Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
AMIDE DERIV,~TIVES AND THEIR THERAPEUTICS USE
The present invention relates to amide compounds, synthesis thereof,
intermediates, salts and solvates thereof, pharmaceutical compositions
containing
them and the use of such compounds and compositions in medicine and therapy,
particularly as central muscle relaxants.
The major limiting side effects of many clinically effective central muscle
relaxant
and anticonvulsants are the induction of sedation and in coordination in the
recipient, which severely limit their usefulness. Examples of such muscle
relaxants
are the cinnamamide derivatives disclosed in EP-A-0381508. Similar side
effects
are found with drugs used in the treatment of anxiety, such as
benzodiazepines.
Although these effects may be transient, patients on such therapy are often
unable
to drive or participate in certain occupations.
It has now surprisingly been found that amides of formula (I) are potent
central
muscle relaxants and have a significantly reduced liability to sedation and
incoordination compared with known agents.
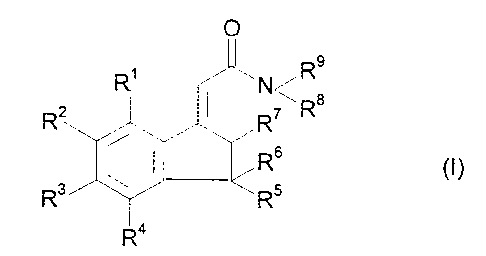
In one aspect the present invention provides the novel compounds of formula
(I)
s
V~Ra
~7
R6
R~
R
wherein R1, R', R3 and R4 are each selected from hydrogen and fluoro and at
least
one and not more than two is fluro;
RS is selected from hydrogen and C,,-C4 alkyl;
CA 02162708 2001-07-10
2
R6 is selected from hydrogen., C~-C4 alkyl and hydroxy; or
R5 and R6 together with the ring carbon form a carbonyl group;
R' is selected from hydrogen and hydroxy;
R8 and R9 are each selected from hydrogen, Cl-C4 alkyl and cyclo(C3 or
C4)alkyl
or together with the nitrogen form a morpholino group; and
and salts and solvates thereon
As used herein,
"C1-C4 alkyl" means a linear or branched chain alkyl group having 1, 2, 3 or 4
carbon atoms:
"cyclo(C3 or C4)alkyl" means a cycloalkyl group having 3 or 4 carbon atoms;
"salts" means base salts formed when, in formula (I), one of Rg and R9 is
hydrogen; and
"solvates" means a combination, in definite proportions, of a compound of
formula
(I) and a solvent therefor.
It will be appreciated that o:he compounds of formula (I) can exist in various
geoisomeric forms and as mixtures thereof in any proportions. The present
invention includes within its scope such geoisomeric forms or mixtures of
geoisomers, including the individual E and Z isomers of the compounds of
formula ( 1 ) as well as mixtures of such isomers, in any proportions.
CA 02162708 2001-07-10
WO 94/26693 PCT/GB94/01003
~1~~~~~
-3-
Included within formula (I) are compounds wherein one or more carbon centers
is/are
chiral. The present invention includes within its scope each possible optical
isomer
substantially free from, i.e., associated with less than 5% of, any other
optical isomer(s), as
well as mixtures of one or more optical isomers in any proportions, including
racemic
mixtures thereof.
It will be evident to a skilled person that certain compounds of formula (I)
can exist in
enantiomeric forms according to the direction of rotation of plane polarized
light when
passed through a sample of the compound. Individual optical isomers as well as
mixtures of
such isomers in any proportions are within the scope of the invention.
As will be appreciated, structural formula (I) is merely a two-dimensional
representation of
the compounds.
Separate groups of compounds, within formula (I), include those wherein
(i) one of Rl, R2, R3 and R4 is fluoro;
(ii) two of RI, R2, R3 and R4 are fluoro;
(iii) Rl is fluoro;
(iv) R2 is fluoro;
(v) R3 is fluoro;
(vi) R4 is fluoro;
(vii) RS is hydrogen;
(viii) RS is CI-C4 alkyl, preferably alkyl having I, 2 or 3 carbon atoms and
more
preferably methyl or ethyl;
(ix) R6 is hydrogen;
4
(x) R6 is C,-C4 alkyl, preferably alkyl having 1, 2 or 3 carbon atoms and more
preferably methyl or ethyl;
(xi) R6 is hydroxy;
(xii) RS and R6 together with the ring carbon form a carbonyl group;
(xiii) R' is hydrogen;
(xiv) R' is hydroxy;
(xv) Rg is hydrogen;
(xvi) Rg is (:,-C4 alkyl, pre:Ferably alkyl having 1, 2 or 3 carbon atoms and
more
preferably methyl, ethyl or isopropyl;
(xvii) R8 i;s cyclo(C3 or C~)alkyl, preferably cyclopropyl;
(xviii) R9 is hydrogen;
(xix) R9 is C,-C4 alkyl, preferably alkyl having l, 2 or 3 carbon atoms and
more
preferably methyl, ethyl or isopropyl;
(xx) R~ is cyclo(C3 or C,;)a.lkyl, preferably cyclopropyl;
(xxi) Rg and R9 together with the nitrogen form a motpholino group;
and salts and solvates thereof.
CA 02162708 2001-07-10
Preferred as a class are compounds wherein the >C=0 group and the benzene ring
are on opposite sides of the e:xo double bond, and salts and solvates thereof.
Individual preferred compaunds within formula (I) include
(E)-2-(6-fluoro-3-methyl-I-in.danylidene)acetamide;
(E)-N-cyclopropyl-2-(6-fluoro-3-methyl-1-indanylidene)acetamide;
(E)-2-(6-fluoro-3,3-dimethyl~-1-indanylidene)-N-methylacetamide;
(E)-N-cyclopropyl-2-(6-fluoro-3-ethyl-1-indanylidene)acetamide;
(E)-N-cyclopropyl-2-(5,6-difluoro-1-indanylidene)acetamide;
(E)-2-(5,6-difluoro-1-indanyl:idene)-N-methylacetamide;
(E)-2-(5,6-difluoro-1-indanyLidene)acetamide;
(E)-2-(5, 7-difluoro-1-indanylidene)acetamide;
(E)-N-cyclopropyl-2-(4,6-difluoro-1-indanylidene)acetamide;
(E)-2-(4,6-difluoro-1-indanyl idene)-N-isopropylacetamide;
(E)-2-(4, 6-difluoro-1-indanylidene)-N,N-dimethylacetamide;
(Z)-2-(4,6-difluoro-2-hydroxy-1-indanylidene)acetamide;
(E)-2-(4,6-difluoro-1-indanyllidene)acetamide;
(E)-2-(6-fluoro- 1-indanylidewe)acetamide;
(Z)-2-(6-fluoro-2-hydroxy- I -indanylidene)acetamide;
(E)-2-(6-fluoro-3,3-dimethyl-l: -indanylidene)acetamide;
(E)-2-(6-fluoro-3-ethyl-1-indanylidene)-N,N-dimethylacetamide;
(E)-2-(6-fluoro-3-hydroxy-1-~indanylidene )acetamide
and salts and solvates thereof'.
Particularly preferred is (E I-2-(4,6-difluoro-1-indanylidene)acetamide,
together
with its salts and solvates.
Preferred salts and solvates a.re those that are pharmaceutically acceptable.
CA 02162708 2001-07-10
WO 94/26693 , . " . ' PCT/GB94/01003
The present invention further provides a compound of formula (I), or a
pharmaceutically
acceptable salt or solvate thereof, for use in the medical treatment of a
mammal including a
human being.
The present invention also provides the use of a compound of formula (I), or a
pharmaceutically acceptable salt or solvate thereof, for the manufacture of a
medicament for
the medical treatment of a mammal including a human being.
The pharmaceutically acceptable salts include ammonium salts; alkali metal
salts, for
example sodium and potassium salts; and alkaline earth metal salts, for
example magnesium
and calcium salts.
Salts that are not pharmaceutically acceptable have utility in the preparation
and/or
purification of the compounds themselves and/or of those salts thereof that
are acceptable,
and/or in non-therapeutic, for example in vitro, applications.
The compounds of formula (I) together with their pharmaceutically acceptable
salts and
solvates are useful in medicine as central muscle relaxants and may thus be
used in the
treatment of conditions associated with abnormally raised skeletal muscle
tone.
They are of especial value in the relaxation of skeletal muscle in spastic,
hypertonic and
hyperkinetic conditions. In particular, they may be used in the treatment and
symptomatic
relief of exertion-induced skeletal muscle spasm, for example, in lower back
pain. They may
also be used in conditions such as spinal cord.injury, parkinsonism, chorea,
arthritis,
athetosis, status epilepticus and tetanus and especially in the relief of
muscle spasm in
conditions such as spasticity, myositis, spondylitis, cerebral palsy,
cerebrovascular disease
and multiple sclerosis. They may also be used as pre-surgical muscle
relaxants.
Compounds of formula (I) together with their pharmaceutically acceptable salts
and solvates
are also useful in the treatment of conditions associated with a conwlsive
state, for example
that following grand mal, petit mal, psychomotor epilepsy or focal seizure.
Compounds of formula (I) together with their pharmaceutically acceptable salts
and solvates
are also useful in the treatment of anxiety; as used herein, this term should
be understood to
include anxiety disorders.
WO 94/26693 PCT/GB94/01003
7_
Anxiety disorders are defined, in the Diagnostic and Statistical Manual of
Mental Disorders
(Third Edition - Revised, 1987, published by the American Psychiatric
Association,
Washington, D.C., U.S.A., see pages 235 to 253), as psychiatric conditions
having
symptoms of anxiety and avoidance behaviour as characteristic features.
Included amongst
such conditions are generalised anxiety disorder, simple phobia and panic
disorder.
Anxiety also occurs as a symptom associated with other psychiatric disorders,
for example
obsessive compulsive disorder, post-traumatic stress disorder, schizophrenia,
mood
disorders and major depressive disorders, and with organic clinical conditions
such as
Parkinson's disease, multiple sclerosis and other physically incapacitating
disorders.
Compounds of formula (I) together with their pharmaceutically acceptable salts
and solvates
are also useful in the treatment of pain, for example that associated with
inflammation
and/or trauma, and have utility as mild and strong analgesics.
Compounds of formula (I) together with their pharmaceutically acceptable salts
and solvates
are also useful in the treatment of inflammatory conditions including
inflammatory arthritic
conditions, for example rheumatoid arthritis, rheumatoid spondylitis,
osteoarthritis and
gouty arthritis; and non-articular inflammatory conditions, for example
herniated/ruptured/prolapsed intervertebral disk syndrome, bursitis,
tendinitis, tenosynovitis,
fibromyalgia syndrome and other inflammatory conditions associated with
ligamentous
sprain and regional musculoskeletal strain. It is particularly notable that
they are less
ulcerogenic than other anti-inflammatory agents such as ibuprofen, naproxen
and aspirin.
The present invention thus also provides a method for the treatment of
a) a condition associated with abnonmally raised skeletal muscle tone;
b) a condition associated with a convulsive state;
c) anxiety;
d) pain; or
e) an inflammatory condition
in a mammal including a human being, the method comprising administering
thereto a
therapeutically effective amount of a compound of formula (I) or a
pharmaceutically
acceptable salt or solvate thereof.
WO 94/26693 PCT/GB94101003
_$_
The compounds of formula (I) and their pharmaceutically acceptable salts and
solvates may
be administered concomitantly with other therapeutic agents for the treatment
of the above-
recited conditions. For conditions associated with abnormally raised skeletal
muscle tone,
such other agents include analgesics such as codeine, acetaminophen,
phenacetin and
ibuprofen. For inflammatory conditions (for example, arthritis) and/or pain,
such other
agents include analgesics, such as codeine, oxycodone, acetaminophen,
phenacetin and
ibuprofen; anti-arthritics, such as methotrexate and azathioprine; and
decongestants, such as
ephedrine and pseudoephedrine.
The compound, salt or solvate (hereinafter together referred to as the active
ingredient) may
be administered by any suitable route including oral, rectal, nasal, topical
(including buccal
and sublingual), vaginal, parenteral (including subcutaneous, intramuscuiar,
intravenous and
intradermal) and transdermal. It will be appreciated that the preferred route
will be
determined by, for example, the condition and age of the recipient and the
identity of the
condition to be treated.
The amount required of the active ingredient depends upon a number of factors
including
the identity of the condition and its severity, the identity of the recipient
and the route of
administration and will ultimately be at the discretion of the attendant
physician.
In general, for each of the above-recited conditions, a suitable dose of the
active ingredient
(estimated as the parent compound) is in the range of 0.05 to 1 OOmg per
kilogram body
weight of the recipient per day, preferably in the range of 0.1 to SOmg per
kilogram body
weight per day, most preferably in the range of 0.5 to 20mg per kilogram body
weight per
day and optimally I to IOmg per kilogram body weight per day. The desired dose
is
preferably presented as two, three, four, five, six or more sub-doses
administered at
appropriate intervals throughout the day.
While it is possible for the active ingredient to be administered alone it is
preferable to
present it as a pharmaceutical composition comprising an active ingredient, as
defined
above, together with an acceptable carrier therefor. Each carrier must be
"acceptable" in the
sense of being compatible with the other ingredients of the composition and
not injurious to
the recipient.
WO 94/26693 PCT/GB94/01003
ms~~o~
_g_
The compositions include those suitable for oral, rectal, nasal, topical
(including buccal and
sublingual), vaginal, parenteral (including subcutaneous, intramuscuiar,
intravenous and
intradermal) or transdermal administration. The compositions may conveniently
be
presented in unit dosage form and may be prepared by any methods well known in
the art of
pharmacy. Such methods include the step of bringing into association the
active ingredient
with the carrier which constitutes one or more accessory ingredients. In
general, the
compositions are prepared by uniformly and intimately bringing into
association the active
ingredient with liquid carriers or finely divided solid carriers or both, and
then if necessary
shaping the product.
Compositions of the present invention suitable for oral administration may be
presented as
discrete units such as capsules, cachets or tablets each containing a
predetermined amount
of the active ingredient; as a powder or granules; as a solution or suspension
in an aqueous
or non-aqueous Liquid; or as an oil-in-water liquid emulsion or a water-in-oil
liquid
emulsion. The active ingredient may also be presented as a bolus, electuary or
paste.
A tablet may be made by compression or molding, optionally with one or more
accessory
ingredients. Compressed tablets may be prepared by compressing in a suitable
machine the
active ingredient in a free-flowing form such as a powder or granules,
optionally mixed with
a binder (e.g. povidone, gelatin, hydroxypropylmethyl cellulose), lubricant,
inert diluent,
preservative, disintegrant (e.g. sodium starch glycollate, cross-linked
povidone, cross-linked
sodium carboxymethyl cellulose), surface-active or dispersing agent. Molded
tablets may be
made by molding in a suitable machine a mixture of the powdered compound
moistened
with an inert liquid diluent. The tablets may optionally be coated or scored
and may be
formulated so as to provide slow or controlled release of the active
ingredient therein using,
for example, hydroxypropylmethyl cellulose in varying proportions to provide
the desired
release profile. Tablets may optionally be provided with an enteric coating,
to provide
release in parts of the gut other than the stomach.
Compositions suitable for oral use as described above may also include
buffering agents
designed to neutralize stomach acidity. Such buffers may be chosen from a
variety of
organic or inorganic agents such as weak acids or bases admixed with their
conjugated salts.
Compositions suitable for topical administration in the mouth include lozenges
comprising
the active ingredient in a flavored basis, usually sucrose and acacia or
tragacanth; pastilles
WO 94/26693 PCT/GB94/01003
4
_ 10-
comprising the active ingredient in an inert basis such as gelatin and
glycerin, or sucrose and
acacia; and mouthwashes comprising the active ingredient in a suitable liquid
carrier.
Compositions for rectal administration may be presented as a suppository with
a suitable
base comprising for example cocoa butter or a salicylate.
Compositions suitable for vaginal administration may be presented as
pessaries, tampons,
creams, gels, pastes, foams or spray formulations containing in addition to
the active
ingredient such carriers as are known in the art to be appropriate.
Compositions suitable for parenteral administration include aqueous and non-
aqueous
isotonic sterile injection solutions which may contain anti-oxidants, buffers,
bacteriostats
and solutes which render the compositions isotonic with the blood of the
intended recipient;
and aqueous and non-aqueous sterile suspensions which may include suspending
agents and
thickening agents, as liposomes or other microparticulate systems which are
designed to
target the compounds to blood components or one or more organs. The
compositions may
be presented in unit-dose or mufti-dose sealed containers, for example,
ampoules and vials,
and may be stored in a freeze-dried (lyophilized) condition requiring only the
addition of the
sterile liquid carrier, for example water for injections, immediately prior to
use.
Extemporaneous injection solutions and suspensions may be prepared from
sterile powders,
granules and tablets of the kind previously described.
Compositions suitable for transdermal administration may be presented as
discrete patches
adapted to remain in intimate contact with the epidermis of the recipient for
a prolonged
period of time. Such patches suitably contain the active ingredient as an
optionally buffered,
aqueous solution of, for example, 0.1 to 0.2M concentration with respect to
the said
compound. As one particular possibility, the active ingredient may be
delivered from the
patch by iontophoresis as generally described in Pharmaceutical Research,
3_(6), 318 (1986).
Preferred unit dosage compositions are those containing a daily dose or unit,
daily sub-dose,
or an appropriate fraction thereof, of active ingredient, for example 1 to 1
S00 mg,
preferably 5 to 1000 mg and most preferably 10 to 700 mg of active ingredient,
estimated as
the parent compound.
n
It should be understood that in addition to the ingredients particularly
mentioned
above the compositions of this invention may include other agents conventional
in
the art having regard to the type of composition in question, for example,
those
suitable for oral administration may include such further agents as
sweeteners,
thickeners and flavouring agents.
The present invention thus also provides a pharmaceutical composition
comprising
a compound of formula (I) or a pharmaceutically acceptable salt or solvate
thereof
together with an acceptable carrier therefor.
The compounds of formula ( 1 ) and their salts and solvates may be prepared in
any
manner known in the art fur the preparation of compounds of analogous
structure,
for example, in accordance with the present invention, by those methods
hereinafter described.
The compounds, salts and solvates may thus be prepared by a process which
comprises
reacting a compound of formmla (II)
7
'6 (n)
t
~5
wherein R' to R' are as hereinbefore defined and Z is a leaving group with an
amine NRBR'' or a suitable derivative thereof. Suitable leaving groups include
halogen atoms such as chlorine or bromine, activated esters (e.g., N-
hydroxysuccinimide, pentafluoro- phenyl, nitrophenyl, 1-hydroxybenzotriazole),
mixed anhydrides(e.g., ethoxycarbonyloxy;l or C~_6 alkoxy (for example,
ethoxy).
The reaction is suitably carried out in an inert organic solvent(e.g.,
dichloromethane) at a temperature of about -20°C - 120°C, and
conveniently at
about 0° to 25°C. Suitable derivatives of the amine include
hydrated and
hydrochloride derivatives, e.g. NH40K NH4C1.
CA 02162708 2001-07-10
12
When Rg and R9 are H the compounds of formula (I) can be prepared by reacting
compounds of formula (II) wherein Z is a halogen atom such as chlorine or
bromine with the amine in hydrated forni, e.g. NH40H, in a suitable organic
solvent(e.g., dichloromethanc:) at a temperature of about O°C to
25°C.
Compounds of formula (I) wherein R6 or R' are hydroxy can be prepared by
reacting compounds of formula II wherein Z is C,_6 alkoxy, for example,
ethoxy,
and the hydroxy group is suitably protected, for example by SiMe2t-Bu; with
the
amine present as the hydrochloride, e.g. NH4C1 in the presence of Me3A1 under
neutral conditions followed by deprotection under neutral conditions with, for
example, pyridinium paratoulenesulfonate(PPTS).
