Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
21 80341
~ -- WO95/18786 PCT~P94/0000l
Method for the Production of E-1-[4 -(2- Dimethylaminoethoxy) -
Phenyl]-1-(3~-Hydroxyphenyl) -2-Phenyl-1-sutene
Field of the Invention
The present invention relates to novel methods for the
production of E-1-[4 -(2- dimethylaminoethoxy) -phenyl]-l-(3 -
hydroxyphenyl) -2-phenyl-1-butene (Droloxifene/INN) in
exceptionally high yield and purity. This compound possesses
valuable therapeutic properties in that it exhibits marked
anti-estrogen effects and is useful in the treatment of
hormone-dependent m~mm~ry tumors.
sackground Art
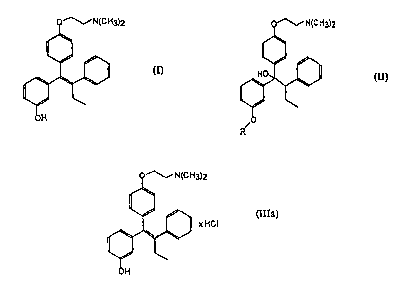
The compound having the formula (I)
o~N(cH3)2
Ih
OH
is disclosed in US 5,047,431. A method for the production of
E-;-[4 -(2- dimethylaminoethoxy) -phenyl]-1-(3 -hydroxyphenyl)
-2-phenyl-1-butene is also described in US 5,047,431 in which
the E/Z-stereoisomer mixture of formula (III) as a free base
2 1 8034 1
~ W095/18786 PCT~P94/00001
o~N(C H3)2
OH
is obtained by dehydrating compounds of the general formula
(II)
o ~N(C H3)2
'. 1~11
R/O
where R is an easily hydrolyzable protecting group
in the presence of dilute hydrochloric acid.- The yield of the
E/Z-isomer mixture of formula (III) in this process is 90%.
The E/Z-isomer mixture of formula (III) is subsequently
isolated, refluxed in concentrated hydrochloric acid, and the
hydrochloride of E-1-[4'-(2- dimethylaminoethoxy) -phenyl]
(3 -hydroxyphenyl) -2-phenyl-1-butene is isolated by
crystallization with a yield of 48~. The E-isomer
hyarochloride is then converted to E-1-[4'-(2-
di~ethylaminoethoxy) -phenyl]-1-(3'-hydroxyphenyl) -2-phenyl-1-
butene by crystallization (yield 96~). The entire process from
cc~pound (II) to compound (I) has a theoretical yield of 41~
and involves a total of two synthetic and three crystallization
steps. The afore-mentioned method is unsatisfactory due to the
21 80341
^ WO95/18786 PCT~P94/00001
low yield of therapeutically active E-1-[4 -(2-
dimethylaminoethoxy) -phenyl]-l-(3 -hydroxyphenyl) -2-phen
butene.
In EP 0313799, the process for the production of E-1-[4 -(2-
dimethylaminoethoxy) -phenyl]-1-(3'-hydroxyphenyl) -2-phen
butene of formula (I) is disclosed in which the carbinol of
formula (IV)
o--~N(CH3)2
is reacted with either sulfuric acid or hydrochloric acid, and
subsequently crystallized to give E-1-[4 -(2-
dimethylaminoethoxy) -phenyl]-1-(3 -hydroxyphenyl) -2-phenyl-1-
butene with yields ranging from 90-96% and purities ranging
from 59.4-99.7%. The procedure disclosed in EP 0313799 has the
disadvantages that the starting compound, the carbinol of
formula (IV) is only obtained with a yield of 90% from the
compound of formula (II) previously described in US 5,047,431.
Thus, the overall yield of the conversion of the compound of
formula (II) to E-1-[4 -(2- dimethylaminoethoxy) -phenyl]-l-
(3 -hydroxyphenyl) -2-phenyl-1-butene is only at-best between
81 and 86%, and the total process involves two synthetic and
three crystallization steps. Furthermore, the purity of E-1-
[4'-(2- dimethylaminoethoxy) -phenyl]-1-(3 -hydroxyphenyl) -2-
phenyl-1-butene obtained by the afore-mentioned methods is
insufficient to allow for direct formulation of E-1-[4 -(2-
dimethylaminoethoxy) -phenyl]-1-(3 -hydroxyphenyl) -2-phenyl-1-
butene into pharmaceutical
21 8034 1
`~ W095/18786 PCT~P94/00001
preparations. Therefore, additional purification steps are
necessary to obtain E-l-[4'-(2- dimethylaminoethoxy) -phenyl]-
l-(3 -hydroxyphenyl) -2-phenyl-l-butene in a form sufficiently
pure to use for the production of a medicinal preparation. The
object of the present invention is to provide methods involving
fewer steps than previously described methods for the
production of E-l-[4'-(2- dimethylaminoethoxy) -phenyl]-l-(3~-
hydroxyphenyl) -2-phenyl-l-butene from compounds of the general
formula (II) which result in an excellent overall yield and
exceptional purity of E-l-[4'-(2- dimethylaminoethoxy) -
phenyl]-l-(3'-hydroxyphenyl) -2-phenyl-l-butene suitable for
direct use in the formulation of pharmaceutical preparation.
