Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 96101830 4 6 7 7 PCTIFR95100916
1
DERIVES DE SILYLMETHYL-ANILINES, LEURS PROCEDES DE FABRICATION ET
LEUR UTILISATION COMME MEDICAMENT
La présente invention a pour objet des dérivés de silylméthyl-anilines,
leurs procédés de fabrication, les compositions pharmaceutiques les
contenant et leur utilisation comme médicaments.
La génération des métabolites oxygénés hautement réactifs est un trait
essentiel de nombreux composatits du métabolisme normal (génération du
métabolisme énergétique par la chaîne de respiration mitochondriale,
détoxication des xénobiotiques par les cytochromes, destruction des
micro-organismes par phagocytose, et même ovulation et fertilisation).
En dépit de l'existence de systèmes de défense antioxydants endogènes
puissants, le débit des espèces oxydantes générées localement peut
fréquemment déborder les défenses naturelles de l'organisme et entraîner
des destructions tissulaires en raison de la péroxydation des lipides
membranaires des cellules et des organites, de la dénaturation des
protéines structurelles et fonctionnelles (enzymatiques) du clivage
radicalaire de brins d'ADN ou d'ARN, et de l'altération des constituants
polvsaccharidiques du tissu interstitiel et des membranes basales.
Il en résulte que cette toxicité locale des radicaux libres et autres dérivés
toxiques de l'oxygène constitue une voie commune à de nombreuses
pathologies telles que les dommages causés par la radiothérapie, les
75 syndromes d'hyperoxygénation (par exemple les dysplasies néonatales
bronchopulmonaires), les réactions inflammatoires (cfr. G. B. Buckley,
Surgery, 113 479, 1993), les maladies auto-immunes, les atteintes toxiques
durant les phases de reperfusions post ischémiques (J. Y. Arigou et al.
Arch. Mal. Coeur, 86, 105-109, 1993 ; N. Kaul et al. J. Pharmacol. Tox.
Methods, 3.0 55-67, 1993), la reperfusion d'organes y compris transplantés,
la carcinogenèse (cfr. H. Sies, Ang. Chem. Int. Ed., 25. 1058-1071, 1986),
l'athérosclérose (cfr. D. Steinberg, New England J. Med. 320, 915-924, 1989
M. Aviram, Atherosclerosis, M, 1-9, 1993) et les processus de vieillissement
au niveau cellulaire (cfr. E. Stadtman, Science, 257 1220, 1992 ; B. Ames et
M. Shigenagu, Ann. N.Y. Acad. Sciences, 85, 1993 ; P.N.A.S., 9a 7915, 1993)
ou neuronal (C. Olanow, TINS, 16. 439, 1993 ; C. Smith et al. Ann. N.Y. Acad.
Sciences; 110, 1993).
WO 96101830 PCT/PR95100916
zf 94677
2
De nombreuses études pharmacologiques et cliniques suggèrent que certains
dérivés antioxydants ou piégeurs de radicaux libres peuvent jouer un rôle
bénéfique dans la prévention et/ou le traitement des pathologies mentionnés
précédemment. A titre d'exemple, çitons la vitamine E ou tocophérols, les
tocotriénols, la vitamine C, le b-carotène, les flavonoïdes tels que la
quercétine, le probucol ou encore l'ebselen. (H. Sies, "Oxidative stress",
Academic Press, 1991 ; A. Ong, L. Packer, "Lipid-soluble antioxydants :
biochemistry and clinical applications", Birkhauser, 1992 ; B. Halliwell,
Drugs, 42 569, 1991 ; M. Santrucek, J. Krepelka, Drugs of the future, 13= 974,
1988).
Certaines amines aromatiques ont également été caractérisées comme
antioxydants. C'est ainsi que par exemple l'aniline et certains de ses dérivés
ont
trouvé une utilité pour retarder le vieillissement oxydatif des caoutchoucs ou
pour protéger certains produits chimiques contre les polymérisations induites
par des oxydants et/ou des radicaux libres. Toutefois, nombre de ces dérivés
n'ont pas trouvé d'application thérapeutique à cause de leur.faible potentiel
antioxydant comparé à leurs effets secondaires toxiques ; c'est ainsi que la
diphenyl-phenylène diamine (DPPD) est capable de protéger l'oxydation des
LDL de lapin in vivo et ainsi de retarder la progression des plaques
athéromateuses, mais son utilisation en clinique humaine est exclue à cause de
ses propriétés mutagènes (C. P. Sparrow et al. J. Clin. Invest., 89= 1885-
1891,
1992).
La demanderesse a présentement découvert de nouvelles silylméthylamines
aromatiques qui possèdent des propriétés pharmacologiques et thérapeutiques
intéressantes puisque ces dérivés se comportent comme des antioxydants
capables de protéger les systèmes biologiques tels que les tissus, les
membranes, les cellules, les protéines structurelles et/ou fonctionnelles et
les
lipides des agressions dues aux radicaux libres et autres dérivés toxiques de
l'oxygène. La très bonne efficacité des composés de l'invention comme
antioxydants, (en particulier leur capacité à protéger les LDL humaines vis-à-
vis des modifications oxydatives induites par le cuivre ou les cellules
endothéliales et leur capacité à piéger les radicaux hydroxylés) associée à
leur
accès facile et peu onéreux par synthèse chimique, à leur stabilité et à leur
WO 96/01830 2194677 PCTIFR95/00916
~
3
faible toxicité, les rend précieux pour le traitement des pathologies dans
' lesquelles une péroxydation joue un rôle initiateur et/ou aggravant.
' Certains dérivés de silylméthylaniines aromatiques sont décrits dans la
littérature. C'est ainsi que des dérivés de N-(dimethylphenylsilyl)-methyl-
fiiran-2-carboxamide ont été décrits comme fungicides (cfr. C.A, 115,
497909), certains dérivés de (silylmethylamino)-nitroarenes ont été décrits
comme matériaux optiques non linéaires (EP 404134) et comme additifs anti-
déflagrants (Combust. Flame, 59 151-165, 1985). Enfm, quelques dérivés ont
été décrits comme intermédiaires de synthèse ou produits secondaires de
réarrangements (J. Organomet. Chem., 113, 115-125, 1976 ; J. Org. Chem.,
-l1 1962-1965, 1976 ; J. Chem. Soc. ; Chem. Commun. 15= 640-641, 1975).
Par ailleurs, l'étude physico-chimique des propriétés de la fonction amine
dans
les silylméthylamines a mis en évidence l'influence du silicium sur leur
potentiel d'oxydation (J. Organomet. Chem., 29. 33-40, 1971) et leur basicité
(J. Am. Chem. Soc., 73 3871, 1951).
Les silylméthylamines aromatiques citées dans la littérature ne sont à aucun
moment décrites comme possédant une activité antioxydante sur des systèmes
biologiques ; cette propriété et ses conséquences thérapeutiques sont
revendiquées dans la présente invention. Tous les dérivés de la présente
invention se différencient donc des dérivés les plus proches de l'art
antérieur
par leur profil pharmacologique et leurs applications thérapeutiques, comme le
prouve l'étude pharmacologique ci-après exemplifiée.
La présente invention a pour objet l'utilisation comme médicament des
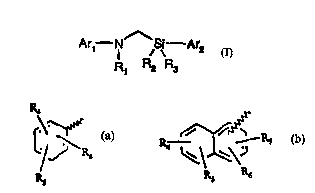
silylméthylamines aromatiques qui répondent à la formule générale (I) :
Ar,-NS\ Ar2
R' R2 R3 (I)
dans laquelle
RI représente un atome d'hydrogène ; un résidu aromatique tel qu'un phényle
ou un naphtyle pouvant être diversement substitué ; un résidu aIlcyle
contenant
de 1 à 20 atomes de carbone en chaine droite ou ramifiée pouvant contenir une
ou plusieurs insaturations ; et/ou des groupes fonctionnels tels que alcool,
WO 96/01830 2194677 PCTIFR95100916
4
éther, thiol, thioéther, halogène (fluor, chlore ou brome), cétone, ester,
amine,
amide, carbamate, carbonate, urée, nitrile, nitro, sulfone, sulfonamide.
R2 et R3, identiques ou différents représentent chacun un résidu aromatique,
un radical hydrogenocarboné linéaire ou ramifié contenant de 1 à 20 atomes de
carbone pouvant contenir une ou plusieurs insaturations et/ou des groupes
fonctionnels.
Arl et Ar2, identiques ou différents, représentent un groupement aryle choisi
parmi les résidus aromatiques suivants :
Ra
yr
X ~ R' ~1 \~J c R,
RS R3 R6 R6
R,,
R'\ /rS I \
R X' X
R6 R' S
R' '
RS R6
dans lesquels la liaison brisée représente une liaison entre le noyau
aromatique
et l'atome d'azote de la formule (I) lorsqu'il s'agit de Arl et une liaison
entre le
noyau aromatique et l'atome de silicium lorsqû il s'agit de Ar2.