Alternatively, compounds o~~ formula (I) wherein R6 or R' are hydroxy can be
prepared from compounds oil formula (I) wherein R6 or R' are H by halogenation
with, for example, N-bromosuccinamide(NBS) followed by hydrolysis with, for
example, silver carbonate (;AgC03). Compounds of formula (I) wherein the R6 or
R' is/are allylic hydroxy can be prepared from cornpourids of formula (I)
wherein
R6 or R' are H by oxidation v~ith, for example.selenium dioxide.
Compounds of formula (IC) wherein Z is a halogen atom can be prepared from
compounds of formula (III)
~H
R:
R5
R3
wherein R'-R' are as hereinbefore defined by reaction with a halogenating
agent
(e.g., oxalyl chloride, or thionyl chloride) in a suitable organic solvent
(e.g.,
benzene, toluene, dichloromethane) optionally in the presense of a catalyst
(for
example DMF) at a temperature of about -20°C to the reflux temperature.
CA 02162708 2001-07-10
13
Compounds of formula (If) wherein Z is alkoxy (e.g., ethoxy) can be prepared
from compounds of formula (III) by reaction with a suitable polar solvent
(e.g., an
organic alcohol such as ethanol) optionally in the presence of a catalytic
amount of
an acid (e.g., tosic acid) at a temperature of about O°C to the reflux
temperature.
Compounds of formula (II) wherein Z is an activated ester (as described
hereinbefore) can be prepared from compounds of formula (III) by reaction with
the phenol or N-hydroxy compound and a carbodiimide(e.g.,
dicyclohexylcarbodiimide or 1-(3dimethylaminopropyl)-3-ethylcarbodiimide) in a
solvent such as dimethylfolur~amide (DMF) or dichloromethane at O°C to
50°C.
Compounds of formula (III) can be prepared by dehydration of compounds of
formula (IV)
R' OH ~ _0H
Rw ..~. ~ ,R'~
R
wherein Rl-R' are as hereinbefore defined by reaction with an appropriate
dehydrating agent(e.g., an acrid such as trifluoroacetic acid) in a suitable
organic
solvent (e.g., dichloromethane) at a temperature of about -20°C to the
reflux
temperature.
Compounds of formula (1(V) can be prepared by saponification of the
corresponding C,_6 alkyl ester, with a base (e.g., sodium hydroxide) in a
suitable
polar solvent (e.g., ethanol) at a temperature of about O°C to the
reflux
temperature or with an aqueous acid (e.g., hydrochloric acid) at a temperature
of
Esters of compounds of formula (IV) having R5 or R' as protected hydroxy
groups, for example, by SiMe,t-Bu, can be dehydrated under neutral conditions
(e.g., Martin Sulfurane, bis[a,,a,-bis(trifluoromethyl)benzenemethanolato]-
diphenylsulfur) to give the corresponding protected hydroxy compounds of
formula (II) and wherein Z is C,_6 alkoxy (e.g., ethoxy).
The esters of compounds of formula (IV) can be prepared from compounds of
formula (V)
CA 02162708 2001-07-10
14
R; ~~ (~
Rs
R5
R
wherein R'-R' are as hereinbefore defined by reaction with hal CHZCOZR
wherein hal is a halogen atom such as chlorine, bromine, or iodine (preferably
bromine), and. R is C, _6 alkyl, (e.g ethyl) in the presence of a metal (e.g.,
zinc,
preferably activated zinc) and a catalytic amount of halogen (e.g., iodine) in
a
suitable organic solvent (e.g., ethyl ether, benzene) at a temperature of
about
O°C to the reflux temperature or by reaction with the lithium salt of
ethyl acetate
in a suitable solvent (e.g., te~trahydrofuran) at a temperature between -100
°C to
room temperature (e.g., -80°C to -70°C).
Compounds of formula (V) having R6 or R' as protected hydroxy groups as
defined above can be prepared from the corresponding unprotected hydroxy
compound of formula (V) by suitable protection under neutral conditions with,
for
example, t-butyl-di-methylsilyl chloride in the presence of a base such as
imidazole.
Compounds of formula(V;) having R6 or R' as hydroxy can be prepared from the
corresponding halogen (e.g:, bromo) compound by hydrolysis under neutral
conditions with, for example, silver carbonate (A~~o,).
Compounds of formula (V) having R6 or R' as allylic alkyl (e.g., methyl) can
be
prepared from the corresponding compounds of formula (V) wherein R6 and/or R'
are H by reaction with a base (e.g., sodium hydride) followed by alkylation
with,
for example, methyl iodide (lV(eI).
Compounds of formula (V) can be prepared from compounds of formula (VI)
CA 02162708 2001-07-10
15
R'
O
2 7
R~ R
w /. ~ Z Rs (VI)
R3r/ ~ R5
wherein Rl-R' and Z are as hereinbefore defined, preferably Z is a halogen
atom
such as chlorine by cyclization in the presence of a lewis acid (e.g.,
aluminum
chloride) in a suitable solvent (e.g., dichloromethane) at a temperature of
about
O°C to the reflux temperature.
Compounds of formula (VI) wherein Z is a halogen atom (e.g., chlorine, or
bromine) can be prepared from the corresponding carboxylic acid by reaction
with
a halogenating agent (e.g., oxalyl chloride or thionyl chloride) either neat
or in a
suitable organic solvent (e.g. methylene chloride or N,N-dimethylformamide) at
a
temperature of about O°C to the reflux temperature.
Compounds of formula (VI) wherein Z is alkoxy (e.g., ethoxy) can be prepared
from compounds of formula (VII)
R'
R7
s
R5
wherein R~-R' are as hereinbefore defined by reaction with a suitable organic
alcohol (e.g., ethanol) optionally in the presence of a catalytic amount of an
acid
(e.g., tosic acid) at a temperature of about O°C to the reflux
temperature.
The carboxylic acids can be prepared by saponification of the corresponding
C,_6
alkyl ester compounds with a base (e.g., sodium hydroxide) in a suitable polar
solvent (e.g., water or ethanol) at a temperature of about O°C to the
reflux
temperature or with an aqueous acid (e.g., hydrochloric acid) at a temperature
of
about O°C to the reflux temperature.
CA 02162708 2001-07-10
16
The carboxyclic acids can be prepared from compounds of formula (VIII)
R 1 C02R
2 l~
(CH )n
. 2 (
R3
RS R6
wherein R and Rl - R6 are as hereinbefore defined and n is 0 or 1 by mono de-
esterification with strong base (e.g., aqueous potassium hydroxide) at the
reflux
temperature.
Compounds of formula (VIII;) can be prepared by reacting a compound of formula
(IX) with a compound of formula (X)
p R1
~(CH2)ri G~ -pR R2
_ i
R3
4 lvig Hal
wherein R, R~ - R6 and n are as hereinbefore defined and Hal is Cl, Br or I
preferably Br in an organic solvent (e.g., anhydrous diethyl ether) and
optionally
in the presence of a copper halide (e.g., copper (I) iodide) at a temperature
of
between -50° C. to the reflux temperature.
Compounds of formula (IX) can be prepared by reacting a compound of formula
(XI) with a compound of formula (XII)
O
~~R
\C _0R C - O
O RS
CA 02162708 2001-07-10
17
wherein R, R' and R6 are as hereinbefore defined and m is 1 or 2 in an organic
solvent (e.g., ethyl ether ar dichloromethane) at a temperature of between
room
temperature and the reflux temperature.
Compounds of formula (X) can be prepared from the corresponding halo (e.g.,
bromo, chlora) compound bar standard techniques well known to those skilled in
the art. The halo compounds themselves can be obtained commercially or
prepared
by methods well known to those skilled in the art ar obtainable from the
chemical
literature.
Alternatively, compounds of formula (IX) can be prepared according to the
procedure of :E.L. Eliel, R.O~. Hutchins, and Sr. M. Knoeber, Organic
Synthesis
Coll. Vol. Vh 442, 1988 with the appropriate modifications readily apparent to
those skilled in the art.
Compounds of formula (XI), and (XII) can be obtained commercially or by
techniques well known to those skilled in the art or readily obtainable from
the
chemical literature.
Alternatively the esters can bc; prepared from compounds of formula (XIII)
R1
2
R3~~~R ~ ~~)
4 R6
wherein R, R' - R'~, R6 and k' are as hereinbefore defined by reduction of the
double bond, e.g., by catalytic reduction with e.g., platinum oxide (Pt02) and
hydrogen, in a suitable organic solvent (e.g., ethanol) at a temperature of
about
20°C to 60°C.
CA 02162708 2001-07-10
18
Compounds of formula (XIII) can be prepared from compounds of formula (XVI)
R1
2
,~ w2
3 w. ~ i
R6
wherein R' - R4, R'' and R' are as hereinbefore defined by esterification with
an
appropriate organic alcohol (e.g., ethanol) aptionally in the presense of a
catalytic
amount of an acid (e.g:, 13(; l, tosic acid, thionyl chloride) at a
temperature of
about 20°C to 60°C.
Compounds of formula (XVI) can also be used to directly prepare the
corresponding unsaturated acrid by reduction of the double bond, e.g., by
catalytic
reduction with e.g., palladium or platinum oxide (Pt02) and hydrogen, in a
suitable
organic solvent (e.g., ethanol) at a temperature of about O°C to the
reflux
temperature.
Compounds of formula (XVI) can be prepared from compounds of formula (XVII)
CA 02162708 2001-07-10
WO 94/26693 PCT/GB94/01003
21~~708
-19-
R1
6
C-R
(~I)
R3~
4
wherein R1 - R4 and R6 are as hereinbefore defined by reaction with
HOOCCHR~COOH
in an organic base(e.g., pyridine) optionally in an organic solvent(e.g.,
dichloromethane)
optionally in a catalytic amount of a base(e.g., piperidine) at a temperature
of about OoC to
the reflux temperature.
Compounds of formula (XVII) and HOOCCHR~COOH can be obtained commercially or
by
methods well known to those skilled in the art or readily obtainable from the
chemical
literature.
Alternatively, compounds of formula (I) can be prepared by reacting
R8RgNCOCH2P0(OR)2 (wherein R,R8,R9 are as hereinbefore defined) with a base
(e.g.,
NaH) in a suitable organic solvent (e.g., THF or DMSO) and reacting the
resultant anionic
species with a compound of formula (VI) or (VIa) respectively at a temperature
of about
0°C to the reflux temperature. The addition of a anionic stablizing
reagent (e.g., potassium
hexamethyldisilizane or a crown ether (e.g., 15-crown-5) can aid the reaction.
The compound RgR9NCOCH2P0(OR)2 can, depending on R, R8 and R9 be obtained
commercially or by methods well known to those skilled in the art or readily
obtainable from
the chemical literature. Alternatively, these compounds can be prepared by
reacting the
appropriate RgR'~NCOCHZZ (wherein Z is as hereinbefore defined) with the
appropriate
P(OR)3 in a suitable organic solvent (e.g., THF) at a temperature of about
0°C to 50°C.
The compound RgR9NCOCH2Z can be prepared by reacting the appropriate R8R9NH
with
ZCH2COZ in a suitable organic solvent (e.g., diethyl ether) at a temperature
of about 0°C
to the reflux temperature.
The compound R8R9NH can be obtained commercially or by methods well known to
those
skilled in the art of preparing amines or readily obtainable from the chemical
literature. The
compound ZCH~COZ can be obtained commercially or by methods well known to
those
20
skilled in the art of preparing such compounds or readily obtainable from the
chemical literature.
Alternatively, compounds ~o f formula (I) can be prepared by reacting
RgR9NCOCH2P~+~(Ph)3C1~-~(wh:erein Rg, R9 and Z are as hereinbefore defined and
Ph
is phenyl) with a suitable base (e.g., NaH) in a suitable organic solvent
(e.g.,
dimethoxyethane) at a temperature of about 0°C to 50°C, and
reacting the resultant
anionic species with a compound of formula (V) at a temperature of about
0°C to the
reflux temperature.
The compound R$R9NCOClHzP~+~(Ph)3CI~-~ can be prepared by reacting the
appropriate RgR9NCOCHZZ with about a 50% molar excess of P(Ph)3
(triphenylphosphine) in a suitable organic solvent (e.g., THF) at a
temperature of
about 20°C to the reflux temperature.
RgR9NCOCH2Z can be prepared as defined hereinbefore.
Alternatively, compounds of formula (I) can also be prepared directly from
compounds o:f formula (III) by reaction with a suitable coupling reagent
(e.g.,dicyclohexylcarbodiimide (DCC) or ethyl chloroformate) followed by
reaction
of the activated ester thus formed with the appropriate amine, HNR8R9.
Alternatively, compounds of formula (I) wherein RS is hydrogen and R6 is
hydroxy
can be prepared from compounds of formula (I) wherein RS and R6 together form
a
carbonyl group by reduction of this carbonyl group using a suitable reducing
agent,
e.g. sodium borohydride in a suitable solvent such as an alkanol (e.g.
ethanol).
The compounds of formula (I) as well as any of the intermediates used in the
preparation of these compounds can be effected with one or more of the
following
optional conversions:
(i) converting a compound of formula (I) or intermediates thereof ~o formed
into
salts thereof;
(ii) when a salt of a compound of fornmla (I) or an intermediate thereof is
formed,
converting the said salt into a compound of formula (I) or an intermediate
thereof.
CA 02162708 2001-07-10
WO 94126693 PCT/GB94/01003
-21 -
The following Examples illustrate the present invention but should not be
construed as a
limitation to the scope thereof.
EXAMPLE 1
Preparation of LE)-2-(6-Fluoro-1-indanvlidene)acetamide
a) Prgparation of 3-(4-Fluorophenvl)propionic Acid
A mixture of 4-fluorocinnamic acid (300.0 g, 1.8 mol, Aldrich) and 5%
palladium on carbon (9.0 g) in ethanol (3 L) was hydrogenated at atmospheric
pressure and room temperature for 4.5 h. The mixture was filtered through
Celite (diatomaceous earth) and the filtrate was concentrated in vacr~o to
give
275.1 g (91%) of 3-(4-fluorophenyl)propionic acid as a white solid, m.p., 86-
88°C;
b) Preparation of 3-(4-Fluoropheny~pro ionyl Chloride
A mixture of 3-(4-fluorophenyl)propionic acid (275.1 g, 1.6 mol) and thionyl
chloride (300 mL, 4.1 mol) was heated to reflux for 3 h, cooled to room
temperature and distilled under aspirator vacuum to give 287.6 g (96%) of 3-(4-
fluorophenyl)propionyl chloride as a pale pink oil, b.p., 120-
122°C/l5mm Hg;
c) P~paration of 6-Fluoro-1-indanone
A solution of 3-(4-fluorophenyl)propionyl chloride (287.6 g, 1.5 mol) in
dichloromethane ( 1.4 L) was added dropwise during 3 h to an ice-cold,
mechanically stirred suspension of aluminum chloride (226.0 g, 1.7 mol,
Aldrich)
in dichloromethane (2.2 L) under nitrogen. The resulting yellowish-black
solution was refluxed for 5 h and allowed to cool to room temperature. The
solution was washed successively with water (2 L), 1N sodium hydroxide (2 L),
water (2 L) and brine (2 L). The organic layer was dried over anhydrous sodium
sulfate, filtered and concentrated to a tan solid (229.1 g, 99%). The solid
was
recrystallized from dichloromethane-hexane to give 215.7 g (93%) of 6-fluoro-1-
indanone as off white crystals, m.p., 57-59°C;
WO 94/26693 PCT/GB94101003
-22-
~1~
d) Preparation of Ethyl 2-(6-Fluoro- I -h dy ro xy-1-indanvllacetate
(i) A mixture of 6-fluoro-I-indanone (S.0 g, 33.3 mmoI), ethyl bromoacetate
(8.3 g, 50.0 mmol, Aldcich), activated zinc powder (3.2 g, 50.0 mmol,
Mallinckrodt; Org. Synth., Coll. Vol. 6, 290, 1988) and a few crystals of
iodine in diethyl ether-benzene ( 1:1, 100 mL) was heated at reflux under
nitrogen for 24 h. The mixture was filtered and the filtrate was
concentrated in vacuo. The residue in diethyl ether was vigorously
stirred with excess dilute ammonium hydroxide, dried and concentrated
to give ethyl 2-(6-fluoro-1-hydroxy-1-indanyl)acetate as an amber oil
(7.6 g, 97%);
(ii) Ethyl acetate (1.8 g, 20 mmol) was added dropwise to a stirred, chilled
(dry ice-acetone bath) 1N solution of lithium bis(trimethylsilyl)amide in
tetrahydrofuran (20 mL, Aldrich) under nitrogen. After 15 min, a
solution of 6-fluoro-1-indanone (3.0 g, 20 mmol) in tetrahydrofuran
(20 mL) was added dropwise and the resulting mixture was stirred for 1h
(dry ice-acetone bath). A 1N solution of hydrochloric acid (20 mL) was
added and the mixture was allowed to warm to room temperature. The
organic phase was separated, dried over anhydrous sodium sulfate,
filtered and concentrated to a pale yellow oil (5.3 g). The mixture was
chromatographed on Silica Gel 60 (silica gel) using a linear gradient of
dichloromethane- hexanes ( 1:1 ) to dichloromethane as eluent. The
fractions containing only ethyl 2-(6-fluoro-I-hydroxy-I-indanyl)acetate
were combined and concentrated in vacuo to give 3.1 g (65%) of a
colorless oil;
e) Preparation of2-(6-Fluoro-I-hydroxv-1-indanvllacetic Acid
A mixture of ethyl 2-(6-fluoro-I-hydroxy-I-indanyl)acetate (44.0 g, 0.18 mol),
1N sodium hydroxide (180 mL) and absolute ethanol (280 mL) was stirred for
18 h at room temperature. The mixture was concentrated in vacuo, diluted with
H20 and extracted with diethyl ether. The aqueous phase was acidified (pH 3)
with dilute hydrochloric acid and extracted with diethyl ether. The diethyl
ether
WO 94/26693 PCT/GB94/01003
- 23 -
layer was washed with brine, dried over sodium sulfate, filtered and
concentrated
in vacuo to give 2-(6-fluoro-1-hydroxy-1-indanyl)acetic acid as an amber oil
(37.7 g, 100%; Note: This compound spontaneously dehydrated upon standing
at room temperature to a mixture of olefins unless immediately reacted with
trifluoroacetic acid);
f) Preparation ofLithium 2-(6-Fluoro-1-hydroxy-1-indanyllacetate.
