Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 94/21608 21573 8 6 PCT/US94/02800
~
-1-
INDOLETETRALINS HAVING DOPAMINERGIC ACTIVITY
BACKGROUND OF THE INVENTION
This invention relates to novel 6,7,8,9-tetrahydro-3H-benz(e)indolamine
derivatives, to therapeutically acceptable acid addition salts thereof, to
processes for
their preparation and intermediates used therein, to methods of using the
derivatives and to pharmaceutical compositions of the derivatives. These
derivatives
exhibit dopamine-receptor stimulating activity in a mammal. Thus, they can be
useful for treating hyperprolactinemia, galactorrhea, amenorrhea, impotence,
schizophrenia, Parkinsonism, diabetes, acromegaly, hypertension and other
central
nervous system disorders which respond to dopamine-receptor stimulation.
A number of 6,7,8,9-tetrahydro-3H-benz(e)indolamine derivatives are known
and described, for eample, L.B. Shagalov et al., Chem. Abstr. 91, 56747 v
(1979) for
Khim. Geterotsikl. Soedin., (3), 360 (1979); L.B. Shagalov et al., Chem.
Abstr. 89,
146703 r (1978) for Khim. Geterotsikl. Soedin., (5), 634 (1978); Derwent
Publications
Ltd., Farmdoc 46000U for Netherlands Patent 7,300,871, published July 30,
1973;
and Derwent Publications Ltd., Farmdoc 24087B for German Offenlegungsschrift
2,740,836, published March 22, 1979. The reported compounds lack the
particular
substituents on the indole ring system which are characteristic of the
compounds of
this invention as well as the various placements of the indole ring structure.
N.J.
Bach and E.C. Kornfeld, U.S. Patent 4,110,339, August 29, 1978, disclose
tricyclic
tetrahydro-2H-benzo(c)pyrroles which are dopamine agonist. These latter
compounds are distinguished most readily from the compounds of this invention
by
having a perfused tricyclic ring system.
European Patent 0055043-B 1 discloses 6,7,8,9-tetrahydro-3H-benz(e) indole
compounds (see, also U.S. Patents 4,510,157 and 4,470,990); however, they lack
the
indole and/or tertiary amine substituents of this invention. Alternate
placement of
the indole ring is not disclosed by this reference.
SUMMARY OF THE INVENTION
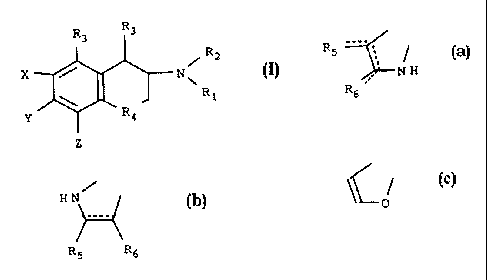
In one aspect the subject invention is directed toward compounds of Formula
I and pharmaceutically acceptable salts thereof:
R3 Rs
R2
X N~
Ri
Y Ra
Z
WO 94/21608 2157~ 86 PCT/US94/02800
-2-
where Z is Rg and X and Y form (a), or X is and Y and Z form (a), (b) or (c)
a)
R5
~NH
R6
b) HN/ w
~
R5 Re
c)
C o
Rl and R2 are independently hydrogen, Cl.g alkyl, Cs.7 cycloalkyl, -CI~-Cs.7
cycloalkyl,
phenyl (optionally substituted with halogen or Cs.g alkyl), -thiophenyl
(optionally
substituted with halogen or Cl.e alkyl), or Cl.g alkyl phenyl;
R. are independently hydrogen, halogen, -0-C1.g alkyl or C1.6 alkyl;
R4 is a valence bond, CH2 or oxygen;
R. and R~ are independently hydrogen, sulfur, -S-Cl., alkyl, halogen,
CON(R3)2,
-COCF3 ,-CO-CI.g alkyl, -CO phenyl, oxygen, -CHO, CN except that when Y and Z
form (b), Rl and R2 are hydrogen or a C1.g alkyl and R3 is hydrogen, then at
least one
of R. and R6 must be other than hydrogen or oxygen. These compounds and
derivatives thereof predominantly exhibit dopamine-receptor stimulating
activity in
mammals.
DETAILED DESCRIPTION OF THE INVENTION
The compounds of this invention are represented by Formula I or a
pharmaceutically acceptable salt thereof as shown above. The compounds provide
a
method for treating mammals, especially humans, suffering from dopamine
generated central nervous system disorders by administering a therapeutically
effective amount of Formula I.
In the structural Formula I, the carbon content of various hydrocarbon-
moieties is indicated by a prefix designating the minimum and maximum
containing
number of carbon atoms in the moiety, i.e., the prefix C'rCj defines the
number of
carbon atoms present from the integer "i" to "j" inclusive. Thus, C'i-Ce alkyl
refers to
straight and branched alkyls of one to six carbon atoms, inclusive, including
isomers
WO 94/21608 215~ ~ 8 6 PCT/US94/02800
~ - : .' . .. ,_ ..
-3-
thereof such as methyl, propyl, ethyl and isopropyl.
Cycloalkyl are three to seven carbon atoms such as cyclopropyl, cyclobutyl,
cyclopentyl and cyclohexyl.
The term "halogen" includes fluoro, chloro, bromo and iodo.
Pharmaceutically acceptable salts means salts useful for administering the
compounds of this invention or useful forms the compounds may take in vitro
and in
vivo and include potassium, sodium, hydrochloride, hydrobromide, hydroiodide,
sulfate, phosphate, acetate, propionate, lactate, mesylate, maleate, malonate,
succinate, tartrate, citrate and the like. These salts may be in hydrated
form.
A pharmaceutical composition is provided by admixing the compound of
Formula I, or or a therapeutically acceptable acid addition salt thereof, with
a
pharmaceutically acceptable carrier.
The exact dosage and frequency of administration depends on the particular
condition being treated, the severity of the condition being treated, the age,
weight,
general physical condition of the particular patient, other medication the
individual
may be taking as is well known to those skilled in the art.
Thus, the subject compounds, along with a pharmaceutically-acceptable
carrier, diluent or buffer, can be administrated in a therapeutic or
pharmacological
amount effective to alleviate the central nervous system disorder with respect
to the
physiological condition diagnosed. The compounds can be administered
intravenously, intramuscularly, topically, transdermally such as by skin
patches,
buccally or orally to man or other vertebrates.
The compositions of the present invention can be presented for
administration to humans and other vertebrates in unit dosage forms, such as
tablets, capsules, pills, powders, granules, sterile parenteral solutions or
suspensions, oral solutions or suspensions, oil in water and water in oil
emulsions
containing suitable quantities of the compound, suppositories and in fluid
suspensions or solutions.
For oral administration, either solid or fluid unit dosage forms can be
prepared. For preparing solid compositions such as tablets, the compound can
be
mixed with conventional ingredients such as talc, magnesium stearate,
dicalcium
phosphate, magnesium aluminum silicate, calcium sulfate, starch, lactose,
acacia,
methylcellulose, and functionally similar materials as pharmaceutical diluents
or
carriers. Capsules are prepared by mixing the compound with an inert
pharmaceutical diluent and filling the mixture into a hard gelatin capsule of
appropriate size. Soft gelatin capsules are prepared by machine encapsulation
of a
WO 94/21608 2157586 PCT/US94/02800
" -4-
slurry of the compound with an acceptable vegetable oil, light liquid
petrolatum or
other insert oil.
Fluid unit dosage forms for oral administration such as syrups, elixirs, and
suspensions can be prepared. The forms can be dissolved in an aqueous vehicle
together with sugar, aromatic flavoring agents and preservatives to form a
syrup.
Suspensions can be prepared with an aqueous vehicle with the aid of a
suspending
agent such as acacia, tragacanth, methylcellulose and the like.
For parenteral administration, fluid unit dosage forms can be prepared
utilizing the compound and a sterile vehicle. In preparing solutions the
compound
can be dissolved in water for injection and filter sterilized before filling
into a
suitable vial or ampoule and sealing. Adjuvants such as a local anesthetic,
preservative and buffering agents can be dissolved in the vehicle. The
composition
can be frozen after filling into a vial and the water removed under vacuum.
The
lyophilized powder can then be sealed in the vial and reconstituted prior to
use.
The compounds of this invention are used to stimulate dopamine receptors in
a mammal in need thereof by administering to the mammal an effective dopamine
receptor stimulating amount of a compound of Formula I or a therapeutically
acceptable acid addition salt thereof. The compounds of this invention are
favorably
used in combination with an effective amount of an agent commonly used in the
treatment of Parkinsonism and related disorders, particularly those selected
from
bromocriptine, lergotrile, levodopa, combination of levodopa and carbidopa, L-
prolyl-
L-leucylglycinamide and L-propyl-N-methyl-D-leucylglycinamide.
The compounds of this invention may be obtained by one of the following
methods described below and outlined in the appropriate charts. For clarity,
the
numerical charts (1-3) describe the last steps leading to the exemplified
final
products while the alphanumerical charts describe routes to desired starting
material to be used in the final steps.
EXAMPLE 1:
7-Di-n-propylamino-6,7,8,9-tetrahydro-lH-benzo[g]indole-2,3-dione (2a, Chart
1)
(R1R2 n-propyl; Rs hydrogen; R4 CH2; YZ (a); R.R. 0).
A solution of 5-amino-2-di-n-propylamino-1,2,3,4-tetrahydronaphthylenee
dihydrochloride (la, Chart 1, preparation see, Stjernlof et al., Eur. J. Med.
Chem.
1993) (35 g, 0.11 mol), chloral hydrate (19.4, 0.12 mol), hydroxylamine
hydro-chloride (23.6, 0.34 mol) and sodium sulfate (119.4 g, 0.84 mol) in 445
mL
water was refluxed for 1 hour under an inert atmosphere. After cooling,
diluted
WO 94/21608 PCT/US94/02800
~- 2157586
-5-
ammonia was added to basify. The aqueous solution was extracted three times
(ethyl acetate). The combined organic extracts were dried (sodium sulfate),
filtered
and evaporated to yield 30 g of the intermediate oxime, which was refrigerated
and
dissolved in freezer cold 90% sulfuric acid (510 mL). The resulting solution
was
stirred at ambient temperature in an inert atmosphere for 0.5 hour and was
then
heated at 80 C for 0.5 hour. The solution was allowed to reach ambient
temperature
within an hour and was then poured on crushed ice. Ethyl acetate and diluted
ammonia (until pH8-9) was added and the mixture was shaken. The aqueous phase
was extracted 4 times (ethyl acetate) and the combined organic phases were
dried
(magnesium sulfate), filtered and evaporated to yield 23.5 g. (71%) of the
reddish oil,
which was sufficiently pure for further synthesis. For analytical and
biological
purposes, smaller amounts of the material was chromatographed on silica
(acetone/methanol, 20:1) to afford an orange solid. m.p. 173-177 C
EXAMPLE 2: Di-n-propyl-(6,7,8,9-tetrahydro-lH-benzo[g]indol-7y1)amine (3a,
Chart 1) (R1R2 n-propyl; R. hydrogen; R4 CH2; YZ (a); R6Rg H).
A solution of
7-di-n-propylamino-6,7,8,9-tetrahydro-lH-benzo[g]indole-2,3-dione, 2a, (Chart
1)
(22.0 g, 73.2 mmol) in dry diethyl ether (100 mL) was added dropwise to a
suspension of lithiumaluminum hydride (9.0 g, 237 mmol) in dry diethyl ether
(600
mL). After stirring overnight at ambient temperature, water (9.0 mL), 15%
sodium
hydroxide (9 mL) and water (27 mL) were consecutively added. The mixture was
stirred for 20 minutes followed by filtration of inorganic material. The
solution was
dried (sodium sulfate), filtered and evaporated to yield 17 g of a blue oil,
which was
purified on silica (acetone/methanol, 20:1) to yield 7.4 g (37%) of the pure
material.
For analytical and biological purposes, the fumarate salt (1/2 fum) was
prepared
which was recrystallized from ethanol/diethyl ether. m.p. 201-205 C.
EXAMPLE 3:
7-Di-n-propylamino-6,7,8,9-tetrahydro-lH-benzo[g]indole-3-carbaldehyde (4a,
Chart
1) (Rl n-propyl; R2 butyl; R. hydrogen; R4 CH2; YZ (a); R. CHO; RB H).
A solution of di-n-propyl-(6,7,8,9-tetrahydro-lH-benzo[g]indol-7yl)amine, (3a,
Chart 1)(0.90 g, 3.33 mmol) in dimethyl formamide (10 mL) was added dropwise
to
an ice-cooled solution of phosphorous oxychloride (1.2 ml, 0.73 g, 4.8 mmol)
under an
inert atmosphere. The solution was stirred for 10 minutes and was then heated
to
50 C. After stirring for 3 hours, the solution was allowed to reach ambient
WO 94/21608 2157586 -6- PCT/US94/02800
temperature, stirred overnight and was then poured on ice. After basification,
the
mixture was heated to 80 C for 10 minutes and then cooled. Extraction three
times
(dichloromethane), followed by drying (magnesium sulfate), filtering and
evaporation
several times with 99% ethanol, yielded 0.82 g (82%) of the material. This
material
was further purified on a silica column (dichloromethane/methanol, 3:1) to
yield 0.53
g (53%) of the pure material as an oil.
EXAMPLE 4: 7-Di-n-propylamino-1,3,6,7,8,9-hexahydro-1H-benzo[g]indole-2-one
(5a, Chart 1) (R1R2 n-propyl; R3 hydrogen; R4 CH2; YZ (a); R. H; R6 0).
A solution of pyridiniumperbromide hydrobromide (560 mg, 1.83 mmol) in
acetic acid (100 mL) was added to a solution of
di-n-propyl-(6,7,8,9-tetrahydro-lH-benzo[g]indol- 7-yl) amine, 3a (Chart 1)
(400 mg,
1.48 mmol) in aqueous (90%) acetic acid while cooling on ice. The temperature
was
then slowly raised to 80 C. The progress of the reaction was followed by GC.
When
the reaction was complete (3 hours), the solution was cooled and evaporated to
an
aqueous residue, which was basified (10% sodium carbonate). This suspension
was
extracted 3 times (ethyl acetate). The combination of the organic phases was
washed
with water, dried (magnesium sulfate), filtered and evaporated to yield a
residue of
390 mg (89%). This material was subjected to a silica column and eluated
(methanol) to afford 170 mg of the pure material, which prior to biological
testing
was treated with 1/2 eq of fumaric acid and recrystallized from methanol. m.p.
136-139 C.
EXAMPLE 5:
Di-n-propyl-(1,6,7,8-tetrahydro-9-oxa-l-azacyclopenta[a]naphthylenee-7-
yl)amine (3b,
Chart 1) (R1R2 n-propyl; R. hydrogen; R4 0; YZ (a); R6R6 H).
Step A. N-(8-Aminochroman-3-yl)-N-propylpropionamide (18a, Chart A).
To an ice-cooled solution of (chroman-3-yl)-N-propylpropionamide 5.5 g (22.3
mmol) in nitromethane (150 mL) was added a mixture of "nitrating acid" (6.2
vol %
nitric acid, 80.6 vol % sulfuric acid, 13.2 vol % water) (13 mL). The solution
was
stirred for 2 hours at 0 C. Additional 4 ml of "nitrating acid" with the same
=
composition as above was added, and the reaction mixture was stirred
overnight.
The reaction mixture was poured on ice and the product was extracted with
dichloromethane. The organic layer was washed with water several times. The
organic layer was separated, dried (magnesium sulfate) and the solvent was
WO 94/21608 2157,386 PCT/US94/02800
~ -..
-7-
evaporated, yielding 4.55 g(70%) of a mixture of
6-nitro-3-(N-Propionyl-N-n-propylamino)chroman (17b, Chart A) and
8-nitro-3-(N-Propionyl-N-n-propylamino)-chroman (17a, Chart A). The crude
mixture
was dissolved in abs. ethanol (300 mL) and hydrogenated in a Parr apparatus
with
Pd/C. for 2.5 hours. The mixture was then filtered and the solvent evaporated,
yielding a mixture of N-(6-amino- and
N-(8-amino-chroman-3-yl)-N-propylpropionamide (18b and 18a respectively). The
regioisomers was separated on a silica column first eluted with ether and then
dichloromethane/methanol (19:1) yielding 1.1 g of 18a (17%).
Step B: N,N-Dipropyl-chroman-3,8-diamine (1b, Chart A and 1).
To a solution of N-(8-amino-chroman-3-yl)-N-propylpropionamide, 18a (Chart
A) (1.1 g, 3.77 mmol) in anhydrous tetrahydrofuran was added lithiumaluminum
chloride (600 mg, 15.8 mmol). The mixture was stirred for 1.5 hours at room
temperature. Excess hydride was quenched by addition of water (0.6 mL), 15%
sodium hydroxide (0.6 mL) and water (1.2 mL). The mixture was filtered and the
solvent evaporated yielding 911 mg (98%) of 2 as an oil. The free base was
converted
to the dihydrochloride with a saturated hydrochloric acid solution in ethanol
yielding 1.2 g of the desired compound as the di-HC1-salt.
Step C: Di-n-propylamino-1,6,7,8-tetrahydro-9-oxa-l-azacyclopenta[a]
naphthylenee-2,3-dione (2b, Chart 1).
This compound was prepared in an analogous manner as for compound 2a
from N,N-dipropyl-chroman-3,8-diamine dihydrochloride, lb (Chart A and 1)(1.2
g,
3.74 mmol) in deionized water (24 mL) treating with chloral hydrate (677 mg,
4.10
mmol), hydroxylamine hydrochloride (920 mg, 13.23 mmol) and anhydrous sodium
sulphate (4.4 g). After usual work-up and extraction (dichloromethane), the
organic
layer was dried and evaporated yielding 1.04 g (3.26 mmol) of the
corresponding
oxime which was treated as described for compound 2a with ice-cooled 90%
sulfuric
acid (165 mL). After usual work-up, 380 mg (34%) of the desired compound was
afforded as a red oil. The crude product was used in the next step without
purification.
Step D: Di-n-propyl-(1,6,7,8-tetrahydro-9-oxa-l-azacyclopenta[a]
naphthylenee-7-yl)amine (3, Chart 1).
To a solution of di-n-propylamino-1,6,7,8-tetrahydro-9-oxa-l-azacyclopenta[a]
WO 94/21608 ~-2157586 PCT/US94/02800
-8-
naphthylenee-2,3-dione, 2b (Chart 1) (380 mg, 1.26 mmol) in dry ether (50 mL)
was
added lithiumaluminum hydride (500 mg, 13.1 mmol) in portions. The mixture was
stirred for 3 hours at ambient temperature. The reaction was quenched by
addition
of water (0.5 mL), 15% sodium hydroxide (0.5mL) and water (1.5 mL). The
mixture
was filtered and the solvent evaporated yielding 197 mg of crude 5 as an oil.
The
product was chromatographed on silica with dichloromethane/methanol (19:1) as
eluant yielding 45 mg (14%) of the desired compound. The amine was converted
into
its neutral fumarate with 10 mg of fumaric acid dissolved in 99% ethanol,
yielding
22 mg (0.5 fumarate).
m.p. 163-165 C (0.5 fumarate).
EXAMPLE 6: 6-Di-n-propylamino-5,6,7,8-tetrahydro-lH-benzo[fJ indole-2,3-dione
(8, Chart 2) (R1R2 n-propyl; Rs hydrogen; R4 CH2; XY indole; R.R. 0).
This compound was prepared from 7-amino-2-di-n-propylamino-1,2,3,4-
tetrahydronaphthylenee, (6, Chart 2) in a similar manner as for compound 2a
(Chart
1). This intermediate compound was prepared according to Stjernlof et al.
(Eur. J.
Med. Chem. 1993 Accepted for publication).
EXAMPLE 7: Di-n-propyl-(5,6,7,8-tetrahydro-lH-benzo[fJindol-6-yl)amine
(lla, Chart 2) (R1R2 n-propyl; R. hydrogen; R4 CH2; XY indole; RaR6 H).
Step A: Di-n-propyl-(6,7,8,9-tetrahydro-3H-benzo[elindol-7-yl)amine (9,
Chart 2) (R1Ra n-propyl; R. hydrogen; R4 CH2; YZ (b); R6R8 H).
This compound (disclosed in EPA 0055043) was synthesized in a similar
manner as compound 3a (Chart 1) from the mixture above (7a and 8a, Chart 2)
yielding an isomeric mixture of 9a and lla (Chart 2). This compound is shown
here
to show the preparation of Example 9, below. Purification on silica
(dichloromethane/methanol, 19:1) afforded the pure isomers. m.p. 112-114 C.
Step B: The title compound was obtained as a second isomer from
foregoing step.
EXAMPLE 8: 7-Di-n-propylamino-6,7,8,9-tetrahydro-3H-benzo
[e)indole-l-carbaldehyde (10a, Chart 2) (R1R2 n-propyl; R3 hydrogen; R4 CH2;
YZ (b);
R6 H; Rs CHO).
This material was prepared in the same manner as described for compound
WO 94/21608 _ 2 15 7 5-8 " PCT/US94/02800
~ . ;
-9-
4a (Chart 1) from di-n-propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)amine,
9a
(Chart 2).
EXAMPLE 9: 6-Di-n-propylamino-5,6,7,8-tetrahydro-lH-benzo
5[fJindole-3-carbaldehyde (12a, Chart 2) (R1R2 n-propyl; R3 hydrogen; R4 CH2;
XY
indole; R6 CHO; R6 H).
