Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02204130 1997-04-30
W O96/18643 PCT/~ o15lo
~.LN~ ANTA~ONT~3T~
The present invention relates to novel compounds having
tachykinin antagonist activity, processes for their production,
pharmaceutical compositions comprising them and their use as
pharmaceuticals or pharmaceutical use.
More particularly the present invention provides in a first
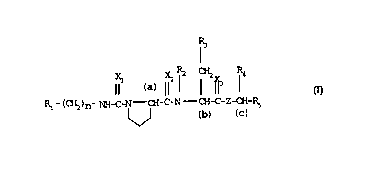
aspect, a compound of formula I
Xl ll2 ~ ¦ If3 ¦4 I)
(CH2)n-NH_C~N - CH-C-N - CH-C-Z-CH-R
~ (b) (C)
wherein
R1 is phenyl mono- or di-substituted by one or two members
selected from the group consisting of halogen, nitro,
cyano, trifluoromethyl, hydroxy, methoxy, hydroxymethyl,
methoxymethyl, methoxycarbonyl, carbamoyl and N-methyl-
carbamoyl,
n is zero or 1,
Xl is oxygen, sulfur or =NCN,
X2 and X3 are each independently oxygen or sulfur,
R2 is hydrogen or methyl,
R3 is phenyl, halo-substituted phenyl, 2-naphthyl,
IH-indol-3-yl or 1-methyl-indol-3-yl,
Z is -N(CH3)- or -CH2-,
R4 is phenyl, 3,5-bis(trifluoromethyl)phenyl or pyridyl and
R5is hydrogen, phenyl 3,5-bis(trifluoromethyl)phenyl or pyridyl,
whereby, when X3 is sulfur, Z is -N(CH3)-,
or acid addition salt thereof.
CA 02204130 1997-04-30
W O96tl8643 PcT/~l95~o~lO
By halogen (and halo) is meant chlorine (chloro), fluorine
(fluoro), bromine (bromo) and iodine (iodo).
When R1 is di-substituted phenyl the substituents may be the same
or different.
1. In a group of compounds of formula I Rl is phenyl mono- or
di-substituted by one or two members selected from the
group consisting of nitro, cyano, trifluoromethyl,
hydroxymethyl, methoxymethyl, carbamoyl and N-
methylcarbamoyl.
2. In a further group of compounds of formula I in accordance
with the present invention n is zero.
2a. When n is zero, R1 is preferably phenyl mono- or di-
substituted by one or two members selected from the group
consisting of nitro, cyano, methoxymethyl, methoxycarbonyl,
carbamoyl and N-methylcarbamoyl (e.g. nitro, cyano,
methoxymethyl, carbamoyl and N-methylcarbamoyl) especially
nitro and methoxymethyl, most especially nitro.
2b. When n is zero, R1 is preferably mono-substituted phenyl, in
particular phenyl mono-substituted at the 2-position.
2c. When n is zero, Rl is most preferably phenyl mono-
substituted at the 2-position by any of the substituents
set forth under 2a above, in particular 2-nitrophenyl and
2-(methoxymethyl)phenyl, especially 2-nitrophenyl.
3. In a yet further group of compounds in accordance with the
present invention n is 1.
3a. When n is 1, R1 is preferably phenyl mono- or di-substituted
by one or more members selected from the group consisting
of halogen, trifluoromethyl, and methoxy, especially
halogen and trifluoromethyl.
3b. When n is 1, R1 is preferably phenyl mono-substituted at the
2-position or di-substituted at the 2- and 6-position.
3c. When n is 1, R1 is most preferably phenyl mono-substituted
at the 2-position or di-substituted at the 2- and 6-
position by any of the substituents set forth under 3a
above, especially phenyl mono-substituted at the 2-
CA 02204130 1997-04-30
W O96/18643 PCTAEP95/04910
position, in particular 2-halo- or 2-trifluoromethylphenyl,
especially 2-chloro- or 2-trifluoromethylphenyl and most
especially 2-chlorophenyl.
In the compounds of formula I
4. Preferably n is zero.
5. Preferably Xl is oxygen or sulfur, especially oxygen.
6. Preferably X2 and X3 are each oxygen.
7. Conveniently R2 is hydrogen.
8. When R3 is halo-subsituted phenyl this is suitably di-halo-
substituted phenyl, in particular 3,4-di-halosubstituted
phenyl. Preferred as halo is chloro, 3,4-di-chlorophenyl
being particularly suitable as R3.
9. Preferably R3 is 2-naphthyl or halo-substituted phenyl, e.g.
as defined under 8 above, in particular 2-naphthyl.
10. Z is preferably -N(CH3)-.
11. When Rs is other than hydrogen R4 and Rs are suitably the
same.
12. Pyridyl as R4 and/or Rs is preferably 2-pyridyl.
13. Preferably R4 is phenyl.
14. Preferably Rs is hydrogen.
15. Most preferably R4 is phenyl and Rs is hydrogen.
The present invention is to be understood as embracing compounds
of formula I in which the meanings of the substituents Rl to Rs~
Xl to X3, n and Z comprise any combination or sub-combination of
the meanings given under formula I and/or under any one or more
of paragraphs 1 through 15 above.
A. In a sub-group of compounds in accordance with the present
lnventlon
Rl is 2-halo- or 2-nitro-phenyl,
n is zero,
X2 and X3 are each oxygen,
R4 is phenyl or pyridyl,
Rs is hydrogen, phenyl or pyridyl and
CA 02204130 1997-04-30
W O96/18643 PcT/~l9s~1510
Xl, R2, R3 and Z have the me~n;ngs hereinbefore given for
formula I.
B. In a further sub-group of compounds in accordance with the
present invention
Rl is a group of formula
R,
wherein Rla is trifluoromethyl, halogen, methoxy or
nitro and
Rlb is hydrogen, trifluoromethyl, halogen,
methoxy or nitro,
n is 1,
X2 and X3 are each oxygen,
R3 is halo-substituted phenyl, 2-naphthyl, IH-indol-3-yl,
or l-methyl-indol-3-yl,
Z is -N(CH3)- and
Xl, R2, R4 and Rs have the meanings hereinbefore given for
formula I
Preferred significances in relation to the sub-groups defined
under A and B above are as indicated under paragraphs 1 through
15 above.
Compounds of formula I in which R4 and/or R5 is pyridyl exist in
free form and in acid addition salt form. The present invention
is to be understood as including both the free compounds of
formula I and their acid addition salts. Suitable
pharmaceutically acceptable acid addition salts for use in
accordance with the present invention include e.g. hydrochloride
salts.
Compounds of the invention comprise two asymmetric carbon atoms
CA 02204130 1997-04-30
W O96/18643 PCTAEP95/04910
[marked (a) and (b) in formula I]. When R4 and Rs are different
and Rs is other than hydrogen, a further asymmetric carbon atom
[(c)] is present. The compounds accordingly exhibit optical
isomerism.
Individual isomers may be obtained in conventional manner, e.g.
by synthesis using optically active starting materials or by
separation of initially obtained isomeric mixtures, for example
employing chromatographic techniques using a chiral support or
by recrystallisation of diastereomeric salt forms.
The present invention is to be understood as embracing both
individual isomers in pure or substantially pure form as well as
mixtures, e.g. racemic and diastereomeric mixtures, unless
otherwise specified.
In formula I each of the carbon atoms (a) and (b) preferably has
the ~S)-configuration. More preferably both carbon atoms (a) and
(b) have the (S)-configuration. Accordingly, in a preferred
aspect the present invention provides a compound of formula I as
hereinbefore defined wherein the carbon atoms (a) and (b) both
have the (S)-configuration in pure or substantially pure form,
e.g. comprising less than 10%, more preferably less than 5%, e.g.
less than 2% of other isomeric forms.
The present invention further provides a process for the
production of a compound of formula I as hereinbefore defined or
acid addition salt thereof, which process comprises reacting a
compound of formula II
R4
H-N - CH-C-N CH-C-Z-CH-~
CA 02204130 1997-04-30
WO96/18643 PCT~5/04910
wherein R2 to Rsl X2, X~ and Z have the me~;ngS hereinbefore
given, with a compound of formula III
Rl~-(cH2)n-N=c=xl (III)
wherein Rl' is phenyl, mono- or di-substituted by one or two
members selected from the group consisting of halogen, nitro,
cyano, trifluoromethyl, protected hydroxy, methoxy, protected
hydroxymethyl, methoxymethyl or methoxycarbonyl, and n and X
have the meanings given for formula I;
when required, deprotecting a compound thus obtained wherein Rl'
is phenyl substituted by protected hydroxy and/or protected
hydroxymethyl and/or transforming a compound thus obtained
wherein Rl' is phenyl substituted by methoxycarbonyl to obtain a
corresponding compound wherein Rl' is phenyl substituted by
carbamoyl or N-methylcarbamoyl;
and recovering the obtained compound of formula I in free or acid
addition salt form.