A mixture of ethyl 2-(6-fluoro-1-hydroxy-1-indanyl)acetate (2.0 g. 8.4 mmol),
1N lithium hydroxide (8.4 mL) and absolute ethanol ( 13.0 mL) was stirred for
18h at room temperature. The mixture was concentrated in vacuo, diluted with
H20 and extracted with diethyl ether. The aqueous phase was concentrated
in vacuo, diluted with toluene ( 100 mL) and concentrated in vacuo to give
lithium 2-(6-fluoro-1-hydroxy-1-indanyl)acetate as a white solid (1.4 g, 77%);
g) Preparation of (E)-2-(6-Fluoro-1-indanylidenelacetic Acid
Trifluoroacetic acid (1.5 mL) was added to a stirred, chilled (ice-methanol
bath)
suspension of lithium 2-(6-fluoro-1-hydroxy-1-indanyl)acetate (0.5 g, 2.3
mmol)
in dichloromethane ( 13.5 mL). After 15 min, the mixture was concentrated
in vacuo and the resulting white solid was recrystallized from aqueous acetone
to
give (E~-2-(6-fluoro-1-indanylidene)acetic acid as white crystals (0.32 g,
73%)
identical to compound of Example 1i by mixed m.p., (203-205°C) and NMR;
h) Preparation of (El-2-(6-Fluoro-1-indanylidenelacetic Acid
Trifluoroacetic acid ( 100 mL) was added to a stirred, chilled (ice-methanol
bath)
solution of 2-(6-Fluoro-1-hydroxy-1-indanyl)acetic acid (37.5 g, 0.18 mol) in
dichloromethane (900 mL). After 1 ~ min, the mixture was concentrated in vacuo
to give (E)-2-(6-fluoro-1-indanylidene)acetic acid as a yellowish-tan solid
(33.0 g, 95%), m.p., 203-205°C;
WO 94/26693 PCT/GB94/01003
-24-
i) Preparation of (El-2-(6-Fluoro-1-indanvlidene)acetvl Chloride
An ice-cold, stirred suspension of (E)-2-(6-fluoro-I-indanylidene) acetic acid
(384 mg, 2 mmol) in benzene (10 mL) was treated with oxalyl chloride (761 mg,
6 mmol) and allowed to warm to room temperature during 1.5 h. The resulting
yellow solution was concentrated in vacuo to give (E)-2-(6-fluoro-1-
indanylidene)acetyl chloride as a pale yellow solid (421 mg, 100%), m.p., 97-
99°C;
j) Preparation of (El-2-(6-Fluoro-1-indanylidene)acetamide
A 29.6% aqueous ammonium hydroxide solution (17.6 mL, 134 mmol) was
added dropwise to a stirred, chilled (ice bath) solution of (E)-2-(6-fluoro-1-
indanylidene)acetyl chloride (14.1 g, 67 mmol) in dichloromethane (16~ mL).
After an hour, the resulting white precipitate was collected by filtration,
dissolved in ethyl acetate (600 mL) and washed with water (3X300 mL). The
ethyl acetate layer was dried over sodium sulfate and concentrated i~r vacuo.
The resulting off white solid was washed with hexane, giving 1 I .6 g (91 %)
of
(E~2-(6-fluoro-1-indanylidene)acetamide, m.p., 180-183°C;
EXAMPLE 2
Preparation of (El-2-(6-Fluoro-1-indan li~dene acetamide
A stirred suspension of (E)-2-(6-fluoro-I-indanylidene)acetic acid (0.5 g, 2.6
mmol) in
dichloromethane ( 10 mL) at -20°C was successively treated dropwise
with ethyl
chloroformate (0.3 g, 2.6 mmol, Aldrich) and triethyiamine (0.3 g, 2.6 mmol,
Eastman).
The mixture was stirred at -20°C for 2 h. A solution of anhydrous
ammonia in
dichloromethane (0.8 M, 12 mL) was added [Note: When aqueous ammonium
hydroxide was used, the mixed anhydride was partially hydrolyzed to the
acid.], the
mixture was stirred for 16 h at room temperature, and subsequently washed
successively
with water, sodium bicarbonate solution, water and brine. The dichloromethane
layer
was dried over sodium sulfate, filtered, and concentrated i~r vacuo to give
0.18 g of a 6:1
mixture of (E)-2-(6-fluoro-1-indanylidene)acetamide and 2-(5-fluoro-1H-inden-3-
yl)acetamide.
WO 94/26693 PCT/GB94/01003
z1 sz ~o~
-25-
EXAMPLE 3
Preparation of,(El-N-Ethyl-2-i(6-fluoro-I-indanylidene)acetamide
This compound was prepared in an analogous manner to that of Example 5 with
replacement of cyclopropylamine in Example 5 with ethylamine (70 wt % in
water). The
chromatography solutions that contained (E)-N-Ethyl-2-(6-fluoro-1-
indanylidene)acetamide were concentrated by spin evaporation in vacuo.
Recrystallization of the residue from dichloromethane-hexanes gave 1.7 g (68%)
of (E)-
N-ethyl-2-(6-fluoro-1-indanylidene)acetamide, m.p. 125-127°C;
EXAMPLE 4
P_Leparation of (E)-N-Cvclopropvl-2-(6-fluoro-1-indanvlideneyacetamide
To an ice-cold stirred solution of (E)-2-(6-Fluoro-1-indanylidene)acetyl
Chloride in
dichloromethane (50 ml) was added cyclopropylamine ( 1.65 g, 28.86 mmol) and
the
reaction was warmed to room temperature overnight. The reaction was evaporated
in vacuo to a solid residue. This residue was dissolved in ethyl acetate (300
ml), washed
with water (75 ml), and the organic layer was concentrated by spin evaporation
in vacuo.
The residue was chromatographed on silica gel using ethyl acetate-hexanes (0:1
to 1:1
gradient) as eluent. Fractions containing only the product were combined and
concentrated by spin evaporation in vacuo. Recrystallization of the residue
from
dichloromethane-hexanes gave 1.6 g (76%) of (E)-N-cyclopropyl-2-(6-fluoro-1-
indanylidene)acetamide as a white powdery solid, m.p. 124-127°C;
(E)-N-Ethyl-2-(6-fluoro-1-indanylidene)-N-methylacetamide was prepared in an
analogous manner with replacement of cyclopropylamine with N-ethylmethylamine
(3.5 mL, 0.025 mol, Aldrich). The residue was chromatographed on silica gel
using
ethyl acetate-hexanes ( 1:5 to 1:2 gradient) as eluent. The chromatography
fractions that
contained (E)-N-ethyl-2-(6-fluoro-1-indanylidene}-N-methylacetamide were
concentrated by spin evaporation in vacuo. Recrystallization of the residue
from ethyl
acetate-hexanes gave 1.32 g (61%) of (E)-N-ethyl-2-(6-fluoro-1-indanylidene)-N-
methylacetamide as a white solid, m.p. 74-77°C,
WO 94/26693 PCT/GB94/01003
_26_
EXAMPLE 5
Preparation of (El-2-(4.6 Difluoro-1-indanylidene)acetamide
t'
a) Preparation of 3-f2. 4-Difluorophenvllpropanoic Acid
A mixture of 2,4-difluorocinnamic acid (30.0g, 0.16 mol, Aldrich) and platinum
oxide hydrate (0.5g, EM Scientific) in 95% ethanol (140 mL) was placed on a
Parr hydrogenation apparatus. After the appropriate amount of hydrogen was
taken up, the catalyst was filtered and the filtrate was concentrated in vacuo
to
give 29.7g (98%) of 3-(2,4-difluorophenyl)propanoic acid as a white solid.
Recrystallization of 1.0g from acetonitrile: water mixtures gave 0.61 g of 3-
(2,4-
difluorophenyl)propanoic acid as a white solid: mp 104-106oC; NMR (DMSO-
d6) ; d 12.2 (br, 1H), 6.98-7.40 (m, 3H), 2.81 (t, ZH), 2.51 (t, 2H).
Anal. Calcd. for C9H8F202 (mw186.15): C, 58.06; H, 4.33.
Found : C, 57.94; H, 4.36.
b) Preparation of 4.6-Difluoro-I-indanone
To a mixture of 3-(2,4-difluorophenyl) propanoic acid (28.7g, 0.15 mol) and
dimethylformamide (5 drops) at ambient temperature was added dropwise oxalyl
chloride (50 mL, Aldrich) . The mixture was stirred at ambient temperature for
18h. The excess oxalyl chloride was removed by distillation in vacno to give 3-
(2,4-difluorophenyl)propionyl chloride. A solution of the 3-(2,4-
difluorophenyl)propionyl chloride in dichloromethane (300 mL) was added
dropwise to a mixture of aluminum chloride (23.4g, 0.18 mol, Aldrich) in
dichloromethane (300 mL) at ice bath temperature. After the addition was
completed, the mixture was refluxed for 3.5h and allowed to come to ambient
temperature overnight. The reaction mixture was poured into ice water (1500
mL), the two phases were separated and the aqueous phase was extracted with
dichloromethane. The combined organic phase was washed successively with
O.1N aqueous sodium hydroxide and saturated sodium chloride solution , dried
over sodium sulfate and concentrated in vacuo to give 21.78 of crude 4,6-
difluoro-1-indanone . Chromatography on silica gel with hexanes:
WO 94/26693 PCT/GB94/01003
~~ ~2~08'
-2~-
dichloromethane (3 :1 ) as eluent gave 10.1 g of a light yellow solid.
Recrystallization of O.Sg from acetone: water mixtures gave 0.2g of 4,6-
difluoro-I-indanone as a white solid: mp 97-99oC; NNtR (CDC13) : d 7.02-7.27
(m, 2H), 3.12 ( t, 2H), 2.76 (m, 2H).
Anal. Calcd. for C9H6F20 (mw 168.14): C, 64.29; H , 3.60.
Found: C, 64.18; H , 3.61.
c) Preparation ofEthyl 2-f4.6-Difluoro-1-hydroxy-1-indany~acetate
A mixture of 4,6-difluoro-1-indanone (12.6g, 0.08 mol), ethyl bromoacetate
(19.0g, 0.11 mol, Aldrich), activated zinc powder (7.5g, 0.11 mol, Aldrich;
Org.
Syn., Coll. Vol. 6, 290, 1988) and a few crystals of iodine in diethyl
etheraoluene
(1:1, 300 mL) was heated at 30-35oC under a nitrogen atmosphere for 24h. A
few more crystals of iodine were added, the temperature was adjusted to 40-
45oC,
and the mixture was kept at that temperature for an additional 24h. The
reaction
mixture was filtered and concentrated in vacuo. The residue was treated with a
mixture of diethyl ether (450 mL), concentrated ammonium hydroxide ( 135 mL)
and water (135 mL). The aqueous phase was separated and extracted with diethyl
ether. The combined organic phase was washed with saturated aqueous sodium
chloride solution, dried over sodium sulfate, filtered and concentrated irr
vacuo to
give 22.7g of crude Ethyl 2-(4,6-difluoro-1- hydroxy-I-indanyl)acetate which
was
Chromatographed on silica gel with dichloromethane:hexanes (9:1 ) as eluent
gave
12.7g (66%) of a yellow oil ; NNiR (CDC13): d 6.67-6.88 (m, 2H), 4.22 (q, 2H),
3.02 (m, 1H), 2.75 (2m's, 3H), 2.31 (m, 2H), 1.28 (t, 3H).
Anal. Calcd. for C13H14F2O3 (mw 256.24): C, 60.93; H, 5.51.
Found: C, 60.68; H, 5.50.
d) Preparation of 2-(4.6-difluoro-1-hydroxy-1-indanyllacetic Acid
A mixture of ethyl 2-(4,6-difluoro-1-hydroxy-1-indanyl)acetate (12.0g , 0.047
mol) and I.ON sodium hydroxide (48 mL, 0.048 mol, Universal Scientific Supply
Co.) in ethanol (75 mL) was stirred for 18h at ambient temperature. The
reaction mixture was concentrated irr vacuo, diluted with water and washed
with
WO 94/26693 PCT/GB94/01003
_ 28 _
diethyl ether. The aqueous phase was neutralized with 1.0N hydrochloric acid
(48 mL, 0.048 mol, Universal Scientific Supply Co.) and extracted with diethyl
ether . The diethyl ether extract was dried over sodium sulfate, filtered and
concentrated in vacuo to give a quantitative yield of crude 2-(4,6-difluoro-1-
hydroxy-1-indanyl)acetic acid. This material was used immediately without
further purification.
e) Preparation of fE)-2-(4.6-difluoro-1-indanylidenelacetic Acid
Trifluoroacetic acid (39.98, 0.35 mol) was added dropwise to a stirred ,
chilled
(ice-methanol bath) mixture of 2-(4,6-difluoro-1-hydroxy-1-indanyl)acetic acid
(11.3g, 0.05 mol) in dichloromethane ( 250 mL). After 35 min. the mixture was
concentrated in vacuo. Dichloromethane was added to the residue and the
mixture was concentrated in vacuo . This procedure was repeated once more to
give 6.4g of crude (E)-2-(4,6- difluoro-1-indanyiidene) acetic acid.
Recrystallization of 0.9g from acetone: water mixtures gave O.15g of (E)-2-
(4,6-
difluoro-1-indanylidene) acetic acid as a white solid: mp 238-239oC; NMR
(DMSO-d6): d12.25 (br , 1H), 7.23-7.65 (m, 2H), 6.46 (t, 1H), 3.20-3.28,
2.97-3.20(2m's, 4H); steady-state nOe: irradiation at 6.46 d, observed 21.6%
nOe at 7.63 d.
Anal. Calcd. for C 11 H8F202 (mw 210.17): C, 62.86; H, 3.84.
Found: C, 62.76; H, 3.86.
fj Preparation of lE)-2-(4.6-difluoro-1-indanylidenelacetyl Chloride
A suspension of (E)-2-(4,6-difluoro-1-indanylidene)acetic Acid (5.498,
0.026mo1) in a mixture of dichloromethane: dimethylformamide (50 mL: 5 drops)
was treated with oxalyl chloride (6.6g 0.052 mol, Aldrich) and allowed to stir
at
ambient temperature for 18h. The resulting solution was concentrated in vacuo
and the residue used without further purification.
WO 94/26693 PCT/GB94/01003
_29_
g) Preparation of !El-2-l4.6-difluoro-1-indanylidenelacetamide
A 30% aqueous ammonium hydroxide solution ( 1.7 mL, 0.026 mol) was added
dropwise to a stirred ,chilled ( ice bath) solution of (E)-2-(4,6-
difluoro-1-indanylidene)acetyl chloride (2.97g, 0.013 mol) in dichloromethane
(50
mL). After 4.5h the mixture was concentrated in vacuo and the residue was
partitioned between 5% aqueous sodium bicarbonate solution and ethyl acetate.
The ethyl acetate solution was washed with saturated aqueous sodium chloride,
dried over sodium sulfate, filtered and concentrated in vacuo. Chromatography
on silica gel with ethyl acetate: hexanes (7:3)as eluent and trituration of
the
resulting solid with pentane gave 1.63g (60%) of (E)-2-(4,6-difluoro-
1-indanylidene)acetamide as a white solid: mp 178-180oC; NMR (DMSO-d6): d
6.94-7.45 (m, 4H), 6.46 (s, 1H), 2.94-3.00, 3.21-3.27 (2m's, 4H); steady-state
nOe: irradiation at 6.46 d, observed 19% nOe at 7.26d.
Anal. Calcd. for C 11 H9F2N0 (mw 209.19): C, 63 .15; H, 4.34; N, 6.70.
Found: C, 63.07; H, 4.36; N, 6.67.
h) P_rgparation of !El-2-!4-chloro-1-indanylidene~acetic Acid
Trifluoroacetic acid (25.1 mL) was added to a stirred, chilled (ice-methanol
bath)
solution of 2-(4-chloro-I-hydroxy-I-indanyl)acetic acid (10.4 g, 0.05 mol) in
dichloromethane (230 mL). After 30 min, the mixture was concentrated
in vacuo. Dichloromethane was added to the residue and the mixture was
concentrated in vacuo to give 6.5 g (68%) of a white solid. Recrystallization
of
0.98 g from acetonitrile: 2-propanol mixtures gave 0.62 g of a white solid:
m.p.,
233-234°C; NMR (DMSO-d6): d12.15 (br, 1H, COOH), 7.30-7.81 (m, 3H, Ar),
6.41 (s, IH, = CH), 3.00-3.06, 3.19-3.22 (2m's, 4H, 2 x CH2); steady-state
nOe;
irradiation at 6.41 d, observed 19.7% nOe at 7.79.
i) Preparation of (E)-2-!4-chloro-I-indanylidene acetyl Chloride
A suspension of (E)-2-(4-chloro-I-indanylidene)acetic acid (5.5 g, 0.03 mol)
in a
mixture of dimethylformamide: dichloromethane (5 drops: SOmL) was treated
with oxalyl chloride (6.6 g, 0.05 mol) and allowed to stir at room temperature
for
WO 94/26693 PCT/GB94/01003
-30-
18h. The resulting solution was concentrated in vacuo and the residue used
without further purification.
j) Preparation of (El-2-(4-chloro-I-inda~lidene)-N-cvclopropvlacetamide
An ice cold solution of (E)-2-(4-choro-1-indanylidene)acetyl chloride (2.95 g,
0.013 mol ) in dichloromethane (30 mL) was treated with cyclopropylamine
( 1.48 g, 0.026 mol, Aldrich) and the mixture was stirred for 4h. The mixture
was
concentrated in vacuo and the residue was taken up in a mixture of ethyl
acetate
and 5% aqueous sodium bicarbonate. The ethyl acetate phase was washed with
5% aqueous sodium bicarbonate, saturated aqueous NaCI and dried (Na~S04).
Filtration and concentration gave 3.5g of crude oil product. Chromatography on
Silca gel with ethyl acetate: hexanes ( I :1 ) as eluent and trituration of
the
resulting solid with hexanes gave 2.46 g (76%) of (E)-2-(4-chloro-1-
indanylidene)-N-cyclopropylacetamide as an off white solid: m.p., 140-
142°C;
NMR (DMSO-d6): d 8.16, d, 1H, NH), 7.29-7.51 (m, 3H, Ar), 6.34 (t, 1 H, _
CH), 2.93-3.07, 3.18-3.31 (2m's, 4H, 2XCH2), 2.72 (m, 1 H, CH), 0.62-0.72,
0.39-0.47 (2m's, 4H, 2XCH2); steady-state nOe: irradiation at 6.34 d, observed
20.3% nOe at 7.46 d.
Anal. Calcd. for C14H14C1N0 (mw 247.72): C, 67.88; H, 5.70; N, 5.65.
Found: C, 67.86; H, 5.74; N, 5.58.
EXAMPLE 6
Preparation of (ESN-C~clopropvl-2-i(4 6 difluoro-1-indanylidenelacetamide
An ice cold solution of (E)-2-(4,6-difluoro-1-indanylidene)acetyl chloride
(2.97 g, 0.013
mol) in dichloromethane (30 mL) was treated with cyclopropylamine (1.48 g,
0.026
mol, Aldrich) and the mixture was stirred for 4h. The mixture was concentrated
in vacuo and the residue was taken up in a mixture of ethyl acetate and 5%
aqueous
sodium bicarbonate. The ethyl acetate phase was washed with 5% aqueous sodium
bicarbonate, saturated aqueous NaCI and dried (Na2S04). Chromatography on
silica
gel with ethyl acetate: hexanes ( 1:1 ) as eluent and trituration of the
resulting solid with
WO 94/26693 PCT/GB94/01003
~ ::
-31 -
pentane gave 1.92 g (59%) of (E)-N-cyclopropyl-2-(4,6-difluoro-1-
indanylidene)acetamide as a white solid: m.p., 156-158°C;
EXAMPLE 7
Preparation of lE~-2-(4-Fluoro-1-indanylidenelacetamide
a) Preparation of Ethyl_ 2-Fluorocinnamate
A solution of 2-fluorocinnamic acid (48.4 g, 0.29 mol, Aldrich) and thionyl
chloride (5 mL) in ethanol (650 mL) was heated to reflux for 48h. The mixture
was concentrated in vacuo. The residue was taken up in ethyl acetate, washed
successively with a 5% aqueous sodium bicarbonate solution, water and brine,
and dried (Na2S04). Filtration and concentration gave 54.25 g (96%) of crude
ethyl 2-fluorocinnamate. This material was used without further purification.
b) Preparation of Ethyl 3-(2-fluorophenyl)propionate
A mixture of ethyl 2-fluorocinnamate (29.25 g, 0.176 mol) and platinum oxide
hydrate (0.25, EM Scientific) in 95% ethanol (150 mL) was placed on a Pan
hydrogenation apparatus and shaken under 2-4 atm at hydrogen pressure. After
the appropriate amount of hydrogen was consumed, the catalyst was removed by
filtration, and the filtrate was concentrated in vacuo to give 29.39 g (99%)
of
crude ethyl 3-(2-fluorophenyl)propionate. This material was used without
further purification.
c) Preparation of 3-(2-Fluorophenvllpropionic acid
A mixture of ethyl 3-(2-fluorophenyl)propionate (25.54 g, 0.130 mol) and a 50%
aqueous solution of sodium hydroxide (30 mL) in water (130 mL) was refluxed
for 2h. After cooling the mixture was washed with diethyl ether (2x100 mL).