This material was prepared in the same manner as described for compound
4a (Chart 1) from 7-di-n-propylamino-6,7,8,9-
tetrahydro-3H-benzo[e]indole-l-carbaldehyde, 10a, (Chart 2)
EXAMPLE 10:
7-Di-n-propylamino-6,7,8,9-tetrahydro-3H-benzo[e]indole-l-carbonitrile (lOb,
Chart
2) (R1R2 n-propyl; R. hydrogen; R4 CH2; XY (b); R5 H; R6 CN).
To an ice-cold solution of
di-n-propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)amine, 9a (Chart 2) (180
mg,
0.67 mmol) in acetonitrile (5 mL) was added dropwise with a solution of
chlorosufonyl isocyanate (60 mL, 0.68 mmol) in acetonitrile (1 mL) over 10
minutes.
After stirring for 1 hour, dimethyl formamide (100 mL) was added and the
reaction
was stirred for an additional 2 hours. Water was added and extraction
(dichloromethane) followed by evaporation of the combined organic extracts
afforded
170 mg (86%) of the crude product. Purification on a silica column
(hexane/ethyl
acetate/methanol) afforded 45 mg (23%) of the pure material.
EXAMPLE 11:
7-[(1R-Phenyl-ethyl)-propyl-amino]-6,7,8,9-tetrahydro-3H-benzo[e]indole- 1,2-
dione
(7b:2, Chart 2) (Rl n-propyl; R2 ethyl phenyl; R. hydrogen; R4 CH2; YZ (b);
R6R6 0)
and 6-[(1R-phenyl-ethyl)-propyl-amino]-5,6,7,8-1H-benzo[flindole-2,3-dione
(8b:2,
Chart 2) (Rl n-propyl; R2 ethyl phenyl; Rs hydrogen; R4 CH2; XY indole; RbR6
0).
Step A: [4-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-phenyl]-acetic acid (19,
Chart
B).
Phthalic acid anhydride (15.1 g, 102 mmol) and 4-amino phenyl acetic acid
(15.2 g, 102 mmol) were dissolved in acetic acid and heated at reflux for 1
hour.
Upon cooling on an ice bath, the product crystallized. The solid was filtered
off,
washed with water, and dried under vacuum overnight. The yield of the title
compound was 24.1 g (86%) as an off white solid.
WO 94/21608 215758 6 PCT/US94/02800
-10-
1H NMR (300 MHz, acetone-ds) d 3.75 (s, 2H), 7.52 (sb, 4H), 7.95 (m, 4H).
Step B: [4-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-phenyl]-acetylchloride (20,
Chart B).
A slurry of [4-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-phenyl]-acetic acid
(19,Chart B) (24.0 g, 85.4 mmol) in dichloromethane (300 ml) was treated with
thionyl chloride (12.4 ml, 170 mmol). After heating at reflux temperature for
3.5
hours, the solvent and excess reagent were removed by evaporation to yield
sufficiently pure product (25.4 g, 99%).
'H NMR (300 MHz, CDC13) d 4.22 (s, 2H), 7.48 (m, 4H), 7.82 (m, 2H), 7.98
(m,2H).
Step C: 2-(6-Oxo-5,6,7,8-tetrahydro-naphthylenee-2-yl)-isoindole-1,3- dione
(21, Chart B).
A slurry of [4-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-phenyl]-acetyl chloride
(20,
Chart B) 25.4 g (85.4 mmol) in dichloromethane (380 ml) was cooled on an ice
bath.
To the stirred slurry was added aluminum chloride 23.7 g (178 mmol) and ethene
gas was bubbled through the mixture for 5 hours. Then the contents of the
reaction
flask were poured into a mixture of water (100 ml), crushed ice (100 g) and
solid
sodium carbonate (20 g). Acetic acid (50 ml) was added and the dichloromethane
phase separated. The aqueous layer was extracted with the same solvent. The
combined extracts were washed with water, saturated sodium hydrogen carbonate
and brine. After drying over magnesium sulfate, the solvent was removed
yielding
23.7 g(95%) of an off white solid. The purity was > 97% (GC).
'H NMR (300 MHz, CDCIs) d 2.57 (t, 2H), 3.12 (t, 2H), 3.62 (s, 2H), 7.30 (m,
3H), 7.80 (m, 2H), 7.95 (m, 2H); 13C NMR (75.4 MHz, CDC1s) d 28.4, 37.8, 44.8,
123.9, 125.3, 125.9, 129.0, 130.2, 131.7, 133.6, 134.5, 137.7, 167.4, 209.8;
MS (EI)
m/e 291 (M+, 100), 249 (91), 115 (68), 76 (61), 104 (37), 117 (33), 263 (28),
77 (20).
Step D: 2-[6-(1R-Phenyl-ethylamino)-5,6,7,8-tetrahydro-naphthylenee-
2-yl]-isoindole-1,3-dione (22, Chart B).
A stirred solution of 2-(6-oxo-5,6,7,8-tetrahydro-naphthylenee-2-yl)-
isoindole-1,3-dione, 21 (Chart B) (19.8 g, 68 mmol) and R -(+)-a-methyl benzyl
amine
(8.50 g, 70 mmol) in 1,2-dichloroethane (350 ml) was treated with acetic acid
(5 ml)
and portionwise additions of sodium triacetoxyborohydride (16.0 g, 75 mmol).
After
stirring at ambient temperature for 6 hours. the reaction mixture was quenched
WO 94/21608 215 7 5 86 PCTIUS94/02800
~ ' - ..
-11-
with saturated sodium carbonate. The organic layer was separated and the
aqueous
layer extracted with 1,2-dichloroethane and diethyl ether. The combined
organic
extracts were dried over magnesium sulfate and the solvents removed. To the
residue was added ethanol (200 ml) followed by HCl saturated ethanol. The
precipitate was filtered and washed with ethanol and diethyl ether. Drying
under
vacuum yielded 20.0 g of the HCl salt. An additional 1.8 g was recovered from
the
mother liquor, giving a total of 21.8 g. (The diasteromer at compound 22
(Chart B)
leads to the synthesis of compound 9a:1 and 9a:2, Chart 2.)
(74%) as a mixture of diastereomers. The GC/MS data below refers to
analysis of the diastereomeric mixture of the free bases. MS (EI) of the less
retained
diastereomer:
m/e 396 (M+, 4), 105 (100), 288 (31), 79 (24), 77 (23), 130 (22), 392 (22),
277
(18) of the more retained diastereomer: 396 (M+, 2), 105 (100), 288 (32), 392
(22), 77
(22), 76 (20), 104 (20), 130 (19).
Step E: N-[6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-1,2,3,4-tetrahydro-
naphthylenee-2-yl]-N-(IR-phenyl-ethyl)-propionamide (23:1 and 23:2, Chart B).
To a stirred slurry of 2-[6-(]R-phenyl-ethylamino)-5,6,7,8-tetrahydro-
naphthylenee-2-yl]- isoindole-1,3-dione hydrochloride, 22 (Chart B) (17.3 g,
40
mmol) in dichloromethane (250 ml) was added triethyl amine (11.7 ml, 84 mmol)
and propionyl chloride (4.90 ml, 56 mmol). The resulting mixture was stirred
at
ambient temperature for 1 hour and then quenched by addition of 10% sodium
carbonate (75 ml) followed by stirring for 0.5 hour. The dichloromethane layer
was
then separated, washed with 10% sodium dihydrogen phosphate, water and then
dried over magnesium sulfate. Removal of the solvent yielded 17.1 g (95%) of
the
product as a yellowish foam. The diastereomeric mixture was then separated
using a
preparative HPLC column (straight phase Si02), eluting with ethyl
acetate/hexane
30/70. The yield of the first eluted isomer (22:1) was 7.28 g and of the later
eluted
(22:2) 7.85 g, both as yellowish foams. The isomeric purities of these
materials as
determined on an analytical HPLC column were 99% and 98% respectively.
(Diastereomer 23:11eads to the synthesis of compound 9a:1, Chart 2, and
23:21eads
to the synthesis of compound 9a:2, Chart 2.)
(23:1):
1sC NMR (75.4 MHz, CDC13) d 9.6, 17.7, 26.7, 28.3, 29.7, 33.0, 52.7, 54.8,
123.6, 123.8, 126.5, 127.0, 127.6, 128.4, 128.9, 129.5, 131.7, 134.2, 137.0,
137.3,
140.1, 167.3, 173.3.
CA 02157586 2004-07-12
-12-
(23:2):
13C NMR (75.4 MHz, CDC13) d 9.6, 17.8, 27.5, 28.5, 29.7, 31.8, 52.6, 54.6,
123.6, 123.8, 126.4, 126.9, 127.6, 128.5, 128.8, 129.5, 131.7, 134.2, 137.1,
140.1,
167.3, 173.4.
Stqp F: N-(6-Amino-1,2,3,4-tetrahydro-naphthylenee-2-yl)-N-(1 R-phenyl-
ethyl)-propionamide (24:2, Chart B).
A solution of N-[6-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)- 1,2,3,4-tetrahydro-
naphthylenee- 2-yl]-N-(IR-phenyl-ethyl)-propionamide, 23:2 (Chart B) (7.0 g,
15.5 mmol) was dissolved in 300 ml ethanol. This solution was treated with
hydrazine
hydrate (0.85 ml, 17 mmol) and heated to 50 C for 1 hour. The solvent and
excess
reagent were then removed by evaporation under vacuum. The residue
was taken up in 1% hydrochloric acid (200 ml). This solution was filtered
through a
celiteTM pad, basified (sodium carbonate) and extracted with dichloromethane.
Additional material was recovered from the celite by extraction with
dichloromethane.
The latter extract was washed with a sodium carbonate solution and combined
with the
former. Drying over magnesium sulfate and removal of the solvent yielded 5.39
g
(106%) as an brownish oil which was > 96% pure by GC. (Diastereomer 24:2 leads
to
the synthesis of compound 9a:2, Chart 2.)
13C NMR (75.4 MHz, CDC13) d 9.7, 18.1, 27.9, 28.6, 29.9, 31.1, 53.0, 54.1,
113.2, 114.7, 126.8, 127.4, 128.1, 128.4, 129.6, 136.7, 140.4, 143.7, 173.6;
MS (El)
m/e 322 (M+, 0.2), 145 (100), 144 (24), 146 (13), 105 (10), 130 (8), 119 (6),
77 (5).
St~G: N-(6-Amino-1,2,3,4-tetrahydro- naphthylenee-2-yl)-N-(1 R-phenyl-
ethyl)-propionamide (24:1, Chart B).
This compound was prepared from 23:1 as described for 24:2 above.
(Diastereomer 24:1 leads to the synthesis of compound 9a: 1, Chart 2.)
13C NMR (75.4 MHz, CDC13) d 9.7, 18.1, 27.2, 28.6, 29.9, 32.4, 53.4, 54.7,
113.2, 114.8, 126.6, 127.1, 127.5, 128.4, 129.7, 137.2, 140.6, 144.1, 173.4;
MS (EI)
m/e 322 (M+, 0.1), 145 (100), 144 (25), 146 (13), 130 (9), 105 (8), 119 (5),
77 (3).
Step H: N-2-(1R-Phenyl-ethyl)-N-2-propyl-1,2,3,4-tetrahydro-naphthylenee-2,6-
diamine (6b:2, Chart B and 2).
To a stirred slurry of lithium aluminum hydride (2.5 g, 66 mmol) in dry
tetrahydrofuran (THF) (300 ml) was added dropwise a solution ofN-(6-amino-
1,2,3,4-
tetrahydro- naphthylenee-2-yl)-N-(1 R-phenyl-ethyl)-propionamide,
2t 575 86
~WD 94/21608 - PCT/US94/02800
24:2 (Chart B) (5.0 g, 15.5 mmol) in THF (100 ml). The mixture was stirred
overnight at ambient temperature and then quenched by the consecutive addition
of
water (2.5 ml), 15% sodium hydroxide (2.5 ml) and water (15 ml). The solution
was
filtered through a celite pad and the solvent removed, yielding 4.96 (104%) of
a
brownish oil, which was used in the following step without further
purification.
{ 'H NMR (300 MHz, CDCIs) d 0.85 (t, 3H), 1.40 (d, 3H), 1.45 (m, 2H), 1.68 (m,
1H), 1.95 (m, 1H), 2.50-2.85 (m, 6H), 2.95 (m, 1H), 3.22 (sb, 2H), 4.08 (q,
1H), 6.38
(d, 1H), 6.45 (dd, 1H), 6.81 (d, 1H), 7.15-7.50 (m, 5H); 13C NMR (75.4 MHz,
CDC1g) d
11.9, 17.9, 24.0, 27.8, 30.2, 33.0, 47.8, 54.5, 57.5, 113.2, 114.8, 126.3,
127.2, 127.6,
128.0, 130.1, 137.3, 143.9, 145.7; MS (EI) m/e 308 (M+, 21), 146 (100), 105
(77), 119
(44), 145 (39), 188 (30), 175 (29), 144 (29), 203 (25), 130 (22).
Step I: N-2-(1R-Phenyl-ethyl)-N-2-propyl-1,2,3,4-tetrahydro-
naphthylenee-2,6-diamine (6b:1, Chart B and 2).
This compound was prepared from 23:1 as described for 6b:2 above.
'H NMR (300 MHz, CDCls) d 0.80 (t, 3H), 1.40 (d, 3H), 1.50 (m, 3H), 1.68 (m,
1H), 2.40-2.75 (m, 6H), 2.95 (m, 1H), 3.33 (sb, 2H,), 3.98 (q, 1H), 6.32 (s,
1H), 6.42
(d, 1H), 6.83 (d, 1H), 7.15 (m, 1H), 7.25 (m, 2H), 7.37 (d, 2H); 13C NMR (75.4
MHz,
CDC13) d 11.8, 19.4, 24.2, 26.9, 30.1, 33.4, 48.2, 54.7, 58.3, 113.2, 112.8,
126.4, 127.0,
127.5, 128.0, 130.1, 137.3, 144.0, 145.9; MS (EI) m/e 308 (M+, 30), 146 (100),
105
(81), 119 (55), 145 (44), 175 (39), 188 (36), 203 (35), 144 (30), 130 (26).
Step J: 7-[(1R-Phenyl-ethyl)-propyl-amino]-6,7,8,9-tetrahydro-3H-
benzo[e]indole-1,2-dione (7b:2, Chart 2) and
6-[(1R-phenyl-ethyl)-propyl-amino]-5,6,7,8-1H-benzo[fJindole-2,3-dione (8b:2,
Chart
2).
A mixture of N-2-(1R-phenyl-ethyl)-N-2-propyl-1,2,3,4-
tetrahydro-naphthylenee- 2,6-diamine as its dihydrochloride, 6b:2 (Chart B and
2)
(6.13 g,16.1 mmol), chloral hydrate (2.92 g, 17.7 mmol), hydroxylamine
hydrochloride (3.54 g, 50.9 mmol) and anhydrous sodium sulfate (17.99 g) was
heated at reflux temperature under a nitrogen atmosphere in water (67 ml) for
1
hour. After cooling to room temperature, the mixture was basified with a 10%
ammonium hydroxide solution (110 ml). The aqueous layer was then extracted
with
several portions of ethyl acetate. The solvent was removed and the resulting
brown
oil (5.5 g) was treated with ice cold 90% sulfuric acid (100 ml). This mixture
was
stirred at -30 C for 1 hour, then gradually warmed to 80 C where it was kept
for 0.5
WO 94/21608 2 15 758 6 -14- PCT/US94/02800
hour. The heat was then removed and the stirring continued for 1 hr. at room
temperature. The contents of the reaction flask were then poured on ice (1000
g), the
pH adjusted to 8-9 with konc. ammonium hydroxide followed by extraction of the
basic aqueous solution with several portions of ethyl acetate. Drying over
magnesium sulfate and removal of the solvent yielded 3.80 g(65%) of a red oil.
'H
NMR analysis of this crude material revealed the regioisomeric composition
(7b:2) to
(8b:2) to be approximately 4:1. This material was used in the subsequent step
without any attempt to separate the isomers.
Stereoisomers of EXAMPLE 11:
7-[(1R-Phenyl-ethyl)-propyl-amino]-6,7,8,9-tetrahydro-3H- benzo[e]indole-1,2-
dione
(5:1, Chart 2) and 6-[(1R-phenyl-ethyl)-propyl-amino]-5,6,7,8-1H
-benzo[flindole-2,3-dione (8b:1,Chart 2).
The synthesis of the mixture of the title compounds was conducted in
accordance with the above procedure 6b: 1, yielding a mixture of regioisomeres
in
similar proportions.
EXAMPLE 12: (1R-Phenyl-ethyl)-propyl-(6,7,8,9-tetrahydro-
3H-benzo[e]indol-7-yl) -amine (9b:2, Chart 2) (Rl n-propyl; R2 ethyl phenyl;
R3
hydrogen; R4 CH2; YZ (b); R5R6 H).
To a stirred suspension of lithium aluminum hydride (3.0 g, 79 mmol) in dry
diethyl ether (200 ml) was added dropwise a solution of a 4:1 mixture of
7-[(1R-phenyl-ethyl)-propyl- amino]-6,7,8,9-tetrahydro-3H-benzo[e]indole-1,2-
dione,
7b:2 (Chart 2) and
6-[(1R-phenyl-ethyl)-propyl-amino]-5,6,7,8-1H-benzo[fJindole-2,3-dione, 8b:2
(Chart 2)
(3.8 g, 10.5 mmol) in a 1:1 tetrahydrofuran/diethyl ether mixture (100 ml).
After the
addition was completed stirring at ambient temperature was continued for 2
hours.
Workup as described for (6b:2, Chart B and 2) yielded 3.2 g of a blue oil
which was
chromatographed on a silica column. The yield of the pure title compound, as
an oil
was 0.66 g (24% based on the content of (7b:2) in the starting material).
'H NMR (300 MHz, CDC13) d 0.83 (t, 3H), 1.42 (d, 3H), 1.50 (m, 2H), 1.85 (m,
1H),
2.10 (m, 1H), 2.45-3.10 (m, 7H), 4.10 (q, 1H), 6.42 (m, 1H), 6.83 (d, 1H),
7.10-7.35 (m,
6H), 7.42 (d, 2H), 7.95 (sb, 1H); 13C NMR (75.4 MHz, CDC13) d 11.8, 18.0,
24.0, 27.1,
27.5, 33.7, 47.8, 54.7, 57.5, 100.4, 108.6, 123.4, 123.9, 126.2, 126.8, 127.5,
127.6,
127.7, 127.9, 133.5, 146.1; MS (EI) m/e 332 (M+, 18), 170 (100), 105 (75), 143
(61),
169 (48), 168 (44), 188 (39), 199 (32), 154 (23), 227 (23), 155 (18), 303
(15).
WO 94/21608 PCT/US94/02800
Stereoisomer of EXAMPLE 12: (1R-Phenyl-ethyl)-propyl-(6,7,8,9-tetrahydro-3H
-benzo[e]indol-7-yl)-amine (9b: 1, Chart 2).
Treatment of a mixture of 7b:1 and 8b:1 as described for the preparation of
9b:2 above yielded the title compound.
'H NMR (300 MHz, CDC1g) d 0.86 (t, 3H), 1.45 (d, 3H), 1.50 (m, 2H), 1.68 (m,
1H),
{ 1.85 (m, 1H), 2.58 (m, 2H), 2.70-3.20 (m, 5H), 4.10 (q, 1H), 6.45 (m, 1H),
6.92 (d,
1H), 7.10-7.32 (m, 6H), 7.42 (m, 2H), 8.08 (sb, 1H); MS (EI) m/e 332 (M+, 20),
170
(100), 105 (65), 143 (46), 199 (38), 169 (37), 188 (37), 168 (36), 227 (27),
162 (20), 154
(17), 303 (17).
EXAMPLE 13: (+)-7-Dipropylamino-6,7,8,9-tetrahydro-3H-benzo[e]indole-1
-carbaldehyde (10a:2, Chart 2) (R1R2 n-propyl; R3 hydrogen; R4 CH2; YZ (b); R6
H; R6
CHO).
S#ep;~ (+)-Propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol- 7-yl)-amine (9e:2,
Chart 2)
--Not a compound of the subject invention-- (Rl H; R2 n-propyl; R3 hydrogen;
R4 CH2; YZ (b); R6R6 H).
To a stirred solution of
(1R-phenyl-ethyl)-propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol- 7-yl)-amine,
9b:2
(Chart 2) (0.60 g, 1.8 mmol) in ethanol was added 0.4 g 10% palladium on
carbon
and 1.0 g ammonium formate. The mixture was stirred at ambient temperature for
1.5 hours. and then filtered through a celite pad. The solvent was removed and
to
the residue was added 10% sodium carbonate solution and ethyl acetate. The
organic layer was separated and the aqueous layer was extracted with ethyl
acetate.
Drying of the combined extracts over magnesium sulfate and removal of the
solvent
yielded 0.39 g(89%) of the title compound as a colorless oil. This procedure
is
shown to provide pathway to Example 17.
'H NMR (300 MHz, CDCls) d 0.95 (t, 3H), 1.57 (q, 2H), 1.72 (m, 1H), 2.20 (m,
1H),
2.73 (m, 3H), 2.90-3.22 (m, 4H), 6.50 (m, 1H), 6.94 (d, 1H), 7.17 (m, 2H),
8.48 (sb,
1H); 13C NMR (75.4 MHz, CDCls) d 11.9, 23.5, 25.4, 29.5, 36.6, 49.0, 54.0,
100.3,
108.9, 123.6, 123.9, 125.6, 126.9, 127.6, 133.8; MS (EI) m/ e 228 (M+, 30),
143 (100),
168 (46), 169 (42), 170 (34), 144 (22), 154 (19), 115 (15), 167 (15), 199
(12), 155 (10),
197 (7); [a]D20 +77.7 (c=1.0, MeOH).
