Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 96/10552 PCT/EP95/03870
- 1 -
PROCESS FOR PREPARING 1,3-ALKANEDIOLS
AND 3-HYDROXYALDEHYDES
This invention relates to a process for preparing
1,3-alkanediols and 3-hydroxyaldehydes by hydroformyla-
ting an oxirane (1,2-epoxide). In particular, the inven-
tion relates to a process for preparing 1,3-propanediol
by hydroformylating ethylene oxide in the presence of a
Group VIII-based hydroformylation catalyst and hydrogena-
ting the hydroformylation product.
The preparation of 1,3-alkanediols like 1,3-propane-
diol (PDO) is disclosed in US-A-3 687 981. The process
comprises the hydroformylation of an oxirane such as
ethylene oxide, in a concentration of more than 15 weight
percent based on the total liquid reaction mixture, in
the presence of a metallic carbonyl catalyst containing a
metal from Group VIII, followed by hydrogenation of the
hydroformylation product. The hydroformylation product of
that process is the cyclic hemiacetal dimer of 3-hydroxy-
propanal (HPA), i.e., 2-(2-hydroxyethyl)-4-hydroxy-1,3-
dioxane. PDO is of particular interest as intermediate in
the production of polyesters for fibres and films.
Despite the publication of this patent in 1972,
fibre-grade polyesters based on PDO are as yet not
commercially available. Separation of the catalyst from
the cyclic hemiacetal produced in US-A-3 687 981, by
phase separation, is complicated and inadequate. As a
result, the cost for preparing polymer-grade PDO is too
high.
In US-A-3 456 017 and US-A-3 463 819 1,3 alkanediols
are prepared directly, with only minor amounts of the
intermediate hydroformylation product, in the presence of
certain phosphine-modified cobalt carbonyl catalysts.
WO 96/10552 ~ ~ ~ '~'. PCT/EP95/03870
- 2 -
Commercialisation of the process of these US patents is
ruled out, due to the excessive amounts of catalyst
employed therein. Also in WO 94/18149 phosphine-modified
cobalt carbonyl catalysts are used. They are used in a
much smaller amount than in the US patents, producing
primarily the 3-hydroxyaldehyde. Although activity of the
phosphine-modified cobalt-carbonyl catalyst described in
the international application is high, improvement re-
mains desirable, particularly in view of the undesirable
co-production of acetaldehyde. Besides, the cost of the
phosphines, which are notoriously difficult to retain
when recycling the catalyst, adversely affect the economy
of that process.
It would be desirable to prepare 3-hydroxyaldehydes
and 1,3-alkanediols selectively and cheaply. It is
therefore an object of the invention to provide an
economical process for the preparation of 3-hydroxy-
aldehydes and 1,3-alkanediols in the presence of a
hydroformylation catalyst which process allows for the
convenient recycle of the catalyst.
Accordingly, the invention provides a process for
preparing 1,3-alkanediols and 3-hydroxyaldehydes by
hydroformylating an oxirane with carbon monoxide and
hydrogen in the presence of one or more Group VIII metal-
based hydroformylation catalysts, which may contain up to
50 moles based on the metal of phosphine-modified
catalysts, and in the presence of an organic solvent,
wherein the concentration of the oxirane at the start of
the reaction is less than 15 percent by weight (wt~)
based on the weight of the total liquid reaction mixture.
The process is preferably carried out at a temperature
below 100 °C.
As a result, an intermediate product mixture is
obtained, essentially composed of starting components and
the 3-hydroxyaldehyde, the latter in an amount that is
WO 96/10552 ~ ~ 2 ~ PCT/EP95l03870
- 3 -
less than 15 wt$ based on the total liquid reaction
mixture. At this concentration, the selectivity towards
the 3-hydroxyaldehyde is high, whereas the catalyst may
be conveniently recycled.
The oxirane comprises an organic compound, two carbon
atoms of which are connected by an oxy linkage as well as
by a carbon-carbon single bond. In general terms, the
oxiranes comprise hydrocarbyl-epoxides, having at least
2, preferably up to 30, more preferably up to 20, most
preferably up to 10 carbon atoms. The hydrocarbyl group
may be aryl, alkyl, alkenyl, aralkyl, cycloalkyl, or even
alkylene; straight chain or branched chain. Suitable
examples of oxiranes include 1,2-epoxy(cyclo)alkanes,
such as ethylene oxide, propylene oxide, 1,2-epoxyoctane,
1,2-epoxycyclohexane, 1,2-epoxy-2,4,4-trimethylhexane and
the like, and 1,2-epoxyalkenes such.as 1,2-epoxy-4-
pentene and the like. Ethylene oxide and propylene oxide
are preferred. In view of the demand for PDO, ethylene
oxide (E0) is the oxirane most preferably used in the
process of the invention.
