Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 0220816~ 1997-06-18
-- 4 --
NUCLEIC ACID BIOSENSOR DIAGNOSTICS
Background of the Invention
The present invention is directed generally to biosensors that are usefui in theidentification and analysis of biologically significant nucleic acids. The biosensors of the
present invention and their applied methods provide a means for the direct analysis of
nucleic acid hybridization and, therefore, have application to a myriad of biological fields
including clinical diagnostics.
The detection and identification of microorganisms is a problem common to
many areas of human and veterinary health. For example, the detection of pathogenic
species such as Salmonella typhimurium, Listeria monocytogenes, and Escherichia coli,
which are c~us~tive agents of major food borne epidemics, is a great concern within the
food industry with respect to the quality and safety of the food supply. In other areas of
human and veterinary health care, detection and identification of infectious diseases
caused by pathogenic microorganisms and viruses is a first step in diagnosis andtreatment. For example, it is estimated that 10-15 million office visits per year are for
the detection and treatment of three major pathogens - Chlamydia ssp., Trichomonas
vaginalis and Gradenerella vaginitis. Infections of these organisms annually effect 3.75
million, 0.75 million and 1.5 million patients, respectively.
Classic~l techniques routinely used for the detection and identification of
microorganisms are often labor intensive, involving plating procedures which require
lengthy analysis times. To illustrate, the method currently employed for the detection of
Listeria monocytogenes in food and feed commodities involves a three stage analysis.
The analysis begins with enrichment of the sample to be analyzed in a nutrient broth for
2 to 4 days. After the enrichment period, plating of the sample onto selective agar
media is done and the sample is allowed to incubate for 2 days in order to obtain
CA 0220816~ 1997-06-18
-- 5
colonies for biotyping and serotyping, which may take as long as 20 days to complete
(McLauchlin et al., 1988, Microbiology Review, 55: 578).
Detection processes based on culturing require analysis times which are too
lengthy for effective monitoring and timely intervention to prevent the spread of
biohazardous materials or treat disease. In addition, although these methods have
been improved over the last decade, the chance of obtaining false negative results is
still considerable, and many microorganisms are difficult to culture. Thus, plating/culture
methods are limited with respect to their sensitivity, specificity, and lengthy analysis
times that are required.
In order to shorten the time required to detect and identify pathogenic bacteria,
viruses and genetic diseases, rapid tests such as enzyme immunoassays (EIA) havebeen developed (Olapedo et al., 1992). Although immunoassay techniques can be very
sensitive and effective, there are practical drawbacks which have restricted the use of
these methods. Such drawbacks include the need for highly skilled personnel, lengthy
analysis and preparation times, and the large quantities of costly reagents that are
required to do such analysis.
With the advent of nucleic acid amplification techniques (the polymerase chain
reaction), the in-vitro amplification of specific sequences from a portion of DNA or RNA
is now possible. Detection of ver,v low numbers of microorganisms has been
demonstrated (Rossen et al., 1991; Golsteyn et al., 1991; Wernars, K., et al., 1991).
The polymerase chain reaction technique is sensitive and specific but involves complex
manipulations in carrying out the tests and is not particularly well-suited for large
numbers of samples. Due to the sensitivity of Polymerase Chain Reaction (PCR)
technology, special rooms or areas for sample preparation and analysis are required to
prevent contamination. In many tests PCR results must be confirmed by additionalhybridization analysis. RNAs are difficult to assay by PCR but are very important for
human viral detection. In general, PCR needs to be automated for acceptance as apractical diagnostic tool. Hybridization methods require as much as three or four days to
CA 0220816~ 1997-06-18
complete results. Although the actual hybridization step can be as short as 18 hours,
the entire detection process of a DNA/DNA hybrid can take as long as three days with a
radioisotope marker.
Thus, there is a great need for simpler, faster and more cost-effective means for
detecting specific biologically important RNA and DNA sequences in the fields ofhuman and veterinary in-vitro diagnostics, food microbiology, and forensic applications.
Biosensors developed to date begin to overcome drawbacks associated with the
current state of the art in detecting and identifying microorganisms. A biosensor is a
device which consists of a biologically active material connected to a transducer that
converts a selective biochemical reaction into a measurable analytical signal
(Thompson et al., 1984. Trends in Analytical Chemistry, 3: 173; Guilbault, 1991,Current Opinion in Biotechnology, 2: 3). The advantages offered by biosensors over
other forms of analysis include the ease of use (by non-expert personnel), low cost,
ease of fabrication, small size, ruggedness, facile interfacing with computers, low
detection limits, high sensitivity, high selectivity, rapid response, and reusability of the
devlces.
Biosensors have been used to selectively detect cells, viruses, other biologically
significant materials, biochemical reactions and immunological reactions by using
detection strategies that involve immobilization of enzymes, antibodies or otherselective proteins onto solid substrates such as quartz and fused silica (for piezoelectric
and optical sensors) or metal (for electrochemical sensors) (Andrade et al., 1990,
Biosensor Technology: Fundamentals and Applications, R. P. Buck, W. E. Hatfield, M.