Stereoisomer of STEP A: (-)-Propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)-
amine (9e:1, Chart 2).
WO 94/21608 PCT/US94/02800
-16-
This compound was prepared as described for 9b: 1 as described for 9e:2 above.
'H NMR (300 MHz, CDC13) d 0.95 (t, 3H), 1.60 (q, 2H), 1.75 (m, 1H), 2.22 (m,
1H),
2.77 (m, 3H), 2.90-3.22 (m, 4H), 6.50 (m, 1H), 6.92 (d, 1H), 7.17 (m, 2H),
8.48 (sb,
1H); "C NMR (75.4 MHz, CDC13) d 11.9, 23.3, 25.3, 29.2, 36.3, 48.9, 54.0,
100.3,
109.0, 123.7, 123.8, 125.3, 126.9, 127.5, 133.8; MS (EI) m/e 228 (M+, 45), 143
(100),
168 (50), 169 (45), 170 (37), 144 (21), 154 (19), 199 (16), 115 (16), 167
(15), 197 (13).
Step B: (+)-Dipropyl-(6,7,8,9-tetrahydro-benzo[e]indol- 7-yl)-amine (9a:2,
Chart 2) --Not a compound of the subject invention-- (R,.R2 n-propyl; R.
hydrogen; R4
CH2; YZ (b); R6R6 H).
To a stirred solution of
(+)-propyl-(6,7,8,9-tetrahydro-3H-benzo[e)indol-7-yl)-amine, 9e:2 (Chart 2)
(0.35 g,
1.53 mmol) in 1,2-dichioroethane (20 ml) was added sodium triacetoxy
borohydride
(0.39 g, 1.83 mmol), propionaldehyde (0.20 ml, 2.8 mmol) and two drops of
acetic
acid. The solution was stirred at ambient temperature for 2 hours. Then the
solvent
was removed, the residue taken up in water and basified (10% sodium
hydroxide).
The liberated amine was extracted with several portions of diethyl ether,
dried over
magnesium sulfate and the solvent removed to yield an oil. The oil was
chromatographed on a silica column, eluting with methanol, yielding 0.35 g
(85%) of
an off white solid.
'H NMR (300 MHz, CDC13) d 0.90 (t, 6H), 1.50 (m, 4H), 1.73 (m, 1H), 2.14 (m,
1H),
2.55 (m, 4H), 2.80-3.15 (m, 4H), 3.20 (dd, 1H), 6.49 (m, 1H), 6.87 (d, 1H),
7.17 (m,
2H), 8.17 (sb, 1H); 1sC NMR (75.4 MHz, CDC1.) d 11.9, 22.2, 25.8, 27.1, 31.8,
52.7,
57.2, 100.5, 108.7, 123.5, 124.1, 126.9, 127.0, 127.8, 133.6; MS (EI) m/e 270
(M+, 9),
170 (100), 143 (47), 168 (30), 169 (26), 241 (24), 154 (19), 171 (14), 155
(14), 144 (14),
115 (10); [a]D'A +64.7 (c=1.0, MeOH).
Stereoisomer of STEP B: (-)-Dipropyl-(6,7,8,9-tetrahydro-benzo[e]indol-7-yl)-
amine (9a:1, Chart 2).
This compound was prepared from 9e:1 as described for 9a:2 above.
1H NMR (300 MHz, CDC13) d 0.91 (t, 6H), 1.52 (m, 4H), 1.72 (m, 1H), 2.15 (m,
1H),
2.55 (m, 4H), 2.80-3.15 (m, 4H), 2.22 (dd, 1H), 6.51 (m, 1H), 6.88 (d, 1H),
7.18 (m,
2H), 8.20 (sb, 1H); 13C NMR (75.4 MHz, CDC1s) d 12.0, 22.2, 25.9, 27.1, 31.8,
52.7,
57.2, 100.5, 108.7, 123.5, 124.1, 126.9, 127.0, 127.8, 133.6; MS (EI) m/e 270
(M+,
17), 170 (100), 143 (42), 241 (33), 168 (21), 154 (14), 171 (14), 144 (12),
155 (12), 167
(9), 115 (8); [a)D20 -62.9 (c=0.17, MeOH).
16WO 94/21608 - 2157586 PCT/US94/02800
-17-
Step C: (+)-7-Dipropylamino-6,7,8,9-3H-benzo[e]indole-l-carbaldehyde (10a:2,
Chart 2) (RiR2 n-propyl; R. hydrogen; R4 CH2; YZ (b); R6 H; R6 CHO).
This compound was synthesized from 9a:2 according to the procedure given
for compound 4a (Chart 1).
'H NMR (300 MHz, CDCI,,) d 0.89 (t, 6H), 1.50 (m, 4H), 1.68 (m, 1H), 2.18 (m,
1H),
2.55 (m, 4H), 2.80-3.10 (m, 3H), 3.20 (m, 1H), 3.60 (m, 1H), 7.02 (d, 1H),
7.19 (d,
1H), 7.89 (s, 1H), 9.97 (sb, 1H), 10.12 (s, 1H); 13C NMR (75.4 MHz, CDC13) d
11.7,
21.6, 25.5, 29.9, 32.8, 52.4, 56.4, 109.5, 120.7, 123.6, 125.9, 129.8, 130.7,
134.7, 135.3,
185.4; MS (EI) m/e 298 (M+, 42), 199 (100), 198 (97), 269 (79), 170 (50), 100
(24).
[a]D20 +59.4 (c=1.0, MeOH).
EXAMPLE 14:
cis-7-Dipropylamino-6-methyl-6,7,8,9-tetrahydro-3H-benzo[e]-indole-1,2-dione
(R1R2
n-propyl; R. CH3; R,, CH2; YZ (b); R6R6 0) and cis-6-dipropylamino-5-Methyl-
5,6,7,8
-tetrahydro-IH-benzo[fJindole-2,3-dione (7d and 8d Chart 2) (R1R2 n-propyl; R.
CH3;
R4 CH2; X1Y indole; R.R. 0).
Step A: 4-Bromo-phenyl acetic acid chloride (25, Chart C).
To a stirred solution of 4-bromo-phenyl acetic acid (25 g, 116 mmol) in
dichloromethane (250 ml) was added thionyl chloride (13 ml, 170 mmol). The
mixture was heated under reflux for 24 hours and then the solvent and the
excess
reagent were removed. The residue was distilled under reduced pressure (110
C/2
mmHg) to yield 19.8 g (73%) of the title compound.
'H NMR (300 MHz, CDCls) d 4.08 (s, 2H), 7.12 (d, 2H), 7.49 (d, 2H).
SteR B: 6-Bromo-3,4-dihydro-2(1H)-naphthyleneone (26, Chart C).
A solution of 4-bromo-phenyl acetic acid chloride, 25 (Chart C) (19.8, 85
mmol) in dichloromethane was cooled on an ice bath. Then the stirred solution
was
saturated with ethene gas and aluminum trichloride (22.0 g, 165 mmol) was
added
in portions. The bubbling of ethene was continued for 5 hours. The mixture was
then poured into and ice 10% hydrochloric acid mixture and the organic layer
separated. The aqueous layer was extracted with dichloromethane and the
combined
extracts were dried over magnesium sulfate. The solvent was removed and the
residue lixiviated with hexane and the solid filtered off. The hexane was
placed in
the cold store to precipitate a second crop of solid. The yield of
sufficiently pure (>
96% by GC) product was 11.0 g(58%) as an off white solid.
WO 94/21608 5 7 8 6 PCT/US94/02800
-18-
1H NMR (300 MHz, CDC13) d 2.55 (t, 2H), 3.05 (t, 2H), 3.51 (s, 2H), 7.00 (d,
1H), 7.36 (d, 1H), 7.39 (s, 1H); MS (EI) m/e 326/324 (M+, 61/59), 182 (100),
184 (95),
115 (48), 103 (41), 117 (37), 116 (26), 77 (20).
Step C: 6-Bromo-l-methyl-3,4-dihydro-lH-naphthylenee-2-one (27, Chart C).
A solution of 6-bromo-3,4-dihydro-2(1H)-naphthyleneone, 26 (Chart C) (3.83 g,
17.0 mmol) and pyrrolidine (2.2 ml, 26 mmol) in benzene (75 ml) was heated at
reflux for 2 hours, (when the formation of the enamine was judged completed by
GC). The benzene and the excess pyrrolidine were removed and the residue
redissolved in dioxane (25 ml). To the dioxane solution was then added methyl
iodide (8.0 ml, 128 mmol) and the mixture was heated at reflux temperature for
18
hours. To this mixture was then added 5% acetic acid (25 ml) and the heating
was
continued for 4 hours, followed by evaporation of the solvents under vacuum (1
mmHg). The residue was taken up in diethyl ether, washed with water and dried
over magnesium sulfate. To this solution was added a small amount of
(2,6-ditert.butyl-4-methyl phenol [BHT]) to prevent autooxidation. Evaporation
of
the solvent yielded 4.1 g(100%) of an oil which was
85% pure by GC analysis, the remainder being unreacted starting material.
This was used in the subsequent step without further purification.
1H NMR (300 MHz, CDC13) d 1.45 (d, 3H), 2.40-2.60 (m, 2H), 2.95-3.15 (m,
2H), 3.45 (q, 1H), 7.07 (d, 1H), 7.38 (m, 2H); MS (EI) m/e 240/238 (M+,
23/24), 116
(100), 115 (88), 117 (56), 196 (54), 198 (54), 197 (26), 195 (22), 91 (21).
Step D: cis-(6-Bromo-l-methyl-6,7,8,9-tetrahydro-naphthylenee
-2-yl)-propylamine (28, Chart C).
To a stirred solution of 6-bromo-l-methyl-3,4-dihydro-lH-naphthylenee-2-one,
27 (Chart C) (4.1 g, 17 mmol), propylamine (1.5 ml, 18 mmol) and 0.5 g BHT in
tetrahydrofuran (THF) (50 ml) was added portionwise sodium
triacetoxyborohydride
(4.5 g, 21 mmol). The mixture was stirred overnight, rigorously protected from
atmospheric oxygen. Then was 10% hydrochloric acid (50 ml) added and most of
the
THF removed. The residual aqueous layer was washed with diethyl ether,
basified
and extracted with ether. Drying over magnesium sulfate yielded 2.4 g of an
oil
which was purified by chromatography on a silica column. The yield of the
title
compound as a colorless oil was 1.56 g (38% based on the content of (27) in
the
starting material).
'H NMR (300 MHz, CDC13) d 0.94 (t, 3H), 1.12 (d, 3H), 1.53 (m, 2H), 1.74 (m,
WO 94/21608 21 57586 PCTIUS94/02800
~
-19-
2H), 2.61 (m, 2H), 2.81 (m, 2H), 2.90 (m, 1H), 3.02 (m, 1H), 6.97 (d, 1H),
7.20 (m,
2H); MS (EI) m/e 283/281 (M+, 39/40), 144 (100), 115 (96), 128 (92), 254 (85),
252
(85), 117 (83), 223 (78), 225 (76).
Step E: cis-(6-Bromo-l-methyl-6,7,8,9-tetrahydro-naphthylenee-2-yl)-
dipropylamine (29, Chart C).
To stirred solution of
cis-(6-bromo-l-methyl-6,7,8,9-tetrahydro-naphthylenee-2-yl)- propylamine, 28
(Chart
C) (1.56 g, 5.55 mmol), propionaldehyde (0.78 g, 13 mmol) and BHT (0.20 g) in
THF
was added portionwise sodium triacetoxyborohydride (3.0 g, 14 mmol). The
mixture
was stirred at ambient temperature for 2 hours and then the solvent was
removed.
The residue was dissolved in 10% hydrochloric acid (50 ml), the solution
washed
with diethyl ether and basified with 15% sodium hydroxide. Extraction with
diethyl
ether, drying over magnesium sulfate and removal of the solvent yielded 1.55 g
(91%) of product as a colorless oil. The purity as determined by GC was > 98%,
the
remainder being BHT.
'H NMR (300 MHz, CDCls) d 0.88 (t, 6H), 1.18 (d, 3H), 1.46 (m, 4H), 1.77 (m,
1H), 1.91 (m, 1H), 2.56 (m, 4H), 2.75-2.95 (m, 3H), 3.05 (m, 1H), 6.97 (d,
1H), 7.22
(m, 2H); 19C NMR (75.4 MHz, ) d 11.9, 18.1, 20.8, 20.9, 29.6, 36.6, 52.8,
59.4, 119.2,
128.7, 131.0, 131.2, 137.9, 141.9; MS (EI) m/e 325/323 (M+, 14/15), 294 (100),
296
(98), 144 (97), 223 (75), 225 (73), 143 (25).
Step F: cis-6-Dipropylamino-5-methyl-5,6,7,8-tetrahydro-naphthylenee-
2-carboxylic acid methyl ester (30, Chart C).
A mixture of
cis-(6-bromo-l-methyl-6,7,8,9-tetrahydro-naphthylenee-2-yl)-dipropylamine, 29
(Chart C) (1.55 g, 4.78 mmol), triethylamine (1.06 g, 10.5 mmol) and methanol
(10
ml) in dimethyl sulfoxide (DMSO) (15 ml) was stirred at ambient temperature
for 20
minutes. Then palladium acetate (0.107 g, 0.48 mmol) and
1,3-bis-(diphenylphosphino)propane (0.196 g, 0.478 mmol) was added and carbon
monoxide (CO) passed through the solution. The mixture was kept under an
atmosphere of CO and heated at 80 C for 2 hours when more triethylamine (1.06
g,
10.5 mmol) and methanol (5 ml) were added. After 6.5 hours the reaction was
judged
complete and the mixture allowed to cool to room temperature. The DMSO was
removed by evaporation under vacuum and the residue treated with 10% sodium
carbonate. The aqueous layer was extracted with several portions of diethyl
ether,
WO 94/21608 21e,~ 7-86 -20- PCT/US94/02800
the combined extracts washed with water followed by drying over magnesium
sulfate. Removal of the solvent yielded 1.24 g (86%) of product as an yellow
oil. This
material was used without further purification.
'H NMR (300 MHz, CDC13) d 0.86.t (6), 1.17 (d, 3H), 1.45 (m, 4H), 1.78 (m,
1H), 1.94 (m, 1H), 2.55 (m, 4H), 2.75-3.00 (m, 3H), 3.12 (m, 1H), 3.88 (3, s),
7.16 (d,
1H), 7.77 (m, 2H); 1SC NMR (75.4 MHz, CDC13) d 11.9, 17.9, 20.7, 21.0, 29.6,
37.2,
51.9, 52.8, 59.2, 126.6, 127.5, 129.4, 129.9, 135.7, 148.5, 167.3; MS (EI) m/e
303 (M+,
13), 274 (100), 203 (82), 126 (55), 128 (52), 115 (45), 171 (43), 129 (42), 98
(27), 200
(27), 143 (26).
Step ~: cis-5-Methyl-N-,N-dipropyl-5,6,7,8-tetrahydro-naphthylenee-
2,6-diamine (6d, Chart C and 2).
To a stirred solution of
cis-6-dipropylamino-5-methyl-5,6,7,8-tetrahydro-naphthylenee-2- carboxylic
acid
methyl ester, 30 (Chart C) (1.20 g, 3.96 mmol) in dichioromethane (60 ml) was
added
concentrated sulfuric acid (25 ml) followed by the portionwise addition of
sodium
azide (3.50 g, 53.8 mmol) (caution, foaming). The mixture was then heated at
reflux
temperature overnight. After cooling the mixture was basified with 15% sodium
hydroxide and the organic layer separated. The aqueous phase was extracted
with
dichloromethane. The combined extracts were dried over magnesium sulfate and
the
solvent removed to yield 1.02 g(99%) as a light brown oil. The purity
according to
GC analysis was > 95%.
'H NMR (300 MHz, CDC13) d 0.85 (t, 6H), 1.22 (d, 3H), 1.45 (m, 4H), 1.75 (m,
1H), 1.83 (m, 1H), 2.55 (m, 4H), 2.76 (m, 2H), 2.86 (m, 1H), 3.02 (m, 1H),
3.48 (sb,
2H), 6.62 (d, 1H), 6.52 (dd, 1H), 6.88 (d, 1H); 19C NMR (75.4 MHz, CDC13) d
11.9,
18.5, 20.8, 21.2, 30.0, 36.1, 52.8, 59.9, 113.5, 114.6, 130.3, 133.3, 136.4,
144.1; MS
(EI) m/e 260 (M+, 6), 133 (100), 160 (64), 144 (14), 130 (13), 231 (13), 161
(8), 117
(7).
Step H: cis-7-Dipropylamino-6-methyl-6,7,8,9-tetrahydro-3H-benzo[e]-
indole-1,2-dione, and
cis-6-dipropylamino-5-Methyl-5,6,7,8-tetrahydro-1H -benzo[f]indole-2,3-dione
(7d and 8d Chart 2).
The regioisomeric mixture of these compounds was prepared as described for
the mixture of 7b:2 and 8b:2. Here the composition of 7d to 8d as determined
by
'HNMR was approximately 6:1. This mixture was used in the subsequent step
WO 94/21608 215 70' 8 6 PCT/US94/02800
-21-
without any attempt to separate the regioisomeres.
EXAMPLE 15: cis-(6-Methyl-6,7,8,9-tetrahydro-3H-
benzo[e]-indol-7-yl)-dipropylamine (9d, Chart 2) (R1R2 n-propyl; R3 CH3; R4
CH2; YZ
(b); R6R6 H), and
cis-(5-methyl-5,6,7,8-tetrahydro-lH-benzo[fJ-indol-6-yl)-dipropylamine (R1R2 n-
propyl;
R3 CH3; R4 CH2; XY indole; R6R6 H).
This product was prepared using the same procedure as described for (9b:2).
Here a 6:1 mixture of
cis-7-dipropylamino-6-methyl-6,7,8,9-tetrahydro-3H-benzo[e]-indole-1,2-dione
and
cis-6-dipropylamino-5-methyl-5,6,7,8-tetrahydro-lH-benzo[f)indole-2,3-dione,
7d and
8d (Chart 2) (1.1 g, 35 mmol) was used, yielding 0.25 g of the pure title
compound
after chromatography of the crude product.
'H NMR (300 MHz, CDClg) d 0.98 (t, 6H), 1.32 (d, 3H), 1.58 (m, 4H), 2.02 (m,
1H),
2.10
(m, 1H), 2.71(m, 4H), 2.98-3.15 (m, 2H), 3.20-3.35 (m, 2H), 6.56 (m, 1H), 7.04
(d,
1H), 7.17 (t, 1H), 7.22 (d, 1H), 8.30 (sb, 1H); 13C NMR (75.4 MHz, CDC13) d
12.0,
18.7, 20.7, 20.9, 27.2, 36.7, 52.9, 59.9, 100.4, 109.0, 123.6, 124.1, 126.7,
126.8, 133.48,
133.51. m/e 284 (M+, 41), 157 (100), 184 (66), 156 (35), 168 (19), 129 (10).
The residual fractions from above chromatography contained
cis-(5-methyl-5,6,7,8-tetrahydro- 1H-benzo[fJ-indol-6-yl)-dipropylamine (lld,
Chart
2). MS (EI) m/e 284 (M+, 52), 184 (100), 255 (63), 157 (63), 156 (35), 168
(22), 129
(11).
EXAMPLE 16:
cis-7-Dipropylamino-6-methyl-6,7,8,9-tetrahydro-3H-benzo[e]-indole-1-
carbaldehyde
(10d, Chart 2) (R1R2 n-propyl; R3 CH9; R4 CH2; YZ (b); R5 H; R6 CHO).
This compound was prepared according to the procedure described for 4a,
(Chart 1).
'H NMR (300 MHz, CDC13) d 0.88 (t, 6H), 1.23 (d, 3H), 1.51(m, 4H), 1.90 (m,
iH),
2.12
(m, 1H), 2.64 (m, 4H), 2.98 (m, 1H), 3.22 (m, 2H), 3.61 (dd, 1H), 7.05 (d,
1H), 7.26 (d,
1H), 7.93 (s, 1H), 10.13 (s, 1H), 10.22 (sb, 1H); 13C NMR (75.4 MHz, CDCls) d
12.0,
18.5, 20.3, 20.9, 30.3, 37.2, 52.6, 59.4, 110.1, 121.1, 123.7, 126.4, 129.1,
135.3, 135.6,
137.3, 185.9; MS (EI) m/e 312 (M+, 8), 213 (100), 212 (25), 184 (23), 100
(15), 156
WO 94/21608 2157586 -22- PCT/US94/02800
(15), 198 (14), 255 (12).
EXAMPLE 17: Di-n-propyl-(6,7,8,9-tetrahydronaphtho[1,2-b]furan-7-yl)-amine.
(15a,
Chart 3) (R1R2 n-propyl; R3 hydrogen; R4 CH2; YZ (c); R$Rs H)
Step A: 6-Di-n-propylamino-5,6,7,8-tetrahydronaphthylenee-l-ol. (13a, Chart
3)
This compound was prepared according to Hacksell et al. (J. Med. Chem.
22:1469 (1979)).
