<'?-O-ethylglycoside of
N-palmitoyl-neuraminic acid amide with arginine were
gathered and evaporated under vacuum. The residue was
dissolved in 50 ml of water and lyophilized. Yield: 50%.
Rf - 0.10, chloroform/methyl alcohol/0.3% CaCl2,
60:40:9.
Example 22
!3-2-O-ethylglycoside of N-palmitovlneuraminic acid amide
with arcrinine
5.56 g (10 mM) of the (3-2-O-ethylglycoside of
N-palmitoyl-neuraminic acid, sodium salt, were
solubilized in 50 ml of anhydrous N,N'-dimethylformamide
at 25 °C; 3.06 g (11 mM) of p-bromophenacyl bromide were
added and the solution was stirred overnight. 18 ml of
anhydrous pyridine and 10.2 g of acetic anhydride were
added and stirring was conducted for 24 hours at 35 °C.
The solution was evaporated under vacuum and the residue
was dissolved with 100 ml of water and extracted three
times with 200 ml of meth~~lene chloride. The organic
phases were washed twice with 50 ml of vuater and then
gathered, anhydrified with anhydrous sodium sulfate, and
evaporated under vacuum. The obtained residue was
solubilized in 50 ml of anhydrous N,N'-dimethylformamide
at 25 °C; 2.64 g (20 mM) of sodium thiophenate were
added and the mixture was stirred for 4 hours. The
solution was evaporated under high vacuum. The residue
was extracted three times with 200 ml of ethyl acetate,
washed with 100 ml of cold 1N HC1 and twice with 50 ml
of water. The organic phases were anhydrified with
anhydrous sodium sulfate, gathered, and evaporated under
vacuum. The residue was dissolved in 30 ml of water and
eluted with water from a column containing 30 ml of a
Dowex*50x8 resin, pyridinium form. The eluate was
lyophilized and then solubil.ized in 100 ml of anhydrous
pyridine; 1.15 g (10 mM) of N-hydroxysuccinimide, 4.13
g (20 mM) of N,N'-dicyclohe:xylcarbodiimide and 11.6 g
Trademark
WO 94/03469 2 ~ 't ~1 b 7 9 P~T/US93/07307
:32
(10 mM) of pyridinium chloride were added at -10 °C.
After 15 minutes, the temperature was raised to 25 °C
and the mixture was stirred. for 5 hours. 4.64 g (15 mM)
of N-nitro-L-arginine benzyl ester hydrochloride
- 5 (prepared according to Bonnaud, Bull. Chim. Farm. 121,
1982) were added at 25 °C. The mixture was stirred
overnight and then evaporated under vacuum. The obtained
residue was dissolved in 60 ml of anhydrous methanol at
25 °C. 100 mg of potassium terbutylate were added, and
the mixture was stirred for 30 minutes. 5 ml of
anhydrous Dowex*50x8 resin, H+ form, were added. The
solution was filtered and evaporated under vacuum. The
residue was dissolved in, 100 ml of a mixture of
n-butanol/water/acetic acid, 4:1:1, and hydrogenated in
an H2 current in the presence of BaS04-supported
palladium. After filtration, the solution was
evaporated, and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/1.5M acetic acid, 110:40:6. The
fractions containing the Q-2=O-ethylglycoside of
N-palmitoyl-neuraminic ac~.d amide with arginine were
gathered and evaporated under vacuum. The residue was
crystallized from a mixture of 100 ml of methanol and
300 ml of ethyl ether. Yield: 60%.
Rf = 0.12, chloroformfmethyl alcohol/H20, 110:40:6.
Example 23
a-2-O-ethylglycoside of N-palmitoyl-neuraminic acid
amide with arainine
5.56 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoyl-neuraminic acid, sodium salt, were
solubilized in 40 ml of wal:er and eluted with water from
a column containing 30 ml of 50x8 Dowex resin,
pyridinium form. The eluate was lyophilized and then
solubilized in 100 ml of anhydrous pyridine. 1.15 g (10
mM) of N-hydroxysuccinimide, 4.13 g (20 mM) of
N,N'-dicyclohexylcarbodiirnide, and 11.6 g (10 mM) of
Trademark
WO 94/03469 21416 7 9 p~/US93/07307
33
pyridinium chloride were added at -10 °C. After 15
minutes, the temperature was raised to 25 °C and the
mixture was stirred for 5 hours. 4.64 g (15 mM) of
N-vitro-L-arginine benzyl ester hydrochloride (prepared
according to Bonnaud, Bull. Chim. Farm. 121, 1982) were
added at 25 °C. The mixture was stirred overnight and
then evaporated under vacuum. The residue was dissolved
in 100 ml of a mixture of n-butanol/water/acetic acid,
4:1:1, and hydrogenated in an H2 current in the presence
of BaS04-supported palladium. After filtration, the
solution was evaporated, and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/1.5M acetic acid,
110:40:6. The fractions containing the
a-2-O-ethylglycoside of N-palmitoyl-neuraminic acid
amide with arginine were gathered and evaporated under
vacuum. The residue was crystallized from a mixture of
40 ml of methanol and 150 ml. of ethyl ether. Yield: 700.
Rf = 0.14, chloroform/methyl alcohol/H20, 110:40:6;
0.32, chlcroform,~methyl alcohol/2.5N NH40H,
60:35:8.
Example 24
Dimethylamide of N-palmitovlneuraminic acid
5.34 g (10 mM) of t:he a-2-O-ethylglycoside of
N-palmitoyl-neuraminic acid were suspended in a mixture
of 100 ml of O.1M H2S04/e~~hanol, 4:1, at 60 °C, and
stirred for 16 hours. The product was extracted once
with 100 ml of ethyl acetate. The organic phases were
washed three times with 50 ml of water, gathered, and
evaporated under vacuum. The obtained residue was
dissolved in 200 ml of anhydrous methanol at 25 °C. 20
ml of anhydrous Dowex 50x8 resin, H+ form, were added.
The mixture was stirred for 2 hours. To the filtered
solution, 4.5 g (100 mM) of dimethylamine were added at
25 °C and the solution wa~> stirred for 24 hours. The
mixture was evaporated, and the residue was purified by
WO 94/03469 PCT/US93/0730°'
34
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/water, 80:20:2. The
fractions containing the dimethylamide of
N-acetyl-neuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 60 ml of a
mixture of water/dioxane, 4:.1, and lyophilized. Yield:
75%.
Rf = 0.63, chloroform/methyl alcohol/H20, 110:40:6.
Example 25
a-2-ethylctlycoside morpholino-propylamide of
N-palmitoylneuraminic acid
5.62 g (10 mM) of ~i-2-ethylglycoside
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methanol; 14.4 g (100 mM) of
N-(3-aminopropyl)-morpholine were added. The mixture was
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 80:10:1. The fractions
containing the compound were gathered and evaporated
under vacuum. The residue was dissolved in 100 ml of
water and lyophilized. Yield: 85%.
Rf = 0.30, chloroform/methyl alcohol/H20, 80:20:2;
0.62, chloroform/methyl alcohol/2.5N NH40H,
110:40:6.
Example 26
Q-2-ethylglycoside morpholino-propylamide of
N-.palmitovlneuraminic acid (malefic acid salt)
5.56 g (10 mM) of ~i-2-O-ethylglycoside
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of pyridine; 2.3 g (20 mM) of pyridinium
chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at 25 °C. 14.4 g (100 mM) of
N-(3-aminopropyl)-morpholine were added. The mixture was
' WO 94/03469 21 g~ 1 ~ 7 9 PCT/US93/07307
stirred overnight at 25 °C. The solution was evaporated
under vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/wate;r, 80:10:1. The fractions
5 containing the(3-2-ethylglycoside morpholino-propylamide
of N-palmitoylneuraminic acid were gathered and
evaporated under vacuum. Th~~ residue was dissolved in
100 ml of water. After adding a stoichiometric amount of
malefic acid, the mixture was lyophilized. Yield: 85%.
10 Rf = 0.30, chloroform/methyl alcohol/H20, 80:20:2;
0.62, chloroform/methyl alcohol/2.5N NH40H,
110:40:6.
