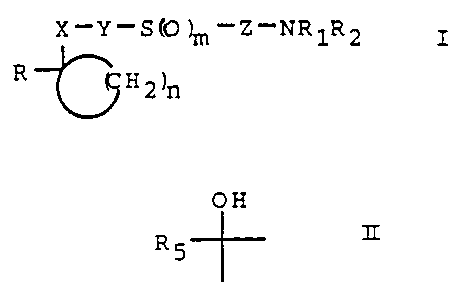Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 94/26704 216 2 7 0 6 PCT~~4/01494
1-arylcycioaikyl sulphides, sulphoxides and sulphones for the treatment of
depression, anxiety and Parkinson's disease
The present invention relates to novel therapeutic
agents, to processes for their preparation, to
pharmaceutical compositions containing them and to their
use in the treatment of depression, anxiety, Parkinson's
disease, obesity, cognitive disorders, seizures,
neurological disorders such as epilepsy, and as
neuroprotective agents to protect against conditions
such as stroke.
The present invention provides compounds of
formula I
X-Y-S (0 ?m-Z-NR1R2
R I
H2~n
and pharmaceutically acceptable salts thereof in which
m is 0, 1 or 2;
n is 2, 3, 4 or 5;
X is carbonyl or a group of formula II
OH
R 5 II
in which R5 is H or an alkyl group containing 1 to 4
carbon atoms;
Y is an alkylene chain containing 1 or 2 carbon
atoms optionally substituted by one or more alkyl groups
containing 1 to 3 carbon atoms;
WO 94/26704 216 2 l 0 6 PCT/EP94/01494
- 2 -
Z is an alkylene chain containing 2 to 5 carbon
atoms optionally substituted by one or more alkyl groups
containing 1 to 3 carbon atoms;
R is phenyl optionally substituted by one or more
halo substituents which are the same or different (for
example fluoro, chloro, bromo or iodo) or R is naphthyl;
and
R1 and R2, which are the same or different, are H,
a straight or branched chain alkyl group containing 1 to
4 carbon atoms, an arylalkyl group in which the alkyl
group contains 1 to 3 carbon atoms, provided that when
R1 is benzyl, R2 is H or methyl.
In preferred compounds of formula I, m is 0, 1 or 2
and n is 3 or 4.
In preferred compounds of formula I, X is carbonyl
or a group of formula II in which R5 is H.
In preferred compounds of formula I, Y is
methylene.
In preferred compounds of formula I, Z is an
alkylene chain containing 2, 3 or 4 carbon atoms
optionally substituted by one or more alkyl groups
containing 1 to 3 carbon atoms. In more preferred
compounds of formula I, Z is an alkylene chain
containing 2, 3 or 4 carbon atoms optionally substituted
by one or more methyl groups.
In preferred compounds of formula I, R is phenyl
substituted by one or two chloro substituents, or R is
naphthyl. In more preferred compounds of formula I, R
is 3-chlorophenyl, 3,4-dichlorophenyl or 2-naphthyl.
WO 94/26704 216 2 7 0 6 PCTIEP94101494
- 3 -
In preferred compounds of formula I, R1 is an alkyl
group containing 1 to 3 carbon atoms or is benzyl, and
R2 is an alkyl group containing 1 to 3 carbon atoms. In
more preferred compounds of formula I, R1 and R2 are
both methyl or ethyl or R1 is benzyl and R2 is methyl.
In especially preferred compounds of formula I, R1 and
R2 are both methyl.
A preferred group of compounds of formula I is
represented by formula III
R3' ~ X-Y-S (0 )m-Z-NR1R2
I~
R / ~H2)n
~J4
and pharmaceutically acceptable salts thereof in which
m, n, X, Y, Z, R1 and R2 are as described above for
formula I;
and R3 is halo (for example fluoro, chloro, bromo or
iodo), and R4 is H or halo (for example fluoro, chloro,
bromo or iodo), or R3 and R4 together with the carbon
atoms to which they are attached form a fused benzene
ring.
In more preferred compounds of formula III, R3 is
chloro and R4 is H, R3 and R4 are both chloro or R3 and
R4 together with the carbon atoms to which they are
attached form a fused benzene ring. In especially
preferred compounds of formula III, R3 is chloro
situated in the 3-substitution position on the phenyl
ring and R4 is H, Rj and R4 are both chloro and are
situated in the 3- and 4- substitution positions on the
phenyl ring respectively, or R3 and R4 together with the
phenyl ring to which they are attached form a 2-naphthyl
group.
' WO 94/26704 PCT/EP94/01494
2~ ~2~1~~
- 4 -
Compounds of formula I and III may exist as salts
with pharmaceutically acceptable acids. Examples of
such salts include hydrochlorides, hydrobromides,
sulphates, methanesulphonates, nitrates, maleates, ~
acetates, citrates, fumarates, tartrates [eg (+)
tartrates, (-)-tartrates or mixtures thereof including
racemic mixtures , succinates, benzoates and salts with
amino acids such as glutamic acid. Compounds of formula
I and III and their salts may exist in the form of
solvates (for example hydrates).
Certain compounds of formula I and III may exist in
more than one crystal form and the present invention
includes each crystal form and mixtures thereof.
It will be appreciated by those skilled in the art
that compounds of formula I and III may contain one or
more chiral centres. When compounds of formula I and
III contain one chiral centre, the compounds exist in
two enantiomeric forms and the present invention
includes both enantiomers and mixtures of those
enantiomers. The individual enantiomers may be obtained
by methods known to those skilled in the art. Such
methods typically include resolution via formation of
diastereoisomeric salts which may be separated, for
example, by crystallisation; via formation of
diastereoisomeric derivatives or complexes which may be
separated, for example, by crystallisation, gas-liquid
or liquid chromatography; via selective reaction of one
enantiomer with an enantiomer-specific reagent, for
example enzymatic esterification, oxidation or
reduction; or via gas-liquid or liquid chromatography in
a chiral environment, for example on a chiral support
for example silica with a bound chiral ligand or in the
presence of a chiral solvent. It will be appreciated
that where the desired enantiomer is converted into
WO 94/26704 PCTIEP94/01494
2162706
- 5 -
another chemical entity by one of the separation
procedures described above, a further step is required
to liberate the desired enantiomeric form.
Alternatively, specific enantiomers may be synthesised
by asymmetric synthesis using optically active reagents,
substrates, catalysts or solvents, or by converting one
enantiomer into the other by asymmetric transformation.
When compounds of formula I and III contain more
than one chiral centre, the compounds may exist in
diastereoisomeric forms. The diastereoisomeric pairs
may be separated by methods known to those skilled in
the art, for example chromatography or crystallisation
and the individual enantiomers within each pair may be
separated as described above. The present invention
includes each diastereoisomer of compounds of formula I
and III and mixtures thereof.
Specific compounds of formula I and III are:-
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[2-(dimethyl-
amino)ethylthio]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[2-(dimethyl-
amino)ethylsulphinyl]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[2-(dimethyl-
amino)ethylsulphonyl]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[2-(diethyl-
amino)ethylthio]ethanone;
2-[2-(N-benzyl-N-methylamino)ethylthio]-1-[1-(3,4-
dichlorophenyl)cyclobutyl]ethanone;
WO 94/26704 ~ ~ 6 2 7 0 6 PCT/EP94/01494
- 6 -
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[2-(dimethyl-
amino)ethylthio]ethanol;
1-[1-t3,4-dichlorophenyl)cyclobutyl]-2-[3-(dimethyl-
amino)propylthio]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-(dimethyl-
amino)propylsulphonyl]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-(dimethyl-
amino)propylthio]ethanol;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-(dimethyl-
amino)-2-methylpropylthio]ethanone;
2-[2-(dimethylamino)ethylthio]-1-[1-(2-naphthyl)cyclo-
butyl]ethanone;
1-[1-(3-chlorophenyl)cyclobutyl]-2-[3-(dimethyl-
amino)propylthio]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[4-(dimethyl-
amino)butylthio]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-(dipropyl-
amino)propylthio]ethanone;
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)-2-methylpropylthio]ethanol;
1-[1-(3,4-dichlorophenyl)cyclopentyl]-2-[3-
(dimethylamino)propylthio]ethanone;
and pharmaceutically acceptable salts thereof in the
form of individual enantiomers, racemates, or other
mixtures of enantiomers.
O 94/26704 216 2 7 0 6 PCTIEP94101494
Specific enantiomeric forms of compounds of formula I
and III are:
(-)-1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanol;
(+)-1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanol;
The present invention also includes pharmaceutical
compositions containing a therapeutically effective
amount of a compound of formula I or III together with a
pharmaceutically acceptable diluent or carrier.
As used hereinafter, the term "active compound"
denotes a compound of formula I or III. In therapeutic
use, the active compound may be administered orally,
rectally, parenterally or topically, preferably orally.
Thus the therapeutic compositions of the present
invention may take the form of any of the known
pharmaceutical compositions for oral, rectal, parenteral
or topical administration. Pharmaceutically acceptable
carriers suitable for use in such compositions are well
known in the art of pharmacy. The compositions of the
invention may contain 0.1-99~ by weight of active
compound. The compositions of the invention are
generally prepared in unit dosage form.
Compositions for oral administration are the
preferred compositions of the invention and these are
the known pharmaceutical forms for such administration,
for example tablets, capsules, granules, syrups,
solutions and aqueous or oil suspensions. The
excipients used in the preparation of these compositions
are the excipients known in the pharmacist's art.