Step B: [5-(2,2-Diethoxy-ethoxy)-1,2,3,4-tetrahydronaphthylenee
-2-yl]-dipropyl-l-amine. (14a, Chart 3)
A solution of 6-di-n-propylamino-5,6,7,8-tetrahydronaphthylenee-l-ol, 13a
(Chart 3) (128 mg, 0.52 mmol) in 20 mL acetonitrile was treated with sodium
hydride (55% in oil) (50 mg, 1.12 mmol). A solution of bromoacetaldehyde
diethylacetal (112 mg, 0.57 mmol) in 5 ml acetonitrile was added and the
mixture
was refluxed for 3 days. After cooling, the reaction mixture was evaporated to
a
residue which was dissolved in water/ethyl acetate. After extraction two
additional
times, the combined organic extracts were dried (magnesium sulfate), filtered
and
evaporated to yield a residue of the raw material as an oil (210 mg, 110%).
This
material was used without further purification in the next step.
MS m/e 363 (M+, 15), 145 (100), 334 (87), 103 (74), 73 (65), 126 (58), 171
(55), 218 (45).
SteR C: Di-n-propyl-(6,7,8,9-tetrahydronaphtho[1,2-b]furan-7-yl)-amine. (15a,
Chart 3)
A solution of [5-(2,2-diethoxy-ethoxy)-1,2,3,4-tetrahydronaphthylenee-2-yl]-
dipropyl-l-amine, 14a (Chart 3) (210 mg, 0.57 mmol) in dichloromethane (20 mL)
was treated with 1,2 eq acetic acid followed by borontrifluoride etherate (330
mg, 2.4
mmol). The reaction mixture was stirred at room temperature for two days and
the
solvent then evaporated. Water was added and the resulting suspension was
basified
(5 M sodium hydroxide). After extraction (three times ethyl acetate) the
solution was
dried (magnesium sulfate, fiitered and evaporated to yield 160 mg (100%) of
the
crude product, which was purified on silica (dichloromethane/methanol, 19:1)
to yield
25 mg (16%) of the pure compound as a colorless oil.
'H NMR (200 MHz, CDC13) d 0.90 (t, 6H), 1.50 (sixt, 4H), 1.70 (oct, 1H), 2.2
WO 94/21608 1 21575V 6 PCT/US94/02800
-23-
(br.d, 1H), 2.55 (t, 4H), 2.8-3.2 (m's, 4H), 3.3 (d of d, 1H), 6.70 (d, J=2.1
Hz, 1H), 6.98
(d, J=8.1 Hz, 1H), 7.34 (d, J=8.0 Hz, 1H), 7.56 (d, J=2.2 Hz, 1H); 13C NMR
(75.4
MHz, CDC13) d 11.8 (CH3), 21.7 (CH2), 23.4 (CHZ), 25.0 (CH2), 31.5 (CH2), 52.6
(CH2),
57.1 (CH), 106.6 (CH), 118.3 (CH), 120.2 (C), 124.3 (CH), 124.4 (C), 132.5
(C), 144.1
5(CH), 153.4 (C); MS m/e 271 (M+, 9), 171 (100), 242 (45), 128 (26), 115 (18),
141
(18), 145 (15).
EXAMPLE 18: S-(-)-7-Dipropylamino-6,7,8,9-tetrahydro-3H-benzo[e]indole-l-
carbaldehyde (10a:1, Chart 2)
This compound was synthesized from 9a: 1 according to the procedure given
for compound 4a (Chart 1)
'H NMR (300 MHz, CDCls) S 0.90 (t, 6H), 1.50 (m, 4H), 1.70 (m, 1H), 2.21 (m,
1H), 2.55 (m, 4H), 2.80-3.10 (m, 3H), 3.20 (m, 1H), 3.60 (m, 1H) 7.04 (d, 1H),
7.18 (d
1H), 7.90 (s, 1H), 9.12 (br s, 1H), 10.15 (s, 1H); 13C NMR (75.4 MHz, CDC13) S
12.0,
22.0, 25.8, 30.2, 33.1, 52.7, 56.6, 109.5, 121.2, 123.8, 126.3, 130.2, 131.1,
134.3, 135.7,
185.5. MS m/z (relative intensity, 70 eV) 298 (M, 43), 199 (100), 198 (91),
269 (81),
170 (45), 100 (18). [a]D20-58.5 (c=1.0, MeOH)
EXAMPLE 19: Di-n-propyl-(6,7,8,9-tetrahydronaphtho[2,1-b]furan-7-yl)-amine
(15b, Chart 3) and Di-n-propyl-(5,6,7,8-tetrahydronaphtho[2,3-b]furan-6-yl)-
amine
(15c, Chart 3)
Step A [6-(2,2-Diethoxy-ethoxy)-1,2,3,4-tetrahydronaphthylene-2-yl]-
dipropyl-l-amine (14b, Chart 3).
Sodium hydride (244 mg, 55-60% in oil, 5.8 mmol) was washed with hexane
two times and then slurried in acetinitrile (20 mL). Dipropyl-(6-hydroxy-
1,2,3,4-
tetrahydronaphthylene-2-yl)-amine (1.26 mg, 5.1 mmol) was added, followed by 2-
bromo-acetaldehyde diethylacetal (1.1 g, 5.6 mmol). The mixture was refluxed
for 2
days, evaporated and redissolved in water/ethyl acetate. The organic solution
was
dried (magnesium sulfate), filtered and evaporated to yield 1.86 g(100%) of
the
desired compound.
Step B Di-n-propyl-(6,7,8,9-tetrahydronaphtho[2,1-b]furan-7-yl)-amine (15b
Chart 3) and Di-n-propyl-(5,6,7,8-tetrahydronaphtho[2,3-b]furan-6-yl)-amine.
(15 c
Chart 3)
To a solution of [6-(2,2-diethoxy-ethoxy)-1,2,3,4-tetrahydronaphthylene-2-yl]-
WO 94/21608 2157586 -24- PCT/US94/02800
dipropyl-l-amine (14 b Chart 3) (1.86 g, 5.1 mmol) and acetic acid (250 }iL)
in
dichloromethane (100 mL) was added borontrifluoride etherate (2.91 g, 2.51 mL,
20.4 mmol). The resulting mixture was stirred over night and evaporated.
Sodium
hydroxide (5 M) and ethyl acetate was added and the mixture was shaken. The
aqueous phase was extracted two additional times using ethyl acetate. The
combined organic extracts were dried (magnesium sulfate), filtered and
evaporated
to a residue of 1.4 g, which was further purified on silica yielding 220 mg
(16%) of
the two isomers.: MS m/z (relative intensity, 70 eV) 271(M+, 28), 171 (100),
242
(79), 128 (19).
EXAMPLE 20: cis-(6S-Methyl-6,7,8,9-tetrahydro-naphto[1,2-b]furan-7R-yl]-
dipropyl-amine (15d, Chart 3).
Step A cis-[5-(2,2-Diethoxy-ethoxy)-1S-methyl-1,2,3,4-tetrahydro-
naphthylene-2R-yl]-dipropyl-amine (14c, Chart 3)
This compound was prepared as described for 14a from cis-(+)-(5-hydroxy-lS-
methyl-1,2,3,4-tetrahydro-naphthylene-2R-yl)-dipropyl-amine (450 mg, 1.72
mmol,
for preparation see Johansson et. al. J. Med. Chem. 1987, 30, 602-611). The
residue
was used without further purification (700 mg); MS m/z (relative intensity, 70
eV))
377 (M+ 34), 348.5 (100), 232 (80), 185 (23), 159 (51).
Step B cis-(6S-Methyl-6,7,8,9-tetrahydro-naphto[1,2-b]furan-7R-yl)-dipropyl-
amine (15d, Chart 3).
This compound was prepared as described for 15a from cis-[5-(2,2-diethoxy-
ethoxy)-1S-methyl-1,2,3,4-tetrahydro-naphthylene-2R-yl]-dipropyl-amine (648
mg,
1.72 mmol). Purification of the crude reaction mixture by flash chromatography
(CH2C12/1VIeOH, 9/1(v/v))afforded 65.8 mg (13.4%) of pure 16d as an oil;
'H NMR (300 MHz, CDC13) S 0.9 (t, 6H), 1.2 (d, 3H), 1.5 (m, 4H), 1.9 (m, 1H),
2.55-
2.75 (m, 4H), 2.8-3.1 (m, 3H), 3.25 (m, 2H), 6.72 (d, J= 2.2 Hz, 1H), 7.0 (d,
J= 8 Hz,
1H), 7.4 (d, J = 8 Hz, 1H), 7.6 (d, J = 2 Hz, 1H); MS m/z (relative intensity,
70 eV)
285 (M+ 36), 256 (100), 185 (96), 158 (36), 128 (16).
EXAMPLE 21: (7,8-Dihydro-6H-1-oxa-as-indacen-7-yl)-dipropyl-amine (15e,
Chart 3)
Step A [4-(2,2-Diethoxy-ethoxy)-indan-2-yl]-dipropyl-amine (14d, Chart 3)
WO 94/21608 215758S PCT/US94/02800
-25-
This compound was prepared as described for 14a from (4-hydroxy-indan-2-
yl)-dipropyl-amine (220 mg, 1.05 mmol). the residue was used without any
further
purification (340 mg); MS m/z (relative intenzity, 70 eV) 349.5 (M+ 6), 321
(22), 320
(100), 103 (10.5), 72 (10).
Step B (7,8-Dihydro-6H-1-oxa-as-indacen-7-yl)-dipropyl-amine (15e, Chart
3))
This compound was prepared as described for 15a from [4-(2,2-Diethoxy-
ethoxy)-indan-2-yl]-dipropyl-amine (340 mg). MS m/z (relative intensity, 70
eV) 257
(M+ 14), 228 (100), 204 (21), 157 (49), 128 (19.6)
EXAMPLE 22: (7,8-Dihydro-6H-3-oxa-as-indacen-7-yl)-dipropyl-amine (15f,
Chart 3) and (6,7-Dihydro-5H-1-oxa-s-indacen-6-yl)-dipropyl-amine (15g, Chart
3)
StepA [5-(2,2-Diethoxy-ethoxy)-indan-2-yl]-dipropyl-amine (14e, Chart 3)
This compound was prepared as described for 14a from (5-hydroxy-indan-2-
yl)-dipropyl-amine (220 mg, 1.05 mmol). The residue was used without any
further
purification (330 mg); MS m/z (relative intensity, 70 eV) 349.5 (M+ 6.8), 321
(22),
320 (100), 133 (28), 103 (10.8), 61 (8.8).
Step B (7,8-Dihydro-6H-3-oxa-as-indacen-7-yl)-dipropyl-amine (15 f, Chart 3)
and (6,7-Dihydro-5H-1-oxa-s-indacen-6-yl)dipropyl-amine (15g, Chart 3)
This compound was prepared as described for 15a from [5-(2,2-Diethoxy-
ethoxy)-indan-2-yl]-dipropyl-amine (340 mg). A mixture of the two regio-
isomers
were obtained in a 1:3 relationship according to gC/MS. Purification of the
crude
reaction mixture by flash chromatography (dichloromethane/methanol, 9/1 (v/v))
afforded 11 mg (13.4%) of the pure mixture as an oil; MS m/z (relative
intensity, 70
eV) 257 (M+ 9.2), 228 (100), 157 (53), 129 (19), 128 (18.6)
EXAMPLE 23: 7-Di-propylamino-1,6,7,8-tetrahydro-9-oxa-1-
azacyclopenta[a]naphthylenee-3-carbaldehyde (4b, Chart 1).
To dry N,N-dimethyl formamide (6.5 mL) at 0 C was added phosphorous
oxychloride (0.24 mL, 2.64 mmol). The solution was stirred in an ice bath for
10
min. Thereafter di-propyl-(1,6,7,8-tetrahydro-9-oxa-l-
azacyclopenta[a]naphthylene-
7-yl)amine (144 mg, 52.9 mmol) dissolved in dry dimethylfomamide (6.5 mL) was
added. The solution was stirred for 10 min at 0 C and for additional 30 min
at
WO 94/21608 2 1~ 75- 8 6 -26- PCT/US94/02800
room temperature. The reaction mixture was placed in an oil bath and stirred
at
50 C for 2h under nitrogen. The solution was allowed to reach room
temperature.
Aqueous sodium hydroxide was added (30 ml, 5%) and the mixture was stirred for
30 min at 50 C. Additional water (25 mL) was added and the mixture extracted
with dichloromethane. The phases were separated and the organic layer was
dried
(sodium sulfate) and evaporated. The crude product was purified using Silica
column chromatography with dichloromethane/methanol (30:1) as eluant, yielding
60
mg (38%) of pure 7-di-propylamino-1,6,7,8-tetrahydro-9-oxa-1-
azacyclopenta[a]naphthylenee-3-carbaldehyde.:
'H NMR (300 MHz, CDC1,,) S 0.91 (t, 6H), 1.46 (m, 4H), 2.53 (m, 4H), 2.96 (d,
2H), 3.27 (m, 1H), 3.93 (t, 1H), 4.42 (d of d, 1H), 6.96 (d, J=8 Hz, 1H), 7.69-
7.80 (m,
2H), 9.1 (s, 1H), 10.0 (s, 1H); 13C NMR (75.4 MHz, CDCls) S 11.7, 21.8, 27.5,
52.8,
53.6, 68.5, 113.7, 116.0, 120.1, 124.2, 125.1, 126.5, 134.4, 140.3, 185.3; MS
m/z
(relative intensity, 70 eV) 300 (M+, 71), 98 (100), 70 (74), 200 (64), 127
(55).
EXAMPLE 24: (f)-Propyl-(6,7,8,9)-tetrahydro-3H-benzo[3]indol-7-yl)-amine
(9e:1, Chart 2)
This compound was prepared from a diastereomeric mixture of 2-[6-(1-
Phenyl-ethylamino)-5,6,7,8-tetrahydro-naphthylenee-2-yl]isoindole-1,3-diones
(8.69 g,
21.9 mmol), using the same reaction sequence as previously described for the
optically active counterparts. The yeild of the title compound, after
chromatography
on a silica column, was 1.11 g.:
'H NMR (300 MHz, CDC1g) S 0.95 (t, 3H), 1.57 (q, 2H), 1.72 (m, 1H), 2.20 (m,
1H), 2.73 (m, 3H), 2.90-3.22 (m, 4H), 6.50 (m, 1H), 6.94 (d, 1H), 7.17 (m,
2H), 8.48
(sb, 1H); '3C NMR (75.4 MHz, CDC1g) S 11.9, 23.5, 25.4, 29.5, 36.6, 49.0,
54.0, 100.3,
108.9, 123.6, 123.9, 125.6, 126.9, 127.6, 133.8; MS (EI) m/z (relative
intensity, 70
eV) 228 (M+, 30), 143 (100), 168 (46), 169 (42), 170 (34), 144 (22), 154 (19),
115 (15),
167 (15), 199 (12), 155 (10), 197 (7).
EXAMPLE 25: Methyl-propyl-(6,7,8,9-tetrahydro-3H-benzo[elindol-7-yl)amine
(9f, Chart 2).
To a solution of propyl-(6,7,8,9-tetrahydro-3H-benzo[3]indol-7-yl)amine (125
mg, 0.54 mmol) and formaldehyde (0.5 mL 37% in water) in tetrahydrofuran (10
mL)
was added sodium triacetoxy borohydride (220 mg, 1.03 mmol). After stirring in
room temperature for 0.5 hr., water and diethyl ether was added followed by 3M
sodium hydroxide to basify the aqueous phase. After shaking and separation the
21575~~
WO 94/21608 ~ PCT/US94/02800
~ .- , ..
-27- -
aqueous phase was extracted two additional times with diethyl ether. The
combined
organic solution was dried (magnesium sulfate) filtered and evaporated to
yield 108
mg (83%) of the pure product as an oil:
'H NMR (300 MHz, CDC13) S 0.95 (t, 3H), 1.62.sxt (2), 1.75 (oct, 1H), 2.2 (br
d, 1H), 2.42 (s, 3H), 2.60 (t, 2H), 2.8-3.1 (m, 4H), 3.2 (d of d, 1H), 6.50
(s, 1H), 6.92
(d, J=8.3 Hz, 1H), 7.16 (m, 1H), 7-18 (d, J=8.3 Hz, 1H), 8.90 (br s, 1H); 13C
NMR
(75.4 MHz, CDC13) S 11.9, 20.5, 25.5, 26.8, 30.8, 37.5, 55.6, 59.7, 100.1,
109.0, 123.6,
123.8, 125.9, 126.8, 127.4, 133.8; MS m/z (relative intensity, 70 eV) 242 (M+,
65),
170 (100), 143 (97), 213 (50), 168 (32), 115 (17).
EXAMPLE 26: Methyl-(1-methylsulfanyl-6,7,8,9-tetrahydro-3H-benzo[e]indol-7-
yl)propylamine (10e, Chart 2).
Sulfuryl chloride (14.0 uL, 14.5 mg, 0.15 mmol) was added to a solution of
dimethyl disulfide (12. 41iL, 20.8 mg, 0.15 mmol) in dichloromethane (3 mL) at
0 C.
After stirring for 15 minutes, this solution was added to an ice-cooled
solution of
methyl-propyl-(6,7,8,9-tetrahydro-3H-benzo[3]indol-7-yl)amine in
dichloromethane
(10 mL). The resulting solution was stirred at room temperature for 2 hours.
Water, followed by 3 M sodium hydroxide was added and the mixture was shaken.
The orgnic phase was dried (magnesium sulfate), filtered and evaporated to
yield a
residue of 55 mg (86%).:
MS m/z (relative intensity, 70 eV) 288 (M+, 100), 216 (94), 259 (58), 207
(58),
174 (37).
EXAMPLE 27: Propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)-(2-thiophen-2-
yl-ethyl)-amine (9g, Chart 2).
Step A N-Propyl-N-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)-2-thiophen-2-
yl-acetamide.
A mixture of propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)amine (99 mg,
0.43 mmol), triethyl amine (200 }iI.) and thiophene-2-acetylchloride (94 }il.,
121 mg,
0.75 mmol) in dichloromethane (15 mL) was stirred for two days. Water was
added
and the mixture shaken. The organic phase was washed (10% sodium carbonate),
dried (magnesium sulfate), filtered and evaportated.:
Ms m/z (relative intensity, 70 eV) 169 (M*-N-propyl-2-tiophenacetamide,
100), 143 (8), 154 (7), 97 (6), 207 (1)
WO 94/21608 215r~-86 - PCTIUS94/02800
1 28-
Step Propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)-(2-thiophen-2-yl-
ethyl)-amine (9g, Chart 2).
This material was slurried in diethyl ether (50 mL). Lithium aluminum
hydride (110 mg, 2.9 mmol) was added inportions. The mixture was then stirred
for
3 hours. The product was completely soluble in diethyl ether in contract to
the
amide. Water (110 }tL) followed by 15% sodium hydroxide (110 }iL) and water
(330
uL) was added. After stirring for 30 minutes, the solid material was filtered
off and
the resulting ethereal solution was evaporated to yield 135 mg (99%) of the
products
as an oil.:
MS m/z (relative intensity, 70 eV) 241 (M+-thiophenemethylene, 68), 170
(100), 154 (11).
F.XAMPLE 28: 7-[Propyl-(2-thiophene-2-yl-ethyl)-amino]-6,7,8,9-tetrahydro-3H-
benzo[eJindole-l-carbaldehyde (10f, Chart 2)
A cooled (-5 C) solution of propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)-
(2-
thiophen-2-yl-ethyl)amine (9g, Chart 2) (94 mg, 0.28 mmol) in N, N-
dimethylformaznide (3 mL) was added to a cooled (-5 C) solution of phosphorous
oxychloride in N,N-dimethylformamide (3 mL). The mixture was stirred at
ambient
temperature for 0.5 hour, then heated at 50 C for 2 hours. After cooling, 15%
sodium hydroxide was added and the mixture was heated at 50 C for 15 minutes
and then cooled. The aqueous mixture was extracted with ethyl acetate and the
organic solution was dried (magnesium sulfate), filtered and evaporated to a
residue
of 84 mg (82%) of the desired material.
MS m/z (relative intensity, 70 eV) 269 (M+-thiophenemethylene, 100), 198
(77), 170 (23), 270 (20), 155 (11)
EXAMPLE 29: Cyclobutylmethyl-propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-
yl)lamine (9h, Chart 2)
teA A Cyclobutanecarboxylic acid propyl-6,7,8,9-tetrahydro-3H-
benzo[e]indol-7-yl)-amide.
A solution of propyl-(6,7,8,9-tetrahydro-3H-benzo[eJindol-7-yl)amine (100 mg,
0.44 mmol), triethyl aminee and cyclobutanecarbonyl chloride (60 mg, 50 mmol)
in
dichloromethane was stirred for 1 hour at room temperature. Water was added
and
the mixture shaken. The organic phase was washed (10% sodium carbonate), dried
WO 94/21608 - 2157586 PCTIUS94/02800
~
-29-
(magnesium sulfate), filtered and evaporated to yield 134 mg (100%) of the
title
compound:
MS m/z (relative intensity, 70 eV) 310 (M+, <1), 169 (100), 55 (10), 143 (8).
Step B Cyclobutylmethyl-propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-
yl)amine (9h, Chart 2).
