Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
I FL/64-20080/A
2 1 97785
~, ~ f ~ ~
Oligonucleotide conjugates, compositions and a method of cleaving ribonucleic acids
The present invention relates to oligonucleotide conjugates with transesterification or
hydrolysis catalysts the oligonucleotide sequence of which is partly non-complementary to a
naturally occurring target ribonucleic acid (target RNA); to a method for the sequence-
specific cleavage of a target RNA under physiological conditions and under the action of the
oligonucleotide associate; to a composition comprising an inert carrier and the oligo-
nucleotide associate; and to the use thereof.
The hydrolytic cleavage of RNA single strands under the catalytic action of metal ions has
already been known for a long time. The cleavage takes place basically in unpaired regions
of the RNA known as "loops". W. J. Krzyzosiak et al. propose in Biochemistry, Volume 27,
pages 5771 to 5777 (1988) the use of lead diacetate for that purpose. G. J. Murakawa et al.
describe in NucleicAcid Research, Volume 17, pages 5361 to 5375 (1989) the use of copper
complexes of 1,10-phenanthroline. J. Ciesiolka et al. disclose in Eur. J. Biochem.,
Volume 182, pages 445 to 450 (1989) europium trichloride for the same purpose for cleaving
tRNAPhe. In J. Am. Chem. Soc., Volume 112, pages 2839 to 2841 (1990), C. S. Chow et al.
use for the same RNA ruthenium and rhodium complexes with phenanthroline ligands. In
Biochemistry, Volume 29, pages 2515 to 2523, L. S. Behlen et al. mention tRNAPhe mutants
with lead diacetate. In addition, N. Hayashi et al. describe in Inorg. Chem., Volume 32,
pages 5899 to 5900 (1993) that lanthanide metal complexes are also suitable for the
cleavage of tRNA.
L. S. Kappen et al. describe in Biochemistry, Volume 32, pages 13138 to 13145 (1993) that
in the oxidative cleavage of single- or double-strand DNA with neocarcinostatin a position-
specific cleavage takes place when, for example, unpaired regions result in a bulge in a DNA
strand. D. Williams et al. disclose in Nucleic Acids Research, Volume 16, pages 11607 to
11615 (1988) that in the hydrolytic reaction of double-strand DNA with copper phen-
anthroline the cleavage takes place preferentially at sites that additionally contain an
unpaired cytidine in the chain.
It has already been described in DE-A-24 51 358 that in the simulated production of inter-
feron with a double-strand (rln.rCn) complex, toxicity is reduced, while the production of
21 ~7785
interferon is retained, when structural disruptions are produced by modifying the rCn chain so
that the rCn chain in the cells is more readily hydrolysed by nucleases. A structural disruption
proposed is the introduction of a nucleotide that impedes pair formation in the complex. It
should also be mentioned that in Inorg. Chem., Volume 32, pages 3983 to 3984 (1993),
K. A. Kolasa points out that RNA in DNA-RNA hybrids are not cleaved by trivalent lanthanide
ons.
It has also been described by D. Magda et al. in J. Am. Chem. Soc., Volume 116, 7439 to
7440 (1994) that conjugates of europium(lll)-texaphyrine and oligonucleotides with DNA
building blocks are capable of cleaving a target RNA, an increase in cleavage of only about
30 % being observed in the region of the texaphyrine complex in the RNA/oligonucleotide
complex. A further disadvantage of those texaphyrine complexes is that in addition hydroxy-
propyl must be bonded in the ligand so as to ensure sufficient solubility. Furthermore, the
imine groups of the ligand are susceptible to hydrolysis, so that the effectiveness in an
aqueous environment declines relatively quickly; that is to say the residence time is too low
for therapeutic applications. In addition, hydrolysis of the ligand liberates the metal and this
can bring about serious toxicity problems and nonspecific cleavage of the RNA. They are
also weak Lewis acids because a charge on the Eu cation is neutralised by a ligand and
therefore a complex having two charges is present. In addition, the described complexes can
be obtained only by procedures that are expensive in terms of synthesis.
WO 94/29316 discloses a method of phosphate ester hydrolysis using conjugates of an
oligonucleotide with a texaphyrine-metal complex. In an example, conjugates are described
which contain dysprosium(lll) as metal and the oligonucleotide sequence of which is so
selected that the bonding of the oligonucleotide sequence to a target RNA brings about in
the latter a "loop" consisting of one or more nucleotides.
It is known that in cells the formation of physiologically harmful polypeptides is brought about
by the gene-controlled formation of mRNA. In order to combat or prevent diseases it is there-
fore desirable to have agents that impede the action of the mRNA. In particular, the mRNA
should be destroyed by irreversible cleavage at a defined site and the information content
should therefore be lost. It is also desirable by a sequence-specific cleavage of RNA chains
to provide fragments that can be used for the more rapid identification of suitable oligo-
21 97785
- 3 -
nucleotides in the "antisense field" for diagnostic purposes (biosensors) or for the treatment
of diseases by affecting metabolic processes in the cell.
Great demands are made of the above-mentioned agents. They must hybridise specifically
with a target RNA and must not affect other DNA and/or RNA molecules present. Inparticular, they must be highly effective even in small amounts and they must be stable with
respect to degradation caused by endogenous defence substances (for example nucleases).
It has now been found that oligonucleotides the sequence of which is only partly comple-
mentary to a target RNA and to which a transesterification catalyst or hydrolysis catalyst is
bonded are highly effective and it is possible to achieve even sequence-specific cleavages in
a target RNA. It has also been found that under comparable reaction conditions considerably
less oligonucleotide transesterification catalyst is required than in the case of a free (that is
to say not bonded to an oligonucleotide) transesterification catalyst. As a result of the
cleavage of the target RNA in the double-strand region, the instability of the RNA/oligo-
nucleotide complex after the cleavage of the RNA is greatly increased and facilitates
breakdown into the free RNA fragments and the free conjugate of oligonucleotide and
hydrolysis or transesterification catalyst. As a result, the conjugate is able to exhibit catalytic
activity and the amounts used can be considerably reduced.
The invention relates to an oligonucleotide of deoxyribonucleotides (NA), unnatural synthetic
nucleotides, or peptide nucleic acids PNA, wherein a transesterification or hydrolysis catalyst
is bonded to the oligonucleotide, and the internal sequence of the oligonucleotide is partly
non-complementary to a naturally occurring target RNA.
Target RNA within the scope of the present invention means that a RNA sequence must be
present in the target. Accordingly, polyribonucleic acids (RNA) may be present. They are
preferably m-RNA (messenger RNA), pre-m-RNA (precursor m-RNA), t-RNA (transfer RNA),
sn-RNA (small nuclear RNA), r-RNA (ribosomal RNA) and viral RNA. However, it is also
possible for mixed sequences of RNA and polydeoxyribonucleic acids (DNA) to be present,
for example the chimeras RNA-DNA (Okazaki fragment). The RNA has a sufficient number
of building blocks for a complex (double strand) to be formed with the oligonucleotide.
21 q7785
- 4 -
Within the scope of the invention, "partly non-complementary" means that the sequence of
the oligonucleotide contains a structural disruption so that base pairing with corresponding
nucleotide building blocks of the target RNA does not occur (for example, base pairing
means the following complementary nucleosides in the target RNA and in the oligo-
nucleotide: A-U, T/U-A, G-C and C-G). In one embodiment, one or more consecutivenucleotide building blocks are absent from the sequence of the oligonucleotide which is
otherwise complementary to the target RNA. As a result, a bulge is formed in the target RNA
which is especially unstable with respect to transesterification and/or hydrolysis. In another
embodiment, the oligonucleotide contains one or more consecutive nucleotide building
blocks that do not form pairs with the corresponding nucleotide building blocks of the target
RNA. As a result of the structural disruption in the double helix in those regions, the RNA is
unstable with respect to transesterification and/or hydrolysis reactions. Preferably from 1 to
10, especially from 1 to 4 and more especially 1 or 2, consecutive nucleotides are absent in
the oligonucleotide. In another embodiment, from 1 to 10, especially from 1 to 4 and more
especially 1 or 2, consecutive non-pairing nucleotide building blocks are present in the oligo-
nucleotide (such structural disruptions are known as "mismatches" and "internal loops").
Within the scope of the invention, "internal sequence" means that, for example, up to 10,
preferably up to 5, especially up to 3 and more especially 1 or 2, of the outer nucleotide
building blocks of the sequence need not be complementary to the target RNA. This may be
advantageous insofar as a transesterification or hydrolysis catalyst bonded at the end of a
sequence may be more mobile and therefore more efficient.
The oligonucleotide may be composed partly or completely of natural DNA building blocks
complementary to the target RNA or it may be composed completely of unnatural synthetic
nucleotides that are likewise complementary to the target RNA, "partly" meaning that in the
oligonucleotide sequence natural DNA building blocks complementary to the target RNA
have been replaced by unnatural synthetic nucleotides that are likewise complementary.
Synthetic building blocks include the modihcations of natural building blocks in the nucleic
base, the furanose ring and/or the bridge groups of the oligonucleotides. Synthetic building
blocks are generally used in order to strengthen the complex bond in duplex structures
and/or to increase the stability of the oligonucleotides with respect to the degradation caused
by, for example, nucleases. A large number of modified nucleosides have become known in
21 ~7785
- 5 -
the field of "antisense technology" for the synthesis or modification of complementary oligo-
nucleotides and are therefore not described in detail here (see, for example, E. Uhlmann et
al., Chemical Reviews, Volume 90, Number 4, pages 543 to 584 (1990)).
Suitable modifications are modifications in the nucleic base moiety (for examplesubstitutions, omission of substituents), in the nucleotide bridge group (for example
modification of the phosphoric acid ester group or the replacement thereof by other bridge
groups) and in the furanose ring (for example substitutions at the 2'-hydroxyl group,
replacement of the furanose oxygen atom, replacement of the furanose ring by mono- or bi-
carbacyclic rings, replacement of the furanose ring by open-chain structures).
The selection and the order of the building blocks in the sequence of the oligonucleotide are
determined by the required duplex formation with a target RNA. The nature and location of
the linkage with the catalyst can also affect the selection and the order of the building blocks.
The non-pairing nucleotides may be natural nucleotides that are so selected that they are
non-complementary to nucleotides in the target RNA (in accordance with the Watson/Crick
definition, for example, pairs such as A-A, U-U, A-G, A-C, G-T, T-U). However, the non-
pairing nucleotides may also be unnatural, synthetic nucleotides. Such nucleotides may be
modified at the nucleotide base, the nucleotide phosphoric acid ester bridge or the furanose
ring. A large number of such modified and synthetic, non-complementary building blocks
have become known and are familiar to the person skilled in the art. In a preferred
embodiment, the oligonucleotide is composed of unnatural complementary nucleotides, the
oligonucleotide containing especially also non-complementary unnatural building blocks.
The number of building blocks in the oligonucleotide is such that hybridisation with the target
RNA takes place. The oligonucleotides may contain, for example, from 5 to 100, preferably
from 5 to 50, especially from 8 to 30 and more especially from 10 to 25, building blocks. The
regions that prevent pair formation with the target RNA (absent or non-pairing nucleotide
building blocks) are arranged preferably in the middle sequence orders of the oligonucleo-
tide, for example between the fourth-last building blocks, or the third-last building blocks, or
the second-last building blocks or the last building blocks of the sequence. In the case of an
oligonucleotide having, for example, 20 building blocks, building blocks are absent or non-
21 q7785
pairing building blocks are present preferably in the region from the fourth to the seventeenthbuilding block. According to the invention, preferred oligonucleotides are those wherein
nucleotides are absent.
The oligonucleotides are preferably composed of nucleosides of the purine series and the
pyrimidine series, especially of 2'-deoxy-2-aminoadenosine, 2'-deoxy-5-methylcytidine, 2'-
deoxyadenosine, 2'-deoxycytidine, 2'-deoxyuridine, 2'-deoxyguanosine and 2'-thymidine.