Umana, E. F. Bowden, Eds., Marcel Dekker Inc., NY, pp. 219; Wise, 1990,
Bioinstrumentation: Research, Developments and Applications, Butterworth Publishers,
Stoneham, MA). However, such sensors are not widely available from commercial
sources due to problems associated with the long-term stability of the selectiverecognition elements when immobilized onto solid surfaces (Kallury et al, 1992,
CA 0220816~ 1997-06-18
Analytical Chemistry, 64: 1062; Krull et al, 1991, Journal of Electron Microscopy
Techniques,18: 212).
An altemative approach is to create biosensors with long-term chemical stability.
One such approach takes advantage of the stability of DNA. With the recent advent of
DNA probe technology, a number of seiective oligomers which interact with the DNA of
important biological species, for instance salmonella, have been identified (Symons,
1989, Nucleic Acid Probes, CRC Press, Boca Raton, FL; Bock et al., 1992, Nature,355: 564; Tay et al., 1992, Oral Microbiology and Immunology, 7: 344; Sherman et al.,
1993, Bioorganic & Medicinal Chemistry Letters, 3: 469). These have been used toprovide a new type of biorecognition element which is highly selective, stable, and can
be easily synthesized in the laboratory (Letsinger et al., 1976, Journal of the American
Chemical Society, 98: 3655; Beaucage et al., 1981, Tetrahedron Letters, 22: 1859;
Alvarado-Urbina et al.,1981, Science, 214: 270) .
Review of the Prior Art
Until recently, the only other research group in existence which has published
work done on the fluorimetric detection of nucleic acid hybridization immobilized onto
optical substrates is that of Squirrell et al. (C.R. Graham, D. Leslie, and D.J. Squirrell,
Biosensors and Bioelectronics 7 (1992) 487-493.) In this work, single-stranded nucleic
acid sequences ranging in length from 16-mer oligonucleotides to 204-base oligomers
functionalized with an aminohexyl linker at the 5' terminus were covalently attached to
optical fiber sections functionalized with 3-aminopropyl triethoxysilane via a
gluteraldehyde linkage. All investigations of nucleic acid hybridization were done by
monitoring fluorescence intensity in an intrinsic mode configuration using
complementary strands which had been previously labeled with a fluorescein moiety.
This yielded a reusable assay system in which signal generation was observed to occur
within minutes and nanomolar detection was achieved. However, this optical sensor
technology developed by Squirrell et al. does not contain a transduction element which
can transduce the binding event in a reagentless manner. For this assay to function,
CA 0220816~ 1997-06-18
-- 8
the target strands must be labeled prior to doing the assay in order for detection,
making this technique unsuitable for practical applications.
Abel and co-workers (Abel, A. P.; Weller, M. G.; Duveneck, G. L.; Ehrat, M. and
Widmer, H. M. Anal. Chem. 1996 68, 2905-2912) of Norvartis Ltd. (formerly Ciba-Geigy
Ltd.) have recently reported an automated optical biosensor system. Their deviceutilizes 5'-biotinylated-1 6-mer oligonucleotide probes bound to an optical fiber
functionalized with avidin to detect complementary oligonucleotides pre-labeled with
fluorescein moieties in a total intemal reflection fluorescence (TIRF) evanescent wave
motif similar to that of Squirrell. Each assay consisted of a 3 minute pre-equilibration,
15 minute hybridization time, 10 minute washing procedure followed by a 5 minuteregeneration cycle (chemical or themmal). A chemical denaturation scheme was
observed to be the preferred embodiment for sensor regeneration as exposure of the
oligonucleotide functionalized optical sensor to temperatures exceeding 52~C caused
irreversible damage to the device, owing to denaturation of the avidin used for
immobilization. This limitation renders the device function labile against sterilization
techniques, such as autoclaving, and also indicates that rigorous cleaning of the sensor
surface, such as by sonication, would also compromise the integrity of the sensor via
denaturation of the affinity pair used to anchor the probe oligonucleotide. In order to
detect nucleic acids not pre-labeled with fluorescein, and to overcome the limitation of
Squirrell, a competitive binding assay was employed by Abel and co-workers. Detection
of the unlabelled analyte was done by pre-treatment of the sensor with fluorescein
labeled "tracer-DNA" followed by monitoring decreases in the fluorescence intensity of
the sensor upon exposure to and subsequent displacement of the tracer-DNA by
complement analyte nucleic acid. The dose-response curves reported by Abel et al.
show a detection limit of 132 pmol (8 x 1013 molecules) for this detection strategy.
However, in addition to high detection limits and the inability of the device to withstand
sterilization, this device cannot be classified as a biosensor technology due to the
necessity for external treatment with tracer-DNA in order to achieve transduction.