Cyclobutanecarboxylic acid propyl-6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)-
amide (134 mg, 0.44 mmol) was dissolved in diethyl ether (20 mL). To this
solution
lithium aluminum hydride (100 mg, 2.6 mmol) was added in portions. The mixture
was stirred for 2 hours. The product was completely soluble in diethyl ether
in
contrast to the amide. Water (100 }iL) followed by 15% sodium hydroxide (100
}iL)
and water (300 }iI.) was added. After stirring for 15 minutes, the solid
material was
filtered off and the resulting ethereal solution was evaporated to yield 108
mg (83%)
of the product as an oil.:
MS m/z (relative intensity, 70 eV) 296 (M+, 43), 170 (100), 241 (50), 267
(36),
143 (28).
EXAMPLE 30: (1-Chloro-6,7,8,9-tetrahydro-3H-benzo[e]indol-7-
yl)cyclobutylmethyl-propylamine. ( lOg, Chart 2)
A solution of cyclobutylmethyl-propyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-
yl)amine 48 mg, 0.16 mmol) and N-chlorosuccinimide (35 mg, 0.26 mmol) in
tetrahydrofuran (5 mL) was stirred at room temperature for 1 day. After
pouring
the reaction mixture into water, extraction (diethyl ether), drying (magnesium
sulfate), filtering, and evaporation yielded 43 mg (80%) of the desired
compound.:
MS m/z (relative intensity, 70 eV) 330 (M, 36), 330 (M+2, 12), 204 (100),
275 (65), 169 (40), 301 (39).
EXAMPLE 31: Dipropylamino-1,3,6,7,8,9-hexahydro-benzo[e]indol-2-one (lOh,
Chart 2).
To a solution of dipropyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)amine (200
mg, 0.74 mmol), water (5 ML) in acetic acid (50 mL) was added dropwise to a
solution of pyridiniumhydrobromide perbromide (310 mg, 0.91 mmol) in acetic
acid
(100 mL). The solution was heated and stirred at 80 C over night. After
cooling the
solution was evaporated to an aqueous residue, which was basified (10% sodium
carbonate) and extracted three times with ethyl acetate. The organic extract
was
dried (magnesium sulfate), filtered and evaporated to yield 210 mg (100%) of
the
WO 94/21608 PCT/US94/02800
2157586 -30-
material.:
MS m/z (relative intensity, 70 eV) 286 (M', 25), 186 (100), 257 (81), 207
(11).
EXAMPLE 32: 1-(7-Dipropylamino-6,7,8,9-tetrahydro-3H-benzo[e]indol-l-yl)-
2,2,2-trifluoro-ethanone (10i, Chart 2).
To an ice-cooled solution of dipropyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-
yl)amine (150 mg, 0.56 mmol) in dimethyl formamide (5 mL) in an inert
atmosphere
was added trifluoroacetic anhydride (300 }iL, 447 mg mg, 2.12 mmol) and the
following mixture was stirred over night at room temperature. Water was added
and the aqueous solution was washed with diethyl ether. After basification (5%
sodium hydroxide), the aqueous solution was extracted two times with diethyl
ether
and one time with ethyl acetate. The combined organic extract was dried
(magnesium sulfate), filtered and evaporated to yield a residue which was
purified
in silica using methanol as eluant. The pure fractions were collected and
evaporated
to yield 130 mg (63%) of the pure material.:
'H NMR (300 MHz, CDC13) 8 0.90 (t, 6H), 1.50 (sxt, 4H), 1.6 (m, 1H), 2.15 (br
d, 1H), 2.55 (t, 4H), 2.8-3.1 (m, 3H), 3.3 (m, 1H), 3.55 (br d, 1H), 7.09 (d,
J=8.4 Hz,
1H), 7.20 (d, J=8.4 Hz, 1H), 8.05 (d, J=1.8 Hz, 1H), 9.5 (br s, 1H); 1gC NMR
(75.4
MHz, CDC13) 8 12.0, 21.8, 257.7, 29.8, 33.5, 52.6, 56.4, 109.4, 112.1, 117.5
(q), 124.4,
127.5, 131.6, 132.1, 135.2, 135.5, 174.8 (q); MS m/z (relative intensity, 70
eV) 366
(M+, 33), 337 (100), 266 (92), 169 (38).
EXAMPLE 33: 1-(7-Dipropylamino-6,7,8,9-tetrahydro-3H-benzo[e]indol-l-
yl)propan-l-one (10j, Chart 2).
A mixture of dipropyl-(6,7,8,9-tetrahydro-3H-benzo[elindol-7-yl)amine (70 mg,
0.26 mmol) and phosphorous oxychloride (48 }iL, 80 mg, 0.52 mmol) in N,N-
dimethyl
propionamide (250 }iI.) was heated at 80 C for 6 hours. After cooling 5 M
sodium
hydroxide was added and the mixture was heated again for 0.5 hour. After
cooling,
the mixture was extracted 3 times with ethyl acetate, dried (magnesium
sulfate),
filtered and evaporated to yield a residue to which 99% ethanol was added.
This
was then evaporated to dryness. A few repetions of the last procedure yielded
a
residue of 86 mg (100%) of the product.:
MS m/z (relative intensity, 70 eV) 326 (M+, 57), 297 (100), 226 (92), 170
(42),
196 (22).
EXAMPLE 34: 1-(7-Dipropylamino-6,7,8,9-tetrahydro-3H-benzo[e]indol-l-
~WO 94/21608 21575 8 6 ' PCT/US94/02800
-31-
yl)ethanone (10k, Chart 2).
A mixture of dipropyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)amine (55 mg,
0.20 mmol) and phosphorous oxychloride (130 pL, 213 mg, 1.39 mmol) in N,N-
dimethylacetamide (200 uL) was heated at 80 C for 4 hours. After cooling 5M
sodium hydroxide was added and the mixture was heated again for 0.5 hours.
After
cooling the mixture was extracted 3 times with ethyl acetate, dried (magnesium
sulfate), filtered and evaporated to yield a residue to which 99% ethanol was
added.
This was then evaporated to dryness. A few repetitions of the last procedure
yielded
a residue of 56 mg (89%) of the product, which was further purified on silica
using
methanol as eluant to give the pure material as a solid.:
m.p. 196-197 (free base); 'H NMR (300 MHz, CDCl3) S 0.90 (t, 6H), 1.50 (sxt,
4H), 1.55 (m, 1H), 2.15 (br d, 1H), 2.5 (t, 4H), 2.55 (s, 3H), 2.8-3.1(m, 3H),
3.3 (m,
1H), 3.55 (br d, 1H), 7.02 (d, J=8.3 Hz, 1H), 7.14 (d, J=8.3 Hz, 1H), 7.77 (d,
J=2.7
Hz, 1H), 8.9 (br s, 1H); 13C NMR (75.4 MHz, CDC1g) 8 12.0 (CH3), 22.0 (CHZ),
25.9
(CH2), 28.9 (CH3), 30.0 (CHZ), 33.5 (CH2), 52.7 (CH2), 56.7 (CH), 109.0 (CH),
120.5
(C), 123.7 (C), 126.5 (CH), 130.7 (C), 131.4 (C), 132.4 (CH), 135.5 (C), 193.1
(C); MS
m/z (relative intensity, 70 eV) 312 (M+, 62), 283 (100), 212 (93), 170 (48).
EXAMPLE 35: 6-Dipropylamino-1,3,5,6,7,8-hexahydro-benzo[f]indol-2-one (12b,
Chart 2).
To a solutionof di-n-Propyl-(5,6,7,8-tetrahydro-lH-benzo[f]indol-6-yl)amine
(30
mg, 0.11 mmol) and water (1 mL) in acetic acid (10 mL) was added
pyridiniumperbromide hydrobromide (40 mg, 0.12 mmol) in acetic acid at room
temperature. The resulting mixture was heated at 80 C over night. After
cooling,
the solution was evaporated to yield an aqueous residue, which was basified
using
3M sodium hydroxide. Extraction (ethyl acetate), drying (magnesium sulfate,
filtering and evaporation yeilded 26 mg (83%) the desired compound:
MS m/z (relative intensity, 70 eV) 286 (M*, 32), 186 (100), 257 (74), 158
(12).
EXAMPLE 36: 1-(6-Dipropylamino-5,6,7,8-tetrahydro-lH-benzoV]indol-3-
yl)propan-1-one (12c, Chart 2).
A mixture of dipropyl-(6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl)amine (60 mg,
0.22mmol) and phosphorous oxychloride (41 }iL, 68 mg, 0.44 mmol) in
dimethylpropionamide (200 uI.) was heated at 80 C for 2 hours. After cooling 5
M
sodium hydroxide (7 mL) was added and the mixture was heated again at 80 C for
15 minutes. After cooling the mixture was extracted 3 times with ethyl
acetate,
WO 94/21608 '~ 157586 -32- PCT/US94/02800 ~
~+
dried (magnesium sulfate), filtered and evaporated to yield a residue to which
99%
ethanol was added. This was then evaporated to dryness. A few repetitions of
the
last procedure yielded a residue of 55 mg (77%) of the product.:
MS m/z (relative intensity, 70 eV) 326 (M+, 43), 226 (100), 297 (92), 170
(25).
EXAMPLE 37: 9-Bromo-6-dipropylamino-5,6,7,8,-tetrahydro-lH-benzo[f]indole-
1,2-dione (8e, Chart 2) and 4-Bromo-7-dipropylamino-6,7,8,9-tetrahydro-3H-
benzo[e]indole-1,2-dione (7e, Chart 2).
Step A N-(6-Dipropylamino-5,6,7,8-tetrahydro-naphthylene-2-yl)-acetamide.
To a cold stirred solution (-0 C) of N,N-dipropyl-1,2,3,4-tetrahydro-
naphthylenee-
2,6-diamine (226 mg, 0.92 mmol) in dichloro methane (5 mL) was added acetyl
chloride (100 pL), 1.37 mmol) followed by triethyl amine (220 }iL), 1.57
mmol). The
mixture was then allowed to reach ambient temperature and was stirred for an
additional 30 minutes. The solvent was removed in vacuo and the residue taken
up
in dilute hydrochloric acid (10 mL). The aqueous solution was washed with
ethyl
acetate (2x10 mL) and then basified with a 15% sodium hydroxide solution.
Extraction with ethyl acetate, drying over magnesium sulfateand removal of the
solvent yielded a pale yellow solid (220 mg, 83%). This material was used in
the
subsequent step without further purification.:
MS (EI) m/z (relative intensity, 70 eV) 288 (M+, 37), 259 (100), 188 (72), 146
(44), 144 (16), 126 (14).
Ste,p B 5-Bromo-N,N-dipropyl-1,2,3,4-tetrahydro-naphthylenee-2,6-diamine
(6e, Chart 2) and 7-Bromo-N,N-dipropyl-1,2,3,4-tetrahydro-naphthylenee-2,6-
diamine
(6f, Chart 2).
A stirred solution of N-(6-Dipropylamino-5,6,7,8-tetrahydro-naphthylene-2-yl)-
acetamide (450 mg, 1.56 mmol) in acetic acid (10 mL) was heated to 90 C. Then
neat bromine (1.1 g, 6.88 mmol) was added in one portion. Stirring was
maintained
for 2 minutes after this addition and then the mixture was cooled. Saturated
aqueous sodium bisulfite was added (10 ml) followed by the addition of 5 M
sodium
hydroxide (50 mL). The aqueous solution was extracted with ethyl acetate
(2x2OmL), dried over magnesium sulfate and evaporated to yield a 4:1 mixture
of
the intermediate N-(3-Bromo-6-dipropylamino-5,6,7,8-tetrahydro-naphthylenee-2-
yl)-
acetamide and N-(1-Bromo-6-dipropylamino-5,6,7,8-tetraydro-naphthylenee-2-yl)-
acetamide as an oil (500 mg). The mixture of bromo acetamides was taken up in
6
~WO 94/21608 21575 86 PCT/US94/02800
-33-
M hydrochloric acid and heated at reflux temperature for 1 hour. After
cooling, the
acidic solution was basified by addition of 5 M sodium hydroxide. The solution
was
then extracted with ethyl acetate (3x20 mL), the combined extracts washed with
brine, drying over magnesium sulfate, and the solvent removed in vacco. The
resulting dark oil consisted of an isomeric mixture of the 7- and 5-bromo
anilines in
a 4:1 ratio (370 mg, 73%). All attempts to separate these isomers failed and
the
mixture was used in the subsequent step without further purification.:
(6e, Chart 2) 'H NMR (300 MHz, CDC13) S 0.88 (t, 6H), 1.45 (m, 4H), 2.05 (m,
1H), 2.45 (m, 4H), 2.6-2.8 (m, 3H), 2.88 (m, 1H), 3.02 (m, 1H), 4.00 (sb, 2H),
6.59 (d,
1H), 6.85 (d, 1H); MS (EI m/z (relative intensity, 70 eV) 324 (M+, 42), 326
(M++2,
41), 224 (100), 226 (96), 295 (88), 297 (86), 145 (58), 144 (38), 130 (20),
199 (12).
(6f, Chart 2). 'H NMR (300 MHz, CDC1g) 5 0.88 (t, 6H), 1.45 (m, 4H), 2.05
(m, 1H), 2.45 (m, 4H), 2.6-2.8 (m, 3H), 2.88 (m, 1H), 3.02 (m, 1H), 3.87 (sb,
2H), 6.49
(s, 1H), 7.13 (s, 1H); MS (EI) m/z (relative intensity, 70 eV) 324 (M+, 42),
326 (M++2,
41), 224 (100), 226 (96), 295 (88), 297 (86), 145 (58), 144 (38), 130 (20),
199 (12).
Step C 9-Bromo-6-dipropylamino-5,6,7,8,-tetrahydro-lH-benzo[fJindole-1,2-
dione (ie, Chart 2) and 4-Bromo-7-dipropylamino-6,7,8,9-tetrahydro-3H-
benzo[e]indole-1,2-dione (7e, Chart 2).:
A 4:1 mixture of 7-Bromo-N,N-dipropyl-1,2,3,4-tetrahydro-naphthylenee-2,6-
diamine and 5-Bromo-N,N-dipropyl-1,2,3,4-tetrahydro-naphthylenee-2,6-diamine
(4.0
g, 12.3 mmol) was converted into its hydrochloride salt by treatment with
ethanolic
hydrochloric acid followed by evaporation of the solvent. The resulting salt
was
dissolved in water (51 mL). To this solution was added chloral hydrate (2.23
g, 13.5
mmol), hydroxyl ammonium chloride (2.71 g, 39 mmol) and anhydrous sodium
sulfate (13.7 g). The mixture was then heated at reflux temperature for 1
hour.
After cooling ammonium hydroxide (72 mL, 3.2%) was added and the aqueous layer
extracted with several portions of ethyl acetate. Drying over magnesium
sulfate and
removal of the solvent in vacuo yielded 4.0 g of a mixture of N-(1-Bromo-6-
dipropylamino-5,6,7,8-tetrahydro-naphthylenee-2-yl)-2-hydroxyimino-acetamide
and
N-( 3-Bromo-6-dipropylamino-5, 6, 7, 8-tetrahydro-naphthylene-2-yl)-2-
hydroxyimino-
acetamide as a dark oil. This oil was dissolved in an ice cold mixture of
sulfuric
acid (61 mL) and water (6 mL). The mixture was stirred at ambient temperature
for
30 minutes and then heated to 80 C for another 30 minutes. After cooling, the
reaction mixture was poured into a crushed ice water mixture (750 mL) and
treated
with concentrated ammonium hydroxide until pH=9. The aqueous layer was
CA 02157586 2001-07-16
-34-
extracted with several portions of ethyl acetate and dried. Evaporation of the
solvent yielded 3.5 g(75%) of a mixture of 9-Bromo-6-dipropylamino-5,6,7,8,-
tetrahydro-lH-benzo[f]indole-1,2-dione and 4-Bromo-7-dipropylamino-6,7,8,9-
tetrahydro-3H-benzo[e]indole-1,2-dione as a red oil.
EXAMPLE 38: (9-Bromo-5,6,7,8-tetrahydro-lH-benzo[flindol-6-yl)-dipropyl
amine (llb, Chart 2) and 4-Bromo-6,7,8,9-tetrahydro-3H-benzo[e]indol-7-yl-
dipropyl-
amine (9i, Chart 2).
A diethyl ether solution (50 mL) of a mixture of 9-Bromo-6-dipropylamino-
5,6,7,8,-tetrahydro-lH-benzo[f]indole-1,2-dione and 4-Bromo-7-dipropylamino-
6,7,8,9-
tetrahydro-3H-benzo[e]indole-1,2-dione (3.5 g, 9.3 mmol) was added dropwise to
a
stirred suspension of lithium aluminum hydride (3.0) g in dry diethyl ether
(100
mL). After the addition was completed, atirring was continued for 2 hours at
ambient temperature. The reaction was then quenched by the consecutive
addition
of water (3.0 mL), 15% sodium hydroxide (3.0 mL) and water (9.0 mL). The solid
was filtered of through a CeliteTM pad and the solvent removed by evaporation
in vacuo
to yield a blue oil. The oil was subjected to chromatography on a silica
column to
yield 400 mg of pure (9-Bromo-5,6,7,8-tetrahydro-lH-benzo[t]indol-6-yl)-
dipropyl
amine. The residual fractions were concentrated and chromatographed on a
preparative HPLC system to yield a small amount of pure (4-Bromo-6,7,8,9-
tetrahydro-3H-benzo[e]indol-70y1)-dipropyl-amine.:
(lib, Chart 2) 'H NMR (300 MHz, CDC13) 8 0.90 (t, 6H), 1.45 (m, 4H), 1.65
(m, 1H), 2.10 (m, 1H), 2.47 (m, 4H), 2.7-3.0 (m, 4H), 3.17 (m, 1H), 6.5 (dd,
1H), 7.16
(dd, 1H), 7.33 (s, 1H), 8.18 (sb, 1H); 18C IVMR (75.4 MHz, CDC13) 8 11.9,
22.1, 26.4,
30.3, 32.9, 52.7, 56.8, 102.9, 106.4, 119.7, 124.4, 126.8,129.3, 130.4, 134.0;
MS (EI)
m/z (relative intensity, 70 eV) 348 (M*,), 350 (M*+2,), 321 (100), 319 (100),
248 (77),
250 (71), 169 (69), 168 (56), 167 (37), 154 (23).
(9i, Chart 2). 'H NMR (300 MHz, CDC1s) 8 0.90 (t, 6H), 1.50 (m, 4H), 1.65 (m,
1H), 2.12 (m, 1H), 2.60 (m, 4H), 2.8-3.2 (m, 5H), 6.56 (m, 1H), 7.10 (a, 1H),
7.22 (m,
1H, 8.26 (ab, 1H); MS (EI m./z (relative intensity, 70 eV) 348 (W, 51), 350 (
M*+2,
49), 321 (100), 319 (98), 248 (76), 250 (72), 169 (48), 168 (45), 167 (32),
154 (18), 223
(17).
EXAMPLE 39: 9-Bromo-6-dipropylamino-5,6,7,8-tetrahydro-lH-benzo[fJindol-3-
carbaldehyde (12d, Chart 2).
A solution of (9-Bromo-5,6,7,8-tetrahydro-lH-benzo[flindol-6-yl)-dipropyl
WO 94/21608 2157586 PCT/US94/02800
-35-
amine (50 mg, 0.14 mmol) in N,N-dimethyl formamide (3 mL) was cooled to 0 C.
To
this solution was added a cooled solution of phosphorus oxychloride (26 }zL,
0.28
mmol) in N,N-dimethyl formamide (1 mL), which had been prepared 20 minutes
previous to use. The mixture was stirred at ambient temperature for 30 minutes
and then for 30 minutes at 50 C an additional 30 minutes. The reaction mixture
was then treated with a 1M sodium hydroxide solution (5 mL) with heating and
stirring maintained for an additional hour. After cooling, the solution was
diluted
with water (25 mL) and extracted with several portions of diethyl ether. The
combined etheral extracts were washed with water, brine, dried over magnesium
sulfate, and the solvent evaporated. The residue was chromatographed on a
silica
column, using methanol as the eluant. This was then repeated with diethyl
ether as
eluant. The yield of the pure title compound was 50 mg (95%).:
'H NMR (300 MHz, CDC19) S 0.87 (t, 6H), 1.46 (m, 4H), 1.68 (m, 1H), 2.10 (m,
1H),
2.48 (m, 4H), 2.7-3.2 (m, 5H), 7.80 (s, 1H), 7.97 (s, 1H), 9.09 (sb, 1H), 9.98
(s, 1H);
13C NMR (75.4 MHz, CDC13) 8 11.9, 21.8, 26.4, 30.2, 32.8, 52.6, 56.5, 106.6,
120.0,
121.1, 123.2, 132.1, 134.0, 134.7, 135.5, 185.3; MS(EI) m/z (relative
intensity, 70 eV)
376 (Mi', 22), 378 (M++2, 22), 347 (100, 349 (94), 276 (70), 278 (65), 197
(44), 168
(30), 250 (13).
EXAMPLE 40: 6-Dipropyl-amino-1, 5, 6, 7-tetrahydro-l-az a-s-indacene-2, 3-
dione
(8f, Chart 2) and 7-dipropyl-amino-3,6,7,8-tetrahydro-l-aza-as-indacene-1,2-
dione (7f,
Chart 2).
Step A: Indan-2-yl-propyl-amine (31, Chart D).
A tetrahydrofuran (150 mL) solution of 2-indanone (11.0 g, 92 mmol), propyl
amine (12 mL, 146 mmol), sodium triacetoxy borohydride (35.0 g, 165 mmol), and
acetic acid (5 mL) was stirred at ambient temperature for 1 hour. The solution
was
then heated at reflux temperature for 2 hours. After cooling, the solvent was
evaporated and the residue taken up in concentrated hydrochloric acid (40 mL).