Example 27
Dimethylaminopropylamide of h1-palmitoyl-neuraminic acid
15 5.34 g (10 mM) of tree a-2-O-ethylglycoside of
N-palmitoyl-neuraminic acid were suspended in a mixture
of 100 ml of O.1M H2S04/etlzanol, 4:1, at 60 °C, and
stirred for 16 hours. The product was extracted once
with 200 ml of ethyl acetate and then twice with 100 ml
20 of ethyl acetate; the organic phases were washed three
times with 50 ml of water, gathered, and evaporated
under vacuum. The obtained residue was dissolved in 200
ml of anhydrous methanol at 25 °C. 20 ml of anhydrous
Dowex 50x8 resin, H+ form, were added. The mixture was
25 stirred for 2 hours. To the' filtered solution 10.2 g
(100 mM) of dimethyl-aminopropylamine were added at 25
C and the solution was stirred for 24 hours. The mixture
was evaporated, and the residue was purified by silica
gel chromatography, using as solvent a mixture of
30 methylene chloride/methyl alcohol/2.5N NH40H, 60:35:8.
The fractions containing the compound were gathered and
evaporated under vacuum. The residue was dissolved in 60
ml of a mixture of water/dioaane, 1:2, and lyophilized.
Yield: 70%.
35 Rf = 0.21, chloroform/methyl alcohol/0.3% CaCl2,
60:40:9.
WO 94/03469 ~ ~ ~'~ ~~ 9 PCT/US93/0730',
36
Examt~le 28
Dimethvlaminopropvlamide of N-palmitoyl-neuraminic acid
(malefic acid salt)
5.34 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoyl-neuraminic acid were suspended in a mixture
of 100 ml of O.1M H2S04/ethanol, 4:1, at 60 °C, and
stirred for 16 hours. The product was extracted once
with 200 ml of ethyl acetate and then twice with 100 ml
of ethyl acetate; the organic phases were washed three
times with 50 ml of water, gathered, and evaporated
under vacuum. The obtained residue was dissolved in 200
ml of anhydrous methanol at 25 °C. 20 ml of anhydrous
Dowex 50x8 resin, H+ form, were added. The mixture was
stirred for 2 hours. To the filtered solution 10.2 g
(100 mM) of dimethyl-aminopropylamine were added at 25
°C, and the solution was stirred for 24 hours. The
mixture was evaporated, and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/2.5N NH40H, 60:35:8.
The fractions containing the compound were gathered and
evaporated under vacuum,. The residue was dissolved in 60
ml of a mixture of water/dioxane, 1:2. A stoichiometric
amount of malefic acid was added, and the mixture was
lyophilized. Yield: 70%.
Rf = 0.21, chloroform/methyl alcohol/0.3% CaCl2,
60:40:9.
Example 29
a-2-O-ethylctlvcoside butylamide of N-palmitoylneuraminic
acid
5.62 g (10 mM) of ,Q-2-O-ethylglycoside
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methanol; 3.66 g (50 mM) of
2-butylamine were added. The mixture was stirred for 5
hours at 40 °C. The solution was evaporated under vacuum
and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
WO 94/03469 PCT/US93/07307
2141679
37
chloride/methyl alcohol/wat:er, 80:10:1. The fractions
containing the a-2-O-ethylglycoside butylamide of
N-palmitoylneuraminic acid ~Nere gathered and evaporated
under vacuum. The residue was dissolved in 60 ml of
dioxane and lyophilized. Yield: 70%.
Rf = 0.71, chloroform/methyl alcohol, 80:20;
0.60, chloroform~'methyl alcohol/2.5N NH40H,
80:20:2.
Examn:Le 3 0
a-2-O-ethvlqlycoside dimethylaminoethylamide of
N-palmitovlneuraminic acid
5.62 g (10 mM) of ~i-2-O-ethylglycoside
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methanol; 4.4 g (50 mM) of
2-dimethylaminoethylamine were added. The mixture was
stirred overnight at 40 °C. The solution was evaporated
under vacuum and the residuf: was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/2..5N NH40H, 80:20:2. The
fractions containing the (3-2-O-ethylglycoside
dimethylaminoethylamide of N-palmitoylneuraminic acid
were gathered and evaporated under vacuum. The residue
was dissolved in 60 ml of water and lyophilized. Yield:
80%.
Rf = 0.11, chloroform/methyl alcohol, 70:30;
0.43, chloroform%methyl alcohol/2.5N NH40H,
110:40:6.
Examp7_e 31
a-2-O-et~lalycoside dinnethvlaminoethylamide of
N-palmitoylneuraminic acid (malefic acid salt)
5.56 g (10 mM) of ~i-2-O-ethylglycoside
N-palmitoylneuraminic acid were solubilized in 50 ml of
pyridine; 2.3 g (20 mM) of pyridinium chloride and 4.12
g (20 mM) of N,N'-dicyclo-he:xylcarbodiimide were added.
The mixture was stirred for 2 hours at 25 °C. 8.8 g (100
WO 94/03469 PCT/US93/0730
38
mM) of dimethylaminoethylamine were added and the
reaction was conducted overnight at 25 °C. The solution
was evaporated under vacuum and the residue was purified
by silica gel chromatography, using as solvent a mixture
of methylene chloride/methyl alcohol/2.5N NH40H,
80:20:2. The fractions containing the
~3-2-O-ethylglycoside dimethylaminoethylamide of
N-palmitoylneuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 60 ml of
water. A stoichiometric amount of malefic acid was added,
and the mixture was lyophilized. Yield: 80%.
Rf = 0.11, chloroform/methyl alcohol, 70:30;
0.43, chloroform/methyl alcohol/2.5N NH40H,
110:40:6.
Example 32
a-2-O-ethylctlycoside dimethylaminoprogylamide of
N-dichloroacetylneuraminic acid
4.34 g (10 mM) of ,Q-2-O-ethylglycoside
N-dichloro-acetylneuraminic acid ethyl ester were
solubilized in 50 ml of anhydrous methanol; 10.2 g (100
mM) of dimethylamino-propylamine were added. The mixture
was stirred for 5 hours at 40 °C. The solution was
evaporated under vacuum and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/water, 40:60:15. The
fractions containing the ~i-2-O-ethylglycoside
dimethyl-aminopropylamide of N-dichloroacetylneuraminic
acid were gathered and evaporated under vacuum. The
residue was dissolved in 60 ml of water and lyophilized.
Yield: 65%.
Rf = 0.37, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
~VO 94/03469 2 ~ 416 l 9 PCT/US93/07307
39
Example 33
~-2-O-ethylglycoside dimethylaminopropylamide of
N-dichloroacetylneuraminic acid (malefic acid salt)
4.34 g (10 mM) of the ~i-2-O-ethylglycoside of_
N-dichloro-acetylneuraminic acid ethyl ester were
solubilized in 50 ml of anhydrous methanol; 10.2 g (100
mM) of dimethylamino-propylamine were added. The mixture
was stirred for 5 hours at 40 °C. The solution was
evaporated under vacuum and the residue was purified by
silica gel chromatography, using as solvent a mixture of
methylene chloride/methyl alcohol/water, 40:60:15. The
fractions containing the ~i-2-O-ethylglycoside
dimethyl-aminopropylamide of N-dichloroacetylneuraminic
acid were gathered and evaporated under vacuum. The
residue was dissolved in 60 ml of water. A
stoichiometric amount of m<~leic acid was added, and the
mixture was lyophilized. '.Field: 65%.
Rf = 0.37, chloroform/methyl alcohol/2.5N NH40H,
40:60:15.
Examx~le 34
~(i-2-O-ethylg~lycoside di~methylaminopro,~ylamide of
neuraminic acid
3.23 g (10 mM) of the ~i-2-O-ethylglycoside of
neuraminic acid ethyl ester, prepared according to
Schauer and Buscher, Biochim. Biophys. Acta 338, 369
(1974), were solubilized in 50 ml of anhydrous methanol;
10.2 g (100 mM) of dimethylamino-propylamine were added.
The mixture was stirred overnight at 40 °C. The solution
was evaporated under vacuum and the residue was purified
by reverse phase chromatography, using as support
Lichroprep* RP 18 (Merck, Darmstadt, Germany) and as
eluant a mixture of methyl alcohol/water, 1:1. The
fractions containing the ~i-2-O-ethylglycoside
dimethyl-aminopropylamide of neuraminic acid were
gathered and evaporated under vacuum. The residue was
*Trademark
WO 94/03469 . PCT/US93/07307
~1~.~~'~ 9 40
dissolved in 50 ml of water and lyophilized. Yield:
60%.