The aqueous phase was chilled in an ice bath, and the pH was adjusted to 3
with
hydrochloric acid . The white precipitate which formed was collected by
filtration, washed repeatedly with water, and dried in a vacuum at 60°C
for 18 h
WO 94/26693 PCT/GB94/01003
-32-
to give 18.66g (85%) of 3-(2-fluorophenyl)propionic acid as a white solid;
m.p.,
72-74°C. This material was used without further purification.
d) Preparation of 4-fluoro-I-indanone
To a mixture of 3-(2-fluorophenyl)propionic acid (18.64 g, 0.111 mol) and
dimethylformamide (5 drops) at room temperature was added dropwise oxalyl
chloride (60 mL). The mixture was stirred at room temperature until gas
evolution had ceased. The excess oxalyl chloride was removed by distillation
to
give 3-(2-fluorophenyl)propionyl chloride. A solution of the 3-(2-
fluorophenyl)propionyl chloride in dichloromethane (230 mL) was added
dropwise to a mixture of aluminum chloride ( 16.25 g, 0.12 mol) in
dichloromethane (230 mL), and the mixture was refluxed for 3.5h. The reaction
mixture was poured into ice water ( 1200 mL), and the two phases were
separated. The dichloromethane phase was washed successively with O.IN
aqueous sodium hydroxide (2x100 mL), water (200 mL), and brine (200 mL),
dried over sodium sulfate, filtered, and concentrated in vacuo. The residual
oil
was chromatographed on silica gel eluting with hexane:dichloromethane (9:1 )
to
give 11.0 g of crude 4-fluoro-1-indanone as a yellow solid. Recrystallization
from acetone : water mixtures gave 8.02 g (48%) of 4-fluoro-1-indanone as a
pale yellow solid: m.p., 71-72°C; NMR (DMSO-d6): d 7.51 (m, 3H, Ar),
3.13
t, 2H, CH2), 2.74 (t, 2H, CH2).
Anal. Calcd. for C9H7F0 (mw 150.152): C, 71.99; H, 4.70.
Found: C, 71.86; H, 4.79.
e) Preparation of Ethvl 2-l4-fluoro-I-hvdroxy-I-indanvllacetate
This compound was prepared in an analogous manner to that described in
Example Sc for ethyl 2-(4,6-difluoro-1-hydroxy-1-indanyl) acetate substituting
4-fluoro-I-indanone (15.53 g, 0.103 mol) for 4,6-difluoro-I-indanone.
Chromatography on silica gel with ethyl acetate:hexanes ( 19:1 ) as eiuent
gave
19.11 g (78%)of ethyl 2-(4-fluoro-1-hydroxy-1-indanyl)acetate which was used
without further purification.
WO 94/26693 ' PCT/GB94/01003
33
fj Preparation of 2-f4-fluoro-I-hydroxy-1-indanyl)acetic Acid
This compound was prepared in a similar manner to that described for 2-(4,6-
difluoro-1-hydroxy-I-indanyl)acetic acid in Example Sd by substituting ethyl 2-
(4-fluoro-I-hydroxy-I-indanyl)acetate (17.35 g , 0.0728 mol) for ethyl 2-(4,6-
difluoro-I-hydroxy-1-indanyl) acetate to give a quantitative yield of crude 2-
(4-
fluoro-1-hydroxy-I-indanyl)acetic acid. This material was used immediately
without further purification.
g) Preparation of lEl-2-(4-fluoro-I-indanylidene)acetic Acid
This compound was prepared in an analogous manner to (E)-2-(4,6-difluoro-I-
indanylidene)acetic acid in Example Se by substituting 2-(4-fluoro-I-hydroxy-I-
indanyl)acetic acid (14.6 g, 0.069 mol) for 2-(4,6-fluoro-I-hydroxy-1-
indanyl)acetic acid to give crude (E)-2-(4-fluoro-1-indanylidene)acetic acid.
Recrystallization from acetonitrile : 2-propanol mixtures gave 6.85 g (52%) of
(E)-2-(4-fluoro-1-indanylidene)acetic acid as a white solid; m.p., 249-
251°C;
h) Preparation of (E)-2-f4-fluoro-1-indanvlidene)acetyl Chloride
This compound was prepared in a similar manner to (E)-2-(4,6difluoro-I-
indanylidene)acetyl chloride in Example Sf by substituting (E)-2-(4-fluoro-I-
indanylidene)acetic acid (5.77 g, 0.03 mol) for (E)-2-(4,6-difluoro-I-
indanylidene)acetic acid. The resulting solution was concentrated »~ vacuo,
and
the residue was used without further purification.
i) Preparation of (E)-2-(4-fluoro-I-indanvlidene)acetamide
An ice cold solution of (E)-2-(4-fluoro-1-indanylidene)acetyl chloride (2.l
1g,
0.01 mol) in dichloromethane (65 mL) was treated with a 30% aqueous solution
of ammonium hydroxide (2.63 ml, 0.02 mol), and the mixture was stirred for
18h. Hexane was added to the mixture, and the solids were collected by
filtration
to give 1.63 g of crude product. Recrystallization from acetonitrile : water
mixtures gave 1.11 g (58%) of (E)-2-(4-fluoro-I-indanylidene)-acetamide as a
white solid; m.p., 198-200oC; .
WO 94/26693 PCT/GB94I01003
-34-
EXAMPLE 8
Preparation of (El-2-N-cvclopropy~4-fluoro-1-indanylideneZacetamide
An ice cold solution of (E)-2-(4-fluoro-1-indanylidene)acetyl chloride (2.11
g, 0.010
mol) in dichloromethane (65 mL) was treated with cyclopropylamine (1.39 mL,
0.02
mol), and the mixture was stirred for 18h. Hexane was added to the mixture,
and the
solids were collected by filtration and washed successively with water and
hexane to give
1.22 g of crude product. Recrystallization from acetonitrile : water mixtures
gave 0.83 g
(36%) of (E)-2-N-cyclopropyl-(4-fluoro-I-indanylidene)acetamide as a white
solid;
m.p., 121-122°C;
EXAMPLE 9
Preparation of (E)-2-(5-fluoro-1-indanvlidenelacetamide
a) Preparation of Ethyl 2-(5-fluoro-I-hvdroxv-I-indanvllacetate
This compound was prepared in an analogous manner to that described in
Example Sc for ethyl 2-(4,6-difluoro-I-hydroxy-1-indanyl) acetate substituting
5-
fluoro-1-indanone (14.77 g, 0.098 mol, Fairfield) for 4,6-difluoro-I-indanone.
Chromatography on silica gel with ethyl acetate: hexanes (9:1 ) as eluent gave
19.56 g (83%) of analytically pure ethyl 2-(5-fluoro-I-hydroxy-I-
indanyl)acetate
as a pale yellow oil; NMR (CDC13): d 6.88-7.30 (m, 3H, Ar), 5.30 (s, 1H, OH),
4.20 (q, 2H, CH2CH3), 2.66-3.08 (m, 4H, 2CH2), 2.30 (t, 2H, CH2Ar),1.28 (t,
3H, CH3).
Anal. Calcd. for C13H15F03 (mw 238.25): C, 65.54; H, 6.35.
Found: C, 65.39; H, 6.33.
WO 94/26693 PCT/GB94/01003
-35-
b) Preparation of 2-l5-fluoro-I-hydroxy-I-indan)r~acetic Acid
This compound was prepared in a similar manner to that described for 2-(4,6-
difluoro-1-hydroxy-1-indanyl)acetic acid in Example Sd by substituting ethyl 2-
(5-fluoro-1-hydroxy-1-indanyl) acetate ( 19.55 g , 0.082 mol) for ethyl 2-(4,6-
difluoro-1-hydroxy-1-indanyl) acetate to give 14.70 g (84%) of crude 2-(5-
fluoro-1-hydroxy-1-indanyl) acetic acid as a white solid. This material was
used
immediately without further purification.
c) Preparation of CEl-2-!S-Fluoro-1-indanylidene)acetic Acid
This compound was prepared in an analogous manner to (E)-2-(4,6difluoro-1-
indanylidene)acetic acid in Example Se by substituting 2-(5-fluoro-1-hydroxy-1-
indanyl)acetic acid(14.70 g, 0.069 mol) for 2-(4,6-difluoro-1-hydroxy-I-
indanyl)acetic acid. Recrystallization from acetonitrile : 2-propanol mixtures
gave 9.05g (68%) of (E)-2-(5-fluoro-1-indanylidene)acetic acid as a white
solid:
m.p., 240-242oC;
d) Preparation of CEl-2-f5-fluoro-1-indanvlidene)ace~yl Chloride
This compound was prepared in a similar manner to (E)-2-(4,6-difluoro-1-
indanylidene)acetyl chloride in Example Sf by substituting (E)-2-(5-fluoro-I-
indanylidene)acetic acid (5.77 g, 0.03 mol) for (E)-2-(4,6-difluoro-1-
indanylidene)acetic acid. The resulting solution was concentrated in vacuo,
and
the residue was used without further purification.
e) Preparation of (E)-2-f 5-fluoro-1-indanylidene)acetamide
This compound was prepared in a similar manner to that described for (E)-2-(6-
fluoro-1-indanylidene)acetamide Example 1 k by substituting (E)-2-(5-fluoro- I
-
indanylidene)acetyl chloride (3.16 g, 0.015 mol) for (E)-2-(6-fluoro-1-
indanylidene)acetyl chloride. Recrystallization from acetonitriie : water
mixtures
gave 1.28 g (44%) of (E)-2-(5-fluoro-1-indanylidene)acetamide as a white
solid;
m.p., 191-193oC;
WO 94/26693 PCT/GB94/01003
k
-36-
EXAMPLE 10
P_rgparation of lEl-N-CYclopropyl-2-f5-fluoro-1-indanvlidene)acetamide
A solution of (E)-2-(5-fluoro-1-indanylidene)acetic acid (0.97 g, 0.005 mol),
I-
hydroxybenzo-triazole (0.68 g, 0.005 mol, Fluka), cyclopropylamine (0.35 mL,
0.005
mol, Aldrich) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
(0.96 g,
0.005 mol, Sigma), which was added last, in dimethylformamide ( 15 mL) was
stirred at
room temperature for 18h, and the solution was concentrated irr vacuo. The
residue was
dissolved in ethyl acetate, washed successively with a 5% aqueous solution of
citric acid
(3x50 mL), a saturated aqueous sodium bicarbonate solution (2 x 50 mL) and
brine, and
dried over sodium sulfate. The solution was concentrated irr vacuo to give
crude (E)-N-
cyclopropyl-2-(5-fluoro-1-indanylidene)acetamide. Chromatography on Silica gel
eluting with hexane : ethyl acetate ( 1 : 1 ) gave 0.52 g (44%) of (E)-N-
cyclopropyl-2-(5-
fluoro-1-indanylidene)acetamide as a white solid: m.p., 137-138°C;
EXAMPLI~ 11
Alternative Preparation of (El-2-(6-Fluoro-1-indanylidenelacetamide
To an ice-cold stirred suspension of NaH (60% dispersion in mineral oil, 12.41
g,
60.25 mmoles, Aldrich) in tetrahydrofuran (30 ml) with 15-crown-5 (3.96 g,
17.98 mmoles, Aldrich) was added under N2, diethyl carbamoylmethylphosphonate
(11.7 g, 59.97 mmoles, K&K-ICN) and 6-fluoro-1-indanone (9.0 g, 59.96 mmol)
respectively in tetrahydrofuran (80 ml ). The mixture was allowed to warm to
room
temperature overnight. The mixture was poured into 200 ice-cold water and
extracted
with three 600 ml portions of diethyl ether. One organic phase was washed
successively
with 200 ml portions of aqueous sodium bisulfate ( 10%) and a saturated sodium
chloride
solution. The organic phase was dried over potassium carbonate, filtered, spin
evaporated irr vacuo and coevaporated with 200 ml dichloromethane to yield a
tacky
solid residue. The residue was chromatographed on Silica Gel 60 using ethyl
acetate:hexane (2:1). Fractions containing (E)-2-(6-fluoro-1-
indanylidene)acetamide
were combined and spin evaporated irr vacuo to give 2.38 g of a yellow solid.
Dilution
of a dichloromethane solution of the crude material with hexane gave 2. I 6 g
( I 8.8%) of
(E)-2-(6-fluoro-1-indanylidene)acetamide, m.p., 178-182° C; NMR (DMSO-
d6); d 7.4-
-37-
7.1 (m, 4H, Ar and NH), 6.88 (br s, 1H, NH), 6.37 (t, 1H, J=2.54 Hz, =CH),
3.22-3.14 (m, 2H, CH2), 2.95-2.89 (m, 2H, CH2); steady-state nOe: irradiation
at d 6.37, significant observed nOe at d 7.33-7.28.
Anal. Calcd for ClIHlOFNO: C, 69.10; H, 5.27; N, 7.33
Found: C, 69.01; H, 5.29; N, 7.28.
EXAMPLE 12
Preparation of (E)2-(6-Fluoro-3,3-dimethyl-1-indanylidene acetamide
a) Preparation of Diethyl Isopropylidenemalonate
Diethyl isopropylidenemalonate was prepared according to the procedure of
E. L. Eliel, R. O. Hutchins, and Sr. M. Knoeber, Organic Synthesis Coll. Vol.
VI, 442, 1988, with following modifications. A mixture of acetone (54 g, 0.93
mol, Mallinckrodt), diethyl malonate (100 g, 0.62 mol, Aldrich), acetic
anhydride (80 g, 0.78 mol, Mallinckrodt), and zinc chloride (12.5 g, 0.78 mol,
Aldrich) was refluxed (90°C oil bath) for 18 h while protected from
moisture.
The reaction solution was diluted with dichloromethane (500 ml) and washed
with cold water (3 x 50 ml). The aqueous washes were combined and
extracted with dichloromethane. All dichloromethane layers were combined
and concentrated by spin evaporation in vacuo. The residual oil was distilled
under vacuum and the fractions boiling at 102 - 138°C at 12 Torr were
combined with the pot residue and heated for 6 h with a 200°C oil bath.
The
dark oil was redistilled to give 40.1 g (32%) of diethyl
isopropylidenemalonate as a clear oil: b.p., 110 -11 S° C/12 mmHg:
b) Preparation of Diethyl 2-(2-(4-Fluorophenvl)-2-methylethyl)malonate
A mixture of 4-fluorophenylmagnesium bromide (82 ml of a 2 N solution in
ethyl ether, 0.164 mol, Aldrich) and copper(I) iodide (0.310 mg, 1.63 mmol,
Aldrich) was stirred for 15 min at -10°C while blanketed with a
nitrogen
atmosphere. To this mixture was added a solution of diethyl
isopropylidenemalonate (29.6 g, 0.148 mol) in anhydrous diethyl ether (250
CA 02162708 2001-08-21
-3 8-
ml) in a thin stream with rapid stirring. The resulting solution was stirred
at
10°C for 2 h, at 25°C for 30 min and then poured with rapid
stirring into 0.5
kg of crushed ice containing 30 ml of 12 N hydrochloric acid. The layers were
separated and the aqueous layer was extracted with ethyl ether (3 x 400 ml).
All ether layers were combined and washed with deionized water (2 x 25 ml),
saturated aqueous sodium bicarbonate (25 ml), and deionized water (25 ml).
The etheral layer was concentrated by spin evaporation in vacuo and the
residue was distilled to give 25.9g (59%) of diethyl 2-(2-(4-fluorophenyl)-2-
methylethyl)malonate as a clear oil (b.p., 140-145°C/0.01 mmHg):
c~ Preparation of 3-(4-Fluorophenyl)-3-methylbu~ric Acid
A solution of diethyl 2-(2-(4-fluorophenyl)-2-methylethyl)malonate (41 g,
0.138 mol) and potassium hydroxide 85% (18.25 g, 0.277 mol, Mallinkrodt)
in 250 ml of deionized water was vigorously refluxed for 4 hours with an
1 SO°C oil bath. After cooling with an ice bath, the solution was
neutralized
with 18 N sulfuric acid (23 ml, 0.414 mol, Mallinkrodt), and extracted with
dichloromethane (4 x 250 ml). The dichloromethane extracts were combined,
washed with water, and concentrated by spin evaporation in vacuo. The
residue was slurried with water and the crystalline product was collected by
filtration to give 24.3 g (90 %) of 3-(4-fluorophenyl)-3-methylbutyric acid,
m.p., 45 - 57°C:
d) Preparation of 3-(4-Fluorophenyl)-3-methylbutyryl Chloride
Oxalyl chloride (46.5 g, 0.367 mol, Aldrich) was added to a solution of 3-(4-
fluorophenyl)-3-methylbutyric acid (24 g, 0.122 mol) at -10°C while
protected
from moisture by a nitrogen atmosphere. The stirnng mixture was allowed to
warm to 25°C and was stirred for 2 h. Fractional distillation gave 26.6
g (76%)
of 3-(4-fluorophenyl)-3-methylbutyryl chloride as a clear oil, b.p., 132 -
138°C:
CA 02162708 2001-08-21
-39-
e) Preparation of 6-Fluoro-3 3-dimethyl-1-indanone.
A solution of 3-(4-fluorophenyl)-3-methylbutyryl chloride (19.0 g, 0.0815
mol) in dichloromethane (100 ml) was added dropwise over 2.5 h to a stirnng
mixture of aluminum chloride (13.57 g, 0.102 mol, Aldrich) in
dichloromethane (200 ml) while protected from moisture by a nitrogen
atmosphere. After stirring 18 h at 25°C, the reaction solution was
poured over
ice (400 g) and the resulting solution was extracted with dichloromethane (2 x
200 ml). The dichloromethane layers were combined, washed with deionized
water (50 ml), and concentrated by spin evaporation in vacuo. The residue
was dissolved in ethyl acetate and washed through a pad of silica gel. The pad
was washed with additional ethyl acetate. Removal of the volatiles from the
combined washes by spin evaporation in vacuo gave 15.2 g (99%) of 6-fluoro-
3,3-dimethyl-1-indanone as a light yellow oil which crystallized on standing,
m.p., 57 - 62°C:
fj Preparation of Ethyl 2-(6-Fluoro-1-hey-3 3-dimethyl-1-indanyl)acetate.
This compound was prepared in a similar manner to ethyl 2-(6-fluoro-1-
hydroxy-1-indanyl)acetate in Example 1d by substituting 6-fluoro-3,3-
dimethyl-1- indanone for 6-fluoro-1-indanone and preparing the activated zinc
by heating zinc dust (Aldrich) with iodine (Aldrich) without solvent. Removal
of the volatiles from the workup solution by spin evaporation in vacuo gave
16.2 g (82%) of ethyl 2-(6-fluoro-1-hydroxy-3,3-dimethyl-1-indanyl)acetate as
a light yellow oil:
CA 02162708 2001-08-21
-40-
g) Preparation of (E)-2-(6-Fluoro-3,3-dimethyl-1-indanylidene)acetic Acid.