Reaction of compounds II with III is suitably performed in an
inert organic medium, e.g. dioxane, at temperatures of from 20~
C to reflux.
Protecting groups of protected hydroxy or hydroxymethyl moieties
comprising Rl' may be any oxy-protecting group as known and
commonly employed in the art of peptide chemistry, for example,
t.butyldimethylsilyloxymethyl. Deprotection may be carried out
in accordance with standard procedures, e.g. as hereinafter
described in relation to EXAMPLES 18 and l9.
Transformation of methoxycarbonyl moieties may also be carried
out in accordance with standard procedures as known in the art,
e.g. by hydrolysis to carboxy, conversion of the carboxy moiety
to a reactive functional derivative, e.g. carbonylhalide or mixed
anhydride moiety, and reaction of this with ammonia or
methylamine, e.g. as hereinafter described in relation to
EXAMPLES l5 and l6.
CA 02204130 1997-04-30
W 096/18643 PcT/~l9s~ol9lo
Starting materials of formula III are known from the art,
commercially available or producible analogously to the known
compounds, e.g. in the case of compounds of formula III wherein
X = =NCN in accordance with the general procedures hereinafter
described in relation to EXAMPLE 4.
Compounds of the formula II may be prepared according to the
following reaction sequence:
2 Cl H2 X C~3 orHalMg-CH -CH
(IV) (Va) (Vb)
R3
1 (a)
R2 CH2
113 ~ R~
RpN CH-C-Z-CR ~ (VI)
Rs
R3 (b)
R2 CH2
HN CH-C-Z-CH ~ (VII)
Rs
Rp_N--CH~C~Rai (C)
(VIII)
13
R2 CH2 /R4
Rp-N - CH-C-N - CH-C-Z-CH \ (IX)
~ (d)
(II)
CA 02204130 1997-04-30
W O96118643 PCTnEPg5/04910
in which ~ represents an amino protecting group, R~ represents
a carboxy activating group, Hal is chlorine, bromine or iodine,
especially bromine, and R2 through R5, X2, X3 and Z have the
meanings hereinbefore given.
Suitable amino protecting groups as ~ include any of those known
and employed in the art of peptide synthesis, t-butoxcarbonyl
(Boc) being, for example, particularly suitable in relation to
formulae (IV) and (VIII).
Reaction of compounds IV with Va and Vb leads to compounds VI
(and hence II) in which Z = -N(CH3)- and -CH2- respectively.
Suitable carboxy activating groups Ra for reaction of IV with Va
as well as process step (c) include mixed anhydride activating
groups, e.g. i-butoxycarbonyloxy. Reaction may be performed in
accordance with any of the techniques known and employed in the
art of peptide chemistry, e.g. in accordance with the general
methods of EXAMPLE lA.2.
Starting materials of the formula Va and Vb are known from the
art or may be prepared analogously to known compounds, e.g. as
hereinafter illustrated in relation to EXAMPLES 5, 20 and 24.
Suitable carboxy activating groups Ra for reaction of IV with Vb
include pyridyl and picolyl ester groups, e.g. 2-pyridyloxy.
Reaction may be carried out in accordance with procedures known
in the art with formation of the Grignard reagent Vb in situ, for
example as hereinafter described in relation to EXAMPLE 28.
Starting materials of formula IV are known or described in the
art or may be produced according to known procedures, for example
by activation of the corresponding N-protected acids, suitably
in situ, for example as hereinafter described in EXAMPLE lA.1.
Alternatively, compounds of formula II wherein R2 is methyl may
be produced proceeding from compounds of formula IV wherein R2 is
CA 02204130 1997-04-30
W O96/18643 PCTnEPgS/04910
hydrogen and carrying out an intermediate methylation, e.g. of
the compound of formula IX prior to step (d), according to the
method of Olsen, J.Org. Chem., 35, 1912-1915 ~1970). This
approach is illustrated in relation to EXAMPLES 27 and 29.
Process steps (b) and (d) are conventional de-protection steps
as commonly practiced on the art of peptide synthesis e.g. as
hereinafter illustrated in EXAMPLE IB.
The following examples are illustrative of the processes for the
production of the compounds of the invention.
EXAMPLE 1:
Preparation of 2-nitrophenylcarbamoyl-[(S)-prolyl]-[(S)-3-(2-
naphthyl)alanyl]-N-benzyl-N-methylamide:
[Formula I: R1 = 2-nitrophenyl; n = zero; X1, X2 and X3 each =
oxygen; R2 = H; R3 = 2-naphthyl, Z = -N(CH3)-; R~ = phenyl and Rs
= Hi both carbon atoms (a) and (b) have the (S)-configuration.]
To (S)-prolyl-(S)-3-(2-naphthyl)alanyl-N-benzyl-N-methylamide
(1.39g) in ethyl acetate (15ml) are added 2-nitrophenylisocyanate
(551 mg). The yellow solution is stirred at room temperature for
1 hour. The reaction mixture is concentrated. The orange residue
is purified by flash chromatography (2:1 ethylacetate: hexane)
to afford a yellow foam. The foam is dissolved in ethyl acetate
(20ml) and dropped slowly into a stirred solution of hexane
(200ml). The yellow precipitate is filtered and dried to yield
the title compound: m.p. = 79-82~C; TLC (silica,
cyclohexane/ethyl acetate 1:2) Rf = 0.36.
EXAMPLE 2:
Preparation of 2-chlorobenzothiocarbamoyl-[(S)prolyl]-[(S)-3-(2-
naphthyl)alanyl]-N-benzyl-N-methylamide.
CA 02204l30 l997-04-30
W O96/18643 PCTnEP95/04910
[Formula I: Rl = 2-chlorophenyl; n = 1; Xl = sulfur; X2 and X3
each = oxygen; R2 = H; R3 = 2-naphthyl; Z = -N(CH3)-; R4 = phenyl
and Rs = H; both carbon atoms (a) and (b) have the (S)-
configuration.]
(S)-Prolyl-(S)-3-(2-naphthyl)-alanyl-N-benzyl-N-mehtylamide
(1.03g) is dissolved in lOml CH2Cl2 with 2-chlorobenzyl
isothiocyanate (455mg), and the solution is stirred at room
temperature for 18 hours. The solvent is removed in vacuo. The
product is purified by flash column chromatography (silica,
cyclohexane/ethyl acetate 1:2) and crystallised from ethyl
acetate, to give fine white needles. These are filtered, and
dried at O.lmmHg/75~C for 18 hours to yield the title compound:
m.p. = 129-131~C,: TLC (silica, cyclohexane/ethyl acetate 1:2)
Rf=0.3.
The following compounds of formula Ia
R Xl ( ) R¦2FH2 / R~
~ ~CH2) n-NH-c-N ~ CH -CO-N-CH-CO-Z-CH
wherein R2 is hydrogen, R3 is 2-naphthyl and Z is -N(CH3)- may be
prepared analogously to example 1 (when n = zero) or example 2
(when n = 1) above.
EXA- Rla n X1 R4 Rs Physical Data
MPLE m.p.(~C)/Rf.
3 2N02- zero S phenyl H 118-121 /0.45~t~
4~ 2N02- zero =NCN phenyl H 92-96 / 0.10('
2N02- zero 0 2-pyridyl 2-pyridyl 90-92 / 0.18(3
6 2N02- zero 0 2-pyridyl H 69-71 / 0.01(3)
CA 02204130 1997-04-30
W 096118643 11 PCT/~l9s~ol9lo
EXA R1' n X, R, Rs Physical Data
MPLE m.p.~C / Rf.