. CA 0220816~ 1997-06-18
g
The prior art with respect to patent literature contains many examples of "sensor"
devices which are based on nucleic acid molecules immobilized on waveguide supports
and transduction strategies based on evanescent excitation. The technology of Gerdt
and Herr (David. W. Gerdt, John. C. Herr "Fiber Optic Evanescent Wave Sensor forImmunoassay", United States Patent No. 5,494,798) describes detection of nucleic acid
hybridization based on alterations in the quantity of light transmitted from one optical
fiber in a coupled fiber system (similar to that of a Mach-Zehnder interferometer~ to the
second fiber of the waveguide system. The quantity of light transferred is a function of
the refractive index of the media on or surrounding the waveguides. Refractive index
alterations affect the penetration depth of the evanescent wave emitted from the first
waveguide into which optical radiation is launched. This standing wave of
electromagnetic radiation subsequently propagates into (and thus transfers optical
radiation to) the second waveguide. Therefore, the device is sensitive to refractive
index alterations occurring within a volume surrounding the first waveguide with a
thickness of ca. one wavelength of the light propagating within that waveguide. One of
the arms of the waveguide may be functionalized with immobilized nucleic acid
molecules which serves to provide selective binding moieties. The change in refractive
index of the thin film of nucleic acids on the first waveguide upon the occurrence of
hybridization with target nucleic acid sequences alters the quantity of light transferred to
the second waveguide, thereby providing a means of signal transduction. Hybridization
events may then be identified based on changes in the output ratios of the two
waveguide arms in the coupled fiber system. One limitation of this technology lies in
the fact that any alterations in refractive index near the surface of the waveguides will
provide alterations in the output ratios of the two fibers. Therefore, non-specific binding
events (such as protein adsorption) will provide false positive results.
In order to avoid the problem of interferents providing false positive results, a
transduction strategy which is sensitive to the structure of the binding pair (i.e.
recognition element and target) is required. The technologies of Fodor, Squirrell (David
James Squirrell "Gene Probe Biosensor Method" International Application Number
PCT/GB92/01698, International Publication Number WO 93/06241, International
CA 0220816~ 1997-06-18
-- 10 --
Publication Date: 1 April 1993.), Sutherland etal. (Ranald Macdonald Sutherland, Peter
Bromley and Bemanrd Gentile "Analytical Method for Detecting and Measuring
Specifically Sequenced Nucleic Acid." European Patent Application Number
87810274.8, Publication Number 0 245 206 A1, Date of Filing: 30 April 1987.),
l lirschfeld (Tomas B. Hirschfeld, "Nucleic Acid Assay Method" United States Patent
Number 5,242,797, Date of Patent: 7 September 1993.), and Abel et al. (Andreas P.
Abel, Michael G. Weller, Gert L. Deveneck, Markus Ehrat, and H. Michael Widmer,
Analytical Chemistry, 1996, 68, 2905-2912.) overcome this limitation by using
fluorescent probes which associate with the binding pair or are attached to selective
binding moieties capable of binding to a portion of the binding pair. These inventions
provide methods to measure nucleic acid hybridization on waveguide surfaces based
on evanescent excitation and TIRF. In each embodiment, an oligonucleotide probe
capable of selective binding to a target sequence is covalently immobilized on awaveguide surface. For the cases of Squirrell and Abel et al., each define two
preferred embodiments for the detection of hybridization events. The first embodiment
of Squirrell and Abel et al. are essentially identical wherein the target nucleic acid is
functionalized with a fluorescently detectable agent (by chemical or enzymatic methods)
as a first step prior to detection. Upon hybridization between the labeled target and
immobilized nucleic acid, the fluorescent agent is then bound in close proximity to the
waveguide surface where it may be excited by evanescent wave formation and
emission from the fluorophore collected and quantitatively measured. In the second
preferred embodiment of Squirrell, hybridization between the immobilized
oligonucleotide and the target sequence is first done. Subsequent to the first
hybridization event, a fluorescently labeled oligonucleotide present in the system may
then undergo hybridization with all or a portion of the remainder of the target sequence
not hybridized with the immobilized sequence. The binding of the third (labeled)oligonucleotide provides a fluorescent species bound in close proximity to the
waveguide which may furnish transduction via evanescent excitation and collection of
the emitted radiation. In the second embodiment of Abel et al., a method for thedetection of nucleic acids not pre-labeled with a fluorescent moiety via a competitive
binding assay is described. Detection of the unlabelled analyte was done by first pre-
CA 0220816~ 1997-06-18
-- 11 --
treating the optical sensor with immobiiized probe nucleic acid with fluorescein labeled
"tracer-DNA". The quantity of tracer-DNA may be monitored via the evanescent
excitation and collection motif. Binding of the analyte could be followed by monitoring
decreases in the fluorescence intensity from the sensor as a function of the
displacement of the tracer-DNA via competitive binding with non-fluorescent analyte
nucleic acid in a dose-response convention.
In the methods of Sutherland et al. and Hirschfeld, transduction of hybridization
events is provided by fluorescent intercalating dyes (e.g. ethidium bromide). Following
hybridization between the single-stranded target and immobilized probe nucleic acids,
intercalant fluorescent dye molecules from solution insert into the base stacking regions
of the immobilized double-stranded nucleic acid. An increase in the fluorescencequantum efficiency, fluorescence lifetime, stokes shift of the fluorescent intercalant
probes often occurs upon association with double-stranded nucleic acid. It is claimed
by the inventors that these enhanced features may be monitored by evanescent
excitation and collection of fluorescence emission.