Tablets may be prepared by mixing the active compound
WO 94/26704 PCT/EP94/01494
with fillers, for example calcium phosphate;
disintegrating agents, for example maize starch;
lubricating agents, for example magnesium stearate;
binders, for example micro-crystalline cellulose or
polyvinylpyrrolidone and other optional ingredients
known in the art to permit tableting the mixture by
known methods. The tablets may, if desired, be coated
using known methods and excipients which may include
enteric coating using for example hydroxypropylmethyl
cellulose phthalate. The tablets may be formulated in a
manner known to those skilled in the art so as to give a
sustained release of the compounds of the present
invention. Such tablets may, if desired, be provided
with enteric coatings by known methods, for example by
the use of cellulose acetate phthalate. Similarly,
capsules, for example hard or soft gelatin capsules,
containing the active compound with or without added
excipients, may be prepared by conventional means and,
if desired, provided with enteric coatings in a known
manner. The contents of the capsule may be formulated
using known methods so as to give sustained release of
the active compound. The tablets and capsules may
conveniently each contain 1 to 500 mg of the active
compound.
Other compositions for oral administration include,
for example, aqueous suspensions containing the active
compound in an aqueous medium in the presence of a non-
toxic suspending agent such as sodium carboxymethyl-
cellulose, and oily suspensions containing a compound of
the present invention in a suitable vegetable oil, for
example arachis oil. The active compound may be
formulated into granules with or without additional
excipients. The granules may be ingested directly by
the patient or they may be added to a suitable liquid
carrier (for example, water) before ingestion. The
W~ 94/26704 216 2 l 0 6 PCT/EP94/01494
_ g _
granules may contain disintegrants, for example an
effervescent couple formed from an acid and a carbonate
or bicarbonate salt to facilitate dispersion in the
liquid medium.
Compositions of the invention suitable for rectal
administration are the known pharmaceutical forms for
such administration, for example, suppositories with
cocoa butter or polyethylene glycol bases.
Compositions of the invention suitable for
parenteral administration are the known pharmaceutical
forms for such administration, for example sterile
suspensions or sterile solutions in a suitable solvent.
Compositions for topical administration may
comprise a matrix in which the pharmacologically active
compounds of the present invention are dispersed so that
the compounds are held in contact with the skin in order
to administer the compounds transdermally.
Alternatively the active compounds may be dispersed in a
pharmaceutically acceptable cream, gel or ointment base.
The amount of active compound contained in a topical
formulation should be such that a therapeutically
effective amount of the compound is delivered during the
period of time for which the topical formulation is
intended to be on the skin.
The compounds of the present invention may also be
administered by continuous infusion either from an
external source, for example by intravenous infusion or
from a source of the compound placed within the body.
Internal sources include implanted reservoirs containing
the compound to be infused which is continuously
released for example by osmosis and implants which may
be (a) liquid such as a suspension or solution in a
WO 94/26704 216 2 7 0 6 PCT/EP94/01494
- 10 -
pharmaceutically acceptable oil of the compound to be
infused for example in the form of a very sparingly
water-soluble derivative such as a dodecanoate salt of a
compound of formula I or III or (b)solid in the form of
an implanted support, for example of a synthetic resin
or waxy material, for the compound to be infused. The
support may be a single body containing all the compound
or a series of several bodies~-each containing part of
the compound to be delivered. The amount of active
compound present in an internal source should be such
that a therapeutically effective amount of the compound
is delivered over a long period of time.
In some formulations it may be beneficial to use
the compounds of the present invention in the form of
particles of very small size, for example as obtained by
fluid energy milling.
In the compositions of the present invention the
active compound may, if desired, be associated with
other compatible pharmacologically active ingredients.
The pharmaceutical compositions containing a
therapeutically effective amount of a compound of
formula I or III may be used to treat depression,
anxiety, Parkinson's disease, obesity, cognitive
disorders, seizures, neurological disorders such as
epilepsy, and as neuroprotective agents to protect
against conditions such as stroke in human beings.
Whilst the precise amount of active compound
administered in such treatment will depend on a number
of factors, for example the age of the patient, the
severity of the condition and the past medical history,
and always lies within the sound discretion of the
administering physician, the amount of active compound
administered per day is in the range 1 to 1000 mg
WO 94/26704 PCT/EP94/01494
2162106
- 11 -
preferably 5 to 500 mg given in single or divided doses
at one or more times during the day.
Compounds of formula I or III may be administered
as a method of treating Parkinson's Disease either alone
or in combination with a dopamine precursor such as
levodopa and/or a dopa decarboxylase inhibitor such as
carbidopa or benserazide.
In yet another aspect, the present invention
provides the use of a compound of formula I or III in
the manufacture of a medicament for use in the treatment
of depression, anxiety, Parkinson's disease, obesity,
cognitive disorders, seizures, neurological disorders
such as epilepsy, and as neuroprotective agents to
protect against conditions such as stroke.
Processes for the preparation of compounds of
formula I will now be described. These processes form a
further aspect of the present invention.
Compounds of formula I in which m is 0, X is
carbonyl and Y is methylene may be prepared by reaction
of a compound of formula IV
COCH2G
R H ) IV
2n
in which G is a leaving group, for example halo, for
example chloro, bromo or iodo, with a compound of
formula V
HS-Z-NR1R2 V
WO 94/26704 2 ~ 6 2 7 ~ ~ PCT/EP94/01494
- 12 -
or a salt thereof in the presence of a base, for example
sodium ethoxide.
Compounds of formula IV in which G is halo may be
prepared by reaction of a compound of formula VI with a
halogenating agent, for example~bromine.
COCH3
R VI
H2)n
Compounds of formula VI may be prepared by reaction
of a compound of formula VII
CN
R ~H2)n VIL
with an organometallic reagent, for example an
organolithium compound of formula CH3Li or a Grignard
reagent such as methylmagnesium iodide, followed by
hydrolysis.
Compounds of formula VII in which n is 2, 3, 4 or 5
may be prepared by reaction of a compound of formula
VIII
R-CH2CN VIIL
with a compound of formula IX
Y- (CH2)n-Y IX ,
in which n is 2, 3, 4 or 5 and Y is a leaving group, for
example bromo, in the presence of a base, for example
sodium hydride, sodium hydroxide or potassium hydroxide,
~ ~ ~~~o~
WO 94/26704 PCT/EP94/01494
- 13 -
optionally in the presence of a phase transfer catalyst,
for example benzyltriethylammonium chloride.
Compounds of formula VII in which n - 3 may be
prepared by the method disclosed in British Patent
Specification 2098602 by selection of the appropriate
starting material.
Compounds of formula V may be prepared by
hydrolysis, for example basic hydrolysis, of a compound
of formula X or a salt thereof.
HN
~S-Z-NR1R2 X
H 2N
Compounds of formula X may be prepared by reaction of a
compound of formula XI
A-Z-NR1R2 XI
in which A is a leaving group, for example chloro, bromo
or iodo, with thiourea.
Compounds of formula XI may be prepared by reaction
of a compound of formula XII
HO-Z-NR1R2 XII
with a halogenating agent, for example thionyl chloride.
Compounds of formula I in which X is a group of
formula II may be prepared by reduction of a compound of
formula I in which X is carbonyl, for example with
sodium borohydride to give compounds of formula I in
which R5 is H, or by reaction of a compound of formula I
WO 94/26704 216 2 7 0 ~ PCT/EP94/01494
- 14 - -
in which X is carbonyl with an organometallic reagent,
for example an organolithium compound of formula RSLi in
which R5 is alkyl to give compounds in which R5 is ,
alkyl.
Compounds of formula I in which m is 1 may be
prepared by oxidising a compound of formula I in which m
is 0 with an oxidising agent, for example magnesium
monoperoxyphthalate.
Compounds of formula I in which m is 2 may be
prepared by oxidising a compound of formula I in which m
is 0 or 1 with an oxidising agent, for example potassium
permanganate.
Compounds of formula I in which m is 0, X is
carbonyl and Y is ethylene may be prepared by the
addition reaction of a compound of formula V with a
compound of formula XIII
CO
X ItI
R (CH2)n
Compounds of formula XIII may be prepared by the
reaction of a compound of formula XIV
X ~1
R ~H2)n
with an oxidising agent, for example manganese dioxide.
~WO 94/26704
216 2 7 0 6 PCTIEP94/01494
- 15 -
Compounds of formula XIV may be prepared by the
reaction of a compound of formula XV
CHO
R ~ H ) XV
2n
with an organometallic reagent, for example a Grignard
reagent of formula CH2=CHMgCl.
Compounds of formula XV may be prepared by
reduction of a compound of formula VII with a suitable
reducing agent, for example diisobutylaluminium hydride,
followed by hydrolysis.
Compounds of formula III may be prepared in a
similar manner to that described for compounds of
formula I.
The therapeutic activity of the compounds of
formula I or III has been indicated by assessing the
ability of the compounds to prevent the ptosis (eye
closure) induced by reserpine in the following manner.