R4, R5, R6 et R7, identiques ou différents peuvent être en différentes
positions
?5 (ortho, meta ou para) sur le noyau aromatique auquel ils sont attachés
représentant chacun un atome d'hydrogène, un résidu aromatique (par exemple
un phenyle pouvant être diversement substitué), un radical alkyle contenant de
wo96101830 2194677
PCT/FB95100916
I à 20 atomes de carbone en chaine droite ou ramifiée [pouvant contenir une
ou plusieurs insaturations et/ou des groupes fonctionnels tels que alcool,
éther,
thiol, thioether, halogène(s), cétone, ester, amine, amide, carbonate,
carbamate, urée, nitrile, nitro, sulfone, sulfonamide], un radical hydroxyle
5 (OH), thiol (SH), alcoxy (OR8), acyloxy (OCOR8), thioéther (SR'g),
trifluorométhyle (CF3), halogène (chlore, fluor, iode ou brome), une triple
liaison diversement substituée, un radical silylé, un nitrile (CN), nitro
(NO2),
un radical oxazolyle, une amine (NR8R9), un carbonyle (COR8), un ester
(C02R8), une amide (NRgCORg), un carbonate (NR8CO2R9), une urée
(NRgCONHRg), une sulfonamide (NR8SO2R9 ou SO2NR8R9) dans lesquels
Rg et Rg, identiques ou différents, représentent un résidu alkyle, aryle ou
alkylaryle pouvant lui-même être diversement substitué.
X et Y, identiques ou différents, représentent chacun un CH2, un atome
d'oxygène ou de soufre ou un résidu NRIO dans lequel RIO représente un
hydrogène, un groupement alkyle ou aryle.
Les composés de formule (I) contenant un ou plusieurs centres asymétriques
présentent des formes isomères. Les racémiques et les énantiomères pures de
ces composés font également partie de cette invention.
L'invention comprend également les sels, solvates (par exemple les hydrates)
et bioprécurseurs de ces composés acceptables pour l'usage thérapeutique.
Parmi les sels acceptables pour l'usage thérapeutique des amines de formule
(1), on citera les sels formés par addition avec des acides organiques ou
minéraux, par exemple les chlorhydrates, bromhydrates, sulfates, sulfonates,
fumarates, maléates, tartrates et succinates.
L'expression bioprécurseurs telle qu'elle est utilisée dans la présente
invention
= 30 s'applique à des composés de formule (I), mais, qui, administrés à un
animal
ou à un être humain sont convertis dans l'organisme en un composé de formule
M.
La présente invention décrit une nouvelle classe d'antioxydants qui se
distingue de l'art antérieur par leur structure chimique puisque les dérivés
faisant partie de la présente invention sont des silylamines aromatiques qui
n'ont jamais été décrites comme pouvant être utiles pour le traitement tant
WO 96l01830 2194677 PCT/FR95100916
~
6
curatif que préventif des pathologies liées à l'oxydation ou aux effets
délétères
de radicaux libres. De plus, la plus grande partie des composés faisant partie
de la présente invention est représentée par des nouvelles molécules dont la
structure et le procédé de fabrication doivent eux-aussi être considérés comme
partie intégrale de la présente invention.
Devront donc être considérs comme faisant partie intégrale de 1` invention,
les
dérivés de formule générale (I)
Ari-NS; Ar2
Ri Rz R3
dans laquelle Arl, Ar2, RI, R2 et R3 sont décrits conune précédemment, si ce
n'est que, :
a) Lorsque Arl représente un phényle, un o-méthyle-phényle, un o-
chlorophényle, un o,p dichlorophényle, un o-méthoxy phényle ou un o-
(hydroxyinéthylphényle) et simultanément que R2 et R3 représentent un
méthyle, et RI représente H, CH3, ou C2H5, alors Ar2 doit être différent de
C6H5.
b) Lorsque Arl, Ar2, R2 et R3 représentent simultanément C6H5, RI doit être
différent d'un méthyle.
c) Lorsque, simultanément, Ar2 et Ar3 représentent C6H5, R2 représente CH3
et RI représente un méthyle ou un éthyle, alors Arl doit être différent d'un
phényle et d'un paranitrophényle.
Les dérivés de formule générale (I) sont généralement préparés par
condensation d'une amine aromatique de formule générale (II) :
WO 96/01830 2194677 PCT/FR95/00916
7
Ar,-N
R'~
(II)
dans laquelle Arl est défini comme précédemment et R'1 est, soit identique à
RI, soit un groupe précurseur de RI qui sera transformé ultérieurement en RI,
avec un halogénométhylsilane de formule générale (III)
ZSi-Ar
2
R3 R4 (III)
dans laquelle R3, R4 et Ar2 sont décrits comme précédemment et Z représente
un groupe portant tel que Cl, Br, I, OMs, OTs, OTf.
Le choix du substituant R' 1 dans le réactif (II) dépendra essentiellement de
la
nature de Rl dans la formule (I).
C'est ainsi que les dérivés de formule (I) dans lesquels Rl représente un
atome
d'hydrogène sont préparés soit directement par condensation des
intermédiaires de formule générale (IIa) :
Arl NH2 (Ira)
dans laquelle Arl est défuù comme précédemment avec un
halogénométhylsilane de formule (III) en présence d'une base forte telle que
par exemple le n-butyllithium, le sec-butyllithium, le diisopropylamidure de
lithium, l'amidure de sodium, dans un solvant aprotique anhydre tel que le
tétrahydrofuranne, l'éther, le diméthoxyethane, éventuellement en présence de
DABCO, TMEDA ou HMPT, à une température comprise entre - 78 et 20 C
selon les techniques et procédés bien connus de l'homme de métier qui, entre
autre, indiquent qu'il est souvent préférable de mélanger la base forte avec
l'amine de formule (I) avant d'introduire l'halogénométhylsilane de formule
(Ila) ; soit indirectement, par condensation initiale d'un dérivé de formule
(lIb) :
Arl - NHCOCF3 (IIb)
WO 96101830 PCT/FR95/00916
~
2194677
8
avec un halogénométhylsilane de formule (III) en présence d'une base telle que
par exemple l'hydrure de sodium ou de potassium dans un solvant aprotique
anhydre tel que le tétrahydrofuranne, ou le diméthylformamide à une
température comprise entre - 20 C et 20 C suivi de la déprotection de la
trifluoroacétamide ainsi formée par réaction avec des réactifs bien connus
pour
ce type de transformation tels que l'hydroxyde de potassium ou l'hydroxyde de
sodium dans le méthanol à une température comprise entre 0 et 50 C ou
encore l'utilisation de réducteurs tels que par exemple l'hydrure de lithium
et
aluminium dans l'éther ou le tétrahydrofuranne ou le borohydrure de sodium
dans un solvant alcoolique tel que l'éthanol.
Une méthode particulièrement appréciée pour la préparation des dérivés de
formule (1) dans lesquels RI représente un substituant alkyle pouvant être
diversement substitué consiste à condenser une amide intermédiaire de
formule générale (IIc)
--
O
Ar~-N k R"i
H (IIc)
dans laquelle Arl est défini comme précédemment et R"I représente un
substituant défuù de telle sorte que - CH2R"I soit équivalent à RI, avec un
halogénométhyle silane de formule (III) en présence d'une base telle que
l'hydrure de sodium ou de potassium, l'amidure de sodium, le
diisopropylamidure de lithium dans un solvant tel que le tétrahydrofuranne, le
DMF, le DMSO, le DME, à une température comprise entre O C et 100 C,
suivi de la réduction de la fonction amide de l'intermédiaire ainsi formé par
les
méthodes bien connues pour ce type de transformation telles que l'utilisation
de l'hydrure de lithium et d'aluminium, dans un solvant tel que le
tétrahydrofuranne ou l'éther anhydre.
Lorsque les intermédiaires de formule générale (III) sont difficilement
accessibles, une méthode complémentaire mais particulièrement appréciée de
préparation des composés de formule générale (I) dans laquelle Arl, RI, R2 et
Ar2 sont définis comme précédemment mais RI représente un hydrogène
consiste à faire réagir un intermédiaire de formule (IV) :
- - -
WO 96/01830 PCT/FR95100916
~ 2194677
9
~1CH3
Ari-N
(IV)
dans laquelle Arl est décrit comme précédemment et R"' 1 représente un
groupe protecteur tel qu'une formamidine (cfr. A. I. Meyer, S. Hellering,
Tetrahedron Lett., 22. 5119, 1981) avec une base forte telle que par exemple
le butyllithium secondaire dans le tétrahydrofuranne ou l'éther en présence de
TMEDA et ensuite de condenser l'organolithien ainsi formé avec un halogeno-
silane de formule générale (V) :
Z.
S\ Ar2
RZ R3 (V)
dans laquelle Ar2, R2 et R3 sont décrits comme précédemment et Z' représente
un halogène tel qu'un chlore, un brome ou un iode. Le produit ainsi formé est
ensuite transformé en composé de formule (I) dans laquelle RI représente un
hydrogène après l'application d'une méthode de déprotection telle que
l'hydrolyse en milieu acide (HCI ou acide trifluoroacétique).
Par ailleurs, les dérivés de la présente invention peuvent également être
préparés par réaction d'une amine aromatique de structure générale (VI)
Arl NHR1 (VI)
dans laquelle Arl et RI sont décrits comme précédemment avec un
chlorométhyl-chlorosilane de structure générale (VII) :
CI^S\ Ar2
CI R3 (VII)
en présence d'un fluorure tel que le fluorure de potassium dans un solvant
aprotique polaire tel que l'acétonitrile à une température entre 40 et 80 C,
suivi
de la réaction du produit ainsi formé avec un orgamétallique de formule
générale (VIII) :
WO 96101830 2 1 ~ ~ ~ ~ PCT/FR95100916
~
R2 - M (VIII)
dans laquelle R2 est décrit comme précédemment et M représente Li, Na, K,
5 MgCI, MgBr ou MgI selon une méthode préalablement décrite pour la
préparation de triméthysilylméthyl-aniline (Cfr. J. Eisch, C.S. Chiv, J.