Rf = 0.1, chloroform/methyl alcohol/0.3% CaCl2,
55:45:10.
Example 35
Q-2-O-ethylctlvcoside dimethylamide of
N-dichloroacetylneuraminic acid
4.34 g (10 mM) of the /3-2-O-ethylglycoside of
N-dichloro-acetylneuraminic acid ethyl ester were
solubilized in 50 ml of anhydrous methanol; 4.5 g (100
mM) of dimethylamine were added. The mixture was stirred
overnight at 40 °C. The so:Lution was evaporated under
vacuum and the residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol/water, 80:10:1. The fractions
containing the ~i-2-O-ethylc~lycoside dimethyl-amide of
N-dichloroacetylneuraminic acid were gathered and
evaporated under vacuum. The residue was crystallized
from 60 ml of methanol a.nd 300 ml of ethyl ether.
Yield: 60%. .
Rf = 0.44, chloroform/methyl alcohol/2.5N NH40H,
110:40:6.
Example 36
a-2-O-ethylcLlycoside ethanolamide of N-palmitoyl-
neuraminic acid
5.48 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoylneuraminic acid ethyl ester were solubilized
in 50 ml of anhydrous methanol; 6.11 g (100 mM) of
ethanolamine were added. The mixture was stirred
overnight at 35 °C. The solution was evaporated under
vacuum and the residue was purified by silica gel.
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol, 90:10. The fractions containing
the a-2-O-ethylglycoside et.hanolamide of N-palmitoyl-
neuraminic acid were gathered and evaporated under
WO 94/03469 2 ~ 't ~16 7 9 ~'CT/US93/07307
~41
vacuum. The residue was dissolved in 60 ml of
dioxane/water, 2:1, and lyophilized. Yield: 85%.
Rf = 0.66, chloroform/methyl alcohol/H20, 110:40:6.
Example 37
Q-2-O-ethylc~lycoside dimethylamide of neuraminic acid
3.23 g (10 mM) of the (3-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized in 50 ml of
water and 11 ml (10 mM) of 1M NaOH were added. The
solution was maintained at :Z5 °C for 30 minutes. 2 m1 of
anhydrous Dowex*50x8 resin, H+ form, were added. The
filtered solution was evaporated under vacuum. The
residue was solubilized in 50 ml of anhydrous pyridine.
2.3 g (20 mM) of pyridinium chloride and 4.12 g (20 mM)
of N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours ai. 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/2.5N NH40H, 110:40:6. The fractions containing
the ~i-2-O-ethylglycoside dimethylamide of. neuraminic
acid were gathered and evaporated under vacuum. The
residue was dissolved in 50 ml of water and lyophilized.
Yield: 55%.
Rf = 0.30, chloroform/rnethyl alcohol/H20, 55:45:10.
Examp:l a 3 8
Q-2-O-ethvlQlycoside dimethvlamide of N-caorylneuraminic
acid
3.23 g (10 mM) of the Q-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
100 ml anhydrous methanol. and 50 ml of anhydrous
methylene chloride. 3.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 3.44 g (20 mM) of capric acid were
added, and the mixture was stirred overnight at 5 °C.
Trademark
WO 94/U34G9 1'Cf/US93/U73U7
2141 bl9
42
After filtration, it was evaporated. The residue was
solubilized in 100 ml of ethanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex* 50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by siluca gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
(3-2-O-ethylglycoside dimeth;ylamide of N-caprylneuraminic
acid were gathered and evaporated under vacuum. The
residue was dissolved in 50 ml of acetone and
precipitated in 20 volumes of n-hexane. Yield: 65%.
Rf = 0.48, chloroform/methyl alcohol/HZO, 80:10:1.
Examx~le 39
Q-2-O-ethvlalvcoside dimethylamide of N-ca~rvlovl-
neuraminic acid
3.23 g (10 mM) of t:he ,Q-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
100 ml anhydrous methanol. and 50 ml of anhydrous
methylene chloride. 8.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimi~de, 2.0 g (20 mM) of
triethylamine, and 2.88 g (20 mM) of caprylic acid were
added and the mixture was stirred overnight at 5 °C.
After filtration, it was evaporated. The residue was
solubilized in 100 ml of ethanol/water, 1:1, and 11 ml
(10 mMj of 1M NaOH were added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex* 50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
~v Trademark
WO 94/03469 PCT/US93/07307
21~~b79
43
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiim~.de were added. The mixture
was stirred for 2 hours at 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
~i-2-O-ethylglycoside dimethylamide of
N-capryloylneuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 50 ml of
acetone and precipitated in 20 volumes of n-hexane.
Yield: 65%.
Rf = 0.61, chloroform/methyl alcohol/H20, 80:20:2.
Examt~le 40
Q-2-O-ethylglycoside dimethylamide of N-olevlneuraminic
acid
3.23 g (10 mM). of t:he ~i-2-O-ethylglycoside of
neuraminic acid ethylester were solubilized at 5 °C in
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. 8.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 5.65 g (20 mM) of oleic acid were
added, and the mixture was stirred overnight at 5 °C.
After filtration, it was evaporated. The residue was
solubilized in 100 ml of et:hanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex*50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 ~) of
N,N'-dicyclohexylcarbodiimi~de were added. The mixture
was stirred for 2 hours at: 25 °C. 4.5 g (100 mM) of
diethylamine were added and. the reaction was conducted
Trademark
~~ WO 94/03469 2 r 416 7 9 E'Cr/US93/07307
44
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture o:E methylene chloride/methyl
alcohol, 9:1. The fractions containing the
R-2-O-ethylglycoside dimetlzylamide of N-oleylneuraminic
acid were gathered and evaporated under vacuum. The
residue was dissolved in 50 ml of tert-butanol and
lyophilized. Yield: 50%.
Rf = 0.42, chloroform/methyl alcohol/H20, 80;20:2.
Examx~ 1 a 41
a-2-O-ethylglycoside dimethylamide of N-valprovl
neuraminic acid
3.23 g (10 mM) of the ~i-2-O-ethylglycoside of
neuraminic acid ethylester were solubilized at 5 °C in
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. E..25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 2.88 g (20 mM) of valproic acid were
added and the mixture was stirred overnight at 5 °C.
After filtration, it was .evaporated. The residue was
solubilized in 100 ml of ethanol/water, l:l, and 11 ml
(10 mM) of 1M NaOH weres added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex* 50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at. 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
(3-2-O-ethylglycoside dimethylamide of
N-valproylneuraminic acid were gathered and evaporated
*Trademark
y0 94/03469 '~ 6 ~ PCT/US93/07307
under vacuum. The residue was dissolved in 50 ml of
acetone and precipitated in 20 volumes of n-hexane.
Yield: 500.
Rf = 0.60, chloroform/methyl alcohol/H20, 80:20:2.
5 Example 42
(3-2-O-ethylglycoside dimethylamide of N-phenylacetvl
neuraminic acid
3.23 g (10 mM) of t:he ~i-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
10 100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. 8.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 2.72 g 1;20 mM) of phenylacetic acid
were added, and the mixturE: was stirred overnight at 5
15 °C. After filtration, it way; evaporated. The residue was
solubilized in 100 ml of et:hanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were: added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex* 50x8 resin, H~ form, were added. The filtered
20 solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at: 25 °C. 4.5 g (100 mM) of
25 diethylamine were added, and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
30 (3-2-O-ethylglycoside dimethylamide of
N-phenylacetylneuraminic acid were gathered and
evaporated under vacuum. The: residue was dissolved in 50
ml of acetone and precipitated in 20 volumes of
n-hexane. Yield: 65%.
35 ,Rf = 0.43, chloroform/methyl alcohol/H20, 80:20:2.
Trademark
m WO 94/03469 2 1 4 1~6 7 9
PCf/US93/07307
46
Examx~le 43
L3-2-O-ethvlglvcoside dimetllylamide of N-miristovl
neuraminic acid
3.23 g (10 mM) of the ~i-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. x;.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimi.de, 2.0 g (20 mM) of
triethylamine, and 4.57 g (20 mM) of miristic acid were
added, and the mixture was stirred overnight at 5 °C.
After filtration, it was evaporated. The residue was
solubilized in 100 ml of ethanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were' added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex* 50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at: 25 °C. 4.5 g (100 mM) of
diethylamine were added and. the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
,Q-2-O-ethylglycoside dimethylamide of
N-miristoylneuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 50 ml of
acetone and precipitated in 20 volumes of n-hexane.
