Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~_ 2170~1
APPLICATION FOR PATENT
~v~lltols: Nir Navot and Nurit Eyal
Title: A MEl~IOD O~ DETERMIN~NG THE PRESENCE AND
QUAN~IFY~G THE NU~ER OF DI- AND
TRINUCLEOTIDE REPEATS AND AN INSTRUMENT AND
KITS THEREFOR
lO This is a con~iml~tion in part of U.S. patent Application No. 08/084,505 filed
July 1st, 1993, which is a con~inll~1;on in part of U.S. patent Application No.
07/919,872 filed July 27th, 1992, now abandoned.
~l~LD AND BACKGROUND OF THE INVENT~ON
The present inYenhon relates to ~e detell,lination of di- and ~inucleotide
repeat mutations in~rolved with increasing number of geneticslly inherited
diseases characterized by the expansion or the amplification of a core di- or
trinucleotide sequences.
More particularly, the present inven~on concems a sensitive method, an
automated ins~ument and kits for unequivocally quantifying the exact number
of di- and trinucleotide repeats in preselected geneac loci.
2s The present method, ins~urnent and kits are also useful for ~ete~.. ;.~; ~g
the number of head to tail repeat sequences of two or more nucleotide bases,
provided that the core sequence consists of no more than ~ree types of
nucleotide bases such æ, for example, ~enine (A), guanine (G) and cytosine
(C) in a core sequence composed of (AAGCGCA)n (n 2 1).
In recent years, increasing number of genetically inherited diseases were-
found to be associated with mutations desigrated unstable trinucleotide repeat
mutations in which a core sequence of three nucleotide bases is exp~n~e~ or
amplified, such that affected individuals and in some cases carriers, co.l~ai
3~ more repe~ an a~)p~elld~r healthy ones, in dle particular DNA locus
implicated with the disease.
2170~Sl
.~ 2
More r~ce.~ , a new cancer gene was discovered and wa~s shown to
cause segJnentC of DNA to be abnorm~11y l~pe~l4~ in pairs and/or triplets in
tumor cells of individuals caT~ying the cancer gene. In this case hundred of
thousands of short units of DNA are copied over and over again, pres...n~bly
s destroying the tumor cells ability to control their growth. This phenomenon issomewhat di~elent than the e~ ion of u~Lstable tnnucleotide ~e~eals in
L~c~ ce~ses since it occurs in many DNA loci within the same cell.
Nevertheless, there is a good reason to believe that both pher~om~n~ are
p op~e~ by similar mech~nicmc. See, Kolata G. Health/Science section, The
lO National Herald Tribune, Thursday, May 13, 1993.
So far, seven genetically tr~ncmitte~l ~lice~ces each involving a unique
genetic locus, have been implicated with trinucleotide repeat mutations. These
include: Fragile XA (A site, Mar~n Bell) syndrome (FRAXA); spinal and
bulbar m11sc 11~r atrophy, SBMA (Kermedy ~ice~ce); Myotonic dystrophy
(CursGhm~nn Steiner~ DM); H11ntington's disease (HD); Spinocerebellar ataxia
type 1 (SCAl); Fragile XE (E site) m~nt~l retardation (FRAXE MR); and
De.~to.ubral pallidoluysian atrophy (DRPLA). Since di- and trinucleotide
l~peals have been observed within or close to a number of additional human
20 genes by gene-bank searches, it is conceivable that di- and trinucleotide
amplifications may be involved in the c~1-s~tion of other genetic ~ice~ces as
well.
Fragile XA syndrome is an X chromosome linked recessive disorder with
2s incomplete penetrance. It is characterized by moderate to severe mental
r. t~ation and other phenotype characteristics, and is one of the most common
forms of mental retardation with an estim~tç~l incitlPnce of 1 in 1250 males andcoll~s~onding 1 in 2500 females (heterozygotes), rendering this disease one of
the most common human diseases and the most common form of f~mili~l
30 ~ tion. Fragile X chromosomes present their unique phenotype when
leukocyte cells carrying them are grown in culture under folate starvation. As
mentioned, ~he fragile X syndrome is char~cteri7~d by incomplete penetrance
hence (l) some males, r~ f~l~d to as no~nal tr~smittin~ males (NTMs), are
clinically nonnal but are infe.led to carry the genetic defect by a position in
3s pedigrees rendering them obligatory carriers; (2) one third of female caITiers
have evidence of mild mPntql ;.Y~ Genetic 1ink~ge studies effected by
restriction L~..~n~ Iength polymorphism analysis of illfolmative pedigrees; and
5omq~ic cell hybrid studies of hqmetçr chromosomes c~ying translocated
2170951
~ 3
se~entc of human fragile X chromosomes in cells grown in culture under
folate starvation, enabled the loc~li7~tion of the fragile X gene to chromosomalband q27.3 on the X chromosome (Xq27.3). Eventually the fragile X defective
gene, ~3esi~tÇ~ fragile X mental reta~dalion 1 (~MRl), was isolated via
5 positional cloning and led to the discovery of a highly polyrnorphic (CGG)n
sequence within its 5' untr~nsl~te~ region. Population and fragile X patients
scfeenillg revealed that healthy individuals are characterized by low numbers ofthe (CGG)n trinucleotide repeat (n = 6-52); carriers are characterized by
metlillm nwnbers of the (CGG)n tnnucleotide repeat (n = 50-200); and affected
o individuals are charactçri7e~ by high numbers of the (CGG)n l~inucleotide
repeat (n = 230-1000). When the (CGG)n tnnucleotide repeats of the FMRl
gene ex~-eeds app~o~;...~tely 230 repeats, the DNA of the entire 5' region of thé
gene l)eCOlUes abnorrnally methylated. This methylation ext~nds u~slle~n into
and beyond the promoter region and results in the transcriptional suppfession oflS the FMRl gene leading to the cessation of the FMRl protein production which
is probably the cause of the phenotype. See Annernieke J.M.H. (1991) Cell,
65:905-914; Pieretti M. (1991) Cell, 66:817-822; Caskey T.C. et al. (1992)
Science, 256:784-788.
Spinal and bulbar muscular atrophy (SBMA), like the fragile X
s~ndrome, is a rare X linked recessive genetic disorder characterized by
~-3ulthood onset of progressive mllsc~ r weakness of upper and lower
e ~ niLies which is secondary to neural degeneration. Affected males have
re~lce~ fertility and excessive development of the male m~mm~ry glands
2S (gynecomastia); female carriers have few or no s~",-ptoms. Genetic linkage
analysis of informative pedigrees enabled the loc~li7~tion of SBMA to
chromosome Xqll-12, the region where the gene encoding the androgen
feCeptO~ (AR) was previously loc~li7e~, rendering this gene a candidate for
SBMA. Studies of the AR gene revealed a highly polymorphic (CAG)n
trimlcleotide repeat, sitll~te~i in exon 1, encoding a variable polyglu~ e
stretch in the AR plole~ urther studies of the AR gene from normal and
SBMA ~ect~d individuals revealed that while low numbers of the (CAG)n
trinl~cleotide repeat (n = 12-34) characterize ap~alelllly healthy individuals,
high numbers of the (CAG)n trinucleotide repeat (n = 40-62) char~cteri7e
3s SBMA ~,cled individuals, while a carrier state is not yet known for this
mnt~tion The infll)pr~ee of the e.~.An-lecl pol~gl,~ .ine tract on ~he AR prote~is not yet established, nevertheless, gain of function, le~tlin~. to ~e SMBA
. 2170951
~_ 4
phenotype is ~uspecled. See, Albert R et al. (1991) Nature, 352:77-79; Caskey
T.C. et al. (1992) Science, 256:784-788.
Myotonic dy~ opl~r (DM) is an autosomal do~ina~t ~ice~ce
s characterized by myotonia, cardiac arrhythmi~c, cataracts, male balding, male
infertility (hypogon~licm)~ and other associated endocrinopathies. The rare
con~nit~l form of DM is associated with profound newborn hypotonia and
m~nt~l ret~rd~t;on. DM has a prevalence of 2.5-5.5 affected per 100,000
individuals. DM was mapped by genetic linkage to chromosome l9ql3.3 and
o the DM gene, ~çsign~te~ myotonin protein kinase (MT-PK), was isolated via
positional cloning and other molecular metho.ls. Further studies revealed a
polyrnorphic (GCT)n trinucleotide repeat situated in the 3' untr~ncl~te~ region
of the MT-PK gene. Analyses of the MT-PK gene from normal and DM
~ccled individuals revealed that while low numbers of the (GCT)n
lS trinucleotide repeat (n = 5-37) characterize ap~ elltly healthy individuals, high
mnnb~r5 of the (GCT)n trinucleotide repeat (n = 100 - >1000) characterize DM
affected individuals, while the carrier state is characterized by medium numbersof the (GCT)n tnnucleotide repeat (n - 50-100). Further studies have revealed
that expansion of the (GCT)n t~inucleotide repeat leads to increased MT-
20 PKmRNA stability, therefore to the production of more MT-PK protein
susl.cctedly le~ling directly or indileclly, to the DM phenotype. See, Fu Y.H.
et al. (1992) Science, 255:1256-1258; Caskey T.C. et al. (1992) Science,
256:784-788.