Organomet. Chem. 358, C I-C5, 1988).
Les intermédiairés silylés de structure (III), (V) ou (VII) sont disponibles
ou
10 préparés par les techniques et méthodes bien connues de la chimie des
organosilylés et sont décrites dans "Silicon reagents in organic synthesis"
(E.W. Calvin, Academic Press, 1988), "The chemistry of organic silicon
compounds" (S. Pataï, Z. Rappoport, J. Wiley, 1989) et "Silicon reagents for
organic synthesis" (W.P. Weber, Springer Verlag, 1983).
Doivent également être considérés comme faisant partie de la présente
invention toutes les méthodes qui permettent de transformer un dérivé de
formule générale (I) en un autre dérivé de formule générale (I).
A titre d'exemple non exclusif, citons le cas de dérivés de formule (I) dans
lesquels P.I (ou éventuellement R4, R5, R6 ou R7) représente un résidu
aliphatique ou aromatique substitué par NH2 : la préparation de ces produits
par réduction d'un précurseur contenant une fonction nitro (NO2) ou amide
(NHCORB) doit également être considéré comme faisant partie de la présente
invention.
On comprendra donc que dans certaines des transformations ci-dessus, il peut
être nécessaire ou souhaitable de protéger des groupes sensibles éventuels de
la molécule en question afm d'éviter des réactions secondaires indésirables.
Ceci peut être réalisé par l'utilisation des groupes protecteurs
conventionnels
tels que ceux décrits dans "Protective Groups in Organic Synthesis" (J.F.
McOwie, Plenum Press, 1973) et dans T.W. Greene, "Protective Groups in
Organic Synthesis" (John Wiley & Sons, 1981). Les groupes protecteurs
peuvent être enlevés lors de toute étape ultérieure appropriée, en utilisant
les
méthodes et techniques également décrites dans les références citées
précédemment.
~ WO 96/01830 2194677 YCT/FR95/00916
11
Lorsque l'on désire isoler un composé selon l'invention à l'état de sel, par
exemple à l'état de sel formé par addition avec un acide, on peut y parvenir
en
traitant la base libre de formule générale (I) par un acide approprié de
préférence en quantité équivalente, ou par le sulfate de créatinine dans un
solvant approprié.
Lorsque les procédés décrits ci-dessus pour préparer les composés de
l'invention donnent des mélanges de stéréoisomères, ces isomères peuvent être
séparés par des méthodes conventionnelles telles que la chromatographie
préparative.
Lorsque les nouveaux composés de formule générale (I) possèdent un ou
plusieurs centres asymétriques, ils peuvent être préparés sous forme de
mélange racéniique ou sous forme d'énantiomères que ce soit par synthèse
énantiosélective ou par résolution. Les composés de formule (I) possédant au
moins un centre asymétrique peuvent par exemple être séparés en leurs
énantiomères par les techniques habituelles telle que la formation de paires
diastéréomériques par formation d'un sel avec un acide optiquement actif tel
que l'acide (-) di-p-toluoyl-l-tartrique, l'acide (+) di-p-toluoyl-tartrique,
l'acide
(+) camphor sulfonique, l'acide (-) camphor sulfonique, l'acide (+)
phénylpropionique, l'acide (-) phénylpropionique, suivie par cristallisation
fractionnée et régénération de la base libre.
D'une façon générale, les composés de formule (I) peuvent être purifiés par
les
méthodes habituelles, par exemple par cristallisation (en particulier lorsque
les
composés de formule (I) sont isolés sous forme de sel), chromatographie ou
extraction.
Les exemples qui suivent illustrent l'invention sans toutefois en limiter la
portée.
WO 96/01830 PCT/FR95/00916
2194677 12
EXEMPLE 1
Synthèse de la ((phényl-diméthyl-silyl)-méthyl] éthyl-phényl amine
(>Nsi
J "
(1)
Etape A: Préparation de la N-acétyl aniline lA
L'anhydrid.e acétique (26 g, 130 mmoles) est additionné lentement à une
solution d'aniline (9,3 g ; 100 mmoles) et de triéthylamine (15,15 g; 150
mmoles) dans 125 ml de dichlorométhane anhydre à 0 C. Après 3 heures à
20 C, le milieu réactionnel est dilué par du dichlorométhane (300 ml) et lavé
avec une solution d'acide chlorhydrique 1 N, puis trois fois par 350 ml d'une
solution aqueuse saturée en chlorure de sodium. La phase organique est séchée
sur sulfate de magnésium, filtrée et évaporée. Le solide obtenu est purifié
par
chromatograplrie sur colonne de silice pour donner 12,15 g de N-acétyl
aniline.
Etape B: Préparation de f(phénvl-diméthyl-silyl)-méthvll acétyl-phényl
amine (1B)
Une solution de N-acétyl aniline (12 g ; 89 mmoles) dans 100 ml de diméthyl
formamide anhydre est ajoutée goutte à goutte à une suspension d'hydrure de
sodium (106 mmoles ; 4,24 g d'une suspension à 60 % dans l'huile qui est
préalablement lavée à l'hexane sec) dans 100 ml de DMF à 0 C. Le milieu
réactionnel est agité pendant 1 heure à 20 C puis le chlorométhyl phényt
diméthyl-silane (25 g ; 135 mmoles) est ajouté au milieu réactionnel qui est
agité à 90 C pendant 16 heures, puis ramené à 20 C et dilué par 500 ml d'éther
et lavé à l'eau. La phase organique est séchée sur sulfate de magnésium,
filtrée
et évaporée.
Une purihication par chromatographie rapide sur colonne de silice permet
d'isoler 17,6 g de l'intermédiaire 1B (62 mmoles ; 70 %).
WO 96/01830 PCT/FR95100916
! 2194677
13
ETAPE C : Préparation du produit 1
Une suspension d'hydrure de lithium et aluminium 1 M dans l'éther (62 ml) est
additionnée lentement à une solution de j(phényI-diméthyl-silyl)-méthyl]
acétyl-phényl amine (17,6 g ; 62 mmoles) dans 65 ml d'éther anhydre à 0 C.
Le milieu réactionnel est agité pendant 1 h à 0 C, dilué par 300 ml d'éther et
traité par un large excès de sulfate de sodium décahydraté puis filtré sur
Célite
et concentré par évaporation de l'éther sous pression réduite. Une
purification
rapide par chromatograpliie sur silice permet d'obtenir 14,85 g (89 %) du
produit souhaité (exemple 1).
IR (Film, cm-1): 2968; 1600; 1502; 1250; I 115; 837.
RMN (CDC13, 200 MHz, ppm):
7,62-7,53, m, 2H; 7,47-7,35, m, 3H; 7,25-7,12, m, 2H; 6,70-6,58, m, 3H;
3,30, q, J=6,8Hz, 2H; 3,02, s, 2H; 1,05, t, J=6,8Hz, 3H; 0,45, s, 6H.
Analyse Elémentaire pour : C 17H23NSi
C H N
Calculées 75,78 8,69 5,20
Trouvées 76,39 8,58 5,06
EXEMPLE 2
Synthèse de la [(phényl-diméthyl-silyl)-méthyl] propyl-phényl amine C.1~1 i
\1
N Si
(2)
Le produit 2 est préparé selon la même méthode préalablement décrite pour la
préparation du produit 1 dans l'exemple 1 mais en partant de 100 mmoles (13
g) d'anhydride propionique.
WO 96/01830 PCTIFR95100916 =
2~ 946~7
14
EXEMPLE 3
Synthèse de la [{(4-méthylthio-phényl) diméthyl-silyl}-méthylj éthyl-
phényl-amine @)
S\
CLN I
Si /
Le dérivé (3) a été préparé selon les modes opératoires décrits dans l'exemple
I mais en utilisant le chlorométhyl- [(4-méthylthio-phényl) diméthyl]-silane
dans l'étape B.
IR (Film, cm-1): 2968; 1597; 1251; 1078; 839.
RMN (CDC13, 200 MHz, ppm):
7,46, A2B2, 2H; 7,30-7,15, m, 4H; 6,67-6,58, m, 3H; 3,29, q, J=6,8Hz, 2H;
3,00, s, 2H:; 2,51, s, 3H; 1,03, t, J=6,8Hz, 3H; 0,35, s, 6H.
Analyse Elémentaire pour : C18H25NSSi
C H N
Calculées 68,53 7,99 4,44
Trouvées 68,64 8,09 4,56
EXEMPLE 4
Synthèse de la [{(4-diméthylamino-phényl)-diméthyl-silyl}-méthylj-éthyl-
phényl amine L)
(>N I Si
~ WO 96101830 2U4 6 7 7 PCTIM5100916
Le dérivé 4 est préparé selon les modes opératoires décrits dans l'exemple 1
mais en utilisant le chlorométhyl-[(4-diméthylamino-phényl)-diméthyl]-silane
dans l'étape B.