Fodor et al. have employed light-directed chemical synthesis to generate
miniaturized, high density arrays of oligonucleotide probes. DNA oligonucleotide arrays
have been fabricated using high-resolution photolithography in combination with solid-
phase oligonucleotide synthesis. This form of DNA chip technology may be used for
parallel DNA hybridization analysis, directly yielding sequence information fromgenomic DNA segments. Prior to sequence identification, the nucleic acid targets must
be fluorescently labeled, either prior to or after hybridization to the oligonucleotide array,
via direct chemical modification of the target strand or by use of an intercalant dye
subsequent to hybridization on the DNA chip. The hybridization pattern, as determined
by fluorescence microscopy, is then deconvolved by appropriate chemometric
processing to reveal the sequence of the target nucleic acid. Rather than focusing on
selective detection of trace quantities of a particular nucleic acid sequence, this
technology has focused on sequence analysis of nucleic acids in suitabiy high copy
number so as to sufficiently occupy the oligonucleotide array.
CA 02208l6~ l997-06-l8
- 12 -
Notwithstanding the indubitable accomplishments of the aforementioned prior
art, there yet exists limitations in these technologies for which further improvements are
most desirous. Although the strategies employed by Sutherland et al. and Hirschfeld
overcome the limitations of Gerdt and Herr with regard to signal origin and the
generation of false positive results, these assay methods are limited by the amount of
signal which can be generated by evanescent excitation. For multimode waveguides,
less than 0.01% of the optical radiation carried within the waveguide is exposed to the
outer medium in the form of an evanescent wave (R.B. Thompson and F.S. Ligler,
"Chemistry and Technology of Evanescent Wave Biosensors" in Biosensors with
FiberoPtics, Eds.: Wise and Wingard, Humana Press Inc., New Jersey,1991, pp.111-138.). In the case where monomodal waveguides are used, ca.10% of the radiat~on
carried by the waveguide is exposed to the outer medium in the form of an evanescent
wave (David. W. Gerdt, John. C. Herr "Fiber Optic Evanescent Wave Sensor for
Immunoassay", United States Patent No: 5,494,798). In the classic total internalreflection fluorescence (TIRF) evanescent wave configuration, the critical angle (~c) for
the waveguide/solution interface (~W~s) is larger than ~c for the waveguide/biological
film interface (~ W/B~, only the evanescent component of the propagated radiation will
enter the biological film. The principle of optical reciprocity states that light coupled back
into a waveguide as a plane wave will be in the same way as the primary process when
a plane wave generates an evanescent wave (Ranald Macdonald Sutherland, Peter
Bromley and Bemanrd Gentile "Analytical Method for Detecting and Measuring
Specifically Sequenced Nucleic Acid" European Patent Application Number
87810274.8, Publication Number 0 245 206 A1, Date of Filing: 30 April 1987, p.13.).
Thus, for the fluorophores excited by evanescent waves created from modes
propagating at or near ~ w/s, none of the fluorescence emission can be coupled back
into the waveguide in the same propagation mode as ~ w/s would be > 90~ (U.J. Krull,
R. S. Brown and E.T. Vandenberg, "Fiber Optic Chemoreception" in Fiber OPtic
Chemical Sensors and Biosensors, vol.2, Ed. O.S. Wolfbeis, CRC Press, Boca
Raton,1991, pp.315-340.). Hence a large portion of the signal would be lost to the
surroundings for systems in which fluorescence emission originates from thin films of a
CA 02208l6~ l997-06-l8
- 13 -
lower refractive index than that of the waveguide onto which they are immobilized. It
has been shown by Love et al that under optimal conditions, only 2% of the lightemitted by the fluorophore in the medium of lower refractive index may be captured and
guided by the fiber tW.F. Love, L.J. Button and R.E. Slovacek, "Optical Characteristics
of Fiberoptic Evanescent Wave Sensors: Theory and Experiment" in Biosensors withFiberoptics Eds.: Wise and Wingard, Humana Press Inc., New Jersey, 1991, pp.139-180.). By using the chemistries (as disclosed in this patent application) for attaching
linker molecules onto optical waveguide supports (preferably optical fibers) and an
automated DNA synthesizer, control over the orientation and a wide range of
oligonucleotide packing densities on the waveguide is afforded. In this way,
immobilized films of oligonucleotides of desired refractive index may be constructed on
waveguide supports so that the oligonucieotide film is made to be an extension of the
waveguide. This intrinsic mode of operation provides a highly efficient means of signal
generation and collection where fluorescence excitation and emission occur within the
waveguide itself, providing an expected enhancement in sensitivity and lowering of
detection limits by six orders of magnitude.
