Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
- 21 96~76
WO 96~03404 PCT~EP95~02953
-- 1 --
Acylimidazopyridines
Field of ~pplication of the invention
The invention relates to novel acylimidazopyridines
which are intended to be used in the pharmaceutical
industry as active compounds for the production of
medicaments.
Known techn;cal hackgrol]nd
European Patent Application EP-A-0 033 094 describes
imidazo[1,2-a]pyridines which in the 8-position carry
an aryl substituent which is preferably a phenyl,
thienyl, pyridyl, or chlorine-, fluorine-, methyl-,
tert-butyl-, trifluoromethyl-, methoxy- or cyano-sub-
stituted phenyl radical. As aryl radicals of particular
interest, EP-A-0 033 094 mentions the radicals phenyl,
o- or p-fluorophenyl, p-chlorophenyl and 2,4,6-tri-
2 D methylphenyl, of which the radicals phenyl, o- or
p-fluorophenyl and 2,4,6-trimethylphenyl are particu-
larly preferred. - European Patent Applications
EP-A-0 204 285, EP-A-0 228 006, EP-A-0 268 989 and
EP-A-0 308 917 describe imidazo[1,2-a]pyridines which
2 5 in the 3-position carry an unsaturated aliphatic
radical, in particular a (substituted) alkynyl radical.
- European Patent Application EP-A-0 266 890 describes
imidazo[1,2-a]pyridines which are substituted in the
8-position by an alkenyl, alkyl or cycloalkylalkyl
radical.
Description of the invention
It has now been found that the compounds described in
greater detail below, which in particular differ from
the compounds of the prior art by the substitution in
the 3- or 8-position, have surprising and particularly
advantageous properties.
21 96~76
WO 96~03404 PCT~EP95~02953
- 2 -
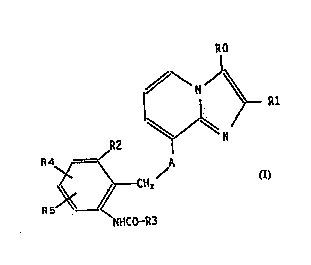
The invention relates to compounds of the formula I
(see attached formula sheetj, in which
R0 is 1-4C-alkyl, hydroxymethyl, halogen or thio-
cyanate,
Rl is 1-4C-alkyl,
R2 is hydrogen, 1-4C-alkyl, 1-4C-alkoxy, halogen or
trifluoromethyl,
R3 is amino, pyridyl, halogen-substituted 1-4C-alkoxy
or substituted 1-4C-alkyl having one or two
identical or different substituents selected from
the group consisting of hydroxyl, oxo, 1-4C-alkyl-
carbonyloxy, carboxyl, halogen, 1-4C-alkoxy, 1-4C-
alkoxycarbonyl, amino, 1-4C-alkylcarbonylamino and
1-4C-alkoxycarbonylamino,
R4 is hydrogen, 1-4C-alkyl, 1-4C-alkoxy, halogen or
trifluoromethyl,
R5 is hydrogen, 1-4C-alkyl, 1-4C-alkoxy or halogen
and
A is O (oxygen) or NH,
and their salts.
1-4C-Alkyl is a straight-chain or branched alkyl
radical having 1 to 4 carbon atoms. Examples which may
be mentioned are the butyl, isobutyl, sec-butyl, tert-
butyl, propyl, isopropyl, ethyl and, in particular, the
methyl radical.
Halogen within the meaning of the invention is bromine,
chlorine or fluorine.
1-4C-Alkoxy is an oxygen atom to which one of the
abovementioned 1-4C-alkyl radicals is bonded. Examples
which may be mentioned are the methoxy and the ethoxy
radicals.
1-4C-Alkylcarbonyl is a radical which, beside the
carbonyl group, contains one of the abovementioned
1-4C-alkyl radicals. An example which may be mentioned
~ . - 2 1 q6076
WO 96~03404 PCT~EP95~02953
in particular is the acetyl radical. A preferred 1-4C-
alkylcarbonyloxy radical which may be mentiolled is the
acetoxy radical.
1-4C-Alkoxycarbonyl is a radical which, beside the
carbonyl group, contains one of the abovementioned
1-4C-alkoxy radicals. Examples which may be mentioned
are the methoxycarbonyl and the ethoxycarbonyl
radicals.
1-4C-Alkylcarbonylamino radicals which may be men-
tioned, for example, are the propionylamino and, in
particular, the acetylamino radicals.
1-4C-Alkoxycarbonylamino radicals which may be men-
tioned, for example, are the methoxycarbonylamino, the
ethoxycarbonylamino and, in particular, the t-butoxy-
carbonylamino radicals.
Suitable salts of compounds of the formula I are pre-
ferably all acid addition salts. Particular mention may
be given to the pharmacologically tolerable salts of
the inorganic and organic acids customarily used in
pharmacy. Pharmacologically nontolerable salts which
can initially be obtained as process products, for
example, in the preparation of the compounds according
to the invention on the industrial scale, are converted
into pharmacologically tolerable salts by the processes
known to the person skilled in the art. Very suitable
are water-soluble and water-insoluble acid addition
salts with acids such as, for example, hydrochloric
acid, hydrobromic acid, phosphoric acid, nitric acid,
sulfuric acid, acetic acid, citric acid, D-gluconic
acid, benzoic acid, 2-(4-hydroxybenzoyl)benzoic acid,
butyric acid, sulfosalicylic acid, maleic acid, lauric
acid, malic acid, fumaric acid, succinic acid, oxalic
acid, tartaric acid, embonic acid, stearic acid,
toluenesulfonic acid, methanesulfonic acid or
21 ~ 076
WO 96~03404 PCT~EP95~02953
3-hydroxy-2-naphthoic acid, the acids being employed in
salt preparation - depending on whetr-her it is a mono-
or polybasic acid and depending on which salt is
desired - in an equimolar quantitative ratio or one
differing therefrom.