5 IR (Film, cm-1): 2968; 1597; 1504; 1111; 839.
RMN (CDC13, 200 MHz, ppm):
7,46, A2B2, 2H; 7,28-7,15, m, 2H; 6,78-6,57, m, 5H; 3,29, q, J=6,8Hz, 2H;
10 3,00, s, 8H; 1,04, t, J=6,8Hz, 3H; 0,34, s, 6H,
Analyse Elémentaire pour : C 18H25N2Si
C H N
15 Calculées 73,02 9,03 8,96
Trouvées 73,16 9,15 9,00
EXEMPLE 5
Synthèse du [{(4-trifluorométhyl-phényl)-diméthyl-silyl}-méthylj-éthyl-
phényl amine (5)
F
F
F
C~N Z
Si (U
Le dérivé L) est préparé selon les modes opératoires décrits dans l'exemple 1
mais en utilisant le chlorométhyl [(4-trifluorométhyl-phényl)-diméthyl] -
silane
dans l'étape B.
IR (Film, cm-1): 2968; 1597; 1504; 1251; 1167; 831.
RMN (CDC13, 200 MHz, ppm):
7,65, A2B2, 4H; 7,28-7,15, m, 2H; 6,67-6,58, m, 3H; 3,29, q, J=6,8Hz, 2H;
3,04, s, 2H; 1,04, t, J=6,8Hz, 3H; 0,41, s, 6H.
WO 96/01530 2194677 pCTffiW5100916
16
Analyse Elémentaire pour : C18H22F3NSi -
C H N
Calculées 64,06 6,57 4,15
Trouvées 64,03 6,63 4,23
EXEMPILE 6
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-éthyl-(4-trifluorométhyl-
phényl)-amine (6)
FF
F
I / N/~Si I /
Le dérivé (6) est préparé selon les modes opératoires décrits dans l'exemple 1
mais en utilisant la 4-trifluorométhyl-aniline à la place de l'aniline dans
l'étape
A.
IR (Film, cm-1): 2968; 1616; 1531;1250; 1113; 815.
RMN (CDCI3, 200 MHz, ppm):
7,53-7,49, m, 2H; 7,42-7,32, m, 5H; 6,60-6,51, m, 2H; 3,29, q, J=6,8Hz, 2H;
3,04, s, 2H; 1,04, t, J=6,8Hz, 3H; 0,36, s, 6H.
Analyse Elémentaire pour : C 18H27F3NSi
C H N
Calculées 64,06 6,57 4,15
Trouvées 64,25 6,68 4,17
WO 96101830 PCTIFR95100916
~ 2194677
17
EXEMPLE 7
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-éthyl-(3-méthoxy-phényl)
amine (7)
~ I I ~
O \ NSi ~
(2)
Le dérivé (7) a été préparé selon les modes opératoires décrits dans l'exemple
I mais en utilisant la 3-méthoxy-aniline au lieu de l'aniline dans l'étape A.
IR (Film, cm-1): 2968; 1610; 1490;1250; 1157; 833.
RMN (CDC13, 200 MHz, ppm):
7,53-7,49, m, 2H; 7,36-7,32, m, 5H; 7,09-7,01, m, 1H; 6,26-6,13, m, 3H, 3,71,
s, 3H; 3,29, q, J=6,8Hz, 2H; 2,97, s, 2H; 1,04, t, J=6,8Hz, 3H; 0,34, s, 6H.
Analyse Elémentaire pour : C18H25NOSi
C H N
Calculées 72,19 8,41 4,68
Trouvées 72,12 8,69 4,73
EXEMPLE 8
Synthèse de la [(phényl-diméthyl-silyl)-méthyl[-éthyl- 5, 6, 7, 8-
tetrahydronaphtylamineM N Si
J "
(s_)
Le dérivé (8) est préparé selon les modes opératoires décrits dans l'exemple 1
mais en utilisant la 5,6,7,8-tetrahydronaphtylamine au lieu de l'aniline dans
l'étape A.
WO 96/01830 2194677 PCTIFR9Sl00916 ib
18
EXEMPLE 9
Synthèse de la [{(2-éthyl-phényl)-diméthyl-silyl}-méthyl]-méthyl-phényl
amine L)
~ \
~ NSi
1 /\ U
Le dérivé (2) a été préparé selon la méthode décrite dans l'exemple I si ce
n'est
que dans l'étape A, l'anhydride acétique est remplacé par l'anhydride formique
et que dans l'étape B, l'électrophile utilisé est le chorométhyl-[(2-éthyl-
phényl)-diméthyl]-silane.
EYEMPLE 10
Synthèse de la [{(4-méthyl-phényl)-diméthyl-silyl}-méthyl]-méthyl (3
1S éthyl)-phényl amine (10)
^ \I
N Si
~ / \ (10
Le dérivé 10 est préparé selon la méthode décrite dans l'exemple 1 si ce n'est
que, dans l'étape A, la 3-éthyl-aniline remplace l'aniline, l'anhydride
formique
remplace l'anhydride acétique et dans l'étape B, l'électrophile utilisé est le
chloromélhyl [-(4-méthyl-phényl)-diméthyl]-silane.
EXEMPLE 11
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-méthyl-napht-1-yl-amine
11
^
N S\
11
WO 96101830 PCT5R95/00916
= 2194677
19
Le composé 11 est préparé selon la méthode décrite dans l'exemple 1 si ce
n'est que l'anhydride acétique est remplacé par l'anhydride formique et
l'aniline
est remplacé par de la 1-naphtylamine dans l'étape A.
EXEMPLE 12
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-méthyl-8-quinolinyl-amine
12
~ i
N I ~ \ I
i j S~
~ N (12)
Le composé 12 est préparé par condensation de la 8-quinolinyl-formamide
avec le chlorométhyl-(phényl-diméthyl)-silane suivi de la réduction de l'amide
selon les méthodes décrites comme étapes B et C dans l'exemple 1.
EYEMPLE 13
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-phényl amine 13
~
I ~ \ I
N Si
H / \ 13
Le composé 13 a été obtenu par réaction de la [(phényl-diméthyl-silyl)-méthyl
]-N-(trifluoroacétyl)-phényl amine (préparée selon les étapes A et B de
l'exemple 1 à partir d'aniline, d'anhydride trifluoroacétique et de
clilorométhyl
(phényl-diméthyl)-silane puis déprotection du dérivé trifluoroacétylé avec de
la potasse alcoolique selon le mode opératoire suivant:
A une solution de potasse (1 g; 17,9 mmole) dans le méthanol (5 ml) est
additionnée à 0 C le dérivé trifluoroacétylé (200 mg; 0,59 mmole) puis le
milieu réactionnel est agité à température ambiante pendant 2 heures. La
solution résultante est alors diluée par de l'ether éthylique (40 ml) et lavée
avec
WO 96/01830 PCT/FR95100916
2194677
=
de l'eau (2 x 15 nd). La phase organique est séchée sur sulfate de magnésium,
filtrée et évaporée.
Une purification par chromatographie rapide sur colonne de silice permet
d'isoler le produit 13 (92 mg; 65 %).
5 IR (Film, cm-1): 3330; 2968; 1606; 1502; 1250; 1115; 839.
RMN (CDC13, 200 MHz, ppm):
7,62-7,53, m, 2H; 7,47-7,35, m, 3H; 7,25-7,12, m, 2H; 6,75-6,61, m, 3H;
10 3,44, brs, 1H; 2,75, s, 2H; 0,45, s, 6H.
Analyse Elémentaire pour : C15H19NSi
C H N
15 Calculées 74,63 7,93 5,80
Trouvées 74,42 7,93 5,65
EXEMPLE 14
Synthèse de la [(phényl-diméthyl-silyl)-méthyll-(3-méthyl-phényl)-amine
20 Ü
aa
NSi H 14
Le dérivé 14 est préparé à partir de la 3-méthyl-aniline, d'anhydride
trifluoroacétique et de chlorométhyl-(phényl-diméthyl)-silane selon la méthode
décrite dans l'exemple 13.
p.î=54 C
IR (KBr, cm-1): 3330; 2968; 1606; 1490;1250; 1113; 833.
RMN (CD,C13, 200 MHz, ppm):
7,59-7,53, m, 2H; 7,41-7,35, m, 3H; 7,09-7,01, m, 1H; 6,53-6,44, m, 3H;
2,72, s, 2H; 2,26, s, 3H; 0,39, s, 6H.
Analyse Elémentaire pour : C16H21NSi
WO 96101830 2 l 94677 PCT/FR95/00916
21
C H N
Calculées 75,23 8,29 5,48
Trouvées 75,28 8,35 5,51
EXEMPLE 15
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-(4-bromo-phényl)-amine
(L5)
Br UI ~
~ ~
N Si
H ~ \ (15)
Le composé 15 est préparé par condensation de la 4-bromo-aniline avec
l'anhydride trifluoroacétique puis avec le chlorométhyl (phényl-diméthyl)-
silane suivi de la déprotection au moyen de l'hydrure de lithium et aluminium
selon les modes opératoires décrits dans l'exemple 1.
IR (Film, cm-1): 3416; 3068; 1597; 1495;1250; 1115; 830.
RMN (CDCI-, 200 MHz, ppm):
7,59-7,53, m, 2H; 7,44-7,38, m, 3H; 7,26, A2B2, 2H; 6,51, A2B2, 2H; 2,94,
s, 2H; 0,41, s, 6H.