A solution of ethyl 2-(6-fluoro-1-hydroxy-3,3-dimethyl-1-indanyl)acetate (16
g, 0.0601 mol) in 1 N sodium hydroxide (60.1 ml, 0.0601 mol) and ethanol (60
ml) was stirred for 20 h. The solution was concentrated to a small volume by
spin evaporation in vacuo, diluted with deionized water (100 ml), and
acidified to pH 3 with 1N hydrochloric acid This biphasic solution was
extracted with dichloromethane (2 x 100 ml). The extracts were combined,
washed with deionized water (20 ml), dried with magnesium sulfate
(Mallinckrodt), and concentrated by spin evaporation in vacuo. The residue
was dissolved in dichloromethane (30 ml), cooled to 0°C, and diluted
with 400
ml of a cold (0°C) solution of trifluoroacetic acid (45 g, Aldrich) in
dichloromethane (400 ml~. After 15 min, the solution was concentrated by
spin evaporation in vacuo and the residue was crystallized by adding hexanes
to give 9.23 g (70%) of (E)-2-(6-fluoro-3,3-dimethyl-1-indanylidene) acetic
acid as a white crystalline solid, m. p., 202 - 203.5°C:
h) Preparation of (E)-2-(6-Fluoro-3,3-dimethyl-1-indan~rlidene)acetyl
Chloride.
To an ice cold, stirred suspension of (E)-2-(6-fluoro-3,3-dimethyl-1-
indanylidene)acetic acid (9.0 g, 0.0409 mol) in dichloromethane (200 ml) was
added oxalyl chloride ( 15.6 g, 0.123 mol, Aldrich). The stirnng suspension
was allowed to warm to 25°C during 2 h. The resulting solution was
concentrated by spin evaporation in vacuo with the addition of
dichloromethane (4 x 75 ml) to give (E)-2-(6-fluoro-3,3-dimethyl-1-
indanylidene)acetyl chloride as an uncharacterized oil. Dichloromethane
(approximately 70 g) was added to dissolve this residual oil and the resulting
solution was divided equally and used without other purification in Examples
12, 13 and 14.
i) Preparation of (E)-2-(6-Fluoro-3 3-dimethyl-1-indan lidene)acetamide
A solution of 30% aqueous ammonium hydroxide (10 ml, 76 mmol,
CA 02162708 2001-08-21
-41-
Mallinckrodt) was added to the solution of (E)-2-(6-fluoro-3,3-dimethyl-1-
indanylidene)acetyl chloride (0.01363 mol) obtained from Example 41 h
diluted with dichloromethane (200 ml) and cooled to 0°C. The biphasic
solution was stirred rapidly and allowed to warm to room temperature over 18
h. The reaction solution was concentrated by spin evaporation in vacuo,
diluted with dichloromethane (200 ml), and washed with 1N aqueous
hydrochloric acid (Mclntosh), a solution of 5% aqueous sodium bicarbonate
(Mallinckrodt), dried with magnesium sulfate (Mallinckrodt), and
concentrated by spin evaporation in vacuo. The residue was chromatographed
on Silica Gel 60 using ethyl acetate-hexanes (1:1), and then ethyl acetate.
Fractions containing (E)-2-(6-fluoro-3,3 dimethyl-1-indanylidene) acetamide
were combined and concentrated by spin evaporation in vacuo .
Recrystallization from dichloromethane-hexanes gave 2.85 g (95%) of {E)-2-
(6-fluoro-3,3dimethyl-1-indanylidine)acetamide as a white crystalline solid,
m.p., 167 - 168°C:
Example 13
Preparation of (E)-2-(6-Fluoro-3,3-dimethyl-1-indanylidene)-N-methylacetamide
This compound was prepared in an analogous manner to Example 15i with the
replacement of the solution of 30% aqueous ammonium hydroxide with a 40%
aqueous solution of methylamine ( 10 ml, Mdrich). Recrystallization from
dichloromethane-hexanes gave 2.89 g (91%) of (E)-2-(6-fluoro-3,3-dimethyl-1-
indanylidene)-N-methylacetamide as a white crystalline solid, m.p., 157 -
158°C:
CA 02162708 2001-08-21
-42-
Example 14
Preparation of (E)-N-CYclo~ropy~6-fluoro-3,3-dimethyl-1-
indanvlidenelacetamide
This compound was prepared in an analogous manner to Example 15i with the
replacement of the solution of 30% aqueous ammonium hydroxide with
cyclopropyl amine (4 ml, Aldrich). Recrystallization from dichloromethane-
hexanes gave 2.86 g (81%) of (E)-N-cyclopropyl-2-(6-fluoro-3,3-dimethyl-1-
indanylidene)acetamide as a white crystalline solid, m.p., 149 - 1
SO°C_
Example 15
Preparation of (E)-2-(6-Fluoro-3-methyl-1-indan lidenelacetamide
a) Preparation of Ethyl 4-Fluorocinnamate
A solution of butyl lithium, 2.5 M in hexanes (159 ml, 0.3975 mol, Aldrich),
was added dropwise over 0.25 hr, with rapid mechanical stirring, to a solution
of triethyl phosphonoacetate (89.2 g, 0.389 mol, Aldnch) in tetrahydrofuran
(800 ml, anhydrous, Aldrich) at <5°C while blanketed with a nitrogen
atmosphere. This solution was stirred for an additional 0.25 hr and cooled to
0
°C with an ice bath and a solution of 4'-fluoroacetophenone (50g, 0.362
mol,
Aldrich) in tetrahydrofuran (50 ml) was then added in one portion. Stirnng
was continued for 18 hr without additional cooling. The solution was then
concentrated to ~100m1 by spin evaporation in vacuo and diluted to 500 ml
with ethyl acetate. After washing with deionized water (3 x 50 ml) this
solution was concentrated by spin evaporation in vacuo. Distillation at
reduced pressure gave 48 g (63%) of ethyl 4-fluorocinnamate as a mixture of
(E) to (Z) isomers (ratio 3:1) contaminated with 16% of triethyl
phosphonacetate as a clear oil, b.p., 138-143°C at 14 Ton:
CA 02162708 2001-08-21
-43-
b) Preparation of Eth~4-Fluorophenyl)but mate
A mixture of ethyl 4-fluorocinnamate (47.5 g, 0.228 mol) and 10% palladium
on carbon (0.85 g, Aldrich) in 95% ethanol was shaken in a Parr hydrogenator
under
2-3 atm of Hz pressure for 1 h. The mixture was filtered and concentrated by
spin evaporation in vacuo. Fractional distillation gave 46.5 g (97%) of ethyl
3-
(4-fluorophenyl)butyrate as a clear oil, b.p., 122 - 128°C: c)
Preparation of 3-
(4-Fluorophenyl)butanoic Acid.
c) Preparation of 3-(4-Fluorophenyl)butanoic Acid
A solution of ethyl 3-(4-fluorophenyl)butyrate (45.3 g, 0.215 mol), 85%
potassium hydroxide ( 14.22 g, 0.215 mol, Mallinckrodt) in 200 ml of
deionized water was refluxed for 2 h, concentrated by spin evaporation in
vacuo, made acidic (pH 3) with 12 N hydrochloric acid (Mallinckrodt), and
extracted with dichloromethane (4 x 200 ml). The dichloromethane layers
were combined, washed with deionized water (50 ml), and concentrated by
spin evaporation in vacuo. The residue was crystallized from
dichloromethane-hexanes to give 34.5 g (88%) of 3-(4-fluorophenyl)butyric
acid as a white crystalline solid:
d) Preparation of 3-(4-Fluorophenyl butyryl Chloride
Oxalyl chloride (71 g, 48.8 ml, 0.560 mol, Aldrich) was added to a mixture of
3-(4-fluorophenyl)butyric acid (34 g, 0.187 mol) in 200 ml of
dichloromethane at -5°C. After stirring for 20 min at this temperature,
the
solution was allowed to warm to 25°C and stirnng was continued for 2 h.
The
volatiles were removed by spin evaporation in vacuo with the addition of
dichloromethane (4 x) during concentration to give 35.1 g (94%) of 3-(4-
fluorophenyl)butyryl chloride as a light yellow oil:
CA 02162708 2001-08-21
-44-
e) Preparation of 6-Fluoro-3-methyl-1-indanone.
This compound was prepared in an analogous manner to Example 15e with
the replacement of 3-(4-fluorophenyl)-3 -methyl-butyryl chloride with 3 -(4-
fluorophenyl)butyryl chloride (35. 1 g, 174.9mmo1). Removal of the volatiles
from the combined washes by spin evaporation in vacuo gave 26.3g (92%) of
6-fluoro-3-methyl-1-indanone as an oil which formed low melting crystals on
standing:
f) Preparation of Ethyl 2-(6-Fluoro-1-hydroxy-3-methyl-1-indanyl)acetate.
This compound was prepared in an analogous manner to Example 15f with the
replacement of 6-fluoro-3,3-dimethyl 1 -indanone with 6-fluoro-3 -methyl-1-
indanone (25g. 140mmo1). Removal of the volatiles from the workup solution
gave 15.0 g (45%) of ethyl 2-(6-fluoro-1-hydroxy-3-methyl-1-indanyl)acetate
as a light tan oil:
g) Preparation of (E)-2-y6-Fluoro-3-methyl-1-indan liy denelacetic Acid.
This compound was prepared in an analogous manner to Example 1 Sg with
the replacement of ethyl 2-(6-fluoro-1-hydroxy-3,3-dimethyl-1-
indanyl)acetate with ethyl 2-(6-fluoro-1-hydroxy-3-methyl-1-indanyl)acetate
(15g, 59. Smmol). Removal of the volatiles from workup gave 9.3 g (76%) of
(E)-2-(6-fluoro-3-methyl-1-indanylidene)acetic acid as a tan solid, m.p., 175 -
177° C:
h) Preparation of (E)-2-(6-Fluoro-3-methyl-1-indanylidenelacetyl Chloride.
This compound was prepared in an analogous manner to Example 1 Sh with
the replacement of (E)-2-(6-fluoro-3,3-dimethyl-1-indanylidene)acetic acid
with (E)-2(6-fluoro-3-methyl-1-indanylidene)acetic acid (9g, 43.6mmo1). The
CA 02162708 2001-08-21
-45-
product residue was dissolved in dichloromethane and used, without
purification, in Example 17, 18, and 19.
i) Preparation of (E)-2-(6-Fluoro-3-methyl-1-indanylidene)acetamide
This compound was prepared in an analogous manner to Example 15i with the
replacement of (E)-2-(6-fluoro-3,3-dimethyl-1-indanylidene)acetyl chloride
with (E)-2-(6-fluoro-3-methyl-1-indanylidene)acetyl chloride (3.26g,
l4.SmmoI). Recrystallization from dichloro- methanehexanes gave 2.39 g
(77%) of (E)-2-(6-fluoro-3-methyl-1-indanylidene)acetamide as a white
crystalline solid, m.p., 149 -151°C:
Example 16
Preparation of (E)-2-(6-Fluoro-3-methyl-1-indanylidene)-N-methylacetamide
This compound was prepared in an analogous manner to Example 16 with the
replacement of (E)-2-(6-fluoro-3,3-dimethyl-1-indanylidene)acetyl chloride
with (E)
2-(6-fluoro-3-methyl-1-indanylidene)acetyl chloride (3.26g, 14. 5mmol).
Recrystallization from dichloro- methanehexanes gave 2.27 g (71 %) of (E)-2-(6
fluoro-3-methyl-1-indanylidene)-N-methylacetamide as a white crystalline
solid,
0
m.p., 168 - 169 C:
Example 17
Preparation of (E)-N-Cyclopropyl-2-(6-fluoro-3 -methyl-1 -
indanylidene)acetamide
This compound was prepared in an analogous manner to Example 17 with the
replacement of (E)-2-(6-fluoro-3,3-dimethyl-1-indanylidene)acetyl chloride
with (E)-
2-(6-fluoro-3-methyl-1-indanylidene)acetyl chloride (3.26g, l4.Smmol).
Recrystallization from dichloromethane-hexanes gave 2.30 g (67%) of (E)-N-
cyclopropyl-2-(6-fluoro-3-methyl-1-indanylidene)acetamide as a white
crystalline
solid, m.p., 132 - 134°C:
CA 02162708 2001-08-21
-46-
Example 18
Preparation of (Z)-2-(6-fluoro-2-hydroxy-1-indanylidene)acetamide Method
a) Preparation of (Z)-2-(2-bromo-6-fluoro-1-indanylidene)acetamide
N-Bromosuccinimide (22.57 g, 126.8 mmoles, Aldrich) and benzoyl peroxide
(1.89 g, 7.8 mmoles, Aldrich) were added to a suspension of (E)-2-(6-fluoro-
1-indanylidene)acetamide (21.00 g, 109.8 mmoles) in carbon tetrachloride
(400mL) and benzene (400mL). The mixture was refluxed under a calcium
chloride drying tube while shining an infrared lamp on it for two hours, after
which time an orange solution formed. The heat and light were removed, and
the solution was stirred at ambient temperature for 18 hours. The mixture was
filtered, and the solids were washed with ethyl acetate. The washings and
filtrate were combined and evaporated in vacuo. The residue was dissolved in
ethyl acetate (800 mL) and washed with water (3 x 200 mL) and brine (200
mL), dried over sodium sulfate, filtered and evaporated in vacuo. The residue
was chromatographed on silica gel eluting first with hexane : ethyl acetate (2
:1) gradually increasing the polarity to hexane : ethyl acetate (1 : 1). The
fractions containing the major spot were combined and evaporated in vacuo to
give a yellow solid which was dried in a vacuum at 70°C for 18 hours to
give
1.022 g (3%) of (Z)-2-(2-bromo-6-fluoro-1-indanylidene)acetamide as a
yellow solid, mp 162-163°C. 1H-NMR
b) A mixture of (Z)-2-(2-bromo-6-fluoro-1-indanylidene)acetamide (5.30 g,
19.25 mmoles) and silver nitrate (10.40 g, 61.18 mmoles, Aldrich) in
dimethoxyethane (265 mL) and water (100 mL) was refluxed for 18 hours.
The mixture was filtered, and the filtrate was diluted with water (700 mL) and
extracted with ethyl acetate (6 x 100 mL). The combined extracts were washed
with water (200 mL) and brine (200 mL), dried over magnesium sulfate,
filtered, and evaporated in vacuo. The residue was chromatographed on silica
CA 02162708 2001-08-21
-47-
gel, eluting with hexane ethyl acetate (2 :1 ), gradually increasing the
polarity
to hexane : ethyl acetate ( 1 : 1 ). The fractions containing the compound
with
R~ 0.18 were combined and evaporated in vacuo to give 1.13 g (28%) of
crude (Z)-2-(6-fluoro-2-hydroxy -1-indanylidene)acetamide as an orange
solid. Recrystallization from ethyl acetate hexane mixtures gave 0.49 g ( 12%)
of (Z)-2-(6-fluoro-2-hydroxy 1-indanylidene)acetamide as an off white solid,
mp 201-202°C;
c) Preparation of (Z)-2-(6-fluoro-2-hydrox~l-indanylidene)acetamide (Method
A suspension of (E)-2-(6-fluoro-1-indanylidene)acetamide (12.00 g, 62.8
mmoles) in dichloromethane (250 mL) was added to a solution of selenium
dioxide (5.20 g, 46.9 mmoles, Aldrich) and tert-butyl hydroperoxide (25 mL,
260.8 mmoles, Aldrich) in dichloromethane (500 mL). The suspension was
stirred at ambient temperature for 3 days. Additional tert-butyl hydroperoxide
( 10 mL, 104.3 mmoles) was added, and the mixture was stirred at ambient
temperature for 18 hours. Additional selenium dioxide (5.00 g, 45.1 mmoles)
was added, and the mixture was stirred at ambient temperature for 18 hours.
Additional tert-butyl hydroperoxide (15 mL, 156.5 mmoles) was added, and
the mixture was stirred at ambient temperature for 18 hours. The mixture was
filtered to remove about one gram of impure product, and the filtrate was
dried over magnesium sulfate, filtered, and evaporated in vacuo. Additional
selenium dioxide (5.00 g, 45.1 mmoles) was added, and the mixture was
stirred at ambient temperature for 18 hours. The mixture was concentrated in
vacuo to 300 mL, hexane was added, and the precipitate was collected by
filtration, washed with hexane, and combined with the solids collected
previously. The combined solids were dissolved in ethyl acetate (700 mL),
washed successively with water (3x100 mL) and brine (100 mL), concentrated
in vacuo to 100 mL, and cooled in an ice bath. The solids were collected by
filtration, and the filtrate was concentrated in vacuo to give a second crop
of
solids. All of the solids were combined and chromatographed on silica gel,
eluting with hexane : ethyl acetate (1: 1). The fractions containing the major
spot were combined and evaporated in vacuo to give 5.80 g of an off white
CA 02162708 2001-08-21
-48-
solid, which was washed with chloroform (3x50 mL) to give 5.43 g (42%) of
(Z)-2-(6-fluoro-2-hydroxy-1-indanylidene)acetamide as a white solid; mp
202-204°C;
Example 19
~Z~-2-(4,6-Difluoro-2-hydroxy-1-indanylidene)acetamide
A suspension of (E)-2-(4,6-difluoro-1-indanylidene)acetamide (10. 0g, 0.05
mol, prepared as in Example 5g in dichloromethane (250 mL) was added
portionwise over a 10 min. period to a mixture of 70% aqueous t-
butylhydroperoxide (19.8 mL, 0.15 mol, Aldrich) and selenium dioxide (3.7g,
0.03 mol, Aldrich) in dichloromethane (500 mL) at ambient temperature. After
18h, additional t-butylhydroperoxide ( 10 mL of a S.OM solution in 2,2,4-
trimethylpentane, 0.05 mol, Aldrich) and selenium dioxide ( 1.8g, 0.02 mol)
were added and the mixture was stirred at ambient temperature. After 18h,
additional t-butylhydroperoxide (10 mL of 70% aqueous solution, 0.08 mol)
and selenium dioxide (3.7g, 0.05 mol) were added and the mixture was stirred
at ambient temperature for 8 days. The resulting solid was filtered off and
washed with dichloromethane to give 5. 85g of crude (Z)-2-(4,6-difluoro-2-
hydroxy-1-indanylidene)acetamide. After 7 days at ambient temperature a
second crop of crude (Z)-2-(4,6-difluoro-2-hydroxy-1-indanylidene)acetamide
was obtained from the filtrate. Column chromatography on silica gel using
ethyl acetate as the eluent followed by a second column chromatography on
silica gel using ethyl acetate:hexanes (3:2) as eluent and trituration of the
resulting solid with pentane gave 2.38g of (Z)-2-(4,6-difluoro-2-hydroxy-1-
indanylidene)acetamide as a pink solid: m.p. 235-237°C.
Example 20
Preparation of (E)-2- 6-fluoro-3 -hydroxy-1-indanylidene)acetamide
CA 02162708 2001-08-21
-49-
a) Preparation of 3-bromo-6-fluoro-1-indanone
A mixture of N-bromosuccinimide (2.76 g, 15.51 mmoles, Aldrich), benzoyl
peroxide (0.41 g, 0.04 mmoles, Aldrich) and 6-fluoro-1-indanone (2.29 g,
15.25 mmoles) in carbon tetrachloride (20 mL) was refluxed under nitrogen
for two hours. The mixture was cooled to ambient temperature, filtered, and
the solids were washed with dichloromethane. The washings and filtrate were
combined, washed successively with 1.0 N sodium hydroxide (2 x 30 mL),
water (2 x 30 mL) and brine (30 mL), and evaporated in vacuo. The residue
was chromatographed on silica gel eluting first with
hexane, gradually increasing the polarity to hexane : ethyl acetate (95:5).