The
acidic solution was washed with diethyl ether and then basified (10 M sodium
hydroxide). Extraction of the aqueous layer with ether, drying over anhydrous
sodium carbonate and removal of the solvent yielded 10.9 g of the title
compound as
a yellow oil (75%).:
'H NMR (300 MHz, CDCIs) 5 0.92 (t, 3H), 1.55 (m, 2H), 2.65 (t, 2H), 2.76 (dd,
2H),
3.18 (dd, 2H), 3.62 (m, 1H), 7.14 (m, 4H); 13C NMR (75.4 MHz, CDC13) 8 11.9,
23.4,
40.0, 50.2, 59.6, 124.6, 126.3, 141.8; MS (EI) m/z (relative intensity, 70 eV)
175 (M+,
WO 94/21608 - ~ 21,,, 7-86 -36- PCT/LJS94/02800
212), 117 (100), 146 (92), 115 (49), 116 (21), 91 (19), 130 (17), 131 (15),
7712.
Step B: N-Indan-2-yl-N-propyl-propionamide (32, Chart 2).
Propionic acid chloride (2 mL, 2.1 g, 23 mmol) was added dropwise to a
solution of indan-2-yl-propylamine (4.0 g, 23 mmol) and triethyl amine in
dichloromethane (50 mL). The reaction mixture was stirred at room temperature
for
2 hours. Aqueous (10%) sodium carbonate was added and the mixture was shaken.
The organic phase was dried (magnesium sulfate), filtered and evaporated to
yield
4.5 g (85%) of the title compound as an oil.:
MS m/z (relative intensity, 70 eV) 231(M+, 2), 116 (100), 146 (8).
Step C: N-(5-Nitro-indan-2-yl)-N-propylpropionamide (34, Chart D) and N-(4-
Nitro-indan-2-yl)-N-propylpropionamide (33, Chart D).
To an ice-cooled solution of N-indan-2-yl-N-propyl-propionamide (4.5 g, 19.4
mmol) in nitromethane was added "nitrating acid" (20.2 mL, see previous
nitrations). The mixture was then stirred at ambient temperature overnight.
Ice
water was added and the mixture was extracted three times (diethyl ether). The
combined organic phases were dried (magnesium sulfate), filtered and
evaporated to
yield a residue (5.34 g, 99%) containing the two regioisomers. This material
was
used directly in the next step without separation of the isomers.:
Step D: N-(5-Amino-indan-2-yl)-N-propylpropionamide (36, Chart D) and N-
(4-amino-indan-2-yl)-N-propylpropionamide (35, Chart D).
To a solution of the isomeric mixture of (N-(4-and-5-Nitro-indan-2-yl)-N-
propylpropionamides, 5.3 g, 19.3 mmol) ammonium formate (33 g) was added under
inert atmosphere Pd/C (1.3 g). The mixture was stirred at ambient temperature
overnight and then filtered through a Celite-pad. The solution was evaporated
and
redissolved in water/diethyl ether. The aqueous phase were extracted two
additional
times with diethyl ether. The combined organic phases was dried (magnesium
sulfate), filtered and evaporated to yield a residue of 3.6 g (76%) containing
the two
isomers. The isomers were separated on sffica using diethyl ether as eluant
giving
3.0 g of N-(5-amino-indan-2-yl)-N-propylpropionamide and 0.5 g of N-(4-Amino-
indan-2-yl)-N-propylpropionamide.:
(36, Chart D) 'H NMR (300 MHz, CDC13) 8 0.82 (t, 3H), 1.15 (t, 3H), 1.55
(sept, 2H),
2.4 (two q, 2H), 2.9 (m, 4H), 3.15 (m, 2H), 3.6 (br s, 2H), 4.65 (q, 0.6H),
5.18 (q,
0.4H), 6.5 (m, 2H), 6.95 (m, 1H); 13C NMR (75.4 MHz, CDC1,,) 8 9.6, 11.2,
11.4, 22.2,
WO 94/21608 2157586 PCT/US94/02800
~
-37-
24.0, 26.7, 26.9, 35.6, 36.0, 36.6, 37.0, 44.2, 46.7, 55.4, 57.8, 111.0,
111.1, 113.6,
113.8, 124.7, 124.9, 129.8, 130.9, 141.4, 142.3, 145.2, 145.5, 173.3, 173.8;
MS m/z
(relative intensity, 70 eV) 246 (M+, 0.2), 131 (100).
(35, Chart D). 'H NMR (300 MHz, CDC13) 8 0.825 (t, 3H), 1.15 (t, 3H), 1.60
(sept,
2H), 2.4 (two q, 2H), 2.7-3.3 (m:s, 6H), 3.6 (br s, 2H), 4.75 (q, 0.5H), 5.20
(q, 0.5H),
6.5 (t, 1H), 6.64 (d, 1H); 13C NMR (75.4 MHz, CDC13) S 9.6, 11.2, 11.5, 22.2,
23.9,
26.8, 27.0, 33.3, 33.4, 36.7, 37.1, 44.3, 47.0, 55.1, 57.4, 112.6, 112.9,
114.4, 124.6,
125.7, 127.6, 128.0, 141.4, 142.3, 142.4, 173.3, 173.9; MS m/z (relative
intensity,
70 eV) 246 (M+, 0.2), 131 (100).
Step E: N,N-Dipropyl-indan-2,5-diamine (6g, Chart D, Chart 2).
A solution of N-(5-amino-indan-2-yl)-N-propylpropionamide (2.6 g, 10.5 mmol)
in dry diethyl ether (50 mL) was added to a slurry of lithiumaluminum hydride
(1.0
g, 26 mmol) in diethyl ether (150 mL) at ambient temperature. After stirring
for 1.5
hours water (1.0 mL) followed by 15% sodium hydroxide (1.0 mL) and water (3.0
mL). After stirring for 10 minutes, the inorganic material was filtered off
and the
resulting solution was evaporated to yield 2.2 g(90%) of the desired product
as an
oil.:
MS m/z (relative intensity, 70 eV) 232 (M', 18), 132 (100), 203 (95), 117
(12).
Step F: 6-Dipropyl-amino-1,5,6,7-tetrahydro-l-aza-s-indacene-2,3-dione (8f,
Chart 2) and 7-dipropyl-amino-3,6,7,8-tetrahydro-l-aza-as-indacene-1,2-dione
(7f,
Chart 2).
N,N-Dipropyl-indan-2,5-diamine was converted into its HCl-salt (giving 2.87
g, 9.5 mmol) and then dissolved in water (39 mL). Chloralhydrate (1.72 g, 10.3
mmol), hydroxylamine hydrochloride (2.08, 30 mmol) and anhydrous sodium
sulfate
(10.5 g) was added. The mixture was refluxed for 1 hour. After cooling,
diluted
ammonia was added for basification. The aqueous mixture was extracted with
ethyl
acetate three times, dried (magnesium sulfate), filtered and evaporated. This
material was cooled and to it was added cooled "wet sulfuric acid" (65 mL of
water/conc sulfuric acid 1:9). The resulting mixture was stirred 0.5 hour at
ambient
temperatue and then heated at 80 C for 1 hour. After cooling, the mixture was
poured on ice and basified using conc aqueous ammonia. The aqueous phase was
extracted several times with ethyl acetate and the resulting organic phase was
dried
(magnesium sulfate), filtered and evaporated to yield 2.47 g (93%) of the
isomeric
mixture.
WO 94/21608 215 75) 8 6 -38- PCTIUS94/02800 ~
EXAMPLE 41: Dipropyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)amine (11e,
Chart 2) and Dipropyl-3,6,7,8-tetrahydro-3-aza-as-indacen-7-yl)amine (9j,
Chart 2).
The isomeric mixture of 6-dipropyl-amino-1,5,6,7-tetrahydro-l-aza-s-indacene-
2,3-dione and 7-dipropyl-amino-3,6,7,8-tetrahydro-l-aza-as-indacene-1,2-dione
(1.38
g, 4.8 mmol) was dissolved in diethyl ether (50 mL) and added dropwise to a
slurry
of lithium aluminum hydride (1.0 g, 26.4 mmol) in diethyl ether (100 mL). The
mixture was stirred at ambient temperature for 1 hour. Water (1.0 mL),
followed by
15% sodium hydroxide (1.0 mL) and water (3.0 mL) was added cautiously and the
mixture was stirred for 10 minutes. The solid material was filtered off and
the
organic solution was evaporated to yield a residue (1.17 g, 95%) containing
the two
regioisomers. This material was subjected to silica column chromatography and
eluted with dusopropyl ether/ethanol (96:4). This material was subjected to
silica
column chromatography and eluted with diisopropyl ether/ethanol (96:4).
Dipropyl-
(3,6,7,8-tetrahydro-3-aza-as-indacen-7-yl)amine (118 mg) was first collected,
followed
by a mixed fraction (176 mg) and dipropyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-
6-
yl)amine (535 mg) giving a total recovery of 829 mg (67%).:
(11e, Chart 2) m.p. 110-112 (free base); 'H NMR (300 MHz, CDC13) 8 0.90 (t,
4H),
1.50 (sxt, 4H), 2.52 (t, 4H), 2.9 (m, 2H), 3.1 (two d, 2H), 3.7 (m, 1H), 6.44
(m, 1H),
7.10 (m, 1H), 7.18 (s.l), 7.40 (s, 1H), 8.06 (br s, 1H); 13C NMR (75.4 MHz,
CDC1$) S
12.1, 20.0, 36.5, 37.0, 53.6, 64.2, 102.2, 106.5, 115.6, 123.5, 127.0, 134.0,
135.3, 136.7;
MS m/z (relative intensity, 70 eV) 256 (M', 19), 227 (100), 156 (89), 129
(19).
(9j, Chart 2). m.p. 105-107 (free base); 'H NMR (300 MHz, CDC13) S 0.94 (t,
6H),
1.58 (sxt, 4H), 2.58 (t, 4H), 3.1 (m, 3H), 3.3 (two d, 1H), 3.9 (pent, 1H),
6.44 (m, 1H),
7.05 (d, J=8.1 Hz, 1H), 7.2 (m.2), 8.5 (br s, 1H); MS m/z (relative intensity,
70 eV)
256 (M+, 20), 227 (100), 156 (78), 129 (17).
EXAMPLE 42: 6-Dipropylamino-1,5,6,7-tetrahydro-l-aza-s-indacen-3-
carbaldehyde (12e, Chart 2).
A solution of dipropyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)amine (100 mg,
0.39 mmol) in dimethyl formamide (5 mL) was added to a solution of phosphorous
oxychloride (200 }1L, 329 mg, 2.14 mmol) in dimethyl formamide (5 mL) at -5 C.
The
mixture was heated at 50 C for 2 hours, poured on ice and treated with 5 M
sodium
hydroxide and heated again for 20 minutes. After cooling, the mixture was
extracted using dichloromethane. The combined organic extracts were dried
(magnesium sulfate), filtered and evaporated to yield a residue of 70 mg,
which was
purified on silica using methanol as eluant, yielding 33 mg (30%) of the pure
WO 94/21608 Fi cl 1575 Q 6 PCT/US94/02800
-39- 0.
compound as an oil.:
'H NMR (300 MHz, CDC19) S 0.90 (t, 6H), 1.5 (m, 4H), 2.55 (t, 2H), 2.8-3.3
(m:s, 4H),
3.65 (m, 1H), 7.20 (s, 1H), 7.75 (s, 1H), 8.1 (s, 1H), 9.55 (s, 1H), 10.0 (s,
1H);
13C NMR (75.4 MHz, CDC13) S 12.0, 19.9, 36.4, 37.1, 53.4, 107.2, 117.0, 119.3,
135.4,
119.3, 123.5, 135.4, 136.3, 137.4, 139.0, 185.2; MS m/z (relative intensity,
70 eV)
284 (M', 11), 255 (100), 184 (40), 156 (18).
EXAMPLE 43: 1-( 6-Dipropylamino-1, 5, 6, 7-tetrahydro-l-az a-s-indacen-3-
yl)propan-1-one (12f, Chart 2).
A solution of dipropyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)amine (53 mg,
0.21 mmol) in N,N-dimethylpropionamide (200 }iL) treated with phosphorous
oxychloride (41 l.iL, 66 mg, 0.44 mmol) and thereafter heated at 80 C for 2
hours.
After cooling 3M sodium hydroxide (7 mL) was added. The resulting mixture was
heated again at 80 C for 15 minutes and then cooled. This aqueous mixture was
extracted with ethyl acetate three times. The combined organic extracts was
dried
(magnesium sulfate), filtered and evaporated to yield a residue, which was
repeatedly redissolved in 99% ethanol and evaporated until the residue showed
a
constant weight of 65 mg (99%) of the desired compound.:
MS m/z (relative intensity, 70 eV) 326 (M+, 42), 226 (100), 207 (43), 170
(25), 196
(20).
EXAMPLE 44: (6-Dipropylamino-1,5,6,7-tetrahydro-l-aza-s-indacen-3-yl)-
phenyl methanone (12 g, Chart 2).
A mixture of dipropyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)amine (31 mg,
0.12 mmol) N,N-dimethylbenzamide (36 mg, 0.24 mmol) and phosphorous
oxychloride (14 }iL, 23 mg, 0.15 mmol) was heated at 85 C overnight. After
cooling,
3M sodium hydroxide (4 mL) was added and the resulting mixture was heated at
85 C for 1 hour. The mixture was cooled and extracted (ethyl acetate) three
times.
The organic solution was dried (magnesium sulfate), filtered and evaporated to
yield
46 mg (100%) of the desired material.: MS m/z (relative intensity, 70 eV) 360
(M+,
12), 331 (100), 105 (38), 260 (31).
EXAMPLE 45: 7-Dipropylamino-1, 6, 7, 8-tetrahydro-l-az a-as-indacene-2, 3-
dione
(2c, Chart 1).
Step A: N,N-Dipropyl-indan-2,4-diamine (la, Chart 1, Chart D).
A solution of N-(4-amino-indan-2-yl)-N-propylpropionamide (0.5 g, 2.0 mmol)
WO 94/21608 PCT/US94/02800
2157580 -40-
in dry diethyl ether (10 mL) was added to a slurry of lithium aluminum hydride
(0.2
g, 5.4 mmol) in diethyl ether (50 mL) at ambient temperature. After stirring
for 1.5
hours water (0.2 mL) followed by 15% sodium hydroxide (0.2 mL) and water (0.6
mL). After stirring for 10 minutes, the inorganic material was filtered off
and the
resulting solution was evaporated to yield 0.42 g(90%) of the desired product
as an
oil.: MS m/z (relative intensity, 70 eV) 232 (M+, 14), 203 (100), 132 (80), 72
(17).
Step B: 7-Dipropylamino-1,6,7,8-tetrahydro-l-aza-as-indacene-2,3-dione (2c,
Chart 1).
N,N-Dipropyl-indan-2,4-diamine was converted into its HCL salt (giving 0.52
g, 1.72 mmol) and dissolved in water (7 mL). Chloralhydrate (312 mg, 1.88
mmol),
hydroxylamine hydrochloride (378 mg, 5.4 mmol) and anhydrous sodium sulfate
(1.91 g) was added. The mixture was refluxed for 1 hour. After cooling,
diluted
ammonia was added for basification. The aqueous mixture was extracted with
ethyl
acetate three times, dried (magnesium sulfate), filtered and evaporated. The
material was cooled and to it was added cooled "wet sulfuric acid" (12 mL of
water/conc sulfuric acid 1:9). The resulting mixture was stirred 0.5 hour at
ambient
temperature and the heated at 80 C for 1 hour. After cooling the mixture was
poured on ice and basified using concentrated aqueous ammonia. The aqueous
phase was extracted several times with ethyl acetate and the resulting organic
phase was dried (magnesium sulfate), filtered and evaporated to yield 0.43
g(83%).:
EXAMPLE 46: Dipropyl-(1,6,7,8-tetrahydro-l-aza-as-indacen-7-yl)amine (3c,
Chart 1).
7-Dipropylamino-1,6,7,8-tetrahydro-l-aza-as-indacene-2,3-dione (0.42 mg, 1.46
mmol) was dissolved in dry diethyl ether (20 mL) and dropped to a slurry of
lithium
aluminum hydride (300 mg, 7.9 mmol) in dry diethyl ether (50 mL). The mixture
was stirred at ambient temperature for 1 hour. Water (300 pL) followed by 15%
sodium hydroxide (300 }iL) and water (900 pL) was added and the mixture was
stirred for 10 minutes. The solid material was filtered off and the organic
solution
was evaporated to yield a residue of 380 mg, which was chromatographed on a
silica
column using diisopropyl ether/ethanol (96:4) giving 175 mg (47%) of the pure
material. The fumaric acid salt was prepared and recrystallized from ethanol.:
m.p. 178-183 (Fumarate); 'H NMR 9300 MHz, CDClg) 8 0.90 (t, 6H), 1.53 (sxt,
4H),
3.0 (m, 2H), 3.1 (m, 2H), 3.9 (pent, 1H), 6.55 (t, 1H), 6.99 (d, 1H), 7.15 (t,
1H), 7.53
WO 94/21608 2 1575 8 6 PCT/US94/02800
-41-
(d, 1H), 7.95 (br s, 1H); '3C NMR (75.4 MHz, CDC13) S 12.0, 20.5, 33.3, 36.5,
53.5,
63.4, 103.2, 116.8, 118.9, 123.2, 123.3, 126.5, 132.7, 136.1; MS m/z (relative
intensity, 70 eV) 256 (M+, 22), 227 (100), 156 (68), 129 (14).
EXAMPLE 47: Benzyl-propyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)amine
(llf, Chart 2) and Benzyl-propyl-(3,6,7,8-tetrahydro-3-aza-as-indacen-7-
yl)amine (9k,
Chart 2).
Step A: N-Indan-2-yl-N-propyl-benzamide (39, Chart D).
To a stirred solution of Indan-2-yl-propyl-amine (20.0 g, 114 mmol) in
dichloro methane (200 mL) triethyl amine (17.4 mL, 125 mmol) was added. To
this
solution was then dropwise added benzoyl chloride (17.6 g, 125 mmol), followed
by
stirring at ambient temperature for 2 hours. The reaction was finally quenched
by
addition of dilute hydrochloric acid (50 mL, 10% HC1). The organic layer was
separated, washed with water and dried over magnesium sulfate. Evaporation of
the solvent yielded 31.5 g(99%) of the title compound as a dark oil.:
MS (EI) m/z (relative intensity, 70 eV) 279 (M', 4), 116 (100), 164 (89), 105
(83), 77
(52), 115 (20), 117 (15), 165 (11).
Step B: N-(5-Nitro-indan-2-yl)-N-propylbenzamide (40, Chart D) and N-(4-
Nitro-indan-2-yl)-N-propylbenzamide (41, Chart D).
To an ice-cooled solution of N-indan-2-yl-N-propyl-benzamide (16.5 g, 59
mmol) in nitromethane (250 mL) was gently added dropwise "nitrating acid" (80
m:;
6.6% nitric acid, 80.3% sulfuric acid and 13.1% water). The resulting mixture
was
stirred for 4 hours and poured on ice water. The mixture was shaken and the
organic solution was dried (magnesium sulfate), filtered and evaporated to
yield
17.6 g (92%) of a mixture containing the two isomers, N-(5-nitro-indan-2-yl)-N-
propylbenzamide and N-(4-nitro-indan-2-yl)-N-propylbenzamide in a 5:1
relationship.:
(40, Chart D) MS m/z (relative intensity, 70 eV) 324 (M+ , 17), 105 (100), 77
(47),
164 (43), 134 (29).
(41, Chart D) MS m/z (relative intensity, 70 eV) 324 (M+, 5), 105 (100), 307
(49), 77
(47), 134 (31) 164 (23).
Step C: N-(5-Amino-indan-2-yl)-N-propylbenzamide (42, Chart D) and N-(4-
Amino-indan-2-yl)-N-propylbenzamide (43, Chart D).
The isomeric mixture containing N-(5-Nitro-indan-2-yl)-N-propylbenzamide
WO 94/21608 21 57586 -42- PCT/US94/02800
and N-(4-Nitro-indan-2-yl)-N-propylbenzamide (17.3 g, 53 mmol) was mixed with
ammonium formate (30 g, 470 mmol) in ethanol (300 mL). Under an inert
atmosphere was added 2.5 g Pd/C and the reaction mixture was stirred at
ambient
temperature overnight and then filtered through a Celite-pad. The solution was
evaporated and redissolved in diethyl ether/water, shaken and the aqueous
phase
was extracted two additional times. The combined organic phases was dried
(magnesium sulfate), filtered and evaporated to yield a residue of 15 g
containing
the two isomers. This material was roughly purified on silica gel column using
diethyl ether as eluant giving 9.1 g (58%) of the two isomers. Of this
material 4.3 g
were further chromatographed in silica using diethyl ether/hexane (3:1) as
eluant,
which yielded 3.1 g of N-(5-nitro-indan-2-yl)-N-propylbenzamide and 1.3 g of a
mixture of both isomers.:
MS m/z (relative intensity, 70 eV) 294 (M+, 0.1), 131 (100), 77 (18), 105
(16).
Step D: N-Benzyl-N-propyl-indan-2,5-diamine (63, Chart 2, Chart D).