Analyse Elémentaire pour : C15H18BrNSi
C H N
Calculées 56,25 5,66 4,37
Trouvées 56,81 5,85 4,39
EXEMPLE 16
Synthèse de la [(phényl-diméthyl-silyl)-méthylj-(3-cyano-phényl)-amine
Ü
WO 96101830 2194677 PCT1FR95100916
22
~
i
N/\Si ~
H
U-6)
Le composé 16 est préparé par condensation de la 3-cyano-aniline
successivement avec l'anhydride trifluoroacétique et le chlorométhyl (phényl-
diméthyl)-silane selon les modes opératoires décrits dans les étapes A et B de
l'exemple 1, suivi de la déprotection au niveau de la trifluoroanilide ainsi
formée par le borohydrure de sodium dans l'éthanol absolu à 20 C pendant 4
heures.
IR (Film, cm-1): 3410; 2968; 2220; 1597; 1479; 1250; 1118; 841.
RMN (CDC13, 200 MHz, ppm):
7,59-7,53, m, 2H; 7,45-7,37, m, 3H; 7,24-7,12, m, 1H; 6,92-6,72, m, 3H;
3,61, brs, 1H; 2,67, s, 2H; 0,40, s, 6H.
Analyse Elémentaire pour : C 16H 18N2Si
C H N
Calculées 72,13 6,81 10,51
Trouvées 72,02 6,87 10,25
EXEMPLE 17
Synthèse de la [((3,4-diméthyl-phényl)-diméthyl-silyl}-méthyll-(3-cyano-
phényl)-amine (17)
\ I ^ I ~
N /5\
N/ I
H U7
Le composé 17 est préparé par la procédure décrite dans l'exemple 16 en
partant du chlorométhyl-(3,4-diméthyl-phényl)-silane.
WO 96/01830 2 1 Q 4 6 ~ ~ PCTIFR95/00916
23
EXEMPLE 18
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-(3-hydroxy-méthyl-phényl)
amine 18
HO
N Si
H 18
Le dérivé 18 a été préparé par condensation initiale de la 3-(formyl)-N-
(trifluoroacétyl)-aniline avec le chlorométhyl-(diméthyl-phényl)-silane (selon
la procédure utilisée dans l'étape B de l'exemple 1) suivi de la réaction de
l'intermédiaire ainsi formé avec un excès de borohydrure de sodium dans le
méthanol.
p.f.=72 C
IR (KBr, cm-1): 3380; 3330; 2968; 1606; 1490; 1250; 1113; 833.
RMN (CDC13, 200 MHz, ppm):
7,59-7,53, m, 2H; 7,41-7,35, m, 3H; 7,09-7,01, m, 1H; 6,53-6,44, m, 3H;
4,78, brm, 2H; 4,57, s, 2H; 2,72, s, 2H; 0,39, s, 6H.
Analyse Elémentaire pour : C16H21NOSi
C H N
Calculées 72,43 8,78 9,38
Trouvées 72,41 8,95 9,35
EXEMPLE 19
Synthèse de la 3-[(phényl-diméthyl-silyl)-méthyl-amino]-éthyl benzylamine
19
WO 96/01830 2194677 PC'S/FR95/00916
24
I ~ I I ~
H N \ NSi
H 19
A une solution du nitrile 16 (15 g, 41,4 mmoles) dans 200 ml de
tétrahydrof.uranne et maintenue à 0 C est ajouté du DiBA1-H (Aldrich, 1,0 M
dans le toluène, 83 ml, 124,3 mmoles, 3éq.). La solution résultante est agitée
pendant 3 heures à 19 température ambiante. 30 ml d'acide chlorhydrique 5N
sont ensuite additionnés et le milieu agité vigoureusement pendant environ 2
heures. La phase aqueuse est ramenée à pH basique avec de la soude
concentrée puis extraite avec de l'acétate d'éthyle. La phase organique est
ensuite lavée avec de l'eau puis séchée sur sulfate de magnésium, filtrée et
évaporée.
L'huile obtenue (9,8 g; 88%) est suffisamment pure pour être directement
utilisable sans purification supplémentaire.
Une solution de cette huile (500 mg ; 3,72 mmoles) et d'éthylamine (1,9M
dans l'éthanol absolu, 19 ml, 1,5éq,) dans 10 ml d'éthanol absolu est agitée
15
heures à température ambiante en présence de tamis moléculaire 3A. Le milieu
est ensuite filtré sur Célite et les volatils évaporés sous vide. L'huile
résultante
est alors rediluée dans 15 ml d'éthanol absolu et la solution refroidie à 0 C.
Du
borohydrure de sodium (283 mg, 7,44 mmoles, 2éq.) est additionné par
portions au milieu réactionnel et l'agitation est encore poursuivie pendant 2
heures à température ambiante. L'éthanol est ensuite évaporée et l'huile
obtenue est diluée dans l'acétate d'éthyle et lavée avec 2 x 10 ml d'eau. La
phase organique est séchée sur sulfate de magnésium, filtrée et évaporée.
Une purification "éclair" de l'huile obtenue sur colonne de silice permet
d'obtenir dans la fraction majoritaire le produit attendu sous la forme d'une
huile jaune claire (300 mg; 27 %).
IR(Film, cm-l): 3414; 3330; 2968; 1606; 1485;1250; 1113; 841.
RMN (CDC13, 200 MHz, ppm):
7,59-7,53, m, 2H; 7,41-7,35, m, 3H; 7,09-7,01, m, 1H; 6,53-6,44, m, 3H;
3,69, s, 2H; 3,43, brs, 1H; 2,75, s, 2H; 2,71, q, J=6,8Hz, 2H; 1,89, brs, 1H;
1,11, t, J=6,8Hz, 3H; 0,39, s, 6H.
WO 96/01830 2194677 PCT/FR95100916
Analyse Elémentaire pour : C 16H21NSi
C H N
Calculées 70,82 7,80 5,16
5 Trouvées 70,32 7,75 5,15
EXEMPLE 20
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-2-naphtylamine (20)
CDa ~
^ ~ i
N Si
10 H / ~ 20)
Le dérivé 20 est préparé par condensation de la N-(tiifluoroacétyl)-2-
naphtylamine avec le chlorométhyl-(diméthyl-phényl)-silane (selon la
procédure décrite dans l'étape B de l'exemple 1) suivi de la déprotection de
15 l'intermédiaire ainsi formé avec de la potasse alcoolique.
IR (Film, cm-1): 3414 ; 3050; 2960; 2802; 1630; 1523; 1250; 833.
RMN (CDC13, 200 MHz, ppm)
20 7,68-7,55, m, 4H; 7,44-7,15, m, 6H; 6,90-6,80, m, 2H; 3,62, brs, 1H; 2,82,
s,
2H; 0,45, s, 6H.
Analyse Elémentaire pour : C 17H2INSi
25 C H N
Calculées 78,30 7,26 4,81
Trouvées 78,73 7,30 4,81
EXEMPLE 21
Synthèse de la [(phényl-diméthyl-silyl)-méthylj-4(phényl-phényl) amine
21)
WO 96/01830
2194677 PCT/FR95/00916
26
N~~Si
H / \ 21)
Le composé 21 est préparé par condensation de la 4-(phényl)-N-
(trifluoroacétyl)-aniline avec le chlorométhyl-(phényl-diméthyl)-silane selon
la
procédure décrite dans l'étape B de l'esemple 1, suivi de la déprotection de
l'aniline par réaction avec le borohydrure de sodium à 20 C dans l'éthanol.
IR (Filin, cm-1): 3414; 3050; 2960; 2802; 1630; 1523; 1250; 833.
RMN (CDCIõ 200 MHz, ppm):
7,68-7,55, m, 4H; 7,44-7,15, m, 6H; 6,90-6,80, m, 2H; 3,62, brs, 1H; 2,82, s,
2H; 0,45, s, 6H
Analyse Elémentaire pour : C21H23NSi
C H N
Calculées 79,44 7,30 4,41
Trouvées 79,58 7,69 4,45
EXEMPLE 22
Synthèse de la [{(4-bromo phényl)-diméthyl-silyl}-méthyl]-4-(phényl-
phényl) amine 22)
Br
caN n Si
H / \ 27)
Le dénivé 22 est préparé selon la même procédure que le dérivé 21 mais en
utilisant le chlorométhyl-(4-bromophényl)-diméthyl-silane comme produit de
départ.
WO 96/01830 2 1 e ~ ~ ~ PCTIFR95l00916
27
EXEMPLE 23
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-3-vinyl-phényl amine 23
\ \ I ^ I i
N Si
H 23 -
Le dérivé 23 est préparé par alkylation de la 3-vinyl-N-(trifluoroacétyl)-
aniline
avec le chlorométhyl-phényl-diméthyl-silane selon la procédure décrite dans
l'étape B de l'exemple 1, suivi de la déprotection de l'aniline par réaction
avec
l'hydrure de lithium et aluminium dans l'éther.
EXEMPLE 24
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-[(3-oxazol-4-yl)-phénylj
amine (24)
~ I I \
\~ ~ \ NSi
N H (L4)
Le dérivé 24 est obtenu par condensation de la 3-(oxazol-4-yl)-N-
(trifluoroacétyl)-aniline avec le chlorométhyl-phényl-diméthyl-silane selon la
procédure décrite dans l'étape B de l'exemple 1 suivi de la déprotection de
l'aniline par réaction avec le borohydrure de sodium dans l'éthanol à 20 C.