Brief Summary of the Invention
Surprisingly, it has been found that the carbinols of the
general formula (II) can be directly converted to the
E/Z-stereoisomer mixtures of formula (III) with remarkably high
yield, and that the isolated E/Z-stereoisomer mixture can be
ccnverted to the biologically active E-l-[4'-(2-
dimethylaminoethoxy) -phenyl]-l-(3'-hydroxyphenyl) -2-phenyl-l-
butene with high yield and exceptional purity. The purity of
the resulting products of the present invention allow for the
direct formulation of pharmaceutical preparations, thereby
circumventing the previous need to further purify E-l-[4'-(2-
dimethylaminoethoxy) -phenyl]-l-(3'-hydroxyphenyl) -2-phenyl-l-
bu;ene for use in manufacturing pharmaceuticals.
Detailed Description of the Invention
Tr.e present invention provides methods for the production of E-
l-[4'-(2- dimethylaminoethoxy) -phenyl]-l-(3'-hydroxyphenyl) -
2-~henyl-l-butene of formula I
- 21 80341
- ~voss/18786 PCT~P94/00001
-
o--~N(cH3)2
,b
f 11
OH
which comprises the steps of
a) heating the compound of formula (II)
O - ~N(cH3)2
,b
where R is an easily hydrolyzable protecting group,
preferably a tetrahydropyranyl group,
at a temperature within the range of 70-80C for a time
within the range of 4-6 hours in the presence of an
organic solvent, preferably 2-propanol, and HCl gas, and
then cooling the reaction to a temperature within a range
of -5 to 5C, preferably 0C, for a time within the range
of 4-6 hours, preferably 5.5 hours, to obtain E/Z-1-[4 -
(2- dimethylaminoethoxy) -phenyl~-1-(3 -hydroxyphenyl) -2-
phenyl-l-butene x hydrochloride of formula (IIIa)
21 80341
95/l8786 PCT~P94/00001
o~N(cH3)2
(IIIa)
1~, 1 )I XHCI
OH
and b) heating the compound of formula(IIIa) at a
temperature within the range of 50-60C for a time within
the range of 10-24 hours in the absence of an organic
solvent and in the presence of 40-S0~ by volume sulfuric
acid, preferably S0~ by volume sulfuric acid, or 32-37~
hydrochloric acid, preferably 37~ hydrochloric acid. When
sulfuric acid is used in step b, the E/Z-stereoisomer
mixture of formula (IIIa) obtained by step a is heated at
a temperature, preferably within the range of 55-60~C for
a time of preferably 14 hours. When hydrochloric acid is
used in step b, the E/Z-stereoisomer mixture of formula
(IIIa) obtained by step a is heated at a temperature,
preferably within the range of 50-55C for a time of
preferably 16 hours. When the ratio of the Z- to the E-
stereoisomer of formula (IIIa) obtained by step a is
greater than 3:7, and preferably greater than 5:1, the
E/Z-stereoisomer mixture of formula (IIIa) is heated at a
temperature preferably within the range of 55-60~C for a
time of preferably 22-24 hours. Thereafter, the reaction
mixture is alkalinized, preferably with a 25~ solution of
ammonia in an organic solvent, such as dichloromethane.
The following examples are set forth as representative and
preferred embodiments of the present invention. These examples
are not to be construed as limiting the scope of the invention
2180341
- -- WO95/18786 PCT~P94/00001
in any manner. It should be understood that variations and
modifications can be made while remaining within the spirit and
scope of the invention.
Example 1
a) 25 parts of 1-[4~-(2-dimethylaminoethoxy)-phenyl] -2-
phenyl-1- [3 -(2-tetrahydropyranyloxy)-phenyl]-n-butan-1-
ol in 150 parts 2-propanol are stirred and heated at a
temperature of 70-80C and hydrochloric gas is introduced.
After ca. 5.5 hours, the suspension is cooled to 0C and
kept at this temperature for 12 hours. The precipitate is
filtered by vacuum and washed with 25 parts 2-propanol.
After drying, 21 parts (96~ theoretical yield) E/Z -1-(4 -
2-dimethylaminoethoxy)-phenyl]-1-(3 -hydroxyphenyl)-2-
phenyl-1-butene x HCl having a content of greater than 70
of the E-stereoisomer (lH-NMR) are obtained; melting
point 215-217C.
lH-NMR- spectrum (CDCl3/DMSO-d6) (100 MHz, chemical shifts
are given in ppm. TMS (~ = 0.0) s = singlet, t = triplet,
q = quartet, m = multiplet):
0.9 (3H, t) CH2C~3
2.4 (2H, q) C~2CH3
2.88 s N(C~3)2/ E-isomer, 2.95 s N(C~3)2/ Z-isomer
3.4 (2H, t) C_2N
4.3 t OC~2/ E-isomer, 4.5 t OC~2/ Z-isomer
6.2 - 7.1 (13 H, m) aromatic protons
6.9 and 12.0 (wide) OH, NH+.