Yield: 60%.
Rf = 0.56, chloroform/methyl alcohol/H20, 80:20:2.
Example 44
13-2-O-ethvlcrlycoside dimethvlamide of N-laurovl-
neuraminic acid
3.23 g (10 mM) of tlae Q-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
Trademark
,CVO 94/03469 214 ~ 6 7 9 P~/US93/07307
47
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. 8,.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimi.de, 2.0 g (20 mM) of
triethylamine, and 4.57 g (20 mM) of lauric acid were
added, and the mixture was stirred overnight at 5 °C.
After filtration, it was evaporated. The residue was
solubilized in 100 ml of et=hanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
Dowex* 50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at. 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
Q-2-O-ethylglycoside dimethylamide of
N-lauroylneuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 50 ml of
acetone and precipitated i.n 20 volumes of n-hexane.
Yield: 60%.
Rf = 0.54, chloroform/methyl alcohol/H20, 80:20:2.
Examp.l a 4 5
Q-2-O-ethylcr~coside dimeth~,rlamide of N-nicotinovl-
neuraminic acid
3.23 g (10 mM) of the a-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. 8"25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 2.46 g (20 mM) of nicotinic acid were
added, and the mixture was stirred overnight at 5 °C.
Trademark
CVO 94/03469 2 ~~ 416 7 9 ~ PCT/US93/07307
48
After filtration, it was evaporated. The residue was
solubilized in 100 ml of ethanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were added. The solution was
maintained at 25 °C for 3C) minutes. 2 ml of anhydrous
Dower 50x8 resin, H+ form; were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
~i-2-O-ethylglycoside dimethylamide of
N-nicotinoylneuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 50 ml of
methanol and precipitated in 20 volumes of
tert-butyl-ether. Yield: 50%.
Rf = 0.27, chloroform/methyl alcohol/H20, 80:20:2.
Examp:L a 4 6
~i-2-O-ethvlcLlycoside dimethvlamide of N-rrimethoxy
benzovlneuraminic acid
3.23 g (10 mM) of the Q-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized at 5 °C in
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. 8.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 4.24 g (20 mM) of trimethoxybenzoic
acid were added, and the mixture was stirred overnight
at 5 °C. After filtration, it was evaporated. The
residue was solubilized in 100 ml of ethanol/water, 1:1,
and 11 ml (10 mM) of 1M NaOH were added. The solution
was maintained at 25 °C for 30 minutes. 2 ml of
anhydrous Dower 50x8 resin,, H+ form, were added. The
Trademark
WO 94/03469 ~ ~ ~ ~ ~ ~,~ PCT/US93/07307
49
filtered solution was evaporated under vacuum. The
residue was solubilized in 50 ml of anhydrous pyridine.
2.3 g (20 mM) of pyridinium chloride and 4.12 g (20 mM)
of N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at 25 °C. 4.5 g (100 mM) of
diethylamine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
~i-2-O-ethylglycoside dimethylamide of
N-trimethoxbenzoylneuraminic acid were gathered and
evaporated under vacuum. The residue was dissolved in 50
ml of methanol and precipitated in 20 volumes of
tert-butyl-ether. Yield: 60%.
Rf = 0.52, chloroform/methyl alcohol/H20, 80:20:2.
Examp7Le 47
a-2-O-ethylql_ycoside pvrrol:idylamide of N-palmitoyl-
neuraminic acid
5.56 g (10 mM) of the a-2-O-ethylglycoside of
N-palmitoylneuraminic acid, sodium salt, were
solubilized in 50 ml of anhydrous pyridine. 2.3 g (20
mM) of pyridinium chloride and 4.12 g (20 mM) of
N,N'-dicyclohexylcarbodiimide were added. The mixture
was stirred for 2 hours at 25 °C. 7.17 g (100 mM) of
pyrrolidine were added and the reaction was conducted
overnight at 25 °C. The solution was evaporated, and the
residue was purified by silica gel chromatography, using
as solvent a mixture of methylene chloride/methyl
alcohol, 9:1. The fractions containing the
(3-2-O-ethylglycoside pyrrolidylamide of
N-palmitoylneuraminic acid were gathered and evaporated
under vacuum. The residue was dissolved in 50 ml of
acetone and precipitated i.n 20 volumes of n-hexane.
Yield: 90%.
Rf = 0.67, chloroform/methyl alcohol/H20, 80:20:2.
-- wo 94/o3a69 21 41 b 7 9
PCT/US93/07307
Examx> 1 a 4 8
Q-2-O-ethylglycoside of N-palmitoylneuraminic acid ethyl
ester
3.23 g (10 mM) of the ~i-2-O-ethylglycoside of
5 neuraminic acid ethyl ester were solubilized at 5 °C in
100 ml of anhydrous methanol and 50 ml of anhydrous
methylene chloride. F3.25 g (40 mM) of
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 5.12 g (20 mM) of palmitic acid were
10 added, and the mixture wasc stirred overnight at 5 °C.
After filtration, it was evaporated. The residue was
solubilized in 100 ml of ethanol/water, 1:1, and 11 ml
(10 mM) of 1M NaOH were added. The solution was
maintained at 25 °C for 30 minutes. 2 ml of anhydrous
15 Dower 50x8 resin, H+ form, were added. The filtered
solution was evaporated under vacuum. The residue was
solubilized in 150 ml of water and the product was
extracted once with 300 ml chloroform and then twice
with 150 ml of chloroform; the organic phases were
20 washed three times with 150 ml of water, gathered, and
evaporated. The residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
chloride/methyl alcohol, 9:1. The fractions containing
the (3-2-O-ethylglycoside of N-palmitoylneuraminic acid
25 were gathered and evaporated under vacuum. The residue
was crystallized from 100 ml of tert-butyl-ether. Yield:
85%.
Rf = 0.44, methylene ~~hloride/methanol, 90:10.
3 0 Examj~ 1 a 4 9
(3-2-O-ethvlcrlycoside of 1~-palmito~rlneuraminic acid
5.65 g (10 mM) of the ~i-2-O-ethylglycoside of
neuraminic acid ethyl ester were solubilized in 100 ml
of ethanol/water, 1:1. 11 ml (10 mM) of 1M NaOH were
35 added. The solution was maintained at 25 °C for 30
minutes. 2 ml of anhydrou;~ Dower 50x8 resin, H+ form,
Trademark
"" WO 94/03469 21416 7 9 PCT/US93/07307
51
were added. The filtered solution was evaporated under
vacuum. Yield: 95%.
Rf = 0.14, methylene chloride/methyl alcohol/H20,
110:10:6.
Example 50
a-2-O-ethylcrlvcoside of N-t~almitovl-neuraminic acid
methyl ester
3.37 g (10 mM) of the a-2-O-ethylglycoside of
neuraminic acid methyl ester were solubilized in 30 ml
of 1M NaOH at 80 °C, and maintained under stirring
overnight. The solution was passed through a column
containing 200 ml of Bio-Rex*70 H+ weakly basic resin,
and then dessicated under vacuum. The residue was
redissolved with 50 ml of anhydrous methanol. 4.13 g (20
mM) of N,N'-dicyclohexylcarbodiimide and 2.3 g of
pyridinium chloride were added, and the mixture was
stirred for 1 hour. After filtration, it was evaporated.
The residue was solubilized at 5 °C in 100 ml of
anhydrous methanol and 50 ml of anhydrous methylene
c h 1 o r i d a . 8 . 2 5 g ( 4 0 m M ) o f
N,N'-dicyclohexylcarbodiimide, 2.0 g (20 mM) of
triethylamine, and 5.12 g (20 mM) of palmitic acid were
added, and the mixture wa;~ stirred overnight at 5 °C.
After filtration, it was evaporated. The residue was
solubilized in 150 ml of water and the product was
extracted once with 300 ml chloroform and then twice
with 150 ml of chlorofor~cn; the organic phases were
washed three times with 150 ml of water, gathered, and
evaporated. The residue was purified by silica gel
chromatography, using as solvent a mixture of methylene
- chloride/methyl alcohol, 9:1. The fractions containing
the a-2-O-ethylglycoside of N-palmitoylneuraminic methyl
ester were gathered and evaporated under vacuum. The
residue was crystallized from 100 ml of
tert-butyl-ether. Yield: '70%.