Male rats of the Charles River CD strain weighing
between 140 and 180 g were randomly separated into five
rats in each cage and supplied with food and water ad
libitum. Eighteen hours prior to initiation of the test
four of the five rats were marked with a pen such that
each rat was individually identifiable; food was then
withdrawn. The following morning, two hours before the
test, the rats were weighed and a semi-randomised code
was used to allocate treatments to rats. The test
commenced by orally administering either:
a) the test compound in solution in deionised water at
a dose volume of 10 ml/kg of body weight, followed
WO 94/26704 PCT/EP94/01494
~~ ~2~06
- 16 -
by immediate intravenous injection of 1 ml/kg of
body weight of reserpine (0.75 mg/kg) in solution
in deionised water containing 238mM citric acid,
1.02 v/v Tween 80 and 0.2~ v/v benzyl alcohol
(treated group);
b) deionised water at a dose volume of 10 ml/kg of
body weight, followed by immediate intravenous
injection of 1 ml/kg of body weight of reserpine
(0.75 mg/kg) in solution in deionised water
containing 238mM citric acid, 1.02 v/v Tween 80
and 0.2~ v/v benzyl alcohol (positive control
group); or
c) deionised water at a dose volume of 10 ml/kg of
body weight, followed by immediate intravenous
injection of 1 ml/kg of body weight of deionised
water containing 238mM citric acid, 1.02 v/v Tween
80 and 0.2~ v/v benzyl alcohol (negative control
group ) .
Three hours later rats were individually placed in
clear perspex boxes (42 x 22 x 22 cm) and observed by a
person who was unaware of the treatment received by each
animal. The degree of ptosis was scored 45 seconds and
75 seconds later using the following observer rating
system: 0 - eye fully open, 1 - eye 1/4 closed, 2 - eye
1/2 closed, 3 - eye 3/4 closed, 4 - eye fully closed. A
mean ptosis score was then calculated for all
identically treated rats usually comprising a group of
eight rats. The mean ptosis score of the negative
control group was then subtracted from the mean ptosis
score of the positive control group to give the ptosis
score induced by reserpine in the absence of the test
compound. The mean ptosis score for each group of
treated rats was determined at more than one dose of
WO 94/26704 2 ~ 6 2 7 ~ 6 pCT~p94/01494
- 17 -
test compound to enable a value for the dose (ED50)
which causes a 50~ prevention of the reserpine-induced
ptosis to be obtained. Examples of compounds which gave
ED50 values of 30 mg/kg or less are given in Table 1.
It is widely understood by those skilled in the art.that
this test is indicative of compounds having
antidepressant activity in humans.
The ability of compounds of formula I or III to
interact with dopamine reuptake sites has been
demonstrated by the following test which determines the
ability of compounds to inhibit dopamine uptake in
vitro.
Striatal tissue from the brains of male Charles
River rats weighing 150-2508 was homogenised in ice-cold
0.32M sucrose (1:10w/v) using a motor driven teflon
pestle (difference in diameter between mortar and pestle
0.5mm). Nuclei and cell debris were removed by
centrifugation at 1,5008 at 4°C for 10 minutes. The
pellet (P1) was discarded and the supernatant
centrifuged at 18,0008 at 4°C for 10 minutes. The crude
synaptosomal pellet (P2) was resuspended in Krebs-
Henseleit buffer (equivalent to 4.2m8 wet weight of
tissue/ml).
Crude synaptosomes were incubated in a shaking
water bath at 37°C for 15 minutes. Aliquots (150,1;
equivalent to 0.625m8 wet weight of tissue/tube) were
then added to tubes containing 275.1 of Krebs-Henseleit
buffer and 50.1 of Krebs-Henseleit buffer (total uptake)
or 50.1 of test compound (10 concentrations ranging from
10-11-10-4M) or 50.1 of GBR 12909 (10-5M; non-specific
uptake). Uptake was initiated by the addition of 25.1
of freshly prepared [3H]dopamine (2.5nM), followed by
CA 02162706 2004-05-03
- 18 -
vortexing and was continued for 5 minutes at 37°C in the
shaking water bath.
Uptake was terminated by filtration under vacuum
through Skatron 11735 filters using a Skatron cell
harvester . Filters were than washed with a-~1 ice-cold
saline. The scored filter paper discs were punched into
vials, scintillation fluid added and radioactivity
- determined by liquid scintillation counting.
The percentage inhibition of specific upt~:e of the
tritiated ligand was calculated for each concentration
of test compound. Inhibition curves were then produced.
The concentration of compounds which gave 50$ inhibition
of specific uptake (IC50~ was obtained from the curve.
The inhibition constant (Ki) was then calculated using
the formula
Ki= IC50
1+([L~/Km)
in which (L1 is the concentration of tritiated ligand
used and Km is the affinity of the uptake site for the
ligand. The Ki values for compounds for formula I and
III are given in Table 1 as the means ~ sem of three
independent determinations.
* Trademark
~WO 94/26704 ~ PCTIEP94101494
- 19 -
TABLE 1
Example No ED50 (mg/kg) Ki(nM)
1 8.5 NT
2 7.7 NT
3 5.9 NT
4 9.7 NT
5 16.4 NT
6 5.7 NT
7 4.2 5.010.5
8 3.4 13.410.1
9 4.8 7.311.6
10 8.7 7.211.0
11 24.1 NT
12 23.4 NT
13 5.9 6.511.0
14 7.3 13.412.1
15 4.0 3.710.4
16 2.2 NT
17 2.5 NT
18 4.9 4.210.4
19 6.9 6.90.6
20 3.7 4.410.2
NT = not tested
WO 94126704 PCT/EP94/01494
- 20 -
The invention is illustrated by the following
Examples which are given by way of example only. The
final product of each of these Examples was ,
characterised by one or more of the following
procedures: gas-liquid or high performance liquid
chromatography; elemental analysis, nuclear magnetic
resonance spectroscopy and infrared spectroscopy.
Example 1
Methylmagnesium iodide was prepared under nitrogen
by dropwise addition of a solution of methyl iodide
(93.8 g) in ether (100 ml), to a stirred suspension of
magnesium turnings (15.9 g) in ether (100 ml) initially
at ambient temperature then, when the exothermic
reaction commenced, at reflux temperature. After the
addition was complete the mixture was stirred for 30
minutes then a solution of 1-(3,4-dichlorophenyl)-
cyclobutanecarbonitrile (100 g) in ether (80 ml) was
added dropwise at ambient temperature. The resulting
suspension was stirred and heated under reflux for 3
hours then stirred at ambient temperature under nitrogen
for 16 hours. The resulting solid was collected by
filtration, washed well with ether, then added in
portions to an ice-cold mixture of water (400 ml) and
concentrated hydrochloric acid (250 ml). The resulting
mixture was heated at 95°C for 1 hour with occasional
stirring then cooled to ambient temperature. The
product was extracted into ether (6 x 150 ml), the
extracts were dried over magnesium sulphate and the
solvent removed in vacuo to give an oil (105 g) which
was distilled to give 1-[1-(3,4-dichlorophenyl)- '
cyclobutyl]ethanone (89.6 g), by 116-118°C/0.13mbar.
A solution of bromine (18 ml) in chloroform (80 ml)
was added dropwise at 10-15°C over 1.5 hours to a
2162706
WO 94/26704 PCT/EP94/01494
- 21 -
stirred solution of the above 1-[1-(3,4-dichlorophenyl)-
cyclobutyl]ethanone (89.6 g) in a mixture of methanol
(120 ml) and chloroform (20 ml). When the addition was
complete, the mixture was stirred at ambient temperature
for 1 hour, then poured onto an excess of ice-water.
The aqueous layer was separated and the product
extracted into dichloromethane (2 x 150 ml). The
combined organic solutions were washed with saturated
aqueous sodium hydrogen carbonate solution (2 x 200 ml)
then with water, dried over calcium chloride and the
solvent removed in vacuo to yield an oil. The oil was
distilled to give 2-bromo-1-[1-(3,4-dichlorophenyl)-
cyclobutyl]ethanone (88.31 g), b.p. 148-154°C/0.66 mbar.
A solution of sodium ethoxide [prepared from sodium
(0.69 g) and ethanol (60 ml)] was added to a stirred
suspension of 2-(dimethylamino)ethanethiol hydrochloride
(2.12 g) in ethanol (30 ml) and the mixture was stirred
at ambient temperature for 1 hour. A solution of 2-
bromo-1-[1-(3,4-dichlorophenyl)cyclobutyl]ethanone
(4.8 g, prepared as described above) in ethanol (30 ml)
was added in one portion, and the mixture was stirred at
ambient temperature for a further 2 hours. The mixture
was then stirred at 50°C for 1 hour, and the solvent
removed in vacuo. The residue was diluted with water
(30 ml) and the product was extracted into ether
(2 x 50 ml). The extracts were dried over magnesium
sulphate, and the solvent removed in vacuo to give an
oil (5.1 g) .
The above oil was dissolved in ether and the
solution saturated with hydrogen chloride. The solvent
was removed in vacuo to give an oil (5.1 g) which was
purified via flash chromatography over silica using
dichloromethane followed by a 1:1 mixture of ethyl
acetate and methanol as eluants. Appropriate fractions
WO 94/26704 PCTIEP94/01494
- 22 -
were combined and the solvents removed in vacuo to give
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[2-(dimethyl-
amino)ethylthio]ethanone hydrochloride as an oil
(2.9 g) .
Example 2
1-[1-(3,4-Dichlorophenyl)cyclobutyl]-2-[2-
(dimethylamino)ethylthio]ethanone (1.7 g, prepared by
basification of the hydrochloride salt obtained in a
similar manner to that described in Example 1) was
dissolved in ethanol (10 ml) and a solution of magnesium
monoperoxyphthalate hexahydrate (1.6 g, 87~ purity) in
water (75 ml) was added. Further ethanol (20 ml) was
added and the mixture was stirred at ambient temperature
for 1 hour.
The solvent was removed in vacuo, the residue
diluted with water and the product extracted into ethyl
acetate. The extract was dried over magnesium sulphate
and the solvent removed in vacuo to give an oil (1.8 g).