2s H~ tin~on's ~ice~se (HD) is a devastating late onset autosomal domin~rlt
neurodegenerative disorder characterized by progressive neurodegeneration
with personality dis~ ce, involuntary movements, cognitive loss and an
inexorable progression to death 15-20 years from time of onset. HD occurs with
a frequency of 1 in 10,000 individuals in most populations of C~lc~si~n
~escr-~-t The HD gene was loc~li7ed to chromosome 4pl6.3 by genetic linkage
analysis with polymorphic DNA markers. Recently, following 10 years of
s;~e rcsed.ch, the defective gene c~lsing HD, ~es~ te~ IT15, was
isolated and a polymorphic (CAG)~, trinucleotide repeat encoding a
poly~l~J~ e stretch, s;~v~te~l in exon 1 of the gene was discovered. It was
3s further found that the (CAG)n trinucleo~de repeat is P~p~n~e~ in HD
chromoso-..es (n = 42-100) as co...l.~. ed with no~nal chromosomes (n = 11-36),
pl. s ~ bly leading to IT15 ~-ot u~s gain of function, suspcdcdly lç~ling to theHD ~hen~,~ype. See, The H~mtin~on's di~e~se collabora~ve lcse~ch group
2170~51
. . ,
. ~_ 5
(1993) CelL 72:971-983; 7luhlk~ C. et al. (1993) Hum. Molec. Genet. 2:1467-
1469.
Spinocerebellar ataxia type 1 (SCAl) is a progressive late onset
5 autosomal ~omin~nt disorder characterized by ataxia, ophthalmoparesis and
vanable degree of motor we~knçss due to neurodegeneration of the cerebellum,
spinal chord and brain stem, leading to complete disability and death 10-20
years after onset. The SCA1 gene was loc~li7e~ to chromosome 6p22-p23 due
to strong genetic !inkage with the highly polymorphic HLA locus and other
0 polymorphic DNA markers. The defective gene c~usin~ SCAl, was isolated in a
yeast artificial chromosome contig and subcloned into cosmids. A polymorphic
(CAG)n trinucleotide repeat encoding a polygl~ -nine stretch, situ~te~ in exon
1 of the SCAl gene was discovered. It was further found that the (CAG)n
trinucleotide repeat is exp~ndefl in SCAl chromosomes (n = 43-81) as
co~ ared wi~ normal chromosomes (n = 19-36), presu,nably leading to SCAl
prot.,~s gain of function, suspectedly leading to the SCAl phenotype. See, Orr
H.T. et al. (1993) Nature Genetics, 4:221-226.
Fragile XE mental let~rdalion (FRAXE MR), like FRAXA is an X
20 chromosome linked recessive disorder with incomplete penetrance. It is
c~acte.i;ced by moderate to severe mental retardation and other phenotype
characteristics. Like FRAXA, FR~XE chromosomes present their unique
phenotype when leukocyte cells carrying them are grown in culture under folate
starvation. Genetic linkage studies enabled the loc~li7~tion of the FRAXE gene
2s to chromosome Xq28. Eventually the FRAXE gene was isolated via positional
cloning and led to the discovery of a highly polymorphic (GCC)n trinucleotide
repeat segrep, ~ with the llise~se. Population and FRAXE patients screening
revealed that healthy individuals are characterized by low numbers of the
(GCC)n trinucleotide repeat (n = 6-25); carriers are characterized by medium
30 numbers of the (GCC)n trinucleotide repeat (n - 116-133); and affected
individuals are characterized by high numbers of the (CGG)n trinucleotide
repeat (n = 200-850). When the (CGG)n trinucleotide repeat of the FRAXE
gene exceeds appro~ tely 200 repeats, the DNA of a CpG island located in
the trinucl~tide lepedls vicinity becomes abnormally methylated, presu",ably
35 leading to the secescion of the FRAXE ~rote,l, production, which is probably
~he cause ofthe plle~ol~rpe. See Knight S.J.L. et al. (1993) Cell, 74:127-134.
2170951
Delltalolubral pallidoluysian atrophy (DRPLA) is a late onset autosomal
do...;~ .t ntulodege~erative disorder, prevalent in Japan, characterized by a
varying combinations of progressive myoclonus, epilepsy, ataxia,
choreo~thetosic and d~menti~ Neuropathological changes consist of combined
~l.eg~nf~ration of the ~e-.tAtc,l~lbal and pallidoluysian systems of the centralnervous system. The ~1ise~se is further characterized by variable penertance,
even in a single family. T.ink~e analysis in DRPLA families enabled to localize
the DRPLA gene to chromosome 12pl2-13. The DRPLA gene was isolated via
- scree.-.ng for (CAG)n unstable trinucleotite repeat that was found to be located
o in exon 1 of the gene, encoding a variable polygl~ ;ne stretch in the DRPLA
protein. It was further found that the (CAG)n trinucleotide repeat is expanded in
DRPLA chromosomes (n = 49-75) as comparcd with normal chromosomes (n =
7-23), presllm~bly le~ding to DRPLA ~lole~,s gain of function, suspectedly
leading to the DRPLA phenotype. See, Nagafuchi S. et al. (1994) Nature
lS Genetics, 6:14-18; Koide R. el al. (1994) Nature Genetics, 6:9-13.
Because of the high frequency, variable penetrance and instability of
Fragile XA syndrome and other genetically inherited disorders associated with
~imlcleotide repeats e~p~ncion~ there is a widely recognized need for, and it
20 would be highly advantageous to have, a low cost method, demanding merely
non skilled personnel for its execution, that enables the efficient and accurate~lete....i~-~tion of the number of repeats in various genes.
Unlike the cornmon gene mutations (e.g., Cystic Fibrosis /~F508), which
2s are stable, that is, they are transmitted nnch~n~ed along the generations of
pedigrees, the situation is somewhat di~erent for the trinucleotide repeat
mutations which are charactcl;,e~l by instability, that is, when the number of
repeats exceeds a threshold value, these mutations have a tendency to expand
and inclllde a greater number of repeats (1) when vertically tr~nsmitted from
30 yale~ to children along genetic traits; and (2) when somatically transmitted to
ght~r cells iTl a given individual, a phenomena ~esiQn~ted somahc instability,
yielding mosaicism.
The two ~pes of instability chalactelizing tTinucleotide repeat mutations
3s will be exemplified herein for the fragile XA syndrome.
Fragile XA unstable alleles are observed in norm~ A~ g males
(NTMs) their as~ to.l,atic ~ ghters and s~ o,nalic male gran~l~hilds.
2170~51
When the number of trinucleotide repeats of such alleles was determined, it was
found to increase along generations, in one example from 82 in the NTM father
to 83 in the as~tQ~ tic d~v~hte~ (90 in a second as~ )to~--At;c ~n~ht~r) to
>200 in the ~ise~seA gr~ndcllildren. The 82, 83 and 90 repeats cor.~ i..g alleles
s are refe.l~d to as pre~ ;on alleles. It was a study of numerous families of this
type that pc...~;Ued a correlation of the phenomenon of anticipation (earlier ages
of onset or severeness in sl)ccessive ge~elalions) and the molecular events of
the (CGG)n eYp~ncion. NTMs carry numbers of CGG repeats outside the range
of normal and bellow those found in affected males. Such males transmit the
0 rc~eals to their prog.,~ with relatively small changes in the repeats number. on
the other hand fem~les who carry similar ~re.r...l~;on alleles are prone to bearplOge~t (male or female) with large e~cr~nsion of the repeats region. Thus, large
CGG amplification associated with fragile XA syndrome appears to be
pre~lon~inA~ a female meiotic event. See, Caskey T.C. et al. (1992) Science,
256:784-788.
Many fragile XA ~liceace~3 individuals were found to be mosaic in respect
with 1he number of the CGG trinucleotide repeats characterizing different cells
in their body, a phenomenon in~lic~ting somatic instability of expanded repeats.
~ stability, characterized by expansion of trinucleotide repeats, is
observed also in DM, HD, FRAXE, DRPLA and SCAl pedigrees. As opposed
to E~E~AXA. DM and FRAXE high risk alleles can expand to similar extent via
both male and female meiosis and to the best of our knowledge somatic
2~ mosaicism wa not yet observed in DM and FRAXE patients. High risk alleles
were not yet found for HD and DRPLA, that is, alleles of these lise~ses are
either ca~ying or not canying the flice~se Never~eless, HD repeats are also
unstable in more than 80% of meiotic tr~ncmi~sions but, on dle other hand, they
are c~a~aclelized by increasing, or alternatively, decrea g numbers of repeats
30 wi~ ~he largest illcr~ase occullulg in paternal tr~ncmicsion (Duyao M. et al.(1993) Nature Genetics, 4:387-392), whereas DRPLA alleles have a tendency to
increase in size along generations. See, Nagafuchi S. et al. (1994) Nature
Genetics, 6:14-18; Koide R. et al. (1994) Nature Gene~cs, 6:9-13.
3s All~,l~ts to correlate the size of trinucleotide repeat mutations and the
sevent~r of t~e ~csoc;s~ netic 3ice~ces were made for f~agile XA syndrome,
myotonic d~a~o~h~ e~.~A~ulubral pallidoluysian allo~Ly and spinocerebellar
ataxia type 1.
