Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
W O 95/29174 21883" 1 ' PCT/EP95/01503
=
1
PROCESS FOR THE DIASTEREOSELECTIVE SYNTHESIS OF
NUCLEOSIDE ANALOGUES
The present invention relates to a diastereoselective process for the
preparation
of optically active cis-nucleoside analogues and derivatives.
Nucleosides and their analogues and derivatives are an important class of
therapeutic agents. For example, a number of nucleoside analogues have
shown antiviral activity against retroviruses such as human immunodeficiency
virus (HIV), hepatitis B virus (HBV) and human T-lymphotropic virus (HTLV)
(PCT publication WO 89/05662 and European Patent publication 0349242 A2).
In particular, 4-Amino-l-(2R-hydroxymethyl-[1,3]oxathiolan-5S-yl)-1 H-
pyrimidin-
2-one, which may be represented by the following formula:
NH2
N
O~N
HOCHa O '
s
(also known as 3TCT"' or lamivudine) and its pharmaceutically acceptable
derivatives, disclosed in International application PCT/GB91/00706,
publication
no. W091/17159, has been described as having antiviral activity, in particular
against retroviruses such as the human immunodeficiency viruses (HIV's), the
causative agents of AIDS (W091/17159) and hepatitis B virus (HBV) (European
Patent Application Publication no. 0474119).
Most nucleosides and nucleoside analogues and derivatives contain at least
two chiral centres (shown as * in formula (A)), and exist in the form of two
pairs
of optical isomers (i.e., two in the cis-configuration and two in the trans-
configuration). However, generally only the cis-isomers exhibit useful
biological
activity. Therefore a general stereoselective synthesis of cis nucleoside
analogues is an important goal.
W O 9529174 21 p() z~y 6 PCT/EP95101503
I (7Ocyr1
2
O Y purine orpyrimidine base
HOCHz
(A)
Different enantiomeric forms of the same cis-nucleoside analogue may,
however, have very different antiviral activities. M M Mansuri et al.,
"Preparation of The Geometric Isomers of DDC, DDA, D4C and D4T As
Potential Anti-HIV Agents", Bioora. Med. Chem. Lett., 1 (1), pp. 65-68 (1991).
Therefore, a general and economically attractive stereoselective synthesis of
the enantiomers of the biologically active cis-nucleoside analogues is an
important goal.
International patent application publication no. W092/20669 discloses a
diastereoselective process for producing optically active cis-nucleoside
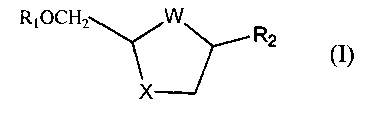
analogues and derivatives of formula (I).
RIOCH2-_~, W\J'Rz (I)
XJ
wherein
W is S, S=O, SO2, or 0;
X is S, S=O, SO2 or O;
R, is hydrogen or acyl; and
R2 is a desired purine or pyrimidine base or an analogue or derivative
thereof;
the process comprising the step of reacting the desired purine or pyrimidine
base or analogue thereof with an intermediate of formula (Ila) or (IIb)
W~,,ML W
(Ila) R3111W,R30.~ ~N+rL (Ilb)
X- X-1/
wherein
R3 is a substituted carbonyl or carbonyl derivative; and
L is a leaving group;
using a Lewis acid of the formula (III)
- - - -
WO 95/29174 2 (Q ~UU2 3ld!'~ LU PCT/EP95101503
= f
3
Ra
RS- ii -RB (III)
~ R
7
wherein
R5, RB and R7 are independently selected from the group consisting of
hydrogen; C7_20 alkyl optionally substituted by fluoro, bromo, chloro, iodo,
C1_6
alkoxy or C6_20 aryloxy; C7_20 aralkyl optionally substituted by halogen,
Ci_2o
alkyl or C1_20 alkoxy C6-20 aryl optionally substituted by fluoro, bromo,
chloro,
iodo, C1_20 alkyl or CI_20 alkoxy; trialkylsilyl; fluoro; bromo; chloro and
iodo; and
R. is selected from the group consisting of fluoro; bromo; chloro; iodo; Cl_Zp
sulphonate esters, optionally substituted by fluoro, bromo, chloro or iodo;
C1_20
alkyl esters optionally substituted by fluoro, bromo, chloro or iodo,
polyvalent
halides; trisubstituted silyl groups of the general formula (R5) (R6) (RO Si
(wherein R5, Rs, and R7 are as defined above); saturated or unsaturated
selenenyl C6_20 aryl; substituted or unsubstituted Cs_20 arylsulphenyl;
substituted or unsubstituted C6-20 alkoxyalkyl; and trialkylsiloxy.