The above oil was dissolved in ethanol and the
solution saturated with hydrogen chloride to give a
solid which was collected by filtration, and
crystallised from ethanol to give 1-[1-(3,4
dichlorophenyl)cyclobutyl]-2-[2-(dimethylamino)ethyl
sulphinyl]ethanone hydrochloride as a white solid
(0.5 g), m.p. 184-185°C.
Example 3
A solution of potassium permanganate (1.2 g) in
water (40 ml) was added to a solution of 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[2-(dimethylamino)ethyl-
thio]ethanone (1.4 g, prepared by basification of the
WO 94/26704 21 ~ 2 7 0 6 PCT/EP94/01494
- 23 -
hydrochloride salt obtained in a similar manner to that
described in Example 1), tetra-n-butylammonium bromide
( 0 .1 g) and acetic acid ( 10 ml ) in toluene ( 30 ml ) , and
the mixture was stirred at ambient temperature for 22
hours. Saturated aqueous sodium hydrogen sulphite
solution was added to the mixture until the purple
colour disappeared and the resulting clear solution was
neutralised by the addition of solid potassium
carbonate. The product was extracted into toluene, the
extracts were dried over magnesium sulphate, and the
solvent removed in vacuo to give an oil (1.7 g).
The above oil was dissolved in ethanol and an
excess of ethereal hydrogen chloride solution was added.
The resulting solution was evaporated to give an oil
which was triturated with dichloromethane to give 1-[1-
(3,4-dichlorophenyl)cyclobutyl]-2-[2-(dimethyl-
amino)ethylsulphonyl]ethanone hydrochloride as a white
solid (0.1 g), m.p. 208-210°C.
Example 4
A solution of sodium ethoxide [prepared from sodium
(0.5 g) and ethanol (40 ml)] was added to a solution of
2-(diethylamino)ethanethiol hydrochloride (1.7 g) in
ethanol (30 ml) and the mixture was stirred at ambient
temperature for 1 hour. A solution of 2-bromo-1-[1-
(3,4-dichlorophenyl)cyclobutyl]ethanone (3.2 g, prepared
in a similar manner to that described in Example 1) in
ethanol (30 ml) was added in one portion and the mixture
was stirred at ambient temperature for 1.5 hours. The
solvent was removed in vacuo and the residue diluted
with water (25 ml). The product was extracted into
ether (2 x 50 ml), the extracts were dried over
magnesium sulphate and the solvent removed in vacuo to
give an oil (3.5 g).
WO 94/26704 PCT/EP94/01494
2162706
- 24 -
The above oil was purified via flash chromatography
over silica using, sequentially, a 1:1 mixture of
dichloromethane and ethyl acetate, ethyl acetate, and a
9:1 mixture of ethyl acetate and methanol as eluants.
Appropriate fractions were combined, and the solvents
removed in vacuo to give an oil., The oil was dissolved
in ether (15 ml) and the solution was saturated with
hydrogen chloride. A cream solid precipitated and was
collected by filtration, washed with a little ether, and
dried in vacuo to yield 1-[1-(3,4-dichlorophenyl)cyclo-
butyl]-2-[2-(diethylamino)ethylthio]ethanone hydro-
chloride (1.6 g), m.p. 109-110°C.
Example 5
A solution of sodium ethoxide [prepared from sodium
(0.2 g) and ethanol (25 ml)] was added to a solution of
2-(N-benzyl-N-methylamino)ethanethiol (1.8 g) in ethanol
(20 ml), and the mixture was stirred at ambient
temperature for 1 hour.
A solution of 2-bromo-1-[1-(3,4-dichlorophenyl)-
cyclobutyl]ethanone (3.2 g, prepared in a similar manner
to that described in Example 1) in ethanol (20 ml) was
added, and the mixture was stirred at ambient
temperature for 3 hours. The solvent was removed in
vacuo and the residue was diluted with water (15 ml).
The product was extracted into dichloromethane, the
extracts dried over calcium chloride and the solvent
removed in vacuo to give an oil. The oil was dissolved
in ethanol and the solution saturated with hydrogen
chloride to give a solid, which was collected by
filtration, washed with a little et:=:.anol, and dried in
vacuo to yield 2-[2-(N-benzyl-N-methylamino)ethylthio]-
1-[1-(3,4-dichlorophenyl)cyclobutyl]ethanone
hydrochloride (1.2 g), m.p. 159-163°C.
WO 94/26704
PCTIEP94101494
- 25 -
Example 6
A mixture of 1-[1-(3,4-dichlorophenyl)cyclobutyl]-
2-[2-(dimethylamino)ethylthio)ethanone (2.0 g, prepared
by basification of the hydrochloride salt obtained in a
similar manner to that described in Example 1) and
sodium borohydride (2.2 g) in propan-2-of (80 ml) was
stirred at ambient temperature for 16 hours.
The resulting suspension was cautiously diluted
with acetone (15 ml), followed by an excess of saturated
aqueous ammonium chloride solution. The resulting
mixture was concentrated in vacuo, the residue was
diluted with water and the product extracted into ether.
The extracts were dried over magnesium sulphate and the
solvent removed in vacuo to give an oil.
The oil was dissolved in ethyl acetate and the
solution saturated with hydrogen chloride. The solution
was diluted with ether. A gum formed and the
supernatant liquor was removed by decantation and
allowed to concentrate at ambient temperature. An oil
was deposited and was separated by decantation of the
liquor. The oil was dissolved in methanol and the
solvent removed by evaporation to give 1-[1-(3,4
dichlorophenyl)cyclobutyl]-2-[2-(dimethyl
amino)ethylthio]ethanol hydrochloride as a colourless
oil (1.55 g).
Example 7
A mixture of 1-chloro-3-(dimethylamino)propane
hydrochloride (200 g), thiourea (98.1 g) and ethanol
(11) was stirred and heated under reflux for 25 hours.
The solution was cooled to ambient temperature and ethyl
acetate added until permanent opalescence was obtained.
WO 94126704 PCTlEP94/01494
2162706
- 26 -
The mixture was stored at 4°C overnight then filtered to
give S-[3-(dimethylamino)propyl]isothiourea
dihydrochloride as a colourless solid. (283 g), m.p.
155-159°C.
S-[3-(Dimethylami;no)propyl]isothiourea
dihydrochloride (283 g) was dissolved in water (340 ml)
and the solution covered with a layer of ether. The
mixture was cooled in ice and 25M aqueous sodium
hydroxide solution (97 ml) was added dropwise. After
the addition the mixture was stirred and heated under
reflux for 2 hours. The solution was cooled to ambient
temperature and the product extracted into ether. The
extract was dried over magnesium sulphate and the
solvent removed in vacuo to give a clear oil (70.6 g), a
sample of which (35 g) was dissolved in ether. The
resulting solution was saturated with hydrogen chloride
to give a colourless solid which was collected by
filtration, washed with ether, and dried in vacuo to
give 3-(dimethylamino)propanethiol hydrochloride (21.2
g), m.p. 103-107°C.
A solution of sodium ethoxide [prepared from sodium
(1.0 g) and ethanol (60 ml)] was added to a suspension
of 3-(dimethylamino)propanethiol hydrochloride (3.5 g)
in ethanol (50 ml), and the mixture was stirred at
ambient temperature for 1 hour. A solution of 2-bromo
1-[1-(3,4-dichlorophenyl)cyclobutyl]ethanone (9.35 g,
prepared in a similar manner to that described in
Example 1 ) in ethanol ( 3 0 ml ) was added in one portion,
and the mixture was stirred at ambient temperature for
25 hours.
The solvent was removed in vacuo to give a solid
residue. Water (30 ml) was added. The product was
extracted into ethyl acetate, the extract was dried over
WO 94/26704 ~ PCT/EP94/01494
- 27 -
magnesium sulphate and the solvent removed in vacuo to
give an oil (10.5 g). The oil was dissolved in ethyl
acetate and the solution was saturated with hydrogen
chloride . The solvent was removed in vacuo to give an
oil (9.1 g), which was purified via flash chromatography
over silica using a 1:1 mixture of ethyl acetate and
methanol as eluant. Appropriate fractions were
combined, and the solvents removed in vacuo to leave an
oil (5 g) .
The above oil was rebasified by addition to an
excess of 5M aqueous sodium hydroxide solution, and the
free base was extracted into ether (2 x 25 ml). The
extracts were washed with water, dried over magnesium
sulphate and the solvent removed in vacuo to give an
oil. The oil was dissolved in ether and saturated with
hydrogen chloride to give a white solid which was
collected by filtration, washed with a little ether, and
dried in vacuo to yield 1-[1-(3,4-dichlorophenyl)
cyclobutyl]-2-[3-(dimethylamino)propylthio]ethanone
hydrochloride (1.6 g), m.p. 115-118°C.
Example 8
A solution of potassium permanganate (3.1 g) in
water (95 ml) was added to a mixture of 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)-
propylthio]ethanone (3.4 g, prepared by basification of
the hydrochloride salt obtained in a similar manner to
that described in Example 7), tetra-n-butylammonium
bromide (0.3 g), acetic acid (25 ml) and toluene
(80 ml), and the mixture was stirred at ambient
temperature for 72 hours. Saturated sodium
metabisulphite solution (~100 ml) was added to the
resulting brown solution until the colour changed to
WO 94/26704 PCT/EP94/01494
2 ~ 627 0 6
_ 2g _
orange, then the mixture was neutralised by the addition
of solid potassium carbonate.