Special preference is given to 2'-deoxyadenosine (A), 2'-deoxycytidine (C), 2'-deoxy-
guanosine (G) and 2'-thymidine (T). Modified building blocks are derived preferably from
natural nucleosides of the purine series and the pyrimidine series, especially from
adenosine, cytidine, guanosine, 2-aminoadenosine, 5-methylcytosine, thymidine and the
afore-mentioned deoxy derivatives. Nucleosides may also be 2'-modified ribonucleosides.
In an especially preferred embodiment of the invention, the oligonucleotide partly comple-
mentary to a target RNA is composed of (1) natural deoxynucleosides, especially from the
group 2'-deoxyadenosine (A), 2'-deoxycytidine (C), 2'-deoxyguanosine (G) and 2'-thymidine
(T), or of complementary unnatural synthetic building blocks, and (2) the characteristic of
being only partly complementary is produced by the absence of preferably from 1 to 4,
especially from 1 to 3 and more especially 1 or 2, building blocks in the otherwise comple-
mentary sequence. Within the scope of the invention special preference is given to those
modified nucleosides which increase the stability of the oligonucleotide with respect to
nucleases.
The oligonucleotide may also comprise sequences of peptide nucleic acids (PNA), the
catalyst preferably being bonded to the nucleic base, the amino end or the carboxyl end. The
nucleic bases are bonded to the amide nitrogen atoms of the peptide sequence. The
complementary sequence may consist of natural or unnatural synthetic amino acid building
blocks, it being possible to obtain the non-complementary characteristic as described above
by the omission of building blocks or by the incorporation of non-complementary building
blocks. The same preferences apply to the structure of the PNA sequence as to the structure
of the oligonucleotides. Examples of PNAs can be found in Science, Volume 254, pages
1497 to 1500.
21 q7785
The transesterihcation and/or hydrolysis catalyst may be bonded, optionally via a bridge
group, to N, S or O atoms in the 3'- or 5'-terminal groups in the oligonucleotide sequence.
The catalysts may, however, also be bonded to C, N or O atoms of nucleic bases in or at the
end of the sequence, to 2'-positions of the furanose ring to O, S or N atoms in or at the end
of the sequence or to O, S or N atoms of the nucleotide bridge group in the sequence. The
nature of the bond depends upon the type of catalyst and the nature of its functional groups.
A catalyst molecule may be bonded, for example, directly or via a bridge group to the oligo-
nucleotide. A bridge group may be, for example, a modified functional group which in turn
may be bonded directly or via a connecting group to the catalyst and/or to the oligonucleo-
tide. The bond to the oligonucleotide may be ionic and, preferably, covalent. The catalysts
may also be bonded to the 6'-carbon atom of a carbacyclic nucleotide analogue.
The bridge group may correspond, for example, preferably to formula I
-X,-X2-X3-(X4)X- (I)
wherein X1 is a direct bond or a bivalent, open-chain or cyclic hydrocarbon group having
from 1 to 22 carbon atoms and being uninterrupted or interrupted by radicals from the group
-S-, -NR-, -C(O)-O- and -C(O)-NR-, or a polyoxaalkylene radical having from 1 to 12 oxa-
alkylene units and 2 or 3 carbon atoms in the alkylene; X2 is -O-, -S-, -NR-, -NH-C(O)-NH-,
-NH-C(S)-NH-, -O-C(O)-NH-, -NH-C(O)-O-, -O-C(O)-O-, -C(O)-O-, -C(S)-O-, -O-C(O)-,
-O-C(S)-, -C(O)-NR-, -RN-C(O)-, -S(O)-O-, -O-S(0)2-, -S(0)2-NR-, -NR-S(O)-,
-P(O)-(OM)-O-, -O-P(O)-(OM)-, -P(O)-(OM)-NR-, -NR-P(O)-(OM)-, -PH(O)-O-, -O-PH(O)-,
-PH(O)-NR- or -NR-PH(O)-; X3 has independently the same meanings as X~ and x is 0
when X3 is a direct bond; X4 iS a bond to an O, N or C atom of a nucleoside building block, or
X4 is -O-P(O)(OM)-O-, -NR-P(O)(OM)-O-, -O-P(O)(OM)-NR- or -NR-P(O)(OM)-NR- when x
is 1 and X3 iS not a direct bond; R is H, C~-C6alkyl, phenyl or benzyl; M is H, C~-C6alkyl,
phenyl or benzyl, an alkali metal cation or an ammonium cation; and x is 0 or 1.
X, contains as a bivalent hydrocarbon group preferably from 1 to 18, especially from 1 to 12,
and more especially from 1 to 8, carbon atoms; and as a polyoxaalkylene radical preferably
from 1 to 6, especially from 1 to 4, oxaalkylene units from the group -CH2-CH2-O- and
-CH2-CH(CH3)-O-. The hydrocarbon group may be, for example, linear or branched C~-C22-
21 ~7785
alkylene, preferably C~-C~8alkylene, especially C~-C~2alkylene and more especially C1-C8-
alkylene; C3-C8cycloalkylene, preferably C5- or C6-cycloalkylene; C6-C12arylene or C7-C12-
aralkylene. Some examples of bivalent hydrocarbon groups are methylene, ethylene, 1,2- or
1,3-butylene, 1,2-, 1,3- or 1,4-butylene, 1,2-, 1,3-, 1,4- or 1,5-pentylene, 1,2-, 1,3-, 1,4-, 1,5-
or 1,6-hexylene, 1,2-, 1,3-, 1,4-, 1,5-, 1,6- or 1,7-heptylene, 1,2-, 1,3-, 1,4-, 1,5-, 1,6-, 1,7- or
1,8-octylene, and the isomers of nonylene, decylene, undecylene, dodecylene, tridecylene,
tetradecylene, pentadecylene, hexadecylene, heptadecylene, octadecylene, nonadecylene
and eicosylene; cyclopentylene, cyclohexylene; naphthylene and especially phenylene;
benzylene and phenylethylene. Some examples of polyoxaalkylenes are ethyleneoxy,bisethyleneoxy, trisethyleneoxy, tetraethyleneoxy and 1,2-propoxy.
R as alkyl contains preferably from 1 to 4 carbon atoms and is preferably methyl or ethyl.
R is especially H.
When M is alkyl, it contains preferably from 1 to 4 carbon atoms and is especially methyl or
ethyl. Preferred alkali metal and ammonium cations are Na+, K+, NH4+ and N(C1-C6alkyl)4+.
A preferred subgroup of bridge groups of formula I are those wherein X1 is a direct bond and
is preferably C1-C4alkylene, phenylene or benzylene, it being possible for the alkylene to be
interrupted by -C(O)-O- or by -C(O)-NH-; X2 is -C(O)-O-, -C(O)-NH-, -NH-C(O)-NH- or
-NH-C(S)-NH-; X3 is C2-C18alkylene, preferably C2-C12alkylene; and X4 is a bond to an O, N
or C atom of a nucleotide building block, or X4 is -O-P(O)(OM)-O-, -NR-P(O)(OM)-O-,
-O-P(O)(OM)-NR- or -NR-P(O)(OM)-NR1- (note: in the case of radicals -O-P(O)(OM)-O-,
-NR-P(O)(OM)-O-, -O-P(O)(OM)-NR- or -NR-P(O)(OM)-NR- the N and O atoms of the
nucleoside building block are integrated into those bridge groups).
Suitable as catalysts bonded to the oligonucleotide are, for example, polypeptides (trans-
ferases/hydrolases), metal salts and metal complexes, the metals preferably being selected
from the subgroups of the Periodic Table of the Elements and from the main group metals
In, Tl, Sn, Pb and Bi. Examples are scandium, yttrium, lanthanum, the lanthanide metals, Ti,
Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Ru, Os, Co, Rh, Ir, Ni, Pd, Pt, Cu, Ag, Au, Zn, Cd and
Hg. Scandium, yttrium, lanthanum, the lanthanide metals, Cu and lead are preferred. Of the
2 1 ~7785
lanthanide metals, Ce, Eu, Gd and Sm are preferred. The metals are preferably in the form
of divalent or trivalent cations.
Suitable anions for the metal salts and metal complex salts can be selected, for example,
from the following group: halide (for example Cl-, Br~ and 1-), the anion of an oxyacid, BF4-,
PF6-, SiF6 and AsF6 .
The anions of oxyacids may be, for example: sulfate, phosphate, perchlorate, perbromate,
periodate, antimonate, arsenate, nitrate, carbonate, the anion of a C1-C8carboxylic acid, for
example formate, acetate, propionate, butyrate, benzoate, phenylacetate, mono-, di- or tri-
chloro- or -fluoro-acetate, sulfonates, for example methylsulfonate, ethylsulfonate, propyl-
sulfonate, butylsulfonate, trifluoromethylsulfonate (triflate), unsubstituted or C,-C4alkyl-,
C,-C4alkoxy- or halo-substituted, especially fluoro-, chloro- or bromo-substituted, phenyl-
sulfonate or benzylsulfonate, for example tosylate, mesylate, brosylate, p-methoxy- or p-
ethoxy-phenylsulfonate, pentafluorophenylsulfonate or 2,4,6-triisopropylsulfonate, and
phosphonates, for example methylphosphonate, ethylphosphonate, propylphosphonate,
butylphosphonate, phenylphosphonate, p-methylphenylphosphonate and benzyl-
phosphonate.
The metal complex catalysts are preferably in the form of metal complex salts with hetero-
organic compounds as complexing agents, the complexing agent being bonded to the oligo-
nucleotide. Numerous complexing agents are known. They may be open-chain or cyclic
organic compounds having hetero atoms selected from the group O, S, N and P. Preference
is given to cyclic or polycyclic organic compounds having a total of from 8 to 26, preferably
from 12 to 20, ring members and from 2 to 12, preferably from 4 to 12 and especially from 6
to 12, hetero atoms. Of the hetero atoms, O and especially N are preferred. Some examples
of complexing agents are crown ethers, cyanines, phthalocyanines, naphthalocyanines,
porphyrines, phenanthrolines, open and cyclised bis- and ter-pyridines, ethylenediamine-
tetraacetic acid and diethylenetriamine pentaacetate.
In a preferred embodiment, the catalytically active oligonucleotides according to the
invention are conjugates of formula ll
21 q7785
- 10-
A-B-oligo (Il),
wherein A is a cyclic or polycyclic metal complex salt, preferably bonded to B via carbon
atoms, with a complexing agent containing at least 12 ring atoms and at least 4 hetero
atoms from the group N and O in the ring and to which there are bonded divalent or trivalent
metal ions selected from the group scandium, yttrium, lanthanum and lanthanide metals; B is
the bridge group of formula I and oligo is an oligonucleotide the internal sequence of which is
partly non-complementary to a target RNA.
Oligo and B are subject to the same preferences as those given above.
The complexing agent may contain up to 22, preferably from 6 to 20, especially from 12 to
20 and more especially from 14 to 20, ring atoms, the ring atoms, apart from the hetero
atoms, preferably being carbon atoms. The number of hetero atoms N and/or O is preferably
from 4 to 12, especially from 4 to 10, and more especially from 4 to 8. In the case of
relatively small ring sizes (for example from 6 to 12 ring atoms), smaller numbers of hetero
atoms are preferred, for example from 4 to 8, especially from 4 to 6. In a preferred subgroup,
the complexing agent contains from 16 to 20 and especially 18 ring atoms and from 6 to 10
and especially 8 N atoms, the other ring members being carbon atoms, and to the ring there
are bonded from 1 to 6 and preferably from 2 to 4 unsubstituted or substituted groups
-CH=CH-CH=CH- in the 1 ,3-position, forming a pyridine group with N atoms of the ring.