7 2NO2- zero S 2-pyridyl H 98-100 t 0.01(3
8 2CI- zero S phenyl H 94-98 / 0.04("
~,
9 4NO2- zero O phenyl H 126-129 / 0.20 ("
2CN- zero O phenyl H 124-126 / 0.25 ~"
11 3CN- zero O phenyl H 105-107 / 0.15("
12 4F- zero O phenyl H 85-87 / 0.28 ("
13 2(CH30CO)- zero O phenyl H 50-60 / 0.23 ~"
14 3NO2 zero O phenyl H 98-101 / 0.25
15~- 2(CH3NHCO)- zero O phenyl H 96-102 / 0.19 (2)
16~- 2(NH2CO)- zero O phenyl H 109-115 / 0.10 (2)
17 2(CH3OCH2)- zero O phenyl H 58-62 / 0.27 ~"
18--- 2HO- zero O phenyl H 98-104 / 0.24 ~'
19' 2(HOCH2)- zero O phenyl H 83-86 / 0.20 (2)
2CI- 1 S 2-pyridyl 2-pyridyl 95-97 / 0.09~3
21 2CI- 1 S 2-pyridyl H 85-87 / o o1(4~
22 2CF3- 1 S phenyl H 175-176/ 0.19~"
23 2CI- 1 O phenyl H 69-73 / 0.18('
24 2CI- 1 S '~ H 125-127 / 0.11~4
CF,
2CI- 1 S phenyl phenyl 102-104 / 0.15~4
26 2Br- 1 S phenyl H 142-143 / 0.45 (1)
The following compounds of formula Ia above wherein Rla is 2NO2-,
R3 is 2-naphthyl, R4 is phenyl and R5 is H may be prepared
CA 02204130 1997-04-30
W O96118643 PCT~Pg5/04910
12
analogously to example 1 (when n = zero) or example 2 (when n =
1) above.
EXAMPLE n X, R2 Z Physical Data
- m.p.~C/Rf.
27 zero ~ CH3- -N(CH3)- 71-74/0.55~2
28 zero O H -CH2- 106-108/0.35(4
29 1 S CH3- -N(CH3)- 84-85/ 0.19(4)
The following compounds of formula Ia above wherein Rz is
hydrogen, Z is -N(CH3)-, R4 is phenyl and R5 is hydrogen may be
prepared analogously to example 1 (when n = zero) or example 2
(when n = 1) above:
EXAMPLE R,- n X, R3 Physical Data
m p ~C/Rf
2NO2- zero O IH-indol-3-yl 96-98/ 0.36(2)
31 2NO2- zero o 3,4-~ ,rophe!lyl 151 -152/0.542)
32 2CI- 1 S 3.4- 'i~ ruphenyl 185-186/0.59(2)
33 2CI- 1 S l-methylindol-3-yl 160-162/0.06(4)
(1) = Silica, cyclohexane/ethyl acetate 1:2
(2) = Silica, cyclohexane/ethyl acetate 1:4
(3) = Silica, CH2Cl2/CH30H 25:1
(4) = Silica, cyclohexane/ethyl acetate 1:1
* For the preparation of the EXAMPLE 4 compound the starting
material of formula III is prepared in situ as follows:
Potassium t-butoxide (lml, lM in tetrahydrofuran) is added to
cyanamide (44mg) in dimethyl formamide (Sml). A white precipitate
forms. The reaction mixture is stirred for 20 mins. at room
temperature. 2-Nitrophenyl isothiocyanate (180mg) is added as a
-
CA 02204130 1997-04-30
W O96118643 PCT~EP95/04910
13
solid and the reaction mixture is stirred for 10 mins. at room
temperature. The mixture is cooled in ice and triethylamine
(0.42ml) added followed by (S)-prolyl-(S)-3(2-naphthyl)alanyl-N-
benzyl-N-methylamide (415 mg) and HsCl2 (300mg). Further reaction
and work-up then proceeds according to example 1.
** The compounds of EXAMPLES 15 and 16 are prepared via the
compound of EXAMPLE 13 employing the following additional
steps:
The product of EXAMPLE 13 is hydrolysed to provide the
corresponding acid in which Rl~ (formula Ia) = 2(HOCO)-. The
acid is then reacted with isobutyl chloroformate and N-
methylmorpholine in ethyl acetate under standard conditions
(temperature maintained below -15~C) whilst dry methylamine
(for the production of the compound of EXAMPLE 15) or
ammonia gas (for the production of the compound of EXAMPLE
16) is slowly introduced into the reaction vessel over a
period of 30 minutes. The obtained raw products are worked
up analogously to the procedures described in EXAMPLE 1.
*** The compounds of EXAMPLES 18 and 19 are prepared via an 0-
protected intermediate, e.g. as follows:
(S)-prolyl-(S)-3-(2-naphthyl)-alanyl-N-benzyl-N-methyl-
amide is reacted with 2-(t.butyldimethylsilyloxy)phenyl
isocyanate/2-(t.butyldimethylsilyloxymethyl)phenyliso-
cyanate analogously to EXAMPLE 1 to yield the EXAMPLE 18
and 19 compounds respectively in t.butyldimethylsilyl-
protected form. Deprotection to yield the EXAMPLE 18 and 19
compounds is effected under standard conditions employing
tetrabutyl ammonium fluoride in tetrahydrofuran.
The starting material for the process of EXAMPLE 1 is prepared
as follows.
E~UUMPLE lA tProc~88 8te~ (~)]
CA 02204130 1997-04-30
W O96118643 PCTnEPgS/04910
14
lA,1 Pre~ration of Boc-(S)-3-(2-na~hthvl)alanine isobutoxv-
formvl ~nh~ride (Formula IV: Rp = Boc, R2 = H, R3 = 2-na~hthvl,
X3 = 0, ~ = i-butoxvcarbonYloxY)
Boc-(S)-3-(2-naphthyl)alanine (480mg) is dissolved in 5ml dry
CH2Cl2 with N-methyl morpholine (170~1, 156mg) and cooled with
stirring under N2 to -15~C, on a salt/ice bath. i-
Butylchloroformate (200,ul, 20mg) in 2ml dry CH2Cl2 is added
dropwise, ensuring that the temp. rPm~ins below -10~C, and the
reaction is stirred for 30 minutes.
lA.2. Pre~aration of Boc-(S)-3-(2-naDhthvl)alanYl-N-benzyl-N-
methv1amide (Formula VI: R~ = Boc, R2 = H, R3 = 2-na~hthvl, X~_~
0, Z = -N(CH3)- R~ = ~henvl, Rs = H)
N-benzylmethylamine (Formula Va) (185mg) is added dropwise in 2ml
dry CH2C12 to the product from EXAMPLE lA.1, again ensuring that
the temperature remains below -10~C, and the reaction is stirred
until complete by TLC. The reaction is diluted to 75ml with
CH2Cl2 and washed with 50 ml dilute aqueous HCl, 50ml water, and
25ml brine. The organic phase is dried over MgSO4, filtered and
the solvent removed in vacuo. The product is purified by flash
column chromatorgraphy (silica, cyclohexane/ethyl acetate 4:1),
to yield the title compound as a colourless foamed solid: TLC
(silica, cyclohexane/ethyl acetate 1:1) Rf = 0.54.
EXA~qPI.E lB tProco~ te~ (b)]
Pre~aration of (S)-3-(2-na~hthvl)alanYl-N-benzyl-N-methYlamide
(Formula VII)
The product of EXAMPLE lA (600mg) is dissolved in 10ml 4.0M HCl
in dioxan, and stirred at room temperature for ca. 30 minutes.
The HCl and dioxan are removed in vacuo, and the residue
dissolved in 100ml water, basified with 2M NaOH(aq.), and
extracted with CH2Cl2 (2x75ml). The organic phase is dried over
MgSOg, filtered and the solvent removed in vacuo, to give the
CA 02204130 1997-04-30
W O96/18643 PCT~P95/04910
title compound as a yellow oil: TLC (silica, CH2Cl2/CH30H/CH3COOH
90:9:1) Rf = 0.43.
E~UUMPLE lC t~ ~t~p (c ) ~
Pre~aration of Boc-(S)-~rolvl-(S)-3-(2-na~hthvl)alanYl-N-benzYl-
N-methvlamide (Eormula IX: Rp = Boc)
The title compound is prepared analogously to EXAMPLE lA. 2 by
reaction of Boc-(S)-proline (310mg) with the product of EXAMPLE
lB (460 mg) and is obtained as a viscous oil: TLC (silica,
cyclohexane/ethyl acetate 1:1) Rf = 0.18.