The hydroformylation reaction is carried out in a
liquid solvent inert to the reactants and products, i.e.,
that is not consumed during the reaction. Upon completion
of the reaction, the liquid solvent facilitates the
separation of the hydroformylation product. The separa-
tion may be carried out by allowing the product to form a
separate layer, as is disclosed in US-A-3 687 981.
However, as discussed below, it is preferred to carried
out the. separation by extraction with an aqueous liquid.
In general, ideal solvents for the hydroformylation
process will (a) exhibit low to moderate polarity such
that the 3-hydroxyaldehyde will be dissolved to a concen-
tration of at least about 5 wt~ under hydroformylation
conditions, while significant solvent will remain as a
separate phase upon extraction with the aqueous liquid,
WO 96/10552 PCTI~P95/03870
- 4 -
(b) dissolve carbon monoxide, and (c) be essentially non-
water-miscible. By "essentially non-water-miscible" is
meant that the solvent has a solubility in water at 25 °C
of less than 25 wt~ so as to form a separate hydrocarbon-
s rich phase upon extraction of the 3-hydroxyaldehyde from
the hydroformylation reaction mixture. Preferably, this
solubility is less than 10 wto, most preferably less than
wt~. The solubility of carbon monoxide in the selected
solvent will generally be greater than 0.15 v/v (1 atm,
25 °C), preferably greater than 0.25 v/v, expressed in
terms of Ostwald coefficients.
The preferred class of solvents are alcohols and
ethers which can be described according to the formula
(1)
R1_O_R2 (1)
in which R1 is selected from hydrogen, a linear,
branched, cyclic or aromatic C1-20 hYdrocarbyl or mono-
or polyalkylene oxide and R2 ~s selected from a linear,
branched, cyclic or aromatic C1-20 hYdrocarbyl, alkoxy or
mono- or polyalkylene oxide, or R1, R2 and O together
form a cyclic ether. The most preferred hydroformylation
solvents can be described by the formula (2)
R3
R4-C-O-R1 (2)
R5
in which R1 is selected from hydrogen or a C1_g hydro-
carbyl and R3, R4 and R5 are independently selected from
a C1_g hydrocarbyl, alkoxy or mono- or polyalkylene
oxide. Such ethers include, for example, tetrahydrofuran,
methyl-t-butyl ether, ethyl-t-butyl ether, ethoxyethyl
ether, phenylisobutyl ether, diethyl ether, diphenyl
ether, and diisopropyl ether. Blends of solvents such as
t-butylalcohol/hexane, tetrahydrofuran/toluene and
tetrahydrofuran/heptane can also be used to achieve the
WO 96/10552 2 PCT/EP95/03870
- 5 -
desired solvent properties. The currently preferred
solvent, because of the high yields of HPA which can be
achieved under moderate reaction conditions, is methyl-t-
butyl ether.
i
The hydroformylation reaction is conducted in the
presence of any metallic carbonyl hydroformylation
catalyst, as long as less than 50 moleo thereof
preferably less than 10 moles thereof are phosphine-
modified. These catalysts are transition metals, particu-
larly those metals of Group VIII of the Periodic Table,
e.g., cobalt, iron, nickel, osmium and complexes de-
scrihed, for example, in US-A-3 161 672. Best results,
however, are obtained when a cobalt-based catalyst is
used, unmodified cobalt carbonyl compounds being
preferred.
The cobalt-based catalyst can be supplied to the
hydroformylation reactor as a cobalt carbonyl such as
dicobaltoctacarbonyl or cobalt hydridocarbonyl. It may
also be supplied in essentially any other form including
metal, supported metal, Raney-cobalt, hydroxide, oxide,
carbonate, sulfate, acetylacetonate, salt of a fatty
acid, or aqueous cobalt salt solution. If not supplied as
cobalt carbonyl, operating conditions should be adjusted
such that cobalt carbonyls are formed, for instance via
reaction with H2 and CO as described in J. Falbe, "Carbon
Monoxide in Organic Synthesis," Springer-Verlag, NY
(1970). Typically, these conditions will include a tem-
perature of at least 50 °C and a carbon monoxide partial
pressure of at least 0.8 MPa (100 psig). For more rapid
reaction, temperatures of 120 to 200 °C should be
employed, at CO pressures of at least 3.5 MPa (500 psig).