The second major improvement provided by our technology is the use of
fluorescent dyes tethered to or otherwise associated with the immobilized
oligonucleotide. Thompson and Krull ({a} M. Thompson and U.J. Krull, Trends in
Analytical Chemistry, 3 (1984) 173-178. {b} M. Thompson and U.J. Krull, Analytical
Chemistry, 63 (1991) 393A-405A.) teach that biosensors may be defined as deviceswhich consists of a biorecognition element and a transduction element. The
biorecognition element may be a biological material capable of participating in highly
selective binding to a target, usually a biologically significant molecule. The transduction
element converts the selective binding reaction into a measurable analytical signal. The
transduction strategy of Gerdt is too non-selective for the technology to be classified as
a biosensor whereas the devices of Fodor, Squirrell, Abel et al., Sutherland et al. and
Hirschfeld do not contain a transduction element at all. In addition to the requirement for
external reagent treatment, in the cases of Fodor, Sutherland et al., and Hirschfeld,
there also exists the extra shortcoming that all intercalant dyes are known or suspected
CA 02208l6~ l997-06-l8
- 14 -
mutagens. Therefore, the troublesome issues of collection and disposal of hazardous
chemical waste exists subsequent to each analysis. By associating the transduction
element with the biorecognition element, the device may function without the need for
extemal reagent treatment and obviated the need to collect and dispose of hazardous
waste. Such a technology then readily lends itself to automated and in-line analysis and
precludes the need for skilled technicians to partake in the analysis procedure or
disposal of waste (provided the sample itself is not biohazardous).
The other advantage provided by the incorporated dye is internal calibration.
More specifically, three key advantages may be realized: 1 ) the associated dye
provides a means to detemmine the quantity of fluorophore and immobilized nucleic acid
on the waveguide; 2) the fluorophore in the presence of single-stranded nucleic acid
provides a baseline signal to which all signals can be referenced, hence providing
meaningful analytical data; and 3) the useful lifetime of the device can be determined
from alterations in the background fluorescence signal from the incorporated
fluorophore over time. Therefore, by including the associated fluorescence
transductiori unit, an internal reference marker and diagnostic tool for the device status
is included as an integral part of the optical biosensor.
CA 02208l6~ l997-06-l8
- 15 -
Summary of the Invention
The present invention concems biosensors for direct detection of nucleic acids
and nucleic acid analogs. The device comprises a light source, a detector, and an
optical element for receiving light from the source and conveying it to an interaction
surface of the optical element. A nucleic acid or nucleic acid analog for a particular
nucleic acid sequence or structure (i.e. which is complementary to the target nucleic
acid(s)), is immobilized onto the interaction surface of the optical element. Fluorescent
ligands are provided that will bind into or onto the hybridized nucleic acid complex and
fluoresce when stimulated by the light source. Subsequent to excitation by
electromagnetic radiation of suitable wavelength bound within the optical element, the
resultant fluorescence is collected within the optical element and guided to the detector
to signal that the target nucleic acid(s) has complexed with the immobilized probe and
thus indicate the presence of the target in the sample. An interaction surface is defined
to mean a surface of the optical element on which nucleic acid is immobilized, and at
which the fluorescent molecules interact with the light.
This invention provides biosensors in which the interaction surface is
functionalized with nucleic acid probe sequences such that the index of refraction of the
immobilized layer (Substrate Linker / Nucleic Acid / Fluorescent Ligand) is equal to or
greater than the refractive index at the surface of the waveguide such that the organic
coating becomes an extension of the waveguide. The index of refraction of the
immobilized layer is dependent, at least in part, on the loading of immobilized
molecules and linkers on the surface and the chemical nature of the immobilized
molecules and any linkers.
Preferred biosensors which offer high-sensitivity and low-detection limits may be
realized by activating the interaction surface of an optical element with substrate linker
molecules of at least about 25A (Angstrom) in length followed by attachment of aselected probe nucleic acid sequence to that linker. (A probe nucleic acid is, at least in
CA 02208l6~ l997-06-l8
- 16 -
part, complementary to a target nucleic acid.) The preferred method for attachment of
the probe nucleic acid to the substrate linker is by in-situ synthesis of the nucleic acid
sequence onto the linker temminus using solid-phase nucleic acid synthesis methods or
routine modifications of thereof. Such methods of in-situ synthesis are particularly
useful for immobilization of nucleic acids of 50 or fewer bases and more particularly
useful for nucleic acids of 30 or fewer bases.
The fluorophore may be tethered to the immobilized DNA, for example, by use of
a hydrocarbon tether. The use of tethered probes can significantly reduce biosensor
response time as the response mechanism is not diffusionally controlled. The
associated fluorophore provides for internal calibration of optical source intensity and
detector drift. It also provides for calibration of photobleaching, and provides for
intemal calibration by monitoring bound against free dye by use of, for example, time-
resolved fluorescence measurements.
The optical element preferably comprises an optical waveguide which also
conveys the fluorescent light to the detector. The optical waveguide preferably conveys
the emitted light by total internal reflection to the detector. The optical waveguide can
comprise an optical fiber, a channel waveguide, or a substrate that confines light by
total internal reflection. The fluorescent molecules preferably provide sufficient Stokes
shift such that the wavelength of the light source and the wavelength of the fluorescent
light are easily separated. The fluorescent molecules can be provided in a solution in
which the optical element is immersed, or by a tether to the nucleic acid that is
immobilized to the linker.