E~UUMPLE lD ~E ~ ~ ~t~p (~) ~
Pre~aration of (S)-~rolYl-(S)-3-(2-na~hthvl)alanyl-N-benzYl-N-
methYlamide (Formula II)
The title compound is prepared analogously to EXAMPLE lB starting
from the product of EXAMPLE lC (740mg) and is obtained as a
colourless foamed solid: TLC (silica, CH2Cl2/CH30H/CH3COOH 90:9:1)
Rf = 0.29.
The product of EXAMPLE lD is also used as starting material for
the production of the compounds of EXAMPLES 2 to 4, 8 to 19, 22,
23 and 26 and the starting materials of formula II for EXAMPLES
6, 7, 21, 25, and 30 to 33 are produced analogously.
The starting materials of formula II for EXAMPLES 5, 20, 24 and
27 to 29 are produced analogously to EXAMPLES lA to lD employing
the following means to obtain the formula Va starting material
and/or adaptations in procedure:
IN REIJ~TION TO EXUU~PLES 5 AIJD 20:
PreDaration of N-methYl-(di-2-~YridYl)methvlamine (Formula Va
R~ and Rc both = 2-~vridyl)
CA 02204130 1997-04-30
W O96/18643 PcT/~l9r~910
16
Di-2-pyridylketone ~2.00g) is dissolved in 20 ml of CH2Cl2.
Heptamethyldisilazan (1.9Og) is added followed by trimethylsilyl
trifluoromethanesulphonate (0.12g) and the reaction mixture is
refluxed for 12 hours. The solvent is removed in vacuo. The crude
oil (2.14g) is dissolved in 20 ml of dry C2HsOH. 0.65 g acetic
acid is added followed by sodium cyanoborohydride (0.68g) and the
reaction mixture is stirred at room temperature for 1 hour. A 1%
solution of KHSO4 is added until the solution is at pH 2 and then
a 2M solution of NaOH is added until the solution is at pH 12.
The C2HsOH is removed in vacuo and the product extracted into
ethyl acetate. The organic layer is dried over MgSO4, filtered
and HCl is bubbled through the filtrate. The obtained title
compound in trihydrochloride salt form is filtered off and dried
for direct further reaction analogously to EXAMPLES lA through
lD.
IN ~ I~TION TO E~UU~PLE 24:
Pre~aration of N-meth~1-3,5-bis(trifluoromethYl)benzvlamide
~Formula Va: R~ = 3,5-b;s(trifluoromethvl)~henYll, R~= H
STEP I N-Boc-3,5-bisttrifluoromethYl)benzvlamine
3,5-bis~trifluoromethyl)benzylamine (5.00g) and N-
(benzyloxycarbonyloxy)succinimide (5.12g) are dissolved in
50ml of tetrahydrofuran and the reaction mixture is stirred
at room temperature for 3 hours. The solvent is removed in
vacuo and the product dissolved in CH2Cl2. The solution is
washed with 50ml H20 and 50ml brine. The organic layer is
dried over MgSO4, filtered and the solvent removed in vacuo
to yield the title compound.
STEP II N-Boc-N-methvl-3 5-bis(trifluoromethyl)-
benzvlamine
The product of STEP II above (6.87g) is dissolved in 50mlof dry tetrahydrofuran and cooled to -78~C. 1.5M LDA in
CA 02204130 1997-04-30
W O96/18643 PCTAEP95/04910
17
tetrahydrofuran (14ml) is added the reaction mixture is
stirred at -78~C for 30 minutes and 2.98g of methyl iodide
is then slowly added. The reaction mixture is stirred at
room temperature for 18h. The solvent is removed in vacuo
and the product purified by flash column chromatography
- (silica, cyclohexane/ethyl acetate 9:1), to yield the title
compound.
S5EP ~II N-methvl-N-3 5-bis(trifluoromethvl)benzvlamine
2.00g of the product of STEP III above is dissolved in
lOOml of C2H~OH and deprotected by adding a catalytic amount
of 10~ palladium on charcoal and placing the solution under
an atmosphere of hydrogen. The catalyst is filtered off
after 4 hours and the solvent removed in vacuo. The
obtained title compound is reacted further analogously to
EXAMPLES lA through lD.
IN R13I-ATION TO E~ S 27 AND 29:
Pre~aration of Boc-(S)-~rolvl-(S)-(N-methvl)-3-(2-na~hthvl)-
alanvl-N-benzvl-N-methvlamide (Formula IX: Rp = Boc, R2 = -CH3,
R3 = 2-na~hthYl Z = -N( CH3 ) -, R~ = ~henYl, Rc = H )
The product of EXAMPLE lC ( 1. 08g) and iodomethane (1.04 ml,
2.38g) are dissolved in 30 ml dimethyl-formamide. Silver oxide
(1.95g) is added, the reaction mixture is heated to 60~C and
stirred until no starting material r~mA; n.s as determined by
analytical HPLC. The reaction mixture is cooled to room
temperature, diluted to 200ml with CHCl3 and washed with 2x lOOml
5% KCN (aq.), 2 x lOOml H20 and 50 ml brine, then dried over
anhydrous MgS04, filtered and the solvent removed in vacuo to
yield the title compound as a pure colourless glass.
The title compound is reacted further analogously to EXAMPLE lD.
CA 02204130 1997-04-30
W O96118643 PCT~P95104910
~N F~U~TION ~ ~UU~P~E 28:
28.A.l. Preparation of Boc-(S)-3-(2-na~hthYl)alanine 2-~YridYl
ester (Formula IV : ~ = Boc, R2 = H. R3 = 2-na~hthyl, R3 =
2-~vr;dvloxv)
Boc-(S)-3-(2-naphthyl)alanine (l.OOg) and 2-hydroxypyridine
(0.33g) are dissolved in Sml of dry pyridine and the solution
cooled to 0~C. Dicyclohexylcarbodiimide (0.72g) is added and the
reaction mixture stirred at 0~C for 6 hours. The solvent is
removed in vacuo and the product dissolved in 2Oml of ethyl
acetate, filtered and the solvent removed in vacuo. The product
is purified by flash column chromatography (silica,
cyclohexane/ethyl acetate, 2:1), to yield the title compound: TLC
(silica, cyclohexane/ethyl acetate 1:1) Rf = 0.41.
28.A. 2. Pre~aration of Boc~1-(S)-na~hthalen-2-vl-methYl-2-oxo-4-
~henvl-butYllamine ~Formula VI: Rp = Boc, R2 = H, R3 = 2-na~hthYl,
Z = -CH2-, R~ = Dhenvl, R5 = Hl
The product of EXAMPLE 29.A.1. (O.85g) is dissolved in lOml of
dry tetrahydrofuran in a flame-dried flask, and the solution is
stirred under nitrogen. The solution is cooled to -78~C and a 2M
solution of phenethylmagnesium bromide (2ml) is slowly added. The
reaction mixture is stirred at -78~C for 1 hour. 20ml of
saturated ammonium chloride solution is added and the product
extracted into ethyl acetate. The organic layer is dried over
MgSO4, filtered and the solvent removed in vacuo. The product is
purified by flash column chromatography (silica, cyclohexane/
ethyl acetate 9:1) to give the title compound: TLC (silica,
cyclohexane /ethyl acetate 4:1) Rf = 0.66.
The title compound is processed further analogously to EXAMPLES
lB through lD.
For larger scale production, the reaction procedures of EXAMPLES
lA through 1 may appropriately be adapted as indicated in the
CA 02204130 1997-04-30
Wo96tl8~3 PCT~5/04910
19
following EXAMPLE 34.
EXANPL~ 34:
Large scale preparation of 2-nitrophenylcarbamoyl-[(S)-prolyl]-
[(S)-3-(2-naphthyl)alanyl]-N-benzyl-N-methylamide:
S ~ P I (- E~U~MPLE (A)
A 0.5-L, 3-necked, round-bottomed flask, equipped with a
mechanical stirrer, digital thermometer, addition funnel,
nitrogen inlet-outlet, and cooling bath is charged with 187,4g
of N-benzylmethylamine and cooled to 1-5~C (internal
temperature). 7.5g of ethyl trifluoroacetate are added dropwise
over a period of 15 minutes while maintA;n;ng an internal
temperature of 1-5~C. The funnel is washed with a total of 7.5ml
of ethyl acetate in three equal portions of 2.5ml each and the
reaction mixture is added. The cooling bath is removed and the
mixture warmed to room temperature (21-23~C) in 30 min. The
mixture is stirred at room temperature for 30 min. and the oil
held.