Preferably the complexing agent contains from 2 to 4 pyridine groups and a further 4 N
atoms in the ring. Preferred metal ions are La, Ce, Nd, Eu and Gd. Preferred anions in the
metal complex salts are halide (Cl, Br~), sulfate, nitrate, PF6, acetate, methylsulfonate,
trifluoromethylsulfonate, carbonate, hydrogen sulfate, hydrogen carbonate and perchlorate.
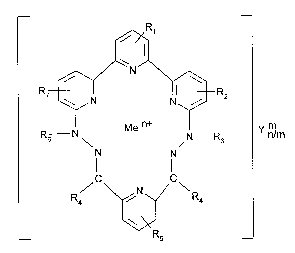
In a very especially preferred embodiment, the conjugates of formula ll are those of
formula lll
- 1 1 - 2 1 q 7 7 8 5
~R1
R7~-- ~ l R2
R6 N\ Me n+ N R y nn/m (111)
N N
Il 11
R / \~ N ~/ \ R
Rs
wherein
Rz and R7 are each independently of the other H, C1-C4alkyl, C1-C4alkoxy, C7-C~2aralkyl or
C6-C~6aryl,
R3 and R6 are each independently of the other H, C1-C4alkyl, C7-C12aralkyl or C6-C.16aryl,
R4 is H, C1-C20alkyl, C5-C8cycloalkyl, C6-C12aryl or C7-C12aralkyl,
Me is a lanthanum, lanthanide metal, yttrium or scandium,
Y is an anion,
n is the number 2 or 3, and
m is the number 1, 2 or 3,
the radicals alkyl, cycloalkyl, aralkyl and aryl being unsubstituted or substituted by C1-C4-
alkoxy, F, Cl, Br, -CN, C1-C4alkyl or by -NO2,
R5 is a radical of formula IV
-B-oligo (IV)
and R1 is H or a substituent or
R5 is H or a substituent and R1 is a radical of formula IV, B and oligo being as defined above,
including the preferences. Suitable, and also preferred, anions for ym- have been mentioned
above. ym- = Cl- is especially preferred.
-12- 21 ~77~5
R2, R3, R6 and R7 as alkyl are preferably methyl or ethyl, as alkoxy preferably methoxy or
ethoxy, as aralkyl preferably benzylene or phenylethylene, and as aryl preferably naphthyl
and especially benzyl. In a preferred embodiment, R2 and R7 are H and R3 and R6 are alkyl.
Especially, R2 and R7 are H and R3 and R6 are C~-C4alkyl and more especially methyl. R2,
R3, R6 and R7 may also be C4-C~2heteroaryl having 0, S, N as hetero atoms. Examples are
pyridyl, thiazolyl, imidazolyl, oxazolyl, furanosyl, pyrrolyl and thiophenyl. They may also be
C~-C4alkylthio, halide, di(C~-C4alkyl)amino, sulfonamide and carboxamide.
R~ and R5 as substituents are preferably C,-C4alkyl, C,-C4alkoxy, C7-C~2aralkyl or C6-C~6-
aryl, C4-C~2heteroaryl having 0, S, N as hetero atoms, C~-C4alkylthio, di(C~-C4alkyl)amino,
halide, sulfonamide and carboxamide.
R~ and R5 are preferably bonded in the p-position to the nitrogen atom of the pyridine ring.
R4 as alkyl contains preferably from 1 to 12, especially from 1 to 8 and more especially from
1 to 4, carbon atoms. Some examples of alkyl are methyl, ethyl and the isomers of propyl,
butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, penta-
decyl, hexadecyl, octadecyl, nonadecyl and eicosyl.
R4 as cycloalkyl contains preferably 5 or 6 ring carbon atoms. Some examples of cycloalkyl
are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclopentyl and cyclooctyl.
R4 as aryl is preferably naphthyl and especially phenyl. When R4 is aralkyl, it is preferably
benzyl or phenylethyl.
A preferred subgroup for R4 is H, C,-C4alkyl, especially methyl, and phenyl or benzyl.
R~ and R5 as alkyl are preferably methyl or ethyl, as alkoxy preferably methoxy or ethoxy, as
aryl preferably naphthyl or phenyl, and as aralkyl preferably phenyl or phenylethyl. Preferably
R, and R5 are H, methyl, ethyl, methoxy or ethoxy.
A preferred subgroup of compounds of formula lll is formed by those wherein R2 and R7 are
H, R3 and R6 are C~-C4alkyl, R4 is H, C~-C4alkyl, phenyl or benzyl, R, is the group
- 1 3 - 2 1 9 7 7 8 5
X~-X2-X3-(X4)x-oligo and R5 is H, methyl or methoxy or R5 is the group X1-X2-X3-(X4)x-oligo
and R~ is H, methyl or methoxy, X~ is a direct bond or C2-C6alkylene, X2 is -O-, -NH-,
-C(O)-O-, -C(O)-NH-, -NH-C(O)-NH- or -HN-C(S)-NH-, X3 is C2-C12alkylene or phenylene, X4
is a bond to an 0, N or C atom of a nucleoside building block, or X4 is -O-P(O)(OM)-O-, x is
O or 1, Me is La, Ce, Nd, Eu or Gd, n is 2 or 3 and m is 1 or 2, Y is Cl, Br, CH3C(O)O, Cl04,
BF4, PF6, F3C-SO3 or tosylate, M is H, Na or K, and oligo is an oligonucleotide radical the
internal sequence of which is only partly complementary to a target RNA and which is
composed of natural deoxyribonucleotide building blocks or unnatural synthetic nucleotide
building blocks, with from 1 to 4 building blocks being absent in comparison with the target
RNA.
Suitable transesterification or hydrolysis catalysts are also nucleases or nuclease fragments,
basic polypeptides, amidine and guanidine derivatives, oligoamines and bisimidazoles. They
can be bonded to the oligonucleotide via the same bridge groups as the metal complexes.
The invention relates also to a process for the preparation of oligonucleotides to which a
transesterification or hydrolysis catalyst is bonded, and the internal sequence of the oligo-
nucleotide is partly non-complementary to a naturally occurring target RNA, and the oligo-
nucleotide is composed of natural deoxyribonucleic acid building blocks or of unnatural
synthetic nucleotide building blocks, wherein a transesterification or hydrolysis catalyst
having a functional group bonded to the basic structure is reacted with the functional group
of a nucleotide building block or a functionally modified group of a nucleoside building block.
Examples of functional groups bonded to the basic structure, optionally via a bridge group
X~, are OH, -SH, -NCO, -NCS, -CN, -O-CH2-OH, -NHR, -C(O)OR, -C(O)SH, -C(O)NHR,
-C(O)Hal with Hal being F, Cl or Br, -C(S)SR, -C(S)NHR, -C(S)OR, -SO3R, -SO2NHR,-SO2CI, -P(O)(OH)2, -P(O)(OH)-NHR, -P(S)(SH)2, -P(S)(SH)-NHR, -P(S)(OH)2,
-P(S)(OH)-NHR, -P(O)(SH)2, -P(O)(SH)-NHR, -P(O)(OH)H, -P(O)(NHR)H, -P(S)(SH)H,
-P(S)(NHR)H, -P(S)(OH)H and -P(O)(SH)H, with R being H, -C,-C6alkyl, -CzH2z-NH2,-CzH2z-SH or -(CzH2zO)y~H and z being a number from 2 to 6 and y being a number from 1
to 20. Examples of functionally modified groups are hydroxyalkoxy or aminoalkoxy which are
bonded optionally via a linker, for example -P(O)OM-O-, to a nucleotide building block. The
functional group may be bonded to the basic structure directly or via a group X~ and the
21 ~77~5
- 14-
group X1 is preferably C~-C12alkylene, C,-C,2alkenylene, C~-C~2alkynylene, C5-C8cyclo-
alkylene, C6-C~2arylene or C7-C~2aralkylene.
The process according to the invention for the preparation of the oligonucleotide conjugates
can be carried out, for example, by dissolving an optionally functionalised oligonucleotide in
a solvent or solvent mixture and then adding the transesterification or hydrolysis catalyst
carrying a functional group and then leaving the reaction mixture to finish reacting, optionally
with stirring. The conjugate formed can then be purified and, if desired, isolated in a manner
known per se.
The reaction temperature may be, for example, from 0 to 120~C, preferably from 20 to 80~C.
The reaction is especially carried out at room temperature.
If the linkage reaction is an esterification, transesterification or amidation reaction, the
carboxylic acid groups in question are activated beforehand in known manner, for example
by reaction with carbodiimides and N-hydroxysuccinimide.
Suitable solvents are, for example, water and polar aprotic solvents which are advant-
ageously water-miscible. Examples of such solvents are alcohols (methanol, ethanol, n- or
iso-propanol, butanol, ethylene glycol, propylene glycol, ethylene glycol monomethyl ether,
diethylene glycol, diethylene glycol monomethyl ether), ethers (diethyl ether, dibutyl ether,
tetrahydrofuran, dioxane, ethylene glycol dimethyl ether, ethylene glycol diethyl ether,
diethylene glycol diethyl ether, triethylene glycol dimethyl ether), halogenated hydrocarbons
(methylene chloride, chloroform, 1,2-dichloroethane, 1,1,1-trichloroethane, 1,1,2,2-tetra-
chloroethane, chlorobenzene), carboxylic acid esters and lactones (ethyl acetate, propionic
acid methyl ester, benzoic acid ethyl ester, 2-methoxyethyl acetate, ~-butyrolactone, ~-
valerolactone, pivalolactone), N-alkylated carboxylic acid amides and lactams (N,N-dimethyl-
formamide, N,N-diethylformamide, N,N-dimethylacetamide, tetramethylurea, hexa-
methylphosphoric acid triamide, N-methyl-~-butyrolactam, N-methyl-~-caprolactam, N-
methylpyrrolidone), sulfoxides (dimethyl sulfoxide, tetramethylene sulfoxide), sulfones
(dimethyl sulfone, diethyl sulfone, trimethylene sulfone, tetramethylene sulfone), tertiary
amines (trimethylamine, triethylamine, N-methylpiperidine, N-methylmorpholine, pyridine),
substituted benzenes (chlorobenzene, o-dichlorobenzene, 1,2,4-trichlorobenzene, nitro-
21 ~7785
- 15-
benzene, toluene, xylene) and nitriles (acetonitrile, propionitrile, benzonitrile, phenylaceto-
nitrile).
The reactants are advantageously used in molar ratios. It is, however, also possible for an
excess of the catalyst or of the oligonucleotide to be used.
For purification it is possible to use customary methods, advantageously, for example,
dialysis, electrophoresis, and chromatographic procedures, such as high-pressure liquid
chromatography (HPLC), reverse HPLC, affinity chromatography, ion exchanger chromato-
graphy and gel chromatography.
The optionally functionalised oligonucleotides to be used can be prepared in a manner
known per se by means of automated synthesisers which are commercially available.
Nucleosides for the synthesis thereof are known, and some are commercially available or
they can be prepared according to analogous procedures.
Transesterification or hydrolysis catalysts having functional groups are known, and some are
commercially available or they can be prepared according to known or analogous
procedures .
The functionalised starting compounds having a basic structure of formula lll are novel. They
are obtainable by condensing a terpyridine of formula V
R,
R7 ~ ) ~ \ ~ R2 (V)
R6 N \ N R
NH2 H2N
- 1 6 - 2 ' 9 7 7 8 5
with a pyridine dialdehyde or pyridine diketone of formula Vl
O O
Il 11
~C \ R4 (Vl)
Rs
in the presence of a salt of formula Vll
M nl(Ym~) (Vll)
wherein R1, R2, R3, R4, R5, R6, R7, Me, Y, n and m are as defined above.