ES{EMPLE 25
Synthèse de la [(phényl-diméthyl-silyl)-méthyll-6-quinolinyl-amine 25)
N
\ \ ~ n 1 i
N /S\
H 25
R'O 96/01830 219 4 6 7 7 PCTaw5100916
28
Le composé 25 est préparé par condensation de la N-(trifluoroacétyl)-6-amino-
quinoline avec le chlorométhyl-diméthyl-phényl-silane selon la procédure
décrite dans l'étape B de l'exemple 1, suivi de la déprotection de l'amine par
réaction avec le borohydrure de sodium dans l'éthanol à 20 C.
EXEMPLE 26
Synthèse de la [(phényl-diméthyl-silyl)-méthyl]-6-benzothiazolyl amine (26)
S >I N/~Si I /
~ / \
H (26)
Le contposé 26 est préparé par condensation de la N-(trifluoroacétyl)-6-
benzothiazolyl amine avec le chlorométhyl-diméthyl-phényl-silane selon la
procédure décrite dans l'étape B de l'exemple 1, suivi de la déprotection de
l'amine par réaction avec le borohydrure de sodium dans l'éthanol à 20 C.
EXEMPLE 27
Synthèse de la [(phényl-diméthyl-silyl)-méthylj-(4-methoxy-phényl)-amine
(U7
UN ~
Si /
H / \ (27)
Une solution de n-butyllithium (62,5 ml d'une solution 1,6 M dans l'hexane,
soit 100 mmoles) est additionnée goutte à goutte à une solution de 4méthoxy-
aniline (11,07 g ; 90 mmoles) dans 100 ml de tétrahydrofaranne anhydre à 0 C
sous atmosphère d'argon. Le milieu réactionnel est agité durant 1 heure à 20 C
puis 25 g de chlorométhyl-diméthyl-phényl-silane dissous dans 100 ml de
tétrahydrofuranne sont introduits dans le milieu réactionnel qui est agité
pendant 20 heures à 60 C. Le milieu réactionnel est ramené à 20 C, dilué dans
~WO 96/01830 2194677 PCT/FR95/00916
29
l'éther éthylique (300 ml) et lavé avec de l'eau, puis trois fois par une
solution
aqueuse saturée en NaC1. La phase organique est séchée sur sulfate de
magnésium, filtrée et concentrée sous pression réduite. Le résidu est distillé
(145-150 C/0,5 mm Hg) pour donner le produit 27 (22 g; 90 %).
IR (Film, cm-1): 3414; 2960; 1512; 1250; 829.
RMN (CDC13, 200 MHz, ppm):
7,64-7,55, m, 2H; 7,40-7,33, m, 3H; 6,66, A2B2, 4H; 3,70, s, 3H; 3,26, brs,
1H; 2,69, s, 2H; 0,4, s, 6H
Analyse Elémentaire pour : C 16H71NOSi
C H N
Calculées 70,80 7,80 5,16
Trouvées 69,81 7,92 4,84
EXEMPLE 28
Synthèse de la 1-[(phényl-diméthyl-silyl)-méthyl]-1-phényl-hydrazine (28)
O I ~
NSi
28
A une solution d'amidure de sodium (520 mg; 13,3 mmoles; 1,2éq.) dans le
tétrahydrofuranne (12 ml) est ajoutée à 0 C de la phénylhydrazine (1,1 ml;
11,1 mmoles). Le milieu résultant est agité 2 heures à la température ambiante
puis de nouveau refroidi à 0 C et le chlorométhyl-diméthyl-phényl-silane (3
ml; 16,6 mmoles; 1,5éq.) est additionné. La solution résultante est chauffée à
60 C pendant 24 heures puis refroidie à 20 C et diluée dans l'éther éthylique
et lavée avec 3 x 50 nil d'eau. La phase organique est séchée sur sulfate de
magnésium, filtrée et concentrée sous pression réduite.
WO 96/01830 2194677 PCTIFR95100916
Une purification sur colonne de silice permet d'obtenir le composé (28) (1,4
g;
50%).
IR (Film, cm-1): 3300; 3067; 1597; 1495; 1250; 841.
5
RMN (CDC13, 200 MHz, ppm):
7,64-7,60, m, 2H; 7,40-7,24, m, 4H; 6,93-6,71, m, 4H; 3,66, brs, 2H; 3,18, s,
2H; 0,45, s, 6H.
Analyse Elémentaire pour : C 15H20N2Si
C H N
Calculées 70,26 7,86 10,92
Trouvées 70,15 7,96 10,93
EXEMPLE 29
Synthèse de la [N-(phényl-diméthyl-silyl)-méthyl]-[N'-(3-méthyl-phényl)]-
aniline .29
aI ~
NSi
à 29
Le dérivé 29 est préparé à partir de la N-(3-méthyl-phényl)-aniline et du
chlorométhyl-(diméthyl-phényl)-silane selon la procédure décrite dans
l'exemple 27.
-
IR (Film, cm-1): 2957; 1593; 1495; 1250; 841.
RMN (CDC13, 200 MHz, ppm):
7,50-7,02, M, 10H; 6,95-6,64, m, 4H; 3,48, s, 2H; 2,21, s, 3H; 0,18, s, 6H.
~ WO 96101830 PCT/FR95/00916
2194677
31
Analyse Elémentaire pour : C22H25NOSi
C H N
Calculées 79,70 7,60 4,22
Trouvées 78,67 7,52 4,25
EXEMPLE 30
Synthèse de la [(phényl-diméthyl-silyl)-méthylJ-(4-méthyl-phényl)-amine
(30)
\ I I ~
N^S~
H 30
Le composé 30 est préparé à partir de 4-méthyl-aniline et du chlorométhyl-
(diméthyl-phényl)-silane selon la procédure décrite dans l'exemple 27.
IR (Film, cm-1): 3412; 2957; 1616; 1520; 1250; I 115; 830.
RIviN (CDCl3, 200 MHz, ppm):
7,59-7,53, m, 2H; 7,44-7,38, m, 3H; 6,97, A2B2, 2H; 6,56, A2B2, 2H; 2,71,
s, 2H; 2,22, s, 3H; 0,39, s, 6H.
Analyse Elémentaire pour : C 16H2INSi
C H N
Calculées 75,53 8,32 5,51
Tronvées 75,72 8,73 5,31
wo 96101930 21/4û t7 Pca/FR95100916 ~
32
EXEMPLE 31
Synthèse de la [[4-méthyl-phényl) diméthyl-silyl]-méthyl]-(2-méthyl-
phényl) amine 31)
NSi I /
.~
1 / \
H 31
Le composé 31 est préparé à partir de la 2-méthyl aniline et du chlorométhyl-
diméthyl-(4-méthyl-phényl)-silane selon la procédure expérimentale décrite
dans l'exemple 27.
EXEMPLE 32
Synthèse de la [{2,6-(diméthyl)-phényi-diméthyl-silyl}]-méthyl]-(3-méthyl
phényl) amine 32) _
H Si
37)
Le composé 32 est préparé par condensation de la 3-méthyl-aniline avec le
chlorométhyl-(2,6-diméthyl-phényl)-diméthyl-silane selon la procédure
expérimentale décrite dans l'exemple 27.
EXEMPLE 33
Synthèse de la [{(4-cyano-phényl) diméthyl-silyl}-méthyl]-3-méthyl phényl
amine 33)
N
I~z
H ~ \ 33)
WO 96/01830 2194677 PCUM5/00916
33
= 'Le composé 33 a été préparé par condensation de la 3-méthyl aniline avec le
chlorométhyl-(4-cyano-phényl)-diméthyl-silane selon la procédure expérimen-
tale décrite dans l'exemple 27.
EXEMPLE 34
Synthèse de la [{(3-méthoxy-phényl)-diméthyl-silyl}-méthylj-3-méthyl
phényl amine 34)
NS
H ~ \ 0
34)
Le dérivé 34 est préparé à partir de la 3-méthyl aniline et du chlorométhyl-(3-
méthoxy-phényl)-diméthyl-silane selon la procédure expérimentale décrite
dans l'exemple 27.
EXEMPLE 35
Synthèse de la [{(1-(naphtyl) diméthyl-silyll-méthyl}-phényi amine 35)
a
N~ S\
Le dérivé 35 est préparé par condensation de l'aniline avec le chlorométhyl-(3-
naphtyl)-diméthyl-silane selon la procédure expérimentale décrite dans
25 l'exemple 27.
PCT/FB95/00916
WO 96/01830 2 1 94677
34
EXEM]PLE 36
Synthèse de la [{(phényl-diméthyl)-silyl}-méthyll-(-4-tertiobutyl-phényl)-
amine (36)
NSi /
/ \
H (36)
Le composé 36 est préparé par condensation de la 4-tertiobutyl-aniline avec le
chlorométhyl-diméthyl-phényl-silane selon la procédure expérimentale décrite
dans l'exemple 27.
EXEMPLE 37
Synthèse de la [{(3-phényl)-phényl-diméthyl)-silyl}-méthyl]-3-(méthoxy
phényl) amine 37)
~ ~
0 N S\
Ü
Le composé 37 est préparé par condensation de la 3-méthoxy-aniline avec le
chlorométhyl-(3-phényl-phényl)-diméthyl-silane selon la procédure
expérimentale décrite dans l'exemple 27.