The process of W092/20669 allows the stereo-controlled synthesis of a racemic
cis-nucleoside analogue from an equimolar mixture of (Ila) and (Ilb), and of a
given enantiomer of a desired cis-nucleoside analogue in high optical purity
if
the starting material is optically pure (Ila) or (Ilb). However, the
W092/20669
process relies on the use of a Lewis acid of formula (III).
There are a number of disadvantages associated with the use of such Lewis
acids. In particular, they are highly reactive and unstable compounds and
there
are therefore hazards associated with their use. Furthermore, they are
expensive and have significant toxic effects. These disadvantages are of
particular importance in relation to the large-scale production of nucleoside
analogues in factory processes.
We have now found that, by judicious selection of the leaving group L in
intermediates (Ila) and (Ilb), the reaction with the purine or pyrimidine
base, or
WO 95/29174 Zl 8CJp zJ0U (_ PCTIEP95/01503
=
4
analogue thereof, can be successfully effected without the addition of a Lewis
acid catalyst, and in particular, without the addition of a Lewis acid of
formula
(III).
The present invention accordingly provides a stereoselective process for
producing cis-nucleoside analogues and derivatives of formula (I)
RiOCH?.--~ W~~Rz (~)
X--lIJ
wherein
W is S, S=O, SO2, or 0;
X is S, S=O, SOz, or 0;
R, is hydrogen or acyl; and
R2 is a purine or pyrimitline base or an analogue thereof;
the process comprising the step of glycosylating the purine or pyrimidine base
or analogue or derivative thereof with an intermediate of formula (IVa) or
(lVb)
(IVa) W'~-,,,~G W
R31iu~õl J"'- R3~/ ~,,,~rG (IVb)
'XJ \X.-//
wherein R3 is a substituted carbonyl or carbonyl derivative; and
G represents halo, cyano or R9S03- where R9 represents alkyl optionally
substituted by one or more halo, or optionally substituted phenyl;
characterised in that the glycosylation reaction is effected without the
addition
of a Lewis acid catalyst.
In a preferred embodiment, the present invention provides a stereoselective
process for producing cis-nucleoside analogues and derivatives of formula (I)
as
previously defined, which process comprises the step of glycosylating the
purine
or pyrimidine base or analogue or derivative thereof with an intermediate of
formula (IVa) or (IVb) as previously defined, characterised in that the
glycosylation reaction is effected without the addition of a Lewis acid of
formula
(III):
W O 95/29174 2188306 PCTIEP95l01503
~ d
R8
I
RS- II -Re (III)
R7
wherein
R5, R6 and R7 are independently selected from the group consisting of
hydrogen; C1-20 alkyl optionally substituted by fluoro, bromo, chloro, iodo,
CI-6
5 alkoxy or C6-20 aryloxy; C7-20 aralkyl optionally substituted by halogen, Cl-
20
alkyl or Cl-20 alkoxy; Cs-2o aryl optionally substituted by fluoro, bromo,
chloro,
iodo, C1-20 alkyl or CI-20 alkoxy; trialkylsilyl; fluoro; bromo; chloro and
iodo;
and
R8 is selected from the group consisting of fluoro; bromo; chloro; iodo; Cl-2a
sulphonate esters, optionally substituted by fluoro, bromo, chloro or iodo; C1-
20
alkyl esters optionally substituted by fluoro, bromo, chloro or iodo,
polyvalent
halides; trisubstituted silyl groups of the general formula (R5) (R6) (RO Si
(wherein R5, R6, and R7 are as defined above); saturated or unsaturated
selenenyl C6-20 aryl; substituted or unsubstituted C6-20 arylsulphenyl;
substituted or unsubstituted C6-20 alkoxyalkyl; and trialkylsiloxy.
