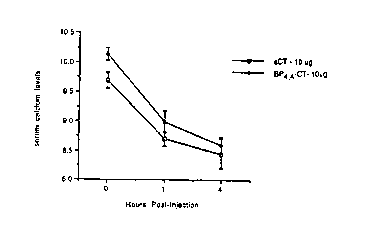Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
X153071
~- WO 94/15962 PCT/US93/12692
1
Description
DERIVATIZED CALCITONINS
Background of the Invention
Bone is a dynamic tissue, and homeostasis in the
adult skeleton requires a balance between the formation of
new bone and the resorption of previously formed bone.
Calcitonin, a peptide hormone secreted by the thyroid and
thymus of mammals, plays an important role in maintaining
bone homeostasis. Calcitonin binds to receptors found on
osteoclasts, cells in the bone tissue which mediate bone
resorption. Calcitonin immobilizes the osteoclast, thus
inhibiting bone resorption with a resultant decrease in
the amount of calcium released by bone into the serum.
This inhibition of bone resorption has been exploited by
using calcitonin as a treatment for osteoporosis.
Calcitonin occurs in the thyroid glands of
mammals and the ultimobranchial glands of lower
vertebrates. Known, naturally occurring calcitonins are
all 32-amino acid polypeptides having an amidated carboxy
terminus and an intramolecular disulfide bond between
cysteine residues in the 1 and 7 positions.
At the present time, salmon calcitonin is
preferred over human calcitonin for treatment of
osteoporosis. The worldwide market for salmon calcitonin
exceeds $500 million annually. Salmon calcitonin has been
shown to be considerably more effective in arresting bone
resorption than human forms of calcitonin. There are
several hypotheses for why salmon calcitonin is more
potent than human calcitonin in treatment of osteoporosis.
These hypotheses include: 1) salmon calcitonin is more
resistant to degradation; 2) salmon calcitonin has a
lower metabolic clearance rate (MCR); and 3) salmon
calcitonin may have a slightly different conformation,
resulting in a higher affinity for bone receptor sites.
S11BSTTTLTtE SHEET (RULE 261
-- WO 94/15962 215 3 0 l 1 PCT/US931I2692 '
2
Despite the advantages associated with the use
of salmon calcitonin for treatment of osteoporosis in
humans, there are also disadvantages. Salmon calcitonin
is administered by injection, a process that many patients
find unacceptable. In addition, some patients develop
antibodies to non-human calcitonin. Therefore, new
analogs of salmon, human or other calcitonins that are
potent inhibitors of bone resorption, less expensive, more
convenient to administer and non-immunogenic are needed.
Summary of the Invention
It is an aspect of the present invention to
provide novel calcitonin derivatives that have enhanced
hypocalcemic activity and/or extended in vivo half-life.
It is a further aspect of the invention to
provide pharmaceutical compositions comprising novel
calcitonin derivatives, as well as methods of reducing
serum calcium in patients by administration of the
pharmaceutical compositions.
Within one aspect, the present invention
provides compounds selected from the group consisting of
S S
R-CONH-Cys-Gly-Asn-Leu-Ser-Thr-Cys-Met-Leu-Gly-Thr-Tyr-
Thr-Gln-Asp-Phe-Asn-Lys-Phe-His-Thr-Phe-Pro-Gln-Thr-Ala-
Ile-Gly-Val-Gly-Ala-Pro-NH2 (Sequence ID Number 1),
S S
R-CONH-Cys-Ser-Asn-Leu-Ser-Thr-Cys-Val-Leu-Gly-Lys-Leu-
Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-
Thr-Gly-Ser-Gly-Thr-Pro-NH2 (Sequence ID Number 2),
and pharmaceutically acceptable salts thereof,
wherein R is substituted or unsubstituted biaryl,
optionally containing 1 or 2 ring nitrogen atoms per ring;
substituted or unsubstituted heterocycloalkyl containing 1
or 2 ring heteroatoms per ring selected from the group
consisting of N, S and O; substituted or unsubstituted
~118STTTiJTE SHEET (RUIE 261
- WO 94/15962 215 3 0 71 PCT/US93/12692
3
bis-heterocycloalkyl containing 1 or 2 heteroatoms per
ring selected from the group consisting of N, S and O; or
substituted or unsubstituted heteroaryl containing 1 to 4
heteroatoms per ring selected from the group consisting of
N, S and O.
Within another aspect, the present invention
provides compounds of the formula R-N-CT, wherein CT is a
calcitonin selected from the group consisting of human,
salmon, eel, rat, porcine, bovine, ovine and chicken
calcitonins and biologically active derivatives and
variants thereof, or a pharmaceutically acceptable salt
thereof; N is an amide linkage; and wherein R is
substituted or unsubstituted biaryl, optionally containing
1 or 2 ring nitrogen atoms per ring; substituted or
unsubstituted heterocycloalkyl containing 1 or 2 ring
heteroatoms per ring, selected from the group consisting
of N, S and O; substituted or unsubstituted bis-
heterocycloalkyl containing 1 or 2 heteroatoms per ring
selected from the group consisting of N, S and O; or
substituted or unsubstituted heteroaryl containing 1 to 4
heteroatoms per ring selected from the group consisting of
N, S and O.
Within a third aspect, the present invention
provides compounds of the formula X-(R-N-CT)n wherein X is
an ion of a transition metal; R is a heterocycloalkyl or
heteroaryl group; CT is a calcitonin selected from the
group consisting of human, salmon, eel, rat, porcine,
bovine, ovine and chicken calcitonins and biologically
active derivatives and variants thereof; N is an amide
linkage; and n - 2 or 3. Pharmaceutically acceptable
salts of these compounds are also provided.