WO 96101830 219 4 b ï 7 PC17FR95/00916
EXEMPLE 38
Synthèse de la [(phényl-méthyl-propyl silyl)-méthyl]-(3-méthyl-phényl)
= amine (38)
5
I ~
NSi
38
Le composé 38 est préparé par condensation de la 3-méthyl-aniline avec le
chlorométhyl-méthyl-propyl-phényl-silane selon la procédure expérimentale
10 décrite dans l'exemple 27.
WO 96101830 21 I, q/~~ PCT/FR95/00916 ~
36
Résultats nharmacoloeigues
L'activité antioxydante des dérivés de la présente invention a été plus
particulièrement démontrée sur des microsomes de foies de rats, suite à une
péroxydation chimique induite par les ions ferriques et sur des LDL humaines.
.L'action inhibitrice des composés de la présente invention vis-à-vis de
l'oxydation des LDL humaines a été démontrée que ce soit à la suite d'une
oxydation chimique par le sulfate de cuivre ou à la suite d'une oxydation
biologique par les cellules endothéliales humaines issues de veines
ombilicales.
1. Inhibition de l'oxydation des microsomes de foie de rat par lésions
ferriques
L'étude a été réalisée selon la technique décrite dans la référence
"Antioxydative properties of harmane and carboline alkaloïds" ; S. Y. H. Tse,
I-T. Mak, B-J. Dickens, Biochem Pharmacol. 42 (3), 459-464, 1991. L'activité
des composés a été testée de façon comparative à la vitamine E. De nombreux
composés de la présente invention ont montré une activité supérieure à la
vitamine E dans ce test comme l'indique quelques exemples dans le tableau ci-
dessous ~
COMPOSES IC50 ( M)
Vitamine E 2,3
Exemple 1 0,5
Exemple 13 0,5
Exemple 14 0,5
Exemple 15 0,7
2. Inhibition de l'oxvdation des LDL humaines par le sulfate de cuivre
Les études ont été effectuées avec des LDL humaines incubées avec du sulfate
de cuivre (10 M) pendant 6 heures à 37 C avec ou sans composé à tester. La
péroxydation lipidique est évaluée par la technique des TBARS comme décrit
WO 96101830 219 467 7 PCTIFW5l00916
37
par C. Breugnot et coll., Biochem. Pharmacol. 40, 1975-1980 (1990).
L'activité anti-oxydante des composés de la présente invention a été étudiée
en
comparaison avec la vitamine E. Les quelques exemples illustratifs repris dans
le tableau ci-dessous montrent que les dérivés de la présente invention ont un
profil anti-oxydant favorable vis-à-vis de la vitamine E:
COMPOSES IC50 ( M)
Vitamine E 7,4
Exemple 14 5
Exemple 15 5
Exemple 7 5
Exemple 18 5
Exemple 19 5
Exemple 20 5
3. Inhibition de l'oxvdation des LDL humaines Dar les cellules endothéliales
de
veine ombilicale humaine en culture
Les LDL humaines sont stockées à 4 C dans un tampon (NaCI 150 mM,
EDTA 0.01%, pH = 7,4) à 4 C. Avant utilisation, un volume de LDL est
dialysé pendant 24 heures à 4 C contre 5 x 1000 volumes de tampon (NaCI
136,8 mM, KCI 2,68 mM, KH2PO4 1,47 mM, Na2HPO4 8,09 mM, pH = 7).
Les LDL sont utilisées aux concentrations finales en protéines (test de
Coomassie, Pierce) de 25 ou 50 g/ml.
Les cellules endothéliales, lignée ECV304 (ATCC CRL-1998) sont cultivées
en puits de 35 mm de diamètre dans un milieu DMEM-HEPES-Glutamax I
supplémenté par un sérum de veau foetal 10%, penicilline-streptomycine 100
UUml - 100 g/ml, et fungizone 2,5 g/ml jusqu'à un état sub-confluent. Les
cellules sont alors incubées 24 heures dans le milieu DMEM-HEPES-
Glutamax 1 contenant ultroser G 2% ; puis, l'oxydation des LDL est
WO 96/01830 2194677 PCT/FR95/00916
38
déclenchée par leur mise en contact avec les cellules pendant 24 heures dans
le
milieu nutritif HAM F-10 (Breugnot C_, Maziere C., Salmon S., Auclair M.,
Santus R., Morliere P., Lenaers A. et Mazière J. C. : Phenothiazines inhibit
copper and endothelial cell-induced peroxidation of low density lipoprotein. A
comparative study with probucol, butylated hydroxytoluene and vitamine E.
Biochemical Pharmacology, 1990, 40:1975-1980).
Le surnageant de culture est collecté, centrifugé (2000 x g, 10 minutes, 4 C),
et la réaction avec l'acide thiobarbiturique (0,12 N, dans un tampon TRIS 10
mM ; pH = 7) est réalisée sur le surnageant après acidification (pH final = 2-
3)
par l'acide trichloroacétique 15 % (Yagi K. : Lipid peroxides and human
diseases. Chernistry and physics of lipids, 1987, 45:337-351). Les
échantillons
sont chauffés 30 minutes à 95 C, les TBARs sont extraits par le butanol-1 et
quantifiés par fluorimétrie (longueurs d'onde d'excitation 515 nm, d'émission
548 nm, spectrofluorimètre Perkin Elmer LS-50B). Les résultats sont exprimés
en runol équivalent malondialdéhyde/mg LDL protéines.
L'activité antioxydante des composés de la présente invention dans ce modèle
a été étudiée en comparaison avec la vitamine E. Comme le montrent les
quelques exemples illustratifs repris dans le tableau ci-dessous, les composés
de la présente invention se sont montrés supérieurs à la vitamine E dans ce
test.
COMPOSES IC50 ( M)
Vitamine E 25
Exemple 1 10
Exemple 5
Exemple 4 5
Exemple 15 3
Exemple 7 5
Ces résultats indiquent clairement la plus grande puissance de certains
dérivés
faisant partie de la présente invention par rapport à la vitamine E comme
WO 96/01830 2t94677 P("r1FR95/00916
39
protecteurs des lipides tels que les microsomes ou les LDL humaines vis-à-vis
des niodifications oxydatives et en particulier lors de l'oxydation biologique
médiée par les cellules endothéliales humaines.
Les modifications oxydatives des LDL apparaissent aujourd'hui clairement
jouer un rôle important dans la genèse et le développement des lésions
vasculaires tels que les athéromes. Dès lors, les propriétés antioxydantes,
notamment au niveau des LDL, des dérivés de la présente invention permettent
leur utilisation comme médicaments dans le traitement et/ou la prévention de
l'athérosclérose incluant ses différentes localisations vasculaires
périphériques,
coronaires ou cérébrales, des maladies dues aux complications vasculaires
liées aux lipoprotéines, mais aussi des pathologies dans lesquelles une
péroxydation lipidique membranaire joue un rôle initiateur et/ou aggravant
telles que les cardiopathies ischénliques, les reperfusions d'organes, y
compris
transplantés, les pathologies ischénùques traumatiques du système nerveux
central ou périphérique, les maladies auto-immunes et les maladies
métaboliques telles que le diabète, et de manière générale toutes les
dyslipidémies.
Par ailleurs, les dérivés de la présente invention se sont montrés
particulièrement efficaces pour neutraliser les radicaux hydroxyles (OH) :
c'est
ainsi que dans un test permettant de déterminer l'aptitude de composés à
piéger
le radical hydroxyl (méthode faisant appel au dosage du radical 2-hydroxy-5,5
diméthyl-1-pyrroline-N-oxyde par RPE selon Miyagawa et Coll., J. Clin.
Biochem. Nutr. 5, l-7, 1988), certains produits de la présente invention se
sont
montrés plus actifs que la vitamine E ou le BHT ; à titre d'exemple, le
tableau
ci-dessous donne les activités comparatives de ces deux produits de référence
et de l'exemple 13 de la présente invention.
COMPOSES CI30 ( M)
Vitamine E 65
BHT 46
Exemple 13 38
WO 96/01830 2194677
7~GlI
Les radicaux hydroxyles constituent une source de radicaux extrèmement
agressifs aux niveaux cellulaires et moléculaires et, dès lors, les composés
de
la présente invention que présentent l'avantage d'être à la fois inhibiteurs
de la
péroxydation de lipides et piégeurs de radicaux libres trouvent aussi leur
utilité
5 pour le traitement et/ou la prévention des maladies inflammatoires aiguës ou
chroniques, ainsi qu'en dermatologie et en dermo-cosmétologie et dans le
traitement de diverses pathologies telles que certaines formes de cancer (en
particulier de tumeurs liées aux radiations ionisantes telles que certains
cancers de la peau), le vieillissement des tissus (en particulier le
vieillissement
10 de la peau), et certaines formes de dégénérescence telles que la maladie de
Parkinson ou d'Alzheimer. A ce titre, divers antioxydants tels que la vitamine
E ont montré un effet protecteur dans un modèle de neurotoxicité induite par
le
MPTP (cf. T. L. Perry et al, Neurosc. Letters, 60, 109, 1985) modèle considéré
comme prédictif de maladies neuro-dégénératives (cf. Neuroscience Facts, 3
15 (23), 89, 1992).