The product was extra:-~ted into ethyl acetate
(3 x 300 ml) (filtration was required for the removal of
interfacial solids), the extracts were combined, dried
over magnesium sulphate and the solvent removed in vacuo
to give a brown oil (3.7 g).
The oil was purified via flash chromatography over
silica using a 4:1 mixture of toluene and triethylamine
followed by a 1:1 mixture of toluene and triethylamine
as eluants. Appropriate fractions were combined and the
solvents rertr ~. ed in vacuo to give a brown oil which
crystallised :.an standing ( 0 .7 g) .
The solid was dissolved in a mixture of hot diethyl
ether (50 ml) and ethyl acetate (8 ml) and the resulting
solution was filtered, cooled and saturated with
hydrogen chloride. The resulting solid was collected by
filtration, dried in vacuo at 40°C for 24 hours then
ground and redried in vacuo at 40°C for a further 24
hours to give 1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylsulphonyl]ethanone hydrochloride as
a white solid (0.4 g), m.p. 168-176°C.
Example 9
A solution of 1-[1-(3,4-dichlorophenyl)cyclobutyl]-
2-[3-(dimethylamino)propylthio]ethanone (4.6 g, prepared
by basification of the hydrochloride salt obtained in a
similar manner to that described in Example 7) in
propan-2-of (75 ml) was added dropwise to a stirred
suspension of sodium borohydride (4.8 g) in propan-2-of
(100 ml) at ambient temperature under a nitrogen
~~'O 94/26704 ~ ~ ~ 2 7 Q 6 PCT/EP94101494
- 29 -
atmosphere, and the mixture was stirred at ambient
temperature for 93 hours.
The resulting suspension was cautiously diluted
with acetone (33 ml) followed by saturated aqueous
ammonium chloride solution (100 ml).
The resulting neutral mixture was concentrated in
vacuo, the residue was diluted with water (100 ml) and
the product extracted into ether (3 x 200 ml). The
extracts were combined, dried over magnesium sulphate
and the solvent removed in vacuo to give a yellow oil
(4 g) .
The product was purified via flash chromatography
over silica using a 9:1 mixture of dichloromethane and
industrial methylated spirits (IMS) followed by a 4:1,
then 1:1 mixture of dichloromethane and IMS as eluants.
Appropriate fractions were combined and the solvents
removed in vacuo to yield 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)propyl-
thio]ethanol as a pale green oil (1.44 g).
Example 10
A mixture of 1-chloro-3-(dimethylamino)-2-
methylpropane hydrochloride (200 g), thiourea (97.3 g)
and ethanol (950 ml) was stirred and heated under reflux
for 72 hours. The solution was allowed to cool and the
solvent was removed in vacuo. The residue was dissolved
in a small volume of ethanol and ether was added until
the first permanent opalescence was observed. The
mixture was stored at 4°C for 16 hours. The solvent was
removed in vacuo to afford a waxy/oily solid which was
dried in vacuo over calcium chloride for 48 hours, then
triturated with propan-2-ol. The resulting solid was
WO 94/26704 216 2 7 0 6 PCT/EP94/01494
- 30 -
collected by filtration, washed with propan-2-ol, and
dried in vacuo to give S-[3-(dimethylamino)-2-
methylpropyl]isothiourea dihydrochloride as a pale brown
solid ( 90 g).
A solution of sodium hydroxide (19.3 g) in water
(20 ml) was added dropwise at 0°C to a stirred solution
of S-[3-(dimethylamino)-2-methylpropyl]isothiourea
dihydrochloride (60 g) in water (100 ml). The stirred
mixture was heated at 95°C for 2 hours and allowed to
cool. The product was extracted into ether (4 x 70 ml),
the combined extracts dried over sodium sulphate and the
solvent removed in vacuo. The residual oil was
dissolved in ether and the solution saturated with
hydrogen chloride. The resulting solid was collected by
filtration, washed with ether, and dried in vacuo over
phosphorus pentoxide for 24 hours to give 3-
(dimethylamino)-2-methylpropanethiol hydrochloride as a
white solid (30 g).
A solution of sodium ethoxide [prepared from sodium
(3 g) in ethanol (300 ml)] was added to a suspension of
3-(dimethylamino)-2-methylpropanethiol hydrochloride
(10.3 g) in ethanol (150 ml) under nitrogen at ambient
temperature, then the mixture was stirred at ambient
temperature for 1 hour. A solution of 2-bromo-1-[1-
(3,4-dichlorophenyl)cyclobutyl]ethanone (20.4 g,
prepared in a similar manner to that described in
Example 1) in ethanol (130 ml) was added and the mixture
was stirred at ambient temperature for 24 hours. The
solvent was removed in vacuo and the residue was diluted
with water (150 ml). The product was extracted into
dichloromethane (4 x 100 ml), the combined extracts were
dried over sodium sulphate and the solvent was removed
_in vacuo. The residue was dissolved in ether and the
solution saturated with hydrogen chloride. The
~O 94/26704
216 2 7 0 6 PCTIEP94/01494
- 31 -
resulting solid was collected by filtration, washed with
ether, and dried in vacuo for 24 hours. The solid
crystallised from a 2:1 mixture of ethyl acetate and
propan-2-of to give 1-[1-(3,4-dichlorophenyl)-2-[3-
(dimethylamino)-2-methylpropylthio]ethanone
hydrochloride as a white solid, (0.55 g), m.p. 136-
137°C.
Example 11
Methylmagnesium iodide was prepared under nitrogen
by dropwise addition of a solution of methyl iodide
(48.3 g) in ether (72 ml) to a stirred suspension of
magnesium turnings (8.2 g) in ether (60 ml) initially at
ambient temperature, then when the exothermic reaction
commenced, at reflux temperature. After the addition
the mixture was stirred at ambient temperature for 30
minutes and a solution of 1-(2-naphthyl)cyclobutane-
carbonitrile (48.2 g) in toluene (100 ml) was added
dropwise at ambient temperature. The resulting mixture
was stirred at ambient temperature for 16 hours.
The resulting solid was collected by filtration,
washed with ether and added in portions to a mixture of
concentrated hydrochloric acid (125 ml) and water
(200 ml). The resulting mixture was heated at ~95°C for
10 minutes, cooled and the product extracted into
toluene (3 x 200 ml). The extracts were washed with
water (200 ml), dried over magnesium sulphate and the
solvent removed in vacuo to give an oil (45 g). The oil
was triturated with petroleum ether (b.p. 40-60°C) to
give a solid which was collected by filtration and dried
in vacuo to yield 1-[1-(2-naphthyl)cyclobutyl]ethanone
(35 g) .
WO 94/26704 216 2 7 0 6 PCT/EP94/01494
- 32 -
A solution of bromine (4.3 ml) in chloroform (20
ml) was added dropwise over 30 minutes at 10-15°C to a
stirred solution of 1-[1-(2-naphthyl)cyclobutyl]-
ethanone (20 g) in a mixture of methanol (20 ml) and
chloroform (30 ml). The mixture was then stirred at ,
ambient temperature for 1.5 hours, poured onto ice-water
(300 ml) and the product extracted into dichloromethane
(3 x 150 ml). The extracts were washed with saturated
aqueous sodium hydrogen carbonate solution, then water,
dried over calcium chloride and the solvent removed in
vacuo to give 2-bromo-1-[1-(2-naphthyl)cyclobutyl]-
ethanone as an oil (24.0 g).
A solution of sodium ethoxide [prepared from sodium
(6.5 g) and ethanol (100 ml)] was added to a suspension
of 2-(dimethylamino)ethanethiol hydrochloride (6.4 g) in
ethanol (10 ml), and the mixture stirred at ambient
temperature for 1 hour. A solution of 2-bromo-1-[1-(2-
naphthyl)cyclobutyl]ethanone (19.5 g, prepared as
described above) in ethanol (50 ml) was added and the
mixture was stirred at ambient temperature for 18 hours.
The solvent was removed in vacuo, and the residue was
diluted with water (200 ml). The product was extracted
into ethyl acetate (2 x 200 ml), and the extracts were
dried over magnesium sulphate and the solvent removed in
vacuo to give an oil (11.0 g). The oil was dissolved in
ethyl acetate and the solution saturated with hydrogen
chloride. The solvent was removed in vacuo to give an
oil (8.5 g) which was triturated with a mixture of
propan-2-ol, ether and ethyl acetate to give a solid.
The solid was collected by filtration and dried in vacuo
to give 2-[2-(dimethylamino)ethylthio]-1-[1-(2-
naphthyl)cyclobutyl]ethanone hydrochloride as a cream
solid (1.7 g), m.p. 95-102°C.
~WO 94/26704
~ 1 ~ 2 7 0 6 pCT/EP94/01494
- 33 -
Example 12
Methylmagnesium iodide (138 ml of 3 M solution in
ether) was added dropwise to a stirred solution of 1-(3-
chlorophenyl)cyclobutanecarbonitrile (53 g) in ether
(100 ml) under nitrogen at 0°C. The mixture was stirred
at ambient temperature for 24 hours. The resulting
solid was collected by filtration, washed well with
ether, then added in portions to an ice-cold mixture of
water (200 ml) and concentrated hydrochloric acid (12S
ml). The resulting yellow suspension was heated at 95°C
for 1 hour with occasional stirring, and then cooled to
ambient temperature. The product was extracted into
ether (5 x 100 ml) and the combined extracts washed with
water (2 x 100 ml), dried over magnesium sulphate and
the solvent removed in vacuo to give an oil which was
distilled to give 1-[1-(3-chlorophenyl)cyclo-
butyl]ethanone (47.5 g), b.p. 108-109°C /2 mbar.