2170~1
For fragile XA, as expected, median IQ score was significantly lower for
fem~les carrying a fully eYp~n~e.l mutation (above 230 repeats) than for
females carrying a plf~ ;on (50-200 repeals) on one of their X
s chromosomes On the other hand, no significant relationship was found between
IQ score and number of CGG repeats, see, Taylor A.K. et al. (1994) JAMA,
271:507-514. Never~heless, it was found that prenatal DNA studies of the
number of CTG tnnucleotide repeats characte~ g myotonic dystrophy alleles
can improve the estim~tion of clinical severity; and that the number of CAG
0 trinucleotide repeats in spinocerebellar ataxia type 1 and de~ tol ubral
pallidoluysian atrophy is correlated with increased progression of the disease
(N~g~filc-lli S. et al. (1994) Nature Genetics, 6:14-18; Koide R et al. (1994)
Nature Genetics, 6:9-13; Orr H.T. et al. (1993) Nature Genetics, 4:221-226).
A~ ls to correlate between the size of trinucleotide repeat mutations
and the age of onset of H~mtin ton's disease resulted in finding a reversed
correlation confined to the upper range of trinucleotide repeat nurnbers (ca. 60-
100 le~eats), see, Andrew S.E. et al. (1993) Nature Genetics, 4:398-403.
Fu~ P .nore, for spinocerebellar ataxia type 1 and ~e~ tolubrâl pallidoluysian
20 allopLy (Nagafuchi S. et al. (1994) Nature Genetics, 6:14-18; Koide R. et al.(1994) Nature Genetics, 6:9-13), â direct correlation between the number of the
(CAG)n trinucleotide repeats expansion and earlier ages of onset was found.
Collectively, these data call for the development of a reliable, accurate
2s and easy to operate di- and trinucleotide repeats quantification method aimed at
post and prenatal ~ ~osis and prognosis.
Three basic methods are Cl~lenlly used to ~ete~mine the number of di-
and trinucleotide r~peat~ in any particular locus, these are: (1) "Southern" blot
30 analysis; (2) in vitro amplification via the Polymerase Chain Reaction (PCR)
and PCR fra~rnPnt size ~letermin~tion; (3) DNA sequencing (usually of PCR
amplified fr~ e-.ls).
"Soulllell," blot analysis for the quantification of di- and tnnucleotide
35 ç~eals is a method based upon: (1) enzymatic cleavage of genomic DNA
ol,la;"ed from ~he e~..;..e~l individual via sequence specific restriction
enymes cleaving the DNA at many sites including ~e fl~nking regions both 5'
and 3' to the DNA region co..l~;n;..g the ~ ;..ed di- or trinucleotide repeats;
2170~1
~ g
(2) gel electrophoresis aimed at size separation of the DNA fragments obtained
under step (l); (3) blotting or transferring the cleaved and size separated DNA
fr~nPntc to a test surface; (4) ~rep~ a labeled probe capable of specific
hybridization with the blotted DNA fragment co..~ g the repeats; (5)
5 hybri~li7ing the labeled probe with the blotted DNA fragments; (6) washing offprobe excess to obtain specific hybri-1ization and to reduce non-specific and
background sigr ~lc; (7) ~etecting positive signals via means dependent upon theprobe labeling ter-~mi~lue employed under step (4); (8) illte.~-cting the results by
~ete- -.inin~ the size of the fragment hybridized to the labeled probe; and finally
o (9) calc~ tin~ the number of di- or tnnucleotide repeats.
"Soulhe.ll" blot analysis for the quantification of di- and trinucleotide
repeats has major drawbacks: (1) the method is primarily dependent upon the
eYict~-nce of suitable restriction enzymes recognition sites in the immediate 5'lS and 3' fl~nking region of the repeats region; (2) gel electrophoresis employed
under "Southern" blot analysis has low resolution capacity for small size
variations, therefore this method is not suitable for monitolil g small variations
in the number of the di- or trinucleotide repeats; (3) "Southern" blot analysis is
not capable of ~ictin~lishing between size variations due to di- or trinucleotide
20 repeats expansion/de-expansion or other molecular events such as the loss or the
formation of a restriction enzyme recognition site/s due to point mutation,
deletions or insertions, yielding a di- or trinucleotide repeats expansion/de-
e~ansion independent length polymorphism; (4) "Southern" blot analysis
dem~n~ls acculate exec-~tion of a multistep procedure, of which most steps
2~ include several complicated steps, are tirne consuming and require highly skilled
personnel for their routine execution, especially gel electrophoresis, blotting and
hybridization; (5) highly skilled personnel are also needed for intel~leling theresults and for calc~lh~ing the di- or trinucleotide repeats number; (6) gel
elecllophoresis, hybridization and washing conditions may vary considerably
30 depending upon fr~nPnt size and sequence, therefore, "Southern" blot analysisr~q ~ircs dilre~,lt calibration of the procedure for any given ~lise~ce; and last but
not least (7) due to its being a mnl1i$tep procedure, "Southern" blot analysis is
not easily applicable for complete autom~ti7~hon.
In vi~ro amplification via the Polymerase Chain Reaction (PCR) and
PCR fr~nen~ size detelll~ation is easier for ex~cnhon as colllp&~d with
"soulhelll" blot analysis and involves less steps, these are: (1) PCR
~m~!ification of the di- or trinucleotide ~eal~ region using PCR plilllCIs ~om
217095i
the 5' and 3' fl~nkin~ regions of the repeats; (2) size ~letçrrnin~tion of thus
ot~t~ined PCR fragments via high resolution gel electrophoresis; and (3)
c~lcnl~tin~ the number of the di- or trinucleotide repeats. See, Erster S.H.
(1992) Hum. Genet., 90:55-61.
s
Although this approach is simpler and therefore easier for routine
eYeclltion it shares some of the drawbacks described for "Southern" blot
analysis, these incl~lde (1) the PCR approach is not capable of ~listin~lichin~
bet~veen size variations due to di- or trinucleotide repeats e~p~nsion/de-
0 eYp~n~ion or other molecular events such as the loss or gain of sequences due todeletions or insertions in the 5' and/or 3' repeats fl~nkin~ regions, yielding a di-
or tnnucleotide repeats expansion/de-expansion independent PCR fr~rnent
length polymorphism; (2) a high resolution gel electrophoresis is required for
resolving small size var,iations in the PCR fr~rnents, this calls for hi~hly skilled
s personnel and therefore not suitable as a routine diagnostics procedure; (3)
highly skilled personnel are also needed for illte.~le~ g the results and for
c~lc~ tin~ the number of the di- or trinucleotide repeats. In addition: (4) the
PCR approach is not suitable for quantifying highly expanded di- or
tlinucleotide repeats, since its amplifying capacity is limited to relatively small
20 ~n.ontc, therefore, in cases where the fragment to be amplified exceeds a
certain size limit the PCR reaction will fail to yield a specific product; and (5)
some of the di- or trinucleotide repeats fonn highly GC rich stretches of DNA
which are not easily amplified via standard PCR protocols.
2~ The most basic method for ~e~ .. ;n~tion of di- or trinucleotide repeats
number is DNA sequencing. The most widely used sequencing method is based
on the dideoxynucleotide chain termin~tion procedure. The technique involves
the incorporation of dideoxynucleotides with the aid of a DNA extension
en~me at the 3'-end of an elong~inf~ DNA chain. Once the dideo~ynucleotide
30 has been incol~olated, f~er elongation of dle chain is blocked. See, Sanger F.
(1981), Science 214, 120~ 1210. Recently automated DNA sequencing
techniques have been developed which provide for more rapid and safer DNA
seql-entcin~ One such approach utili7~s a set of four chain terrnin~tin~
fluor~scçntly labeled dideoxynucleotides. See Chehab, F.F., et al. (1989), Proc.3s Na~d. Acad. Sci. (USA) 86, 9178 9182; Prober, J.M., et al. (1987), Science 238,
336 341; Smith, L.M., et al. (1980), Nature 321, 674 678). In this metho~
succinyl fluo,.,sce~n dyes are used. Each dideoxynucleotide receives a dif~lent
dye of di~,ellt al)sol~Lion and emission characteristics. Thus, DNA molecules
2170951
11
l~bele-l with each of the di~cre,lt dideoxynucleotides may be ~ tin~ hed from
one another. Using these dideoxynucleotides, it is possible to sequence a DNA
seg~f-.t by carrying out a single reaction in which all four of the di~rerellllyl~bel~d dideoxynucleotides are added together into a single reaction llu~ , and
s the res~llting labeled oligonucleotide fr~n~nte may then be resolved by
polyacrylamide gel electrophoresis in a single sequencing lane on the gel. The
gel is then sc~nne~l by a fluorimeter capable of distingnishing the di~rerellt
fluorescent labels. The sequence of the diLrerent labels along the lane is then
tr~ncl~ted into ~e sequence of the tested DNA se~rnP-nt
DNA seq~lencing as a method for ql-~ntification of di- or trinucleotide
repeat numbers has few major drawbacks, these are: (1) a high voltage and high
resolution gel electrophoresis is required for resolving the single stranded DNAnested fragments obtained during the sequencing reaction, differing from each
5 other merely by one nucleotide base, this calls for highly sk~led personnel and
therefore not suitable as a routine diagnostics procedure; (2) some of the di- or
~mlcleotide repeats form highly GC rich stretches of DNA which are not easily
sequenced via standard sequencing protocols; and (3) the sequencing approach
is not suitable for quarl~ir~ying large di- or trinucleotide rcpedls since it is limited
20 by the resolution power of the sequencing gel.
It is an object of the present invention to provide a simple, reliable, rapid,
highly accurate and easy to operate di- and trinucleotide repeats quantificationm~,~o~ aimed at post and prenatal diagnosis and prognosis which do not require
2s electrophoresis or similar separation according to size as part of its
methodology.