The
fractions containing the major spot were combined and evaporated in vacuo to
give a 2.30 g (66%) of 3-bromo-6-fluoro-1-indanone as a yellow oil which
was used without further purification.
b) Preparation of 3 -hydroxy-6-fluoro-1-indanone
A mixture of 3-bromo-6-fluoro-1-indanone (2.50 g, 10.0 mmoles) and silver
carbonate (4.19 g, 15.2 mmoles, Aldrich) in dimethoxyethane (85 mL) and
water (65 mL) was stirred overnight at ambient temperature. The mixture was
filtered through a pad of celite, and the filtrate was diluted with water (500
mL) and extracted with ethyl acetate (4 x 100 mL). The combined extracts
were washed with water (100 mL) and brine (75 mL), dried over sodium
sulfate, filtered, and evaporated in vacuo to give 2.80 g (quantitative) of
crude
3-hydroxy-6-fluoro-1-indanone which was used without further purification.
Chromatography of 0.41 g on silica gel, eluting with hexane: ethyl acetate (3
4) gave 0.050 g of analytically pure 3-hydroxy-6-fluoro-1-indanone as a tan
solid, mp 73-76°C;
c) Preparation of 3-((tert-butyldimethylsilyl)oxy)-6-fluoro-1-indanone
A solution of 3-hydroxy-6-fluoro-1-indanone (4.09 g, 24.6 mmoles) in
dimethylformamide (10 mL) was added to a solution of tert-butyldimethylsilyl
chloride (4.60 g, 30.5 mmoles, Aldrich) and imidazole (4.22 g, 62.0 mmoles,
CA 02162708 2001-08-21
-50-
Aldrich) in dimethylformamide (20 mL). The solution was stirred at ambient
temperature for 18 hours and evaporated in vacuo. The residue was dissolved
in dichloromethane (200 mL) and washed with water (6 x 75 mL) and brine
(100 mL), dried over magnesium sulfate, filtered and evaporated in vacuo.
The residue was chromatographed on silica gel eluting with hexane : ethyl
acetate (95 : S). The fractions containing the major spot were combined and
evaporated in vacuo, and the residue was dried in a vacuum at ambient
temperature for 18 hours to give 4.14 g (60%) of 3-((tert-
butyldimethylsilyl)oxy-6-fluoro-1-indanone as a white solid, mp 56-
58°C;
d) Preparation of Ethyl 2-(3-((tert-butyldimethvlsilyl)oxy)-6-fluoro-1-h day-1-
indanyl) acetate
A solution of ethyl acetate (1.00 mL, 10.3 mmoles) and lithium
diisopropylamine [This salt was prepared from diisopropylamine (1.41 mL,
10.0 mmoles, Aldrich) and n-butyl lithium (4.00 mL of a 2.5 M hexane
solution, 10.0 mmoles, Aldrich)], in tetrahydrofuran ( 1 S mL) was stirred at -
78°C under nitrogen for 15 minutes. A solution of 3-((tert-
butyldimethylsilyl)oxy)-6-fluoro-1-indanone (2.80 g, 10.0 mmoles) in
tetrahydrofuran (15 mL) was added dropwise over a 7 minute period, and the
solution was stirred at -78°C under nitrogen for 1.5 hours. A solution
of
ammonium chloride (1.60 g, 30.0 mmoles) in water (9 mL) was added, and
the resulting suspension was allowed to warm to ambient temperature. The
layers were separated, and the aqueous layer was extracted with ether (2 x 100
mL). The organic extracts were combined and washed successively with water
(100 mL) and brine (100 mL), dried over magnesium sulfate, filtered and
evaporated in vacuo. The residue was chromatographed on silica gel eluting
with hexane : ethyl acetate (98:2), gradually increasing the polarity to
hexane
ethyl acetate (4 :l). The fractions containing the major spot were combined
and evaporated in vacuo, and the residue was dried in a vacuum at ambient
temperature for 18 hours at 60°C to give 2.86 g (78%) of ethyl 2-(3 -
((tert-
butyldimethylsilyl)oxy)-6-fluoro-1-hydroxy-1-indanyl) acetate as a clear oil;
CA 02162708 2001-08-21
-51-
e) Preparation of (E -Ethyl 2-(3 -((tert-butyldimethylsilyl)oxv)-6-fluoro 1 -
indanylidene) acetate
A solution of ethyl 2-(3 -((tert-butyldimethylsilyl)oxy)-6-fluoro 1 -hydroxy-1-
indanyl) acetate (2.80 g, 7.6 mmoles) was added to a solution of bis[2,2,2-
trifluoro-1-phenyl-1-(trifluoromethyl)-ethoxy]diphenylsulfurane (6.30 g, 9.4
mmoles, Fluka) in dichloromethane (50 mL) under a nitrogen atmosphere.
The solution was stirred at ambient temperature for 35 minutes and poured
into water (500 mL). The organic layer was separated, washed with brine (250
mL), dried over magnesium sulfate, filtered and evaporated in vacuo. The
residue was chromatographed on silica gel eluting with hexane ethyl acetate
(99 :1). The fractions containing the major spot (and also a minor impurity)
were combined and evaporated in vacuo to give 2.68 g (quantitative) of crude
(E)-ethyl 2-(3-((tert-butyldimethylsilyl)oxy)-6-fluoro-1-indanylidene) acetate
as a yellow oil which was used without further purification;
f) Preparation of (E -_) 2-(3-((tert-butyldimethylsilyl)oxy)-6-fluoro-1-
indanylidene
acetamide
A solution of dimethylaluminum amide was prepared by adding trimethyl
aluminum (6.5 mL of a 2.0 M toluene solution, 13.0 mmoles, Aldrich) to a
solution of ammonium chloride (0.695 g, 13.0 mmoles) in dichloromethane
(25mL) under a nitrogen atmosphere and stirring for 45 minutes at ambient
temperature. This solution of dimethylaluminum amide (13.0 mmoles) was
added to a solution of (E)-ethyl 2-(3 ((tent-butyldimethylsilyl)oxy)-6-fluoro-
1-
indanylidene) acetate ( 1.190 g, 3.4 mmoles) in dichloromethane (60 mL)
under a nitrogen atmosphere. The mixture was stirred at ambient temperature
for 30 minutes and refluxed for 18 hours. After cooling to ambient
temperature and then in an ice bath, the mixture was quenched by dropwise
addition of 0.5 N hydrochloric acid until gas evolution ceased. The solution
was diluted with water (50 mL), the layers were separated, and the aqueous
layer was extracted with dichloromethane (75 mL). The organic layers were
combined, washed successively with water (75 mL) and brine (75 mL), dried
over magnesium sulfate, filtered and evaporated in vacuo. The residue was
CA 02162708 2001-08-21
-52-
recrystallized from dichloromethane: hexane mixtures to give 0.321 g (29%)
of (E)-2-(3-((tert-butyldimethylsilyl)oxy)-6-fluoro-1-indanylidene) acetamide
as a white solid, mp 160-165°C;
g) Preparation of (E)-2-(6-fluoro-3-hydroxy-1-indanylidene)acetamide
A solution of (E)-2-(3 ((tent-butyldimethylsilyl)oxy)-6-fluoro-1-indanylidene)
acetamide (1.80 g, 5.6 mmoles) and pyridinium p-toluenesulfonate (0.85 g,
3.4 mmoles, Aldrich) in ethanol (65 mL) was heated at 55-68°C for 3.5
hours
under a nitrogen atmosphere and evaporated in vacuo. The residue was
dissolved in ethyl acetate (150 mL) and washed successively with water (2 x
150 mL) and brine (150 mL), dried over magnesium sulfate, filtered and
evaporated in vacuo. The residue was chromatographed on silica gel eluting
with ethyl acetate, gradually increasing the polarity to ethyl acetate:
ethanol
(95 : 5). The fractions containing the major spot were combined and
evaporated in vacuo, and the residue was dried in a vacuum at 80° C for
18
hours to give 0.72 g (62%) of (E)-2-(6-fluoro-3-hydroxy-1-
indanylidene)acetamide as a white solid, mp 166-1 68°C;
Example 21
Preparation of (Z)-~2,3-dihydroxy-6-fluoro-1-indanylidene) acetamide
a) Preparation of (Z)-~2,3-dibromo-6-fluoro-1-indanylidene)acetamide
N-Bromosuccinimide (49.37 g, 277.4 mmoles, Aldrich) and benzoyl peroxide
( 1.60 g, 6.6 mmoles, Aldrich) were added to a suspension of (E)-2-(6-fluoro-1-
indanylidene)acetamide (17.68 g, 92.5 mmoles) in carbon tetrachloride (335
mL) and benzene (335 mL). The mixture was refluxed under a calcium
chloride drying tube for four hours, after which time an orange solution
formed. The heat was removed, and the solution was stirred at ambient
temperature for 18 hours. The mixture was filtered, and the solids were
washed with ethyl acetate. The washings and filtrate were combined and
evaporated in vacuo. The residue was dissolved in ethyl acetate (800 mL) and
CA 02162708 2001-08-21
-53-
washed with water (3 x 200 mL) and brine (200 mL), dried over sodium
sulfate, filtered and evaporated in vacuo. The residue was chromatographed
on silica gel eluting with hexane : ethyl acetate (2 : 1). The fractions
containing the compound with Rf--- 0.4 in hexane: ethyl acetate ( 1 : 1 ) were
combined and chromatographed again on silica gel eluting with hexane : ethyl
acetate (2 :1). The fractions containing the compound with R~ 0.4 in hexane
ethyl acetate (1 : 1) were combined and evaporated in vacuo to give a solid
which was washed with hexane and dried in a vacuum at 50°C for 18 hours
to
give 1.35 g (4%) of (Z)-2-(2,3-dibromo-6-fluoro-1-indanylidene)acetamide as
a yellow solid, mp 158-163°C (decomposed).
b) A mixture of (Z)-2-(2,3-dibromo-6-fluoro-1-indanylidene)acetamide (0.54 g,
1.55 mmoles) and silver carbonate (0.56 g, 2.03 mmoles, Aldrich) in
dimethoxyethane ( 15 mL) and water (30 mL) was refluxed for 6 hours. The
mixture was stirred overnight at ambient temperature and refluxed again for 6
hours. The mixture was diluted with water (100 mL) and extracted with ethyl
acetate (6 x 30 mL). The combined extracts were washed with water (100 mL)
and brine (100 mL), and evaporated in vacuo. The residue was
chromatographed on silica gel, eluting with hexane: ethyl acetate (2 : 1 ).
The
fractions containing the compound with Rf--- 0.15, eluting with ethyl acetate,
were combined and evaporated in vacuo to give 0.12 g (35%) of crude (Z) 2-
(2,3-dihydroxy-6-fluoro-1-indanylidene) acetamide as a beige solid.
Recrystallization from ethyl acetate : hexane mixtures gave 0.037 g (11%) of
(Z)-2-(2,3-dihydroxy-6-fluoro-1-indanylidene)acetamide as an off white solid
which was shown by 1H-NMR to be a mixture (85 : 15) of diastereomers, mp
212-220°C; 1H-NMR (DMSO-d6): d 7.82 (d, 2H), 7.28-7.76 (m, 3H), 6.96
(s,
1H), 6.80 (s, 0.15H), 6.54 (s, 0.85H), 6.51 and 6.12 (m, 0.3H), 5.92 (d,
1H),4.81 (m, 1.7H); steady-state nOe: irradiation at 6.47d, observed 20% nOe
at 7.38d.
EXAMPLE 22
CA 02162708 2001-08-21
-54-
Preparation of (El-2-(6-Fluoro-3 -oxo-1-indanylidene)acetamide
a) Preparation of (E)-Ethyl-3-fluorocinnamate
This compound was prepared in an analogous manner to Example 18a with
the replacement of 4'-fluoroacetophenone with 3-fluorobenzaldehyde (33. 6g,
0.3mmo1 Aldrich). Distillation gave 32.85g (56%) of (E)-ethyl 3-
fluorocinnamate in 5 fractions (b.p., 140 - 1 S 5°C at 15 Torr) which
were
equally contaminated with approximately 13% of triethyl phosphonoacetate.
This material was used without additional purification. 1H NMR (DMSO-d6 ):
d 7.66-7.47 (m, 2H), 7.45-7.40 (m, 1 H), 7.27-7.20 (m, 1 H), 6.70 (d, 1 H, JHH
=
l6Hz), 4.18 (q, 2H, JHH = 7.2Hz), 4.10-3.96 (m, 0.78H), 3.77 (d, 0.26H, JPH =
21.3 Hz), 1.24(t, 3H, JHH = 7.0 Hz), 1.24- 1.14(m, 1.17H).
b) Preparation of Diethyl 2-carbethoxy-3-(3-fluorophenyl)~lutarate
Sodium metal (0.388g, 0.0169 mol) was stirred in diethyl malonate (15.28 g,
0.0953 mol, Aldrich) under a nitrogen atmosphere at 120°C for 0.33 hr.
To the
resulting solution was added (E)-ethyl 3-fluorocinnamate (16.4g, 0.0845 mol)
and stirnng was continued for 7 hrs at the same temperature. The dark
solution was cooled, dissolved in dichloromethane (500 ml) and made acidic
with 30 ml of 1N aqueous hydrochloric acid (Macintosh). The volatiles were
removed from the resulting froth by spin evaporation in vacuo and the residue
was dissolved in ethyl acetate. This solution was washed with 5% aqueous
sodium bicarbonate until neutral, water, and the volatiles were removed by
spin evaporation in vacuo. Distillation gave 20 g of a material boiling
between
130 - 185°C at 0.150 Torr. Redistillation gave 14.72 g (44%) of diethyl
2-
carbethoxy-3-(3-fluorophenyl)glutarate as a clear liquid: b.p., 155 -
160°C at
0.1 Torr;
c) Preparation of 3-(3-Fluorophenyl)~lutaric acid
To a hot solution of sodium hydroxide (19.15 g, 0.479 mol) in water (50 ml)
was added a solution of diethyl 2-carbethoxy-3-(3-fluorophenyl)glutarate (18.
CA 02162708 2001-08-21
-ss-
8g, O.OS32 mol) in ethanol (36 ml). The resulting slurry was refluxed for 5
hrs.
The mixture was poured into icewater and the ethanol was removed by spin
evaporation in vacuo. The residual aqueous solution was acidified with
concentrated hydrochloric acid (12 N) and the solution (200 ml) was extracted
with ethyl acetate (3 x 300 ml). The ethyl acetate layers were combined,
washed with water (s0 ml) and the volatiles were removed by spin
evaporation in vacuo to give a solid that was recrystallized from
dichloromethane and hexanes to give 9.3 g (77%) of 3-(3-
fluorophenyl)glutaric acid a white solid; m.p., 126 -127.s°C;
d) Preparation of 2~6-Fluoro-3-oxo-1-indanyl)acetic acid
Polyphosphoric acid (39.6g, Aldrich) and 3-(3-fluorophenyl)glutaric acid
(6.6g, 0.0292 mol) were combined and the mixture heated with an oil bath at
120°C for 10 min. The now red solution was cooled to approximately
60°C
and water (approximately 100 ml) was added dropwise, with efficient stirring.
The resulting precipitate was collected and washed with water.
Recrystallization from dichloromethane and hexanes gave s.3g (87%) of 2-(6-
fluoro-3-oxo-1 indanyl)acetic acid: m.p., l s0 - 1 s 1 °C;
e) Preparation of 2-(6-Fluoro-3-oxo-1-indanyl)acetyl chloride
Oxalyl chloride (4.s g, 0.03s mol, Aldrich) was added to an ice cold stirring
mixture of 2-(6-fluoro-3-oxo-1-indanyl)acetic acid (s.0 g, 0.024 mol) in
dichloromethane (200 ml) under a nitrogen atmosphere. The mixture was
allowed to warm to room temperature and stirnng was continued for 48 hrs.
The volatiles were removed from the solution by spin evaporation in vacuo
with the addition of dichloromethane (3 x s0 ml) to give 2-(6-fluoro-3-oxo-1-
indanyl)acetyl chloride which was used without purification or analysis.
f) Preparation of 2-(6-Fluoro-3-oxo-1-indanyl)acetamide
CA 02162708 2001-08-21
-56-
A solution of 2-(6-fluoro-3-oxo-1-indanyl)acetyl chloride (prepared from
0.024 mol of 2-(6-fluoro-3-oxo-1-indanyl)acetic acid) in dichloromethane
(150 ml) was cooled to 0°C and stirred rapidly while 50 ml of ammonium
hydroxide, 28-30%, was added. The resulting mixture was allowed to warm to
room temperature and stirnng was continued for 18 hr. The volatiles from this
mixture were removed by spin evaporation in vacuo and the residue was
dissolved in dichloromethane (250 ml) and washed with water (3 x 50 ml).
The dichloromethane phase was then slurned with Silica Gel 60 and the
volatiles were removed by spin evaporation in vacuo. This silica was then
applied to a column of Silica Gel 60 (51 x 400 mm) wet with dichloromethane
and the product was removed by elution with methanol:dichloromethane
(3:97) to give, after recrystallization from methanol, 2.4 g (48%) of 2-(6-
fluoro-3-oxo-1 indanyl)acetamide as a yellow solid: m.p., 150-152°C
g) Preparation of (E)-2-(6-Fluoro-3-oxo-1-indanylidene)acetamide
A mixture of 2-(6-fluoro-3-oxo-1-indanyl)acetamide (0.750g, 0.0036 mol), N-
bromosuccinimide (0.750g, 0.0042 mol, Aldrich), benzoyl peroxide (0.270g,
0.0011 mol, Aldrich) in tetrachloromethane (37 ml) and benzene (37 ml) was
stirred while heating with an oil bath at 120°C for 20 min. This
reaction was
combined with a similarly run reaction (except 0.0024 mol scale). The
solution was slurried with Silica Gel 60 and the volatiles were removed by
spin evaporation in vacuo. This silica gel was then applied to a column of
Silica Gel 60 (51 x 450mm) wet with dichloromethane and the product was
removed by elution with methanol:dichloromethane (3:97). Alter the volatiles
were removed from the combined fractions containing product by spin
evaporation in vacuo, the residue was recrystallized from methanol to give 0.8
lOg (58%) of (E)-2-(6-fluoro-3-oxo-1-indanylidene)acetamide: m.p.,
235°C
(dec.);
CA 02162708 2001-08-21
-57-
Example 23
Preparation of (E)-N-Cyclopropyl-2-(6-fluoro-3-ethyl-1-
indanylidene)acetamide
a) Preparation of Ethyl 3-(4-fluorophenyl)pentenoate
A solution of butyllithium, 1.6M in hexanes (230m1,0.368 mol, Aldrich) was
added dropwise over 0.5 hr, with rapid mechanical stirnng, to a solution of
triethyl phosphonoacetate (78.9g, 0.351 mol, Aldrich) in tetrahydrofuran
(800m1, anhydrous, Aldrich) at <5°C while blanketed with a nitrogen
atmosphere. This solution was stirred for an additional 0.25 hr and cooled to
-5°C with a methanol:ice bath and a solution of 4'-fluoropropiophenone
(50g,0.328 mol, Aldrich) in tetrahydrofuran (SOmI) was then added in one
portion. Stirring was continued for 18 hr without additional cooling. The
solution was concentrated to a golden yellow sludge by spin evaporation in
vacuo and diluted to IOOOmI with ethyl acetate. After washing with deionized
water (3 x 100 ml) this solution was concentrated by spin evaporation in
vacuo.. Distillation at reduced pressure gave 45.65g (63%) of ethyl 3-(4-
fluorophenyl)pentenoate as a mixture of (E) and (Z) isomers (ratio 1:1)
contaminated with 30% of triethyl phosphonoacetate as a clear liquid, b.p.