A 12-L, 4-necked, round-bottomed flask, equipeed with a
mechanical stirrer, digital thermometer, addition funnel,
nitrogen inlet-outlet, and cooling bath is charged with 394,0g
of Boc-(S)-3-(2-naphthyl)alanine, and 5,6L of ethyl acetate. The
solution is cooled to -15~C (internal temperature) and 174,4g of
4-methylmorpholine are added over a period of 5 minutes. The
addition funnel is washed with 25ml of ethyl acetate and this is
added to the reaction mixture. The mixture is stirred for 10
minutes and a solution of 172.Oml (181.12g) of isobutyl
chloroformate in 125ml of ethyl acetate is added over a period
of 30 minutes while maintaining an internal temperature of -14
to -16~C. The addition funnel is washed with 50ml of ethyl
acetate in two equal portions of 25ml each and this is added to
the reaction mixture. The suspension is stirred at -14 to -15~C
for an additional 30 minutes. A solution of above prepared N-
CA 02204130 1997-04-30
W O 96/18643 PCT/~s~l~J5lo
benzylmethylamine in 125ml of ethyl acetate is added at a
constant rate, over a period of 40 minutes, while maint~ining an
internal temperature of -14 to -15~C. The addition funnel is
washed with 50ml of ethyl acetate in two equal portions of 25ml
each and this is added to the reaction mixture. After stirring
at the same temperature for an additional 1 hour, a solution of
34.4ml (36.22g) of isobutyl chloroformate in 25ml of ethyl
acetate is added over a period of 10 minutes while maint~;n;ng
an internal temperature of -14 to -16~C. The addition funnel is
washed with lOml of ethyl acetate in two equal portions of 5ml
each and this is added to the reaction mixture. The suspension
is stirred at -14 to -16~C for an additional 15 minutes. A
solution of 37.4g of N-benzylmethylamine (pretreated with 1.5g
of ethyl trifluoroacetate as above) in 25ml of ethyl acetate is
added at a constant rate, over a period of 10 minutes, while
maintaining an internal temperature of -14 to -15~C. The addition
funnel is washed with lOml of ethyl acetate in two equal portions
of 5ml each and this is added to the reaction mixture. The
reaction mixture is warmed to room temperature (21-22~C) over a
period of 1 hour. The reaction mixture is stirred at room
temperature (21-22~C) for an additional 1 hour. 2.5 L of water
are added and stirring is continued for 5-10 minutes. The organic
layer is separated and washed with 1.876 L of lN hydrochloric
acid followed by 1.8 L of water. The organic layer is washed with
1.5 L of 5% aqueous sodium bicarbonate. The resulting organic
layer is washed with 1.5 L of water, followed by 1.0 L of brine
and filtered in a Buchner funnel with suction to obtain 6.1 L of
a solution of Boc-(S)-3-(2-naphthyl)alanyl-N-benzyl-N-methylamide
in ethyl acetate. This solution is held overnight at room
temperature under nitrogen for the next step.
STEP II ~-- Ea~P~E lB)
A 12-L, 4-necked, round-bottomed flask, equipped with a
mechanical stirrer, digital thermometer, addition funnel, drying
tube, and cooling bath is charged with a solution of 455.75g of
hydrochloric acid gas in 2.2 L of ethyl acetate. The solution is
CA 02204130 1997-04-30
Wo96/18~3 PCT~5/04910
21
cooled to 10~C (internal temperature) and 6.1 L of the crude
product of STEP I in ethyl acetate is added over a period of 25
to 30 minutes while maintA;n;ng an internal temperature below
20~C. The mixture is warmed to room temperature (22-23~C) and
stirred at this temperature for an additional 3 hours. The
reaction mixture is concentrated under reduced pressure (40-45~C,
100 to 110 mm Hg) until 5.0 L of solvent is collected, cooled to
20-22~C and stirred for 15-30 minutes. The solid is collected by
filtration in a Buchner funnel with suction and the solid washed
with a total of 1.2 L of ethyl acetate in four equal portions of
300 ml each. The solid is dried at 50-55~C (762mm Hg) for 24
hours to obtain a constant weight of pure (S)-3-(2-
naphthyl)alanyl-N-benzyl-N-methylamide hydrochloride. Purity:
98.4% (by HPLC); [a]D: +24.8~ (c=l,methanol).
STEP III (-- E~UU~P~E IC)
A 5-L, 4-necked, round-bottomed flask, equipped with a mechanical
stirrer, digital thermometer, addition funnel, and cooling bath
is charged with 332.0 g of the product of STEP II and 2.0 L of
isopropyl acetate. The suspension is cooled to 10-12~C (internal
temperature) using an ice-water bath. 1.4 L of 5% aqueous sodium
hydroxide are added with efficient stirring over a period of 10
minutes while maintaining an internal temperature of 10-12~C. The
mixture is warmed to 21-22~C in 30 minutes. The organic layer is
separated and washed with 0.7 L of water followed by 0.25 L of
brine. The organic layer is dried over lOOg of anhydrous sodium
sulfate and filtered in a Buchner funnel with suction. The solids
are washed with a total of 90 ml of isopropyl acetate in three
equal portions of 30 ml each. The organic layer is concentrated
under reduced pressure (40-lOOmbar; 43-45~C) until no further
solvent distills to obtain 0.35 L of (S)-3-(2-naphthyl)alanyl-N-
benzyl-N-methylamide (free base) as an oil. This is held.
A 12-L, 4-necked, round-bottomed flask, equipped with a
mechanical stirrer, digital thermometer, addition funnel,
nitrogen inlet-outlet, and cooling bath is charged with 205.4g
CA 02204130 1997-04-30
Wo96/18643 PCT~5/04910
22
of Boc-(S)-proline, and 3.2 L of ethyl actate. The mixture is
stirred for 5 minutes to obtain a solution. 125.6g of 4-
methylmorpholine are added over a period of 10 minutes while
maint~;n;ng an internal temperature of 20-22~C. The addition
funnel is washed with 25 ml of ethyl acetate and this is added
to the reaction mixture. The solution is cooled to -15~C
(internal temperature) and a solution of 132.9g of isobutyl
chloroformate in 75 ml of ethyl acetate is added over a period
of 25 to 30 minutes while maint~;n;n~ an internal temperature of
-14 to -16~C. The addition funnel is washed with 60 ml of ethyl
actate in three equal portions of 2Oml each and this is added to
the reaction mixture. The suspension is stirred at -14 to -15~C
for an additional 30-35 minutes. A solution of the previously
prepared 0.35 L of (S)-3-(2-naphthylalanyl-N-benzyl-N-methylamide
in 0.35 L of ethyl actate is added at a constant rate of -10
ml/minute over a period of 70 minutes while maintaining an
internal temperature of -14 to -15~C. The addition funnel is
washed with a total of 75 ml of ethyl acetate in three equal
portions of 25 ml each and this is added to the reaction mixture.
The reaction mixture is warmed to room temperature (21-22~C) over
a period of 1 hour. The reaction mixture is stirred at room
temperature (21-22~C) for an additional 1 hour. 3.0 L of water
are added at 21-23~C and the whole is stirred for 5-10 minutes.
The organic layer is seprated and washed with 1.5 L of lN
hydrochloric acid followed by 1.5 L of water. The resulting
organic layer is washed sequentially with 1.5 L of 5% aqueous
sodium bicarbonate, 1.5 L of water, and 1.0 L of brine. The
organic layer is filtered in a Buchner funnel with suction to
obtain 3.93 L of a solution of Boc-(S)-prolyl-(S)-3-(2-
naphthyl)alanyl-N-benzyl-N-methylamide. This solution is held
overnight at room temperature under nitrogen for the next step.
S~EP IV (- E~U~MP~E IC)
A 12-L, 4-necked, round-bottomed flask, equipped with a
mechanical stirrer, digital thermometer, addition funnel, drying
tube, and cooling bath is charged with a solution of 337.3g of
CA 02204130 1997-04-30
W O96/18643 PCT~P95/04910
~3
hydrochloric acid gas in 1.63 L of ethyl acetate. The solution
is cooled to 6~C (internal temperature) and 3.93 L of the crude
product solution of STEP III is added over a period of 25 to 30
minutes while maint~;n;n~ an internal temperature below 20~C. The
addition funnel is washed with a total of 180 ml of ethyl acetate
in three equal portions of 60ml each and this is added to the
reaction mixture. The mixture is warmed to room temperature (22-
23~C) and stirred at this temperature for an additional 2 hours.