It is another object of the present invention to provide a diagnostic kit
and an automated instrument to be used for carrying out the above method of
30 the invention.
2170~51
` 12
SUMMARY OF THF. ~VENTION
Acco,ding to the ~resenl invention, there is provided a method, named
Trinucleotide Repeats Quallti~ication (TriQ), for ~ete-,.,;.-in~ the number of di-
s and trinncleotide repeals associated with various liise~ses.
The method of the invention dep~o-n~ls upon counting s~ccessive steps of
incorporation of two to three types of primer extension units, depending on the
core repeat sequence, to a 3'-end of an oligonucleotide primer annealed to a
0 sin~le stranded nucleic acid sequence template preferably 3' of the di- or
trim~clsotide repeats region, said counting being terrnin~te~ at the incorporation
of an additional type of primer ext~oncion unit, cont~ining a detection moiety,
capable of base pairing unth a nucleotide base located 5' of the repeats region,said nucleotide base is the first nucleotide base that is not identical with thes nucleotide bases in the core sequence of the repeats in said nucleic acid
template.
One of the applications of the current invention is to enable quick
~lGte-,..;..~l;on of the number of di- or trinucleotide repeats in genetic loci
20 co.~f~ such repeats.
In the broad application of the method of the invention the addition of
prirner eYt~ncion units to a 3'-end of the oligonucleotide plimer is carried outsin~ rly, that is, primer extension units complementary to the di- or
2s trinucleotide repeats core sequence are added at the 3'-end of the elong~ting oligonucleotide primer one after the other. Also according to the broad
application of the method of the invention the detection moiety cont~ining
p,ill1er eytçncion unit that is complelucnt~y to a nucleotide base located 5' ofthe repeat~ region, which is the first nucleotide base not identical with the
30 nucleotide bases in the core sequence of the repeats, is present in all-
i,lcol~or~Lion steps.
In a plefe.le~ use of this application of the present invention for the
~et~ ;on of the mlmber of trimlcleotide repeats in genetic loci CO,,I~;"il~g
3s such lcp~t~, the addition of primer extension units to a 3'-end of the
oli~om~cleQtide primer is carried out in pairs followed by a single primer
P.l~ n unit addition, or ~lt~om~tively~ vice versa, a single primer extension
unit ad~lition is followed by the addition of a pair.
-. 2170951
Also accor&lg to the plefelled use of this application of the method of
the invention a detection moiety co..~ g primer eYtencion unit that is
comple...e-.~ r to a nucleotide base located 5' of the repeats region, said
nucleotide bace is the first nucleotide base not identical with the nucleotide
bases in the core sequence of the repeats, is present only in some or all steps
prcccA;n~ the incolyolalion of a primer extçncion unit complem~nt~ y to the
first nucleotide base in the core sequence of the di- or trinucleotide r~eals.
0 Another use of this application of the cullenl invention enables
genotyping of an individual, that is, to determine the genotype of an individualat any DNA locus co--~ g di- or trinucleotide repeats, that is to qua~ the
number of re~eats cont~ined in any of said locus allele.
Yet another use of this application of the cull~,nt invention enables the
dete....i..~tion of the number of head to tail repeat sequences comprised of twoor more core nucleotide bases, provided that the core repeat sequence consists
of no more than three types of nucleotide bases.
According to features of prefe~led embodiments of the invention
described below suitable detection moieties of primer extension units include
those facilitating direct or indirect detection and which are pc~ anendy
conjugated to any location at the primer extension unit, or alternatively,
removable or destructible. Detecting dhe detection moiety, whether direcdy or
2~ indirectly, may be carried out in the reaction chamber or in a dirrelellt chamber
depending whether the detection moiety is removable or not.
AccGrding to still further fealures in the described preferred
embo-lim~nts, the e~erlcion moiety is a deoxyribonucleohde, such as dATP,
dCTP, dGTP, dTTP and dUTP.
The oligonucleotide primer may be of any suitable length. Time and
e~l ellsc considerations tend to shit yrefer~llce toward shorter oligonucleotide
which is still sufficiently long to ensure high sequence specificity while at the
3~ same time e ~Iclll ;1l~ rapid, easy and accuratc l~reparalion.
The sample of genetic m~ten~l being tested by the method of invention
may be in the form of RNA or DNA.
--- 2170951
14
The e~tension moiety may, for example, be ~ rl-ed to any suitable
dçtech-n moiety, such dS a radioactive label, e.g., 32p and various fluorescent
labels. Another example involves nucleotides having a detection moiety
5 attacllment which may function for indirect detection.
Accord~g to furdler fealu~es of ~fe.l. d embo~lime~tc described below
re~e,e..lc are collected and are reused in further steps.
Also accor~ing to the present invention, there is provided a diagnostic kit
for quantifying the number of di- or trinucleotide repeats, concictin~ (a) any
number of suitable oligonucleotide primers (b) two or three primer ç~tencion
units; (c) further one or two primer extension units of a type not included under
(b), said primer extencion UllitS co~ a detection moiety; (d) a template
lS depen~lPnt extencion en~yrne; and (e) at least one buffer.
Also according to the present invention, there is provided an automated
i,lsl~ ent suitable for executing the steps concistin~ the mel;hod of the
invention.
BRIEF DESCRIPTION OF THE DRAWINGS
The invention is herein described, by way of example only, with
refclence to the accol,lpanying drawings, wherein:
2~
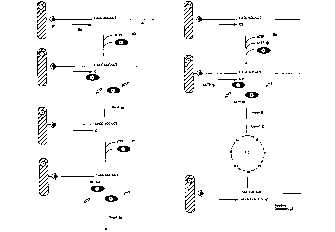
FIG. 1 is a scll~m~ic outline of fealules of a method aimed at q~ ir~ing
the number of CAG trinucleotide repeats in examined nucleic acid sequence.
FIG. 2 is a schPm~tic oudine of features of modified and more efficient
30 methods aimed at ~ ir~ing the nllmber of CAG trinucleotide repeats in-
,A.n;ne~ nucleic acid sequence.
FIG. 3 is a sçhPm~tic ou~dine of fealules of a method aimed at
Cim~ eous q.~ ification of ~e number of CAG trinucleotide le~)edls in two
3s alleles each c~--4;--c a difrerent number of said l~peals.
FIG. 4 is a sr-h~ oudine of fca~ s of an i~l.u~nent suitable for
eY~c~ the steps consistinp dle method of the inventio~
- 2170951
n~SCRIPTION OF THF. PREFER~F.n EMBODIMENTS
The present invention is of a novel method for dete~ninin~. t~.e number of
5 di- or trinucleotide lcpeals associated with vanous ~i~e~cec
The principles and operation of a method according to the present
invention may be better understood with leferc~ce to the drawings and the
accG.~ying descliylion.
The plese.~t invention will be described in more detail with emphasis on
a method for qu~ liLcation of the number of trinucleotide repeats in genes,
which re~eals are associated with genetic disorders. While this application of
the method of the invention is presently ylefe~led~ this is by no means the onlyapplication of the invention as will no doubt be appreciated by those skilled inthe art. For example, the method has various other applications including, but
not li~ c.1 to, the detection of specific genetic sequences in samples such as
nucleotide repeats characterized by a core sequence consisted of two or three
types of nucleotides; those associated with certain genetic or other diseases and
20 pathogenic microorg~nisms for example bacteria and viruses; in testing of
c-"i1~; and in forensic medicine; cancers; and plant polymorphism.
As for quantifying the nurnber of trinucleotide repeats in genes
associated with trinucleotide repeats expansion the method of the invention
25 depends upon counting sllccessive steps of incorporation of two to three types
of primer extension units, depending on the core repeat sequence, to a 3'-end ofan oligonucleotide primer annealed to a single stranded nucleic acid sequence
template preferably 3' of the trinucleotide repeats region, said counting being
telmin~te~l at the inco"~oralion of an additional type of primer extension unit,30 CO.~ p a det~ction moiety, capable of base pairing with the first nucleotide
base of a type that is not included among the nucleotide bases in the core
sequence of the repeat in said nucleic acid template.