A solution of bromine (9.9 ml) in dichloromethane
(50 ml) was added dropwise at 10-15°C over 3 hours to a
stirred solution of 1-[1-(3-chlorophenyl)cyclobutyl]-
ethanone (38 g) in a mixture of methanol (75 ml) and
dichloromethane (15 ml). When the addition was
complete, the mixture was stirred at ambient temperature
for 2.5 hours, then poured onto an excess of ice-water.
The aqueous layer was separated and the product
extracted into dichloromethane (3 x 90 ml). The
combined organic solutions were washed with saturated
aqueous sodium hydrogen carbonate solution (2 x 100 ml)
and water (100 ml), dried over calcium chloride and the
solvent removed in vacuo to leave 2-bromo-1-[1-(3-
chlorophenyl)cyclobutyl]ethanone as an oil (47 g).
A solution of sodium ethoxide [prepared from sodium
(5.3 g) and ethanol (500 ml)] was added to a stirred
~~b2T06
WO 94!26704 PCTIEP94101494
- 34 -
suspension of 3-(dimethylamino)propanethiol
hydrochloride (16.2 g, prepared in a similar manner to
that described in Example 7) in ethanol (250 ml) under
nitrogen and the mixture .,was stirred at ambient
temperature for 1 hour. A~~ 'solution of 2-bromo-1- [1- (3- ,
chlorophenyl)cyclobutyl]ethanone (30 g) in ethanol
(130 ml) was added in one portion, and the mixture was
stirred at ambient temperature for a further 24 hours.
The solvent was removed in vacuo, and the residue was
diluted with water (200 ml). The product was extracted
into dichloromethane (4 x 100 ml). The combined
extracts were dried over magnesium sulphate and the
solvent removed in vacuo to afford a red/brown oil (31
!)
A sample (8 g) of the above oil was dissolved in
ether and the solution saturated with hydrogen chloride.
The solvent was removed in vacuo and the residue
triturated with ether. The resulting solid was
collected by filtration and crystallised from propan-2-
ol/ether to give 1-[1-(3-chlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanone hydrochloride as a
white solid (1.7 g), m.p. 66-68°C.
Example 13
4-(Dimethylamino)butanol (88.5g) was added dropwise
at 0°C over 2 hours to stirred thionyl chloride (93.4g)
then the mixture was stirred at ambient temperature for
1 hour and poured into ethanol (500m1). The stirred
solution was heated under reflux for 10 minutes, then
the solvent was removed in vacuo. The solid residue '
crystallised from ethanol as a white solid which was
collected by filtration, washed with ethanol, and dried
in vacuo at ambient temperature for 24 hours to give 1-
2162706
~O 94/26704 PCTIEP94/01494
- 35 -
chloro-4-(dimethylamino)butane hydrochloride as a white
solid (115g), m.p. 100-105°C.
A stirred mixture of 1-chloro-4-(dimethylamino)-
butane hydrochloride (115g), thiourea (51.9g) and
ethanol (500m1) was heated under reflux for 24 hours
then allowed to stand at 4°C for 24 hours. The
resulting solid was collected by filtration, washed with
ether, and dried in vacuo at ambient temperature for 24
hours to give S-[4-(dimethylamino)butyl]isothiourea
dihydrochloride as an off-white solid (140g), m.p. 179-
182°C.
A solution of sodium hydroxide (16.4g) in water
(16.5m1) was added dropwise under nitrogen at 0°C to a
stirred solution of S-[4-(dimethylamino)butyl]-
isothiourea dihydrochloride (51g) in water (60m1), then
the mixture was heated at 95°C for 2 hours and allowed
to cool to ambient temperature. Water (100m1) was
added, and the product was extracted into ether (50m1),
dichloromethane (3 x 50m1), and ether (2 x 50m1). The
combined organic solutions were dried over magnesium
sulphate, then the solvents were removed in vacuo. The
residue was dissolved in ether and the solution was
saturated with hydrogen chloride. The resulting solid
was collected by filtration and dried in vacuo at
ambient temperature to give 4-(dimethylamino)butanethiol
hydrochloride as a white solid (21g) which was used
without further purification.
A solution of sodium ethoxide [prepared from sodium
(1.6g) and ethanol (175m1)] was added at ambient
temperature under nitrogen to a stirred suspension of 4-
(dimethylamino)butanethiol hydrochloride (5.5g) in
ethanol (75m1) and the mixture was stirred at ambient
temperature for 1 hour. A solution of 2-bromo-1-[1-
~1627~6
WO 94/26704 PCT/EP94/01494
- 36 -
(3,4-dichlorophenyl)cyclobutyl]ethanone (llg,prepared in
a similar manner to that described in Example 1) in
ethanol (40m1) was added, and the mixture was stirred at
ambient temperature for 48 hours. The solvent was
removed in vacuo, and the residue was diluted with water
(200m1). The product was extracted into dichloromethane
(4 x 90m1) then the combined extracts were dried over
sodium sulphate, and the solvent removed in vacuo. The
residual oil was purified via column chromatography over
silica using a 9:1 mixture of toluene and triethylamine
as eluant. Appropriate fractions were combined, and the
solvents were removed in vacuo. The residue was
dissolved in ether, and the solution was saturated with
hydrogen chloride. The resulting solid was collected by
filtration, washed with ether, dried in vacuo over
phosphorus pentoxide for 24 hours, and reczystallised
from a 4:1 mixture of ethyl acetate and ethanol. The
resulting solid was collected by filtration, washed with
ethyl acetate, and dried in vacuo at ambient temperature
over phosphorus pentoxide for 48 hours to give 1-[1-
(3,4-dichlorophenyl)cyclobutyl]-2-[4-(dimethylamino)-
butylthio]ethanone hydrochloride as a white solid
(5.E-~~, m.p. 129-130°C.
Ex° 14
.~-(Dipropylamino)propanol (32.9g) was added
dropwise a4 0°C over 1.5 hours to stirred thionyl
chloride (15.7m1). When the addition was complete, the
mixture was stirred at ambient temperature for 2 hours
then poured into ethanol (250m1). The stirred mixture
was heated under reflux for 10 minutes, then the solvent
was removed in vacuo to leave 1-chloro-3-(dipropylamino)
-propane hydrochloride as an off-white solid (42g) which .
was used without purification.
-WO 94/26704
2 ~ 6 ~ 7 0 6 PCT~~4/01494
- 37 -
A stirred mixture of 1-chloro-3-(dipropylamino)-
propane hydrochloride (42g), thiourea (15.5g) and
ethanol (250m1) was heated under reflux for 24 hours
then allowed to cool to ambient temperature. Ethyl
acetate was added until a faint opalescence was
observed, then the mixture was stored at 4°C for 24
hours. After this time it had deposited an oil which
was isolated by decantation of the bulk of the solvent
followed by removal of residual solvent in vacuo. The
oil was triturated with ethanol, the solvent was removed
by decantation, and the residue was dried in vacuo at
ambient temperature for 24 hours to give a pale brown
solid. The ethanol solution decanted from the oil was
concentrated in vacuo, and the residue was triturated
with ethanol as described above to give a second crop of
pale brown solid. The ethanol solution remaining after
isolation of the second crop was concentrated in vacuo
and the residue was triturated with propan-2-of to give
a third crop of solid. The three crops were combined to
give S-[3-(dipropylamino)propyl]isothiourea
dihydrochloride as a pale brown solid (51g), m.p 143-
145°C.
M Aqueous sodium hydroxide solution (11m1) was
added dropwise at 0°C under nitrogen to a stirred
25 solution of S-[3-(dipropylamino)propyl]isothiourea
dihydrochloride (40g) in water (100m1), then the mixture
was stirred at 95°C for 2 hours and allowed to cool to
ambient temperature. The product was extracted into
ether (4 x 70m1), the extracts were dried over magnesium
sulphate, and the solvent was removed in vacuo. The
residue was dissolved in ether, and the solution was
saturated with hydrogen chloride to give a small amount
of white solid which was collected by filtration. The
filtrate was concentrated in vacuo, and the residue was
combined with the white solid and dissolved in ethanol.
. WO 94/26704 PCT/EP94/01494
216~~'0~ . - 38 -
The solution was saturated with hydrogen chloride and
the solvent was removed in vacuo to leave crude 3
(dipropylamino)propanethiol hydrochloride (18.3g) as a
colourless semisolid which was used without
purification.
A solution of sodium ethoxide [prepared from sodium
(1.6g) and ethanol (175m1)] was added at ambient
temperature under nitrogen to a stirred suspension of
the crude 3-(dipropylamino)propanethiol hydrochloride
(7g) in ethanol (75m1), then the mixture was stirred at
ambient temperature for 1 hour. A solution of 2-bromo-
1-[1-(3,4-dichlorophenyl)cyclobutyl]ethanone (10.6g,
prepared in a similar manner to that described in
Example 1) in ethanol (40m1) was added, and the mixture
was stirred at ambient temperature for 24 hours. The
solvent was removed in vacuo, the residue was diluted
with water (100m1), and the product was extracted into
dichloromethane (4 x 75m1). The extracts were dried
over sodium sulphate, the solvent was removed in vacuo,
and the residue was purified via column chromatography
over silica using a 19:1 mixture of toluene and
triethylamine as eluant. Appropriate fractions were
combined and the solvents were removed in vacuo to leave
1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dipropylamino)propylthio]ethanone as a pale yellow oil
(5.5g).