The compounds disclosed above are combined with
a pharmaceutically acceptable carrier to produce
pharmaceutical compositions. These compositions are
useful for, ~nte~ G_'~:~, reducing serum calcium in a
patient.
S118ST1TUTE SHEET (RULE 261
2153011
CVO 94/15962 PCT/US93/1269'- N
4
List of Abbreviations
DMSO Dimethylsulfoxide
BHA Benzhydrylamine
MBHA 4-methylbenzhydrylamine
FMOC 9-fluorenylmethoxycarbonyl
BOC t-butyloxycarbonyl
Trit Trityl
But t-butyl
PMC 2,2,5,7,8-pentamethylchroman-6-sulfonyl
BOP Benzotriazolyl-N-oxytrisdimethylaminophos-
phonium hexafluorophosphate
PyBOP Benzotriazole-1-yl-oxy-tris-pyrrolidino
phosphonium hexafluorophosphate
PyBrOP Bromo-tris-pyrrolidino-phosphonium
hexafluorophosphate
HBTU [2-(1H-benzotriazole-1-yl)1,1,3,3,-
tetramethyl uronium hexafluorophosphate
HOBt 1-hydroxybenzotriazole
DIEA Diisopropylethyl amine
Et3N Triethylamine
DMF Dimethylformamide
NMP N-methylpyrrolidone
HPLC High performance liquid chromatography
AAA Amino acid analysis
DCM Methylene chloride
SCT Salmon calcitonin
TFA trifluoroacetic acid
DIC Diisopropyl-carbodiimide
DCC dicyclohexylcarbodiimide
FAB-MS Fast Atom Bombardment - Mass Spectroscopy
Brief Description of the Drawings
Figures 1, 2 and 3 represent the results of an
assay measuring serum calcium levels using a compound that
is representative of the present invention. BP4~4-CT (ZP
SlIBSrITITTE SHEET (RULE 261
-. W'O 94/15962 215 3 0 71 PCT/US93/12692
1) is the derivatized calcitonin compound and SCT is
salmon calcitonin.
Detailed Description of the Invention
The present invention provides new derivatized
calcitonin molecules. These molecules are provided in
both monomeric and multimeric forms. The derivatized
calcitonins of the present invention provide various
advantages over currently available calcitonins, including
higher specific activity, increased half-life, increased
stability and/or reduced immunogenicity.
The derivatized calcitonins of the present
invention include derivatized human, salmon, eel, rat,
porcine, bovine, ovine and chicken calcitonins. Salmon
and, in particular, human calcitonins are preferred. The
calcitonins may be derived from the native, wild-type
molecules or may be derived from modified forms of
calcitonin having biological activity. A variety of
modified calcitonins are known in the art, including
calcitonins having amino acid substitutions (e. g. U.S.
Patents Nos. 4,824,936; 4,764,589; 4,663,309 and
4,658,014), deletions (e. g. U.S. Patents Nos. 4,820,804;
4,764,591; 4,639,511; 4,605,514 and 4,537,716) and
calcitonins containing D-amino acid substitutions (U. S.
Patent No. 4,652,627). The term "biologically active" is
used herein to denote calcitonins that exhibit bone
resorption inhibiting activity. Hypocalcemic activity and
receptor-mediated stimulation of cAMP are indicators of
bone resorption inhibiting activity of calcitonins.
The molecules of the present invention are
characterized by a derivatized amino terminus, formed by
combining a calcitonin with a cyclic, polycyclic or
heterocyclic moiety selected from the group consisting of
substituted or unsubstituted biaryl, optionally containing
1 or 2 ring nitrogen atoms per ring; substituted or
unsubstituted heterocycloalkyl containing 1 or 2 ring
SU9STtTIJTE SHEET (RULE 261
WO 9 215 3 0 71 PCT/US93/1269?
6
heteroatoms per ring selected from the group consisting of
N, S and O; substituted or unsubstituted bis-
heterocycloalkyl containing 1 or 2 heteroatoms per ring
selected from the group consisting of N, S and 0; and
substituted or unsubstituted heteroaryl containing 1 to 4
heteroatoms per ring selected from the group consisting of
N, S and O, wherein the cyclic, polycyclic or heterocyclic
moiety is joined to the N-terminal cysteine residue of the
calcitonin via an amide linkage. Thus, within one aspect
of the present invention, a calcitonin is reacted with a
carboxylic acid of one of the above-described moieties to
yield an N-terminal derivatized calcitonin.
As used herein, the terms cyclic, polycyclic,
heterocyclic, heterocycloalkyl and equivalents thereof are
used to denote ring structures having three or more atoms
per ring. In general, these structures will have fewer
than nine atoms per ring, preferably five, six or seven
atoms per ring, although structures containing twelve or
more ring atoms may be used. The terms biaryl, heteroaryl
and equivalents thereof are used to denote aromatic ring
structures having five or more atoms per ring, preferably
five to eight ring atoms, more preferably five or six ring
atoms per ring.
Within the present invention, the cyclic,
polycylic, and heterocyclic moieties include fused ring
structures containing from 2-3 rings per fused ring group.
Preferred fused ring structures include biaryl moieties
having the structure
S118STmJTE SHEET (RULE 261
-' ~i'O 94/15962 215 3 0 71
PCT/US93/12692
. ...,..