In order to better unde~ d the embodiment of the present invention,
35 1~fc~ ce is made to Figure 1, which is a schematic depiction of a method aimed
at quantiijring the numb~- of CAG trinucleotide ~pcals in e~A~..;.~e~ nucleic
acid sequence which inclndes: (a) if ~e nucleic acid of inte~s1 is not single
stranded, ~eating the sample cont~inin~ the nucleic acid to obtain u~lpaired
- - 2170~51
nudeotide bases S~3A~ . the repeats and fl~nking regions; (b) under
hybri~i7~tion conditions, contActing the ul~pai~ed nucleotide bases with an
oligom~rleoti~e primer, having a sequence which is complçmçnt~ry to a stretch
of nucleotide bases sit~l~te~ p~fe~ably 3' of the repeats region in the e~ ed
s single strand sequence, ~I~;fe,ably, the 3'-end of said oligonucleotide primer is
annealed to the first nucleotide 3' of the l~eals region; (c) providing means toensure that at least the ç~r~mined nucleic acid and the oligonucleotide primer are
. confined to a reaction ch~mber at all further steps; (d) the template primer
hybrid is contacted with a primer extension unit which is capable of base
o pairing with the first nucleotide base in the core sequence of the repeats, dGTP
in the given example, and a template dependent extension enzyme; (e) after a
sl~itable incllbation time, non-incorporated primer extension units are
climin~teA preferably washed away; (f) the template primer hybrid, now said
primer elongated by one unit, is contacted with a primer extension unit which islS capable of base pairing with the second nucleotide base in the core sequence of
the repeats, dTTP in the given example, and a template dependent extension
enzyme; (g) after a suitable incubation time non-incorporated primer extension
units are elimin~te~l p~efelably washed away; (h) the template primer hybrid,
now said primer elongated by one additional unit, is contacted with a primer
20 e~rt~n.cion unit which is capable of base pairing with the third nucleotide base in
the core sequence of the re~eal~, dCTP in the given example; a detection moiety
co.~t~i..in~ primer extension unit which is capable of base pairing with a
nucleotide base 5' of the repeats region, said nucleotide base being the first
nucleotide base of a type not included among the nucleotide bases in the core
2~ sequence of the trinucleotide repeats, dATP$ in the given example; and a
template dependent extension enzyme; (i) after a suitable incubation time non-
incorporated primer extension units are elimin~te~ preferably washed away; (.i)
detccling for the presence of the detection moiety cont~ining primer extension
unit; (k) steps (d) to (i) are 1~ pedted until detecting the detection moiety of the
30 primer e~ sion unit which is capable of base pairing with a nucleotide base 5'
of the lepe~b region, said nucleotide base being the first nucleotide base of a
type not incll~decl arnong the nucleotide bases in the core sequence of the
tr mlcleo1ide repeats, dATP* in the given example; (1) the number of repeats as
stated under O enables to calculate the number of trinucleotide repeats, CAG
3s in ~e given example, ~llel~;rol~e, enables the de~e~llunation of the exact
lepelilion number.
- 21709Sl
1~
In order to better understand the preferred embodim~nt of the present
invention, refe~ellce is made to Figure 2 which is a sch~m~hc depiction of
modified and more efficient methods aimed at quantif~ing the number of CAG
trinucleo~tide le~)faLs in e~ .;..ed nucleic acid sequence which includes: (a) if
s the n~lc.leic acid of illteleal is not already single stranded, treating a sample
co..tA;~.;ng the nucleic acid to obtain unl)aired nucleotide bases sp~nnin~ the
epeàls and fl~nking regions; (b) under hybridization conditions, cont~cting the
~paired nucleotide bases with an oligonucleotide primer, having a sequence
which is complemPnt~ry to a stretch of nucleotide bases situated preferably 3' of
o the reped~s region in the e~mined single strand nucleic acid template,
l.reff,ably, the 3'-end of said oligonucleotide primer is annealed to the first
nucleotide 3' of the repea~ region; (c) providing means to ensure that the
eY~mined nucleic acid and the oligonucleotide primer are confined to a reaction
chamber at all further steps; (d) the template primer hybrid is contacted with
IS primer eytp-ncion units which are capable of base pairing with the first and
second nucleotide bases in the core sequence of the repeats, dGTP and dTTP in
the given example, and a template depPn~ent extension enzyme; alternatively
(d~ the template pnmer hybrid is contacted with a primer extension unit which
is capable of base paillng with the first nucleotide bases in the core sequence of
20 the repeals, dGTP in the given example, and a template dependent extension
enzyme; (e) after a suitable incubation time non-incorporated primer extension
units are elimin~te~l preferably washed away; (f) the template primer hybrid,
now said primer elongated by two units, is contacted with a primer extension
unit which is capable of base painng with the third nucleotide base in the core
2s sequence of the repeats, dCTP in the given example; a detection moiety
col-~ai~ -g primer e~rtPnSion unit which is capable of base pairing with a
nucleotide base 5' of the r~eals region, said nucleotide base being the first
nucleotide base of a type not included among the nucleo~de bases in the core
sequence of the trin-lcleotide r~peal~, dATP* in the given example; and a
30 template dependent e~ sion enzyme; alternatively (~) the template prirner
hybrid, now said primer elongated by one unit, is contacted with pnmer
Ç~f~cion units which are capable of base pairing with the second and third
nucleotide bases in the core sequence of the .epeals, dTTP and dCTP in the
given example; a detection moiety co..~ primer extension unit which is
3s capable of base ~ g with a nucleotide base 5' of the repeats region, said
nucleotide base being the first nucleotide base of a type not inchlde~ among thenucleotide bases in the core sequence of the trinucleotide lepeals, dATP* in thegiven eYP~nple; and a template llepen~ent eAI~-.sion enzyme; (g) after â suitable
- 2170951
~_ 18
incllb~tion time non-inco1po1ated primer extension units are elimin~te~l
pfefe1ably washed away; (h) detecting for the presence of the detection moiety
co~ . primer cYt~nsion unit; (i) steps (d) to (h) are rel~eA~ until detecting
the detection moiety of the primer eYt~nsion unit which is capable of base
s pairing with a nucleotide base 5' of the repeats region, said nucleotide base
being the first nucleotide base of a type not included arnong the nucleotide
bases in the core sequence of the trinucleotide re~eals, dATP* in the given
example; (i) the number of 1e~eals as stated under (i) enables to calculate the
number of trinucleotide repeats, CAG in the given example, therefore, enables
o the ~let~ ~ .. .i..~*on of the exact repclilion number.
The human genome contains two alleles of each gene. Each of the alleles
is located on one of the chromosomes m~king up a pair of homologue
chromosomes. Alleles characterized by triple repeats are very polymorphic by
lS nature, therefore most individuals contain non-identical alleles for each of their
trinucleotide repeats contAinin,~ genes, said alleles differ from one another bythe number of trinucleotide repeats they contain. As explained, for diagnostic
and prognostic pu~.~oses, it is important and crucial to dete11nille the exact
number of tnnucleotide repeats cont~in~o~3 in both alleles of the exarnined gene.
20 In order to better understand the prefe11ed embodiment of the present invention,
r~fere11ce is made to Figure 3 which is a schematic depiction of a method aimed
at siml)lt~neous quantification of the number of a CAG trinucleotide repeats in
two alleles each contains a different number of said repeats. According to this
embodiment, steps of incorporating pTimer extension units complementary to
2s the core sequence of the repeats are performed as detailed above until a
detection of the detection moiety cont~ining~ primer extension unit which is
capable of base pairing with a nucleotide base 5' of the repeats region, said
nucleotide base being the first nucleotide base of a type not included arnong the
nucleotide bases in the core sequence of the trinucleotide repeats, is made, and30 the m~itude of the signal recorded. Counting these steps enables to determine~e number of trinllcleotide repeats in the allele cont~ining the lower number ofel~eals. After records have been made, steps of incorporating primer extension
units co~nplçmentAry to the core sequence of the repeats are continue~ as
detailed until fuIther detection of the detection moiety co~tAin;np~ primer
3s e~t~ nsion unit which is capable of base pairing with a nucleotide base 5' of the
r~peats region, said nucleotide base being the first nucleo~de base of a type not
inr,~ le~ among the nucleotide bases in the core sequence of the trinucleotide
1~,~ts. Co~ting these steps and adding the reslllte~ number to the former
. 2170951
. ~ ,9
enables to ~ete~...;..e the number of trinucleotide repeats in the allele con~ gthe greater number of repeats.
Under this desc~ ion, the ~etection moiety of the primer e~t~ncion unit
s is non-removable, thus, as the second signal is obtained a s~n~ g of signals is
made. E~refelably a removable ~etection moiety may be used and removed after
letectin~ the signal from the allele cont~ p less ~pedtS, ~ltçrn~tively~ a
di~e.ent kind of detection moiety may be used, hence, the detection of the
second signal is simpler since in both cases the second detection, as the first, is
o based on all or non-detection.
The embo~ime~ts of the present invention are also useful for dete~ g
the nllmber of head to tail repeat sequences of two or more nucleotide bases,
provided that the core sequence consists of no more than three types of
lS nucleotide bases.
Methods according to l~lefe.led embodiments of the present invention
enjoy a nnmber of advantages relative to the prior art:
First, these are high resolution methods capable of a precise and
unambiguous det~,...i.~tion of the number of trinucleotide repeats in a selectedlocus contained in a genetic material sample and is therefore suitable for
monitoring small variations in trinucleotide repeat nurnbers.
2s Second, the methods according to preferred embodiments of the present
invention is c~p~ble of rlictin~lichin~ between size variations due to
trim)cleotide lelpedls e~rA~siorl/de expansion or other molecular events such as~e loss or formation of a restriction enzymes recognition site, or such as the
loss or gain of sequences due to deletions or insertions.
Third, these methods do not include any kind of gel electrophoresis or
other size based separations and therefore may be easily ~ntom~ted diminichin~
the r~ e~,ent for highly skilled personnel for their routine execution.
Four~, ordir~y persormel are sufficient for intel~lcling the results and
for calclll~tin~ the trinucleotide sepeals number.
.- 2170951
Finally, the high GC contellt of some of the trinucleotide repeats create
gel rnigration artifacts due to the formation of strong secondary structures. Since
~e afo~-ne~l;oned methods of the invention aimed at the de~ hon of
trinucleotide repeals number are gel electrophoresis indepen~ent these ar~facts,s attributed to gel electrophoresis dependent methods, are cilcu~l-rented.