The reaction mixture is concentrated under reduced pressure (40-
44~C, 80 to llOmm Hg) until 4.7 L of solvent is collected. The
resulting 0.66 L of an oil is dissolved in 1.4 L of water and
extracted with 1.0 L of ethyl acetate. The organic layer is
extracted with 0.2 L of water. The aqueous layers are combined
and transferred to a 5-L, 4-necked, round-bottomed flask,
equipped with a mechanical stirrer, digital thermometer, addition
funnel, and colling bath. The aqueous layer is cooled to 15~C
(internal temperature) using an ice-water bath and a precooled
solution (20-21~C) of 120g of sodium hydroxide in 1.2 L of water
is added to it over a period of 20-30 minutes while maintaining
an internal temperature below 18~C (pH should be 9-10). The
mixture is warmed to room temperature (21-23~C) in 10 minutes and
extracted with 3.0 L of isopropyl acetate. The organic layer is
separated and the aqueous layer extracted with a total of 1.0 L
of isopropyl acetate in two equal portions of 0.5 L each. The
combined organic layers are washed with 0.7S L of water followed
by 0.5 L of brine. The organic layer is dried over 125g of
anhydrous soldium sulfate and filtered in a Buchner funnel with
suction. The solids are washed with a total of lOOml of isopropyl
acetate in two equal portions of 50ml each to obtain 5.02 L of
(S)-prolyl-~S)-3-(2-naphthyl)-alanyl-N-benzyl-N-methylamide. This
solution is held under nitrogen for the next step.
STEP V (- E~UUMP~E 1)
A 12-1, 4-necked, round-bottomed flask, equipped with a
mechanical stirrer, digital thermometer, addition funnel,
nitrogen inlet-outlet, and cooling bath is charged with 5.02 L
CA 02204130 1997-04-30
W O96118643 PCTnEP95/04910 24
of a solution of the product of STEP IV in isopropyl acetate. The
solution is cooled to 10-11~C (internal temperature) in an ice-
water bath (bath temperature 6-7~C) and a solution of 156g of 2-
nitrophenyl isocyanate in 0.5 L of isopropyl acetate is added
over a period of 20 to 30 minutes while maint~;n;ng an internal
temperature below 17-18~C. The addition funnel is washed with a
total of 50 ml of isopropyl acetate in two equal portions of 25ml
each and this is added to the reaction mixture. The mixture is
warmed to room temperature (22-23~C) and stirred at this
temperature for an additional 1 hour. The reaction mixture is
filtered and concentrated under reduced pressure (40-45~C, 70 to
lOOmm Hg) until no more solvent distills. The ~0.64 kg of crude
product is dissolved in 0.5 L of ethyl acetate/hexane mixture
(60:40 v/v) by heating at 40~C (bath temerpature), cooled and
loaded onto a chromatography column contA;ning 8.5 kg of silica
gel. The column is eluted until the liquid level reaches the
silica gel. The flask is washed with a total of 0.9 L of ethyl
acetate/hexane mixture (60:40 v/v) in three equal portions of 0.3
l each and loaded onto the column. Each time the column is eluted
until the li~uid level reaches the silica gel. The column is
eluted with 36.5 L of ethyl acetate/hexane mixture (60:40 v/v)
and then with 38 L of ethyl acetate. Fractions 16-24 containing
the product are combined and solvents evaporated (39-44~C, 70-
llOmm Hg) until no solvent distills. The resulting oil is
suspended in 1.8 L of ethanol (190 proof) and solvents evaporated
~39-44~C, 70-llOmm Hg). The residue is dissolved in 3.1 L of
ethanol (190 proof) by heating (bath temperature 40-45~C). The
resulting 3.6 L solution is cooled to 29-30~C (internal
temperature) and added to 13 L of water, which is precooled to
7-8~C (internal temperature, bath temperature is O to -2~C) in
a 12-L, 4-necked, round-bottomed flask, equipped with a
mechanical stirrer, digital thermometer, addition funnel,
nitrogen inlet-outlet, and cooling bath, over a period of 30
minutes while maintaining an internal temperature of 7-9~C. The
addition funnel is washed with a total of 10 ml of ethanol (190
proof) in two equal portions of 50 ml each and this is added to
the suspension. The suspension is stirred at the same temperature
CA 02204130 1997-04-30
W O 96/18643 PCTnEP95/04910
for an additional 35 minutes and the solid collected by
filtration over a polypropylene pad filter in a Buchner funnel
with suction. The solid is washed with a total of 3 L of water
in three equal portions of 1 L each. The solid is dried in a kilo
plant SS-vacuum tray dryer on a polyethylene liner sheet in a
tray at 42-43~C (6.18 psia or ca. 319mm Hg) to obtain a constant
weight (44 hours) of the product compound 2-nitrophenylcarbamoyl-
[(S)-prolyl]-[(S)-3-(2-naphthyl)alanyl]-N-benzyl-N-methylamide
as a yellow solid. Purity: 99.4% (by HPLC 951068) [a]2QD: -59.8~
(c=l,methanol).
Compounds of formula I and their pharmaceutically acceptable acid
addition salts, hereinafter referred to collectively as "AGENTS
OF THE INVENTION", exhibit tachykinin antagonist activity. More
particularly AGENTS OF THE INVENTION exhibit potent antagonist
activity at the NK-l tachykinin (substance P) receptor. AGENTS
OF THE INVENTION are accordingly useful as pharmaceuticals, e.g.
as hereinafter further set forth.
Binding affinity for the NK-l receptor may be demonstrated by
ability to displace [3H]-substance P binding, e.g. as indicated
by the following test method:
TEST I
DisDlacement of ~3Hl-substance P bindinq from mebranes from Cos-7
cells transfected with cloned human NK-l rece~tor (hNK-IR).
Pre~aration of membranes containinq hNK-lR
Transient expression of recombinant DNA in Cos-7 cells and the
subsequent harvesting of the cells is performed analogously to
standard techniques (Sambrook et al., 1989; Kriegler 1990).
Membranes are prepared from the transfected Cos-7 cells by
homogenisation at 10 000 rpm for 30 seconds, using a Kinematica
homogeniser. The resultant suspension is centrifuged for 30 min.
CA 02204130 1997-04-30
W O96/18643 PCT/~9s~ 9lo
26
at 28 000 xg. The pellet is washed a further two times by
resuspension in Tris-HCl (50 mM, pH 7.4) and re-centrifugation.
The final pellet is resuspended at 2-8 mg protein/ml in Tris-HCl
(50 mM, pH 7.4), cont~;n-ng 5% glycerol and 500 ul aliquots are
frozen rapidly on dry-ice.
~3Hl Substance P bindin~ to hNK-lR rece~tor containin~ membranes
Membranes prepared as above are maintained in suspension at
-70~C. Binding assays are performed in 1.2 ml micronic
polypropylene tubes containing in a final volume of 0.5 ml:
binding buffer (composition in ,ugm~l: chymostatin, 2; leupeptin,
4; bacitracin, 40, 2mM MnCl2, 0.1% bovine serum albumin, 20mM
Hepes, pH7.4); 400ul membrane suspension (0.019 + 0.003 mg
protein per tube); 50 ul 6 nM [3H]Substance P and 50 ul 50%
dimethyl sulfoxide (to define total), 50ul CP96,345 (Snider et
al., 1991) (lOuM) (to define non-specific binding) or 50 ul
concentration of test compound at varying. 10 mM stocks are made
of test compounds in 100% dimethyl sulfoxide (DMSO). This stock
is further diluted to lmM in 50% DMSO before use. Six
concentrations of each test compound are used to give inhibition
curves. All assays are done in triplicate. Specific binding to
NKl receptors is defined as the difference between that found in
total bound tubes and that found in non specific binding tubes.
Reaction is initiated with the addition of the radioligand and
incubated at 24~C for 45 minutes. Reaction is terminated by the
addition of 500ul of ice cold Tris-HCl buffer (50 mM, pH 7.4).
The binding mixture is rapidly filtered over Whatman GF/B filter
sheets ~pre-soaked in 0.3% polyethyleneimine for 2-3 hours at
room temperature). The tubes and filters are washed 6 times with
lml of ice cold wash buffer. Radioactivity bound to the filters
is estimated using liquid scintillation in a Canberra Packard
TopCount. Microscint-40 is the liquid scintillant used. Binding
parameters are calculated by the method of Munson and Rodbard,
1980 using LIGAND.