Addition of high surface area activated carbons or
zeolites, especially those containing or supporting
platinum or palladium metal, is known to accelerate the
formation of cobalt carbonyls.
WO 96/10552 ~ ~ ~ PCT/EP95103870
6
The catalyst is preferably maintained under a stabi-
lising atmosphere of carbon monoxide, which also provides
protection against exposure to oxygen. The most economi-
cal and preferred catalyst activation and reactivation
(of recycled catalyst) method involves converting the
cobalt salt (or derivative) under H2/CO in the presence
of the catalyst promoter employed for hydroformylation.
The conversion of Co2+ to the desired cobalt carbonyl is
carried out at a temperature within the range of 75 to
200 °C, preferably 100 to 140 °C and a pressure within
the range of 7.0 to 34.6 MPa (1000 to 5000 psig) for a
time preferably less than about 3 hours. The preforming
step can be carried out in-a pressurised preforming
reactor or in-situ in the hydroformylation reactor.
The amount of Group VIII metal present in the
reaction mixture will vary depending upon the other
reaction conditions, but will generally fall within the
range of 0.01 wtg to 1 wt~, preferably 0.05 to 0.3 wt~,
based on the weight of the reaction mixture.
The hydroformylation reaction mixture will preferably
include a catalyst promoter to accelerate the reaction
rate. The promoter will generally be present in an amount
within the range of 0.01 to 0.6 moles per mole of Group
VIII metal.
Suitable promoters include sources of mono- and
multivalent metal cations of weak bases such as alkali,
alkaline earth and rare earth metal salts of carboxylic
acids. Suitable metal salts include sodium, potassium and
caesium acetates, propionates and octoates~ calcium
carbonate and lanthanum acetate. The currently preferred
metal salt is sodium acetate. ,
Also suitable are lipophilic promoters such as lipo-
philic mono- or dihydroxyarenes, lipophilic tertiary
amines or arsines, or lipophilic phosphine oxides
respectively arsine oxides, which accelerate the rate of
WO 96/10552 ~ ~ ~ 2 ~ PCT/EP95103870
hydroformylation without imparting hydrophilicity (water
solubility) to the active catalyst. As used herein,
"lipophilic" means that the promoter tends to remain in
the organic phase after extraction of HPA with water.
Suitable lipophilic mono- or dihydroxyarenes include
those represented by formulae (3) and (4):
C6R50H (3) C6R4(OH)2 (4)
in which each R group is independently selected from
hydrogen, a halide, a linear, branched, cyclic or
aromatic C1_25 hydrocarbyl, alkoxy or mono- or poly-
alkylene oxide, or in which two or more R groups together
form a ring structure. Examples include phenol, nonyl-
phenol, methylphenol, butylphenol, isopropylphenol, 2,2-
bis(4-hydroxyphenyl)propane, naphthol, hydroquinone,
catechol, dihydroxynaphthalenes and dihydroxyanthracenes.
Excellent results have been achieved with phenol and
nonylphenol, which are hence preferred.
Suitable lipophilic amines and arsines include those
represented by formulae (5) and (6):
NR'3 (5) AsR'3 (6)
in which each R' group is independently selected from a
linear, branched, cyclic and aromatic C1-25 hYdrocarbyl,
alkoxy or mono- or polyalkylene oxide, or in which two or
more of the R' groups together form a ring structure.
Such arsines include triphenylarsine and triethylarsine.
Examples in which two or more of the R' groups together
form a ring structure include pyridine and substituted
pyridines described by formula (7):
A1
~C C
//
A~-C AS
l~)
WO 96/10552 ~ ~ ~ ~ ~ ~ PCT/EP95/03870
- g _
in which each of the A groups is independently selected
from hydrogen or a linear, branched, cyclic or aromatic
C1-25 hYdrocarbyl, two or more of which may form a ring
structure. Substituted pyridines wherein A1 and A5 are
both bulky groups, such as t-butyl, are not preferred.
The lipophilic tertiary amine is preferably a non-
chelating amine of conjugate acid having a pKa in the
range of 5 to 11. Such lipophilic tertiary amines include
dimethyldodecylamine, pyridine, 4-(1-butylpentyl)pyri-
dine, quinoline, isoquinoline, lipdine and quinaldine.
The preferred amine is nonylpyridine.