Compounds to be emphasized are those of the formula I,
in which
R0 is 1-4C-alkyl, hydroxymethyl or halogen,
R1 is 1-4C-alkyl,
R2 is 1-4C-alkyl or halogen,
R3 is amino, pyridyl, halogen-substituted 1-4C-alkoxy
or substituted 1-4C-alkyl having a substituent
selected from the group consisting of hydroxyl,
oxo, 1-4C-alkylcarbonyloxy, halogen, 1-4C-alkoxy,
amino, 1-4C-alkylcarbonylamino and 1-4C-alkoxy-
carbonylamino,
R4 is hydrogen,
R5 is hydrogen and
A is O (oxygen) or NH,
and their salts.
Compounds to be particularly emphasized are those of
the formula I, in which
R0 is methyl, hydroxymethyl or chlorine,
R1 is methyl,
R2 is 1-4C-alkyl, chlorine or fluorine,
R3 is amino, chlorine-substituted 1-4C-alkoxy or
substituted 1-4C-alkyl having a substituent
selected from the group consisting of hydroxyl,
oxo, 1-4C-alkylcarbonyloxy, chlorine, 1-4C-alkoxy,
amino, 1-4C-alkylcarbonylamino and 1-4C-alkoxy-
carbonylamino,
R4 is hydrogen,
R5 is hydrogen and
A is 0 (oxygen) or NH,
and their salts.
21 ~6076
WO 96~03404 PCT~EP95~02953
Preferred compounds are those of the formula I, in
which
R0 is methyl or hydroxymethyl,
Rl is methyl,
5 R2 is 1-4C-alkyl,
R3 is amino,l-hydroxyethyl,l-acetoxyethyl,
l-oxoethyl,acetoxymethyl, 2-oxopropyl,
2-hydroxypropyl,l-butoxycarbonylaminomethyl,
acetaminomethyl, aminomethyl, 3-chloropropyl,
methoxymethyl or 2-chloroethoxy,
R4 is hydrogen,
R5 is hydrogen and
A is O (oxygen) or NH,
and their salts.
Exemplary compounds according to the invention are
listed in Table 1 which follows:
Table 1
2D
Compounds of the formula I (see attached formula sheet)
with Rl=CH3, R4=H, R5=H and the following further
substituent meanings:
R0 R2 R3 A
F CH3 CH(OH)CH3 NH
F CH3 CH(OH)CH3 O
F Cl CH(OH)CH3 NH
F Cl CH(OH)CH3 O
Cl CH3 CH(OH)CH3 NH
Cl CH3 CH(OH)CH3 O
Cl Cl CH(OH)CH3 NH
Cl Cl CH(OH)CH3 O
F CH3 NH2 NH
F CH3 NH2 ~
F Cl NH2 NH
F Cl NH2 ~
21 ~6076
WO 96~03404 PCT~EP95~02953
Continuat;on of Table 1
R0 R2 R3 A
Cl CH3 NH2 NH
Cl CH3 NH2 ~
Cl Cl NH2 NH
Cl Cl NH2 ~
CH3 ClCH(OH)CH3 NH
CH3 ClCH(OH)CH3 O
CH20H ClCH(OH)CH3 NH
CH20H ClCH(OH)CH3 O
and the salts of the compounds mentioned in the table.
Compounds of the formula I can each have a chiral
center in the substituent R3. The invention therefore
includes under the chiral compounds both the pure
enantiomers and their mixtures in any mixing ratio,
including the racemates.
The invention further relates to a process for the
preparation of the compounds of the formula I and their
salts. The process comprises
a) for the preparation of compounds of the formula I
in which R0 is hydroxymethyl, reducing compounds
of the formula II (see attached formula sheet), in
which Rl, R2, R3, R4, R5 and A have the meanings
indicated above, or
b) introducing the radical R3-CO- into compounds of
the formula III (see attached formula sheet), in
which R0, Rl, R2, R4, R5 and A have the meanings
indicated above, in a suitable manner and if
desired then converting the radical R3 into
another radical R3, or
2 1 ~6076
WO 96~03404 PCT~EP95~02953
-- 7 --
c) for the preparation of compounds I in which R3 is
amino, reacting compounds of the formuia III (see
attached formula sheet), in which R0, R1, R2, R4,
R5 and A have the meanings indicated above, with
alkali metal cyanate,
and if desired then converting the compounds I obtained
into their salts, or if desired then liberating the
compounds I from salts of the compounds I obtained.
The reduction of the compounds II is performed in a
manner customary per se to the person skilled in the
art. It is carried out in inert solvents, e.g. lower
aliphatic alcohols, e.g. using suitable hydrides, such
as, for example, sodium borohydride, if desired with
addition of water.
The introduction of the radical R3-C0- into the
compounds III is carried out in a manner familiar
per se to the person skilled in the art, for example as
described in the following examples or analogously
using those processes such as are described in European
Patent Application EP-A-0 308 917.
The reaction according to process variant c) is like-
wise carried out in a manner familiar per se to the
person skilled in the art, such as is known for the
preparation of urea derivatives from amines, for
example by addition of an aqueous solution of an alkali
metal cyanate (e.g. potassium cyanate) to a solution or
suspension of the compound III in the acid.
The person skilled in the art is familiar on the basis
of his expert knowledge with the reaction conditions
which are specifically necessary for carrying out the
process.
21 ~6076
.
WO 96~03404 PCT~EP95~02953
-- 8
The isolation and purification of the substànces
according to the invention is carried out in a manner
known per se, for example, in such a way that the
solvent is distilled off in vacuo and the residue
obtained is recrystallized from a suitable solvent or
subjected to one of the customary purification methods,
such as, for example, column chromatography on suitable
support material.
Acid addition salts are obtained by dissolving the free
base in a suitable solvent, e.g. in water, in a
chlorinated hydrocarbon, such as methylene chloride or
chloroform, a lower aliphatic alcohol (ethanol,
isopropanol), a ketone, such as acetone, or an ether,
such as THF or diisopropyl ether, which contains the
desired acid, or to which the desired acid is then
added.
The salts are obtained by filtration, reprecipitation,
precipitation with a nonsolvent for the addition salt
or by evaporation of the solvent. Salts obtained can be
converted by basification, e.g. with aqueous ammonia
solution, into the free bases, which in turn can be
converted into acid addition salts. In this manner,
pharmacologically nontolerable acid addition salts can
be converted into pharmacologically tolerable acid
addition salts.