A solution of N-(5-amino-indan-2-yl)-N-propylbenzamide (3.0 g, 10.2 mmol) in
diethyl ether (50 mL) was added to a slurry of lithium aluminum hydride (1.4,
36.9
mmol) in diethyl ether (60 mL). After stirring overnight, water (.14 mL), 15%
sodium hydroxide (1.4 mL) and water (4.2 mL) was added in consecutive order.
The
mixture was stirred for 0.5 hour and then filtered through a Celite pad.
Subsequent
evaporation yielded 2.60 g(91%) of the desired material.:
'H NMR (300 MHz, CDC13) 8 0.86 (t, 3H), 1.50 (sxt, 2H), 2.48 (t, 2H),2.8-3.05
(m:s,
4H), 3.52 (br s, 2H), 3.65 (s, 2H), 3.72 (pent, 1H), 6.50 (d, J-7.9 Hz, 1H),
6.56 (s, 1H),
6.97 (d, J=7.9 Hz, 1H), 7.2-7.5 (m, 5H); 1gC NMR (75.4 MHz, CDC1s) 8 11.8,
20.0,
34.9, 36.0, 52.9, 55.3, 62.3, 111.3, 113.4, 124.8, 126.5, 128.0, 128.5, 132.0,
140.6,
143.2, 144.8; MS m/z (relative intensity, 70 eV) 280 (M+, 21), 251 (100), 91
(48), 132
(42), 222 (22), 120 (17).
Step E: 6-(Benzyl-propyl-amino)-1,5,6,7-tetrahydro-l-aza-s-indacene-2,3-dione
(8g, Chart 2) and 7-(Benzyl-propyl-amino)-3,6,7,8-tetrahydro-l-aza-as-indacene-
1,2-
dione (7g, Chart 2).
A mixture of N-benzyl-N-propyl-indan-2,5-diamine hydrochloride (3.5 g, 9.9
mmol), chloral hydrate (1.8 g, 11 mmol), hydroxylamine hydrochloride (2.2 g,
32
mmol) and dry sodium sulfate (11 g) was refluxed for 1.5 hours. After cooling
the
mixture was basified using diluted ammonia. The resulting mixture was
extracted
(ethyl acetate), dried (magnesium sulfate), filtered and evaporated to a
residue of 3.9
CA 02157586 2001-07-16
-43-
g. This material was cooled and treated with a cooled solution of water (7 ml)
in
conc. sulfuric acid (63 mL). The resulting mixture was heated at 80 C for 45
min.
After cooling, the mixture was poured on ice and basifled using conc. ammonia
in
water. Extraction (ethyl acetate), drying (magnesium sulfate), filtering and
evaporation yielded 3.17 g(96%) of the desired regioisomers:
Step F: The isomeric mixture of 6-(benzyl-propyl-amino)-1,5,6,7-tetrahydro-l-
aza-s-indacene-2,3-dione and 7-(Benzyl-propyl-amino)-3,6.7,8-tetrahydro-l-aza-
as-
indacene-1,2-dione (3.17 g, 9.5 mmol) in dry diethyl ether (50 mL) was added
dropwise over a period of 20 min to a slurry of lithium aluminum hydride in
dry
diethyl ether (150 mL) and stirred over night at ambient temperature. Water (2
mL) followed by 15% sodium hydroxide (2 mL) and water (6mL) was added and the
mixture was stirred for 2 hrs. The solid material was filtered off and the
organic
solution was evaporated to yield a residue of 2.75 g. This material was
subjected to
silica column chromatography, eluating with hexane fraction/diethyl ether
(3:1).
Pure benzyl-propyl-(3,6,7,8-tetrahydro-3-aza-a.s-indacen-7-yl)amine (250 mg)
followed
by a mixed fraction (110 mg) and benzyl-propyl-(1,5,6,7-tetrahydro-l-aza-s-
indacen-
6-yl)amine (920 mg) was collected as oils giving a total recover of 1.28 g (44
%):
(11f, Chart 2) 'H NMR (300 MHz, CDC13) 5 0.85 (t, 6H), 1.51. (sxt, 4H), 2.50
(t, 4H), 2.95-3.2 (m, 4H), 3.70 (s, 2H), 3.75 (pent, 1H), 6.55 (m, 1H), 7.10
(t, 1H),
7.15-7.45 (m, 7H), 8.0 (br s, 1H); 'C NMR (75.4 MHz, CDC13) S 11.9, 20.0,
35.7,
36.2, 53.1, 55.5, 63.1, 102.2, 106.5, 115.6, 123.5, 126.5, 127.0, 128.1,
128.6, 134.1,
135.3, 136.8, 140.8; MS m/z (relative intensity, 70 eV) 304 (M', 26), 275
(100), 156
(39), 246 (31), 91 (32).
(9k, Chart 2). 'H NMR (300 MHz, CDCIa) 8 0.90 (t, 6H), 1.56 (sxt, 4H), 2.56
(t, 4H), 3.05-3.35 (m, 4H), 3.70 (a, 2H), 3.95 (pent, 1H), 6.48 (s, 1H), 7.06
(d, Je8.3
Hz, 1H), 7.15-7.50 (m, 7H), 8.18 (br s, 1H); 18C NMR (75.4 MHz, CDCI,) 8 12.0,
20.3,
34.4, 36.0, 53.0, 55.4, 62.3, 100.6, 109.2, 118.6, 124.2, 124.7, 126.6, 128.1,
128.6,
132.7, 133.2, 135.0, 140.8; MS m/z (relative intentity, 70 eV) 304 (M', 22),
275 (100),
156 (40), 207 (37), 246 (31), 91 (30).
EXAMPLE 48: Propyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)-amine (11g,
Chart 2.
To a solution of benzyl-propyl-(3,6,7,8-tetrahydro-3-aza-as-indacen-7-yl)amine
(9k, Chart 2) (0.87 g, 2.86 mmol) in ethanol (50 mL) was added Pd/C (0.5 g,
inert
atmosphere) and ammonium formate (1.5 g, 24 mmol). The mixture was heated at
50 C for 30 min., cooled and filtered through a Celite-pad. The organic
solution was
WO 94/21608 PCT/US94/02800
2157586 -44-
evaporated and the residue dissolved in water and basified (10% sodium
carbonate).
Extraction with diethyl ether, drying (magnesium sulfate), filtering, and
evaporation
yielded 0.45 g (74%) of the title compound.
'H NMR (300 MHz, CDCl3) S 0.95 (t, 3H), 1.4 (br s, 1H), 1.55 (sxt, 2H), 2.67
(t, 2H), 2.83 (m, 2H), 3.25 (m, 2H), 3.65 (pent, 1H), 6.45 (m, 1H), 7.11 (m,
1H), 7.18
(s, 1H), 7.43 (s, 1H), 8.3 (br s, 1H); 13C NMR (75.4MHz, CDC13) 8 11.9, 23.5,
39.5,
40.0, 50.2, 60.6, 102.1, 106.8, 115.9, 123.6, 127.1, 133.8, 135.4, 136.4; MS
m/z
(relative intensity, 70 eV) 214 (M+, 100), 145 (97), 156 (91), 185 (77), 155
(71), 130
(59).
EXAMPLE 49: Cyclopropylmethyl-propyl-(1,5,6,7-tetrahydro-l-aza-s-indacen 6-
yl)-amine ( llh, Chart 2).
Step A: Cyclopropanecarboxylic acid propyl-(1,5,6,7-tetrahydro-l-aza-s-
ubdaceb-6-yl)-amide.
To a stirred solution of propyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)-
amine
(11g, Chart 2) (75 mg, 0.35 mmol) in dichloromethane (5 mL) was added
triethylamine (0.1 mL), followed by the addition of cyclopropanecarbonyl
chloride (40
}iL, 46 mg. 0.44 mmol). The mixture was stirred at ambient temperature for 2
hrs.
The reaction was quenched by the addition of 10% aqueous sodium carbonate and
the mixture stirred. The organic phase was washed with dilute hydrochloric
acid,
dried (megnesium sulfate and concentrated to yield 100 mg (100%) of the
desired
compound.
Step B: Cyclopropylmethyl-propyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)-
amide (100 mg, 0.35 mmol) in dry diethyl ether was added in small porions
lithium
aluminum hydride (90 mg, 2.37 mmol). The reaction mixture was stirred over
night
and then quenched by the consecutive addition of water (90 }iL), 15% sodium
hydroxide (90 }iL) and water (270 pL). After stirring for 15 min., the solid
material
was filtered off and the solvent removed in vacuo to yield a residue of 78 mg
(83%)
of the title compound.
MA m/z (relative intensity, 70 eV) 268 (M, 30), 239 (100), 156 (42), 155 (23),
210
(20) 192(9).
EXAMPLE 50: 1-[6-(Cyclopropylmethyl-propyl-amino)-1,5,6,7-tetrahydro-l-aza-
s-indacen-3-yl]-propan-l-one (12h, Chart 2.)
To a solution of cyclopropylmethyl-propyl-(1,5,6,7-tetrahydro-a-aza-s-indacen-
6-yl)amine (llh, Chart 2) (62 mg, 0.23 mmol) in N,N-dimethyl propionamide (200
uL) was added phosphorous oxychloride (40 }iL, 66 mg, 0.43 mmol). The mixture
CA 02157586 2001-07-16
-45-
was heated at 80 C for 1 hr. Then 15% sodium hydroxide (7mL) was added and the
mixture was heated for an additional 15 min. After cooling, the aqueous
mixture
was extracted with ethyl acetate. The organic extract was dried (magnesium
sulfate)
and concentrated to yield 74 mg (100%) of the desired material.
MS m\z (relative intensity, 70 eV) 324 (M', 20), 295 (100), 212 (24), 253
(16),
182 (15), 154 (9).
EXAMPLE 51: Cyclopropylmethyl-propyl-(3-propyl-1,5,6,7-tetrahydro-l-aza-s-
indacen-6-yl)-amine (12i, Chart 2).
To a stirred solution of 146-(cyclopropylmethyl-propyl-amino)-1,5,6,7-
tetrahydro-1-aza-s-indacen-3-yl]-propan-1-one (12h, Chart 2) (58 mg, 0.18
mmol) in
dry diethyl ether (4 mL) was added in small portions lithium aluminum hydride
(40
mg, 1.06 mmol). The reaction mixture was stirred over night and then quenched
by
the consecutive addition of water (40 pL), 15% sodium hydroxide (40 }tL) and
water
(120 uI.). After stirring for 15 min., the solid material was filtered off and
the
solvent removed in uacuo to yield a residue of 28 mg (50%) of the title
compound.
MS m\z (relative intensity, 70 eV) 310 (M', 27), 281 (100), 198 (33), 168
(219), 252 (15), 155 (8).
EXAMPLE 52: [2-(4-Fluoro-phenyl)-ethyl]-propyll-(1,5,6,7-tetrahydro-l-aza-s-
indacen-6-yl)-amine (11i, Chart 2).
To a stirred solution of propyl-(1,5,6,7-tetrahydro-l-aza-s-indacen-6-yl)amine
(11 g, Chart 2) (90 mg, 0.42 mmol) in dichloromethane (5 mL) was added
triethylamine (0.1 mL), followed by the addition of 4-fluoro-phenyl-acetyl
chloride
(75 mg, 0.43 mmol). The mixture was stirred at ambient temperature for 2 hrs.
The reaction was quenched by the addition of 10% aqueous sodium carbonate and
the mixture stirred. The organic phase was washed with dilute hydrochloric
acid,
dried (magnesium sulfate and concentrated to yield 140 mg (96%) of the desired
compound.
MS m\z (relative intensity, 70 eV) 348 (M', 0.2), 155 (100), 109 (8), 129 (3).
Steg$: [2-(4-Fluoro-phenyl)-ethyl]-propyl-(1,6,6,7-tetrahydro-l-aza-s-indacen-
6-yl)-amine ((11i, Chart 2).
To a stirred solution of 2-(4-fluoro-phenyl)-ethyl]-propyl-(1,5,6,7-tetrahydro-
l-
aza-s-indacen-6-yl)-acetamide (140 mg, 0.40 mmol) in dry diethyl ether (4 mL)
was
added in small portions lithium aluminum hydride (150 mg, 3.95 mmol). The
reaction mixture was stirred for 2 hrs. and then quenched by the consecutive
addition of water (150 pL), 15% sodium hydroxide (150 }i.L) and water (450
}iL).
After stirring for 15 min., the solid material was filtered off and the
solvent removed
WO 94/21608 8 6 -46- PCTIUS94/02800 '
in vacuo to yield a residue of 108 mg (81%) of the title compound.
MS m\z (relative intensity, 70 eV) 334 (M+, 2), 227 (100), 156 (58), 228 (17),
155 (16), 129 (13).
EXAMPLE 53: N,N-Diethyl-2-(6-{[2-(4-fluoro-phenyl)ethyl]propylamino}-1,5,6,7-
tetrahydro-l-aza-s-indacen-3-yl)-2-oxo-acetamide (12j, Chart 2).
An ice-cooled solution of [2-(4-fluoro-phenyl)-ethyl]-propyl-(1,5,6,7-
tetrahydro-
1-aza-s-indacen-6-yl)-amine (11i, Chart 2) (100 mg, 0.30 mmol) in dry diethyl
ether
(1 mL) was treated dropwise with oxalyl chloride (76 }iL, 110 mg, 0.87 mmol).
A
precipitate was formed and more diethyl ether (1 mL) was added to facilitate
stirring, which was continued for 0.5 hr. Then, a solution of diethyl amine
(300 uL,
218 mg, 3.0 mmol) in dry diethyl ether (0.5 mL) was added. Stirring at ambient
temperature was continued for 2 hrs. Finally, saturated sodium hydrogen
carbonate
was added and the organic layer separated. The aqueous layer was extracted
with
ethyl acetate. The combined organic phases were dried (magnesium sulfate),
filtered
and evaporated to yield a residue (67 mg) of crude material consisting mainly
of the
title compound.
MS m\z (relative intensity, 70 eV) 461(M+, 2), 354 (100), 283 (28), 182 (26),
355 (25), 154 (13), 72 (10).
Pregaration 1 trans-4-Benzyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (54 Chart E).
Ste A: trans-Trifluoro-methanesulfonicacid 4-benzyl-1,2,3,4,4a,5,6,lOb-
octahydro-benzo[[f3quinolin-7-yl ester (44, Chart E).
A solution of trans-7-hydroxy-4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-
benzo[fJquinoline (3.3 g, 13.47 mmol, (for preparation see Wikstr6m et al., J.
Med.
Chem., 1982, 25, 925-931), triethyl amine (2.04 g, 20.2 mmol) in 300 mL of
dichloromethane was cooled to -30 C. Then triflic anhydride (5.7 g, 20.2 mmol)
in
mL of dichloromethane was added dropwise. The reaction mixture was allowed
to reach room temperature and was then stirred for 2 hrs at 25 C. The reaction
mixture was quenched with cold water, the organic layer separated and washed
with
30 2 portions of cold 5% HCl-solution. Following a wash of the organic portion
with brine and drying (magnesium sulfate), the solvent was removed under
reduced
pressure and the residue was chromatographed on a silica column with
methanol:dichloromethane (1:19 (v/v)) as eluant. The solvents from the
collected
fractions containing pure 44 were evaporated yielding a pale oil 4.87 g (85%).
MS m\z (relative intensity, 70 eV) 425 (M, 42), 292 (78), 264 (21), 91 (100).
Step B: trans-4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[f]quinolin-7-
CA 02157586 2001-07-16
-47-
carboxylic acid methyl ester (46 Chart E).
A mixture of compound trans-trifluoro-methanesulfonic acid 4-benzyl-
1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-yl ester (3.1 g, 7.3 mmol),
triethyl
amine (2 ml, 14.6 mmol), pethanol (12.3 ml, 0.28 mol), palladium acetate (50
mg,
0.22 mmol), 1.3-bis-(diphenylphosphino)propane (90 mg, 0.22 mmol) in dimethyl
sulfoxide (25 mL) was stirred at room temperature for 15 min or until all
particles
were dissolved (Dolle et al., J. Chem. Soc. Chem. Commun., 1987, 904-905). A
stream of CO(g) was passed through the solution for 4-5 min. and then the
reaction
vessel was placed under a alightly positive pressure of carbon monoxide (g) (1
atm.).
The temperature was increased to 70 C (oil bath). After 6 h GC analysis
revealed a
complete absence of the starting triflate and indicated trans-4-benzyl-
1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-carboxylic acid methyl ester
as the
major product. The reaction mixture was allowed to cool to room temperature.
Water (200 mL) was then added and the aqueous solution was extracted with 5
portions of diethyl ether. The combined organic phases were washed with water
until neutral, dried (magnesium sulfate) and evaporated. The residue was
chromatographed on a silica column using methanol:dichloromethane (1:19 (v/v))
as
eluant. The solvents from the collected fractions containing pure 46 were
evaporated yielding 1.9 g (78%).
MS m\z (relative intensity, 70 eV) 335 (M' 100), 268 (20), 244 (23), 91 (50).
Step C: trans-(4-Benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[f)quinolin-7-yl)-
methanol (48 Chart E).
A solution of trans-4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[f)quinolin-7-
carboxylic acid methyl ester (1.9 g, 5.67 mmol) in 50 mL of dry diethyl ether
was
cooled to 0 C. Solid lithium aluminum hydride (1.5 g, 39.5 mmol) was then
added in
small portions. The mixture was stirred for 1 hour at 0 C. The reaction was
then
terminated by the addition of water (1 mL), 15% sodium hydroxide solution (1
mL)
and finally water (3 mL). The resulting mixture was filtered through a pad of
Celite, dried (magnesium sulfate) and evaporated to dryness. The residue was
chromatographed on a silica column with methanol:dichloromethane (1:19 (v/v))
as
eluant. The solvents from the collected fractions containing pure 48 were
evaporated yielding 1.5 g(86%) as an oil.
MS m\z (relative intensity, 70 eV) 307 (M' 100), 230 (10), 158 (15), 91 (47).
Step D: trans-4-Benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[flquinolin-7-
carbaldehyde (50 Chart E).
To a solution of trans-(4-benzyl-1,2,3,4,4a,5,6,lOb-octahydro-benzo[f7-
quinolin-
WO 94/21608 2 15 r~ 8 g~ PCT/US94/02800
( i9 -48-
7-yl)-methanol (1.5 g, 4.88 mmol) in 80 mL of dichloromethane was added
pyridinium chlorochromate (1.5 g, 7.44 mmol) in small portions. The mixture
was
stirred for 1 hr. at room temperature and then quenched with cold 10% sodium
carbonate solution (50 ml), the layers separated and the waterphase extracted
with
2 portions of dichloromethane (100 ml). The combined organic phases were dried
(magnesium sulfate) and the solvent was removed under reduced pressure. The
residue was chromatographed on a silica column with methanol:dichloromethane
(1:19 (v/v)) as eluant. The solvents from the collected fractions containing
pure 50
were evaporated yielding 1.2 g(80%) as an oil.
MS m\z (relative intensity, 70 eV) 305 (M+ 100), 214 (16), 159 (20), 129 (17),
91 (61).
Step E: trans-2-Azidomethyl-3-(4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-
benzo[fJquinolin-7-yl)-acrylic acid methyl ester (52 Chart E).
Trans-4-benzyl-1,2,3,4,4a,5,6, lOb-octahydro-benzo[f3quinolin-7-carbaldehyde
(1.4 g, 4.59 mmol), ethyl azidoacetate (2.7g, 20.9 mmol), diethyl ether (10
ml), and
methanol (30 ml) were cooled to -5 C (Henn et al., J. Chem. Soc. Perkin Trans.
I,
1984, 2189-2196). Sodium methoxide (3.89 ml, 18.4 mmol, of a 25% w/w solution
in
methanol) was slowly added. The reaction was allowed to reach ambient
temperature and stirred for 1 hr. The mixture was cooled to 0 C and saturated
aqueous ammonium chloride added. The reaction was rapidly partioned between
ether and 10% aqueous sodium carbonate. The ethereal layer was washed with
water and brine and the resulting organic phase dried (magnesium sulfate) and
evaporated to dryness. The residue was used without any further purifications
(1.85
g, 100%).
Step F: trans-4-Benzyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (54 Chart E).
trans-2-Azidomethyl-3-(4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-
7-yl)-acrylic acid methyl ester (1.85 g) and toluene (30 ml) were brought to
reflux for
2 hrs. The solution was cooled and the solvent was evaporated to dryness.
Purification of the crude reaction mixture by flash chromatography
(toluene:ethyl
acetate, 4/1 (v/v)) afforded 0.8 g (46%) of pure 54 as an oil.
MS m\z (relative intensity, 70 eV) 374 (M' 100), 342 (9), 224 (8), 159 (7), 91
(13).
Preparation 2 cis-4-Benzyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (55 Chart E).
Step A: cis-Trifluoro-methanesulfonic acid 4-benzyl-1,2,3,4,4a,5,6,10b-
WO 94/21608 PCTIUS94/02800
= ,,. . . .
-49-
octahydro-benzo[fJquinolin-7-yl ester (45, Chart E).
This compound was prepared as described for 44 from cis-7-hydroxy-4-benzyl-
1,2,3,4,4a,5,6,10b-octahydrobenzo[flquinoline (6.45 g, 22 mmol, for
preparation see
Wikstrom et al., J. Med. Chem., 1982, 25, 925-931). Purification of the crude
reaction mixture by flash chromatography (dichloromethane/methanol, 19/1
(v/v))
afforded 7.8 g (83%) of pure 45 as an oil.
MS m\z (relative intensity, 70 eV) 425 (M+ 18), 292 (91), 264 (13), 91 (100).