It will be appreciated that, if the glycosylation step is carried out using an
equimolar mixture of intermediates (IVa) and (lVb), a racemic mixture of cis-
nucleoside analogues will be obtained. However, it is preferred that
glycosylation is effected using an optically pure compound of formula (IVa) or
(lVb), thereby producing the desired cis-nucleoside analogue in high optical
purity.
A "nucleoside" is defined as any compound which consists of a purine or
pyrimidine base linked to a pentose sugar.
As used herein, a "nucleoside analogue or derivative" is a compound
containing a 1,3-oxathiolane, 1,3-dioxolane or 1,3-dithiolane linked to a
purine
or pyrimidine base or an analogue thereof which may be modified in any of the
~ following or combinations of the following ways: base modifications, such as
addition of a substituent (e.g., 5-fluorocytosine) or replacement of one group
by
an isosteric group (e.g., 7-deazaadenine); sugar modifications, such as
WO 95/29174 PCT/EP95/01503
2188306
6
substitution of hydroxyl groups by any substituent or alteration of the site
of
attachment of the sugar to the base (e.g., pyrimidine bases usually attached
to
the sugar at the N-1 site may be, for example, attached at the N-3 or C-6 site
and purines usually attached at the N-9 site may be, for example, attached at
N-7).
A purine or pyrimidine base means a purine or pyrimidine base found in
naturally occurring nucleosides. An analogue thereof is a base which mimics
such naturally occurring bases in that its structure (the kinds of atoms and
their arrangement) is similar to the naturally occurring bases but may either
possess additional or lack certain of the functional properties of the
naturally
occurring bases. Such analogues include those derived by replacement of a
CH moiety by a nitrogen atom, (e.g., 5-azapyrimidines such as 5-azacytosine)
or conversely (e.g., 7- deazapurines, such as 7-deazaadenine or 7-
deazaguanine) or both (e.g., 7-deaza, 8-azapurines). By derivatives of such
bases or analogues are meant those bases wherein ring substituents are either
incorporated, removed, or modified by conventional substituents known in the
art, e.g., halogen, hydroxyl, amino, Cl-6 alkyl. Such purine or pyrimidine
bases,
analogues and derivatives are well known to those skilled in the art.
As used herein, halo means bromo, chloro, fluoro or iodo.
As used herein, unless otherwise stated, alkyl means straight, branched or
cyclic saturated hydrocarbon groups, or mixtures thereof.
Optionally substituted phenyl means unsubstituted phenyl or phenyl substituted
by one or more CI-6alkyl, nitro, amino, halo or cyano groups.
Preferably R2 is a pyrimidine base. More preferably R2 is cytosine or
5-fluorocytosine.
R3 is a carbonyl linked to hydrogen, hydroxyl, trialkylsilyl, trialkylsiloxy,
Ci-30 alkyl, C7-30 aralkyl, C1-30 alkoxy, Cl-30 alkylamine (secondary or
tertiary),
C1-30 alkylthio; C6-20 aryl; C2-20 alkenyl; C2-20 alkynyl;
or R3 is 1,2-dicarbonyl, such as
WO 95/29174 2188306 PCT/EP95/01503
~
7
0o
CHj C-C-
optionally substituted with Cl-6 alkyl or C6-20 aryl;
or R3 is an anhydride, such as
0 0
II II
cH, C-o-c-
optionally substituted with Cl$ alkyl or C6-20 aryl;
or R3 is an azomethine linked at nitrogen to hydrogen, Cl-20 alkyl or CI-10
alkoxy
or CI-20 dialkylamino and at carbon to hydrogen, CI-20 alkyl, or Cl-2Q alkoxy;
or R3 is a thiocarbonyl (C=S) substituted with hydroxyl, Cl-2p alkoxy, or C1-
20
thiol.
Preferably R3 represents a group -C(=O)OR4 where R4 represents an optionally
substituted alkyl group. Preferably R4 represents a chiral auxiliary.