R ~ / \ X
X3\X/Xl Y2\Y/Y
wherein X, X1, X2, X3, Y, Y1, Y2 and Y3 are individually C
or N and R' is linear C1-Cg alkyl, branched C1-C12 alkyl,
nitro, hydroxyl, carboxyl, trifluoromethyl, carboxylamide,
sulfhydryl, cyano, halo, alkoxy, ester or H.
A second group of preferred fused ring
structures includes the structure
X Y
R'
X4 Xs Y5 Y4
wherein X4 and Y4 are individually C, N, O or S, X5, X6,
Y5 and Y6 are individually C or N subject to the
limitations that when X4 is S or O, X5 must be C and when
Y4 is S or O, Y5 must be C and R' is as defined above. A
third group of preferred fused ring structures includes
heterocycloalkyl moieties having the structure
(n~R~
Y X
wherein X is N, O or S; Y is C or N; n=1 or 2; and R' is
as defined above. A fourth group of preferred fused ring
SUBSTiTLTrE SHEET (RULE 26b
V'O 94/15962 215 3 0 ~ 1 8 PCTIUS93I12692
structures includes bis-heterocycloalkyl moieties having
the structure
R'
X~Xl Y-Y1
~R'
10
wherein X, X1, Y and Y1 are individually N, O or S and R'
is as defined above. A fifth group of preferred fused
ring structures includes heteroaryl moieties having the
structure
/Y
R'
A
or the structure
R'
X
Y
wherein X is C, O, S or N; Y, Z and A are individually C
or N and R' is as defined above.
A preferred class of calcitonin derivatives
includes those modified by the addition of a compound of
the formula:
SUBSTtflITE SHEET (RULE 261r
~.___ __ .__._ __._~
WO 94/15962 PCT/US93/12692
215~d~1 9
R'
R'
\ \
X1 Y1
wherein X1 and Y1 are individually C or N and R' is linear
C1-Cg alkyl, branched C1-C12 alkyl, nitro, hydroxyl,
trifluoromethyl, carboxyl, carboxylamide, sulfhydryl,
cyano, halo, alkoxy, ester or H. Particularly preferred
members of this class include those modified by the
addition of the compounds:
R'
R'
T\
or N
N
R'
R'
A second preferred class of calcitonin
derivatives includes those modified by the addition of a
compound of the formula:
R'
12 X2
(n)
wherein n=1 or 2, X2 is N, 0 or S, Y2 is C or N and R' is
as defined above. Particularly preferred members of this
class include those modified by the addition of the
compound:
SUBSTitIJTE SHEET (RULE 26b
WO 94/15962 PCT/US93112691
~ 1530 1 1~
i
N
R'
H
A third preferred class of calcitonin
derivatives includes those modified by the addition of a
compound of the formula:
R'
X3 Y3
R'
wherein X3 and Y3 are individually N, O or S, and R' is as
defined above. Particularly preferred members of this
class include those modified by the addition of the
compound:
R'
N N
R' H H
A fourth preferred class of calcitonin
derivatives includes those modified by the addition of a
compound of the formula:
~Ya
X4
A--
R'
SlJBSTTTIJfE SKEET (RULE 26b
~. ~..r
___.._ ___ _ _ _ _ ..
WO 94/15962 215 ~ ~ ~ ~ PCTIUS93/12692
11
wherein Y4 is O, S or N; X4, Z and A are individually C or
N, subject to the limitation that X4 is not N when Y4 is O
or S; and R' is as described above. Particularly
preferred members of this class include those modified by
the addition of the compounds:
H H
R. R.
or
N N
A fifth preferred class of calcitonin
derivatives includes those modified by the addition of a
compound of the formula:
X5
/
R'
Ys
wherein X5 is N, Y5 is C or N, and R' is as described
above. Particularly preferred members of this class
include those modified by the addition of the compounds:
N
R' or R.
N N
A sixth preferred class of calcitonin
derivatives includes those modified by the addition of a
compound of the formula:
SlJ9SnZ11TE SHEET (RULE 261
2153011 12
Xs
R
Ys
wherein X5 is N, Y5 is C or N, and R' is as described
above.
Within the compounds described above, those in
which R' is C02H or H are particularly preferred.
The calcitonin derivatives of the present
invention can be synthesized by solid phase or solution
phase methods conventionally used for the synthesis of
peptides (see, for example, Merrifield, R.B. J. Amer.
Chem. Soc. 85: 2149-2154, 1963; Birr, C. Aspects of the
Merrifield Peptide Synthesis, Springer-Verlag, Heidelberg,
1978; Carpino, L.A., Acc. Chem. Res. 6:191-198, 1973; Kent
S.B., Ann. Rev. Biochem. 57:957-989, 1988; Gregg et al.
Int. J. Peptide Protein Res. 35:161-214, 1990; The
Peptides, Analysis. Synthesis. Bioloay, Vol. 1. 2. 3, 5:
Gross, E and Meinhofer, J. eds., Acad. Press, New York,
1979; and Stewart et al., Solid Phase Peptide Synthesis.
2nd. ed. Pierce Chem. Co., Rockford, I1, 1984.