~Rf = 0.65, methylene chloride/methanol, 90:10.
Trademark
"''WO 94/03469 2 1 .416 l 9 PCT/US93/07307
52
Example 51
a-2-O-ethylqlycoside of N-palmitoylneuraminic acid
sodium salt
5.47 g (10 mM) of the a-2-O-ethylglycoside of
-neuraminic acid methyl ester were solubilized in 100 ml
of methanol/water, 1:1. 11 ml (10 mM) of 1M NaOH were
added. The solution was maintained at 25 °C for 30
minutes. 2 ml of anhydrous Dower 50x8 resin, H+ form,
were added. The filtered solution was evaporated under
vacuum. Yield: 95%.
Rf - 0.25, chloroform/methyl alcohol/H20,
110:10:6.
Example 52
13-2-O-ethylcLlycoside of N-dichloroacetvlneuraminic acid
ethyl ester
3.23 g (10 mM) of the (3-2-O-ethylglycoside of neuraminic
acid ethyl ester were solubilized in 50 ml of anhydrous
pyridine at 25 °C; 14.3 g (100 mM) of methyl
dichloroacetate were~added. The mixture was stirred for
24 hours and then evaporated under vacuum. The residue
was purified by silica gel chromatography, using as
solvent a mixture of: methylene chloride/methyl
alcohol/H20, 80:10:1. The fractions containing the
/3-2-O-ethylglycoside of N-dichloroacetylneuraminic ethyl
ester were gathered and evaporated under vacuum. The
residue was crystallized from a mixture of 50 ml of
methanol and 200 ml of ethyl ether. Yield: 85%.
Rf =0.49, chloroform/meth.anol, 80:20.
BIOLOGICAL STUDIES
The antineuronotoxic activities of the new amides
of the neuraminic acids of the present invention are
demonstrated by the following experimental studies
conducted with the ~i-2-O-ethylglycoside of the
dimethylamide of N-palmitoylneuraminic acid of the
formula:
Trademark
WO 94/03469 ~ ~ ~~ ~ ~ ~'"~ ~ PCT/US93/07307
53
HO
0 HJ ~ y0/'~ 0
/N
H / \G"~~
identified as ND37.
Examble 53
Antineuronotoxic effect of 1VD37 in vitro on cerebellar
crranule cells : protective effect on exocrenous
glutamate induced neurotoxic;itv
MATERIALS AND METHODS
Cell cultures
Primary cell cultures of cerebellar granule cells
were prepared from 6 day old Sprague-Dawley rats.
Neurons were grown in 35 mm plates for 11-13 days and
maintained in a humid environment (95% air and 5o CO2).
Cultures (3x106 cells/plate) are mainly formed by
granule cells (95%) with a small amount (5%) of glial
cells (Gallo V. et al.: Selective release of glutamate
from cerebellar granule cells differentiating in
culture. Proc. Natl. Acad. Sci. USA 79, 7919-23, 1982).
Glial proliferation was prevented by arabinofuranoside
cytosine.
Derivative ND37 was solubilized at a concentration
of 1x10-2 M in dimethylsul:Eoxide (1% DMSO) and then
diluted at various concentrations in Locke's solution
(154 mM NaCl/5.6 mM KC1/ 3.6 mM NaHC03/2.3 mM CaCl2/1 mM
MgCl2/5.6 mM glucose/5 mM HEl?ES, pH 7.4). The following
concentrations were tested: 5x10-6 M, 1x10-5 M, and 2x10-
5
M.
Description of the exogenous ctlutamate neurotoxicitv
model
The cell culture medium was aspirated from the
plates (and correctly maintained) . Plates were washed (3
x 2 ml) with Locke's solution, and solutions (1.5 ml)
WO 214 i 6 l ~ PCT/US93/073Q7
54
containing the compound to be tested (concentrations
from 5x10-6 M to 2x10-5 M) were added and incubated for
2 hours in an incubator at :37 °C (5% C02).
Treated cells were washed (3 x 2 ml) with Locke's
solution + 10% fetal calf serum, then washed (3 x 2 ml)
with Locke' s solution without Mg++ . 100 ~,M glutamate ( 1 . 5
ml) in Locke's solution (-Mg++) or culture medium
(controls) were added. Incubation with glutamate was
performed for 15 minutes ai. room temperature (27 °C) .
Glutamate was removed, th~~ plates were washed with
Locke's solution (2 x 2 ml), and then incubated in
presence of the starting medium (correctly maintained)
for 24 hours at 37 °C in an incubator (5% C02). At the
end of the incubation, cellular viability was assayed
via quantification by the MTT colorimetric test (Mosmann
T.: Rapid colorimetric assay for cellular growth and
survival: application to proliferation and cytotoxicity
assays. J. Immunol. Meth. 65, 55: 63, 1983 and modified
according to Skaper S.D. et al.: Death of cultured
hippocampal pyramidal neurons induced by pathological
activation of N-methyl-D-aspartate receptors is reduced
by monosialogangliosides. J. Pharm. Exp. Ter. 259,1
452-457, 1991) . Data are e:Kpressed as % survival. The
significance was calculated according to the Dunnett
test.
Results
The obtained results (Table 1) show that:
- ND37 has a marked antineuronotoxic activity:
the presence of the free compound in the incubation
medium during the exposure t:o the glutamate toxin is not
necessary. The neuroprotect:ive effect of ND37 is very
high: at a concentration of 1x10-5 M there is a
protection of about 63%, and the highest protection
(about 83%) is reached at ~~ concentration of 2x10-5 M.
WO 94/03469 21416 "~ 9 PC'f/US93/07307
Table 1. Antineuronotoxic effect of ND37 in cerebellar granule
cells: protective effect glutamateinduced
on exogenous
neurotoxicity
Groups MTT % survival
values
1) control 0.195 f 0.016100
2) glutamate 0.075 0.009 38 ~ 5
3) glutamate + ND37 (5x100.110 t 0.01537 ~ 5
6 M)
(1x10 5 M) 0.123 0.006 * 63 3
(2x10-5 M) 0.326 0.016 * 83 6
Significance (Dunnett's
test)
* p < 0.01 (vs group
2)
5 Example 54
Antineuronotoxic effect of ND37 in vitro in cerebellar
ctranule cells : protective e:Efect on exocrenous glutamate-
induced neurotoxicity durincr cotreatment of cells with
the active compound
10 MATERIALS AND METHODS
Cell cultures
Primary cerebellar granule cells were prepared from
8 day old rats (Zivic Mi:Ller, Pittsburgh, PA, USA).
Neurons were cultivated in :35 mm plates for 7-8 days and
15 maintained in a humid environment (95% air and 5% C02).
Cultures (3x106 cells/plate) are mainly formed by
granule cells (95%) with a small amount (5%) of glial
cells (Gallo V. et al.: Se:Lective release of glutamate
from cerebellar granule cells differentiating in
20 culture. Proc. Natl. Acad. Sci. USA 79, 7919-23, 1982).
Glial proliferation was prEwented by arabinofuranoside
cytosine.
Derivative ND37 was solubilized at a concentration
of 1x10-2 M in dimethylsu7.foxide (1% DMSO), and then
WO 94/03469 PCT/US93/07307
2141619
56
diluted at various concentrations in Locke's solution
(154 mM NaCl/5.6 mM KC1/ 3..6 mM NaHC03/2.3 mM CaCl2/1 mM
MgCl2/5.6 mM glucose/5 mM HEPES, pH 7.4). The following
concentrations were tested: 5x10-6 M, 1x10-5 M, and 2x10
- 5 5 M.
Description of the exocte:nous glutamate neurotoxicitv
model
The cell culture medium was aspirated from the
plates (and correctly maintained) . Plates were washed (3
x 2 ml) with Locke's solution without Mg++, and solutions
(1.5 ml) containing the compound to be tested
(concentrations from 5x10-6 M to 2x10-5 M) were added and
incubated for 2 hours in an incubator at 37 °C (5% C02).