The term "chiral auxiliary" describes an asymmetric molecule that is used to
effect the chemical resolution of a racemic mixture. Such chiral auxiliaries
may
possess one chiral centre such as a-methylbenzylamine or several chiral
centres such as menthol. The purpose of the chiral auxiliary, once built into
the
starting material, is to allow simple separation of the resulting
diastereomeric
mixture. See, for example, J Jacques et al., Enantiomers. Racemates and
Resolutions, pp. 251-369, John Wiley & Sons, New York (1981).
Preferably the chiral auxiliary R4 will be selected from (d)-menthyl, (I)-
menthyl,
(d)-8-phenylmenthyl, (I)-8-phenylmenthyl, (+)- norephedrine and (-)-
norephedrine. More preferably R4 is (I)-menthyl, or (d)-menthyl, most
preferably
(I)-menthyl.
Preferably W is 0.
Preferably X is S.
WO 95/29174 Lr} 1885y06 PCT/EP95/01503
8
Preferably G represents halo such as Cl, Br or I, more preferably Cl,
The intermediates of formulae (lVa) and (lVb) may be isolated or they may
conveniently be generated in situ.
Suitably the intermediates of formulae (IVa) and (lVb) are generated from the
corresponding trans alcohols of formulae (Va) and (Vb):
(Va) R31u11,.l R3 W nOH (Vb)
\X~OH XJ
wherein R3, W and X are as previously defined, or from the epimeric cis
alcohols of formulae (Vc) and (Vd):
R91ii,,,W % OH Ra'~( 'W OH
X-~ (Vc) (Vd)
by reaction with a reagent, suitable to introduce the group G.
Suitable reagents for introducing the group G will be readily apparent to
those
skilled in the art and include halogenating agents such as, for example oxalyl
bromide. Preferred halogenating agents are Vilsmeier-type reagents, which
may conveniently be generated in situ by reaction of an N,N-disubstituted
amide, such as dimethylformamide (DMF), and a halogenating agent such as an
oxalyl halide, e.g. oxalyl chloride, a thionyl halide, e.g. thionyl chloride,
a
phosphorus halide, e.g. phosphorus trichloride or phosphorus oxychloride, an
alkyl or phenyl sulphonyl halide or anhydride. The halogenation reaction is
suitably effected under conventional conditions.
The intermediate of formula (lVa) or (lVb) is reacted with a silylated purine
or
pyrimidine base, conveniently in a suitable organic solvent such as a
hydrocarbon, for example, toluene, a halogenated hydrocarbon such as
dichloromethane, a nitrile, such as acetonitrile, an amide such as
dimethylformamide, an ester, such as ethyl acetate, an ether such as
CA 02188306 2004-06-21
WO 95/29174 P'CT/EP95101503
9
tetrahydrofuran, or a ketone such as acetone, or a mixture thereof, preferably
at
elevated temperature, such as the reflux temperature of the chosen solvent.
Silylated purine and pyrimidine bases may be prepared as described in
W092/20669, for
example by reacting the purine or pyrimidine base with a silylating agent such
as t-butyldimethylsilyl triflate, 1, 1, 1, 3, 3, 3-hexamethyldisilazane,
trimethylsilyl
triflate or trimethyisilyl chloride, with acid or base catalyst, as
appropriate.
Suitable methods are described in detail in the accompanying examples.
The cis-nucleoside analogue obtained from the reaction of the compound of
formula (I) with the purine or pyrimidine base or analogue thereof may then be
reduced to give a specific stereoisomer of formula (I). Appropriate reducing
agents will be readily apparent to those skilled in the art and include, for
13 example, hydride reducing agents such as lithium aluminium hydride, lithium
borohydride or sodium borohydride. We have found that stereointegrity is
maintained using sodium borohydride in the presence of a phosphate or borate
buffer, for example dipotassium hydrogen phosphate, as the reducing agent.