Initial protein experiments with human NK-1 receptor transfected
-
CA 02204130 1997-04-30
W O96/18643 ~CTAEP95/04910
27
Cos-7 cell membranes show that the specific binding of [3H]
Substance P increase in parallel with protein concentration up
to 80-100 lug/assay tube. Routinely the protein concentration is
19 + 3 ~ug/assay tube. At this concentration, the specific binding
of [3H] Substance P is routinely >70% of total binding and 3% of
total radioactivity added to the incubation medium.
The association of [3H] Substance P to human NK1 receptor/Cos-7
membranes is rapid, reaching equilibrium at 20 min. and being
stable upto 90 min. at room temperature. Binding is measured at
45 minutes in all subsequent assays.
Saturation curves for [3H] Substance P binding to human NKl
receptor/Cos-7 cell membranes are measured after 45 min. of
incubation at room temperature. The equilibrium dissociation
constant (KD = 85 + 12 pM) and number of binding sites (B~ = 537
+ 139), is estimated by non-linear iterative curve-fitting of at
least three data sets, simultaneously, for each transfection,
using LIGAND (Munson et al., 1980) and the arithmetic mean
calculated across all ten transfections.
References:
Kriegler et al. (1990): Gene Transfer and Expression. A
laboratory manual, Stockton Press.
Munson et al. (1980): Anal. Biochem. 107:220.
Sambrook et al.: (1989) Molecular Cloning: A laboratory manual
(2nd edition), Cold Spring Harbour Laboratory Press. Refs.
Snider et al. (1991): Science 251: 435-436.
AGENTS OF THE INVENTION are active in displacing [3H] Substance
P in this test method at concentrations of the order of from Ki
= about 0.01 to about 10.0 nM.
Pharmacological, e.g. analgesic, utility of AGENTS OF THE
INVENTION as NK-1 receptor antagonists can also be demonstrated
in accordance with standard test models for examples as follows:
CA 02204130 1997-04-30
W O96/18643 PCT~EPgS/04910
28
TRCT T I: ~yp ~ T.t~R.CT2- Mr~nlZT.
Test groups of 6 male Dunkin-Hartley guinea pigs (ca. 250g)
receive 100~1 of 1% carrageenan injected intraplantar.
Mechanical hyperalgesia is measured employing a Ugo Basile
Analgesymeter (250g max. applied to the paw) and the withdrawal
threshold determined as the first signs of distress in the
animal. Thermal hyperalgesia is determined by placing ~nim~ls in
a perspex box, applying ramp heat stimulus to the plantar surface
of the paw and measuring the latency to paw withdrawal
[Hargreaves et al., Pain 32, 77-88 (1988)]. Withdrawal thresholds
to mechanical and thermal stimuli are measured in both inflamed
and non-inflamed paws.
Thermal/mechanical hyperalgesia is measured 24 hours after
carrageenan injection. Test substance, i.e. AGENT OF THE
INVENTION in 10% DMSO in tragacanth (1%), is then administered
p.o. at varying dosage and thermal/mechanical hyperalgesia re-
measured after a further 3 hours.
In the above test method AGENTS OF THE INVENTION are found to be
active in reducing mechanical hyperalgesia at dosages of the
order of from about 0.1 to about 5.0 mg/kg p.o. and thermal
hyperalgesia at dosages of the order of from about 0.5 to about
5.0 mg/kg p.o..
AGENTS OF THE INVENTION are accordingly useful as
pharmaceuticals, e.g. as tachykinin particularly NK-1 (substance
P), antagonists, e.g. for the treatment of diseases or clinical
conditions characterised by or having an aetiology comprising
excessive or undesirable substance P mediated activity.
In particular they are useful as analgesics or anti-nociceptive
agents for the treatment of pain of various genesis or aetiology.
They are also useful as anti-inflammatory or anti-oedemic agents
for the treatment of inflammatory reactions, diseases or
conditions.
CA 02204130 1997-04-30
W O96/18643 PCTnEP95/04910
29
In relation to their analgesic activity and in contrast with
other tachykinin, e.g. NK-l, antagonists known from the art,
AGENTS OF THE INVENTION have surprisingly been found to have
marked or enhanced activity following oral administration. They
have also and in contrast with other tachykinin, e.g. NK-l,
antagonists known from the art, been found to have marked anti-
nociceptive action upon the central nervous system following
systemic administration, i.e. they readily penetrate the CNS.
Having regard to their analgesic/anti-inflammatory profile,
AGENTS OF THE INVENTION are in particular useful for the
treatment of inflammatory pain, hyperalgesia and, especially
chronic pain, e.g. severe chronic pain. They are, for example
useful for the treatment of pain, inflammation and/or oedema
consequential to trauma, e.g. burns, sprain, fracture or the
like, as well as surgical intervention, e.g. for the treatment
of post-operative pain. They are further useful for the treatment
of inflammatory pain of diverse genesis, e.g. for the treatment
of arthritis and rheumatic diesease, teno-synovitis, vasculitis,
and rheumatic joint pain, e.g. rheumatid arthritis, as well as
for the treatment of gout.
AGENTS OF THE INVENTION are further useful for the treatment of
pain associated with angina, renal or billiary colic and
menstruation.
AGENTS OF THE INVENTION are also useful for the treatment of
pain associated with migraine. They are further useful as anti-
ematic agents, for the treatment of emesis, e.g. emesis
consequential to chemotherapy, poisons, pregnancy or migraine,
as well as for the treatment of incontinence and gastrointestinal
disorder such as retard emptying of the stomach, dyspepsia,
esophageal reflux and flatulence.
AGENTS OF THE INVENTION are further useful in the treatment of
chronic or obstructive airways disease, e.g. for the control or
prevention of bronchial oedema, pulmonary mucosal secretion or
CA 02204130 1997-04-30
W O96/18643 PCTnP9S/04910
airways hyperreactivity, e.g. for use as therapeutic or
prophylactic agents in the treatment of asthma. AGENTS OF THE
INVENTION are useful for the treatment of atopic and non-atopic
asthma, e.g. for the treatment of allergic asthma, exercise
induced asthma, occupational asthma, asthma following bacterial
infection and drug-induced, e.g. asprin induced, asthma as well
as of wheezy infant syndrome.
Further inflammatory or obstructive airways diseases to which the
present invention is applicable include pneumoconiosis (an
inflammatory, commonly occupational, disease of the lungs,
frequently accompanied by repeated inhalation of dusts) of
whatever type or genesis, including, for example, aluminosis,
anthracosis, asbestosis, chalicosis, ptilosis, siderosis,
silicosis, tabacosis and, in particular, byssinosis.
Yet further inflammatory or obstructive airways diseases and
conditions for which the ACTIVE AGENTS may be used include adult
respiratory distress syndrome (ARDS), chronic obstructive
pulmonary or airways disease (COPD or COAD), and bronchitis.
ACTIVE AGENTS may also be used for the treatment of allergic and
vasomotor rhinitis.
AGENTS OF THE INVENTION are further indicated for use in the
treatment of
- disorders of the central nervous system, in particular anxiety
states, for example in the treatment of anxiety, depression,
psychosis, schizophrenia, panic attack, phobias such as
agrophobia, stress related somatic disorders and addiction
disorders such as alcoholism or cocaine abuse;
- neurodegenerative disorders such as dementia, including senile
dementia, Alzheimer's disease and Down's syndrome;
- demyelinating diseases such as MS, ALS and other
neuropathological disorders, for example peripheral
CA 02204130 1997-04-30
W O96118643 PCTnEP95/04910 31
neuropathy, e.g. diabetic and chemotherapy induced neuropathy;
AGENTS OF THE INVENTION are yet further indicated for use in the
treatment of diseases or conditions associated with dysfunction
of the immune system, e.g. autoimmune diseases, in particular
where these are associated with inflammatory, oedemic or
nociceptive event. Particular diseases or conditions in this
category include, for example autoimmune haematological disorders
(including e.g. haemolytic anaemia, aplastic anaemia, pure red
cell anaemia and idiopathic thrombocytopenia), systemic lupus
erythematosus, polychondritis, sclerodoma, Wegener
granulamotosis, dermatomyositis, chronic active hepatitis,
myasthenia gravis, psoriasis, Steven-Johnson syndrome, idiopathic
sprue, autoimmune inflammatory bowel disease (including e.g.
ulcerative colitis and Crohn's disease) endocrine opht~lmopathy,
Graves disease, sarcoidosis, multiple sclerosis, primary billiary
cirrhosis, juvenile diabetes (diabetes mellitus type I), uveitis
(anterior and posterior), keratoconjunctivitis sicca and vernal
keratoconjunctivitis, interstitial lung fibrosis, psoriasis,
psoriatic arthritis and glomerulonephritis (with and without
nephrotic syndrome, e.g. including idiopathic nephrotic syndrome
or m;n;m~l change nephropathy) as well as vasculitis. AGENTS OF
THE INVENTION may also be useful as immuno-suppressant or
immunosuppressive adjuvents, e.g. for use in conjunction with
other immunosuppressive, e.g. cyclosporin or immunosuppressive
macrolide therapy, for the suppression of allograft rejection,
for example following allogenic e.g. allogenic kidney, liver,
corneal, heart, lung or heart-lung transplantation.