Treated cells were washed (3 x 2 ml) with Locke's
solution + 10% fetal calf serum, then washed (3 x 2 ml)
with Locke's solution without Mg++. 1.5 ml of Locke's
solution (-Mg++) or 50 ~.M of glutamate ~ the test
compound (concentrations ',between 1 and 4x10-5 M) in 1.5
ml of Locke's solution (-Mg++) were added. The incubation
was conducted for l5~minwtes (37 °C). Glutamate and the
compound were removed. The plates were washed with
Locke's solution (2 x 2 ml), and then incubated in the
presence of the starting medium (correctly maintained)
for 24 hours at 37 °C in an incubator (5% C02). At the
end of the incubation, cellular viability was assayed
via quantification by the fluorescein diacetate (FDA)
and propidium iodide (PI) colorimetric test (Manev H. et
al.: Glutamate induced neuronal death in primary
cultures of cerebellar granule cells: protection by
synthetic derivatives of endogenous sphingolipids; J.
Pharm. Exp. Ther. 252,1 9:19-427, 1990). Monolayers were
washed with Locke's solution and stained for 3 minutes
at 22 °C -with a solution containing 36 ~M FDA and 7 ~M
of PI. The stained cells where immediately analyzed
using a standard fluorescence microscope for
epiillumination (Vanox Olympus, 450 nm excitation, 520
nm emission). FDA, a non polar ester, crosses cell
Trademark
WO 94/03469 2 ~ 6 '~ ~ PCT/US93/07307
57
membranes and is hydrolyzed. by intracellular esterases
with the consequent production of a yellow greenish
color. Neuronal damage influences the FDA-induced color
and allows the permeation of PI, which is a polar
compound capable of interacting with nuclear DNA,
producing a brilliant red fluorescence.
After glutamate treatment, some neurons can
degenerate and detach from the plates. The loss of cells
was estimated by comparing the number of intact or
degenerated neurons in a well defined field, which was
photographed before and after 24 hours after application
of glutamate. The percent of surviving neurons in 4
representative fields (magnification 40x) of each
monolayer was determined by a researcher unaware of the
experimental conditions, evaluating the FDA/PI color,
and calculating it as follows:
FDA positive cel:Ls
___________________________.______________ x 100
FDA positive + PI positive ~~ loosened cells
Data are expressed as % of surviving cells.
Significance was calculated using the Dunnett's test.
Results
The data in Table 2 show that the neuroprotective
effect of ND37 on glutamate 'toxicity is immediate, i.e. ,
ND37 protects neurons at a dose of 10-40 ~.M, even if
administered contemporaneously with the application of
the toxin. This shows the rapid mode of action of ND37.
WO 94/03469 ~ PCT/U593/0730'
1 '~ 9 58
Table Antineuronotoxic effect of ND37 during
2: cotreatment of
exogenous glutamate on cerebellar granule
cells (pro-
tective effect)
Groups ~ surviving cells N = 4
(average + s.e.) FDA/PI test
1) control 89 t 7.0
2) glutamate 22 _+ 1.5
3) glutamate + ND37 (1x10 S M) 34 _+ 2.0
(2x10 5 M) 70 + 6.0
(4x10-5 M) 88 ~ 4.5
Significance (Dunnett's test)
* p < 0.01 (vs group 2)
Example 55
In vivo effect of ND37 on cerebral damage induced by
intracerebroventricular (icv) infection of
N-methyl-D-aspartate in neonatal rats
MATERIALS AND METHODS
Description of the model
Experiments were performed on 7 day old neonatal
rats weighing ca. 13 grams. At the 7th day, animals,
after ether anesthesia, were lesioned by icv injection
of 25 nmoles/~.1 of N-methyl-D-aspartate (NMDA), Sigma,
St. Louis, MO, USA, according to the method described by
McDonald et al. (McDonald J.W. et al.: Neurotoxicity of
N-methyl-D-aspartate is markedly enhanced in developing
rat central nervous system. Brain Res. 459, 200-203,
1988). The excitotoxin was solubilized in saline, and
the pH was brought to 7 . 4 by adding 1N NaOH . The NMDA
injection (25 nmoles/~,1) was performed slowly (2
minutes) at the level of the right lateral ventricle
utilizing a Hamilton syringe. Saline was administered to
control animals (1 ~.1 icv).
The compound was administered subcutaneously (sc)
after suspension in 1% DMSO (experiment No. l) or in 0.5%
tragacanth (experiment No.2). The compound was tested at
the following doses: 1-3-5 mg/kg sc.
WO 94/03469 ~ ~ ,~'~ (~ PCT/US93/07307
59
The treatment was conducted performing two
administrations:
- 1 hour before NMD;A injection;
- immediately after NMDA injection.
Experimental
crroups
1. (n = 8) saline (1 ~l) + saline
(1 ~,1)
2. (n = 14) NMDA (25 nmo:Les/~,l)+ saline (1 ~.1)
3. (n = 15) NMDA (25 nmoles/~,1)+ ND37 (1 mg/kg/ml)
4. (n = 15) NMDA (25 nmoles/~1)+ ND37 (3 mg/kg/ml)
5. (n = 15) NMDA (25 nmoles/~,1)+ ND37 (5 mg/kg/ml)
The number of animals in each experimental
utilized
group (corresponding to the total number of animals,
i.e., experiment 1 + exp eriment according to
2,
Table 3) is indicated brackets.
in
Parameters
Animals were sacrificed on the 12th day (i.e., 5
days after NMDA injection) for the evaluation of the in
toto brain weight, defined :in mg.
The statistical significance was evaluated using
Dunnett's test.
Results
The obtained results ('.Cable 3) show that:
- treatment with ND:37 is effective in reducing
the brain damage induced by the excitotoxin (evaluated
as the lowering of total brain weight;
- ND37 is significat:ively effective (p < 0.01)
at a dose of 1 mg/kg sc.
WO 94/03469 PCT/US93/073(17
~1 ~~6~ 9
Table 3 Antineurotoxic effect of ND37 in vivo_ evaluation of the
protection of cerebral damage (total brain weight) induced
by NMDA in neonatal rat
Experiment n.l (vehicle = DMSO 1%)
Groups Compounds Doses Total brain
weight
(mg/kg) (mg)
1. 4) control + saline 1017
(n
=
2. 7) NMDA + saline 841
(n
=
3. 8) NMDA + ND 37 1 960
(n
=
4. 8) NMDA + ND 37 3 968 *
(n
=
5. 8) NMDA + ND 37 5 959
(n
=
Experiment n.2
Groups Compounds Doses Total brain
weight
(mg/kg) (mg)
1. 4) control + saline 1017
(n
=
2. 7) IAA + saline 841
(n
=
3. 7) NMDA + ND 37 1 1019
(n
=
4. 7) NMDA + ND 37 3 977
(n
=
5. 7) NMDA + ND 37 5 976 *
(n
=
* p < 0.01 vs lesioned not treated (group 2)
(In all experiments the standard deviation is less than 5%).
In brackets is the indication of the number of animals.
Compounds under examination were administered at doses of 1-3-5 mg/kg
sc 1 hours before NMDA injection (25 nmoles/1 ul) and immediately
after NMDA. Brain weight is expressed in mg.
Modulatorv in vitro effect of ND37 on release and/or
uptake of excititatorv neurotransmitter in cerebellar
5 Qranule cells: determination of glutamate and aspartate
content in t~otassium induced depolarization
MATERIALS AND METHODS
Cell cultures
Primary cell cultures of cerebellar granule cells
10 were prepared from 8 day old Sprague-Dawley rats.
Neurons were grown in 35 mm plates for 11-13 days
and maintained in a humid environment (95% air and 5%
C02). Cultures (3x106 cells/plate) are mainly formed by
granule cells (95~), with a small amount (5%) of. glial
15 cells (Gallo V. et al.: Selective release of glutamate
from cerebellar granule cells differentiating in
culture. Proc. Natl. Acad. Sci. USA 79, 7919-23, 1982).
214:16'9
WO 94/03469 PCT/US93/07307
61
Glial proliferation was prevented by arabinofuranoside
cytosine.
Derivative ND37 was sol.ubilized at a concentration
of 10 M in dimethylsulfoxide (1% DMSO), and then diluted
at various concentrations (0.1 - 1 - 10 - 20 ~,M) in
Locke's solution (154 mM NaCl/5.6 mM KC1/ 3.6 mM
NaHC03/2.3 mM CaCl2/1 mM MgC7.2/5.6 mM glucose/5 mM HEPES,
pH 7.4) .