Suitable phosphine oxides and arsine oxides include
those represented by formulae (8) and (9):
O=PR"3 (8) 0=AsR"3 (9)
in which each R" group is independently selected from a
halide, a linear, branched, cyclic or aromatic C1-25
hydrocarbyl, alkoxy or mono- or polyalkylene oxide, or in
which two or more R groups together form a ring struc-
ture. Such phosphine oxides include triphenylphosphine
oxide, tributylphosphine oxide, dimethylphenylphosphine
oxide and triethylphosphine oxide. The currently
preferred phosphine oxide is triphenylphosphine oxide.
It is generally preferred to regulate the concentra-
tion of water in the hydroformylation reaction mixture,
as excessive amounts of water reduce the selectivity
towards the 1,3-alkanediols and 3-hydroxyaldehydes below
acceptable levels and may induce formation of a second
liquid phase. At low concentrations, water can assist in
promoting the formation of the desired cobalt carbonyl
catalyst species. Acceptable water levels will depend
upon the solvent used, with more polar solvents generally
more tolerant of higher water concentrations. For
example, optimum water levels for hydroformylation in
methyl-t-butyl ether solvent are believed to be within
the range of 1 to 2.5 wt~.
WO 96/10552 PCT/EP95/03870
g _
The hydrogen and carbon monoxide will generally be
introduced into the reaction vessel in a molar ratio
within the range of 1:2 to 8:1, preferably 1:1.5 to 5:1.
The reaction is carried out under conditions effec-
tive to produce a hydroformylation reaction mixture
comprising a major portion of the 3-hydroxyaldehyde and a
minor portion of by-product, if any. Moreover, the level
of 3-hydroxyaldehyde in the reaction mixture is prefer-
ably maintained at less than 15 wto, preferably 5 to 10
wt~. (To provide for solvents having different densities,
the concentration of 3-hydroxyaldehyde in the reaction
mixture can be expressed in molarity, i.e., less than
1.5M, preferably within the range of 0.5 to 1M.).
Suitable, the reaction is carried out at a oxirane
concentration that is less than 12 wt~.
Generally, the hydroformylation reaction is carried
out at elevated temperature less than 100 °C, preferably
60 to 90 °C, most preferably 75 to 85 °C, and at a
pressure within the range of 3.5 to 34.6 MPa (500 to 5000
psig), preferably (for process economics) 7.0 to 24.2 MPa
(1000 to 3500 psig), with higher pressures generally
imparting greater selectivity. The concentration of 3-
hydroxyaldehyde in the intermediate product mixture can
be controlled by regulation of process conditions such as
oxirane concentration, catalyst concentration, reaction
temperature and residence time. In general, relatively
low reaction temperatures (below 100 °C) and relatively
short residence times within the range of 20 minutes to 1
hour are preferred.
In the practice of the invention method, it is possi-
ble to achieve 3-hydroxyaldehyde yields (based on the
oxirane conversion) of greater than 80$. For instance,
with the hydroformylation ofEO in the presence of a
cobalt carbonyl, formation of more than 7 wt$ HPA in the
dilute hydroformylation product mixture, at rates greater
WO 96/10552 ~ ~ ~ ~ PCTIEP95/03870 ~;
- 10 -
than 30 h-1 are achievable. (Catalytic rates are referred
to herein in terms of "turnover frequency" or "TOF" and 1
are expressed in units of moles per mole of cobalt per
hour, or h-1.) Reported rates are based on the observa-
tion that, before a majority of the oxirane, here EO, is
converted, the reaction is essentially zero-order in EO
concentration and proportional to cobalt concentration.
As mentioned above, separation of the hydroformyla-
tion product mixture is carried out economically most
attractively by extraction with an aqueous liquid.
Preferably the aqueous liquid is water. The amount of
water added to the hydroformylation reaction product
mixture will -generally be such as to provide a weight
ratio of wate-r: mixture within the range of 1:1 to 1:20,
preferably 1:5 to 1:15. The addition of water at this
stage of the reaction may have the additional advantage
of suppressing formation of undesirable heavy ends.
Extraction with a relatively small amount of water
provides an aqueous phase which is greater than 20 wt~
3-hydroxyaldehyde, preferably greater than 35 wt~ 3
hydroxyaldehyde, permitting economical hydrogenation of
the 3-hydroxyaldehyde to the 1,3-alkanediol. The water
extraction is preferably carried out at a temperature
within the range of 25 to 55 °C, with higher temperatures
avoided to minimise condensation products (heavy ends)
and catalyst disproportionation to inactive, water-
soluble Group VIII metal (e.g., cobalt) compounds. In
order to maximise catalyst recovery discussed supra, it
is preferred to perform the water extraction under 0.5 to
1.5 MPa (50 to 200 psig) of carbon monoxide at 25 to
55 °C.