The process can be carried out, for example, as follows: the compounds of formulae V, Vl
and Vll, preferably in equivalent amounts, are dissolved in a solvent and then reacted with
one another at elevated temperatures. It is advantageous also to use condensation
catalysts, for example concentrated mineral acids, especially hydrochloric acid, or acidic ion
exchangers. It may be advantageous to add water-binding agents or to remove the water of
reaction from the reaction mixture.
The reaction temperature may be, for example, from 40 to 220~C, preferably from 50 to
1 50~C.
The solvents used are advantageously organic polar aprotic solvents. Such solvents have
been mentioned above.
The metal salts of formula Vll are generally known and most are commercially available.
The preparation of the novel compounds of formula V containing a functional group can be
carried out analogously to the procedure described by E. C. Constable in Polyhedron,
21 q7785
- 17-
Volume 7, No. 24, pages 2531 to 2536 (1988), functional groups optionally being provided
with protecting groups.
Most of the compounds of formulae V and Vl with or without functional groups are known or
they can be prepared in accordance with known or analogous procedures. Compounds of
formula Vl wherein R4 is H, R5 is C2-C~8alkylene-X5 and X5 is -C(O)-OR, -C(O)-NHR, -SO2-R
or -SO2-NHR, and R is H or C1-C6alkyl, are novel and can be obtained as follows: with
palladium catalysis a corresponding 3-halo-pyridine-1,5-dicarboxylic acid ester is alkenylated
with an alkene of the formula CH2=CH-C~-C~6alkylene-carboxylic acid ester, the alkene
group is hydrogenated, for example catalytically, then hydrogenation to the corresponding
1,5-dihydroxymethylpyridine-alkylcarboxylic acid ester is carried out, and the hydroxymethyl
groups are oxidised to aldehyde groups and optionally the ester group is hydrolysed to the
carboxylic acid group or the ester group is amidated to the carboxylic acid amide.
Compounds of formula Vl wherein R4 is H or C1-C12alkyl, R5 is C2-C18alkylene-X5 and X5 is
-C(O)-OR, -C(O)-NHR, -SO2-R or -SO2-NHR, and R is H or C1-C6alkyl, are novel and can be
obtained as follows: with palladium catalysis a corresponding 3-halo-1,5-dihydroxymethyl-
pyridine protected, for example, by acetyl, (obtainable by reduction of the corresponding 3,5-
dicarboxylic acid methyl ester) is alkenylated with an alkene of the formula CH2=CH-C~-C16-
alkylene-carboxylic acid ester, the alkene group is hydrogenated, for example catalytically,
the hydroxyl groups are deprotected and optionally the compound is oxidised to the corres-
ponding 3,5-pyridinealdehyde the aldehyde groups of which can be C1-C,2alkylated, for
example with Grignard reagents, optionally the ester group is hydrolysed to the carboxylic
acid group or the ester group is amidated to the carboxylic acid amide, and the secondary
alcohol groups are oxidised to keto groups.
The oligonucleotides according to the invention are excellently suitable for the cleavage,
especially the sequence-specific cleavage, of RNA sequences, it being necessary to use
only surprisingly small amounts because of their capacity for catalytic action.
The invention relates also to a method of cleaving the phosphate nucleotide bridge of
ribonucleic acids under physiological conditions and under the action of a synthetic trans-
esterification and/or hydrolysis catalyst, in which method (a) the target RNA is complexed
21 ~7785
- 18-
with an oligonucleotide the internal sequence of which is partly non-complementary to the
target RNA and to which a transesterification or hydrolysis catalyst is bonded, and (b) then
allowed to react and cleaved.
The method according to the invention can be carried out in vivo by administering the oligo-
nucleotides or in vitro by combining a target RNA and an oligonucleotide to be used accord-
ing to the invention.
Physiological conditions are familiar to the person skilled in the art and include, for example,
carrying out the method in an aqueous medium and in a pH range of from 5 to 9, preferably
from 5 to 8 and especially from 5 to 7.5, it being possible for the aqueous medium to contain
further inert constituents, for example salts of alkali metals or alkaline earth metals, and
especially buffer systems.
The method may be carried out at a temperature of, for example, from 0 to 100~C. preferably
from 20 to 50~C and especially from 30 to 40~C.
In the method according to the invention, the cleavage is carried out with transesterification
of the phosphate bridge bond to form a fragment having a 2',3'-cyclic phosphate terminal
group and a further fragment having a 5'-hydroxyl terminal group. The cyclic phosphate can
then be hydrolysed further.
The oligonucleotides according to the invention can be used as medicaments. In addition,
the oligonucleotides according to the invention have a high degree of stability with respect to
degradation by nucleases. Especially surprising is their excellent pairing with complementary
nucleic acid strands of the RNA type. In addition, they exhibit unexpectedly high cellular
uptake. The oligonucleotides according to the invention are therefore suitable especially for
antisense technology, that is to say for inhibiting the expression of undesirable protein
products by binding to appropriate complementary nucleotide sequences of mRNA
(EP 266 099, WO 87/07300 and WO 89/08146). They can be used in the treatment of
infections and diseases for example by blocking the expression of bioactive proteins at the
nucleic acid stage (for example oncogenes). The oligonucleotide fragments prepared
according to the invention are suitable also as diagnostic agents and can be used as gene
1 9 2 1 9 7 7 8 5
probes for detecting viral infections or genetically caused diseases by selective interaction at
the stage of the single- or double-strand nucleic acids.
The invention relates also to the use of oligonucleotides prepared according to the invention
as diagnostic agents for detecting viral infections or genetically caused diseases.
The invention relates also to the oligonucleotides according to the invention for use in a
therapeutic method for the treatment of diseases in warm-blooded animals, including human
beings, by the inactivation of nucleotide sequences in the body. The dosage on administra-
tion to warm-blooded animals of approximately 70 kg body weight may be, for example, from
0.01 to 1000 mg per day. Administration is made preferably in the form of pharmaceutical
compositions parenterally, for example intravenously or intraperitoneally. For parenteral
administration there are suitable especially aqueous solutions of a water-soluble active
ingredient, for example a water-soluble physiologically acceptable salt, or aqueous suspen-
sions of such active ingredients, the solutions or suspensions comprising viscosity-
increasing agents, for example sodium carboxymethylcellulose, sorbitol and/or dextran, and
optionally stabilisers. The active ingredient, optionally together with excipients, may also be
in the form of a Iyophilisate and can be made into a solution prior to administration by the
addition of suitable solvents.
The invention relates also to an aqueous composition and especially a pharmaceutical
composition based on an aqueous solution or suspension, comprising an effective amount of
an oligonucleotide according to the invention alone or together with other active ingredients,
water as pharmaceutical carrier, preferably in a significant amount, and optionally excipients.
The pharmacologically effective oligonucleotides according to the invention can be used in
the form of parenterally administrable compositions or in the form of infusion solutions. Such
solutions are preferably isotonic aqueous solutions or suspensions, it being possible, for
example in the case of Iyophilised compositions that comprise the active ingredient alone or
together with a carrier, for example mannitol, for such solutions or suspensions to be made
up prior to use. The pharmaceutical compositions may be sterilised and/or may comprise
excipients, for example preservatives, stabilisers, wetting agents and/or emulsifiers,
solubilisers, salts for regulating the osmotic pressure and/or buffers. The pharmaceutical
21 97785
- 20 -
compositions, which may if desired comprise other pharmacologically active substances, for
example antibiotics, are prepared in a manner known per se, for example by means of
conventional dissolving or Iyophilising procedures, and comprise approximately from 0.1 %
to 90 %, especially from approximately 0.5 % to approximately 30 %, for example from 1 %
to 5 %, active ingredient(s). The conjugates according to the invention may also be
administered by means of inhalation or in a liposomal form of administration.
The conjugates according to the invention can also be used for diagnostic purposes or as
molecular-biological tools as sequence-specific endoribonucleases.
The drawings show by way of example various possible structures of a hybrid of an anti-
sense oligonucleotide conjugate according to the invention and a target RNA molecule, the
structure of the conjugate in each case being so selected that in the hybrid at least one
unpaired nucleotide occurs on the target RNA.
Fig. 1 shows diagrammatically a hybrid of a target RNA (line labelled "5' ") and an antisense
oligonucleotide (line labelled "3' "), to which according to the invention a complex (labelled
"Ln") is bonded as transesterification or hydrolysis catalyst (so-called conjugate), the
bonding site of the complex being located within the antisense oligonucleotide sequence.
The numbering indicated relates to the nucleotide building blocks of the target RNA, the
numbering being such that the nucleotide of the target RNA that is complementary to the
nucleotide of the antisense oligonucleotide to which the complex is bonded is designated "0".
The numbering then continues upwards (+1, +2 etc.) in the 3'-direction and downwards (-1,
-2 etc.) in the 5'-direction of the target RNA. An unpaired nucleotide on the target RNA
occurring as a result of the structure of the antisense oligonucleotide (it is also possible for
several unpaired nucleotides to occur) is shown as a bulge and in the present instance is
located, starting from position 0, in the 3'-direction (here: at position +2) on the target RNA.
Fig. 2 shows diagrammatically a hybrid of a target RNA and an antisense oligonucleotide
conjugate according to the invention, an unpaired nucleotide being located, starting from
position 0, in the 5'-direction on the target RNA (in this case at position -2). Otherwise the
definitions given in relation to Fig. 1 apply in corresponding manner.
-21 - 2 1 97785
Fig. 3 shows diagrammatically a hybrid of a target RNA and an antisense oligonucleotide
conjugate according to the invention, in which the bonding site of the complex is located at
the end of the antisense oligonucleotide, an unpaired nucleotide being located, starting from
position 0, in the 5'-direction on the target RNA (in this case at position -3). Otherwise the
definitions given in relation to Fig. 1 apply in corresponding manner.
The following Examples illustrate the invention.
A) Preparation of starting compounds for the terpyridine-lanthanide complexes
Example A1: Preparation of terpyridine-bis-hydrazino compounds
(a) With cooling with an ice bath, 40 ml of a 2N aqueous potassium hydroxide solution are
added to a solution of 6-acetyl-2-bromopyridine (100 mmol) in 200 ml of methanol. After the
addition of the appropriately substituted benzaldehyde (400 mmol) the cooling bath is
removed and the mixture is stirred for 4 hours at room temperature. The product is filtered
off, washed three times with water and twice with cold methanol and dried under a high
vacuum .
In accordance with that procedure, compounds a.1 (R1: phenyl-4-OCH3; MS 317.7) and a.2
(R,: phenyl-4-NO2; MS 333.6) are prepared.
(a)
11 0
~N
Br
(b) The a,~unsaturated carbonyl compound from (a) (30 mmol), 1-(2-bromopyridylcarbonyl-
methyl)pyridine iodide (12.1 9, 30 mmol) and ammonium acetate (13.9 9, 180 mmol) are
placed in a flask, and 100 ml of acetic acid are added thereto. The mixture is boiled at reflux.
- 22 - 2 1 ~ 1 7 8 5
After 2 hours the mixture is cooled to room temperature and filtered and the product so
obtained is dried under a high vacuum.
In accordance with that procedure, compounds b.1 (R1: phenyl-4-OCH3; MS 497.1) and b.2
(R~: phenyl-4-NO2; MS 512) are prepared.
At room temperature under an argon atmosphere, lithium aluminium hydride (22 mmol) is
added in portions to a solution of titanium tetrachloride (30 mmol) in 75 ml of tetrahydrofuran
(abs.). The resulting suspension is stirred at room temperature for 20 minutes and then
cooled to 0~C. Compound b.2 (10 mmol) is added and the suspension is stirred at room
temperature for 30 minutes. After the careful dropwise addition of 50 ml of water at 0~C,
25 ml of a 25 % aqueous ammonia solution are added. 150 ml of chloroform are added to
the mixture and filtration is carried out over Celite. The aqueous phase is separated off and
extracted three times with chloroform. All the organic phases are combined, washed once
with water, dried over sodium sulfate and concentrated. In accordance with that procedure,
compound b.3 (R1: phenyl-4-NH2; MS 482.5) is prepared.