Description of exogenous glutamate neurotoxicitv model
The cell culture medium was aspirated from the
plates. Plates were washed. (3 x 2 ml) with Locke's
solution, and solutions (750 ~,1) containing the compound
to be tested (concentrations from 0.1 to 20 ~.M) were
added and incubated for 2 hours in an incubator at 37
°C (5% C02) both in the presence and absence of
different depolarizing concentrations of KC1 (between 5
and 50 mM) . The incubation medium was gathered, filtered
through a 0.2 ~ filter, and processed (220 ~,1) for the
analysis of the amino acid content.
Parameters
The glutamate and aspartate content (expressed as
~,M) was evaluated by HPLC (high pressure liquid
chromatography), according to Bidlingmeryer B.A. et al.,
J. Chromatogr. 336, 93-104, 1984.
Results
The obtained results show that ND37 is able to
diminish in cerebellar granule cells, in a dose-
dependent manner (Table 4), the potassium-induced
increase of glutamate and aspartate (Table 5). The
compound, administered at closes up to 20 uM, did not
modify the basal levels (controls) of extracellular
glutamate and aspartate (Table 5), whereas it reduced by
ca. 30% the extracellular glutamate induced by KC1
potassium already at a dose of 0.1 ~M (Table 4). ND 37
WO 94/03469 PCT/US93/073~
62
completely abolished the glutamate increase induced by
KC1 in the culture medium at a dose of 20 ~,M (Table 5).
20 ~M ND73 completely abolished the extracellular
increase of glutamate and aspartate induced by KC1 (15,
25, 30, and 50 mM). The extracellular values for
glutamate and aspartate in cultures exposed to KC1 in
the presence of ND37 were between 0.01 and 0.28% of the
values obtained without the compound (Table 5).
The possible mechanism of action of ND37 may reside
in the release and/or uptake of endogenous compounds.
Table 4: Dose dependent inhibitory action of ND37 (0.1 - 1 - 10 -
uM) on extracellular glutamate increase induced by KC1
in primary cerebellar granule cells.
Extracellular $ KC1
Groups glutamate 50 mM
content
(uM)
1) controls 0.04 t 00
2) KCl (50 17.40 t 0.19 100
mM)
3) KC1 (50 + ND37(0_1 12.60 0.57 72
mM) uM)
4) KC1 (50 + ND37(1 13.30 t 1.33 76
mM) uM)
5) KCl (50 + ND37(10 5.60 2.37 * 32
mM) uM)
6) KC1 (50 + ND37(20 0.11 ~ 0.09 * 0.01
mM) uM)-
Triplicate (mean s.e.)
experiments +
* p c 0.01 group
vs. 2
(Dunnett's
test)
~ ..,WO 94/03469 214 ;~ ~; ~ g
PCT/US93/07307
63
Table 5: Effect of ND37 (20 uM) on extracellular glutamate and
aspartate content in cerebellar granule cells in depo-
larizing conditions induced by different concentrations
of KC1 (15 - 50 mM).
Extracell_ % Extracell. % corresp.
corresp.
Groups glutamate KC1 aspartate KC1
content (uM) content
(uM)
1)controls 0.354 0.020
2)controls
+
ND37 (20 0.362 0.039
uM)
3)KC1 (30 23.115 :100 1.016 100
mM)
KC1 (50 +
mM)
ND37 (20 0.318 * 0.01 0.063 0.06
uM)
4)KC1 (50 10.863 :100 0.621 100
mM)
KC1 (50 +
mM)
ND37 (20 0.246 * 0.02 0.078 0.13
uM)
5)KC1 (25 5.195 :100 0.386 100
mM)
KC1 (50 +
mM)
ND37 (20 0.330 * 0.06 0.107 0.28
uM)
6)KC1 (15 2.459 100 0.484 100
mM)
KC1 (50 +
mM)
ND37 (20 0.899 * 0.34 0.046 0.09
uM)
Triplicate
experiments
* p < 0.01 (Dunnet's test)corresponding treated
vs group
only
with KC1 (in all experiments:>tandarddeviation than 5%)
less
Examp7Le 56
Electrophysioloctical characterization of neurons
treated with ND37: absence of an effect on Qlutamate-
stimulated cationic channels.
Materials and Methods
Primary cell cultures of cerebellar granule cells
were prepared from 8 day old rats (Zivic Miller,
Pittsburgh, PA, USA).
Neurons were grown on 35 mm plates for 7-8 days and
maintained in a humid environment (95% air and 5% C02).
Cultures (2.5x106 cells/plate) were mainly formed by
granule cel is (95%) , Grith a small amount (5%) of glial
cells (Gallo V. et al.: Selective release of glutamate
from cerebellar granule cells differentiating in
culture. Proc. Natl. Acad. Sci. USA 79, 7919-23, 1982).
WO 94/03469 PCT/US93/073~'
~14~~~ 9
64
Glial proliferation was prevented by arabinofuranoside
cytosine.
Primary cell cultures of cortical neurons were
prepared from one day old neonatal mice (Zivic Miller,
Pittsburgh, PA, USA) (Bertolino and Vicini: Mol.
Pharmacol. 34, 98-103, 1988).
Cultures were grown on 35 mm plates, to which was
added 10 ~g/ml of poly-L-lysine, at a density of 5x105
cortical neurons/plate. Cultures were prepared on basal
Eagle medium (Gibco) containing loo fetal calf serum
(Gibco), 25 mM KC1, 2 mM glutamine, and 100 ~g/ml
gentamycine . Glial proliferation was prevented by adding
1 ~M arabinofuranoside cytosine 24 hours after seeding.
Cortical neurons were cultivated for 3 weeks.
Derivative ND37 was solubilized at a concentration
of 1x10-2 M in dimethylsulfoxide (1% DMSO), and then
diluted at various concentrations in Locke's solution
(-Mg++) (154 mM NaCl / 5.6 mM KC1 / 3.6 mM NaHC03 / 2.3
mM CaCl2 / 5 . 6 mM glucose / 5 mM HEPES , pH 7 . 4 ) . ND3 7
was analyzed at concentrations of 20 and 30 ACM.
Description of electrophysiological measurements of
glutamate-related cationic channel activity in
cerebellar granule neurons and cortical neurons in
culture
Experiments were performed at room temperature
utilizing the following medium 145 mM NaCl, 1 mM CaCl2,
5 mM glucose, and 5 mM HEPES/NaOH at pH 7.4, and patch
pipettes, close to the preparation, containing 145 mM
CsCl2, 1 mM CaCl2, 11 mM ethyleneglycol (~i-aminoethyl
ether) bis N,N'-tetraacetic acid, 10 mM HEPES/Cs(OH)2 at
pH 7.2. Plates were continually perfused at a flow rate
of 1 ml/min.
a) "Sealed" registrations on whole cells (Hamill
O.P. et al., "Improved patch-clamp techniques for
high-resolution current recording from cells and
_yy0 94/03469 2141 ~6 /~ ~ PCT/US93/07307
E~ 5
cell-free membranes patches", Pfluegers Arch. 391,
85-100,1981) were performed on cerebellar granule cells
or cortical neurons from neonatal rats and grown in
cultures for 1 or 2 weeks, respectively.
Glutamate was released. under pressure (2 - 6 psi)
from glass micropipettes (with tips having a greater
delivery capacity than that, 4 - 6 ~M, of patch
pipettes) on the cell bodies of voltage clamped neurons
(maintenance voltage - 40 mV).
b) The activation by glutamate of single channels
was externally recorded on membrane portions prepared
from cerebellar granule cells pretreated with ND37 for
30 minutes at 37 °C. Micropipettes, filled (1 ~,M) with
glutamate, were positioned close to the membrane
portions and were also utilized in order to drip the
neurotransmitter. Single channel currents were
registered according to the method described by
Bertolini and Vicini ("Voltage-dependent block by
strychnine of N-methyl-D-aspartic acid-activated
cationic channels in rat cortical neurons in culture",
Mol. Pharmacol. 34, 98-103, 1988).
Registration on the whole cell
Glutamate (50 ~,M), able to activate currents
directed inside, was released by pressure on
voltage-clamped cell bodies of cerebellar and cortical
neurons . Because these experiments were performed in the
absence of Mg++, the respon;ae to glutamate was probably
mediated by the NMDA and non-NMDA glutamate receptor.
The combined administration of ND37 (30 ~.M) and
glutamate (50 ~,M), does not influence the control
response, which is not different from the oontrols (mean
200 pA at voltage - 50 mV) in the three different
conditions in 3 cerebellar <~nd 5 cortical preparations.