The above described embo-liments of the present invention may be made
more efficient in term of costs by reusing materials, that is, after each step of the
ones described, re~nts are collected and are reused in further steps. A re-
o concentration and purification procedures may be needed before reuse of thesereaction reagents is made.
The specific application of the inventive method for the quantification of
trinucleotide repeals in genes known to carry such repeats is presently a
IS pl~;felled embodiment. In this application the method may be used as a
diagnostic assay to determine the number of trinucleotide repeats present in
individuals su~el~lg from, or showing syrnptoms of, diseases known to be
caused by eXI~nsion of such repeats in or close to specific genes. The method
may also be applied for simultaneous screening of apparently healthy
20 individuals to dele~ e whether any of them are carriers of any such repeats.
This is the case, for example, in the well elucidated Hnntin~on's disease in
which lice~ce~1 individuals have e~cp~n~e~ tnnucleotide repeats in at least oneallele encoding ~e IT15 gene. Furdlermore, the method may also be applied for
scf~enil~g embryos by analyzing sarnples of arnniotic fluid cells to detç~ ;ne
2s whether the embryos have any known trinucleotide repeats expansion in one or
two or none of the alleles encoding genes known to be involved in specific
genetic diseases in which such expansions are involved.
The genetic material to be analyzed may, in principle, be any RNA or
30 DNA obt~ ed from the tissues or body fluids of hllm~ns ~nim~lc or plants or
obtained ~om cultures of microor~nicmc or hllm~n animal or plant cells or
nucleic acid synthesi7e~l by çYtçnsion enzyme. The genetic material may
~h~ tively be obtained from non-living sources suspected of con~ ng matter
from living or~ ,-.. sources, as may be the case when applying the method in
3s forensic medicine for de~clil,g and identifying specific nucleotide sequencesese~t in or on samples of clo~ing, Çu~n.lure, weapons and other items found
at the scene of a crime. In this instance, the genetic material obtained is usuatly
- 21709~1
21
in the form of DNA, since any RNA in such samples would normally have been
degraded by ribonucleases.
The sample of nucleic acids can be drawn from any source and may be
s natural or synthetic. The sample of nucleic acids may be made up of
deoxyribonucleic acids, ribonucleic acids, or copolymers of deoxyribonucleic
acid and ribonucleic acid. The nucleic acid of i~t~esl can be synthesi7e~
enzymatically in vitro, or synthPsi7e-l non-enymatically. The sample
col-t~ the nucleic acid or acids of in~ere~l can also comprise extr~g~nomic
0 DNA from an or~nism RNA tr~nscripts thereof, or cDNA p~epared from RNA
s~ ts thereo Also, the nucleic acid of i~e~sl can be synthesi7e~ by ~e
polymerase chain reaction.
The e~..i..ed nucleic acid can be made single stranded by using
lS appro~,liate ~en~t lring conditions, which may include heat, aLkali, formamide,
urea, glyoxal, enzymes, and combinations thereof.
Ex~min~*on of nucleic acids obtained from two or more individuals can
be made simlllt~neously, for initial screening purposes. Furthermore, some
20 genes co.~t~ trinucleotide repeats which share identical repeat core sequence,
nevertheless the trinucleotide repeats fl~nking regions differ substantially in
their nucleotide base sequences. Therefore it is also possible to quantify the
number of trinucleotide repeats in two or more genetic loci simultaneously.
2s The oligonucleotide primers can be any length or sequence, can be DNA
or RNA, or any modification thereof. It is necess~ry, however, that the length
and sequence of the oligonucleotide ~)lilllels be chosen to op~mize the
specificity of the hybridization to the target sequences of ll~tere~l.
Time and expense considerations tend to shift preferellce toward shorter
oligonucleotides which are still sufficiently long to ensure high sequence
specificity while at the same time en~-mng rapid, easy and acculate l,lepa,al on.
The oligomlcleotide primer may be any suitable species, preferably an
3s oligodeoxyribonucleotide, an oligoribonucleotide, a p~oteil- nucleic acid or a
copolymer of deoxyribonucleotides, p~otelll nucleic acids and ribonucleotides.
The olipom~cleo~de primer can be either natural or synthetic. The
- 2170~51
~ 22
oligonucleotide primer can be synthesized enzymatically in vivo, enzymatically
in vitro, or non-en_ymatically in vi~ro.
In addition, the oligonucleotide primer must be capable of hybridiing or
S ~nne~ling with a stretch of nucleotide bases present in the nucleic acid of
u~terc~l preferably 3' of the trinucleotide repeats region to be quantified. Oneway to accomplish the desired hybridization is to have the template dependçnt
primer be sul~ lly complementary or fully complem~nt~ry to the known
nucleotide bases sequence preferably 3' of the trinucleotide repeats region to be
10 q1~ntified. For convenience, in some in~t~nces the 3'-end of the oligonucleotide
cr may overlap part of the repeat sequences. Furthennore, the
oligonucleotide primer may be suitable for annealing with any of the strands in
the e~-..;..ed nucleic acid and the types of primer çxtçn~ion units used changedaccordi~gly.
The single stranded examined nucleic acid and the oligonucleotide
primer should be confined to a reaction chamber throughout the experimerlt~l
steps in the method of the invention. This could be achieved, for example, by
immobili7inP any of the mentioned molecules to a solid support. The
20 imrnobilization may be effected by binding the molecules to the solid supportvia (1) multiple ionic interactions between any of said molecules and the solid
support; (2) multiple covalent bindings between the molecules and the solid
~u~oll, (3) direct single point coupling of the molecules to the solid support (4)
indirect single point coupling of the molecules to the solid support via an
25 anchoring moiety conjugated to the molecules and a complement anchonng
sites athched to the solid S~ppOll, e.g., biotin conjugated to the single stranded
eY~ e-l nucleic acid or ~ltern~hvely the oligonucleotide primer or both and
avidin, sLIeplavidin or antibiotin antibody are attached to the solid support; or
magnetic beads are conjugated to the single stranded e~mined nucleic acid or
30 ~ sl;~tely the oligonucleotide pn~ner or both and the solid support is a
magret or elec~.ol..agnet. (5) indirect multiple points coupling of the molecules
to the solid support via anchoring moieties conjugated to the molecules at
multiple sites and a complement anchoring sites ~ ed to the solid support. A
single point coupling of the e~c~mined nucleic acid, or ~ltenl~tively~ the
3s oligor.l~cle ~ide primer or both to the solid support, whether direct as under (3);
or indirect as under (4), is the ~lefe,.ed methodology since it ma~imi7es the
accessibility of other reaction components to these molecules and form less
steric constrains.
2170951
di~ on, the confinement of both the examined nucleic acid and the
oligonucleotide primer to the reaction chamber may be effected by e~ll~pil1g
these molecules using a porous membrane with a molecular weight cut off that
5 will facilitate the elimin~tion of primer ext~n~ion units via sepa~a~i~e filtration
and will, at the same time, retain the above mentioned molecules. While by
immobilizing the mentioned molecules to a solid support the template
depPn~ent eYt~ncion enzyme is discarded along with the pnmer extencion units
and therefore fresh enzyme should be added after each step, the enzyme, due to
10 its high molecular weight, is retained upon e,lllappi~lg the molecules using a
porous membrane with a molecular weight cut off that will facilitate the
el i~ ;on of primer extension units via separative filtration.
Any suitable extension moiety of the primer extension units may be used.
5 The extension moiety however should contain a 3'-OH group enabling further
elongation of the oligonucleotide primer. The extension moiety of the primer
e~t~nCion units may be deoxyribonucleotides, ribonucleotides or their 3'-OH
co..tA;n;.-~ analogs. Preferably, the extension moiety is a deoxyribonucleotide,such as dATP, dCTP, dGTP, dTTP and dUTP.
The elimin~tion of non-incorporated primer extension units from the
reaction chamber, prece~lin~ further incorporation steps as delineated above,
may be effected via physically removing said primer extension units from the
reaction chamber by, for example, filtration as mentioned, or alternatively, by
25 destroying said primer extension units chernically or enzyrnatically wi~in the
reaction chamber, for example by aLkaline phosphatase that will dephosphorilate
said primer extension units, rendering them inapplicable for enzyrnatic
incorporation.
Different versions of the methods for ~el~.. i"inp the number of
trinucleotide repeats in a nucleic acid of interest are possible. In one version, the
t~-nlpl~te is deoxyribonucleic acid, the primer is an oligodeoxyribonucleotide,
oligoribonucleotide, protein nucleic acid, or a copolymer of
deox~Tibomlcleotides, l~lole~n nucleic acids and ribonucleotides, and the
3s templ~t~ de~çn~Pnt enzyme is a DNA extension enzyme.
In a second version, ~e template is a ribonucleic acid, the primer is an
olip~eox~ribon-l~leo1ide, oligoribonucleotide, p,oteill nucleic acid or a
2170g51
~_ 24
copolymer of deo~yribonucleotides, protein nucleic acids and ribonucleotides,
and the template dependent enzyme is a reverse transcriptase.
In a third version, the template is a deoxyribonucleic acid, the primer is
s an oligoribonucleotide, and the enzyme is an RNA extension enzyme.
In a fourth version, the template is a ribonucleic acid, the primer is an
oligoribonucleotide, and the template dependent enzyme is an RNA replicase.