The starting compounds II can be prepared in a manner
known per se, for example by reaction of the compounds
IV with the compounds v (see attached formula sheet),
in which R1, R2, R3, R4, R5 and A have the meanings
indicated above and X is a suitable leaving group, e.g.
a halogen atom (preferably chlorine or bromine), or
analogously using those processes such as are des-
cribed, for example, in European Patent Applications
EP-A-0 268 989 or EP-A-0 308 917.
21 96076
WO 96~03404 PCT~EP95~02953
The starting Gompounds III are disclosed in
EP-A-0 308 917 or they can be prepared in a manner
analogous to that described there.
For example, the starting compounds III can be prepared
by reduction in a manner known per se from the corres-
ponding nitro compounds.
The following examples serve to illustrate in greater
detail the preparation of the compounds according to
the invention. In particular, the examples also serve
to describe by way of example the preparation of the
compounds of the formula I and the preparation of
selected starting compounds. Likewise, further com-
pounds of the formula I and further starting compoundswhose preparation is not described explicitly can be
prepared in a manner which is analogous or in a manner
which is familiar per se to the person skilled in the
art using customary process techniques. The
abbreviation RT stands for room temperature, h stands
for hour(s), min stands for minute(s), m.p. for melting
point and dec. for decomposition.
Examples
Final products
.
1. 8-~2-(2(S)-Acetoxypropionylamino)-6-methylbenzyl-
amlnol-2.3-dimethylimidazorl,2-alpyridine
O-Acetyl-L-lactoyl chloride (0.34 g, 2.25 mmol), dis-
solved in anhydrous dichloromethane (5 ml), is added
dropwise at RT to a solution of 8-(2-amino-
6-methylbenzylamino)-2,3-dimethylimidazo[1,2-a]pyridine
(0.56 g, 2.2 mmol) in anhydrous dichloromethane
(25 ml). The solution is subsequently stirred at RT for
2 h and then extracted with aqueous sodium hydrogen
carbonate solution (3 x 15 ml). The organic phase is
21 96076
WO 96~03404 PCT~EP95~02953
- 10 -
dried over magnesium sulfate and concentrated. The
residue is purified by chromatography on silica gel
(eluent: toluene/dioxane = 9:1). After concentration of
the fractions of Rf = 0.2 and crystallization from
5 diisopropyl ether, the title compound (0.76 g, 96~) is
obtained as a colorless crystallizate. M.p. 177-178~C,
specific rotation (589 nm, 22~C): -37~C (c = 1, CHC13).
2. 8-~2-(2(R)-Acetoxypropionylamino)-6-methylbenzyl-
aminol-2.3-dimethylimidazo~1 2-alpyridine
According to the procedure indicated in Example 1,
O-acetyl-D-lactoyl chloride and 8-(2-amino-
6-methylbenzylamino)-2,3-dimethylimidazo[1,2-a]pyridine
15 give the title compound of m.p. 177-179~C. Specific
rotation (589 nm, 22~C): +40~C (c = 1, CHCl3).
3. 8-~2-(2(S)-Acetoxypropionylamino)-6-methylbenzyl-
aminol-3-formyl-2-methylimidazo~1 2-alpyridine
2D
According to the procedure indicated in Example 1,
O-acetyl-L-lactoyl chloride and 8-(2-amino-6-methyl-
benzylamino)-3-formyl-2-methylimidazo[1,2-a]pyridine
give, after crystallization of the crude product from
25 ethyl acetate/diisopropyl ether, the title compound of
m.p. 132-136~C.
4. 8-~2-(2(R)-Acetoxypropionylamino)-6-methylbenzyl-
am;nol-3-formyl-2-methylim;dazo~1 2-alpyridine
3D
According to the procedure indicated in Example 1,
O-acetyl-D-lactoyl chloride and 8-(2-amino-6-methyl-
benzylamino)-3-formyl-2-methylimidazo[1,2-a]pyridine
give, after chromatography on silica gel (eluent:
35 toluene/dioxane = 9:1) and crystallization from ethyl
acetate/cyclohexane, the title compound of m.p. 131-
134~C.
21 ~607~
Wo 96/03404 - 11 - PCT/EP95/02953
5. 8-~2-(2(S)-Hy~roxyprop;ony1 ~m; no)-6-methylh~nzyl
~m;nol -~ 3-~methyl;m;~orl 2-~l~yridine
A solution of 8-[2-(2(S)-acetoxypropionylamino)-
6-methylbenzylamino]-2,3-dimethylimidazo[1,2-a]pyridine
(0.70 g, 1.77 mmol) in methanol (15 ml) is treated with
sodium methoxide (0.2 ml of 30~ strength solution in
methanol) and stirred at RT for 30 min. Water (20 ml)
is then added and the methanol is distilled off on a
lo rotary evaporator. The precipitate deposited in the
aqueous residue is filtered off, washed with water and
dried in vacuo. The crude product is purified by
chromatography on silica gel (eluent: ethyl
acetate/methanol = 9:1). After concentration of the
fractions of Rf = 0.25 and crystallization from ethyl
acetate/diisopropyl ether, the title compound (0.5 g)
is isolated as a beige crystallizate. M.p. 185-186~C,
specific rotation (589 nm, 22~C): -41~C (c = 1, CHCl3).
6. 8-~ (R)-~y~roxyprop;onyl ~m; no)-6-methylbenzyl-
aminol-2 3-~;met~ylimidazo~l 2-alpyr;~;ne
According to the procedure indicated in Example 5,
8-[2-(2(R)-acetoxypropionylamino)-6-methylbenzylamino]-
2,3-dimethylimidazo[1,2-a]pyridine and sodium methoxide
give the title compound. M.p. 178-180~C, specific
rotation (589 nm, 22~C): +37~C (c = 1, CHCl3).