According to the process of the invention, as well as the process described in
W092/20669, the final compound is typically obtained as a solution in a polar
solvent, such as an aqueous solvent. This presents a practical problem in that
compounds of formula (I) have a high solubility in polar media, making their
efficient isolation from such media difficult. We have now found that
compounds of formula (I) may be efficiently isolated from solution in polar
solvents by formation of a salt having poor aqueous solubility. If desired,
the
water-insoluble salt may subsequently be converted to the free base, or to a
different salt thereof by conventional methods. We have further found that the
salicylate salt is particularly suitable for this purpose.
The present invention thus provides a process as previously described further
comprising the step of isolating the compound of formula (I) as a water-
insoluble
salt, especially a salicylate salt.
CA 02188306 2004-06-21
WO 95/29174 PCT/EP95/01503
Salicylate salts of compounds of formula (I) are within the scope of the
pharmaceutically acoeptable derivatives described and claimed in European
Patent Application publication no. 0382526 and publication no. W091/17159,
but are not specifically disclosed therein. Such salts are therefore novel and
5 form a further aspect of the present invention.
In a further or altemative aspect, the present invention provides salicylate
salts
of compounds of formula (I), or hydrates thereof.
10 In particular, we have found that formation of the salicylate salt of 4-
amino-l-
(2R-hydroxymethyl-[1,3]oxathiolan-5S-yl)-1 H-pyrimidin-2-one (lamivudine,
3TC710) affords considerable advantages for the isolation of that compound
from
polar solvents.
In a preferred embodiment the invention therefore provides 4-amino-l-(2R-
hydroxymethyl)-[1,3]oxathiolan-5S-yi)-1 H-pyrimidin-2-one salicylate, or
hydrates
thereof.
The salicylate salt of lamivudine is a pharmaceutically acceptable salt and as
such it and its hydrates may be used as antiviral agents as described in
W091 /17159.
The salicylate salt of lamivudine or its hydrates may be formulated as a
pharmaceutical composition as described in W091 /17159.
The salicylate salts of compounds of formula (I) may be prepared by treating a
solution containing a compound of formula (1) with salicylic acid. Suitable
solvents include for example, water and polar organic solvents such as ethers,
for example tetrahydrofuran or dioxan and alcohols, for example methanol and
ethanol, or mixtures of solvents, in particular mixtures containing an organic
solvent and water.
The salicylate salts are conveniently converted, if desired, to the
corresponding
free bases by treatment with a base, suitably a tertiary amine such as, for
example triethylamine.
WO 95/29174 2180306 PCT/EP95/01503
=
11
Other suitable water-insoluble salts and methods for their preparation and
conversion to free bases will be readily appreciated by those skilled in the
art.
Intermediate alcohols (Va) and (Vb) and the epimeric cis alcohols (Vc) and
(Vd)
may be prepared by the methods described in W092/20669, for example, by
reduction of the corresponding carbonyl compounds or by condensation of an
aldehyde of formula R3-CHO, or a derivative thereof, with hydroxyacetaldehyde
or mercaptoacetaldehyde, or suitable derivatives thereof. Further details of
the
preparation of such alcohols may be found in the accompanying examples.
Compounds of formulae (Va) and (Vb) are key intermediates for the preparation
of enantiomerically pure cis-nucleoside analogues or derivatives, according to
the process of the invention. The absolute stereochemistry of the groups R3, W
and X in (Va) or (Vb) is preserved in the resulting cis-nucleoside analogue or
derivative of formula (I).
Reactions for the preparation of alcohols of formulae (Va) and (Vb) and their
cis
epimers (Vc) and (Vd) typically result in the formation of mixtures of
isomers.
When compounds of formulae (Va) or (Vb) are isolated by crystallisation from
mixtures containing their enantiomers and/or their cis stereoisomers, the
yield
may be limited by the proportion of the desired isomer (Va) or (Vb) present in
solution.
We have now found that crystallisation of the trans isomers (Va) and (Vb) is
favoured over the crystallisation of the corresponding cis isomers (Vc) and
(Vd).
Where R3 is an achiral moiety, a 1:1 mixture of the trans isomers (Va) and
(Vb)
may be crystallised from mixtures of the cis and trans isomers (Va), (Vb),
(Vc)
and (Vd).