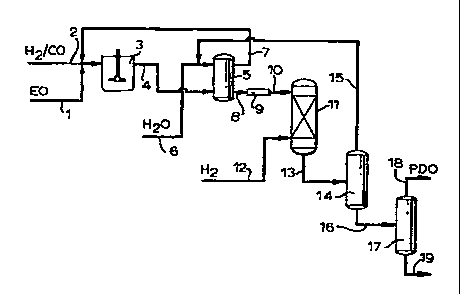
The invention process can be conveniently described
by reference to Figure 1. By way of example, the hydro-
formylation of EO as oxirane will be described. Separate
or combined streams of EO (1), carbon monoxide and
WO 96/10552 ~ ~ ~ PCT/EP95/03870
-. 11 -
hydrogen (2) are charged to hydroformylation vessel (3),
which can be a pressure reaction vessel such as a bubble
column or agitated tank, operated batch-wise or in a
continuous manner. The feed streams are contacted in the
presence of an unmodified cobalt-based catalyst, i.e., a
cobalt carbonyl compound which has not been prereacted
with a phosphine ligand.
Following the hydroformylation reaction, the hydro-
formylation reaction product mixture (4) containing HPA,
the reaction solvent, PDO, the cobalt catalyst and a
minor amount of reaction by-products, is passed to ex-
traction vessel (5), to which an aqueous liquid, gener-
ally water and optionally a miscible solvent, are added
via (6) for extraction and concentration of the HPA for
the subsequent hydrogenation step. Liquid extraction can
be effected by any suitable means, such as mixer-setters,
packed or trayed extraction columns or rotating disk
contactors. Extraction can if desired be carried out in
multiple stages. The water-containing hydroformylation
reaction product mixture can be passed to a settling tank
(not shown) for resolutioninto aqueous am organic
phases.
The organic phase containing the reaction solvent and
the major portion of the cobalt catalyst can be recycled
from the extraction vessel to the hydroformylation reac-
tion via (7). Aqueous extract (8) is optionally passed
through one or more acid ion exchange resin beds (9) for
removal of any cobalt catalyst present, and the de-
cobalted aqueous product mixture (10) is passed to
hydrogenation vessel (11) and reacted with hydrogen (12)
yn the presence of a hydrogenation catalyst to produce a
hydrogenation product mixture (13) containing PDO. The
hydrogenation step may also revert some heavy ends to
PDO. The solvent and extractant water (15) can be
recovered by distillation in column (14) and recycled to
WO 96/10552 ~ ~ PCT/EP95/03870
- 12 -
the water extraction process via a further distillation
(not shown) for separation and purge of light ends. PDO-
containing stream (16) can be passed to one or more
distillation columns (17) for recovery of PDO (18) from
heavy ends (19) .
The process of the invention permits-the selective
and economical synthesis of PDO at moderate temperatures
and pressures without the use of a phosphine ligand for
the hydroformylation catalyst. The process involves
preparation of a reaction product mixture dilute in HPA,
then concentration of this HPA by water extraction for
subsequent hydrogenation of HPA to PDO.
Comparative example 1
This experiment illustrates the hydroformylation of
ethylene oxide (E0) catalysed by a phosphine-modified
cobalt catalyst derived from dicobaltoctacarbonyl.
A 300 ml stirred reactor was chargedwith 0.878
dicobaltoctacarbonyl, 1.338 bis(1,2-diphenylphosphino)-
ethane, 0.1258 sodium acetate trihydrate, 0.518 2-ethyl-
hexanoic acid, and 147.28 "NEODOL" 23 (trademark), a
blend of C12 and C13 alcohols. Reactor contents were
heated to 165 °C under 1:1 H2:C0 synthesis gas for 1
hour, with agitation at 1000 rpm, to preform the active
catalyst. The reactor temperature was decreased to 90 °C,
and 208 EO (i.e., 11.8 wt~) were injected via a "blow-
case" vessel charged with 10.4 MPa (1500 psig) synthesis
gas. The reactor pressure was topped to 10.4 MPa (1500
psig). Reactor pressure decreased over time as a result
of hydroformylation of EO substrate. The reactor was
refilled to 10.4 MPa (1500 psig) with 1:1 H2:C0 upon a
decrease in pressure to 9.1 MPa (1300 psig). In this
manner, the uptake of synthesis gas could be monitored as
a function of time, to follow the course of the reaction. ,
Samples of the reaction mixture were periodically
withdrawn into chilled n-propanol containing an internal
WO 96/10552 2 ~ 0 13 2 ~ PCT/EP95/03870
- 13 -
standard (toluene or ethyl acetate) for analysis by
capillary gas chromatography (with flame ionisation
detector). The analysis indicated an 87g conversion of EO
in 3 hours, to give 10 weight percent 3-hydroxypropanal
(HPA) intermediate, with some minor hydrogenation to 1,3-
propanediol (PDO). This result corresponds to an
effective reaction rate of 15 moles of HPA formed per
mole of Co catalyst per hour (TOF). Apparent selectivity
to acetaldehyde, expressed as the molar ratio of acetal-
dehyde to the sum of HPA and acetaldehyde, was 27~.