7. 8-~2-(2(S)-Hy~roxyprop;onyl ~m; no)-6-methylh~n~yl-
~m; nol-3-hydroxymethyl-2-methylim;dazo~1,2-
~lpyr;~;ne
A suspension of 8-[2-(2(S)-acetoxypropionylamino)-
6-methylbenzylamino]-3-formyl-2-methylimidazo-
[1,2-a]pyridine (0.51 g, 1.25 mmol) in anhydrous
methanol (40 ml) is stirred at RT for 2 h with sodium
methoxide (0.1 ml of a 30~ strength solution in
methanol). Sodium borohydride (0.55 g, 1.4 mmol) is
2 1 96076
WO 96/03404 - 12 - PCT/EP95/02953
then added to the yellow solution and it is stirred at
RT fGr a furth~r hour. Water (50 ml) is then added and
the methanol is distilled off on a rotary evaporator.
The precipitate deposited in the aqueous residue is
filtered off, washed with water and dried in vacuo. The
crude product is purified by chromatography on silica
gel (eluent: ethyl acetate/methanol 20:1 to 10:1).
After concentration of the fractions of Rf = 0.3 and
crystallization from ethyl acetate/cyclohexane, the
title compound (0.26 g, 47~) is obtained as a beige
powder. M.p. 164-168~C (dec.).
8. 8- r2- (2(R)-Hy~roxyprop; onyl ~m;no)-6-~met~ylhen~yl-
aminol-3-hy~roxymethyl-2-methyl;m;da~o r 1 2-
~lpyri~;ne
According to the procedure indicated in Example 7,8-[2-(2(R)-acetoxypropionylamino)-6-methylbenzylamino]-
3-formyl-2-methylimidazo[1,2-a]pyridine, sodium methox-
ide and sodium borohydride give the title compound as abeige powder. M.p. 166-170~C.
9a. 8- r2 - (2-~hloroacetylam;no)-6-methylh~n~yl~m;nol-
2 3-~;methyl;m;dazo rl 2-alpyr;~;ne
A solution of 8-(2-amino-6-methylbenzylamino)-
2,3-dimethylimidazo[1,2-a]pyridine (0.28 g, 1 mmol) in
anhydrous dichloromethane (15 ml) is treated with 1.1
equivalents of chloroacetyl chloride and stirred at RT
for 2 h. It is then extracted with aqueous sodium
carbonate solution (3 x 15 ml). The organic phase is
washed with water (15 ml), dried over magnesium sulfate
and concentrated. After crystallization from ethyl
acetate/diisopropyl ether, the title compound (0.35 g,
84~ of theory is isolated as a beige solid. M.p. 159-
162~C.
2 ! 9 6076
WO 96/03404 - 13 - PCT/EP95/02953
According to the procedure described in Example 9a,
reaction of 8-(2-amino-6-methylbenzylamino)-2,3-
dimethylimidazo[1,2-a]pyridine or of 8-(2-amino-
6-methylbenzylamino)-3-chloro-2-methylimidazo[1,2-a]-
pyridine gives the following compounds:
9b. 2,3-Dimethyl-8-[6-methyl-2-(2-oxopropionylamino)-
benzylamino]imidazo[1,2-a]pyridine. M.p. 156-158~C
(crystallization from ethyl acetate/diisopropyl
ether).
9c. 8-[2-(2-Acetoxyacetylamino)-6-methylbenzylamino)-
2,3-dimethylimidazo[1,2-a]pyridine. M.p. 96-98~C.
9d. 8-[2-(2-Chloroethoxycarbonylamino)-6-methylbenzyl-
amino]-2,3-dimethylimidazo[1,2-a]pyridine hydro-
chloride. Extraction with water instead of with
sodium carbonate solution. M.p. 199-202~C (crys-
tallization from ethyl acetate).
9e. 8-[2-(2-Chloroethoxycarbonylamino)-6-methylbenzyl-
amino]-3-chloro-2-methylimidazo[1,2-a]pyridine.
M.p. 136-139~C.
9f. 8-[2-(4-Chlorobutyrylamino)-6-methylbenzylamino]-2,3-
dimethylimidazo[1,2-a]pyridine. M.p. 138-140~C.
9g. 8-[2-(4-Chlorobutyrylamino)-6-methylbenzylamino]-
3-chloro-2-methylimidazo[1,2-a]pyridine. M.p. 130-
132~C.
9h. 8-[2-(2-Methoxyacetylamino)-6-methylbenzylamino)-2,3-
dimethylimidazo[1,2-a]pyridine. M.p. 130-131~C.
lOa. 8-r2-(2-Acetyl ~m; noacetyl ~m; n~) -6-met~yl h~n~yl ~m; n~l -
2.3-~ m~t~y] 1 m; ~o ~1 . 2 -~1 pyr;~; n~
A solution of N-acetylglycine (0.36 g, 3.0 mmol) in
anhydrous dichloromethane (20 ml) is treated at -10~C
with N-methylmorpholine (0.34 g, 3.0 mmol) and stirred
for 1 h. Isobutyl chloroformate (0.4 ml, 3.0 mmol),
dissolved in 5 ml of dichloromethane, is then added
dropwise and the mixture is again stirred at -10~C for
1 h. A solution of 8-(2-amino-6-methylbenzylamino)-2,3-
- 2 ! 9 6076
WO 96/03404 PCT/EP95/02953
- 14 -
dimethylimidazo[1,2-a]pyridine (0.43 g, 1.5 mmol) in
dichloromethar.e (30 ml) is then added dropwise. The
solution is then warmed to RT, stirred for 20 h and
then extracted with water (4 x 50 ml). The organic
phase is dried over magnesium sulfate and concentrated.
The residue is chromatographed on silica gel (eluent:
ethyl acetate/methanol 10:1). The fractions of
Rf= 15 are concentrated and crystallized from ethyl
acetate/diisopropyl ether. The title compound (0.14 g,
26~) is isolated as a beige solid. M.p. 192-195~C.
According to the procedure described in Example lOa,
reaction of various carboxylic acids with isobutyl
chloroformate and the appropriately substituted
imidazopyridines gives the following compounds:
lOb. 8-[2-(2-Acetylaminoacetylamino)-6-methylbenzyl-
amino]-3-formyl-2-methylimidazo[1,2-a]pyridine.