The
use of solid phase methodology is preferred. Briefly, an
N-protected C-terminal amino acid residue is linked to an
insoluble support such as divinylbenzene cross-linked
polystyrene, polyacrylamide resin, Kieselguhr/polyamide
(pepsyn K), controlled pore glass, cellulose,
polypropylene membranes, acrylic acid-coated polyethylene
rods or the like. Cycles of deprotection, neutralization
(in the case of BOC chemistry, vide infra) and coupling of
successive protected amino acid derivatives are used to
link the amino acids from the C-terminus according to the
amino acid sequence. Preferred solid supports are
divinylbenzene cross-linked polystyrene resins, which are
commercially available in a variety of functionalized
forms, including chloromethyl resin, hydroxymethyl resin,
w WO 94115962 215 ~ 0 71 PCT/US93/12692
13
paraacetamidomethyl resin, benzhydryl amine (BHA) resin,
p-methylbenzhydrylamine (MBHA) resin, oxime resins, 4-
alkoxybenzyl alcohol resin, 4-(2',4'-
dimethoxyphenylaminomethyl)-phenoxymethyl resin, 2,4-
dimethoxybenzhydrylamine resin, and 4-(2',4'-
dimethoxyphenyl-FMOC-aminomethyl)-
phenoxyacetamidonorleucyl-MBHA resin (Rink amide MBHA
resin). BHA, MBHA and Rink amide MBHA resins are
particularly preferred, since they can directly provide C-
terminal amides after cleavage of the peptide chain from
the resin. A particularly preferred resin for use within
the present invention is the Rink amide MBHA resin
(available from Nova Biochem, La Jolla, CA). A preferred
protecting group for the a-amino group of the amino acids
is acid-labile t-butyloxycarbonyl (BOC). BOC is
deprotected using trifluoroacetic acid (TFA) in a suitable
solvent, such as methylene chloride. The resultant TFA
salt is neutralized with a base, such as diisopropylethyl
amine (DIEA) or triethylamine (Et3N), then coupled with
the protected amino acid derivative. Another preferred
protecting group for a-amino acids is base-labile 9-
fluorenylmethoxycarbonyl (FMOC). Suitable protecting
groups for the side chain functionalities of amino acids
chemically compatible with BOC and FMOC groups are well
known in the art. When using FMOC chemistry, the
following protected amino acid derivatives are preferred:
FMOC-Cys(Trit), FMOC-Ser(But), FMOC-Asn(Trit), FMOC-Leu,
FMOC-Thr(Trit), FMOC-Val, FMOC-Leu, FMOC-Gly, FMOC-
Lys(Boc), FMOC-Gln(Trit), FMOC-Glu(OBut), FMOC-His(Trit),
FMOC-Tyr(But), FMOC-Arg(PMC), and FMOC-Pro. The amino
acid residues can be coupled by using a variety of
coupling agents and chemistries known in the art, such as
direct coupling with DIC, DCC or BOP; via preformed
symmetrical anhydrides; via active esters such as
pentafluorophenyl esters; or via preformed HOBt active
esters. Activation Y:ith HBTU ([2-(1H-Benzotriazole-1-
S118STtfUTE SHEET (RULE 261
2153071 14
yl),1,1,3,3-tetramethyluronium hexafluorophosphate]) in
the presence of hydroxybenzotriazole (HOBt) is preferred.
The solid phase method can be carried out
manually, although automated synthesis on a commercially
available peptide synthesizer (e. g. Applied Biosystems
431A or the like) is preferred. In a typical synthesis,
FMOC-Rink-amide MBHA resin is treated with 20% piperidine
in NMP to remove the FMOC group. After washing the resin
with NMP, the first amino acid (the C-terminal FMOC-Pro)
is loaded on the resin using the HBTU/HOBt method.
Successive deprotection (with 20% piperidine/NMP) and
coupling cycles according to ABI FastMoc# protocols (ABI
user bulletins 32 and 33, Applied Biosystems Inc.) are
used to build the whole peptide sequence.
Before attaching the N-terminal modifying group,
the identity and integrity of the peptide are established,
such as by deprotecting and cleaving a small portion of
the FMOC-peptide resin with Reagent K (0.75 g cystalline
phenol, 0.25 ml ethanedithiol, 0.5 ml thioanisole, 0.5 ml
2o dionized water, 10 ml TFA) or the like. The peptide is
precipitated and washed with ether, purified by reverse
phase HPLC and characterized by amino acid analysis and
mass spectroscopy.
The amino terminus of the calcitonin is then
derivatized. When using FMOC chemistry, the N-terminal
FMOC group of the peptide resin is typically deprotected
with 20% piperidine in DMF for 20-30 minutes. The resin
is filtered, washed thoroughly with DMF and DCM, and
dried. The carboxylic derivatives to be used for N
terminal modification (to introduce R groups) can be
coupled to the N-terminal amino group of the resin-linked
peptide by the same activating procedures used in peptide
synthesis, such as carbodiimide-mediated coupling; mixed
anhydride; symmetrical anhydride; preformed active ester;
acyl chloride; BOP, PyBOP, or PyBrOP-mediated coupling
with or without HOBt; or variations and improvements
Trademarks
WO 94/15962 215 3 0 71 PCT/US93/12692
thereof known in the art (see, for example, Coste et al.,
Tetrahedron Lett. 31: 669, 1990 and Coste et al.,
Tetrahedron Lett. 31: 205, 1990). Coupling can be done
manually or by automated means. A particularly preferred
5 method is to first pre-activate the carboxylic acid
derivative by reacting it with HOBt and DIC in a mixture
of DCM and DMF at room temperature to form the HOBt ester
of the carboxylic acid derivative. This mixture is then
added to the deprotected peptide-resin, and the suspension
10 is shaken at room temperature. The reaction is followed
by Kaiser test. The resin is then filtered, washed with
DMF and DCM, and dried.