Example 1
A 300 ml stirred batch reactor was charged under
nitrogen with 0.878 dicobaltoctacarbonyl, 1.5g toluene
(internal marker) 1.5g undecanol (second marker), and
1478 methyl-t-butyl ether (MTBE). The nitrogen atmosphere
was flushed with H2 before the reactor was filled to 8.3
MPa (1200 psi) with 1:l CO/H2. Reactor contents were
heated to 80 °C for 45 minutes, before injection of 20g
E0, with simultaneous increase in reactor pressure to
10.3 MPa (1500 psi) at an H2/CO ratio of 2.3. The
concentration of EO at the start of the reaction was 11.7
wt~. Reactor contents were sampled and analysed. Forma-
tion of 2.7 wt~ HPA was observed after 30 minutes, for a
rate of 20.2 h-1.
Example 2
The conditions of Example 1 were repeated with
addition of 0.5g of dimethyldodecylamine and injection of
12g EO (i.e., 7.4 wt~). Sampling after 45 minutes of
reaction indicated formation of 5.7 wt~ HPA, for a rate
of 31 h-1. This corresponds to a 1.5-fold rate increase
over that observed in the absence of promoter. The
reaction was continued until formation of 10 wt~ HPA at
virtual complete conversion of ethylene oxide.
Following reaction, the mixture was cooled to 25 °C
and extracted with 30g deionised water under 2.1 MPa (300
WO 96/10552 ~ PCT/EP95/03870
- 14 -
psi) C0. The mixture was then transferred to a separation
vessel under 0.7 MPa (100 psi CO). Separation yielded
30.758 of a lower aqueous layer containing 24.0 wt~ HPA,
and an upper organic solvent layer containing 1.0 wt~
HPA. Colorimetric analysis of upper and lower layers
indicated 94~ of the cobalt catalyst to reside in the
upper solvent layer, demonstrating separation of a major-
ity of cobalt catalyst from a majority of HPA product.
Comparative example 2
This experiment illustrates separation of HPA from
the cobalt hydroformylation catalyst by distillation.
113.458 of EO hydroformylation reaction product contain-
ing 14.328 of HPA intermediate were diluted with 50.18 of
tetraethylene glycol dimethylether. The mixture was
distilled via a short-path batch still at 10 mm Hg under
a slow nitrogen purge at a distillate bottoms temperature
ranging from 66 to 108 °C. Distillate fractions were
collected and were found by gas chromatographic analysis
to contain 6.328 HPA. No HPA was evident in the remaining
bottoms sample, which exhibited a significant increase in
components heavier than HPA. Total HPA recovery was thus
44o with the remainder degraded to heavy ends.
This experiment demonstrates the problems inherent in
thermal separation of highly-reactive HPA intermediate
from the reaction mixture. More than half the HPA inter-
mediate was degraded during the separation.
Example 3 _. .
This invention experiment demonstrates separation and
concentration of HPA by water extraction. 1507.68 of EO
hydroformylation reaction product (MTBE solvent with
sodium acetate promoter at 0.2 Na/Co) containing 6.0 wt~ w
HPA intermediate were water extracted at 25 °C under 0.8
MPa (100 psig) nitrogen in a stirred reactor with 2988 of
deionised water, giving 400.58 of a lower layer contain-
ing 20.8 wt~ HPA intermediate (3.5-fold concentration).
WO 96/10552 ~ 2 0 1 3 2 3 PCT/EP95/03870
- 15 -
Overall HPA material balance from gas chromatographic
analysis of feed, upper phase and lower phase indicated
complete recovery of HPA within g.c. experimental error.