b) 3 parts of E/Z-1-[4 -(2- dimethylaminoethoxy) -phenyl]
(3 -hydroxyphenyl) -2-phenyl-1-butene x HCl (isomer
mixture) are stirred into 25 parts 37~ hydrochloric acid
and the suspension is heated at 50C for 16 hours with
vigorous stirring. Subsequently the suspension is cooled
2 1 8034 1
- - WO95/l8786 PCT~P94m000l
and alkalinized by the addition of 15 parts ice and 50
parts dichloromethane with 25~ ammonia. The organic phase
is washed several times with water. After the removal of
the organic solvent 2.6 parts (95~ theoretical yield) of
E-1-[4 -(2-dimethylaminoethoxy) -phenyl]-1-(3 -
hydroxyphenyl) -2-phenyl-1-butene having a content of 100
of the E-stereoisomer (HPLC) remain. Crystals from
acetone have a melting point of 164C.
Exa~ple 2
a) 25 parts of 1-[4 -(2-dimethylaminoethoxy)-phenyl] -2-
phenyl-l- [3 -(2-tetrahydropyranyloxy)-phenyl]-n-butan-1-
ol in 150 parts 2-propanol are stirred and heated at a
temperature of 70-80C and hydrochloric gas is introduced.
After ca. 5.5 hours, the suspension is cooled to 0C and
kept at this temperature for 12 hours. The precipitate is
filtered by vacuum and washed with 25 parts 2-propanol.
After drying, 21 parts (96~ theoretical yield) E/Z -1-(4 -
2-dimethylaminoethoxy)-phenyl]-1-(3 -hydroxyphenyl)-2-
phenyl-1-butene x HCl having a content of greater than 70
of the E-stereoisomer (lH-NMR) are obtained; melting
point 215-217C.
lH-NMR- spectrum (CDC13/DMSO-d6) (100 MHz, chemical shifts
are given in ppm. TMS (~ = 0.0) s = singlet, t = triplet,
q = quartet, m = multiplet):
0.9 (3H, t) CH2C_3
2.4 (2H, q) C~2CH3
2.88 s N(C~3)2/ E-isomer, 2.95 s N(C~3)2/ Z-isomer
3.4 (2H, t) C~2N
4.3 t OC~2/ E-isomer, 4.5 t OC~2/ Z-isomer
6.2 - 7.1 (13 H, m) aromatic protons
6.9 and 12.0 (wide) OH, NH+ .
- _ W095/18786 2 1 8 0 3 4 l PCT~P94/0000l
b) 6 parts of E/Z-1-[4 -(2- dimethylaminoethoxy) -phenyl]-1-
(3 -hydroxyphenyl) -2-phenyl-1-butene x HCl (isomer
mixture) are stirred into 33 parts 50~ by volume sulfuric
acid and heated at 55C for 14 hours with vigorous
shaking. After the addition of lO parts water and 80
parts toluene, the reaction mixture is alkalinized with
25~ ammonia. After washing with water, the organic phase
is concentrated by vacuum distillation and the resulting
suspension is crystallized from toluene. The precipitate
is filtered by vacuum and washed with 6 parts toluene.
After drying, 5.3 parts (97~ theoretical yield) of E-1-
[4 -(2-dimethylaminoethoxy) -phenyl]-1-(3'-hydroxyphenyl)
-2-phenyl-1-butene having a content of lOO~ of the E-
stereoisomer (HPLC) remain. Crystals from acetic acid
ethylester have a melting point of 164C.
Example 3
3 parts of E/Z-1-[4 -(2- dimethylaminoethoxy) -phenyl]-1-
(3 -hydroxyphenyl) -2-phenyl-1-butene x HCl having a
content of more than 90~ of the Z-stereoisomer are stirred
into 30 parts 50~ by volume sulfuric acid and heated at a
temperature of 55-60C for 24 hours with vigorous shaking.
Subsequently the reaction mixture is cooled and
alkalinized by the addition of 8 parts water and 20 parts
dichloromethane with 25~ ammonia. The organic phase is
washed with water. After the removal of the organic
solvent by vacuum distillation, 2.3 parts (-83~ theoretical
yield) of E-1-[4 -(2-dimethylaminoethoxy) -phenyl]-1-(3 -
hydroxyphenyl) -2-phenyl-1-butene having a content of
99.8~ of the E-stereoisomer (HPLc) remain. Crystals from
ethanol have a melting point of 164C.
2 1 80341
_ WOgS/l8786 PcT~Ps4/oooo
Example 4
A pharmaceutical preparation containing E-1-[4 -(2-
dimethylaminoethoxy) -phenyl]-1-(3 -hydroxyphenyl) -2-
phenyl-1-butene can be prepared by mixing 111 g, mannitol,
15 g, corn starch and 6 g, alginic acid, 20.0 g, and
finely powdered E-l-[4~-(2-dimethylaminoethoxy) -phenyl]-
1-(3'-hydroxyphenyl) -2-phenyl-1-butene, granulating the
mixture and drying the granules. After carefully mixing
the granules with 0.75 g, methyl cellulose and 1.5 g,
magnesium stearate, the mixture is compressed into one
thousand tablets, each containing 20 mg, active
ingredient.