M.p. 150-154~C.
lOc. 8-[2-(2-tert-Butyloxycarbonylaminoacetylamino)-
6-methylbenzylamino]-2,3-dimethylimidazo[1,2-a]-
pyridine. M.p. 172-174~C.
lOd. 8-[2-(2-tert-Butyloxycarbonylaminoacetyl-
amino)6-methylbenzylamino]-3-formyl-
2-methylimidazo[1,2-a]pyridine. M.p. 112-116~C.
lOe. 2,3-Dimethyl-8-(2-[(pyridine-2-carbonyl)amino]-
-6-methylbenzylamino)imidazo[1,2-a]pyridine.
M.p. 178-179~C.
lOf. 2,3-Dimethyl-8-(2-[(pyridine-3-carbonyl)amino]-
6-methylbenzylamino)imidazo[1,2-a]pyridine.
M.p. 119-121~C.
11. 8-~2-(2-~m;roacetyl~m;no)-6-met~ylhenzyl~m;nol-
~ 3-~;methyl;m;~zo~1.2-alpyr;~;ne
8-[2-(2-tert-Butyloxycarbonylaminoacetylamino)-
6-methylbenzylamino]-2,3-dimethylimidazo[1,2-a]pyridine
(0.44 g, 1 mmol) is added in portions at RT to a
21 9~076
Wo 96/03404 - 15 - PCT/EP95/02953
solution of anisole (2 ml) in trifluoroacetic acid
~10 ml) and the mixture is ~tirred for 3Q min. The
reaction mixture is then added to 2 N sodium hydroxide
solution (50 ml) and stirred for 15 min at 4~C. The
precipitate is filtered o,ff, washed with water,
precipitated with stirring in diisopropyl ether and
dried. The title compound (0.29 g, 85%) is isolated as
a beige solid. M.p. 134-136~C.
12. 8-r2-(2-Am;no~cetyl~m;~o)-6-methylhenzyl~m; nol-
3-formyl-2-methyl;m;~zo rl . 2 -~1 pyri~;ne
According to the procedure indicated in Example 11,
starting from 8-[2-(2-tert-butyloxycarbonylaminoacetyl-
amino)-6-methylbenzylamino]-3-formyl-2-methylimidazo-
[1,2-a]pyridine gives the title compound as a beige
solid. M.p. 149-151~C.
13. 8-r2-(2-Am;~o~cetyl~m;~o)-6-meth,ylh~n7,yl~m;nol-
3-hy~roxymethyl-2-methyl;mi~zo r1 . 2-alpyr;~;ne
A solution of 8-[2-(2-aminoacetylamino)-6-methylbenzyl-
amino]-3-formyl-2-methylimidazo[1,2-a]pyridine (0.24 g,
0.68 mmol) in anhydrous ethanol (25 ml) is treated with
sodium borohydride and stirred at RT for 30 min. Water
(50 ml) is then added and the ethanol is distilled off
on a-rotary evaporator. The precipitate in the aqueous
residue is filtered off, washed with water and dried.
The solid is then again precipitated with stirring from
diisopropyl ether and dried again. The title compound
(0.29 g, 85~) is isolated as a beige solid. M.p. 134-
136~C.
14. 8-r2-(2-Acetyl~m;~oacetyl~m;~o)-6-,methylhen7,yl-
~m;nol-3-~y~roxymethyl-2-methyl;m;~zo rl . 2-al-
I?Yrl~lne
2~ 9607~
WO 96/03404 PCT/EP95/02953
- 16 -
According to the procedure indicated in Example 13,
startins from 8-[2-(2-acetylaminoacetylamino)-6-methyl-
benzylamino]-3-formyl-2-methylimidazo[1,2-a]pyridine
gives the title compound as a beige solid. M.p. 162-
167~C.
15. ~ 3-D;methyl-8-r6-methyl-2-(3-oxobl~tyryl~m;no)-
benzyl ~m; nol;m;~70 rl 2-alpyr;~ne
A solution of diketene (0.7 ml of 50~ strength solution
in acetone, 3.5 mmol) in 2 ml of acetone is added drop-
wise at RT to a suspension of 8-(2-amino-
6-methylbenzylamino)-2,3-dimethylimidazo[1,2-a]pyridine
(1.0 g, 3.5 mmol) and 4-dimethylaminopyridine (10 mg)
in anhydrous acetone (30 ml). The mixture is stirred at
RT for a further 1 h. The solvent is then distilled off
on a rotary evaporator and the residual oil is
recrystallized from ethyl acetate. The title compound
(0.31 g, 10~) is isolated as a beige product. M.p. 133-
135~C.
16. 2.3-D;methyl-8- r 6-methyl-2-(3-oxobutyryl~m;no)-
ben7yl0xyl;mi~zo r 1 . 2 -alpyr;~;ne
According to the procedure described in Example 15,
starting from 8-(2-amino-6-methylbenzyloxy)-2,3-dimethyl-
imidazo[1,2-a]pyridine, diketene and 4-dimethylamino-
pyridine gives the title compound (49~) as a beige powder.
M.p. 174-175~C.
17. 3-Formyl-2-methyl-8-r6-~ethyl-2-(3-oxohutyryl-
am;no)h~n7,yloxyl;m;dazo rl, 2-alpyr;~lne
According to the procedure described in Example 15,
starting from 8-(2-amino-6-methylbenzyloxy)-3-formyl-2-
methylimidazo[1,2-a]pyridine, diketene and 4-dimethyl-
aminopyridine gives the title compound (49~) as a beige
powder. M.p. 174-175~C.
2 1 9~076
WO 96/03404 - 17 - PCT/EP95/02953
18. 8-~2-(3-~y~roxyhl]tyryl~m;no)-6-meth~ylh~n7,yloxyl-
3-hy~rcxy~et~yl-~-m~thyl;~; d~70 rl, 2-~1 ~yridine
A solution of 3-formyl-2-methyl-8-[6-methyl-2-(3-oxo-
butyrylamino)benzyloxy]imidazo[1,2-a]pyridine (0.8 g,
2.1 mmol) in tetrahydrofuran (80 ml) and ethanol
(80 ml) is treated with sodium borohydride (80 mg) and
stirred at RT for 30 min. It is then neutralized with
acetic acid, the solvent is distilled off and the
residue is chromatographed on silica gel (eluent: ethyl
acetate/methanol = 10:1). The fractions of Rf = 0.15
are concentrated and crystallized from ethyl acetate.