The upper layer following water extraction contained
0.14 wt~ cobalt, or 65$ of the initially-charged
catalyst.
This experiment demonstrates the catalyst and product
recovery advantages of the invention PDO preparation
method. Separation of HPA from the reaction mixture was
very efficient and selective. The use of water and low
temperatures avoided the degradation of HPA shown in
Comparative example 2. The method also allows concentra-
tion of HPA for more efficient hydrogenation and final
recovery. In addition, a significant fraction (650) of
the cobalt catalyst was readily separated from aqueous
HPA product, making efficient recycle of catalyst with
reaction solvent possible.
Example 4
A 300-ml stirred batch reactor was charged under
nitrogen with 0.878 dicobaltoctacarbonyl, 1.5g toluene
(internal marker), 2g deionised water and 1468 MTBE. The
nitrogen atmosphere was flushed with H2~ and the reactor
was filled to 4.2 MPa (600 psig) H2 and then to 8.4 MPa
(1200 psig) with 1:1 CO/H2. Reactor contents were heated
to 80 °C for one hour, and lOg of EO (6.2 wt~) was then
injected, with simultaneous increase in reactor pressure
to 10.4 MPa (1500 psig) via addition of 1:1 CO/H2. Reac-
tor contents were sampled and analysed at approximately
40~ and nearly 100 conversion of EO, which occurred
within two hours. At approximately 40~ conversion, 3.3
wt~ HPA had been formed at a rate of 18 h-1.
Example 5
Example 4 was repeated in the absence of added water
and with addition of 0.148 of sodium acetate trihydrate
as promoter, added at a ratio Na/Co of 0.2. The concen-
WO 96110552 ~ ~ ~ ~ ~ ~ PCT/EP95/03870
16
tration of EO at the start of the reaction was 6.3 wt~.
HPA was formed at a rate of 41 h-1. After cooling and
addition of 30g deionised water for extraction, 77~ of
the cobalt catalyst remained with the upper solvent ,
layer. 23~ of the cobalt was extracted with the aqueous
product. This fraction corresponds approximately to the.
amount of sodium acetate added to promote the reaction.
Examples 6 to 11
These experiments illustrate the effectiveness of
lipophilic promoters such as phenol, nonylphenol,
hydroquinone, 4-(1-butylpentyl)pyridine, triphenylarsine
and triphenylphosphine oxide both to accelerate the
hydroformylation reaction and to permit the recycle of
essentially all the cobalt catalyst in the organic phase
following water extraction of product HPA. Example 4 was
repeated with addition of respectively 0.12 g of phenol
(Example 6), 0.258 of nonylphenol (7), 0.148 of hydro-
quinone (8), 0.278 4-(1-butylpentyl)pyridine (9), 0.4g
triphenylarsine (10) or 0.4g triphenylphosphine (11) as
promoter, for-a ratio of 0.25 moles promoter per mole of
cobalt (11 at 0.26) and a concentration of EO at the
start of the reaction in the range of 6.2 to 6.3 wt~.
Reactor contents were sampled and analysed at approxi-
mately 50°s conversion and at virtual complete conversion.
Following the reaction, the mixture was cooled to
room temperature. About 30g of deionised water were added
for extraction of product under 1.5 MPa (200 psig) syn-
thesis gas. After 30 minutes, mixing was terminated and
an aqueous product layer containing HPA was isolated.
Both layers were analysed.
Results of these experiments are summarised in the
Table. From the table it may be learned that the use of a
promoter provides a rate increase over that observed in
the absence of a promoter in Example 4. Recycle of the
cobalt catalyst with the organic layer represents a
WO 96/10552 ~ ~ ~ ~ ~ PCT/EP95/03870
- 17 -
substantial reduction in cobalt loss relative to that
observed with sodium acetate promotion in Example 5.
WO 96110552 ~ ~ ~ ~ PCT/EP95/03870
_ Z8 _
r-i M O l0I1 tn O ~ M I~ N t
~ O 01
V'01 CON (~ N ri ~-iO
M M M N
O t!7al O al t~ O IW -t~' N Ol
Q'O 01~- I M .-iO
t'
M M M N
01 I~O m O 01 ~ tnC t!7
-.-r
c!'N O C M r-i~-i N
tn M M N
00 t!7N 00Ol c1'~'tn ~-1d' 01a1
~ r-.~'--p1
crN 00d1 W' Q' ~-i
M N M N O
lfl~' OJlfllO M h c-1I~ alOl
~ O ,-ial
<t'ri 01O tn t!W -i
M M M N O
l0 l~O1 ~'lflM O ~ ~'00 01l0
~ .