The derivatized peptide is cleaved from the
resin and deprotected by treatment with TFA containing
15 appropriate scavengers. Many such cleavage reagents, such
as reagent K and others, may be used. The modified
peptide is separated from the resin by filtration and
isolated by ether precipitation. Further purification may
be achieved by conventional methods such as gel filtration
and reverse phase HPLC.
The disulfide bond between the two cysteines at
positions 1 and 7 of calcitonin is formed according to
conventional methods,, such as by aerial oxidation or
oxidation with potassium ferricyanide or DMSO (Tam et al.
J. Am. Chem. Soc. 113:6657-6662, 1991). A particularly
preferred method is oxidation with DMSO. After the
oxidation reaction is complete, as monitored by HPLC or
Ellman's reagent, the peptide is isolated and purified by
HPLC and analyzed for purity by analytical HPLC and mass
spectrometry.
Within another aspect of the invention,
multimeric calcitonins are provided. A calcitonin
derivatized with a heterocycloalkyl or heteroaryl group is
combined with an ion of a transition metal to form a
complex. Preferred calcitonin derivatives in this regard
are those modified by the addition of a linear bicyclic
SUBSTtTITtE SHEET (RULE 261
~fO 9415962 215 3 0 71 16 PCT~S9311269~
moiety containing two ring nitrogen atoms, such as a
bipyridine or bipyridine-like moiety. For example, salmon
calcitonin derivatized with dicarboxy-bipyridine forms a
3:1 molar complex with Fe2+. Other metals useful in this
regard include Cr3+, Fe3+, Mn2+, Co2+~ Co3+, Ni2+, Cu2+~
Cu+ and Zn2+. A preferred method for preparing complexes
is by titration of modified calcitonins with an aqueous
solution of an appropriate metal salt.
Calcitonin derivatives according to the present
invention may be in the form of pharmaceutically
acceptable salts, especially acid-addition salts including
salts of organic acids and mineral acids. Examples of
such salts include salts of organic acids such as formic
acid, acetic acid, propionic acid, glycolic acid, lactic
acid, pyruvic acid, oxalic acid, succinic acid, malic
acid, tartaric acid, citric acid, benzoic acid, salicylic
acid and the like. The acid-addition salts of the basic
amino acid residues are prepared by treatment of the
peptide with the appropriate acid or mineral according to
procedures well known to those skilled in the art, or the
desired salt may be obtained directly by lyophilization
out of the appropriate acid.
The compounds of the present invention have
hypocalcemic activity and are useful within human and
veterinary medicine for the reduction of serum calcium and
regulation of bone metabolism. The compounds of the
present invention may be used, for example, in the
treatment of osteoporosis, Paget's disease,
hyperparathyroidism, osteomalacia, idiopathic
hypercalcemia of infancy and other conditions. The
calcitonin derivatives can also be used to inhibit gastric
secretion in the treatment of acute pancreatitis and
gastrointestinal disorders.
Pharmaceutical compositions are administered at
daily to weekly intervals. An "effective amount" of such
a pharmaceutical composition is the amount that provides a
SllBSTmJTE SHEET (RULE 26b
_____. ~__ __ __~ .._._~ ~ _____._ .__.__.____~_~___ t
__ 2 7 5 3 0 l 1
clinically significant reduction in serum calcium,
inhibition of bone resorption, inhibition of gastric
secretion or other effect. Such amounts will depend, in
part, on the particular condition to be treated, age,
weight, and general health of the patient, and other
factors evident to those skilled in the art. The
concentration of therapeutically effective doses of the
calcitonin derivatives can vary widely depending on the
indication and are well known in the art. For example,
therapeutic doses for the treatment of osteoporosis
generally range from 50-150 International Units (I.U.).
Potency is estimated by comparing the hypocalcemic effect
in rats with that of a standard preparation and is
expressed in International Units, as described in the
International Reference of Preparation, distributed by the
National Institute for Biological Standards and Control,
Holly Hill, London. Compounds having significantly
enhanced half-lives may be administered at lower doses.
The calcitonin derivatives and their
pharmaceutically acceptable salts are formulated with a
pharmaceutically acceptable carrier for parenteral, oral,
nasal, rectal or transdermal administration according to
conventional methods. Formulations may further include
one or more diluents, fillers, emulsifiers, preservatives,
buffers, excipients, etc. and may be provided in such
forms as liquids, powders, emulsions, suppositories,
liposomes, transdermal patches, tablets, etc. One skilled
in this art may formulate the compounds of. the present
invention in an appropriate manner, and in accordance with
accepted practices, such as those disclosed in Remington's
Pharmaceutical Sciences, Gennaro, ed., Mack Publishing
Co., Easton, PA, 1990.
The following examples are offered by way of
illustration, not limitation.
2153071
18
Example 1
A. Synthesis of salmon calcitonin:
Linear protected salmon calcitonin (SCT) was
synthesized by solid phase methodology on an Applied
Biosystems, Inc. (Foster City, CA) 431A peptide
synthesizer using FMOC chemistry and HOBt and HBTU
activation methodology. Rink amide MBHA resin (Nova
Biochem, La Jolla, CA) with 0.43 mM substitution (58o mg)
was used. All the couplings were > 98.5 %. At the end of
the synthesis the resin was thoughly washed with NMP and
CH2C12 and dried. A small portion (27 mg) of the resin-
linked FMOC-SCT was deprotected and cleaved in 10 ml of
reagent K for 2.5 hr. The resin was filtered and solution
evaporated. The residue was chromatographed on a Vydac# C-
4 (2.2 x 25 cm) HPLC column using a gradient of 0-60% B in
40 minutes (Solvent A: 0.1% TFA in water and Solvent B:
0.1% TFA in acetonitrile) to provide pure FMOC-SCT.