The title compound (0.52 g, 65~) is isolated as a beige
solid. M.p. 168-169~C.
l9. 2,3-D;methyl-8-(6-meth~yl-2-l]re;dobenzyl ~m; no) -
;m;~7.0r1,~-alpyr;d;ne
A solution of potassium cyanate (0.33 g, 4 mmol) in
8 ml of water is slowly added dropwise at RT to a
suspension of 8-(2-amino-6-methylbenzylamino)-2,3-di-
methylimidazo[1,2-a]pyridine (0.56 g, 2 mmol) in 80~
strength acetic acid (25 ml). The solution is stirred
at RT for a further 2 h. Water (80 ml) is then added
and the mixture is extracted with ethyl acetate
(4 x 75 ml). The organic extracts are washed with water
(50 ml), dried over magne-sium sulfate and concentrated.
The residue is crystallized from diisopropyl
ether/ethyl acetate. The title compound (0.18 g, 28~)
is isolated as a beige solid. M.p. 280~C (dec.).
20. 3-Formyl-~-methyl-8-(6-methyl-~-ure;~ohenzyl-
~m; no); m; d~zo ~1 . 2 -~1 pyr;~;ne
According to the procedure indicated in Example 19,
starting from 8-(2-amino-6-methylbenzylamino)-3-formyl-
2-methylimidazo[1,2-a]pyridine and potassium cyanate
- 21 96376
WO 96~03404 PCT~EP95~02953
- 18 -
gives the title compound (94~) as a beige powder.
M.p. 291-292~C.
21. 3-Hydroxymethyl-2-methyl-8-(6-methyl-2-ureido-
benzylamino)imidazorl,2-alpyridine
A solution of 3-formyl-2-methyl-8-(6-methyl-2-ureido-
benzylamino)imidazo[1,2-a]pyridine (0.27 g, 0.8 mmol)
in ethanol (20 ml) is treated with sodium borohydride
(30 mg, 0.8 mmol) and stirred at RT for 1 h. Water
(50 ml) is then added and the ethanol is distilled off
on a rotary evaporator. The precipitate in the aqueous
residue is filtered off, washed with water and dried in
vacuo. The crude product is purified by chromatography
on silica gel (eluent: dichloromethane/methanol
10:1). After concentration of the fractions of Rf = 0.3
and crystallization from diisopropyl ether, the title
compound (0.11 g, 41~) is isolated as a beige solid.
M.p. 247~C (dec.).
22. 3-Chloro-2-methyl-8-(6-methyl-2-ureidobenzyl-
amino)imidazo~1,2-alpyridine
According to the procedure indicated in Example 19,
starting from 8-(2-amino-6-methylbenzylamino)-3-chloro-
2-methylimidazo[1,2-a]pyridine and potassium cyanate
gives the title compound (72~) as a beige powder.
M.p. 265-267~C.
Start'ng compounds
A1. 8-(2-tert-Butoxycarbonylamino-6-methylbenzyl-
amino)-2,3-dimethylimidazorl,2-alpyridine
5.5 g of sodium iodide and 8.0 g of sodium carbonate
are added at RT to a solution of 8-amino-2,3-
dimethylimidazo[1,2-a]pyridine (4.8 g) and 2-tert-
butoxycarbonylamino-6-methylbenzyl chloride (9.2 g) in
21 96076
WO 96~03404 PCT~EP95~029S3
- 19 -
acetone (250 ml) and the mixture is then heated to
boiling under reflux for 6 h. After cooling the
solution to RT and concentrating, the residue is
dissolved in a mixture of 200 ml of ethyl acetate and
200 ml of water and the organic phase is separated off.
After three further extractions with 100 ml of ethyl
acetate in each case, the combined organic phases are
dried over magnesium sulfate and then concentrated. The
title compound crystallizes as a slightly yellow solid.
Chromatographic purification on silica gel (eluent:
toluene/dioxane = 20:1) and recyrstallization from
diisopropyl ether gives 7.1 g (62~) of the title
compound of m.p. 149-152~C.
A2. 8-(2-tert-butoxyc~rbonylamino-6-methylbenzyl-
amino)-3-formyl-2-methylimidazo~1 2-alpyridine
Starting from 8-amino-3-formyl-2-methylimidazo[1,2-a]-
pyridine (4.0 g), 2-tert-butoxycarbonylamino-6-methyl-
benzyl chloride (7.0 g), sodium iodide (4.1 g) andsodium carbonate (6.1 g) in acetone (250 ml)
analogously using the process of Example Al gives,
after chromatography on silica gel (eluent:
toluene/dioxane 9:1) and recrystallization from
diisopropyl ether, 7.3 g (81~) of the title compound of
m.p. 210-212~C.
A3. 8-(2-Amino-6-methylbenzylamino)-2,3-dimethyl-
imidazo~1 2-alpyridine
Method A:
A solution of 8-(6-methyl-2-nitrobenzylamino)-2,3-
dimethylimidazo[1,2-a]pyridine (61 g) in methanol
(5.5 l) is hydrogenated at RT and under atmospheric
pressure for 1.5 h in the presence of 15 g of palladium
on active carbon (5~) as catalyst. After filtering off
the catalyst and concentrating, the residue is
21 96076
WO 96/03404 PCT/EP95/02953
- 20 -
dissolved in boiling ethyl acetate (2.7 l). After
cooling to RT, 51 g (82~) of the title compound of m.p.
206-208~C are isolated.
Method B:
6.7 g of 8-(2-tert-butoxycarbonylamino-6-methylbenzyl-
amino)-2,3-dimethylimidazo[1,2-a]pyridine are added at
25-30~C in portions to a mixture of trifluoroacetic
acid (30 ml) and anisole (3 ml). After stirring at RT
for 30 minutes, the solution is poured into 100 ml of
ice-water and then treated with 75 ml of 6 N sodium
hydroxide solution. The precipitate is filtered off and
purified chromatographically on silica gel (solvent:
toluene/dioxane = 8:1). Recrystallization from ethyl
acetate gives 3.1 g (62~) of the title compound of m.p.
206-208~C.