c1'~ 61ri CO 00.--I Ol
N M M r-i Ol O
t!7 O O M t
N t
.-I O
V~ M
M O
O
V' M N
o~ dP
-- 'O 'LS
Ls N N
U
N ct5 ~ cif
r-I U r-I U
c~
.t-~3 v7 ~-iU r1
O 3
da-O O -t~~
cn O O o~ O cd O
fi
FCb b" 3 ~
> D .~-~,-aw ccsc~ --~, ~ O 3
~ ~
f U ~ ~ ~ x U U
U ~ U
dt~~ C~ ~ O -~ -.-i
~
x H t=~ 3
WO 96/10552 ~ ~ PCT/EP95103870
- 19 -
Example 12
This example illustrates hydrogenation of aqueous HPA
obtained from water extraction of the product of EO
hydroformylation. 333.48 of extract containing 20 wt% HPA
were added to a 500 ml autoclave reactor containing 5.078
of a powdered supported nickel hydrogenation catalyst
(Calsicat E-475SR, 50% Ni). The reactor was charged with
7.0 MPa (1000 psig) H2 and heated to 60 °C for 3 hours.
At this time, gas chromatographic analysis indicated 99%
conversion of HPA, at 93% selectivity to PDO (moles PDO
formed divided by moles HPA consumed) and 3% selectivity
to propanol. The reaction temperature was increased to
90 °C for one hour, after which an HPA conversion in
excess of 99% was indicated, at an apparent selectivity
of 99% PDO and 3.5% propanol. Heating was continued for
one additional hour at 110 °C to further improve the
selectivity towards PDO by reversal of heavy ends formed
during hydroformylation or early hydrogenation.
Example 13 -
In order to examine the role of the promoter, a
series of reactions was carried out in a small-scale
reactor fitted with optics for in situ infrared analysis.
In the first reaction, 80 mg (0.234 millimoles) of
recrystallised (from CH2C12) dicobaltoctacarbonyl were
added to 17 ml of dried and distilled MTBE in the 30 ml
reactor bottom fitted with a ZnS (45) infrared crystal.
The top was closed onto the unit and the reactor assembly
was removed from the dry box. The inert atmosphere was
replaced with carbon monoxide by alternately pressurising
the reactor to 1.5 MPa (200 psig) with CO and then
depressurising the vessel to atmospheric pressure for a
total of 3 cycles. The unit was finally pressurised to
1.5 MPa (200 psig) with C0. The unit was then heated to
80 °C and the pressure in the reactor was adjusted to 2.7
MPa (375 psig) with pure C0. 1.28 (27 millimoles) of EO
'' WO 96110552 ~ PCTIEP95/03870
- 20 -
(i.e., 8.5 wt~) were added to the reactor with hydrogen
gas pressure, bringing the total pressure inside the unit
to 11.1 MPa (1600 psig) to produce a 3:1; H2:C0 gas cap. s
Infrared spectra were recorded at 3 minute intervals to
monitor the progress of the~reaction. The pressure in the
unit dropped due to gas consumption and synthesis gas
(1:1) was added as required to maintain the total
pressure in the reactor between approximately 10.8 and
10.4 MPa (1550 and 1500 psig). A reactor profile of
pressure and temperature data-was measured digitally via
transducer and thermocouple.
The second reaction was carried out in a like manner
except that 16 mg (0.096 millimoles) of sodium octoate
was also added to the reaction mixture. Again, the
concentration of EO at the start of the reaction was 8.5
wt~. The rate of HPA formation was calculated from syn-
thesis gas consumption and checked against the appearance
of aldehyde at 1724 cm 1 and the disappearance of the EO
band in the infrared spectrum at 870 cm 1. The TOF of the
reaction in the absence of a promoter was 15 h 1 and in
the presence of sodium octoate the TOF was 41 h 1. At the
beginning of the reaction, the infrared spectrum of the
catalyst region (2300-2000 cm 1) displayed patterns
characteristic of dicobaltoctacarbonyl. The reaction run
in the absence of a promoter showed no change in this
region of the infrared over the course of the reaction.
In contrast, the reaction run with the promoter changes
rapidly producing a pattern characteristic of the cobalt
acyl complex in addition to the patterns from dicobalt-
octacarbonyl. This indicates that the promoter changes
the rate determining step in the reaction cycle, result-
ing in a faster overall reaction rate.