The template dependent enzyme prefe.ably is confined to the reaction
charnber. This could be achieved as described above by using a porous
membrane with a molecular weight cut off that upon filtration will enable the
elimin~tion of primer extension units but will retain the enzyme. Alternatively,the enzyrne may be linked via a long and flexible linking chain to a solid
lS support, said solid support used also for immobilization of the exarnined nucleic
acid and/or the oligonucleotide primer. The linking chain, being long and
flexible, will allow the collision of the enzyme with its substrates, a prerequisite
for catalysis. In both cases the addition of costly fresh enzyrne in every
incorporation step, as described above, is elimin~te~l
The nucleic acid of i,lte,est may contain non-natural nucleotide analogs
such as deoxyinosine or 7 deaza 2' deoxyguanosine. These analogs destabilize
DNA duplexes and could allow a primer annealing and extension reaction to
occur in a double stranded sample without completely sep~aling the strands.
2s
Any suitable detection moiety of the primer extension unit which is
capable of base pairing with a nucleotide base 5' of the repeats region, said
nucleotide base being the first nucleotide base of a type not included arnong the
nucleotide bases in the core sequence of the trinucleotide repeats, may be used.30 FullL~ ore, the detection moiety of the prirner extension unit should have
physical and chemical prop~lies which do not interfere with its er~natic
addition to the 3'-OH group of the elongated oligonucleotide p~irner. The
~letection moiety of ~e primer extension unit may facilitate the direct or indirect
detection of its presence. For indirect detection the detection moiety of the
3s primer ~(f ~c;on unit may include a molecule of a type selected from the group
concictin~ of enymes, catalysts, haptens, antibodies, sub~llatcs, coenzymes and
ch~nil~....;.~scence. ~efe.dbly the ~etection moiety of the primer extension
. 2170951
unit f~cili~t~ the direct detection and may include a molecule of a type selected
from the group consisting of fluorescence and radioactivity.
The detection moiety may be conjugated at any position to the primer
s eYtension unit. Furthermore, the detection moiety may be of a kind that is
rernovable or destructible by chemical, physical or enzymatic l-nanipulation.
Removable or distractible detection moieties of the primer extencinn unit which
is capable of base pairing with a nucleotide base 5' of the repeats region, saidnucleotide base being ~he first nucleotide base of a type not included among the10 nucleotide bases in the core sequence of the trinucleotide repeats are preferably
used, for reasons explained above, when the simultaneous quantification of two
alleles each co.~t~;l.;..g a different number of tnnucleotide repeats is desired.
The detection of the detection moiety, to a very large extent, is
lS dependent upon its chemical and physical properties. Any suitable detection
approach may be selected. In a case where the detection moiety of the primer
extension unit is non-removable, the detection, whether direct or indirect, is
prefe.~dbly carried out in the reaction camber. On the other hand, in a case
where the detection moie~ of the primer extension unit is removable, the
20 detection~ whether direct or indirect, may be carried out in a di~erent chamber.
The ongoing research to determine the genetic basis for diseases and the
advent of technologies such as the Polymerase Chain Reaction (PCR) has
resulted in the discovery and complete sequencing of so far seven genes in
25 which trinucleotide repeats expansion would lead to either no expression of the
gene product or e~ ession of a product which is qualitatively or qu~llitalively
impa~ed and thereby res~ltin,~ in a disease. Since trinucleotide repeats have
been observed within or close to a number of human genes by gene bank
se~ches, it is conceivable that trinucleotide amplification may be involved in
30 the ~lls~tion of other genetic ~lice~ses as well. There is thus an ever expanding
field of application of the above method of the invention.
Since di- and trinucleotide repeats are highly polyrnorphic, that is for
each gene characte.~d by such repeats at least few dozen alleles L~l~,g in
35 their lepedls nurnber exist, the method of the invention may also be applied in
the field of forensic medicine in which polymoIphism in specific genes can be
dete.~ ncd in, for e~a."plc, blood or semen samples obtained at the scene of a
crime and the results used to indicate whether or not a particular sl~spect was
2170951
~_ 26
involved in the crime. Similarly, the aforesaid dete~ ation may also be used
to dete~ e whether a certain male individual is the father in cases of disputed
pA t~
s There is evidence that cerhin cancers may be the result of di- and
trinucleotide-eYpAncion in many gene targets, therefore, the present invention
may be used as a cancer early diagnostic and prognostic tool.
Another application of the present methods is the detection of
microor~AnicmC in a sample on the basis of the presence of specific sequences
in the sample. For eYAmrle, an individual suspected of being infected by a
rnicroorgAnicm such as a bacteria or virus, can be tested by using an
oligonucleotide primer which anneal only with a specific bacterial and/or viral
DNA sequences and not with sequences present in the examined individual. The
oligonucleotide primer's sequence is selected to enable its hybridization 3' of a
stretch of nucleotide bases composed of one to three types of nucleotide bases
followed by a di~lent type of nucleotide base in the examined sequence and a
procedure similar to the ones described above is carried out. Detecting the
detection moiety of the primer extension unit that is complementary to said
di~re,lt type of nucleotide base is an indication of the presence of the
c~A~n;~.ed genetic material in the sample. One example of such an application isin the screening of individuals for the presence of the AIDS virus.
The invention will now be further illustrated by the following examples:
EXAMPLES
Oligonucleotide primers and suitable detection moiety containing primer
e~tension units aimed at the Quantification of trinucleotide repeats in the
seven genes known to carry said repeats
As mentioned, so far seven genetic ~iceAses each involving a unique
genetic locus have been irnplicated with trinucleotide repeat mutations. These
include: Fragile XA (A site, Martin Bell) syndrome (FRAXA); Kennedy ~i~e~ce
3s (spinal and bulbar muscular allOp~ly, SBMA); Myotonic dystrophy
(Cu~sckn~nn Steiner~ DM); ~lmtin~Qn'S disease (HD); Spinoce~bellar ataxia
~pe 1 (SCAl); Fragile XE (E site) mental retardation (FRAXE MR); and
Dc~ olublal paUidoluysian alloph~r (DRPLA).
- 21709Sl
27
Table I lists six of these ~ise~ses which are known to result from
~inucleotide ~epe~ls e~pallsion; ~ropliate 18 mer oligonucleotide primers, the
upper primer being suitable for hyblidization with a (-) strand therefore is
s suitable for hybridization with the (-) strand of single stranded DNA, whereasthe lower primer being suitable for hybridization wi~h a (+) strand therefore issuitable for hybri~li7~tion with the (+) st~and of single stranded DNA or
enzymatically ~anscribed RNA; and detection moiety cont~inin~ pIimer
e~lel-cion units suitable for del~ .,.;.-;--~ ~he number of trinucleo~de repeats as
o described above.
Table I:
Disease and Primers: Extension
(repeats core units:
sequence):
Fragile XA 5' AGGGGGCGTGCGGCAGCG 3' dT/ATP
(CGG)n 5' CGGGCGCTCGAGGCCCAG 3' dT/ATP
Kennedy disease 5' GCCAGTTTGCTGCTGCTG 3' dTTP
(CAG)n 5' CCTGGGGCTAGTCTCTTG 3' dATP
Myotonic dystrophy 5' GTCCTTGTAGCCGGGAAT 3' dATP
(GCT)n 5'ATGGTCTCTCATCCCCCC3' dTTP
Huntington's disease 5' GAGTCCCTCAAGTCCTTC 3' dTTP
(CAG)~, 5'CGGCGGTGGCGGCTGTTG3' dATP
Spinocerebellar 5' CCGGGACACAAGGCTGAG 3' dTTP
ataxia ~pe 1 5' CTGCTGCTGGATGCTGATG 3' dATP
(CAG)n
D~llL~tolu~lal 5' CACCACCAGCAACAGCAA 3' dTTP
3s pallidoluysian 5' CCCAGAGTTTCCGTGATG 3' dATP
dt~ h,,t
(CAG)n
2170~51
~_ 28
Methods aimed at the quantification of trinucleotide repeats
EXAMPLE 1
s Figure 1 illustrates a method aimed at quantifying the number of CAG
trinucleotide repeats in e~c~mined nucleic acid sequence containing 3 said
repeats followed by a thyrnine residue (---CAG CAG CAG T---). The sample
co.-~ in~. the nucleic acid of illte.esL is treated to obtain unpaired nucleotide
bases sp~ the repeats and fl~nkin~ regions (a). An oligonucleotide primer
o is annealed to the nucleic acid template under hybridization conditions,
preferably the 3'-end of said oligonucleotide primer is annealed to the first
nucleotide 3' of the repeats region (b). The exarnined nucleic acid is attached to
a solid support (referred to as "S" in the Figure) indirectly via a single pointcoupling consicted of anchoring moiety conjugated to the molecules and a
complement anchoring site attached to the solid support (c). l~e template
prirner hybrid is contacted with a dGTP primer exte~sion unit which is capable
of base pairing with the first nucleotide base in the core sequence of the repeats,
and a template dependent extension enzyme, referred to as "E" in the Figure (d).After a suitable incubation time, non-incorporated dGTP primer extension unit
is washed away along with the extension enzyme (e). The template primer
hybrid, now said primer elongated by one guanine residue, is contacted with
dTTP primer extension unit which is capable of base pairing with the second
nucleotide base in the core sequence of the repeats, and a template dependent
eAlel.sion enzyme (f). After a suitable incubation time non-incorporated dTTP
primer eYt~ncion units are washed away (g). The template primer hybrid, now
said prirner elongated by guanine followed by thirnine residues, is contacted
wi~ dCTP primer extension unit which is capable of base pairing with the third
nucleotide base in the core sequence of the repeats, a dATP detection moiety
col-t~ primer extension ur~it, efe~led to as "*" in the Figure, which is
capable of base pairing with a nucleotide base 5' of the repeats region, said
nucleotide base being the first nucleotide base of a type not included arnong the
nucleotide bases in the core sequence of the ~inucleotide repeats, and a
templ~te depend~nt extension enzyme O. after a suitable incubation tirne, non-
inc~ oraled dCTP and dATP* primer eYtencion units are washed away (i).