In the practice of the present invention, the light source can be any suitable
source such as a gas laser, solid state laser, semiconductor laser, a light emitting
drode, or white light source. The detector can be any suitable detector such as a
photomultiplier tube, an avalanche photodiode, an image intensifier, multi-channel
plate, or semiconductor detector. The biosensor system can be a multi-wavelength,
CA 02208l6~ l997-06-l8
- 17 -
multi-fluorescent system. The light coupling of the system can also be modified to allow
a multitude of disposable biosensors to be analyzed either sequentially or in parallel.
The biosensor system of the present invention can be constructed and used to
detect each of a mixture of target nucleic acids (for example, Chlamydia and Gonorrhea
in urogenital infections or E. coli and Salmonella during food processing). This may be
done by using a plurality of fluorophores (which, for example, fluoresce at different
wavelengths), each of which is tethered to an immobilized nucleic acid probe that is
characteristic of or specific for detection of a given species or strain. In this example,
the observed wavelength(s) of fluorescence emission will then be specific for
hybridization of a given target nucleic acid to its complementary immobilized probe.
The biosensors of the present invention have an improved detection limit and
sensitivity with respect to the prior art and are shown to be stable over prolonged
storage and severe washing and sterilization conditions. Sensors stored over 1 year in
vacuo, in 1:1 ethanol/water solutions, absolute ethanol, or dry at -20~C provide identical
response characteristics to those freshly prepared. Adsorbed fluorescent contaminants
accumulated through storage can be removed (as confirmed through fluorescence
microscopy investigations) by sonicating the biosensors in 1:1 ethanol/water where the
sensitivity of the device has consistently been observed to increase by a factor of c.a.
2.5 from this pre-treatment with respect to that of freshly prepared biosensors not
cleaned before use. Unlike those of the prior art (e.g. Abel et al.), the optical
biosensors of the present invention have also shown to be thermally stable wherein
device function is maintained after sterilization by autoclaving (20 minutes, 120~C, 4
atmospheres over-pressure). The ability to clean and sterilize a biosensor device so
that it may be usable in an on-line configuration and/or in clinical applications is a
significant advantage yet realized only by the technology reported herein. Biosensors of
this invention also allow for more rapid sample analysis with improved response time for
signal generation.
CA 02208l6~ l997-06-l8
- 18 -
The present invention also provides a recyclable or disposable biosensor for
detecting a target nucleic acid, which biosensor includes an optical element forreceiving and conveying light to an interaction surface of the optical element and
nucleic acid, for a particular nucleic acid sequence which is complementary to the target
nucleic acid, immobilized onto the interaction surface of the optical element. The
recyclable or disposable biosensor preferably comprises an optical waveguide, which
preferably conveys the light by total intemal reflection to the interaction surface of the
optical waveguide when the organic coating is of equal or higher refractive index in
comparison to the surface of the waveguide. The optical waveguide preferably
comprises an optical fiber. Fluorescent molecules are provided in a solution in which
the recyclable or disposable biosensor is immersed that will bind upon hybridization of
the immobilized nucleic acid with complementary target nucleic acid and fluoresce
when stimulated by light. Altematively, the fluorescent molecules are provided bound
by a tether to the immobilized nucleic acid.
The present invention provides biosensors for direct analysis of nucleic acid
hybridization by use of an optical substrate such as an optical wafer or an optical fiber,
and nucleic acids or nucleic acid analogs which have been immobilized onto the optical
substrate. Generation of a fluorescence signal upon hybridization to complementary
nucleic acids and nucleic acid analogs in a sample may be achieved in a number of
different ways. Biosensors of this invention are sufficiently sensitive to directly detect
very small quantities of target nucleic acids in a sample without the need to employ
nucleic acid amplification methods such as PCR techniques. Biosensors of this
invention can have detection limits for target nucleic acids below 1 o6 molecules.
The optical biosensor comprises nucleic acid strands or nucleic acid analogs of a
specific selected sequence immobilized onto activated optical supports. The selected
irrimobilized sequences are capable of binding to target sequences, including
sequences characteristic of and selective for viruses, bacteria, or other microorganisms
as well as of genetic disorders or other conditions. Biosensors having such
characteristic or selective immobilized sequences are useful for the rapid screening of
CA 0220816~ 1997-06-18
-- 19 --
genetic disorders, viruses, pathogenic bacteria and in biotechnology applications such
as the monitoring of cell cultures and gene expression. One important avenue which
has been widely ignored by the nucleic acid biosensor community is the investigation of
multi-stranded (~ 3) nucleic acid formation. For example, triple-helical oligonucleotides
have been reported to offer potential use as: sequence-specific artificial nucleases ({a}
Moser, H.E.; Dervan, P.B. Science, 1987,238,645. {b} Strobel, S.A.; Doucettestamm,
L.A.; Riba, L.; Housman, D.E.; Dervan, P.B. Science, 1991,254,1639.), DNA-binding
protein modulators/gene expression regulators ({a} Cooney, M.; Czernuszewicz, Postel,
E.H.; Flint, S.J.; Hogan, M.E. Science, 1988,241,456. {b} Durland, R.H.; Kessler, D.J.,
Gunnel, S., Duvic, M.; Pettit, B.M.; Hogan7 M.E.; Biochem., 1991,30,9246. {c} Maher,
L.J.; Dervan, P.B.; Wold, B.; Biochemistr~, 1992, 31, 70. {d} Maher, L.J. BioEssays,
1992, 14, 807. {e} Maher, L.J. Biochemist~, 1992, 31, 7587. {f} Duvalvalentin, G.;
Thoung, N.T.; Hélène, C. Proc. Nat. Acad. Sci. USA, 1992,89,504. {g} Lu, G.; Ferl,
R.J. Int. J. Biochem., 1993, 25, 1529.), materials for genomic mapping ({a} Ito, T.,
Smith, C.L.; Cantor, C.R. Proc. Natl. Acad. Sci. U.S.A., 1992,89,495. {b} Ito, T., Smith,
C.L.; Cantor, C.R. Nucleic Acids Res. 1992,20,3524.), and highly selective screening
reagents to detect mutations within dupiex DNA (Wang, S.H., Friedman, A.E., Kool,
E.T. (1995) Biochemist~34, 9774-9784.). The present invention can also be used to
detect the formation of multi-stranded nucleic acid hybrids (for example, formation
triple-helical nucleic acids), and therefore could, for example, operate to monitor the
effectiveness, dose dependence and intracellular concentration of nucleic acid
pharmaceuticals used in gene therapy applications or as an assay to identify multi-
strand formation associated with any of the aforementioned potential applications
associated with triple-helical oligonucleotides.