Retention time of the peptide was 36.38 min. The peptide
was characterized by amino acid analysis and mass
spectometry (ms 3656.33) to establish the integrity of the
salmon calcitonin link to the resin.
B. Preparation of Bibvridine-modified calcitonin lZP-1
or BP4,4-CT)
2,2'-Bipyridine-4,4'-dicarboxylic acid (0.2 mM)
was dissolved in 3 ml of 5% diisopropylethylamine (DIEA)
in CH2C12 in a flask equipped with a stir bar and drying
tube. N-Hydroxybenzotriazole (HOBt) (0.5 mM) and
diisopropylcarbodiimide (DIC) (0.5 mM) were added to form
the HOBt diester; after 45 minutes of stirring, the
solution had cleared and turned faint yellow. SCT-resin
(100 mg) was treated with 20% piperidine in DMF to remove
the N-terminal protecting group. The deprotected SCT-
resin was added to the solution of 2,2'-bipyridine
carboxylic acid HOBt diester. The mixture was kept
Trademark
WO 94/15962 ~ 15 3 0 71 PCT/US93/12692
19
shaking overnight. A negative Kaiser test for free NH2
group indicated that coupling was complete. The SCT-resin
was washed with DMF and CH2C12.
The reduced form of bipyridine-modified SCT was
cleaved from the resin with reagent K, and purified by
reverse phase HPLC. A mixture of acetonitrile and water
containing O.la trifluoroacetic acid was used as HPLC
solvent. A linear gradient (20% CH3CN/water to 800
CH3CN/water in 20 minutes) was used to elute the peptide.
The major peak was collected and lyophilized. The
resulting peptide was oxidized by first dissolving the
peptide in to aqueous ammonium bicarbonate solution
containing 14% DMSO, at peptide concentration of 5 mg/ml.
After the mixture was held overnight at room temperature
the product was isolated and purified by reverse phase
HPLC. The yield was 15 mg. The purified peptide was
characterized using mass spectroscopy and AAA. FAB-MS
gave m/e - 3660 (MH+). AAA gave Asx 2.20 (2), Glx 3.43
(3), Ser 4.16 (4), Gly 3.38 (3), His 0.98 (1), Arg 1.19
(1), Thr, 4.96 (5), Pro 2.24 (2), Tyr 1.21 (1), Val 1.00
(1), Cys 0.86 (2), Leu 4.54 (5), Lys 2.22 (2).
Circular dichroism spectroscopy was used to
study the conformation of the calcitonin derivatives and
their Fe(II) complexes in aqueous buffer solution. The
apparent molecular weight of bipyridine modified
calcitonins was determined by gel-filtration and
sedimentation equilibrium experiments in the presence and
absence of Fe(II). The formation constants and
dissociation rates of the trimeric calcitonin-Fe(II)
complexes was determined by UV spectroscopy.
C. Preparation of Nicotinic acid-modified calcitonin
(ZP-5 or Nic-SCT)
Nicotinic acid (3.7 x 10-5 mol) was dissolved in
a few drops of DMF. DIC (3.7 x 10-~ mol) was then added.
SlIBSTlTITtE SHEET (RULE 26b
-- ~ 153071 20
The mixture was shaken at room temperature for
approximately 10 minutes, after which the deprotected SCT-
resin was added. The coupling reaction was followed using
the Kaiser Test. The suspension was shaken overnight.
The Nic-SCT was cleaved from the resin by mixing
reagent K (1.65 ml TFA, 0.112 ml 88% phenol, 0.098 ml H20,
0.1 ml thioanisole, 0.05 ml ethanedithiol) with the Nic-
SCT resin. The reaction mixture was shaken at room
temperature for 2.5 hours, and then the mixture was
filtered to remove the resin. The peptide was
precipitated in cold ether, and collected by
centrifugation. The precipitate was washed with ether
three times and dried in a dessicator for approximately
one hour. The dried peptide was dissolved in 0.5 ml H20
and then applied to a Sephadex# G-15 column. The column
was eluted with 5% acetic acid. The peptide was detected
by absorbance at 270 nm. Fractions containing the peptide
were pooled and lyophilized.
The Nic-SCT was analyzed by reversed phase HPLC
using a C4-analytical column (Vydac, Hesperia, CA). The
HPLC was monitored at 270 nm. The eluant was acetonitrile
and water containing 1% TFA. A linear gradient, from 20%
to 80% acetonitrile in 20 minutes, was used to elute the
Nic-SCT. The major peak (55% acetonitrile) was collected
and lyophilized. The total amount of peptide was
approximately 18 mg.
The Nic-SCT (18 mg) was oxidized with DMSO to
form an intramolecular disulfide bond. The disappearance
of the reduced from of Nic-SCT was followed by Ellman~s
reagent. The inital concentration was 2 mg/ml in buffer
solution (2% NH4HC03, pH 8.0) containing 10% DMSO. The
mixture was stirred overnight. The HPLC of the oxidized
Nic-SCT showed a single peak, and the retention time ( 59 %
acetonitrile) was longer than that of the reduced form
(55% acetonitrile). The oxidized Nic-SCT was found to be
Trademark
«'O 94/15962 ~ 1 5 3 ~ ~ ~ PCT/US93/12692
21
>95o pure based on HPLC. The total amount of pure peptide
was 17 mg.