~N \~ (b)
Br Br
The appropriate dibromoterpyridine compound from (b) (10 mmol) is dissolved in 30 ml of
methylhydrazine and heated under reflux for 17 hours. After cooling to room temperature,
the mixture is concentrated and the residue is taken up in 20 ml of methanol. The product is
filtered off and dried under a high vacuum.
In accordance with that procedure, compounds c.1 (R1: phenyl-4-OCH3; MS 427) and c.2
(R,: phenyl-4-NH2; MS 412.5) are prepared.
2' 97785
- 23 -
The methoxy compound c.1 (10 mmol) is suspended in 100 ml of chloroform and, with
cooling with an ice bath for 20 minutes, a 1 molar solution of boron tribromide (50 mmol) in
methylene chloride is added. The suspension is heated under reflux for 5 days. After cooling
to room temperature, the mixture is poured into 300 ml of ice-water, acidified with 200 ml of
2N aqueous hydrochloric acid. Affer extraction with ether (twice), the aqueous phase is
adjusted to pH 9.0 with 10 % aqueous sodium carbonate solution and stirred for 30 minutes.
The precipitated product c.3 (R1: phenyl-4-OH; MS 413.5) is filtered off and dried under a
high vacuum.
11
~\ N \/ ~ (c)
~ N N~
H3C NH2 H2N CH3
Example A2: Preparation of 3-[4'-(2',6'-diformylpyridine)]propionic acid
(a) 3.5 g of 4-bromopyridine-2,6-carboxylic acid dimethyl ester, 390 mg of tritolylphosphine,
9.3 ml of acrylic acid tert-butyl ester, 7.1 ml of triethylamine, 30 ml of dimethylformamide and
287 mg of palladium acetate are mixed and heated at 110~C. After 90 minutes the reaction
mixture is cooled to room temperature, diluted with ether/methylene chloride (1:1) and
extracted by shaking with NH4CI/H2O. The organic phase is dried with Na2SO4, concentrated
using a rotary evaporator and dried under a high vacuum.
C H N
calculated: 59.81 5.96 4.36
found: 59.8 6.0 4.1
-24- 21 97785
250 mg of palladium on active carbon (5 %) and 2.5 g of the compound obtained above are
dissolved in 250 ml of methanol and hydrogenated overnight at room temperature under an
H2 atmosphere. The product is filtered through Hyflo, and the filtrate is concentrated using a
rotary evaporator and dried at room temperature under a high vacuum.
C H N
calculated: 59.43 6.55 4.33
found: 59.3 6.6 4.3
5.0 9 of the compound obtained above are dissolved in 50 ml of methanol and 50 ml of tetra-
hydrofuran. After cooling to 0~C,1.1 9 of NaBH4 are added. After 50 minutes a further 1.1 9
of NaBH4 are added and after 130 minutes a further 0.5 9 of NaBH4 is added. After a total of
165 minutes the reaction mixture is heated to room temperature. The mixture is cooled to
0~C. After 3.5 hours a further 1.1 9 of NaBH4 are added. After 6 hours the mixture is
concentrated to a volume of 60 ml. A saturated ammonium chloride solution is then added
dropwise, extraction is carried out four times with CH2CI2, and the organic phases are
washed once with ammonium chloride solution, dried with Na2SO4, filtered and
concentrated.
C H N
calculated: 62.90 7.92 5.24
found: 63.0 7.9 5.2
19.8 9 of the compound obtained above are dissolved in 300 ml of dioxane. Then 16.2 9 of
selenium dioxide are added. The reaction mixture is heated at 100~C and stirred, and after
45 minutes cooled to room temperature. After a further two hours' stirring the reaction
mixture is filtered and concentrated using a rotary evaporator.
C H N
calculated: 63.87 6.51 5.32
found: 64.14 6.53 5.43
4.7 9 of the compound obtained above are added to 17.2 ml of ice-cold trifluoroacetic acid.
After conversion to the acid the mixture is concentrated at 0~C.
-25- 21 ~7785
C H N
calculated: 57.97 4.38 6.76
found: 57.55 4.21 6.61
(b) 5 g of 4-bromopyridine-2,6-dicarboxylic acid dimethyl ester are dissolved in 175 ml of
tetrahydrofuran at room temperature. Then 75 ml of methanol are added. The mixture is
cooled to 0~C; 3.44 g of sodium borohydride are added in portions over a period of
45 minutes and the mixture is allowed to rise to room temperature. After 1 hour 30 ml of
acetone are added dropwise within a period of 10 minutes. The reaction mixture is heated
under reflux for 1 hour. The reaction mixture is then concentrated to dryness using a rotary
evaporator. The residue is stirred at room temperature into 50 ml of pyridine. 0.1 9 of 4-
dimethylaminopyridine is added thereto and the mixture is then cooled to 0~C. 34.4 ml of
acetic anhydride are added dropwise within a period of 30 minutes. The suspension is
allowed to rise to room temperature. 50 ml of tetrahydrofuran are added. After being stirred
overnight at room temperature, the reaction mixture is filtered and washed twice using 50 ml
of tetrahydrofuran each time. The filtrate is concentrated using a rotary evaporator. 4-Bromo-
2,6-di(acetoxymethyl)pyridine is obtained by crystallisation (melting point: 66-69~C).
0.982 g of 4-bromo-2,6-di(acetoxymethyl)pyridine, 1.5 g of 3-(tributylstannyl)-acrylic acid
ethyl ester and 176 mg of palladium tetrakis(triphenylphosphine) are dissolved in 25 ml of
dioxane and heated at 90~C. After 90 minutes the reaction mixture is cooled. The solid
product is separated off and recrystallised from hexane/ethyl acetate.
MS 321 (M ).
2.74 g of the compound obtained above and 70 mg of Wilkinson's catalyst are dissolved in
150 ml of benzene. 12.2 ml of triethylsilane are added and the solution is heated at reflux.
270 mg of catalyst triethylsilane in excess are added in portions within a period of one hour.
The product is purified by chromatography.
MS 323.
267 mg of sodium are dissolved in 50 ml of ethanol. 7.2 ml of that solution are added to a
solution of 1.845 9 of the compound obtained above in 35 ml of ethanol. After being stirred
for 2.5 hours at room temperature the reaction mixture is filtered through silica gel and the
-26- 21 97785
filtrate is concentrated to dryness. The product is dried overnight under a high vacuum.
NMR (CDCI3) ~ 7.0 (2H,s), 4.7 (4H,s), 4.1 (2H,q), 2.9 (2H,t),1.2 (3H,t).
1.27 g of the compound obtained above are dissolved in 30 ml of dioxane. 714 mg of
selenium dioxide are added thereto. The reaction mixture is heated and after 2 hours filtered
through cotton wadding. The filtrate is concentrated to dryness. The residue is taken up in
ethyl acetate/methylene chloride (5 %) and filtered over silica gel.
'H-NMR (CDCI3) ~ 10.1 (2H,s), 8.0 (2H,s), 4.1 (2H,q), 3.1 (2H,t), 2.7 (2H,t), 1.2 (3H,t).
13 ml of a previously prepared solution (0.949 g of copper bromide.dimethyl sulfide in 10 ml
of ether, cooled to 0~C, 5.9 ml of methyllithium added) are added at 0~C to a solution of
45 ml of ether and 350 mg of the compound obtained above. After being stirred for 5.5 hours
at room temperature the mixture is cooled to 0~C. 2 ml of glacial acetic acid in 8 ml of ether
are added and 8 ml of ethanethiol. After stirring overnight at room temperature, 60 ml of
water are added and the mixture is extracted by shaking four times with methylene chloride.
The organic phase is predried with Na2SO4, filtered, concentrated using a rotary evaporator
and purified by chromatography. MS 266 (M+H)+
0.5 ml of DMSO is added at -78~C to a solution of 4.5 ml of methylene chloride + oxalyl
chloride. After 15 minutes the solution is added to a solution of 193 mg of the compound
obtained above in 4 ml of methylene chloride. After two hours at -78~C, 1.5 ml of triethyl-
amine are added. After 30 minutes' stirring at 0~C, 15 ml of water are added and the mixture
is extracted by shaking four times with diethyl ether. The organic phase is predried with
Na2SO4, concentrated using a rotary evaporator and purified by chromatography.
C H N
calculated: 63.87 6.51 5.32
found: 63.96 6.55 5.45
0.464 g of the compound obtained above and 5 ml of 4N HCI are heated together at 50~C
After 90 minutes the reaction mixture is cooled to room temperature and diluted with ice-
water. The crystalline product is obtained.
21 97785
C H N
calculated: 61.27 5.57 5.95
found: 61.2 5.5 6.2
B) Preparation of the terpyridine-lanthanide complexes
Example B1:
(a) 1 mmol of the respective terpyridine-bis-hydrazino compound from Example A1(c) is
taken up in 60 ml of absolute methanol under argon; the lanthanide(lll) acetate (1 mmol) is
added and the mixture is heated under reflux for 10 minutes. There are then added in
succession to that solution 1.2 mmol of the appropriate 2,6-dicarbonyl compound and
5 mmol of concentrated aqueous hydrochloric acid. The mixture is boiled for 2 days. After
cooling to room temperature, the product is filtered off and dried under a high vacuum.
In accordance with that procedure, compounds 1.1 to 1.28 of Table 1 are prepared.
(b) 1 mmol of the respective terpyridine-bis-hydrazino compound from Example A1 (c) is
taken up in 60 ml of absolute methanol under argon; the lanthanide(lll) chloride (1 mmol) is
added and the mixture is heated under reflux for 10 minutes. There are then added in
succession to that solution 1.2 mmol of the compound obtained in Example A2(a). The
mixture is boiled overnight. After cooling to room temperature, the solvent is removed and
the product is obtained by recrystallisation from dimethyl sulfoxide and toluene.
In accordance with that procedure, compounds 1.29 to 1.32 of Table 1 are prepared.
-28- 21 97785
Table 1:
11
~N N~
Ln ¦ 3 Cl
H C/N\N ~N~cH
R4/~ ~ R4
R5
Comp. No. Ln3+ R1 R4 R5 molar mass [M-CI-]
calc./found
1.1 La Ph-H H H 706/705
1.2 La Ph-OH H H 722.4/722.3
1.3 La Ph-OCH3 H H 736.4/735.6
1.4 La Ph-NH2 H H
1.5 La Ph-H CH3 H 734.4/734.4
1.6 La Ph-OH CH3 H 750.4/750.9
1.7 La Ph-OCH3 CH3 H 764.5/765.0
1.8 La Ph-NH2 CH3 H 749.5/749.5
1.9 Eu Ph-H H H 719.4/718.9
1.10 Eu Ph-OH H H 735.4/735.8
1.11 Eu Ph-OCH3 H H 749.5/749.3
1.12 Eu Ph-NH2 H H 734.5/734.5
1.13 Eu Ph-H CH3 H 747.5/747
1.14 Eu Ph-OH CH3 H 763.5/763.7
21 q7785
- 29 -
1.15 Eu Ph-OCH3 CH3 H 777.5/777.3
1.16 Eu Ph-NH2 CH3 H 762.5/762.5
1.17 Ce Ph-NH2 CH3 H 750.7/749.3
1.18 Pr Ph-NH2 CH3 H 751.4/750.9
1.19 Nd Ph-NH2 CH3 H 754.8/752.7
1.20 Gd Ph-NH2 CH3 H 767.8/766.3
1.21 Tb Ph-NH2 CH3 H 769.5/768.7
1.22 Dy Ph-NH2 CH3 H 773.1/773.2
1.23 Ho Ph-NH2 CH3 H 775.5/774.4
1.24 Er Ph-NH2 CH3 H 777.8/776.8
1.25 Tm Ph-NH2 CH3 H 779.5/778.8
1.26 Yb Ph-NH2 CH3 H 783.6/783.0
1.27 Lu Ph-NH2 CH3 H 785.5/784.7
1.28 Y Ph-NH2 CH3 H 699.4/698.1
1.29 La H HCH2CH2COOH
1.30 Eu H HCH2CH2COOH
1.31 La Ph-H HCH2CH2COOH *
1.32 Eu Ph-H HCH2CH2COOH**
Ph: 4-phenylene
* C H N Cl
calculated t+ 2DMSO):45.81 4.16 11.55 10.96
found: 45.3 4.3 11.8 10.6
** C H N Cl
calculated (+2DMSO+4H20):42.11 4.58 10.62 10.07
found: 42.2 4.6 10.6 9.5
Example B2: Preparation of isothiocyanate derivatives
A solution of the corresponding complex of Table 1 is added to a suspension of 4.4 mmol of
sodium hydrogen carbonate and 3.5 mmol of thiophosgene in 4 ml of chloroform. The
mixture is stirred vigorously at room temperature for 2.5 hours. The chloroform phase is
30- 21 97785
separated off and washed once with water. All the aqueous phases are combined and dried.