Example 15
A solution of fumaric acid (0.65g) in hot ethanol
(20m1) was added to a solution of 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)
propylthio]ethanone (2.1g, prepared by basification of
the hydrochloride salt obtained in a similar manner to
that described in Example 7) in ether (10m1) and the
O 94/26704
216 ~ ~ ~ 6 PCT/EP94/01494
- 39 -
mixture was allowed to stand at 4°C for 96 hours. No
solid precipitated, so the solvents were removed _in
vacuo to leave a brown oil which was triturated with
petroleum ether (b.p. 40-60°C). The resulting solid was
collected by filtration, washed with ether, dried _in
vacuo at ambient temperature for 18 hours, and
recrystallised from a 3:2 mixture of ethyl acetate and
petroleum ether (b.p. 60-80°C). The resulting solid was
collected by filtration, washed with petroleum ether
(b. p. 60-80°C), and dried in vacuo at ambient
temperature for 18 hours to give 1-[1-(3,4-dichloro-
phenyl)cyclobutyl]-2-[3-(dimethylamino)propylthio]
ethanone fumarate as a white solid (1.1g), m.p. 100-
103°C.
Example 16
Sodium borohydride (3.2g) was added in portions at
0°C under nitrogen to a stirred solution of 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)-
propylthio]ethanone (16 g, prepared by basification of
the hydrochloride salt obtained in a similar manner to
that described in Example 7) in methanol (200m1) then
the mixture was stirred at ambient temperature for 7
days and diluted with water (350m1). The product was
extracted into dichloromethane (4 x 100m1) and the
extracts washed with water (100m1) and saturated aqueous
sodium chloride solution (100m1), dried over sodium
sulphate and the solvent removed in vacuo to leave a
green oil. The oil was resolved via preparative scale
chiral high performance liquid chromatography to give
(-)-1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanol as an oil (3.5g),
[oc]Dt - -8.615°(C=l; ethanol) and (+)-1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)
WO 94/26704 PCT/EP94/01494
21627 06
- 40 -
propylthio]ethanol as an oil (3.4g), [oc]Dt - +9.740°
(C=1; ethanol).
A solution of citric acid (1.77g) in hot ethanol
(10m1) was added to a solution of (-)-1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)-
propylthio]ethanol (3.42g) in ether (30m1), and the
mixture was stored at 4°C for 18 hours. The resulting
solid was collected by filtration, washed with ether and
dried _in vacuo at ambient temperature for 24 hours, to
give (-)-1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanol citrate as a white
solid (3.9g), m.p.109-115°C, [oc]Dt= -10.17°(C=1;
methanol).
Example 17
A solution of citric acid (1.73g) in hot ethanol
(10m1) was added to a solution of (+)-1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)
propylthio]ethanol (3.298, preparation described in
Example 16) in ether (30m1) and the mixture was stored
at 4°C for 18 hours. The resulting solid was collected
by filtration, washed with ether and dried in vacuo at
ambient temperature for 24 hours, to give (+)-1-[1-(3,4
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)
propylthio]ethanol citrate as a white solid (3.8g),
m.p.109-115°C, [oc]Dt =+10.88°(C=1; methanol).
Example 18
A stirred mixture of 1-chloro-3-(dimethylamino)
propane hydrochloride (305.7g), thiourea (150g) and
ethanol (1530m1) was heated under reflux for 24 hours
then cooled to ambient temperature. Ethyl acetate was
added until a faint opalescence was observed, then the
2~ X2706
~WO 94/26704 PCT/EP94101494
- 41 -
mixture was stored at 4°C for 72 hours. The resulting
solid was collected by filtration, washed with ethyl
acetate, and dried in vacuo at 40°C to give S-[3
(dimethylamino)propyl]isothiourea dihydrochloride as a
white solid (403g), m.p. 155-157°C.
A solution of sodium hydroxide (250g) in water
(250m1) was added at <25°C over 10 minutes to a stirred
solution of ~S-[3-(dimethylamino)propyl]isothiourea
dihydrochloride (731g; prepared in a similar manner to
that described above) in water (880m1), then the mixture
was stirred at 95°C for 3 hours and cooled to 10°C. The
product was extracted into dichloromethane (4 x 500m1),
the extracts were combined, and the solvent was removed
in vacuo. The residue was disolved in ether (11), the
solution was decanted from a white solid residue, and
the solvent was removed in vacuo to leave a colourless
oil (362.7g). The oil was added dropwise over 20
minutes at <15°C to stirred 5M hydrochloric acid
(650m1), then the mixture was concentrated in vacuo at
70°C to give a white solid. The solid was diluted with
propan-2-of (11) and the solvent was removed in vacuo,
then the residue was diluted with toluene (11) and the
solvent was removed in vacuo. The residue was
triturated with ether (11) and the resulting solid was
collected by filtration, washed with ether, and dried in
vacuo at 40°C for 3 days and at ambient temperature over
phosphorus pentoxide for 5 days to give 3-
(dimethylamino)propanethiol hydrochloride as a white
solid (405.9g), m.p. 81-84°C.
A solution of 1-(3,4-dichlorophenyl)cyclo-
butanecarbonitrile (163.6g) in ether (130m1) was added
dropwise over 0.5 hours at ambient temperature under
nitrogen to stirred methylmagnesium iodide (3M solution
in ether; 300m1), then the mixture was heated under
WO 94/26704 PCT/EP94/01494
2~ 62~ Ob
- 42 -
reflux for 1 hour, diluted with ether (100m1), heated
under reflux for a further 2.5 hours, then stirred at
ambient temperature for 18 hours. The resulting solid ,
was collected by filtration, washed well with ether, and
added in portions at <20°C to a stirred mixture of ,
concentrated hydrochloric acid (410m1) and water
(650m1) . The mixture was heated at 95°C for 0.5 hours
with occasional stirring then cooled to ambient
temperature. The product was extracted into
dichloromethane (3 x 200m1), the combined extracts were
dried over magnesium sulphate, and the solvent removed
in vacuo to leave 1-[1-(3,4-dichlorophenyl)-
cyclobutyl]ethanone as a dark red oil (172.78) which was
used without purification.
A solution of bromine (96m1) in chloroform (427m1)
was added dropwise at 10-15°C over 1.5 hours to a
stirred solution of 1-[1-(3,4-dichlorophenyl)cyclobutyl]
ethanone (477.28; prepared in a similar manner to that
described above) in a mixture of methanol (643m1) and
chloroform (107m1). After the addition was complete,
the mixture was stirred at ambient temperature for 3
hours, then poured into ice-water (21). The aqueous
layer was separated and washed with dichloromethane (3 x
500m1), then the combined organic solutions were washed
with saturated aqueous sodium hydrogencarbonate solution
(2 x 400m1) and water (500m1), dried over calcium
chloride, and the solvents were removed in vacuo to
leave 2-bromo-1-[1-(3,4-dichlorophenyl)cyclobutyl]
ethanone as an orange oil (593.58) which was used
without purification.
A solution of 3-(dimethylamino)propanethiol
(235.68; prepared by basification of the hydrochloride
salt) in ethanol (11) was added dropwise at ambient
temperature to a stirred solution of sodium ethoxide
~WO 94/26704 ~ PCTIEP94/01494
- 43 -
[prepared from sodium (50g) and ethanol (21)], then the
mixture was stirred at ambient temperature for 1 hour.
A solution of 2-bromo-1-[1-(3,4-dichlorophenyl)-
cyclobutyl)ethanone (817.48; prepared in a similar
manner to that described above) in ethanol (1.51) was
added and the mixture was stirred at ambient temperature
for 18 hours. The solvent was removed in vacuo, and the
solid residue was diluted with water (21). The product
was extracted into dichloromethane (3 x 500m1), the
combined extracts were dried over magnesium sulphate,
and the solvent was removed in vacuo. The residue was
disolved in ether, the solution was decanted from an
insoluble gum, and the solvent was removed in vacuo to
leave an orange oil (7138). The oil was dissolved in
petroleum ether (b.p.60-80°C) (3.51), charcoal and
magnesium sulphate were added, the mixture was filtered
(celite), and the solvent was removed in vacuo to leave
a pale orange oil (7138). The oil was added at <20°C to
a stirred mixture of concentrated hydrochloric acid
(255m1) and water (1750m1) then the mixture was
concentrated in vacuo at 50°C. The residue was
repeatedly diluted with toluene and concentrated in
vacuo until all of the water had been removed, then the
residue was triturated with ether (2.51). The ether was
removed by decantation, and the residue was dissolved in
ethyl acetate (2.51). Ether (51) was added, and the
resulting solid was collected by filtration, washed with
ether, suspended in ethyl acetate (2.51), collected by
filtration, washed with ethyl acetate, and dried in
vacuo at 50°C to give crude 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)-
propylthio]ethanone hydrochloride as a cream solid -
(425g) .
21 ~2~ 0 6
WO 94/26704 PCT/EP94/01494
- 44 -
Repetition of this experiment on approximately i~
scale gave further crude product as a cream solid
(313g) .
The two crops of impure solid were combined,
dissolved in water (2.51), and basified to pH9 by
addition of solid sodium carbonate. The free base was
extracted into dichloromethane (3 x 500m1) and the
solvent was removed in vacuo The resulting gum was
partitioned between water and ethyl acetate; an emulsion
formed, so the. mixture was filtered (Celite), then the
organic layer was separated, dried over sodium sulphate
and the solvent was removed in vacuo to leave a brown
oil (638g). The oil was purified in portions (50g) by
filtration through silica using a 9:1 mixture of
dichloromethane and methanol as eluant. Appropriate
fractions were combined and the solvents were removed in
vacuo to give a pale orange oil (484.3g) . The oil was
added at <20°C to a stirred mixture of concentrated
hydrochloric acid (173m1) and water (415m1), and the
resulting mixture was concentrated in vacuo. The
residue was dried by repeated dilution with toluene and
removal of the solvent in vacuo, then the resulting gum
was dissolved in ethyl acetate (500m1) and diluted with
ether (2.51). The resulting solid was collected by
filtration, washed with ether and dried in vacuo at 45°C
to give 1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanone hydrochloride as a
cream solid (507.1g).