D. Preparation of Pycolinyl-modified calcitonin (ZP 6 or
Pic-SCT)
Pic-SCT was synthesized by a procedure similar
to that described for Nic-SCT. Picolinic acid (3.7 x
10-5 mol) was coupled to the deprotected SCT-resin (100
mg) as described above. The Pic-SCT was deprotected and
cleaved from the resin using reagent K as described above.
The Pic-SCT was purified by HPLC using a linear gradient
from 20% to 80% acetonitrile in 20 minutes. The major
peak (60% acetonitrile) was collected and lyophilized.
The toal amount of peptide was approximately 20 mg. The
purified Pic-SCT was then oxidized as generally described
for Nic-SCT. The HPLC of the oxidized form showed a
single peak, and the retention time (61% acetonitrile) was
almost the same as that of the reduced form. The oxidized
Pic-SCT was found to be 90% pure based on HPLC (17 mg).
The Pic-SCT is further purified by additional HPLC runs.
E. Preparation of 2-Pvrazinoyl-modified calcitonin (ZP 7
or 2-Pyr-SCT)
2-Pyr-SCT was synthesized by a procedure similar
to that described for Nic-SCT. 2-Pyranizinecarboxylic
acid (3.7 x 10-5 mol) was coupled to the deprotected SCT-
resin (100 mg) as described above. The 2-Pyr-SCT was
deprotected and cleaved from the resin using reagent K as
described above. The 2-Pyr-SCT was purified by HPLC using
a linear gradient from 20% to 80o acetonitrile in 20
minutes. The major peak (59% acetonitrile) was collected
and lyophilized. The toal amount of peptide was
approximately 20 mg. The purified 2-Pyr-SCT was then
oxidized as generally described for Nic-SCT. The HPLC of
SUBSTITU'T'E SHEEP (RULE 261
__ 2153071 22
the oxidized form showed a single peak, and the retention
time (61% acetonitrile) was slightly longer than that of
the reduced form. The oxidized 2-Pyr-SCT was found to be
>95% pure based on HPLC (18 mg).
F. Preparation of Additional Modified Calcitonins
2,2'-bipyridine-5,5'-dicarboxylic acid (ZP-2 or
BP5,5-SCT), 4-4'-biphenyldicarboxylic acid (ZP-3 or BPhe-
SCT), isonipecotic acid (ZP-8 or isonip-SCT), DL-
pipecolinic acid (ZP-9 or pip-SCT) and nipecotic acid (ZP-
10 or nip-SCT) modified salmon calcitonins were
synthesized by procedures essentially the same as that
described for Nic-SCT. The compounds were purified by
HPLC as described above and assayed as described below.
Example 2
Characterization on BP4,4-CT: Ability to Bind Calcitonin
Receptor
The ability of the derivatized BP4~4-CT .molecule
to bind the calcitonin receptor was assayed by measuring
the ability of the molecule to increase levels of cyclic
AMP in mammalian cells expressing either a recombinant
human calcitonin receptor or an endogenous hamster
calcitonin receptor.
The human calcitonin receptor was prepared as
follows: A plasmid, designated pHOLLEX (deposited as an
E. coli strain XL-1 blue transformant with the American
Type Culture Collection (12301 Parklawn Dr., Rockville,
MD) on September 1, 1992 under accession number 69067),
containing a DNA construct capable of directing the
expression of a human calcitonin receptor was used to
transfect cell line BHK/KZ10-20-48 (as disclosed in
U.S. Patent No. 5,622,839).
BHK/KZ10-
20-48 is a BHK cell line transfected with a plasmid
E'
2153071 23
containing a luciferase gene whose expression is dependent
upon a cyclic AMP response element (CRE), which induces
expression of downstream coding sequences in the presence
of cyclic F,MP. Thus, stimulation of the cyclic AMP
pathway, for example by the binding of ligand to the
calcitonin receptor, results in expression of the
luciferase gene. The vector without the calcitonin
receptor was used to transfect the cell line BHK/KZ10-20-
48 for a negative control. The transfectants were grown
l0 in a selective growth medium (Dulbecco~s Modified Eagle
Medium (DMEM) with 5% serum) containing both 500 ug/ml
6418-neomycin and 250 nM methotrexate.
A BHK cell line expressing the endogenous
hamster calcitonin receptor was transfected with plasmid
KZ10-20-48 to provide the CRE-inducible luciferase
receptor described above. The transfected cells were
cultured in selective growth medium containing both 500
~Cg/mL 6418-neomycin and 250 nM methotrexate.
Cells expressing the calcitonin receptor were
assayed in triplicate for the induction of the CRE-linked
luciferase response to salmon calcitonin (SCT) and
derivatized calcitonins at concentrations ranging from
10-13 M to 10-6 M. MICROLITE#flat bottom tissue culture
plates (Baxter Scientific Products, Chicago, IL) were set
up such that each well contained 2 x 104 cells in 100 ul of
selection media, and the cells were grown overnight. SCT
and GF43 were prepared at 2X the final assay concentration
to give 10-13 to 10-6 M in growth medium. Induction was
initiated by removing old medium from the wells and adding
100 ul of fresh growth medium and 100 ~tl of either 2X
solution in triplicate sample wells. Uninduced luciferase
levels were determined in triplicate wells to which 100 ~tl
of DMEM containing 10% fetal calf serum was added. The
plates were incubated for four hours at 37°C, 5% C02 to
allow induction of luciferase. Following induction, the
media were removed, and the wells were washed once with
Trademark
~.~ ii
2153071 24
200 ~11/well PBS. After the wash, 20 ~tl of a 1:5 dilution
(in sterile water) of the stock Cell Culture Lysis Reagent
(Luciferase# Assay System, Promega~ Corp., Madison, WI) was
added to each well, and the plates were incubated for 15
minutes at room temperature. The plates were transferred
to a Labsystems Luminoskar~' microtiter luminometer
(Labsystems Inc., Morton Grove, IL) which added 40 ~1 of
Luciferase# Assay Substrate (Luciferase# Assay System,
Promega)# mixed the reaction for three seconds and
integrated the luciferase signal for two seconds per well.