Accordingly, the present invention provides, in a further or alternative
aspect, a
method for enhancing the yield of the trans isomers (Va) and (Vb) from a
mixture of the trans and cis isomers, which method comprises treatment of the
mixture of trans and cis isomers, at least partially in solution, with an
agent
WO 95/29174 2138J 06 PCT/EP95/01503
12
capable of effecting interconversion of the isomers without complete
suppression of the crystallisation of the trans isomers.
We have further discovered that, where R3 is a chiral moiety, a single trans ~
enantiomer of formula (Va) or (Vb) may be selectively crystallised from a
mixture
of stereoisomers.
Thus, for example, compounds of formula (Va) wherein R3 represents -C(=0)RA,
where Ra is 1-menthyl, can be selectively crystallised from a mixture of
stereoisomers, in particular a mixture containing alcohols (Va), (Vb) and the
epimeric cis alcohols (Vc) and (Vd).
Similarly, compounds of formula (Vb) wherein R3 represents -C(=O)R4, where R4
is d-menthyl, can be selectively crystallised from a mixture of stereoisomers,
in
particular a mixture containing alcohols (Va), (Vb) and the epimeric cis
alcohols
(Vc) and (Vd).
Therefore, in a preferred aspect, the present invention provides a method for
enhancing the yield of a single enantiomer of formula (Va) or (Vb) from a
mixture of isomers, which method comprises treatment of the mixture of
isomers,
at least partially in solution, with an agent capable of effecting
interconversion of
the isomers without complete suppression of the crystallisation of the desired
single enantiomer (Va) or (Vb).
Agents capable of effecting interconversion of the isomers without complete
suppression of the crystallisation of the trans isomers include, for example,
alcohols, such as, for example, methanol, ethanol, n-propanol, i-propanol, n-
butanol, i-butanol, t-butanol, and organic bases, in particular tertiary
amines, for
example, pyridine and triethylamine and Hunig's base. A preferred agent is
triethylamine.
The interconversion of isomers may be effected in any suitable solvent or
mixture of solvents which does not otherwise react with the alcohols of
formulae
(Va) or (Vb) or their cis isomers, under conditions of concentration and
temperature which permit crystallisation of the desired isomer or isomers and
WO 95/29174 PCTlEP95f01503
=
13
which do not cause significant degradation of the desired isomer or isomers.
Suitable solvents may include for example, aliphatic or aromatic hydrocarbons,
ethers, esters and chlorinated hydrocarbons. The interconversion will
preferably
be effected at a temperature of about -20 to 120 C, more preferably in the
range of about -10 to 80'C, such as about 00 to 50 C.
It will be appreciated by those skilled in the art that selection of solvent,
temperature, interconversion agent and, particularly, the quantity of the
interconversion agent is best conducted as an integrated exercise dependent on
the nature of the groups R3, X and W present in the isomers. However, when an
organic base is used as the interconversion agent, the preferred quantity is
generally less than two mole-equivalents based on the total of all isomers of
(Va) and (Vb) present.
Further guidance as to preferred reaction conditions may be gained from the
accompanying examples.
The interconversion of isomers may be conducted separately from the
preparation of the isomeric mixture; however, it is conveniently conducted
concomitantly with that preparation.
The interconversion procedure may also be used to increase the isomeric purity
of isolated (Va) or (Vb).
By means of the interconversion process, the isolated yield of the desired
isomer (Va) or (Vb) may be enhanced to greater than 50% of theory (based on
formation of all stereoisomers), typically to between about 60% and about 90%
of theory; but it is not ruled out that yields approaching 100% of theory may
be
obtained.
A particularly preferred embodiment of the process of the present invention
using I-menthol as chiral auxiliary is represented in Scheme 1 and is
described
, in detail in the accompanying examples, which are to be construed as
illustrative of the invention and not as limiting thereof.
W O 95129174 L1%~~.,3U(U L PCT/EP95/01503
UU
14
Scheme I
0II 0
0 OH O OH
LO/
OH S NH2
O J I
L N~
0 JI.' ~Ci O \NJ
S 0~
S-i
(not isolated)
NHz NH2
OH
N N
CO=H
OO~N ~ ~~ pO~N
HO HO \ J _ - - - -
~~~ H20 S
The invention is further illustrated by the following non-limiting examples.