WO 94/03469 PCT/US93/07z~'
66
Results
In order to evaluate the activities of ND37 on
glutamate response, 10 different experiments were
performed. In cerebellar granule cell membrane
preparations, high conductivity, glutamate-activated (50
pS) cationic channels are prevalent. Pretreatment of
cells for 30 minutes with 20 ~.M ND37 does not influence
the channel conductivity and the frequency of their
opening (Table 6). According to the single channel
registration, no kinetic and conductivity variations
were observed after ND37 treatment. The inside currents
activated by glutamate (50 ~,M) given contemporaneously
with ND37 (30 ~.M) were not different from those
activated only by glutamate.
Table 6: Effect of ND37 on glutamate controlled cationic channels
activity in cerebellar granule neurons and cortical
1 5 neurons in culture.
Single channel current registration
Scerebellar granule neurons)
Group
n = 10 Channel conductivity Opening frequence
(pS) (opening/sec)
1) Glutamate (1 mM) SO 4.5 + 2.9
2) Glutamate + ND37 (20 uM) 50 4.1 + 2.1
Conclusions
ND37, a compound protecting neurons from glutamate
receptor-mediated toxicity, does not seem to produce
this effect blocking the ionotropic glutamate receptors,
as can be seen from the absence of effects of cationic
glutamate stimulated channels.
Example 57
The neuritogenic activity of the new compounds of
the present invention can be shown by experiments
~WO 94103469 2 ~ 4 ~ s 7 ~ PCT/US93/07307
67
performed with the aforesaid compound ND37, and with the
2-ethylglycoside of N-:palmitoyl-neuraminic acid
dimethylaminopropylamide, which will be subsequently
referred to as ND35.
MATERIALS AND METHODS
Cell cultures
C1300 mouse neuroblast:oma cells, Neuro-2a clone,
(American Cell Type Culturfs Collection, Bethesda, MD)
were grown at a density of 10,000 cells/well in a
culture medium containing Dulbecco's modified Eagle
medium (DMEM, Flow), 10% fetal calf serum (FCS, batch IP
02, Seromed), penicillin (100 units per ml, Irvine), and
L-glutamine (2 mM, Sigma). Cells were incubated at 37
°C for 24 hours, and medium was removed and substituted
with 350 ~,1 of fresh medium plus compounds to be tested.
Tested compounds and their solubilization
Compounds ND35 and ND37 were solubilized at a
concentration of 1x10-2 M in dimethylsulfoxide (1% DMSO) .
For the different compounds, progressive dilutions in
culture medium were performed (concentrations from 1x10-5
to 1x10-4 M).
Parameters
NeuritoQenic activity (cell number with neurites,
optical microscopy)
Culture plates were incubated with compounds to be
tested and analyzed using a phase contrast microscope
(250x). Nine fields with prefixed coordinates were
chosen and photographed. Then the total number of cells
and those with neurites (length at least double the cell
diameter) were counted in blind in each photograph. The
percentage of cells with ne:urites was determined after
counting of at least 100 cells (Facci L. et al.:
Promotion of neuritogenesis in mouse neuroblastoma cells
by exogenous ganglioside GM7_. J. Neurochem. Raven Press,
New York, 299-305,1984).
WO 94/03469 ' PCT/US93/073°'
~r
68
Results
The obtained results (Table 7) show that
derivatives ND35 and ND37 both induce neuritogenesis in
vitro. In particular, under the tested experimental
conditions, it was shown that:
- the neuritogenic effect was already
significant at a concentration of 5x10-5 M (p < 0.01),
with the highest efficacy (about 480 of cells with
neurites) at a concentration of 1x10-4 M.
Table 7: Neuritogenic effect of ND35 and ND37 in N2a neuroblastoma
cells.
Compounds (concentrations) Cells with neurites (%)
Control 2 _+ 3
ND37 (1x10 4) 48 _+ 9
(5x10 5) 27 _+ S
(2.5x10 5) 8 _+ 4
(1x10 5) 5 + 3
ND35 (1x10 4) 39 _+ 7 *
(5x10 5) 28 _+ 7 *
(2.5x10 5) 7 _+ 3
(1x10 5) 7 + 2
* p < 0.01 vs. control (Dunnett's test)
-~..WO 94/03469 ~ ~ ~ ~ ~ ~ PCT/US93/07307
69
CONCLUSIONS
The foregoing results show a good pharmacological
profile of the compounds according to the present
invention. Their antineuronotoxic and modulatory effects
on extracellular levels of e:r~cititatory neurotransmitter
amino acids should be noted in particular.
Owing to their antineuronotoxic activity, the new
derivatives of neuraminic: acid can be used in
pathologies related to the excitotoxic effect of
excititatory amino acids. It has been shown that these
amino acids, e.g., glutami.c acid and aspartic acid,
besides their important functions in several
physiological processes such as, for example,
synaptogenesis and plasticity, are involved in the
ethiogenesis and/or evolution of different pathologies
related to neuronal evolution and/or death. Although
neuronal damage can have several causes, neuronal
disfunctions excite a cascade of cellular events, such
as the activation of Ca++ ion-dependent enzymes, the
influx of Ca++ ions, and the activation of second
messengers, which cause neuronal death. Damage to the
nervous system due to excititatory amino acids is
present in ischaemia, h~~rpoxia, epilepsy, trauma,
compressions, metabolic disfunctions, aging, and
toxic-infective and chronic neurodegenerative diseases,
like Alzeimer's and Huntingt:on's diseases.
Because of the modu7.atory effect of the new
derivatives on the processes of release and/or uptake,
and of the increase in the intracellular space, of
neurotransmitter amino acids, these new compounds have
therapeutic relevance in neuro-psychiatric disorders,
where the pathological event: derives from the imbalance
of the aforesaid processes. Owing to the fact that the
protective action of the derivatives of invention
against the toxicity of exci.titatory amino acids occurs
through the activation of glutamate receptors, the use
of these compounds does not. have the disadvantages of
.. ,0 2141 a ~ 9
other known derivatives, which block these receptors
(see Olney J.W. et al.: "Pathological changes induced
in cerebrocortical neurons by phencyclidine and related
drugs", Science 224, 1360--1362, 1989; Olney J.W. et
al.: "NMDA antagonist neu:rotoxicity: mechanism and
prevention", Science 254, 1515-1518, 1991).
Finally, the new compounds of the present
invention, because of their neuritogenic activity, are
valuable in therapies related to the recovery of
nervous functions in pathologies associated with
neuronal damage, like peripheral neuropathies.
PHARMACEUTICAL APPLICATIONS OF THE COMPOUNDS
OF THE PRESENT INVENTION
Objects of the pre:>ent invention also include
pharmaceutical preparations having, as active
ingredients, one or more of the new aforesaid
derivatives and, particularly, those especially
mentioned or those described in the foregoing examples.
These pharmaceutical preparations can be used
for oral, rectal, parentera.l, local or intradermic use.
They can therefore be in solid or semisolid form, e.g.,
pills, tablets, gelatineous soft capsules, capsules,
soft gelatin suppositories, etc. For parenteral use,
it is possible to use formulations for int:ramuscular,
subcutaneous, or transdermic use, or suitable for
infusions or intravenous injections, and they can
therefore be prepared as solutions of active components
or as a lyophilized powder of the active components,
eventually to be added to one or more excipients or
pharmaceutically acceptablE~ solvents, which are usable
for the aforesaid purpose, and which are osmolar with
physiological fluids. For local application, spray
preparations, e.g., nasal sprays, can be employed, or
ointments for topical use, or plasters for transdermal
administration can be used..
,1 2141679
The preparations of the present invention can be
used both in man and animals.. Preferably, they contain
between about 0.01 and 10~ of the active components
for solutions, sprays, ointments and creams, and
between 1~ and 100, preferably between 5$ and 50$ of
the active component, for so7lid form preparations.
The dosage will vary according to the
indication, the desired ei=fect, and the route of
administration. For therapeutic administration, or for
prophylaxis by the parenteral route, the dosage varies
preferentially between 0.05 and 10 mg per kg body
weight per day, and especially between 0.05 and 2 mg
per kg body weight per day.
The invention being thus described, it will be
obvious that the same may be varied in many ways. Such
variations are not to be regarded as a departure from
the spirit and scope of the invention, and all such
modifications as would be obvious to one skilled in the
art are intended to be included within the scope of the
following claims.