The fold induction of luciferase for each compound was
calculated as follows:
Fold induction = Induced signal - Uninduced sictnal
Uninduced signal
Results of the assays are summarized in the
Table.
Table
Hamster HCT ZP- ZP- ZP- ZP- ~ZP- ZP- ZP- ZP- ZP-
CT 1 2 3 5 6 7 8 9 10
rece for
% SCT 5.9 73 125 341 71 98 155 106 147 152
EC50 x
103
Human CT HCT ZP- ZP- ZP- ZP- ZP- ZP- ZP- ZP- ZP-
rece for 1 2 3 5 6 7 8 9 10
% SCT 345 97 ~ 199 450 112 140 158 ~ ~ 243
146 230
EC50
These data show that derivatized calcitonins ZP-
1, ZP-2, ZP-5, ZP-6, ZP-7, ZP-8, ZP-9 and ZP-10 have an
EC50 (half maximal activity) similar to salmon calcitonin.
and have greater activity than human calcitonin. ZP-3 has
potency similar to human calcitonin.
Trademark
r__
215371 25
Example 3
In vivo Effects ef 3P-~T on serum ~alcium ~eveis
The biological activity of bipyridine-calcitonin
in vivc was measured as the ability of BP4~4-CT to lower
serum calcium in mice as compared to the action of salmon
calcitonin. Six-week old Swiss-Webster male mice (12 to
18 grams) were given single, subcutaneous injections of
BP4,4-CT, salmon calcitonin or vehicle. Blood samples for
serum calcium measurements were collected from orbital
sinus punctures at timed intervals of 0, 1 and 4 hours
after injection. Serum calcium was analyzed using a Dacos
Excel# Analyzer (Coulter Electronics Company, Hialeah,
Fla.). The compounds were given at the following doses
with eight mice randomly assigned to each of the following
groups:
Group No. Dose
1 vehicle 0.9% saline
2 BP-CT 100 ng/ml
3 BP-CT 1 ~g/ml
4 BP-CT 10 ~tg/ml
5 SCT 100 ng/ml
6 SCT 1 ~tg/ml
7 SCT 10 ~tg/ml
Figures 1, 2 and 3 demonstrate that equivalent
amounts of BP4~4-CT and salmon calcitonin equally
decreased serum calcium levels in a dose-dependent manner.
Although certain embodiments of the invention
have been described in detail for purposes of
illustration, it will be readily apparent to those skilled
in the art that the compositions and methods described
herein may be modified without departing from the spirit
and scope of the invention. Accordingly, the invention is
not limited except as by the appended claims.
# Trademark
-.~. WO 94/15962 215 3 0 l 1 PCT/US93/12692 ~~
26
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: ZymoGenetics, Inc.
4225 Roosevelt Way N.E.
Seattle
WA
US
98105
University of Washington
Seattle
WA
US
98105
(ii) TITLE OF INVENTION: DERIVATIZED CALCITONINS
(iii) NUMBER OF SEQUENCES: 2
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: ZymoGenetics, Inc.
(B) STREET: 4225 Roosevelt Way, N.E.
(C) CITY: Seattle
(D) STATE: WA
(E) COUNTRY: USA
(F) ZIP: 98105
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC comxatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.25
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE:
(C) CLASSIFICATION:
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: Sawislak, Deborah A.
(B) REGISTRATION NUMBER: 37,438
(C) REFERENCE/DOCKET NUMBER: 92-20PC
SUBSTINfE SHEET (RULE 26b
2153011
WO 94/15962 PCT/US93/12692
27
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: 206-547-8080 ext 427
(B) TELEFAX: 206-548-2329
(2) INFORMATION FOR SEQ ID N0:1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 32 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ix) FEATURE:
(A) NAME/KEY: Modified-site
(B) LOCATION: 32
(D) OTHER INFORMATION: /label= amidated
/note= "C-terminal proline residue is amidated"
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:1:
Cys Gly Asn Leu Ser Thr Cys Met Leu Gly Thr Tyr Thr Gln Asp Phe
1 5 10 15
Asn Lys Phe His Thr Phe Pro Gln Thr Ala Ile Gly Ual Gly Ala Pro
20 25 30
s
(2) INFORMATION FOR SEQ ID N0:2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 32 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ix) FEATURE:
(A) NAME/KEY: Modified-site
(B) LOCATION: 32
(D) OTHER INFORMATION: /label= amidated
/note= "C-terminal Proline residue is amidated."
SU8ST~1T~E SHEET (RULE 261
215 3 0 71 PCT/US93/12692
27/1
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:2:
Cys Ser Asn Leu Ser Thr Cys Val Leu Gly Lys Leu Ser Gln Glu Leu
1 5 10 15
His Lys Leu Gln Thr Tyr Pro Arg Thr Asn Thr Gly Ser Gly Thr Pro
20 25 30
SIJBSTfTIT~E SHEET (RULE 26b
r. _ _..,~-_~__..._.... .. _._.~_ ~.~.~.. ..r ____ . I