CA 02208165 1997-06-18
- 20 -
Brief Description of the Drawings
~ ~ ~ + o~O
conden~ 3~n
O O
HO~O--~ O ,O
O O
OH
DMTo ~Base
coupling and activation
OH
DMTO-- Base
~O~
It2N~ H2N~ H2N H2N
> ~ ~
~ ~ ~ 1~
--S~O--Si--O--Si--O--S~
derivdti~tion
Sur~ace of
Optical Fibre
DMTo Base
/~sl-o~N~~b'~~ ~ ~~
1~ O O
-- Surfare of NH2
Figure 1 (a). Synthetic scheme of Arnold et al. used to activate the giass or fused silica
surfaces with long chain aliphatic spacer molecules terminated with 5'-0-
dimethoxytrityl-2'-deoxythymidine.
CA 02208l65 l997-06-l8
- 21 -
NO2
NO2 NO2
~) SOCI, ~ H2N--(CHz)3--Si(OEt)
N b E
IC--N H
C--OH C--C I ~ <
O ~ <~
Si(OEt)3
H2 ~ +
?
H
Z rt / H ~, T o I u e ~ n e
(C 2H s)3 N
/ F u s e d S i I i c a
C--NH C--NH
O < O
--O--S i--O-- --O--S i--O--
O O
si I ~ I
Figure 1(b). Synthetic scheme of Brennan et al. used to create alkylamine substrate
linker molecules on hydroxylated fused silica surfaces.
- 2 2
HO,
0~
O~
H E G, HO~
H+
Fused Silica Surface ~ ~
o O o o 8 ~
Figure 1 (c). Synthetic scheme of Maskos and Southern used to functionalize
hydroxylated fused silica surfaces with GOPS followed by extension with HEG.
HO~
O~
0
O~ ~O~
0~ ~O 0
O~ ~~ 0~
O O o
~ HO < HO ~ HO ~
--si ~o~S .o,si o .si ~o,Oi,o,si-- --si ~o~S ,o,si,o si 'o OI o ,si--
Figure 1 (d). Possible closed loop structure formation as a consequence of the
synthesis scheme used in Fig. 1 (c).
CA 02208165 1997-06-18
- 23 -
0
H3C O ~ = C H3 H3C 0~30c H3
0~ ~~
NaH H~CO--~ tOCH3
0~ ~ O
O
0~
~'~ HO~
O O~
--s i o ,si O ,si-- o
Figure 1(e). Synthetic scheme used to extend GOPS functionalized substrates withDMT-HEG via a base catalyzed mechanism.
OCH~ H~CC~ 30CH, II~CO~ OcH~
Me--S~l o3
Pyridine o Pyridine o~
o~ o
oO~ oJ
H O
O=S=O OH O OH
Me ~o-Si~o~si~o-si~
Fused Silica Surface +
\~ OH OH OH
~0 ,si~o,si~o,s
Figure 1(f). Synthetic scheme used to covalently link DMT-HEG onto hydroxylated
fused silica surfaces via activation with methanesulfonyl chloride.
CA 02208165 1997-06-18
- 24 -
¢~O~V~R
Figure 2. The phenoxyacetyl protecting group used for exocyclic amine (R) protection
on nucleoside phosphoramidite synthons.
H~C~35--a t~ ~ ~ ~H~
o
H,C~RI~
N02
ni~n~ ~ ~ ~
O~N~--4 H~N~NH2
-- --~H~ Al (~). NiCI ~H20, ~N~ rOCH~
OCH~
2 C~3 ~ p~ndin~
~ \
OC~ \
DMr--11~MT D~T~N~3NH--DIUT
~ ~~ OCH~
Figure 3(a). Synthetic scheme used to create a hydrocarbon-tethered analogue of
Ethidium Bromide.