The products 2.1 to 2.15 of Table 2 so obtained are used further without further purification.
Table 2:
Comp. No. Ln3+R~ R4 R5 molarmass [M-CI]
calc. /found
2.1 Ce Ph-NCS CH3 H 792.7/792.7
2.2 Pr Ph-NCS CH3 H 793.5/791.2
2.3 Gd Ph-NCS CH3 H 809.9/807.4
2.4 Tb Ph-NCS CH3 H 811.5/811.7
2.5 Dy Ph-NCS CH3 H 815.1/815.9
2.6 Ho Ph-NCS CH3 H 817.5/816.2
2.7 Er Ph-NCS CH3 H 819.9/819.0
2.8 Tm Ph-NCS CH3 H 821.5/820.1
2.9 Yb Ph-NCS CH3 H 825.6/826.4
2.10 Lu Ph-NCS CH3 H 827.6/825.5
2.11 Y Ph-NCS CH3 H 741.5/740.2
2.12 La Ph-NCS CH3 H 791.5/792.1
2.13 Eu Ph-NCS CH3 H 804.6/804.7
2.14 La Ph-NCS H H
2.15 Eu Ph-NCS H H
C) Preparation of the amino-oligonucleotides
About 30 mg of the 'controled pore glass' (CPG) solid phase are weighed into a Standard
Applied Biosystem reaction vessel for a 1.5 llmol synthesis. The CPG solid phase (1) carries
the protected 3'-building block (in the Example, dC) of the amino-oligonucleotide to be
synthesised.
- 31 - 2 1 9 7 7 8 5
"~
N o C(CH3)3
~J i ~J (1)
I
OCH3 o
~0
HN~
CPG O
For the oligomerisation, phosphorus amidites (6), (7), (8) and (9) are used.
~ , r(cH3)~ (6)
OCH3 O
,P OCH2CH2CN
N(CH(CH3)2)2
-32- 2 1 97785
~ (7)
OCH3 o
P--OCH2CH2CN
N(CH(CH3)2)2
N ~ ~' ' 'NH
OCH3 O
P--OCH2CH2CN ~
C(CH3)3
N(CH(CH3)2)2
- 33 -
21 ~7785
~l ~
blN N (9)
OCH3 o
P--OCH2CH2CN
N(CH(CH3)2)2
For the later linkage of the metal complexes via the amino function, separate phosphorus
amidites (10), (11), (12) and (13) are used.
~C N H
~3 \ N O (10)
OCH3 o ~~ F F
/ P \ , F ~/
N(CH(CH3)2)2 OCH2CH2CN ~ ,~
HN
- 34 -
21 97785
O
OCH3 o \--~
OCH2CH2CN \ O
N(CH(CH3)2)2 NH~
F~ F
NH (12)
P--OCH2CH2CN
N(CH(CH3)2)2
- 35 -
/O NH ~ 2 1 9 7 785
P--OCH2CH2CN \ ¦ (13)
N(cH(cH3)2)2
H3CO ~ ~ ~
The synthesis cycles are carried out using the Syntheseautomat 394 by Applied Biosystem
with a modification (coupling time of the phosphorus amidites of the deoxy series (6), (7), (8)
and (9) is 2 minutes, that of the amidites (10) and (11) is 10 minutes, (12) is 5 minutes and
(13) is 40 minutes; (13) is used in 100-fold excess) in accordance with the standard protocol
of the Applied Biosystem company (User Manual Version 2.0 (1992) 1.0 ~mol cycle,Appendix 1-41).
Further commercially available reagents used are:
0.1 M phosphorus amidite
tetrazole/acetonitrile: 4 %, 96 %
tert-butylphenoxyacetic acid anhydride/pyridine/tetrahydrofuran: 10 %, 10 %, 80 %
N-methylimidazole/tetrahydrofuran: 16%, 84%
trichloroacetic acid/dimethylchloromethane: 2%, 98%
iodine/water/pyridine/tetrahydrofuran: 3%, 2%, 20%, 75%
The following amino-oligonucleotides are synthesised:
(821) 5'-GTA GAC TGG CGA GAT* CGG CAG TCG GCTAG-3',
wherein T* is
o
o O NH2
-36- 2~ q7785
in which T is thymine,
(823) 5'-GTA GAC TGG CGA GAT* CGG CAG TCG GCT AG-3',
wherein T is
~'0 NH2
o
I
in which T is thymine,
(940) 5'-GTA GAC TGG CGA GAT CGG CAG T*CG GCT AG-3',
wherein T* is
o
H N ~ ~ N H2
and
3~- GA TCG GCT GAC GGC TAG AGC--O ~ ~ o ~''~'~~~' ~ N H2
(691) / ~
O O
D) Preparation of the terpyridine-lanthanide-oligonucleotide conjugates
Example D1: Preparation of conjugates in which the oligonucleotide is bonded to the
terpyridine moiety of the lanthanide complex
(a) 0.2 mg of the respective amino-oligonucleotide is dissolved in 150 1ll of pyridine/water/-
triethylamine (90:15:1). After the addition of 1 mg of the appropriate isothiocyanato complex
37 2 1 ~7785
of Table 2, the mixture is left to stand at room temperature for 1 hour. The reaction mixture is
dialysed once against a 0.1 molar potassium chloride solution and three times against water.
Purification of the product by reversed-phase HPLC (gradient: from 0 % to 30 % acetonitrile
in 0.05M triethylammonium acetate in 90 minutes) on a Nucleosil~-C18-column or by ion
exchanger HPLC (gradient: 10 minutes 20 % 1M potassium chloride solution and 80 %
20mM potassium phosphate solution pH 6.0 containing 20 % acetonitrile; then in the course
of 60 minutes to 80 % potassium chloride solution) at 60~C on a PVDI.4000A column, 5 ~Lm,
yields the pure conjugates 3.1 to 3.13, 3.18 and 3.21 of Table 3.
Example D2: Preparation of conjugates in which the oligonucleotide is bonded to the
pyridine moiety of the lanthanide complex
3.3,umol of dicyclohexylcarbodiimide and 3.3 ~lmol of N-hydroxysuccinimide are added to a
solution of 3 ~mol of the corresponding carboxylic acid derivative 1.29 to 1.32 in 200 ~l of
dimethyl sulfoxide and the mixture is left to stand at room temperature for 16 hours. After the
addition of 100 ~mol of N,N-diisopropylethylamine, 0.2 mg of the corresponding amino-
oligonucleotide is added. After four days at room temperature, the reaction mixture is
dialysed twice against 50mM triethylammonium hydrogen carbonate and twice against
water. Purification by reversed-phase HPLC (see (a)) yields compounds 3.14 to 3.17, 3.19,
3.20 and 3.22 to 3.25 of Table 3.
-38- 21 97785
Table 3:
N
~N N~
Ln 3+
R4 ~N \ R4
R8
Comp. No. Ln R4 Rg R8 MM RT
3.1 La CH3Ph-691 H 7082/7093
3.2 Eu CH3Ph-691 H 7095/7090
3.3 Ce CH3Ph-691 H 27.4
3.4 Pr CH3Ph-691 H 42.5*
3.5 Gd CH3Ph-691 H 27.0
3.6 Tb CH3Ph-691 H 27.7
3.7 Dy CH3Ph-691 H 27.6
3.8 Ho CH3Ph-691 H 26.7
3.9 Er CH3Ph-691 H 27.2
3.10 Tm CH3Ph-691 H 27.5
3.11 Yb CH3Ph-691 H 28.8
3.12 Lu CH3Ph-691 H 27.9
3.13 Y CH3Ph-691 H 27 4
3.14 Eu H phenyl A-6917064/7086
3.15 La H phenyl A-6917051/7072
39 21 97785
3.16 Eu H H A-691
3.17 La H H A-691
3.18 Eu CH3 Ph-821 H 9843/9853
3.19 La H phenyl A-821 9800/9800
3.20 Eu H phenyl A-821 9813/9839
3.21 Eu CH3 Ph-823 H 9842/9861
3.22 La H phenyl A-823 35.6*
3.23 Eu H phenyl A-823 9811/9826
3.24 La H phenyl A-940 9757/9829
3.25 Eu H phenyl A-940 9770/9794
MM: molar mass calculated/found
RT: ion exchanger HPLC retention time (minutes)
Ph-691: -phenyl-N(H)C(S)-oligo 691
Ph-821: -phenyl-N(H)C(S)-oligo 821
Ph-823: -phenyl-N(H)C(S)-oligo 823
A-691: -CH2CH2C(O)-oligo 691
A-821: -CH2CH2C(O)-oligo 821
A-823: -CH2CH2C(O)-oligo 823
A-940: -CH2CH2C(O)-oligo 940
* reversed-phase HPLC retention time (minutes)
E) Preparation of the target RNA
The oligonucleotides designated "target RNA" are for preparative reasons for the most part
chimeric molecules that consist partly of deoxyribonucleic acid (DNA) building blocks
(labelled "d") and partly of ribonucleic acid (RNA) building blocks (labelled "r"). The nucleo-
tides of a target RNA that after hybridisation are unpaired with a corresponding antisense
oligonucleotide are located in the ribonucleic acid moiety thereof.
2 1 ~7785
Example E1:
Synthesis of 5'd(CTA GCC GAC TGC) r(CGA UGA CUC GCC AC), RNA E1.
About 30 mg of the 'controled pore glass' (CPG) solid phase are weighed into a Standard
Applied Biosystem reaction vessel for a 1.5 ~mol synthesis. The CPG solid phase (1) carries
the protected 3'-building block (in the Example, rC) of the RNA to be synthesised.
OCH,
\~lN ~ CH (1)
OCH3 Si
HN ~J~ CH3CH3
CPG O
For the oligomerisation, phosphorus amidites (2) to (9) are used.