140-146°C at aspirator pressure:
b) Preparation of Ethyl 3-(4-Fluorophenyl)valerate
A mixture of ethyl 3-(4-fluorophenyl)pentenoate (45.65 g,0.137 mol) and 10%
palladium on carbon (0.86 g, Aldrich) in 95 % ethanol was shaken under 4
atm hydrogen pressure in a Parr hydrogenator for 1.5 hr. The mixture was
filtered and concentrated by spin evaporation in vacuo. Fractional
distillation
gave 38.95g (63%) of ethyl 3-(4-fluorophenyl)valerate as a clear oil
contaminated with 29% triethyl phosphonoacetate, b.p., 133-142°C at 17
mm
Hg:
CA 02162708 2001-08-21
-5 8-
c) Preparation of 3-(4-Fluro~henyl)valeric acid
This compound was prepared in an analogous manner to Example 18c with
the replacement of 3-(4-fluorophenyl)butyrate with ethyl 3-(4-
fluorophenyl)valerate (38.95 g 0.144 mol, containing 29% triethyl
phosphoneacetate) and using an excess of 85% potassium hydroxide (18.05 g,
0.273 mol, Mallinckrodt). The dichloromethane layers were combined,
washed with deionized water (50 ml) and concentrated by spin evaporation in
vacuo. The residue was crystallized from hexanes to give 23.47 g (83%) of 3-
(4-flurophenyl)valeric acid as a white crystalline solid: NMR (DMSO-db): d
12 (s, 1H), 7.28-7.24 (m, 2H), 7.14-7.08 (m, 2H), 2.91-2.89 (m, 1H), 2.64-
2.41 (m, 2H), 1.66-162 (m, 1H), 1.56-1.51 (m, 1H), 0.71 (t, 3H, J=7.3 Hz).
d) Preparation of 3-(4-Fluorophenywaleroyl chloride
This compound was prepared in an analogous manner to Example 18d with
the replacement of 3 -(4-fluorophenyl)butyric acid with 3-(4
fluorophenyl)valeric acid (23.47g, 0.120 mol). The volatiles were removed by
spin evaporation in vacuo with the addition of dichloromethane (6 x 250 ml)
during concentration to give 25.25 g (98%) of 3-(4-fluorphenyl)valeroyl
chloride as a golden yellow liquid:
NMR (DMSO-db): d 7.3-7.22 (m, 2H), 7.15-7.06 (m, 2H), 2.98-2.73 (m, 1H),
2.66-2.38 (m, 2H), 1.74-1.37 (m,2H), 0.70 (t, 3H, J=7.2 Hz).
e) Preparation of 3-Ethyl-6-fluoro-1-indanone
This compound was prepared in an analogous manner to Example 18e with
the replacement of 3-(4-fluorophenyl)butyryl chloride with 3-(4-
fluorophenyl)valeroyl chloride(25.27g, 0.118 mol). The dichloromethane
extractions were combined, washed with deionized water (100 ml) and
concentrated by spin evaporation in vacuo The residue was chromatographed
on Silica Gel 60 using a step gradient going from hexanes to ethyl acetate:
CA 02162708 2001-08-21
-S 9-
hexanes/1:1. Fractions containing 3-ethyl-6-fluoro-1-indanone were combined
and concentrated by spin evaporation in vacuo with dichloromethane (2 x 150
ml) added during concentration to give 17.488 (83%) of 3-ethyl-6- fluoro-1-
indanone as a canary yellow syrup: NMR (DMSO-d6): d 7.74-7.7 (dd, 1H,
JHF=8.4 Hz, JHH=4.8 Hz), 7.6-7.53 (ddd, 1H, JHF=9.0 Hz, JHH=9.0 Hz and 2.7
Hz), 7.37 (dd, 1H,JHF=7.8 Hz, JHH= 2.4 Hz), ~3.3(m, 1H, partially obscured by
water), 2.88 (dd,lH, Jgem=19.2 Hz, J=7.6 Hz), 2.39 (dd, 1H, Jgem=19.2 Hz,
J=2.4Hz), 1.98-1.90 (m, 1H), 1.54-1.44 (m, 1H), 0.90 (t, 3H, J=7.3 Hz).
f) Preparation of cis and traus Ethyl 2-(3-ethyl-6-fluoro-1-hydroxy-1-
indanyl)acetate.
This compound was prepared in an analogous manner to Example 18f with
replacement of 6-fluoro-3-methyl-1-indanone with 3 -ethyl-6-fluoro-1-
indanone (17.3 g, 0.097 mol). Removal of the volatiles from the workup
solution gave 25.178 (97%) of cis and traps ethyl 2-(3-ethyl-6-fluoro-1-
hydroxy-1-indanyl)acetate as a golden yellow oil: NMR (DMSO-db): d 7.23-
7.21 (m, l H), 7.13-7.06 (m, 2H), 5.48 (s, 1 H), 4.0 (q, 2H, J = 7.2 Hz), 2.90-
2.82 (m, 1H), 2.80-2.73 (m, 1H), 2.70-2.55 (m, 2H), 2.04-1.9 (m,lH), 1.83-
167 (m, 1H), 1.46-1.28 (m,lH), 1.11 (t,3H, J=7.1 Hz), 0.95 (t, 3H, J=7.3 Hz).
g) Preparation of (E~-2-~3 -Ethyl-6-fluoro-1-indanylidene)acetic acid
This compound was prepared in an analogous manner with Example 188 with
the replacement of ethyl 2-(6-fluoro-1-hydroxy-3-methyl-1-indanyl)acetate
with ethyl 2-(3 -ethyl-6-fluoro-1-hydroxy-1-indanyl)acetate (24.858, 0.093
mol). Removal of the volatiles from the workup gave a beige residue.
Recrystallization from dichloromethane-hexanes gave 12.91 g (63%) of (E)-2-
(3-ethyl-6-fluoro-1-indanylidene)acetic acid as a white crystalline solid,
m.p.,
145-148°C:
h) Preparation of (E)-2-(3-ethyl-6-fluoro-1-indan lidene)acetyl chloride.
This compound was prepared in an analogous manner to Example 18h with
CA 02162708 2001-08-21
-60-
replacement of (E)-2-(6-fluoro-3-methyl-1-indanylidene) acetic acid with (E)-
2-(3-ethyl-6-fluoro-1-indanylidene)acetic acid (5.7g. 25.88mmo1). The
product residue was dissolved in dichloromethane and used without
purification in Example 26.
i) Preparation of~(E)-N-Cyclopropvl-2-(6-fluoro-3 -ethyl-1-
indanylidene)acetamide.
This compound was prepared in an analogous manner to Example 20 with the
replacement of (E)-2-(6-fluoro-3-methyl-1-indanylidene)acetyl chloride with
(E)-2-(3-ethyl-6-fluoro-1-indanylidene)acetyl chloride (3.1 8g, 13.3 mmol).
Recrystallization from dichloromethane-hexanes gave 2.24g (65%) of (E)-N-
cyclopropyl-2-(6-fluoro-3-ethyl-1-indanylidene)acetamide as a white
crystalline solid, mp.,143-147°C:
Exam 1p a 24
Preparation of (E) -N-Cyclopro~yl-2~6-fluoro-3-propyl-1-
indan ly)acetamide.
a) Preparation of 4' fluorobutyrophenone
Aluminum chloride (139g, 1.04 mol) was added to a solution of butyryl
chloride (55.45g, 0.520 mol, Aldrich) in dichloromethane (SOOml) stirnng
under a nitrogen atmosphere at 25°C. A solution of fluorobenzene (50.1
g,
0.521 mol, Aldrich) in dichloromethane was added and stirnng was continued
for 18h The reaction solution was poured over ice and extracted with
dichloromethane (3 x 400m1). The combined dichloromethane extractions
were washed with deionized water (2 x 250 ml), 1.0 N hydrochloric acid
(SOOmI), saturated sodium bicarbonate solution (2 x SOOmI) and deionized
water ( 4 x 250 ml),concentrated by spin evaporation in vacuo. This material
was combined with material from a similar preparation (using 0.26 mol of
CA 02162708 2001-08-21
-61-
fluorobenzene) for distillation. Distillation at reduced pressure gave 69.27g
(53%) of 4'-fluorobutyrophenone as a pale yellow liquid which later partially
crystallized, b.p. 108-1120C at 30 millitorr:
NMR (DMSO-d6): d 8.03 (q, 2H, J= 9.0 Hz and 5.6 Hz), 7.31 (t, 2H, J= 8.9
Hz), 2.97 (t, 2H, J- 7.OHz), 1.65-1.55 (m, 2H), 0.91 (t, 3H, J=7.3 Hz).
b) Preparation of Eth~4-fluorophenvl)hexenoate
A solution of butyllithium, 2.5M in hexanes (166 ml, 0.416 mol, Aldrich) was
added dropwise over 0.5 hr, with rapid mechanical stirring, to a solution of
triethyl phosphonoacetate (93.2g, 0.416 mol, Aldrich) in tetrahydrofuran
(700m1, anhydrous,Aldrich) at <5°C while blanketed with a nitrogen
atmosphere. This solution was stirred for an additional 0.25 hr and cooled to -
5°C with a methanol:ice bath and a solution of 4'-fluorobutyrophenone
(69 g,
0.416 mol, Aldrich) in tetrahydrofuran (150m1) was then added in one
portion. Stirnng was continued for 18 hr without additional cooling. The
solution was concentrated to a dark camel sludge by spin evaporation in vacuo
and diluted to 600 ml with deionized water. The aqueous solution was
extracted with dichloromethane (5 x 500 ml) and the dichloromethane was
concentrated by spin evaporation in vacuo. Distillation at reduced pressure
gave 58.5g (60%) of ethyl 3-(4-fluorophenyl) hexenoate as a mixture of (E)
and (Z) isomers (ratio 1:1) as clear liquid, b.p. 140-150°C at
aspirator
pressure.
c) Preparation of Ethy~4-Fluoro~henyl hexanoate
A mixture of ethyl 3-(4-fluorophenyl)hexenoate (58.12 g, 0.246 mol) and 10%
palladium on carbon (1.1 g, Aldrich) in 95 % ethanol was shaken in a Parr
hydrogenator under a pressure of 2-4 atm of hydrogen for 0.75 hr. The
mixture was filtered and concentrated by spin evaporation in vacuo to give
58.4 g (99.6%) of ethyl 3-(4-fluorophenyl)hexanoate as a clear liquid, NMR
(DMSO-d6): d 7.27-7.20 (m, 2H), 7.12-7.04 (m, 2H), 3.91 (q, 2H, J=7.2Hz),
2.99 (m, 1H), 2.66-2.59 (m, 1H), 2.52-2.44 (m, 1H, partially obscured by
DMSO), 1.59-1.46 (m, 2H), 1.13-1.07 (m, 2H), 1.02 (t, 3H, J= 7.2Hz), 0.78 (t,
CA 02162708 2001-08-21
-62-
3H, J= 7.5 Hz).
d) Preparation of 3-(4-Fluorophenyl)hexanoic acid
This compound was prepared in an analogous manner to Example 65c with
the replacement of 3 -(4-fluorophenyl)valerate with ethyl 3(4-fluorophenyl)
hexanoate (58 g, 0.243 mol). The dichloromethane layers were combined,
washed with deionized water (250 ml) and concentrated by spin evaporation
in vacuo. The residue was coevaporated with hexanes (200 ml) to give 46.81 g
(92%) of 3-(4-fluorophenyl)hexanoic acid as a pale yellow oil:
e) Preparation of 3-(4-Fluorophenyl)hexanoyl chloride
This compound was prepared in an analogous manner to Example 18d with
the replacement of 3-(4-fluorophenyl)butyric acid with 3-(4-
fluorophenyl)hexanoic acid (46.5g, 0.222 mol). The volatiles were removed
by spin evaporation in vacuo with the addition of dichloromethane (5 x 250
ml) during concentration to give 50.01 g (99%) of 3-(4-fluorophenyl)hexanoyl
chloride as a golden yellow liquid:
fj Preparation of 6-fluoro-3-propyl-1-indanone
This compound was prepared in an analogous manner to Example 18e with
the replacement of 3-(4-fluorophenyl)butyryl chloride with 3-(4-fluorophenyl)
hexanoyl chloride (49.95 g, 0.218 mol). The dichloromethane extractions were
combined, washed with deionized water (250 ml) and concentrated by spin
evaporation in vacuo. The residue was coevaporated with dichloromethane
(100 ml) to give 41.26 g (98%) of 6-fluoro-3-propyl-1-indanone as a golden
yellow syrup:
g) Preparation of cis and traps Ethyl 2 ~6-fluoro-1-hydroxy-3-propel-1-
indanyllacetate.
This compound was prepared in an analogous manner to Example 18f with
CA 02162708 2001-08-21
-63-
replacement of 6-fluoro-3 -methyl-1-indanone with 6-fluoro-3 -propyl-1-
indanone (40.75 g, 0.212 mol). Removal of the volatiles from the workup
solution gave 57.48 g (97%) of cis and trans ethyl 2-(6-fluoro-1-hydroxy-3-
propyl-1-indanyl)acetate as a golden yellow oil:
h) Preparation of (E)-2-(6-Fluoro-3 ~propyl-1-indan~rlidene)acetic acid
This compound was prepared in an analogous manner with Example 18g with
the replacement of ethyl 2-(6-fluoro-1-hydroxy-3-methyl-1-indanyl)acetate
with ethyl 2-(6-fluoro-1-hydroxy-3-propyl-1-indanyl)acetate (57.12 g, 0.204
mol). Removal of the volatiles from the workup gave a golden yellow residue.
The residue was slurried in hexanes to give 24.49 g (51 %) of (E)-2-(6-fluoro-
3-propyl-1-indanylidene)acetic acid as a white crystalline solid, m.p., 141-
144°C:
NMR (DMSO-d6):
i) Preparation of (E)-2-(6-Fluoro-3-propyl-1-indanylidenJ)acetyl chloride.
This compound was prepared in an analogous manner to Example 18h with
replacement of (E)-2-(6-fluoro-3-methyl-1-indanylidnene)acetic acid with (E)-
2-(6-fluoro-3 -propyl-1-indanylidene) acetic acid (15.01 g, 64. 07mmo1). The
product residue was dissolved in dichloromethane and used without
purification in Example 27j.
j) Preparation of (E)-N-C~propyl-2-(6-fluoro-3-propel-1-
indanylidene)acetamide.
This compound was prepared in an analogous manner to Example 20 with the
replacement of (E)-2-(6-fluoro-3-methyl-1-indanylidene)acetyl chloride with
(E)-2-(6-fluoro-3-propyl-1-indanylidene)acetyl chloride (3.26 g, 0.013 mol).
The volatiles were removed by spin evaporation in vacuo to give a golden
yellow oil. The oil was chromatographed on Silica Gel 60 with a step gradient
CA 02162708 2001-08-21
-64-
ofhexanes to ethyl acetate-hexanes (1:1). Fractions containing (E)-N-
cyclopropyl-2-(6-fluoro-3-propyl-1-indanylidene)acetamide were combined
and concentrated by spin evaporation in vacuo with hexanes (4 x 250m1)
added during concentration to give 2.09 g (59%) of (E)-N-Cyclopropyl-2-(6-
fluoro-3-propyl-1-indanylidene)acetamide as a white powdery solid, m.p. 94-
97°C:
Example 25
Preparation of (Z)-2-(6-Fluoro-1-indanylidene~acetamide
A solution of (E)-2-(6-fluoro-1-indanylidene)acetamide (20g,104.6 mmol) in
dichloromethane:methanol (3: 1) (1000m1) was irradiated by an Canrad-Hanovia
quartz, mercury-vapor photochemical immersion lamp, 450 wattts (Ace Glass,
7825-
35) for
O.Sh. The volatiles were removed by spin evaporation in vacuo to give a beige
residue. This residue was chromatographed on Silica Gel 60 using a step
gradient
going from ethyl acetate: hexanes(l:l) to ethyl acetate:ethanol(1 :1).
Fractions
containing (Z)-2-(6-fluoro-1-indanylidene)acetamide were combined and
concentrated by spin evaporation in vacuo. The resulting solid was slurried in
hexanes
to give 7.52g (37%) of (Z)-2-(6-fluoro-1-indanylidene)acetamide as a white
crystalline solid, m.p., 175-177°C:
Example 26
Preparation of (E)-~4 6-Difluoro-3-oxo-1-indanylidene)acetamide
a) Preparation of 2-4-Dicarbethox~3,5-difluor~henyl)-5-hydroxy-5-methyl-
1-cvclohexanone
Slightly warm, liquid 3,5-difluorobenzaldehyde (5.0g, 0.0352 mol, Aldrich),
95% ethanol (1.75 ml), and piperidine (0.7 ml) were added, with stirring, to
ethyl acetoacetate (9.2g, 0.0704 mol, Aldrich). The solution was stirred until
homogeneous and was then placed in a water bath to control the slightly
CA 02162708 2001-08-21
-65-
exothermic reaction. After 4 hrs, the crystalline mass was dissolved in warm
dichloromethane (100 ml). Dilution with hexanes (300 ml) gave a turbid
solution. After standing for 24 hrs, the crystalline product was collected by
filtration and washed with hexanes to give 8.0g (59%) of 2,4-dicarbethoxy-3-
(3, 5-difluorophenyl)-5-hydroxy-5-methyl-1-cyclohexanone: m.p., 185 -
186°C;
b) Preparation of 3-(3,5-Difluoropheny~lutaric acid
To a hot (95°C) solution prepared from sodium hydroxide (322g, 8.1
mol) and
deionized water (322 ml) was added a mixture of 2,4-diacetyl-3-(3,5
difluorophenyl)-5-hydroxy-5-methyl-1 -cyclohexanone (43 g, 0.112 mol) in
ethanol (322 ml) with rapid stirring. The resulting mixture was refluxed for 4
hrs using an oil bath at 140°C. The ethanol was removed by spin
evaporation
in vacuo and the resulting slurry was cooled with an ice bath, and
concentrated hydrochloric acid (12N) was added to adjust the pH to
approximately 1. The precipitate was dissolved by the addition of water, and
this aqueous solution was extracted with ethyl acetate (total volume 1500 ml).
The ethyl acetate extracts were combined, washed with water, dried with
MgS04, and the volatiles were removed by spin evaporation in vacuo.
Recrystallization of the residue from dichloromethane and hexanes gave 9.
l Og (33%) of 3-(3,5-difluorophenyl)glutaric acid: m.p., 170 - 172°C;
c) Preparation of 2-(4. 6-Difluoro-3 -oxo- 1 -indanyl)acetic acid
This compound was prepared in an analogous manner to that of Example 22d
with the replacement of 3-(3-fluorophenyl)glutaric acid with 3-(3,5
difluorophenyl)glutaric acid (9.028, 36.9mmo1) and an increase of the heating
time from 10 min to 30 min. Chromatography of the collected product on a
column of Silica Gel 60 (51 x 450 mm) with methanol: dichloromethane
(4:96) gave a material which was recrystallized from water to give 1.93 g
(23%) of 2-(4,6-difluoro-3-oxo-1-indanyl)acetic acid: m.p., 170 - 172
°C;
d) Preparation of 2-(4 6-difluoro-3-oxo-1-indanyl acet~chloride
CA 02162708 2001-08-21
-66-
This compound was prepared in an analogous manner to that of Example 22e
with the replacement of 2-(6-fluoro-3-oxo-1-indanyl)acetic acid with 2-(4,6-
difluoro-3-oxo-1-indanyl)acetic acid (3.858, 0.017 mol). The 2-(4,6-difluoro-
3-oxo-1-indanyl)acetyl chloride thus prepared was used without additional
purification or analysis.
e) Preparation of 2-(4,6-Difluoro-3-oxo-1-indanyl)acetamide
This compound was prepared in an analogous manner to that of Example 22f
with the replacement of 2-(3-fluoro-3-oxo-1-indanyl)acetyl chloride with 2-
(4,6 difluoro-3-oxo-1-indanyl)acetic acid (4.28, l7mmol). After
chromatography, recrystallization twice from dichloromethane:hexanes gave
2.88 (77%) of 2-(4,6 Difluoro-3-oxo-1-indanyl)acetamide: m.p., 155 -
157°C;
f) Preparation of (E)-2-(4,6-Difluoro-3-oxo-1-indanylidene)acetamide
A mixture of 2-(4,6-difluoro-3-oxo-1-indanyl)acetamide (1 .0g, 0.0044 mol),
N-bromosuccinimide (0. 9508, 0.00533 mol), 2,2'-azobis(2-
methylpropionitrile) (0.3508, 0.00213 mol, Kodak), tetrachloromethane (SO
ml) and benzene (SO ml)was heated with an oil bath at 120°C for 1 hr.