All
temperatures are in degrees centigrade. DMSO means dimethyl sulphoxide.
Example 1
4-Amino-1-(2R-hydroxymethyl-f 1 3loxathiolan-5S-yl)-1 H-pyrimidin-2-one
(a) (2R 5R)-5-Hydroxy-f1 3loxathiolane-2-carboxylic acid, 2S-isopropyl-5R-
methyl-I R-cyclohexyl ester
WO 95/29174 2188306 PCT/EP95l01503 -
=
A mixture of 1-menthyl glyoxylate hydrate (25g) and acetic acid (2.5mL) in
toluene (125mL) was stirred and heated to reflux. Water was removed by
azeotropic distillation via a Dean-Stark trap. The resulting solution of 1-
menthyl
5 glyoxylate was concentrated by distillation under reduced pressure
collecting ca
70mL distillate, and then cooled to 20-25 . The volume was adjusted to 75mL
by adding ca 15mL toluene, dithianediol (8.25g) was added, and the mixture
heated at reflux for about lh. The mixture was cooled to about 800, and
clarified. The filtrate was cooled to 0-5 , and a solution of triethylamine
(1.5mL)
10 in hexane (150mL) was added over ca 1.25h at 0-5 . The resulting suspension
was stirred at 0-5 for about 6h, then the product isolated by filtration. The
product was washed with a mixture of toluene and hexane (1:3, 2x50mL), and
dried in vacuo at 40-45 to constant weight.
15 (b) (2R,5R)-5-(4-Amino-2-oxo-2H-ovrimidin-l-vl)- r1,31oxathiolane-2-
carboxvlic
acid, 2S-isopropvl-5R-methvl-1 R-cvclohexvl ester
A solution of (2R,5S)-5-chloro-[1,3]oxathiolane-2-carboxylic acid, 2S-
isopropyl-
5R-methyl-1 R-cyclohexyl ester was prepared as follows:
A solution of (2R,5R)-5-hydroxy-[1,3]oxathiolane-2-carboxylic acid, 2S-
isopropyl-5R-methyl-1 R-cyclohexyl ester (300g) in dichloromethane (3000mL)
containing methanesulphonic acid (0.7mL) was treated with dimethylformamide
(85mL), cooled to ca 8 and thionyl chloride (80mL) added over ca 10min. The
resultant solution was stirred at 10-15 for ca 1.5h, then concentrated by
distillation under atmospheric pressure (over ca 1.5h), collecting ca 2.1L
distillate. The solution was cooled to 20-25 .
A solution of silylcytosine was prepared as follows:
A suspension of cytosine (115.5g), methanesulphonic acid (0.7mL) and
hexamethyldisilazane (242mt) was heated in toluene (290mL) at reflux until a
clear solution was obtained (ca 1.5h).
WO 95129174 PCTIEP95101503
2188306
16
The solution of silylcytosine was treated with triethylamine (145mL), the
solution
of (2R,5S)-5-chloro-[1,3]oxathiolane-2-carboxylic acid, 2S-isopropyl-5R-methyl-
1 R-cyclohexyl ester added maintaining a gentle reflux, washing in with
dichloromethane (300mL). The resulting mixture was heated at reflux for 4h,
and added to a mixture of triethylamine (73mL) and water (1200mL) held at 30-
350, over ca 1.5h. The resulting suspension was stirred for ca 45min, then
hexane (1200mL) added over ca 10min at 30-35 . The suspension was stiired
at ambient temperature overnight, then filtered. The solid was washed with
water (2x600mL) and isopropyl acetate (2x600mL), and dried in vacuo at 40-45
to constant weight. 'HNMR (D6-DMSO) Sõ 0.75 (3H,d); 0.89(d), 0.9(m), 0.91(d),
1.0-1.2(m) (9H); (9H,m); 1.43, 1.50 (2H,m); 1.67 (2H,m); 1.9-2.0 (2H,m); 3.14
(1 H,dd); 3.55 (1 H,dd); 4.69 (1 H,dt); 5.70 (1 H,s); 5.80 (1 H,d), 6.36 (1
H,dd), 7.28
(brs), 7.33 (brs) (2H); 7.97 (1 H,d)
(c) 4-Amino-1 -(2R-hvdroxvmethvl-r1.31oxathiolan-5S-vl)-1 H-pvrimidin-2-one
monosalicylate
A solution of dipotassium hydrogen phosphate (137g) in water (150mL) was
stirred at ca 20 , and (2R,5R)-5-(4-amino-2-oxo-2H-pyrimidin-l-yl)-
[1,3]oxathiolane-2-carboxylic acid, 2S-isopropyl-5R-methyl-lR-cyclohexyl ester
(100g) added. IMS (750mL) was added, and the suspension stirred for 10min.