~\ C(CH3)3 (2)
OCH3 ~ O-Si(CH3)2-C(cH3)3
P--OCH2CH2CN
N(CH(CH3)2)2
- 41 - 2 1 9 7 7 8 5
~3~0C ~,
N/~O (3
OCH3 ~ o-Si(CH3)2-C(CH3)3
P--OCH2CH2CN
N(CH(CH3)2)2
N ~ NH (4)
OCH3 O O-si(cH3)2-c(cH3)3
OCH2CH2CN C(CH3)3
N(CH(CH3)3)2
2 1 ~7785
O~/ C(CH,),
OCH3 O O-si(cH3)2-c(cH3)3
P--OCH2CH2CN
N(CH(CH3)2)2
\~ ~ r(CH3), (6)
OCH3 O
P--OCH2CH2CN
N(CH(CH3)2)2
-
43 21 97785
~
,J ~ ,~
OCH3 O
P--OCH2CH2CN
N(CH(CH3)2)2
N~ (8)
OCH3 O
OCH2CH2CN C(CH3)3
N(CH(CH3)3)2
-44- 21 97785
N ~ N (9)
OCH3 o
P--OCH2CH2CN
N(CH(CH3)2)2
The synthesis cycles are carried out using the Syntheseautomat 394 by Applied Biosystem
with a modification (coupling time of the phosphorus amidites of the ribo series is
10 minutes) in accordance with the standard protocol of the Applied Biosystem company
(User Manual Version 2.0 (1992) 1.0 ~mol cycle, Appendix 1-41).
Further commercially available reagents used are:
0.1 M phosphorus amidite
tetrazole/acetonitrile: 4 %, 96 %
tert-butylphenoxyacetic acid anhydride/pyridine/tetrahydrofuran: 10 %, 10 %, 80 %
N-methylimidazole/tetrahydrofuran: 16%, 84%
trichloroacetic acid/dimethylchloromethane: 2%, 98%
iodine/water/pyridine/tetrahydrofuran: 3%, 2%, 20%, 75%
The following substrate RNA is synthesised: title RNA E1
(b) Separation from the solid phase (CPG) and deprotection of the base: 800 ~LI of ammonia-
saturated ethanol are added to the solid phase (1.5 ~mol synthesis) and incubated at room
temperature overnight. The ammonia-saturated ethanol is prepared from one part ethanol
and three parts 33% ammonia. After the incubation, the ammonia-saturated ethanolic
solution is decanted off, the CPG is washed with ammoniacal ethanol and the combined
solutions are Iyophilised.
21 97785
(c) Deprotection of the tert-butyl-dimethylsilyl (TBDMS) protecting group: 800 ~l of 1 M
tetrabutylammonium fluoride/tetrahydrofuran (TBAF/THF) solution are added to theIyophilised sample. The sample is mixed intensively for 30 minutes. Incubation is carried out
for 24 hours at room temperature with the exclusion of light.
The RNA is mixed with 50mM triethylamine hydrogen carbonate (TAHC) solution pH 7.0
(1 + 1) and dialysed directly at 4~C (water has Nanopure~quality).
(d) Dialysis: Dialysis is carried out 3 times against 7.5mM TAHC solution pH 7Ø (The
solution is prepared using Nanopure~quality water, adjusted to pH 7.0 with CO2 and
precooled to 4~C.) The sample is Iyophilised and taken up in diethyl-pyrocarbonate-treated
[Sambrook, Fritsch, Maniatis, Molecular Cloning, A Laboratory Manual, Second Edition, Cold
Spring Harbor Laboratory Press (1989)] and autoclaved H20 (DEPC-H20). An aliquot is
used for determining the concentration at 260 nm. Further procedures with RNA are always
carried out under RNase-free and foreign-metal-ion-free conditions.
(e) 5'-Terminal labelling of the substrate RNA with 32[p] ~-ATP: For the enzymatic kinase
reaction, 100 pmol of RNA from the above synthesis protocol are incubated in a volume of
20 ~l at 37~C for 20 minutes.
The reaction solution contains 0.5 1ll of T4-polynucleotide kinase (Promega, 10 units/lll), 2 ~l
of kinase buffer (50mM Tris-HCI pH 7.5, 10mM MgCI2, 5mM 1,4-dithio-DL-threitol, 0.1mM
spermidine) and 0.5 1ll of 33[P] ~-ATP (Amersham, >1000 Ci/mmol, 10 ~lCi/~
138 ~l of Tris-HCI/EDTA (1 OmM/1 mM, pH 7.5), 2 ~l of glycogen (35 mg/ml) and 40 ~l of
NH4CH3COO (10M) are then added. After the addition of 600 ~l of ethanol, the sample is
cooled for 30 minutes at -20~C and then centrifuged for 20 minutes at 4~C. The pellet is
Iyophilised; 15 1ll of application buffer (0.025 % bromophenol blue, 0.025 % xylene cylanol in
a 1:1 mixture of 80 % formamide and 7M urea, 20mM citric acid, 1 mM EDTA) are added and
the mixture is denatured for 1 minute at a temperature of 95~C, immediately placed on ice
and for the purpose of gel-electrophoretic separation placed into a 1.0 cm x 1 mm bag. The
gel-electrophoretic separation is carried out for 2.5 hours at 55 Watt after a pre-run of
40 minutes at 55 Watt.
- 46 - 2 I q 7 7 8 5
(f) Purification and isolation of the kinased substrate RNA: For the gel-electrophoretic
separation of the kinase reaction, a 12 % polyacrylamide gel (1 mm x 30 cm x 40 cm) is
prepared. The polymerisation reaction is carried out in 170 ml. For that purpose, 51 ml of
acrylamide solution (40 % acrylamide/bisacrylamide 10:1),17 ml of TBE buffer (0.89M
tri(hydroxymethyl)aminomethane, 0.89M boric acid, 0.02M ethylenediaminetetraacetic acid)
and 71.4 9 of urea are mixed with the corresponding amount of H2O. The polymerisation is
started with 170111 Of ammonium peroxydisulfate solution (25 % w/v) and 170 ~LI of TEMED
(N,N,N',N'-tetramethylethylenediamine). The gel can be used after 1 hour.10-fold diluted
TBE buffer is used as elution buffer.
After the gel-electrophoretic separation, the kinased RNA is detected by means of an over-
laid X-ray film and excised from the gel. In an electro-elution apparatus (Schleicher and
Schuell) the RNA is eluted from the piece of gel by the application of 100 V (3.3 Wcm).
10-fold diluted TBE buffer is used as elution buffer.40111 Of NaCH3COO (3M pH 5.2) and
1 ml of ethanol are added to the isolated RNA in 360 ~l of eluate. The sample is cooled at
-20~C for 20 minutes and then centrifuged at 4~C for 20 minutes. The pellet is lyophilised
and taken up in 30 ~l of H2O. The solution is analysed in a scintillation counter in accordance
with the Czerenkow protocol and adjusted to 12 000 cpm/~l.
Example E2:
The procedure is as in Example E1 and target RNA "RNA E2" having the following sequence
is prepared:
5'd (CTA GCC GAC TG) r(CCG AUC UCA AG) d(CCA GTC TAC).
Examples E3 to E31:
Analogously to Example E1 there are prepared further target RNA molecules E3 to E30 and
a target DNA molecule E31 the structures of which are shown in Chapter F in Tables 4, 6
and 8.
F) Application Examples (RNA cleavage)
The cleavage behaviour of various antisense oligonucleotide conjugates according to the
invention with respect to various target RNAs is investigated. The cases shown in Figures 1,
47 21 ~7785
2 and 3 differ from one another according to the position of the bonding of the complex to the
antisense oligonucleotide sequence in question and according to the position of the
mismatch (unpaired nucleotides) that occurs in the hybrid of target RNA and antisense
oligonucleotide conjugate in relation to.
Example F1:
This Example relates to the cleavage of a target RNA by an antisense oligonucleotide
conjugate according to the invention, as illustrated in Fig. 3.
For the gel-electrophoretic separation and identification of the RNA products after the
cleavage reaction, a 12 % Long Ranger@~ gel (AT Biochem., modified polyacrylamide gel)
(0.4 mm x 30 cm x 40 cm) is prepared. The polymerisation reaction is carried out in 90 ml.
For that purpose, 21 ml of Long Ranger~ solution (50%),11 ml of TBE buffer (0.89M tri-
(hydroxymethyl)aminomethane, 0.89M boric acid, 0.02M ethylenediaminetetraacetic acid)
and 37 g of urea are mixed with the corresponding amount of H2O. The polymerisation is
started with 450 !ll of ammonium peroxydisulfate solution (10 % w/v) and 45 ~l of TEMED.
The gel can be used after 1 hour.16.66-fold diluted TBE buffer is used as elution buffer. The
separation takes place within a period of 75 minutes at 60 Watt.
After the gel-electrophoretic separation, the labelled cleavage products (RNA oligomers) are
detected or counted by means of an overlaid X-ray film or by means of a Phosphorimager~
The cleavage reaction is carried out in a volume of 10 ~I.
Example of a concentration series:
1 ~l of oligonucleotide conjugate (10 ~M), or corresponding dilutions (final concentration
1 ~M, 750nM, 500nM, 250nM,100nM, 50nM,10nM,1 nM and 0.5nM), 4 1ll of Tris-HCI buffer
(50mM pH 7.4 at 37~C) and the corresponding amount of H20 are pipetted into 1 ,ul of
substrate RNA (12 000 cpm). The mixture is heated at 85~C for 1 minute and then
incubated at 37~C for 16 hours. The reaction is stopped by the addition of 5 ~l of application
buffer (0.025 % bromophenol blue, 0.025 % xylene cylanol in a 1:1 mixture of 80 %
formamide with 7M urea, 20mM citric acid and 1 mM EDTA). For the gel-electrophoretic
21 97785
- 48 -
separation, 7.5 1ll of the sample are denatured for 1 minute at 95~C, immediately placed on
ice and placed into a gel bag.
Example of a time series:
1 ~1 of oligonucleotide conjugate (10~1M), 4 ~l of Tris-HCI buffer (50mM pH 7.4 at 37~C) and
the corresponding amount of H20 are pipetted into 1 ~ll of substrate RNA/DNA (12 000 cpm).
The mixture is heated at 85~C for 1 minute and then incubated at 37~C for 2h, 8h, 16h, 40h
and 64h. The reaction is stopped by the addition of 5 ~ll of application buffer (0.025 %
bromophenol blue, 0.025 % xylene cylanol in a 1 :1 mixture of 80 % formamide with 7M urea,
20mM citric acid and 1 mM EDTA). For the gel-electrophoretic separation, 7.5 1ll of the
sample are denatured for 1 minute at 95~C, immediately placed on ice and placed into a gel
bag.
The substrate RNA concentration is estimated as a 25-fold excess as follows:
With 100 pmol of crude product of RNA and a yield of 10 % in the gel purification, in
accordance with the protocol described the final concentrations are 0.04 IlM of substrate
RNA and 1 ~M of oligonucleotide conjugate in the reaction mixture.
If the terpyridine-lanthanide complex alone is used as comparison, 400 ilM of complex are
required in order to achieve approximately the same cleavage as 40 nM of oligonucleotide
conjugate. That is a 10 000-fold excess of complex with respect to oligonucleotide conjugate.
In the concentration series, a cleavage of 40 ~M of substrate RNA/DNA with 40 nM of oligo-
nucleotide conjugate is demonstrated after 16h at 37~C.
Cleavage products in the cleavage with compound 3.2 from Table 3 with RNA E1:
<20% of the starting material 5'd(CTA GCC GAC TGC) r(CGA UGA CUC GCC AC) are
uncleaved.
Main cleavage products (~ 80%):
5'd(CTA GCC GAC TGC) r(CGA UGcp)
5'd(CTA GCC GAC TGC) r(CGA Ucp)
5'd(CTA GCC GAC TGC) r(CGcp)
49 21 97785
Further cleavage products (~ 5%):
5'r(CUA GCC GAC UGC CGA UCU CGCcp)
5'r(CUA GCC GAC UGC CGA UCU Ccp)
(cp = 2',3'-cyclophosphate).
Example F2:
This Example relates to the cleavage of a target RNA by an antisense oligonucleotide
conjugate, as illustrated in Fig. 1.
The procedure is as in Example E1 using compound 3.20 from Table 3 and the target RNA
RNA E2.
Cleavage products:
<5% of the starting material 5'd(CTA GCC GAC TG) r(CCG AUC UCA AG) d(CCA GTC
TAC) are uncleaved.