A4. 8-(2-Amino-6-methylhenzyl~m;no)-3-formyl-2-met~yl-
im;dazo~ lpyr;dine
Starting from 8-(2-tert-butoxycarbonylamino-6-
methylbenzylamino)-3-formyl-2-methylimidazo[1,2-a]-
pyridine (3.6 g), trifluoroacetic acid (15 ml) and25 anisole (5 ml) according to the procedure described for
Example A3 (Method B) gives, after chromatography on
silica gel (eluent: toluene/dioxane = 9:1) and
crystallization from ethyl acetate/cyclohexane, 2.3 g
(76~) of the title compound of m.p. 230-234~C.
B1. 8-(2-Am;no-6-methylhenzyl~m;no)-3-chloro-2-methyl-
;~;dazor1,2-~lpyr;~;ne hydrochlor;de
A solution of 3-chloro-2-methyl-8-(6-methyl-2-nitro-
benzylamino)imidazo[1,2-a]pyridine (2.0 g, 6 mmol) in
methanol (175 ml) and dioxane (175 ml) is treated with
platinum-on-carbon catalyst (5~ strength) and
hydrogenated at RT under atmospheric pressure for 2 h.
21 ~6076
WO 96/03404 PCT/EP95/02953
- 21 -
After 2 h, 2 N hydrochloric acid (5 ml) is added and
~he mixture ls hydrogenated under the same conditions
again for 1 h. The catalyst is then filtered off, the
filtrate is adjusted to pH 8.5 using 2 N sodium
hydroxide solution and the solvent is distilled off on
a rotary evaporator. The residue is dissolved in
boiling ethyl acetate (400 ml). After cooling to RT,
diisopropyl ether (250 ml) is added and, to complete
crystallization, the mixture is stirred at 4~C for 30
min. The precipitate is then filtered off with suction,
washed with diisopropyl ether and dried in vacuo. The
title compound (1.66 g, 92~) is isolated as a beige
solid. M.p. 243-246~C.
B2. 3-Chloro-~-met~yl-8-(6-methyl-2-n;troben~yl~m;no)-
im;~zorl.2-~lpyr;~;ne
Starting from 8-amino-3-chloroimidazo[1,2-a]pyridine
(9.26 g), 6-methyl-2-nitrobenzyl chloride (10.5 g),
sodium carbonate (13.7 g) and sodium iodide (8.55 g) in
acetone (380 ml) according to the procedure indicated
in Example A1 gives, after chromatography on silica gel
(eluent: toluene/dioxane = 20:1) and crystallization
from ethyl acetate/cyclohexane, 10.6 g (63~) of the
title compound of m.p. 142-144~C.
B3. 8-Am;no-3-chloro;m;dazo~1,2-alpyr;~;ne
A suspension of 3-chloro-2-methyl-8-pivaloylamino-
imidazo[1,2-a]pyridine (4.0 g, 15 mmol) in 60~ strength
sulfuric acid (25 ml) is stirred at 100~C for 1 h.
After cooling to RT, water (100 ml) is added and the
mixture is adjusted to pH 10 using 10 N sodium
hydroxide solution. It is then extracted with ethyl
acetate (3 x 50 ml). The combined organic extracts are
washed with water (50 ml), dried over magnesium sulfate
and concentrated. The residue is taken up in boiling
toluene, and the solution is clarified with silica gel
2! 96076
WO 96/03404 PCT/EP95/02953
- 22 -
and crystallized. The title compound is isolated as a
beige solid Yield 1.9 g (70~ ! m.p. 126-127~C.
B4. 3-~hloro-2-methyl-8-p;v~loylam;no;m;~zo~1.2-al-
pyr;~;ne
5.0 g (18.6 mmol) of 2-methyl-8-pivaloylaminoimidazo-
[1,2-a]pyridine hydrochloride, prepared from 8-amino-2-
methylimidazo[1,2-a]pyridine and pivaloyl chloride,
m.p. 229-230~C, are dissolved in glacial acetic acid
(20 ml) and chlorine gas is slowly passed in at 15~C
until the reaction has ended according to TLC checking
(about 20 min). The solvent is then distilled off, the
residue is taken up in ethyl acetate/water (in each
case 30 ml), and the mixture is rendered basic using
saturated sodium hydrogencarbonate solution and
extracted. It is then extracted again with ethyl
acetate (3 x 30 ml). The combined organic extracts are
washed with water (50 ml), dried over magnesium sulfate
and concentrated. The residue is purified by
chromatography on silica gel (eluent: toluene/dioxane =
9:1). Concentration of the fractions of Rf = O . 2 gives
the title compound (4.1 g, 83~) as a colorless solid.
M.p. 117-118~C.
Commerc;al ut;l;ty
The compounds of the formula I and their salts have
useful pharmacological properties which make them com-
mercially utilizable. In particular, they have a markedinhibition of gastric acid secretion and an excellent
gastric and intestinal protective action in warm-
blooded m~mm~l S. The compounds according to the
invention are distinguished here by a high selectivity
of action, a comparatively long duration of action, a
good enteral activity, the absence of signi~icant side
effects and a large therapeutic breadth.
2t 96076
WO 96~03404 PCT~EP95~02953
- 23 -
"Gastric and intestinal protection" in this connection
is understood as meaning the prevention and tLeatment
of gastrointestinal diseases, in particular gastro-
intestinal inflammatory diseases and lesions (such as,
for example, gastric ulcer, duodenal ulcer, gastritis,
hyperacidic or medicament-related functional
gastropathy), which can be caused, for example, by
microorganisms (e.g. Helicobacter pylori), bacterial
toxins, medicaments (e.g. certain antiinflammatories
and antirheumatics), chemicals (e.g. ethanol), gastric
acid or stress situations. The compounds according to
the invention in this case also have an intrinsic
action against the microorganism Helicobacter pylori.
The compounds according to the invention surprisingly
prove clearly superior to the compounds known from the
prior art in their excellent properties in various
models in which the antiulcerogenic and the anti-
secretory properties are determined. On account of
these properties, the compounds of the formula I and
their pharmacologically tolerable salts are outstand-
ingly suitable for use in human and veterinary
medicine, where they are used, in particular, for the
treatment and/or prophylaxis of disorders of the
stomach and/or intestine.
The invention therefore further relates to the com-
pounds according to the invention for use in the
treatment and/or prophylaxis of the abovementioned
diseases.