AGENTS O~ THE INVENTION are yet further indicated for use in the
treatment of allergic diseases or conditions, e.g. of the skin,
eye, naso-pharynx or gastro intestinal tract, in particular where
such disease or condition is associated with inflammatory,
oedemic or nociceptive reactions. Examples of such diseases or
conditions include, for example, exzema, hypersensitivity
disorders such as poison ivy allergy, contact dermatitis,
conjunctivitis, vernal conjunctivitis, keratoconjunctivitis
CA 02204130 1997-04-30
W O96/18643 PCT~P95/04910
32
sicca, urtharia and other eczemoid dermatoses.
AGENTS OF THE INVENTION are also useful in the treatment of
disorders of blood flow caused by vasodilation and vasospastic
diesease such as angina, migraine and Reynaud's disease.
In addition to the foregoing AGENTS OF THE INVENTION have also
been found to possess P-glycoprotein blocking activity. AGENTS
OF THE INVENTION are accordingly further indicated for use as
adjuvent or co-therapy with drug substances of other therapeutic
category for example:
- for increasing or enhancing effectiveness of, or increasing or
enhancing sensitivity to, other chemotherapeutic drug therapy,
in particular anti-microbial (e.g. anti-bacterial, anti-viral,
antifungal or anti-protozoal) chemotherapy, chemotherapy for
AIDS and, especially, anti-cancer or anti-tumor (e.g. anti-
neoplastic or cytostatic) chemotherapy. They are accordingly
indicated for use, e.g. as a means of reducing regular
chemotherapeutic dosage levels, for example, in the case of
anti-neoplastic or cytostatic drug therapy, as a means of
decreasing overall drug toxicity and, more especially, as a
means of reversing or reducing resistance, including both
inherent and acquired resistance, to chemotherapy;
- to enable or potentiate other drug therapy directed at the
central nervous system, e.g. to enhance drug penetration of
the blood-brain barrier, for example to enable, increase or
enhance other psychotropic drug therapy, e.g. for
administration in conjunction with other analgesic or
psychomotor stimulatory or depressant agents or agents, for
example, for treatment of neurodegenerative disease including
Parkinson's disease, Alzheimer's disease and so forth as well
as chemotherapy to be directed at tumor of the brain;
- as antiparasitic, particularly antiprotozoic, agents, e.g.,
particularly against organisms of the genera Toxo~lasma (e.g.,
Toxoplasma gondii) and Plasmodia (e.g., Plasmodium
falciparum).
CA 02204130 1997-04-30
W O 96/18643 PCT~Pg5/04910
33
For the above indications the dosage of AGENTS OF THE INVENTION
will, of course, vary depending upon, for example, the host, the
mode of administration and the nature and severity of the
condition being treated as well as the relative potency of the
particular AGENT OF THE INVENTION employed. However, in general,
satisfactory results in ~nim~ls, e.g. for the treatment of pain,
migraine and emesis, are indicated to be obtained at daily
dosages of from about 0.1 to about 10 mg/kg p.o.. In larger
m~mm~ls, for example humans, an indicated daily dosage is in the
range of from about 7.0 to about 700 mg/day p.o., e.g. ca.
100mg/day p.o. conveniently ~ministered once or in divided doses
up to 4 x per day or in sustained release form, e.g. for the
treatment of pain, migraine and emesis. Oral dosage forms
accordingly suitably comprise from about 1.5 to about 150 or 700
mg e.g. from about 25 to 100 mg AGENT OF THE INVENTION admixed
with an appropriate pharmaceutically acceptable diluent or
carrier therefor.
Having regard to their relatively low solubility AGENTS OF THE
INVENTION for oral administration are suitably formulated in a
composition comprising a hydrophilic phase (e.g. propylene
glycol/ethanol) a hydrophobic phase (e.g. vegetable oil mono-di-
triglycerides such as commercially available under the registered
trade mark MAISINE) and a surfactant (e.g. a polyoxyhydrogenated
vegetable oil such as commercially available under the registered
trade mark CREMOPHOR). Formulations for i.v. administration may
be prepared by dissolution of the selected AGENT OF THE INVENTION
in ethanol together with an appropriate surfactant, e.g.
CREMOPHOR RH 40. The following example is illustrative of the
preparation of galenic forms suitable for oral administration:
COMPONENT QUANTITY/UNIT DOSE
1. AGENT OF THE INVENTION, e.g.
compound of EXAMPLE 1. 100.00 mg
2. Propylene glycol 94.70 mg
3. Corn oil-mono-di-triglycerides,
e.g. MAISINE 319.90 mg
CA 02204130 1997-04-30
WO96/18643 PCT~5/04910
34
COMPONENTQUANTITY/UNIT DOSE
4. Polyoxyl 40 hydrogenated castor oil,
e.g. CREMOPHOR RH 40 383.70 mg
5. Ethanol, dehydrated 94.70 mq
Total 993.00 mg
Component 4 is warmed at 40~C until liquified. Components 2, 3
and 5 are added and the whole mixed in conventional manner until
a clear solution is obtained. Component l in finely divided form,
e.g. compound of EXAMPLE l (free base, amorphous) pin-milled, if
required at low temperature, is added to the obtained solution
and the whole mixed until a clear solution is obtained. The
product is suitable for use as a solution for drinking.
Alternatively the composition may be put up in soft or hard-
gelatin encapsulated form, e.g. with each capsule containing 50
or l00mg component l.
AGENTS OF THE INVENTION may alternatively be administered, e.g.
topically in the form of a cream, gel or the like for example for
the treatment of conditions of the skin as hereinbefore described
or by inhalation, e.g. in dry powder form, for example for the
treatment of obstructive or inflammatory airways disease, or by
any other appropriate route, e.g by injection or infusion.
The preferred AGENT OF THE INVENTION is the product or EXAMPLE
l. In one series of experiments an established ED50 for this in
TEST II above is of the order of 0.73 + 0.09 mg/kg p.o. for
mechanical hyperalgesia and of 1.75 + 0.64 mg/kg p.o. for thermal
hyperalgesia. An estimated EDso for aspirin in the same test
method is of the order of ca. 30 mg/kg for mechanical and ca. l00
mg/kg for thermal hyperalgesia. Indicated oral dosages for the
EXAMPLE l compound as an analgesic agent will thus be of the
order of l/40th to l/50th of those clinically employed using
asprin.
A further preferred AGENT OF THE INVENTION is the product of
EXAMPLE 17. In one series of experiments in accordance with TEST
CA 02204130 1997-04-30
W O96118643 PCTAEP95/04910
II above the ED50 for this compound is found to be of the order
of 1.0 mg/kg p.o. for mechanical hyperalgesia. Indicated oral
dosages for the EXAMPLE 17 compound as an analgesic agent will
thus be of the order of 1/30th of those clinically employed using
asplrin .
In accordance with the foregoing the present invention also
provides:
1) An AGENT OF THE INVENTION for use as a pharmaceutical, e.g.
for use as an NK-1 (substance P) antagonist, for example
for use in any of the particular indications hereinbefore
set forth, in particular for use as an analgesic, anti-
inflammatory or anti-oedemic agent or for use in treating
allergic conditions or reactions, e.g. rhinitis, or in
treating emesis;
2) A pharmaceutical composition comprising an AGENT OF THE
INVENTION as active ingredient together with a
pharmaceutically acceptable diluent or carrier therefor;
and
3) A method for the treatment of any of particular indication
hereinbefore set forth in a subject in need thereof which
comprises administering an effective amount of an AGENT OF
THE INVENTION to said subject.