Main cleavage products (~. 95%):
5'd(CTA GCC GAC TG) r(CCG AUC UCA Acp)
5'd(CTA GCC GAC TG) r(CCG AUC UCAcp)
5'd(CTA GCC GAC TG) r(CCG AUC UCcp)
Example F3:
This Example relates to further investigations into the cleavage of various target RNAs by
various antisense oligonucleotide conjugates, as illustrated in Fig. 1.
Table 4 below shows the structures of the target RNAs used. Nucleotides printed in bold
type are complementary to that nucleotide of the antisense oligonucleotide conjugate to
which the complex is bonded. Underlined nucleotides are unpaired in the hybrid of target
RNA and conjugate (mismatch). Double underlining in the case of target RNA E17 means in
each case that as a result of the sequence chosen, two adjacent nucleotides in the under-
lined region are unpaired but it is not possible to determine unambiguously which nucleo-
tides are involved. The position of the conjugate, especially of the complex, in relation to the
target RNAs is also shown in diagrammatic form.
-50- 21 97785
Table 4: Structures of various target RNAs
antisense
oligo- ~J
nucleOtide 3,
conjugate
(diagram.)
target
RNA
E3 5' d (CTA GCCGAC TGC) r(CGAUCU CUUG) d(C CAG TCT AC)
E4 5' d (CTA GCCGAC TGC) r(CGACUCU CG) d(C CAG TCT AC)
E5 5' d (CTA GCCGAC TGC) r(CGAUGCU CG) d(C CAG TCT AC)
E6 5' d (CTA GCCGAC TGC) r(CGAUCGU CG) d(C CAG TCT AC)
E7 5' d (CTA GCCGAC TGC) r(CGAUCU GCG) d(C CAG TCT AC)
E8 5' d (CTA GCCGAC TGC) r(CGAUCU CUG) d(C CAG TCT AC)
E9 5' d (CTA GCCGAC TG) r(CCGAUCU CAG) d(C CAG TCT AC)
E10 5' d (CTA GCCGAC TG) r(CCGAUCU _CG) d(C CAG TCT AC)
E11 5' d (CTA GCCGAC TG) r(CCGAUCU CG UC C) d(AG TCT AC)
E12 5' d (CTA GCCGAC TG) r(CCGAUCU CG CG C) d(AG TCT AC)
E13 5' d (CTA GCCGAC TG) r(CCGAUCU GACG) d(C CAG TCT AC)
E14 5' d (CTA GCCGAC TG) r(CCGAUCU gcG) d(C CAG TCT AC)
E15 5' d (CTA GCCGAC TG) r(CCGAUCU AGCG) d(C CAG TCT AC)
E16 5' d (CTA GCCGAC TG) r(CCGAUCU _CG) d(C CAG TCT AC)
E17 5' d (CTA GCCGAC TG) r(CCGAUCU CUCG) d(C CAG TCT AC)
E18 5' d (CTA GCCGAC TG) r(CCGAUCU CGAG) d(C CAG TCT AC)
E2 (s.a.) 5' d (CTA GCC GACTG) r(C CGA UCU C_G) d(C CAG TCT AC)
E19 5' r CUA GCC GACUGC CGA UCUAACGC CAG UCU AC
s.a. = see above
For the cleavage, the procedure is as in Example F1, but using the antisense oligonucleotide
conjugates (see also Table 3) and target RNAs (see Table 4) shown in Table 5 below.
Table 5 shows the main cleavage products of the target RNA in question. By way of
example, the entry "+5A" in the cleavage of target RNA E2 by conjugate 3.22 means that
cleavage takes place between nucleotides +5A and +6A.
-51 - 2 1 97785
Table 5: Main cleavage products of the cleavage of various target RNAs by various
antisense oligonucleotide conjugates
Conjugate3.18 3.21 3.19 3.20 3.22 3.23
target
RNA
(mismatch
indicated)
E3 +5U, +3U, * +3U +3U -1 G
+5U,+6U +6U +6U
E4 -1 G +2U * -1 G -1 G -1 G
+1C
E5 -1 G, +2G * -1 G -1 G -1 G
+2G OA
E6 -1 G, +3G * +4U * *
+3G +3G
E7 +5C +5C no +5C * *
+4G cleavage
E8 +5U +5U * -1G * *
+5U
E9 * +5A * +4C * *
+5_ +4C
E10 * +5C, +5C +5C * *
+4A +3U
E11 * no * +6U * *
+6U
E12 * no * +7G * *
+7G
E13 +4G, +4G * +3U, * +4G,
+4G, +5A +5A +3U +4G,+5A, +5A
~6C +6C,
-52- 2 1 ~7785
E14 -1 G +3U, +5G +5G +5G, +5G
+4G, +5G +4G -1 G
E15 +3U,+4 +3U +5G +5G +5G, +5G,
+4_, +5G A+5G, +3U +3U,
+6C -1 G
E16 +4A, +3U, +3U, +3U, +3U, -1G,
+4_, +5_ +5A +4A +4A, +4A, +4A +3U,
+5A +5A +4A+5A
E17 -1G +3U * -1G no -1G
in the region cleavage
of +1 U to
+6C
E18 +5G, +5G +5G +5G +5G -1G,
+5G, +6A +6A +4C,
+5G
E2 +5A, +5A +4C, see +5A +4C,
+5_, +6A +6A +5AExample +5A,
F2 +6A,-1 G
E19 +4A, * +3U, +3U, * -1G,
+4A, +5_ +3U +4A, +4A, +3U,
+5A +5A +4A+5A
*: not investigated
Example F4:
This Example relates to further investigations into the cleavage of various target RNAs by
various antisense oligonucleotide conjugates, as illustrated in Fig. 2.
Table 6 below shows the structures of the target RNAs used. The remarks made in
connection with Table 4 apply.
2 1 q7785
- 53 -
Table 6: Structures of various target RNAs
antisense
ollgo- ~
nucleotide 3, \
conJugate
(diagram.)
target RNA
E4 5' d (CTA GCCGAC TGC)r(CGACUCU CG) d(C CAG TCT AC)
E5 5' d (CTA GCCGAC TGC)r(CGAUGCU CG) d(C CAG TCT AC)
E6 5' d (CTA GCCGAC TGC)r(CGAUCGU CG) d(C CAG TCT AC)
E7 5' d (CTA GCCGAC TGC)r(CGAUCU GCG) d(C CAG TCT AC)
E8 5' d (CTA GCCGAC TGC)r(CGAUCU CUG) d(C CAG TCT AC)
E9 5' d (CTA GCCGAC TG) r(C CGA UCU CAG) d(C CAG TCT AC)
E10 5' d (CTA GCC GACTG) r(C CGA UCU_CG) d(C CAG TCT AC)
E20 5' d (CTA GCC GACTG) r(C CGA UCAUCG) d(C CAG TCT AC)
E12 5' d (CTA GCC GACTG) r(C CGA UCU CG CG C) d(AG TCT AC)
E13 5' d (CTA GCC GACTG) r(C CGA UCU_CG) d(C CAG TCT AC)
E14 5' d (CTA GCC GACTG) r(C CGA UCUGGCG) d(C CAG TCT AC)
E15 5'd (CTA GCC GACTG) r(C CGA UCUAGCG) d(C CAG TCT AC)
E16 5'd (CTA GCC GACTG) r(C CGA UCUAACG) d(C CAG TCT AC)
E18 5' d (CTA GCC GACTG) r(C CGA UCUCGAG) d(C CAG TCT AC)
E2 5' d (CTA GCC GACTG) r(C CGA UCUCAAG) d(C CAG TCT AC)
E19 5' r CUA GCC GAC UGC CGA UCU_CGC CAG UCU AC
For the cleavage, the procedure is as in Example F1, but using the antisense oligonucleotide
conjugates (see also Table 3) and target RNAs (see Table 6) shown in Table 7 below.
Table 7 shows the main cleavage products of the target RNA in question (see also the
remarks relating to Table 5).
54- 21 97785
Table 7: Main cleavage products of the cleavage of various target RNAs by an antisense
oligonucleotide conjugate
Conjugate 3.25
target
RNA
(mismatch
indicated)
E4 -1 OG
-8C
E5 -1 OG
-7G
E6 -6G
-6G
E7 -6U,
-5G -7C,
-8U
E8 -5C,
-4U -6U,
-7C
E9 -5C,
4_ -4A
E1 0 -6U
-5A
E20 -7C
-6A
E1 2 -2G,
-2G -3C
E 13 -5A,
-5A, -6G -6G
2 1 q7785
E1 4 -7U
-5G, -6G
E1 5 -5G,
-5G, -6A -6A
E1 6 -5A,
-5A, -6A -6A
E1 8 -3G,
-4_,-5G 4A
E2 ~A,
-4A, -5A -5A
E1 9 -6A,
-5_, -6_ -7U
Example F5:
This Example relates to further investigations into the cleavage of various target RNAs by
various antisense oligonucleotide conjugates, as illustrated in Fig. 3.
Table 8 below shows the structures of the target RNAs used. A control DNA (E31 ) is also
used. The remarks made in connection with Table 4 apply.
Table 8: Structures of various target RNAs
antisense
oligo- ~J
nucleotide 3,
conjugate
(diagram.)
target
RNA
E21 5'd (CTA GCC GAC TGC) r(CGAC UCU CGC CAC)
E22 5'd (CTA GCC GAC TGC) r(CGA UGCU CGC CAC)
E23 5'd (CTA GCC GAC TGC) r(CGAG UCU CGC CAC)
E24 5'd (CTA GCC GAC TGC) r(CGACC UCU CGC CAC)
21 ~7785
- 56 -
E1 (s.a.) 5' d(CTA GCCGAC TGC) r(CGAUGACU CGC CAC)
E14 5' d(CTA GCCGACTG)r(C CGA UCUGG CG) d(CCA GTC TAC)
E2 5' d(CTA GCCGACTG)r(C CGA UCUC_ G)d(CCAG TCT AC)
E25 5' d(CTA GCCGAC TGC CGA)r(U ACU CGC CAC)
E26 5' d(CTA GCCGAC TGC) r(CGAUGGCU CGC CAC)
E27 5' d(CTA GCCGAC TGC) r(CGAUAGCU CGC CAC)
E28 5' d(CTA GCCGAC TGC) r(CGAUCUCU _GC CAC)
E29 5' d(CTA GCCGAC TGC) r(CGAU_CU CGC CAC)
E30 5' rCUA GCCGAC UGC CGA UGACU CGC CAC UCU AC
control
DNA:
E31 5' d(CTA GCCGAC TGC CGATGACT CGC CAC)
s.a. = see above
For the cleavage, the procedure is as in Example F1, but using the antisense oligonucleotide
conjugates (see also Table 3) and target RNAs (see Table 8) shown in Table 9 below.
Table 9 shows the main cleavage products of the target RNA in question (see also the
remarks relating to Table 5).
Table 9: Main cleavage products of the cleavage of various target RNAs by various
antisense oligonucleotide conjugates
Conjugate 3.2 3.14
target RNA
(mismatch
indicated)
E21 -1 C, -1 C
-5C
E22 -5U, *
-4G
E23 -7G, +2C +2C
-5G
21 q7785
- 57 -
E24 -1 C -2U
-5C, -6C
E1 see -5G
-4A, -5G Example F1
E14 * OG,-1 C,
-2G,-3G -2G
E2 * -1 A,
-1 _,-2A -2A
E25 -5G -5G,
-4_, -5G -6U
E26 -6U +2C,
-4_, -5G -5G
E27 -6U -6U
-4G, -5A
E28 -1 C -2U
in the region
of-6U to
-1C
E29 -5U -5U
-4A
E30 -5G,-6U, *
-4A, -5G -7A
E31 no no
-4A,-5G cleavage cleavage
*: not investigated