1-[1-(3,4-Dichlorophenyl)cyclobutyl]-2-[3-
(dimethylamino)propylthio]ethanone hydrochloride (480g)
was suspended in an excess of saturated aqueous sodium
hydrogencarbonate solution, the mixture was stirred at
ambient temperature for 0.5 hours, then the free base
was extracted into dichloromethane. The extracts were
WO 94/26704 PCT/EP94/01494
~~62106
- 45 -
dried over sod~.um sulphate and the solvent was removed
in vacuo to leave a brown oil (415g). The oil was
dissolved in ether (1700m1) and added to a solution of
citric acid (210g) in hot ethanol (3200m1), then the
mixture was allowed to cool to ambient temperature and
was stored at 4°C for 48 hours whereupon it deposited a
pale brown solid. The supernatent liquor was removed by
decantation, the residue was diluted with ethanol
(300m1), and the mixture was warmed gently to loosen the
crystalline mass. The product was collected by
filtration, washed with ether, dried in vacuo, and
recrystalised from ethanol. The resulting solid was
collected by filtration, washed with ethanol, and dried
in vacuo at 50°C for 4 hours to give 1-[1-(3,4-
dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)
propylthio]ethanone citrate as a cream solid (410g),
m.p. 103-105°C.
Example 19
The mother liquors remaining after isolation of the
product described in Example 10 were concentrated in
vacuo and the residue was diluted with water and
basified by the addition of 5M aqueous sodium hydroxide
solution. The product was extracted into ether and the
extracts were washed with water, dried over magnesium
sulphate and the solvent removed in vacuo to leave 1-[1-
(3,4-dichlorophenyl)cyclobutyl]-2-[3-(dimethylamino)-2-
methylpropylthio]ethanone as a pale yellow oil. A
sample (4g) of this oil was dissolved in methanol (40m1)
and sodium borohydride (0.8g) was added in portions
under nitrogen at 0°C. The mixture was stirred at
ambient temperature for 7 days then diluted with water
(120m1). The product was extracted into dichloromethane
(4 x 50m1) then the combined extracts were washed with
water (50m1) and saturated aqueous sodium chloride
WO 94/26704 PCT/EP94/01494
- 46 -
solution (50m1), dried over sodium sulphate and the
solvent removed in vacuo to give a colourless oil
(4.2g) .
The oil was dissolved in ether and the solution
saturated with hydrogen chloride to give a solid which
was collected by filtration, washed with ether and dried
in vacuo over phosphorus pentoxide for 72 hours. The
resulting white solid was hygroscopic, so it was
dissolved in water and basified by the addition of
saturated aqueous sodium hydrogencarbonate solution.
The product was extracted into dichloromethane (3 x
50m1) and the combined extracts were washed with water,
dried over sodium sulphate and the solvent removed in
vacuo to leave 1-[1-(3,4-dichlorophenyl)cyclobutyl]-2-
[3-(dimethylamino)-2-methylpropylthio)ethanol as a pale
green oil (3g) .
Example 20
1,4-Dibromobutane (106m1) was added dropwise over 1
hour at 70-80°C under nitrogen to a stirred mixture of
3,4-dichlorophenylacetonitrile (150g),
benzyltriethylammonium chloride (2g) and 50~ aqueous
sodium hydroxide solution (300m1). When the addition
was complete the mixture was stirred at 70-80°C for 2
hours, then cooled to ambient temperature. Ether
(400m1) and water (200m1) were added and the layers were
separated. The aqueous layer was washed with ether (2 x
200m1), then the combined organic solutions were dried
over magnesium sulphate and the solvent removed in
vacuo. The residue was distilled to give 1-(3,4- T
dichlorophenyl)cyclopentanecarbonitrile as a pale yellow
oil (135g), b.p. 132-140°C/0.4 mbar.
~WO 94/26704 PCT/EP94/01494
- 47 -
Methylmagnesium iodide (3M solution in ether;
100m1) was added dropwise at 0°C under nitrogen to a
. stirred solution of 1-(3,4-dichlorophenyl)
cyclopentanecarbonitrile (48g) in ether (100m1), then
the mixture was stirred at ambient temperature for 24
hours. The resulting solid was collected by filtration,
washed with ether, and added in portions to an ice-cold
mixture of water (200m1) and concentrated hydrochloric
acid (125m1). The mixture was heated at 95°C for 1
hour, then allowed to cool to ambient temperature. The
product was extracted into ether (5 x 100m1), and the
combined extracts were washed with water (2 x 100m1),
dried over magnesium sulphate, and the solvent removed
in vacuo. The residue was distilled to give 1-[1-(3,4-
dichlorophenyl)cyclopentyl]ethanone as a pale yellow oil
(31.9g), b.p. 124-128°C/0.5 mbar.
A solution of bromine (6.1m1) in dichloromethane
(50m1) was added dropwise over 3 hours at 10-15°C under
nitrogen to a stirred solution of 1-[1-(3,4-
dichlorophenyl)cyclopentyl]ethanone (31.9g) in a mixture
of methanol (60m1) and dichloromethane (10m1), then the
mixture was stirred at ambient temperature for 2.5 hours
and poured into an excess of ice-water. The aqueous
layer was separated and washed with dichloromethane (3 x
100m1), then the combined organic solutions were washed
with saturated aqueous sodium hydrogencarbonate solution
(2 x 100m1) and water (100m1), dried over calcium
chloride, and the solvent removed in vacuo. The residue
was distilled in vacuo, and the fraction of b.p. >174°C/
1.3 mbar was collected and redistilled. Material of
b.p. >182°C/2.6 mbar in this second distillation was
collected and redistilled to give 2-bromo-1-[1-(3,4-
dichlorophenyl)cyclopentyl]ethanone as a pale yellow oil
(11.8g), b.p. 156-162°C/0.4 mbar.
WO 94/26704 216~~ ~ ~ PCTIEP94/01494
- 48 -
A solution of sodium ethoxide [prepared from sodium
(1.4g) and ethanol (175m1)] was added at ambient
temperature under nitrogen to a stirred suspension of 3-
(dimethylamino)propanethiol hydrochloride (4.5g,
prepared in a similar manner to that described in
Example 7) in ethanol (75m1); then the mixture was
stirred at ambient temperature for 1 hour. A solution
of 2-bromo-1-[1-(3,4-dichlorophenyl)cyclopentyl]ethanone
(10.5g) in ethanol (40m1) was added, and the mixture was
stirred at ambient temperature for 24 hours. The
solvent was removed in vacuo, the residue was diluted
with water (100m1), and the product was extracted into
dichloromethane (4 x 75m1). The extracts were dried
over sodium sulphate, the solvent was removed in vacuo,
and the residue was purified via column chromatography
over silica using a 19:1 mixture of toluene and
triethylamine as eluant. Appropriate fractions were
combined and the solvents removed in vacuo to leave a
pale brown oil (7g). The oil was dissolved in ether,
the solution was saturated with hydrogen chloride, and
the solvent was removed in vacuo. The residue was
triturated with ether, and the resulting solid was
collected by filtration, washed with ether, and dried in
vacuo at ambient temperature over phosphorus pentoxide
for 48 hours to give 1-[1-(3,4-dichlorophenyl)
cyclopentyl]-2-[3-(dimethylamino)propylthio]ethanone
hydrochloride as a white solid (5.6g), m.p. 77-80°C.
Example 21
The use of compounds of the present invention in
the manufacture of pharmaceutical compositions is
illustrated by the following description. In this
description the term "active compound" denotes any
compound of the invention but particularly any compound
~WO 94/26704
2 T ~ 2 7 0 6 PCT~~4/01494
- 49 -
which is the final product of one of the preceding
Examples.
a) Capsules
In the preparation of capsules, 10 parts by weight
of active compound and 240 parts by weight of lactose
are de-aggregated and blended. The mixture is filled
into hard gelatin capsules, each capsule containing a
unit dose of part of a unit dose of active compound.
b) Tablets
Tablets are prepared from the following
ingredients.
Parts by weicrht
Active compound 10
Lactose 190
Maize starch 22
Polyvinylpyrrolidone 10
Magnesium stearate 3
The active compound, the lactose and some of the
starch are de-aggregated, blended and the resulting
mixture is granulated with a solution of the polyvinyl
pyrrolidone in ethanol. The dry granulate is blended
with the magnesium stearate and the rest of the starch.
The mixture is then compressed in a tabletting machine
to give tablets each containing a unit dose or a part
of a unit dose of active compound.
c) Enteric coated tablets
WO 94/26704 PCT/EP94/01494
2~ 627 06
- 50 -
Tablets are prepared by the method described in
(b) above. The tablets are enteric coated in a
conventional manner using a solution of 20~ cellulose
acetate phthalate and 3~ p diethyl phthalate in
ethanol:dichloromethane (1:1).
d) Suppositories
In the preparation of suppositories, 100 parts by
weight of active compound is incorporated in 1300 parts
by weight of triglyceride suppository base and the
mixture formed into suppositories each containing a
therapeutically effective amount of active ingredient.