La plus grande activité inhibitrice de processus oxydatifs observée avec de
nombreux dérivés de la présente invention par rapport à la vitamine E les rend
donc particulièrement utiles pour le traitement etlou la prévention des
maladies
neurodégénératives.
La présente invention a également pour objet les compositions
pharmaceutiques contenant comme principe actif un composé de formule
générale I ou un de ses sels acceptables pour l'usage pharmaceutique, mélangé
ou associé à un excipient approprié. Ces cômpositions peuvent revêtir, par
exemple, la forme de compositions solides, liquides, d'émulsions, lotions ou
crèmes.
Comme compositions solides pour administration orale, peuvent être utilisés
des comprimés, des pilules, des poudres (capsules de gélatine, cachets) ou des
granulés. Dans ces compositions, le principe actif selon l'invention est
mélangé
à un ou plusieurs diluants inertes, tels que aniidon, cellulose, saccharose,
lactose ou silice, sous courant d'argon. Ces compositions peuvent également
comprendre des substances autres que les diluants, par exemple un ou
plusieurs lubrifiants tels que le stéarate de magnésium ou le talc, un
colorant,
un enrobage (dragées) ou un vernis.
WO96/01830 219 467 7 PCT5it95/00916
41
Comme compositions liquides pour administration orale, on peut utiliser des
solutions, des suspensions, des émulsions, des sirops et des élixirs
pharmaceutiquement acceptables contenant des diluants inertes tels que l'eau,
l'éthanol, le glycérol, les huiles végétales ou l'huile de paraffine. Ces
compositions peuvent comprendre des substances autres que les diluants, par
exemple des produits mouillants, édulcorants, épaississants, aromatisants ou
stabilisants.
Les compositions stériles pour administration parentérale, peuvent ëtre de
préférence des solutions aqueuses ou non aqueuses, des suspensions ou des
émulsions. Comme solvant ou véhicule, on peut employer l'eau, le
propylèneglycol, un polyéthylèneglycol, des huiles végétales, en particulier
l'huile d'olive, des esters organiques injectables, par exemple l'oléate
d'éthyle
ou autres solvants organiques convenables.. Ces compositions peuvent
également contenir des adjuvants, en particulier des agents mouillants,
isotonisants, émulsifiants, dispersants et stabilisants. La stérilisation peut
se
faire de plusieurs façons, par exemple par filtration aseptisante, en
incorporant
à la composition des agents stérilisants, par irradiation ou par chauffage.
Elles
peuvent également être préparées sous forme de compositions solides stériles
qui peuvent être dissoutes au moment de l'emploi dans de l'eau stérile ou tout
autre milieu stérile injectable.
Les compositions pour administration rectale sont les suppositoires ou les
capsules rectales qui contiennent, outre le produit actif, des excipients tels
que
le beurre de cacao, des glycérides semi-synthétiques ou des
polyéthylèneglycols.
Les compositions pour administration topique peuvent être par exemple des
crèmes, lotions, collyres, collutoires, gouttes nasales ou aérosols.
Les doses dépendent de l'effet recherché, de la durée du traitement et de la
voie d'administration utilisée ; elles sont généralement comprises entre 0,001
g
et 1 g (de préférence comprises entre 0,005 g et 0,25 g) par jour de
préférence
par voie orale pour un adulte avec des doses unitaires allant de 0,1 mg à 500
mg de substance active, de préférence de 1 mg à 50 mg.
WO 96101830 2194677 PCT/FR95100916
42
D'une façon générale, le médecin déterminera la posologie appropriée en
fonction de l'âge, du poids et de tous les autres facteurs propres au sujet à
traiter. Les exémples suivants illustrent des compositions selon l'invention
[dans ces exemples, le terme "composant actif' désigne un ou plusieurs
(généralement un) des composés de formule (I) selon la présente invention]
Comprimés
On peut les préparer par compression directe ou en passant par une granulation
au mouillé. Le mode opératoire par compression directe est préféré mais il
peut ne pas convenir dans tous les cas selon les doses et les propriétés
physiques du composant actif.
A - Par compression directe
mg pour 1 comprimé
composant actif 10,0
cellulose microcristalline B.P.C. 89,5
stéarate de magnésium 0 5
100,0
On passe le composant actif au travers d'un tamis à ouverture de maille de 250
m de côté, on mélange avec les excipients et on comprime à l'aide de
poinçons de 6,0 mm. On peut préparer des comprimés présentant d'autres
résistances mécaniques en modifiant le poids de compression avec utilisation
de poinçons appropriés.
B - Gran:ilation au mouillé
mg pour un comprimé
composant actif 10,0
lactose Codex 74,5
amidon Codex 10,0
amidon de maïs prégélatinisé Codex 5,0
stéarate de magnésium 0 5
Poids à la compression 100,0
On fait passer le composant actif au travers d'un tamis à ouverture de maille
de
250 m et on mélange avec le lactose, l'amidon et l'amidon prégélatinisé. On
humidifie les poudres mélangées par de l'eau purifiée, on met à l'état de
granulés, on sèche, on tamise et on mélange avec le stéarate de magnésium.
WO 96/01830 =y t ry q/ 7^J PCT/FR95/00916
43
Les granulés lubrifiés sont mis en comprimés conune pour les formules par
= compression directe. On peut appliquer sur les comprimés une pellicule de
revêtement au moyen de matières filmogènes appropriées, par exemple la
méthylcellulose ou l'hydroxy-propyl-méthyl-cellulose, selon des techniques
classiques. On peut également revêtir les comprimés de sucre.
Capsules
mg pour une capsule
composant actif 10,0
*amidon 1500 89,5
stéarate de magnésium Codex 0 5
Poids de remplissage 100,0
*une forme d'amidon directement compressible provenant de la fuine
Colorcon Ltd, Orpington, Kent, Royaume Uni.
On fait passer le composant actif au travers d'un tamis à ouverture de maille
de
250 m et on mélange avec les autres substances. On introduit le mélange dans
des capsules de gélatine dure n 2 sur une machine à remplir appropriée. On
peut préparer d'autres unités de dosage en modifiant le poids de remplissage
et,
lorsque c'est nécessaire, en changeant la dimension de la capsule.
Sirop
mg par dose de 5 ml
composant actif 10,0
saccharose Codex 2750,0
glycérine Codex 500,0
tampon )
arôme )
colorant ) q.s.
préservateur )
eau distillée 5,0
On dissout le composant actif, le tampon, l'arôme, le colorant et le
préservateur dans une partie de l'eau et on ajoute la glycérine. On chauffe le
restant de l'eau à 80 C et on y dissout le saccharose puis on refroidit. On
combine les deux solutions, on règle le volume et on mélange. Le sirop obtenu
est clarifié par filtration.
WO 96101830 PCT1FR95100916
2194677
44
Suppositoires
Composant actif 10,0 mg
*Witepsol H15 complément à 1,0 g
*Marque commercialisée pour Adeps Solidus de la Pharmacopée Européenne.
On prépare une suspension du composant actif dans le Witepsol H15 et on
l'introduit dans une machine appropriée avec moules à suppositoires de 1 g.
Liquide pour administration par injecrion intraveineuse
gn
composant actif 2,0
eau pow- injection Codex complément à 1000,0
On peut ajoutet du chlorure de sodium pour régler la tonicité de la solution
et
régler le pH à la stabilité maximale et/ou pour faciliter la dissolution du
composant actif au moyen d'un acide ou d'un alcali dilué ou en ajoutant des
sels tampons appropriés. On prépare la solution, on la clarifie et on
l'introduit
dans des ampoules de dimension appropriée qu'on scelle par fusion du verre.
On peut également stériliser le liquide pour injection par chauffage à
l'autoclave selon l'un des cycles acceptables. On peut également stériliser la
solution par filtration et introduire en ampoule stérile dans des conditions
aseptiques. La solution peut être introduite dans les ampoules en atmosphère
gazeuse.
Cartouches pour inhalation
g/cartouche
composant actif micronisé 1,0
lactose Codex 39,0
Le composant actif est micronisé dans un broyeur à énergie de fluide et mis à
l'état de fines particules avant mélange avec du lactose pour comprimés dans
un mélangeur à haute énergie. Le mélange pulvérulent est introduit en capsules
de gélatine dure n 3 sur une machine à encapsuler appropriée. Le contenu des
cartouches est administré à l'aide d'un inhalateur à poudre.
Aérosol sous pression à valve doseuse
mg/dose pour 1 boite
composant actif micronisé 0,500 120 mg
acide oléique Codex 0,050 12 mg
loWO 96101830 2194677 PCTIFR95l00916
trichlorofluorométhane pour usage
pharmaceutique 22,25 5,34 g
dichlorodifluorométhane pour usage pharmaceutique 60,90 14,62 g
5 Le composant actif est micronisé dans un broyeur à énergie de fluide et mis
à
l'état de fmes particules. On mélange l'acide oléique avec le
trichlorofluorométhane à une température de 10-15 C et on introduit dans la
solution à l'aide d'un mélangeur à haut effet de cisaillement le médicament
micronisé. La suspension est introduite en quantité mesurée dans des boîtes
10 aérosol en aluminium sur lesquelles on fixe des valves doseuses appropriées
délivrant une dose de 85 mg de la suspension ; le dichlorodifluorométhane est
introduit dans les boites par injection au travers des valves.