3~ Det~c~ for the presence of the detection moiety (*) co~ dATP primer
c~ ;on unit (j). Steps (d) to (j) are repeated altogether dlree tirnes until
~et~ the detection moiety (*) of the dATP primer e~tension unit (j3). The
2170~51
~_ 29
number of repeats equals the nurnber of the CAG trinucleotide repeaes, thereforeenabling ~etemtin~thon of the exact repetition number.
EXAMPLE 2
s
Figure 2 illustrates a modified and more efficient method aimed at
.lu~~ ing the number of CAG trinucleotide re~eals in ex~tmined nucleic acid
sequence co..~ 3 said repeats followed by a thimine residue (---CAG CAG
CAG T---). The sample contStining the nucleic acid of il~teles~ is treated to
o obtain ullpaired nucleotide bases sp~.-n;..~ the repeats and fl~tnkin_ regions (a).
An oligonucleotide primer is annealed to the nucleic acid terr,plate under
hybridization conditions, preferably the 3'-end of said oligonucleotide primer is
stnnestled to the first nucleotide 3' of the repeats region (b). The examined
nucleic acid is attached to a solid support (S) indirectly via a single point
lS coupling conci~te~l of anchoring moiety conjugated to the molecules and a
comple~e~t anchoring site attached to the solid support ~c). The template
primer hybrid is contacted with dGTP and dTTP primer extension units which
are capable of base pstiring with the first and second nucleotide bases in the core
sequence of the repeats and a template dependent extension enzyme (E) (d). The
20 template primer hybrid, now said primer elong~ted by a guanine and a thimine
residues, is contacted with a dCTP primer extension lmit which is capable of
base p~tirin~ with the third nucleotide base in the core sequence of the repeats, a
dATP detection moiety cont~tinin_ prirner extension unit (*) which is capable
of base pairing with a nucleotide base 5' of the repeats region, said nucleotide2s base being the first nucleotide base of a type not included among the nucleotide
bases in ~e core sequence of the trinucleotide repeats, and a template
depe,ldellt e~l~nsion enzyme (f). After a suitable incubation time non-
inco~ ated dCTP and dATP* primer extencion units are washed away (g).
Dete~!;np for the presence of the dATP* detection moiety contStining~ primer
30 ~ .c;on unit (h). Steps (d) to (h) are repeated altogether three times until-~ehc~ g the detection moiety (*) of the dATP primer extension unit (h3). The
mlmb~r of re~ed~ equals the number of the CAG trinucleotide repeats, therefore
enabling ~et~ sttion of the exact repetition number.
3s EXAIVIPLE 3
Figure 3 illuallales a metho-l aimed at simnltstneous qu~ltirlcation of the
n~lmb~ of CAG trinucleotide le~,cats in two alleles the first co.~tSt;~ g one and
~_ 30 2170~51
the secon~l two said repeals followed in both cases by a thimine residue. Steps
of incorporating pli ~c~ eYtPncion units complementary to the core sequence of
the repeats are ~e ro-...ecl as detailed above until a ~letection of the detection
moiety cor.~ primer extension unit (A*) which is capable of base pairing
s with a nucleotide base 5' of the repeats region, said nucleotide base being the
first nucleotide base of a type not included among the nucleotide bases in the
core sequence of the tTinucleotide repeats, is made and the magl~itude of the
signal recorded (*). Counting these steps enables to ~lete~ ;..e the number of
trinucleotide repeats in the allele co~ g the lower number of repeats. After
0 records have been made, steps of incorporating primer eYtension units
complementary to the core sequence of the repeats are cont;nl~e~ as detailed
until further detection of the detection moiety co..~ g primer extension unit
(A*) which is capable of base pairing with a nucleotide base 5' of the repeats
region, said nucleotide base being the first nucleotide base of a type not
IS included among the nucleotide bases in the core sequence of the tnnucleohde
repeats is made and the ma~itl~de of the additive signal ($*) or a di~erent
signal, denoted as a closed box in the Figure, is recorded. Counting these steps(1 step in the given example) and adding the resulted number to the folmer (1
step in the given example) enables to ~ete~ ;..e the number of trinucleotide
20 repeats in the allele cont~ining the greater number of repeats (2 in the given
example).
A diagnostic kit for quantifying trinucleotide re~eat mutahons
EXAMPLE 4
A dia~ostic kit for carIying out a preferred embodiment of ~e methods
rdil~g to the present invention detailed above may contain ~e following
CO--c! ;l ~)entc
a) any number of suitable oligonucleotide prirners;
b) one to three ~ nc~ extension units;
3s c) Fur~er one or more primer extension units of a type not included under
b), said primer ~ .sion units co.~ p a detection moiety;
2170951
31
d) a suitable template depPn~ent e~tencion enzyrne for car~ing out the
primer eAlension unit incorporation, or extension, steps of the method;
e) suitable buffer/s in aqueous solution for carrying out the ~nne~lin~
s ext~nsion, wash and detection steps of the method; and
When the kit is to be used for Fragile XA; Kennedy disease; Myotonic
d~.LIophy; ~l~ntin~on's disease; Spinocerebellar ataxia type l; Fragile XE
m~nt~l retardation; and De.llalol,lbral pallidoluysian atrophy, it may contain,
o for example, any one or all of the specific oligonucleotide primers listed in
Tables I above for quantifying the tnnucleotide repeats eYr~nsions occurring
in these genes. When the kit is to be used in the screening for the presence of
one or all of the various listed genetic ~ise~ses it may contain any suitable
number of the oligonucleotide primers in any suitable combination. Different
lS combinations of primers may be included in kits for different intended
populations. When the kit is to be used forensic or paternity typing it may
contaul any combination of specific oligonucleotide ~ els, each designed to
quanlif~ a particular trinucleotide repeats contained in any of the mentioned orother genes. Depending on the circumstances, all of the kits may also contain
20 any nl-mber of additional oligonucleotide primers suitable for determinin~. the
presence or absence of a DNA sequence corresponding specifically to the
presence of a pathogen, for example, the presence of the AIDS virus.
Accordu~gly, one kit may be used for testing any number of genes or gene, and
this only requires that the kit contain a number of the specific oligonucleotide2s primers, all the other components of the kit being the same in all cases.
While the invention has been described with respect to a limited
nllmbPr of embodiments, it will be appreciated that many variations,
modifications and other applications of the invention may be made.
An ulsllul~,cnt for q,lalllifying trinucleotide repeat mutations
EXAMPLE S
3s Figure 4 i~ atcs an aulo-~ate~ instrument suitable for eYecllhnp. thesteps in the method of the present invention for the ~let~ ...;n~1;on of the number
of trinllcl~olide r~pea~ co~ led in specific genetic loci. The steps of the
reaction are carried out in â reaction chamber (1) to which reagents may be
2170~51
32
added or elimin~te~ via controllable valved tubing. Within the reaction chamber
included is a solid support (2) suitable for immobilizing the nucleic acid of
~l~resl, the oligonucleotide primer or both. ~ltern~tively or additionally the
reaction chamber outlet cont~ins a porous membrane (3) facilit~ting the
s retention of at least the nucleic acid of illtere~L and the oligonucleotide primer
while facilit~tin~ the sepdlali.re filtration of primer extension units. Delector
device (4) aimed at the detection of the detection moiety contained by the
prima e~cten~ion unit which is capable of base p~irin~ with a nucleotide base S'of the rcpe~ region, said nucleotide base being the first nucleotide base of a
o type not included among the nucleotide bases in the core sequence of the
trinucleotide repeats is located at the reaction chamber in cases where said
detection moiety is of an non-removable type, alternatively, the detector device(5) may be located elsewhere, for exarnple at the oudet, in cases where said
detection moiety is of a removable type. The reaction chamber further contains
lS two to three (6) inlets each connected to a reservoir (7) collt~inin~ one to two
types of the primer extension units capable of base pairing with the first (lst),
second (2nd) and third (3rd) nucleotide base in ~e core sequence of the
tnnucleotide repeats. The second or ~ird of said reservoir, depending upon the
specific application, further contains a detection moiety cont~ining primer
20 ~Ytension unit (1:)) which is capable of base painng with a nucleotide base 5' of
the repeats region, said nucleotide base being the first nucleotide base of a type
not included among ~e nucleotide bases in the core sequence of the
trinucleotide repeats. Further reservoirs and inlets for the a.lministration of
mater als such as a suitable template dependent enzyme; reaction buffer, wash
2s bu~er/s, detection buffer/s (sensitizer) and ~e like materials may also be
included (8). The reaction chamber and any reversoire may be temperature
controlled (9). Tubing connecting the reaction chamber oudet with any of dle
reservoirs may be added as well as a reconcentration (10) and/or purification
(11) device aimed at ~e reuse of discarded materials. The instrument may be
30 G~ ated m~n~ y or preferably automatically for example the instrument
o~c~alion may be controlled by a built in or external computer.