A solution of sodium borohydride (20g) in water (200mL) containing sodium
hydroxide solution, 25% w/w (2mL) was added over 70min, keeping the
temperature in the range 15-30 . The addition funnel was rinsed with water
(50mL), and the mixture stirred at 15-30 until the reaction was judged
complete
by HPLC (150min). The mixture was allowed to settle, and the lower aqueous
layer discarded. The pH of the organic phase remaining was adjusted to 4-4.5
with conc. hydrochloric acid (27mL), whilst maintaining the temperature in the
range 20-25 . The addition funnel was rinsed with water (20mL), then the pH of
the solution adjusted to 6.8-7.2 with 2M sodium hydroxide solution (110mL).
The addition funnel was rinsed with water (20mL), and the reaction mixture was
transferred to a distillation vessel, washed in with water (50mL), and the
solution heated to reflux. The solution was concentrated to ca 6.45vol under
atmospheric pressure, then cooled to 20-25 .
W O 95R9174 2188306 PCT/EP95101503
=
17
Menthol was removed by extraction with toluene (500mL, 2 x 200mL), the
aqueous phase was diluted with water (255mL) then treated with salicylic acid
(36g), washing in with water (40mL). The mixture was heated to give a solution
(at 71 ), then cooled to 58 . The solution was seeded with authentic
lamivudine
salicylate, then cooled to 5-10 over ca 4h. The suspension was stirred for 1h
at this temperature, then filtered. The product was washed with water (1 x
100mL, 2 x 200mL), and dried in vacuo at 45-50 to constant weight. 'HNMR
(Ds-DMSO) Sõ 3.11 (dd), 3.45 (dd) (2H); 3.77 (2H,m); 5.20 (1H,m); 5.82 (IH,d);
6.22 (1 H,m); 6.91 (2H,m); 7.48 (1 H,m); 7.62 (2H,br); 7.80 (1 H,dd); 7.92 (1
H,d).
(d) 4-Amino-1-(2R-hvdroxymethvl-f 1.31oxathiolan-5S-yl)-1 H-pvrimidin-2-one
4-Amino-1-(2R-hydroxymethyl-[1,3]oxathiolan-5S-yl)-1 H-pyrimidin-2-one
monosalicylate (66.7g) was stirred with IMS (470mL), and heated to 70-75 to
give a solution. The solution was clarified into a crystallisation vessel, and
rinsed in with a further 170mL IMS. Triethylamine (26mL) was added, and the
solution distilled until 280mL remained. The solution was cooled to 70 over
20
min, seeded, then isopropyl acetate held at 60 (600mL) added over 2.25h,
maintaining the temperature above 55 . The mixture was cooled to room
temperature overnight, then cooled to 8-10 and stirred for lh. The product
was
isolated by filtration (transferred to the filter with 30mL isopropyl
acetate),
washed with isopropyl acetate (2 x 130) and dried in vacuo at 40-45 , to
constant weight. 'HNMR (D6-DMSO) 5H 3.10 (1H,dd); 3.39 (1H,dd); 3.72
(2H,m); 5.15 (1H,t); 5.29 (1H,t); 5.72 (1H,d); 6.19 (1H,dd); 7.17 (1H, brs);
7.22
(1 H,brs); 7.80 (1 H,d).
.