The
reaction solution was diluted with dichloromethane, slurried with Silica Gel
60, and the volatiles were removed by spin evaporation in vacuo. This silica
was then applied to a column of Silica Gel 60 (51 x 450 mm) wet with
dichloromethane and the product was removed by elution with
methanol:dichloromethane (2:98). The volatiles were removed by spin
evaporation in vacuo to give 0.6138 of a residue. This residue was
recrystallized from methanol to give 0.3028 (31 %) of (E)-2-(4,6-difluoro-3-
oxo-1-indanylidene)acetamide: m.p., 250°C (dec.);
Example 27
Preparation of (E)-2-(4-6-difluoro-3 -hydroxy-1-idanylidene)acetamide
CA 02162708 2001-08-21
-67-
A suspension of (E)-2-(4,6-difluoro-3-oxo-1-indanylidene)acetamide (0.100 g,
0.45 mmol) and sodium borohydride (0.017 g, 0.45 mmol) in 95% ethanol was
stirred at ambient temperature for 2 hours. The mixture was cooled in an ice
bath and quenched with 0. 1N hydrochloric acid (3 mL). The ethanol was
evaporated in vacuo, and the residue was dissolved in ethyl acetate (50 mL),
washed successively with water (2 x30mL) and brine (30 mL), dried over
sodium sulfate, filtered, and evaporated in vacuo. The residue was washed
successively with cold ethyl acetate, hexane, and diethyl ether to give 0.046
g
(45%) of (E)-2-{4,6-difluoro-3-hydroxy-1-indanylidene)acetamide as a white
solid; mp 232-237°C (dec.)
1H-NMR(DMSO-d6): 87.06-7.42 (m, 4H), 6.49 (s, IH), 5.52 (d, 1H), 5.30
(m, l H), 3.44 (m, l H), 3.03 (d, 1 H).
The following compounds were prepared by methods similar to those of the
indicated Examples
CA 02162708 2001-08-21
-68-
Ex Compounds M.p.(C) Method
No (Ex.
No.)
28 (E)-2-(6-Fluoro-3-ethyl-1-indanylidine)-N-methylacetamide141-143 26
29 (E)-2-(6-Fluoro-3-ethyl-1-indanylidine)acetamide163-166 15
30 (E)-2-(6-Fluoro-3-ethyl-1-indanylidine)-N-isopropylacetamide127-130 26
31 (E)-2-(6-Fluoro-3-ethyl-1-indanylidine)-N,N-dimethylacetamide79-82 26
32 (E)-2-(6-Fluoro-3-propyl-1-indanylidine)-N-methylacetamide105-107 26
33 (E)-2-(6-Fluoro-3-propyl-1-indanylidine)-N,N-dimethylacetaamide95-97
26
34 (E)-2-(6-Fluoro-3-propyl-1-indanylidine)-N-isopropylacetamide108-110 26
35 (E)-2-(6-Fluoro-3-propyl-1-indanylidine)acetamide167-169 15
36 (E)-4-(2-(6-Fluoro-1-indanylidine)acetyl)moipholine133-136 4
37 (E)-2-(6-Fluoro-1-indanylidine)-N-isopropylacetamide143-145 4
38 (E)-2-(6-Fluoro-1-indanylidine)-N-methylacetamide201-205 4
39 (E)-N-Cyclobutyl-2-(6-fluoro-1-indanylidine)acetamide137-139 4
40 (E)-2-(6-Fluoro-1-indanylidine)-N-propylacetamide82-84 4
41 (E)-2-(6-Fluoro-1-indanylidine)-N,N-dimethylacetamide74-77 4
42 (E)-N-Cyclopropyl-2-(5,6-difluoro-1-indanylidine)acetamide169-171 4
43 (E)-2-(5,6-Difluoro-1-indanylidine)-N-methylacetamide209-211 4
44 (E)-2-(5,6-Difluoro-1-indanylidine)acetamide165-167 1
45 (E)-2-(5,7-Difluoro-1-indanylidine)acetamide161-162 1
46 (E)-N-Cyclopropyl-2-(5,7-difluoro-1-indanylidine)acetamide145-147 4
47 (E)-2-(5,7-Difluoro-1-indanylidine)-N-methylacetamide193-195 4
48 (E)-2-(4,6-Difluoro-1-indanylidine)-N-isopropylacetamide167-170 4
49 (E)-2-(4,6-Difluoro-1-indanylidine)-N,N-dimethylacetamide105-106 4
50 (E)-2-(4,6-Difluoro-1-indanylidine)-N-ethylacetamide130-132 4
51 (E)-N-Ethyl-2-(4,6-difluoro-1-indanylidine)-N-methylacetamide96-98 4
52 (E)-2-(4,7-Difluoro-1-indanylidine)acetamide167-169 5
53 (E)-2-(4,5-Difluoro-1-indanylidine)-N,N-dimethylacetamide195-197 5
54 (E)-N-Cyclopropyl-2-(4,5-difluoro-1-indanylidine)acetamide135-137 4
55 (E)-N-Cyclopropyl-2-(4,7-difluoro-1-indanylidine)acetamide134-136 4
56 (E)-2-(4,6-Difluoro-1-indanylidine)-N-methylacetamide181-183 4
CA 02162708 2001-08-21
-69-
Pharmaceutical Compositions
In the following Examples 57 to 61 the "Active Ingredient" is a compound of
formula (I) or a pharmaceutically acceptable salt or solvate thereof.
Example 57
Tablet Compositions
The following compositions A, B and
C are prepared by wet granulation
of the
ingredients with a solution of povidone,followed by of magnesium
addition stearate
and compression
m /tablet m /t~ ablet
(a) Active ingredient 250 250
(b) Lactose B.P. 210 26
(c) Povidone B.P. 15 9
(d) Sodium Starch Glycollate 20 12
(e) Magnesium Stearate 5 3
500 300
CA 02162708 2001-08-21
-70-
Composition B
m./tablet m /tg ablet
Active ingredient 250 250
(b) Lactose 150 -
(c) Avicel PH 101 60 26
(d) Povidone B.P. 15 9
(e) Sodium Starch Glycollate20 12
(f) Magnesium Stearate 5 3
500 300
Composition C
mg/tablet
Active ingredient 100
Lactose 200
Starch 50
Povidone 5
Magnesium Stearate 4
359
The following compositions, D and E, are prepared by direct compression of the
admixed ingredients. The lactose in composition E is of the direct compression
type
(Dairy Crest -"Zeparox").
CA 02162708 2001-08-21
-71-
Composition D
mg/tablet
Active ingredient 250
Pregelatinized Starch NF 15 150
400
Composition E
mg/tablet
Active ingredient 250
Lactose 150
Avicel 100
500
Composition F (Controlled Release Formulation
The composition is prepared by wet granulation of the ingredients (below) with
a
solution of povidone followed by the addition of magnesium stearate and
compression.
m /t
(a)Active ingredient 500
(b)Hydroxypropylmethylcellulose 112
(Methocel K4M Premium)
(c)Lactose B.P. 53
(d)Povidone B.P. 28
(e)Magnesium Stearate 7
700
CA 02162708 2001-08-21
-72-
Example 58
Capsule Compositions
Composition A
A capsule composition is prepared by admixing the ingredients of Composition
D in Example 57 above and filling into a two-part hard gelatin capsule.
Composition B (infra) is prepared in a similar manner.
Composition B
mg/capsule
(a) Active ingredient 250
(b) LactoseB.P. 143
(c) Sodium Starch Glycollate 25
(d) Magnesium Stearate 2
_ 420
Composition C
m.~/capsule
(a) Active ingredient 250
(b) Macrogol 4000 B.P. 350
600
CA 02162708 2001-08-21
-73-
Composition D
mg/capsule
Active ingredient 250
Lecithin 100
Arachis Oil 100
450
Capsules of composition D are prepared by dispersing the active ingredient in
the
lecithin and arachis oil and filling the dispersion into soft, elastic gelatin
capsules.
Composition B
mg/capsule
(a)Active ingredient 100
(b)Lactose 300
(c)Magnesium Stearate 2
(d)Sodium Lauryl Sulfate 2
(e)Sodium Starch Glycollate 50
(f)Talc, USP 25
479
A capsule composition is prepared by micronizing the active ingredient using a
GEM-T Type 1047 Jet Mill and admixing with the remaining ingredients of
Composition B and filling into a two-part hard gelatin capsule.
Composition F (Controlled Release Capsule)
The following controlled release capsule composition is prepared by extruding
ingredients a, b and c using an extruder, followed by spheronization of the
extrudate and drying. The dried pellets are then coated with release-
controlling
membrane (d) and filled into a two-piece, hard gelatin capsule.
CA 02162708 2001-08-21
-74-
mg/capsule
(a) Active ingredient 250
(b) Microcrystalline Cellulose 125
(c) Lactose B.P. 125
(d) Ethyl Cellulose 13
513
Example 59
Injectable Composition
Active ingredient 0.200 g
95% Ethanol and PEG 400, 1:1 ratio
Sterile water q.s. to 10 mL
The active ingredient is dissolved in 95% Ethanol and PEG 400 (1:1). The
batch is then made up to volume with the water and filtered through a sterile
micropore filter into a sterile 10 mL amber glass vial (type 1 ) and sealed
with
sterile closures and overseals.
CA 02162708 2001-08-21
-75-
Example 60
Syrup
Active ingredient 0.25 g
Sorbitol Solution 1.50 g
Glycerol 2.00 g
Sodium Benzoate 0.005 g
Flavor, Peach 17.42.3169 0.0125 mL
Purified Water q.s. to 5.00 mL
The active ingredient is dissolved in a mixture of the glycerol and most of
the
purified water. An aqueous solution of the sodium benzoate is then added to
the
solution, followed by addition of the sorbitol solution and finally the
flavor.
The volume is made up with purified water and mixed well.
Example 61
Suppository
mg/suppositor
Y
Active ingredient 250
Hard Fat, B.P. (Witepsol H15 - Dynamit NoBel) 1770
2020
One-f fth of the Witepsol H 15 is melted in a steam jacketed pan at
45°C
maximum. The active ingredient is sifted through a 200 ~,M sieve and added to
the molten base with mixing, using a Silverson fitted with a cutting head,
until
smooth dispersion is achieved. Maintaining the mixture at 45°C, the
remaining
Witepsol H 15 is added to the suspension and stirred to ensure a homogenous
CA 02162708 2001-08-21
-76-
mix. The entire suspension is passed through a 250 ~,M stainless steel screen
and, with continuous stirring, is allowed to cool to 40°C. At a
temperature of
38°C to 40°C, 2.02 g of the mixture is filled into suitable, 2
mL plastic molds.
The suppositories are allowed to cool to room temperature.
Example 62
Pessaries
mg/pessary
Active ingredient 250
Anhydrate Dextrose 380
Potato Starch 363
Magnesium Stearate 7
1000
The above ingredients are mixed directly and pessaries prepared by direct
compression of the resulting mixture.
Example 63
Central Muscle Relaxant Activity
Central muscle relaxant activity of compounds of formula (I) was determined
using a Straub tail test based on that described by K.O. Ellis and J.F.
Carpenter,
Neuropharmacol, 13, 211 (1974).
The Straub tail test result is reported as an ED50 in mg/kg. The ED50 is
defined as the dose of compound administered, which prevents Straub tail in
50% of mice. The compound is administered by oral gavage 60 min. prior to
scoring.
CA 02162708 2001-08-21
The side effect potential of these compounds was determined using the mouse
rotorod test as described by G.D. Novak and J.M.-Zwolshei,
J.Pharmacological. Methods, 10, 175 (1983). Rotorod result is reported as
EDSp in mg/kg. The ED50 is the dose which causes 50% of the animals to fail
to maintain position on a cylinder rotating at 11 r.p.m.
Antagonism of morphine-induced Straub tail reflects central muscle relaxant
efficacy while failure in the rotorod test reflects sedation and
incoordination.
Determination of the ratio of rotorod failure to antagonism of morphine-
induced Straub tail is a means of assessing side effect liability of central
muscle
relaxants (G.D. Novak, Drug Dev Res 2, 383 (1982).
Compound of Straub Tail Rotorod Rotorod/Straub Tail
Example No_ .p oED50 ~~50
mg/kg. mg/kg_
1 51 88 1.7
54 79 1.5
Example 64
Anticonvulsant Activity
Anticonvulsant activity of compounds of formula (I) was determined using a
method described by Mehta et al., J.Med.Chem 24 ,465 (1981).
The anticonvulsant activity is reported as an ED50 in mg/kg. The ED50 for
protection against maximal electroshock-induced convulsions was the dose
which prevented hind limb extension in 50% of the animals. The ED50 for
protection against Metrazol-induced convulsions was the dose which prevented
convulsions in 50% of the animals.
CA 02162708 2001-08-21
Example 65
Anxiolvtic Activity
Anxiolytic activity of compounds of formula (I) was measured using the
method of Geller and Seifier, J.Psychopharmacolgia, l, 482 (1960) as modified
by Pollard and Howard, Psychopharmacology, 62, 117 ( 1979). Clinically
efficacious anxiolytics increase punished responding. The anxiolytic activity
of
the compound is reported as the lowest dose necessary to produce a significant
increase in punished responding in rats(MED).
Example 66
Antiinflammatory Activity
Compounds of formula (I) possess anti-inflammatory activity as demonstrated
using a modification of the standard carrageenan pleurisy assay as described
by
R. Vinegar, J.F., Traux, and J.L. Selph(Pro. Soc. Exp. Biol Med. 143:711-714,
1973). The rats used in these experiments were Lewis males, weighing 160-180
g, assigned to groups consisting of 5 animals. Test compounds were given to
fasted rats by oral gavage 0.5 hr prior to intrapleural injection of 50 mg
carrageenan. After 4 hr, the pleural exudate was collected and the edema
volume and cell number were determined. ED50 values were estimated by
linear regression analysis, and represent the doses at which a given drug
produced 50% inhibition of carrageenan-induced cell accumulation and edema
formation within the rat pleural cavity.
Compound of .p oED50 m~/kg
Example No. Cells Edema
1 21 20
16 12
CA 02162708 2001-08-21
-79-
Example 67
Established Adiuvant Arthritis
Compounds of formula (I) also exhibit chronic anti-inflammatory activity as
evidenced by inhibition of established adjuvant-induced polyarthritis in the
rat.
The procedures for this test have been described in detail by R. Vinegar, J.F.
Truax, J. L. Selph, A. Lea, and P.R. Johnston (J. Immunopharmacol. 1:497-
520, 1979). The rats used in these studies were female Lewis rats whose
starting weight was 190 ~ 10 g. Arthritic rats were assigned to treatment
groups
consisting of six animals each. Fed rats were dosed by oral gavage starting on
day 21 post adjuvant injection; therapy was continued until day 28. The
incidence and severity of arthritic lesions were assessed using a modification
of
the scoring procedure described by H.L.F. Currey and M. Ziff(JExp. Med.
121:185-203, 1968). Briefly, the bilateral joints were scored for erythema,
edema, and ankylosis as outlined below:
Joint Evaluated Arbitrary Joint Score (rank)
Ki Lett
ht
Wrist 0-4 0-4
Ankle 0-4 0-4
Tarsus 0-4 0-4
Metacarpals 0-4 0-4
Metatarsals 0-4 0-4
The maximal possible score per rat was 40. Experimental results were analyzed
by one-way ANOVA, followed by osp t hoc comparisons of treatment effects
versus untreated arthritic control using the Newman-Keuls test. The percent
inhibition of each drug-treated group was calculated from the mean relative to
the arthritic control. Compound of Example No.S significantly (p<0.01 )
lowered arthritic scores on days 22, 25, and 27 in rats with established
adjuvant
arthritis dosed b.i.d. with 50 mg/kg. Spleen weight and plasma fibrinogen were
measured postmortem on day 27 and were also significantly reduced (p<0.01).
CA 02162708 2001-08-21
-80-
Example 68
Mild Analgesia
Compounds of formula (I) possess mild analgesic activity as demonstrated
using a modification of the trypsin -induced rat hind limb hyperalgesia assay
as
described by R. Vinegar, J.F. Truax, J.L. Selph and P.R. Johnston (J.
Pharmacol, Meth., 23:51-61, 1990). The rats used in these studies were Lewis
male, weighing 160-180 g. and assigned to groups consisting of 5-6 animals.
Test compounds were given to fasted rats by oral gavage 0.5 hours' prior to
the
subplantar injection of 250 mg trypsin in one hind limb. One hour later the
rats
were evaluated for hyperalgesia using an F-shaped mechanical force clamp on
the injected hind limb metatarsal area. Latency (seconds) to the algesic
response (vocalization or flight) was determined, with 4 seconds being the
maximum latency allowed. ED50 values were estimated by linear regression
analysis and represent the dose at which a given drug extended the latency
response to produce 50% inhibition using the formula: (4 sec. - Control
Latency) - (4 sec. - Test Latency)/4 sec. -Control Latency X 100.
Compound of Example No. .p oED_50. mg/kg
4.0
Example 69
Stron~gesia
Compounds of formula (I) possess strong analgesic activity as demonstrated
using the phalanges algesic assay [a modification of the trypsin -induced rat
hind limb hyperalgesia assay as described by R. Vinegar, J.F. Truax, J.L.
Selph
CA 02162708 2001-08-21
-81-
and P.R. Johnston (J. Pharmacol. Meth. 23: 51-61, 1990)]. The rats used in
these studies were Lewis male weighing 160-180 g and assigned to groups of
5-6 animals. The phalanges algesic assay is an algesic test (no hyperalgesia)
in
which test compounds were given to fasted rats by oral gavage. One hour later
an F-shaped mechanical force clamp was applied to the phalanges of one hind
limb which initiated an algesic response(vocalization or flight). Latency
(seconds) to the algesic response was determined with 3 seconds maximum
allowed time. ED50 values were estimated by linear regression analysis and
represent the dose at which a given compound extended the latency response to
produce 50% inhibition using the formula: (3 sec. - Control Latency) - (3 Sec.
-
Test Latency)/3 sec. -Control Latency X 100.
Compound of Example No. .p oED_50. mg/kg
22
Example 70
Toxicity data
(i) Compound of Example 1
Single doses ( 15, 45, 100 or 250 mg/kg) were administered by oral
gavage to groups of four non-fasted male CD-1 mice (Charles River).
The maximum tolerated dose was greater than 250 mg/kg as there were
no deaths within the seven days post dosing.
(ii) Compound of Example 5
Single doses (5, 15, 40, 100, 250, 500 or 1000 mglkg) were
administered by oral gavage to groups of four non-fasted male CD-1
mice (Charles River). The maximum tolerated dose was greater than
1000 mg/kg as there were no deaths within the seven days post dosing.
CA 02162708 2001-08-21