The invention likewise comprises the use of the com-
pounds according to the invention for the production of
medicaments which are employed for the treatment and/or
prophylaxis of the abovementioned diseases.
21 96076
WO 96~03404 PCTtEP95~02953
- 24 -
The invention furthermore comprises the use of the com-
pounds according to the invention ror tne treatment
and/or prophylaxis of the abovementioned diseases.
The invention further relates to medicaments which con-
tain one or more compounds of the formula I and/or
their pharmacologically tolerable salts.
The medicaments are prepared by processes known per se,
which are familiar to the person skilled in the art. As
medicaments, the pharmacologically active compounds
according to the invention (= active compounds) are
either employed as such, or preferably in combination
with suitable pharmaceutical auxiliaries or excipients,
in the form of tablets, coated tablets, capsules,
suppositories, patches, (e.g. as TTS), emulsions sus-
pensions or solutions, the active compound content
advantageously being between 0.1 and 95~ and it being
possible to achieve by the appropriate choice of the
auxiliaries and excipients a pharmaceutical admini-
stration form exactly suited to the active compound
and/or to the desired onset of action (e.g. a
sustained-release form or an enteric form).
The person skilled in the art is familiar on the basis
of his expert knowledge with the auxiliaries and
excipients which are suitable for the desired pharma-
ceutical formulations. Beside solvents, gel-forming
agents, suppository bases, tablet auxiliaries and other
active compound excipients, for example, antioxidants,
dispersants, emulsifiers, antifoams, flavor corrigents,
preservatives, solubilizers, colorants or, in par-
ticular, permeation promoters and complexing agents
(e.g. cyclodextrins) can be used.
The active compounds can be administered orally,
parenterally or percutaneously.
21 96076
WO 96~03404 PCT~EP95~02953
- 25 -
In general, it has proven advantageous in human
medicine to administer the active compound~s) in the
case of oral administration in a daily dose of approxi-
mately 0.01 to approximately 20, preferably 0.05 to 5,
in particular 0.1 to 1.5, mg/kg of body weight, if
appropriate in the form of several, preferably 1 to 4,
individual doses to achieve the desired result. In the
case of a parenteral treatment, similar or (in par-
ticular in the case of intravenous administration of
the active compounds), as a rule, lower doses are used.
Any person skilled in the art can easily fix the
required optimum dose and manner of administration of
the active compounds in each case on the basis of his
expert knowledge.
If the compounds and/or salts according to the inven-
tion are to be employed for the treatment of the above-
mentioned diseases, the pharmaceutical preparations can
also contain one or more pharmacologically active con-
stituents of other pharmaceutical groups, such asantacids, for example aluminum hydroxide, magnesium
aluminate; tranquilizers, such as benzodiazepines, for
example diazepam; spasmolytics, such as, for example,
bietamiverine, camylofin, anticholinergics, such as,
for example, oxyphencyclimine, phencarbamide; local
anesthetics, such as, for example, tetracaine,
procaine; and optionally also enzymes, vitamins or
amino acids.
Emphasis is to be given in this connection in par-
ticular to the combination of the compounds according
to the invention with pharmaceuticals which inhibit
acid secretion, such as, for example, H2 blockers (e.g.
cimetidine, ranitidine), H+/K+ ATPase inhibitors (e.g.
omeprazole, pantoprazole), or furthermore with so-
called peripheral anticholinergics (e.g. pirenzepine,
telenzepine) and also with gastrin antagonists with the
aim of potentiating the main action in an additive or
21 96r)76
WO 96~03404 PCT~EP95~02953
- 26 -
superadditive sense and/or of eliminating or of lower-
ing the side effects, or furthermore the cGmbination
with substances having antibacterial activity (such as,
for example, cephalosporins, tetracyclines, nalidixic
acid, penicillins or alternatively bismuth salts) for
the control of Helicobacter pylori.
Pharmacology
The excellent gastric protective action and the gastric
acid secretion-inhibiting action of the compounds
according to the invention can be detected in investi-
gations on animal experimental models. The compounds
according to the invention investigated in the model
mentioned below have been provided with numbers which
correspond to the numbers of these compounds in the
examples.
Testlng the secretion-inhibiting action on the perfused
rat stomach
Table A which follows shows the effect of the compounds
according to the invention after intravenous admini-
stration on the pentagastrin-stimulated acid secretion
of the perfused rat stomach in vivo.
TabIe A
No. DoseInhibition of acid secretion
(~mol/kg) (~)
i .v .
3 96
6 3 94
19 3 100
21 96076
WO 96~03404 - 27 - PCT~EP95~02953
Methodo~ogy
After tracheotomy, the abdomen of anesthetized rats (CD
rats, female, 200-250 g; 1.5 g/kg i.m. urethane) was
opened by a median upper abdominal incision and a PVC
catheter was fixed transorally in the esophagus and a
further one via the pylorus in such a way that the tub-
ing ends just projected into the gastric lumen. The
catheter leading from the pylorus led outwards via a
side opening in the right abdominal wall.
After thorough irrigation (about 50-100 ml), warm
physiologicaI NaCl solution at 37~C was continuously
passed through the stomach (0.5 ml/min, pH 6.8-6.9;
Braun-Unita I). In the effluate in each case collected
at an interval of 15 minutes, the pH (pH meter 632,
glass electrode EA 147; ~ = 5 mm, Metrohm) and, by
titration with a freshly prepared 0.01 N NaOH to pH 7
(Dosimat 665 Metrohm), the secreted HCl were
determined.
The stimulation of gastric acid secretion was effected
by continuous infusion of 1 ~g/kg (= 1.65 ml/h) of i.v.
pentagastrin (left femoral vein) for about 30 min after
the end of the operation (i.e. after determination of 2
initial fractions). The substances to be tested were
administered intravenously in 1 ml/kg of liquid volume
min after beginning the pentagastrin continuous
infusion.
The body temperature of the animals was kept at a con-
stant 37.8-38~C by infrared irradiation and heating
pads (automatic, continuous regulation by means of a
rectal temperature sensor).
The table indicates the dose which led to a maximum
inhibition of the acid secretion by about 100~.