Step B: cis-4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-
carboxylic
acid methyl ester (47 Chart E).
This compound was prepared as described for 46 from cis-trifluoro-
methanesulfonic acid 4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-
yl ester
(3.7 g, 8.7 mmol). Purification of the crude reaction mixture by flash
chromatography (dichloromethane/methanol, 19/1 (v/v)) afforded 2.0 g (69%) of
pure
47asanoil.
MS m\z (relative intensity, 70 eV) 324 (M' 100), 258 (20), 244 (23), 91 (79).
Step C cis-(4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-yl)-
methanol (49 Chart E)
This compound was prepared as described for 48 from cis-4-benzyl-
1,2,3,4,4a,5,6,10b-octahydro-benzo[flquinolin-7-carboxylic acid methyl ester
(1.75 g,
5.22 mmol). Purification of the crude reaction mixture by flash chromatography
(dichloromethane/methanol, 19/1 (v/v)) afforded 1.4 g (88%) of pure 49 as an
oil;
MS m/z (relative intensity, 70 eV) 307 (M+ 100), 230 (14), 158 (12), 91 (47).
Step D cis-4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-
carbaldehyde (51 Chart E)
This compound was prepared as described for 50 from cis-(4-benzyl-
1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-yl)-methanol (1.6 g, 5.21
mmol).
Purification of the crude reaction mixture by flash chromatography
(dichloromethane/methanol, 19/1 (v/v)) afforded 1.45 g (92%) of pure 51 as an
oil;
MS m/z (relative intensity, 70 eV) 305 (M+ 100), 214 (18), 159 (19), 129 (17),
91(58).
Step E cis-2-Azidomethyl-3-(4-benzyl-1,2,3,4,4a,5,6,10b-octahydro-
benzo[fJquinolin-7-yl)-acrylic acid methyl ester (53 Chart E)
WO 94/21608 -50- PCT/US94/02800
This compound was prepared as described for 52 from cis-4-benzyl-
1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-carbaldehyde: (1.2 g, 3.93
mmol). The
residue was used without further purification (1.56 g, 99%).
Step F cis-4-Benzyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (55 Chart E)
This compound was prepared as described for 54 from cis-2-Azidomethyl-3-(4-
benzyl-1,2,3,4,4a,5,6,10b-octahydro-benzo[fJquinolin-7-yl)-acrylic acid methyl
ester
(1.56 g). The residue was used without further purification (1.35 g, 91%);
MS m/z (relative intdensity, 70 eV) 374 (M+ 100), 342 (14, 224 (8), 159 (6),
91
(13).
Preparation 3 trans-2,3,4,5,6,7,10,17-Octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (56 Chart E)
To a solution of trans-4-benzyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (0.8 g, 2.13 mmol)
in
methanol (15 ml, plus some few drops of toluene) were added ammonium formate
(0.84 g, 8.52 mmol) and Pd/C (125 mg) under an Ar atmosphere. The mixture was
refluxed for 1.5 hours and then filtered through a pad of Celite. The solvents
were
evaporated under reduced pressure and the residue redissolved in
dichloromethane
(50 ml). The organic mixture was washed several times with 10% aqueous
solution
of sodium carbonate, dried (magnesium sulfate) and evaporated to dryness (630
mg).
The residue was used without further purification:
MS m/z (relative intensity, 70 eV) 284 (M+ 100), 283 (21), 252 (15), 241 (30),
209 (18).
Preparation 4 cis-2,3,4,5,6,7,10,17-Octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (57 Chart E)
This compound was prepared as described for 56 from cis-4-Benzyl-
2,3,4,5,6, 7,10,17-octaydro-lH-4,17-diaza-cyclopenta[a]phenanthrene-16-
carboxylic
acid methyl ester (0.8 g). The residue was used without furthe rpurification:
MS m/z (relative intensity, 70 eV) 284 (M+ 100), 241 (28), 227 (36), 207 (43),
195 (31).
Preparation 5 trans-4-Propyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (58 Chart E)
=WO 94/21608 Z 9 ~ PCT/US94/02800
-51-
A mixture of trans-2,3,4,5,6,7,10,17-Octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (0.63 g),
propionaldehyde
(0.28 ml) and acetic acid (0.2 ml) were dissolved in tetrahydrofuran (30 ml).
To the
mixture, sodium triacetoxy borohydride (0.74 g) was added in one portion, and
the
resulting suspension was stirred at room tempeorature for 12 hours. The
mixture
was then evaporated to dryness, redissolved in dichloromethane (50 ml) and
washed
several times with 10% aqueous solution of sodium carbonate. The organic phase
was dried (magnesium sulfate), filtered and evaporated to dryness.
Purification of
the crude reaction mixture by flash chromatography (dichloromethane/methanol,
12/1(v/v)) afforded 0.5 g (69%, based on 54) of pure 58 as an oil;
'H NMR (300 MHz, CDC13) 8 0.9 (t. 6H), 1.2 (m, 1H), 1.5-1.8 (m, 4H), 2.2-3.2
(m, 9H), 3.95 (s, 3H), 7.2-7.3 (m, 3H), 9.1 (s, 1H); MS m/z (relative
intensity, 70 eV)
326 (M+ 35), 297 (100), 265 (16), 133 (17), 84 (29); Analysis calc'd for
C2OH26N2O2x
0.25 H20: C, 72.6; H, 8.1; N, 8.5; found: C, 72.1; H, 7.6; N, 8Ø
Preparation 6 cis-4-Propyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene-16-carboxylic acid methyl ester (59, Chart E)
This compound was prepared as described for 58 from cis-2,3,4,5,6,7,10,17-
octahydro-lH-4,17-diaza-cyclopenta-[a]phenanthrene-16-carboxylic acid methyl
ester
(0.47 g):
MS m/z (relative intensity, 70 eV) 326 (M+ 33), 298 (22), 297 (100), 265 (14),
133 (10), 84 (19).
Preparation 7 trans-4-Propyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta[a]phenanthrene (60, Chart E).
To a solution of trans-4-propyl-2,3,4,5,6,7,10,17-octahydro-lH-4,17-diaza-
cyclopenta-[a]phenanthrene-16-carboxylic acid methyl ester (180 mg, 0.55 mmol)
in
methanol (3 ml) was added 3 N sodium hydroxide (2 ml). The mixture was brought
to reflux for 2 hours and then acidified (pH ffi 2) with 10% aqueous solution
of
hydrochloric acid. The solvent was removed and azeotroped with absolute
ethanol
in vacuo. The remaining residue was redissolved in pyridine (5 ml) and
refluxed for
2 hours and then cooled. The solution was evaporated to yield a residue (150
mg)
containing the title compound and the corresponding carboxylic acid;
MS m/z (relative intensity, 70 eV) 268 (M+ 42), 240 (19.7), 239 (100), 168
(11), 84 (32)
CA 02157586 2001-07-16
-52-
Table 1. Effects on DOPA and 5-HTP accumulation in reserpine - pretreated
rats. Also shown are the in vitro affinities for brain dopamine (D2,
SpiperoneTM) and
serotonin (5-HT1A, 8-OH-DPAT) receptor sites in non-pretreated rats.
Example DOPA ED50 (umol/kg, 5-HTP ED50 In vitro binding Ki nM
sc.) (pmol/kg,
sc.)
Stri. Cortex. Limbic D2-spip. 5HT1A
1 P(78%, 50) I(50) I(50) 3520* 9930
2 P(48%, 50) I(50) P(61%, 50) 320 1120
4 I(50) I(50) 5.8 15900 25300
5 P(26%, 3.1) I(50) P(62%, 50) 270 178
7A** 0.3 I(12.5) P(72%, 3.1) 50 500
7A(-)** P(31%, 25) P(73%, 25) P(68%, 25) 257 1453
7A(+)** 0.12 P(67%, P(80%, 12.5) 35 NT
12.5)
7 P(48%, 50) I(50) I(50) 4200 340
8 0.015 P(57%, 0.2) I(50) 16 2600
8(+) P(60%, 0.2) 1(0.2) 1(0.2) NT NT
9 P(37%, P(85%, 1(12.5) 278* NT
12.5) 12.5)
10 0.004 P(79%, 3.1) 1(3.1) 10.1* NT
16 P(36%, 60) I(60) P(50) 924* NT
17 1.0 P(79%, 1(33.3) 205* NT
33.3)
15 P(35%, 50) P(88%, 50) P(52%, 50) 164* 124
* Data from binding experiments using D2-CHO cells and RacloprideTM as the
labeled
ligand.
** Not a compound of the subject invention.
The animals were pretreated with reserpine (5 mg/kg, s.c.) 18 hours before
the experiment. Test drugs were administered and immediately thereafter the
motron activity was measured in photocell motility boxes. The rats were
injected
with the decarboxylase inhibitor NSD 1015 (100 mg/kg, s.c.) and killed 30
minutes
later. The accumulation of DOPA in the striatum, limbic and areas and that of
~WO 94/21608 2~5 ry5 S e PCTIUS94/02800
-53- f U
5-HTP in the limbic forebrain was taken as a measure of the DA and 5-HT
synthesis
rates, respectively. The accumulation of DOPA in the cortex area was taken as
a
measure of NA synthesis rate. DA receptor agonists are known to decrease the
DOPA accumulation via an activation of feedback mechanisms while DA receptor
antagonists are inactive in reserpine-pretreated animals. The same theory is
valid
for 5-HT receptor agonists and antagonists. Dose-response curves were
constructed
for each compound (4-5 dose levels, n = 4) and the half-maximal decrease
(ED50)
was calculated.
The maximal decrease of DOPA in striatum was found to be 80% and 50% for
that of 5-HTP in the limbic region. I = inactive; no significant effect at the
highest
dose (shown in brackets). P = partial decrease; maximal decrease was not
reached at
the highest dose (shown in brackets). The % decrease from control is also
shown.
NT= not tested.
WO 94/21608 PCT/US94/02800 =
21575) O 6 -54-
U
z
0 0
O N
_ - x
z
N
O
z
o o 00
o o
- ~ / z
z
8-Z o
I-)-z
0 0
~ / ~
WO 94/21608 2 15 75 8 6 PCT/US94/02800
= -55-
~
z
0
o
N
z--~
o x
o -~
- .~
u
o
z xz
o N \ /
~ - z
0
z--~
o
o ~
~
_ 00
0
z--~
o
~ -
~ ~-z
~--z
o ~
p
o 0 z-~
0
z
z
N
SUBSTITUTE SHEET (RULE 26)
WO 94/21608 215758 6 -56- PCT/US94/02800
U
V
-~--z
o x
N
~. ~
V
z z
o N
V
CY] ~
z
~
O m
0
_ - ~
0
~. ~ u
~ ~ x
~WO 94/21608 5 7 58 6 PCTIUS94/02800
_57.
C ~
z z 0
8-
0 z z
C
z ozo z z
C*4
o xz" z
C
z
cc
cc o x x
zx -~
-~
M M
- - ~ ~j foo,
z
N
Lfl)
z/\ CY) zo '
~ z
i
O
OH OSO2CF3 CO2CH3 CH2OH 00
Bz / I Bz
N ~ N, N,Bz N,Bz
44 trans 46 trans 48 trans
.~~
45 cis 47 cis 49 cis 00
CO2CH3 H3CO2C
CHO N3~
HN
Bz ~
N Bz N Bz N
~ . =
-~ -~
50 trans 52 trans 54 trans
51 cis 53 cis 55 cis
H3CO2C H3CO2C
..~ -- *trans HN HN HN
NH N-n-Pr N-n-Pr 56 trans 00- 58 trans 60 Chart E A
57 cis 59 cis 00
~
WO 94/21608 2157586 PCT/US94/02800
-59-
Lz u
x-z o
x
x M
z
a-z
r~-z
x ,t
N x
z
z
0 x
0 0
g-z
g z
WO 94/21608 PCT/US94/02800
-60-
N
cd
U
~ ~ .
a x ~ P4 x~
a / ~ >,
~ ' -
u
, ~ z 'Jy
L~ z
~ x
~ x
a a z
~ Z a x
ao
+ ,,
~ o -
~,
x
N
N
WO 94/21608 215708 6 PCT/US94/02800
PJ"
vVo
-~-z~
a x
,
o
.~
0 o-~
C
-~ ~
N
X
WO 94/21608 21C7 J 8 6 - PCT/US94/02800
t3 62-
Key for the charts
Chart 1
la R=n-Propyl, X=-CH2
l b R-n-Propyl, X--O-
1 c R-n-Propyl, Xsvalence bond
2a R-n-Propyl, X--CHZ
2b R-n-Propyl, X--O-
2c R-n-Propyl, X-valence bond
3a R-n-Propyl, X--CHZ
3b R=n-Propyl, X--O-
3c R-n-Propyl, X-valence bond
4a R-n-Propyl, X--CH2
4b R=n-Propyl, X--O-
5a R-n-Propyl, X--CH2
Chart 2
6a R,=RZ=n-Propyl, R3-H, X--CH2-, Yl=H, Y2-H
6b: 1 R,-n-Propyl, RZ-1-R-Phenyl-ethyl, R3-H, X--CH2-, Y1-H, Y2-H
6b:2 R,-n-Propyl, RZ-1-R-Phenyl-ethyl, R3-H, X--CH2-, Y,=H, Y2-H
6c R,Rz=-(CH2)4 , R3=H, X--CH2-, Y,=H, YZ-H
6d R,=R2-n-Propyl, R3-CHõ X--CH2-, Y,=H, Y2-H
6e R,-R2=n-Propyl, R3-CH3, X--CH2-, Y1=Br, YZ-H
6f R,-R2-n-Propyl, R3=CH31 X--CH2-, Yl-H, Y2-Br
6g R,-RZ=n-Propyl, R3-H, X=valence bond, Y1-H, Y2-H
6h R,-n-Propyl, R2-benzyl, R3-H, X-valence bond, Y1-H, YZ-H
WO 94/21608 2157586 PCT/US94/02800
~
-63-
7a R,=RZ=n-Propyl, R,-H, X--CH2-,Y-H
7b:1 R,=n-Propyl, RZ=1-R-Phenyl-ethyl, R,-H, X--CH2-,Y-H, leading to 9a: 1
7b:2 R,=n-Propyl, R2=1-R-Phenyl-ethyl, R,-H, X=-CH2-,Y=H, leading to 9a:2
= 7c R,RZ=-(CH2)4 , R3=H, X--CH2-,Y-H
7d R,=RZ=n-Propyl, R,=CHõ X=-CH2-,Y=H
7e R,=R2=n-Propyl, R,=CHõ X=-CH2-,Y=Br
7f R,=R2=n-Propyl, R3-H, X-valence bond,Y=H
7g R,=n-Propyl, RZ=benzyl, R,-H, X-valence bond, Y=H
8a R,=R2=n-Propyl, R3-H, X=-CH2-,Y=H
8b: 1 R,=n-Propyl, R2=1-R-Phenyl-ethyl, R,-H, X=-CH2-,Y=H
8b:2 R,=n-Propyl, RZ=1-R-Phenyl-ethyl, R,-H, X=-CH2-,Y=H
8d R,=R2=n-Propyl, R,=CH3, X=-CH2-,Y=H
8e R,=RZ=n-Propyl, R3=CH3, X=-CH2-,Y=Br
8f R,=R2=n-Propyl, R,=H, X-valence bond,Y-H
8g R,=n-Propyl, R2=benzyl, R,-H, X=valence bond, Y=H
9a R,=RZ=n-Propyl, R3-H, X--CH2-, Y=H
9a:1 R,=R2=n-Propyl, R3-H, X=-CH2-, Y-H, (-)-enantiomer of 9a
9a:2 R,=R2=n-Propyl, R3-H, X=-CH2-, Y-H, (+)-enantiomer of 9a
9b:1 R,=n-Propyl, R2= 1 -R-Phenyl-ethyl, R,=H, X--CH2-, Y-H, leading to 9a:1
9b:2 R,=n-Propyl, R2=1 R-Phenyl-ethyl, R,=H, X=-CH2-, Y-H, leading to 9a:2
9c R,R2=-(CH2); , R,=H, X=-CH2-, Y=H
9d R,=R2=n-Propyl, R,=CHõ X--CH2-, Y=H
9e:1 R,=n-Propyl, R2=R3=H, X--CH2-, Y=H, leading to 9a:1
9e:2 R,=n-Propyl, R2=R3=H, X--CH2-, Y=H, leading to 9a:2
9f R,=n-Propyl, RZ=methyl, R,=H, X=-CH2-, Y-H
9g R,=n-Propyl, RZ=2-thiophenethyl, R3-H, X--CH2-, Y=H
9h R,=n-Propyl, RZ=cyclobutylmethyl, R3-H, X--CH2-, Y-H
9i R,=RZ=n-Propyl, R,=H, X--CH2-, Y=Br
9j R,=RZ=n-Propyl, R,-H, X-valence bond, Y-H
9k R,=n-Propyl, R2=benzyl, R,=H, X-valence bond, Y-H
WO 94/21608 2157PCT/US94/02800
~U~ -64-
l0a:l R,-R2=n-Propyl, R,-H, R4-CHO, RS-H, X--CH2-, Y-H
10a:2 R,-RZ-n-Propyl, R3-H, R4-CHO, RS=H, X--CH2-, Y-H
(+)-enantiomer of 10a
lOb R,=RZ=n-Propyl, R,=H, R,=CN, X$-CH2-, Y=H
10d R,-R2=n-Propyl, R,-CH,, R4-CHO, X--CH2-, Y-H
10e R,-n-Propyl, R2-methyl, R3-H, R4-SMe, X--CH2-, Y-H
lOf R,=n-Propyl, RZ-2-thiophenethyl, R3-H, R,-CHO, X--CH2-, Y-H
lOg R,-n-Propyl, RZ-cyclobutylmethyl, R3-H, R4=CHO, X--CH2-, Y-H
10h R,-RZ-n-Propyl, R,-H, R4-H, X--CH2-, Y-H
l0i R,-RZ=n-Propyl, R3-H, R,-COCF3, X--CH2-, Y-H
lOj R,-RZ-n-Propyl, R3-H, R4-COEt, X--CH2-, Y-H
10k R,-R2-n-Propyl, R,-H, R4-COMe, X--CH2-, Y-H
11 a R,-RZ-n-Propyl, R3-H, X--CH2-, Y-H
llb R,-RZ-n-Propyl, R,-H, X--CH2-, Y-Br
lld R,-RZ-n-Propyl, R3-CH3, X--CH2-, Y-H
l le R,-Rz-n-Propyl, R,-H, X-valence bond, Y-H
llf R,-n-Propyl, RZ-benzyl, R3-H, X-valence bond, Y-H
llg R,-n-Propyl, R2-H, R,-H, X-valence bond, Y-H
i lh R,-n-Propyl, R2-cyclopropylmethyl, R3-H, X-valence bond, Y-H
11 i R,-n-Propyl, RZ-(4-Fluoro-phenyl)ethyl, R3-H, X-valence bond, Y-H
12a R,-RZ-n-Propyl, R3=H, R4=CHO, X--CH2-, Y-H
12b R,-RZ-n-Propyl, R3-H, R4-H2, X--CH2-, Y-H
12c R,-RZ-n-Propyl, R3-H, R,-COEt, X--CH2-, Y-H
12d R,-RZ-n-Propyl, R3-H, R4-CHO, X--CH2-, Y-Br
12e R,-RZ-n-Propyl, R3-H, R4-CHO, X-valence bond, Y-H
12f R,-RZ-n-Propyl, R,=H, R,=COEt, X-valence bond, Y-H
12g R,-RZ-n-Propyl, R3-H, R4=COPh, X-valence bond, Y-H
12h R,-n-Propyl, RZ-cyclopropylmethyl, R3-H, R4-COEt, X=valence bond, Y-H
12i R,-n-Propyl, RZ=cyclopropylmethyl, R3-H, R4=n-Propyl, X-valence bond, Y-H
12j R,=n-Propyl, R2=(4-Fluoro-phenyl)ethyl, R3-H,
R,-n-Propyl, X-valence bond, Y-H
~WO 94/21608 2157 ~ g G PCT/US94/02800
-65-
Chart 3
13a R,-n-Propyl, RZ-H, X=-CHZ-, a--O-
13b R,-n-Propyl, RZ=H, X=-CHZ-. b--O-
13c R,-n-Propyl, RZ-Me, X--CH2-, a--O-
13d R,-n-Propyl, RZ-Me, X-valence bond, a--O-
13e R,-n-Propyl, R2-Me, X-valence bond, b--O-
14a R,-n-Propyl, RZ-H, X--CH2-, a--O-
14b R,-n-Propyl, R2-H, X--CHZ-, b--O-
14c R,-n-Propyl, RZ-Me, X--CHZ-, a--O-
14d R,-n-Propyl, RZ-Me, X-valence bond, a--O-
14e R,-n-Propyl, RZ-Me, X-valence bond, b--O-
15a R,-n-Propyl, R2-H, X--CHZ-, a--O-, b--C-
15b R,-n-Propyl, RZ-H, X--CHz , b--O-, a--C-
15c R,-n-Propyl, RZ-H, X--CHZ-, b--O-, c--C-
15d R,-n-Propyl, RZ-Me, X--CH2-, a--O-, b--C-
15e R,-n-Propyl, R2-Me, X-valence bond, a--O-, b--C-
15f R,-n-Propyl, R2-Me, X-valence bond, b--O-, a--C-
15g R,-n-Propyl, R2-Me, X-valence bond, b--O-, c--C-
--