CA 02208165 1997-06-18
- 25 -
O~c~ pyridille O~N ~ ~
HO,~ ~ b~
~N~ ~ ~O~N~ ~
~0~5,G Me~l ~ ~OH ~
~3
pyridine NaH ~\ ~ ,~ A A A
H O O O O O O O
Me
N ~ ~ e
/--\ A /~ \
\=/ O O O O O O O--~
Remove CB2 ¦ ~ ~
and DMT~L T
OMe
C l~
Me Cl--P~
DMT-CI ¦ N _<
H~ ~ DuT TBAF ~ , ~N~DMT ~C-N
A A A A A /~ ~ O O O O O O O_
Ne
Figure 3(b). Synthetic scheme used to create a polyether-tethered phosphoramidite
analogue of Ethidium Bromide.
- -
CA 02208165 1997-06-18
- 26 -
Step 1.
CH3 1
~ I ~1 A ~
CH3
Step 2.
CH3
~N~_SH CH31 in acetone~ S--CH3 ~ ~S--CH3
Step 3.
+ ~5--CH3 EtOH ~N ~1
CH3
Step 4.
CA 02208165 1997-06-18
- 27 -
Me- -Cl + DMT-HEG
\
Me-'-O O O O O O O-DMT
(Mes-HEG-DMT)
Me~ OH ,Me NaH Me~ ,Me
Me~N ~ N~Me pyridine Me,N ~ N'M e
M es-H E G -DMT pyridine
Me
Me
/~--~\o/----\o~~--~\o/~--~\o/~--~\O O--DMT
~N Me
Me
CA 02208165 1997-06-18
- 28 -
Step 5.
CH3
~N~N Ao/ \oA AoAo
~ dioxane ~ H3C' ~J~N CH3
A A ~ / \ ~\
DMT--O O O O O O CH3
,~,-N -C H3 C H3 ~ C H3 N ~.
~o)~N ~,N ~1~ N ,N~G<o ~J
Figure 3(b). Synthetic scheme used to create a poiyether-tethered analogue of the bis-
intercalative fluorescent probe YOYO-1. Removal of the DMT protecting group followed
by treatment with ~-cyanoethyl-N,N-diisopropyl phosphityl chloride will yield the tethered
YOYO-1 phosphoramidite synthon.
CA 02208165 1997-06-18
- 29 -
Rotating ~Video
Mirror =J Monitor
(;~< ~ ~ SIT Camera f~
A Computer
< ~ Ar+Laser
Dichroic Mirror ~/ ~ (488 nm)
(495 nm Cut~R) r
J ,a-- EB and ssDNA Solutions were added
~ ~ using a Microsyringe
Objective ~1 ~",~ /(0.2micronfilter)
Optical Fiber ~ ~ 1
j ~_ = ~1~ ' .
Immobilized Single ~ ~ / L
Strandsof DNA
HybridizaUon Buffer
~_ Syringe
Free Single Stranded
'~~ ~Compiement DNA/RNA
'~ = MagneticS~rrer
3 mL Plastic Cuvette
To Waste Stopcock
Figure 4(a). Schematic diagram of one embodiment of an apparatus used to measurefluorescence intensity from optical fibers coated with immobilized DNA.
CA 02208165 1997-06-18
- 30 -
Pholomultiplier
__ ~Tube
~~ Fused Silica Optical Fiber ~ ¦
(Jacket and Cladding Removed)~
__ ~
Fiber Coupler JL i~
_~ ~Sample Outlet Fiber Coupler ~X
~nlet
Sensing Fit~er ~ ~ Ther~ s~or I ~ ¦
,~ ,~ ~ GradientSilvered ¦ Inter~srence
Stainless Steel ~ [
Chamber /' L
Th~,.,.. ' '.;~. Temperature ~ ~ ~\
Controller ~ ~ _ Shutter
Ar~j,L ~sr~.l i42i n~ cuL.I~
l~ff~f~fbfbf~f~f~'V~fl '' ~c~
i, :. PC Equipped with
~ - ~ Interfacing Hardware
Autosampler ~ Peristaltic Pump ~ J J
Figure 4(b). Schematic diagram an example of a dedicated instrument for analysis of
nucleic acid samples by the fiber-optic nucleic acid biosensor of the present invention.
CA 02208l65 l997-06-l8
- 31 -
Dir hroic Mirror l r: ~ lJ,'
(495 nm Cut~F) \~C ~
Ar~ Laser ~488 nm output) ~ ~ Lens
~ V
C
DeliveryFiber~
Fiber Coupler
Sample Outlet
Sample Inlet ~ ~ Thermistor \
Sensing Fiber ~Stainless Stee~
~ ; ~ Chamber ~~~ \
~Temperature Controlled
liybridi -tion Cell Holder ~ J
Glycol Solution To and From a f~
Tc. "~ e r, u~l .," ", IdLI.3 Circulating Bath / ~/
PC Equiped with an A/D Board
Figure 4(c). Schematic representation of a biosensor system in which light from a
suitable source is directed through a dichroic mirror beam splitter and focused onto a
fiber or waveguide coupler and then into an optical fiber having single-stranded nucleic
acid bound to the surface thereof, and in which any resultant fluorescent light travels
back through the coupler, and passes through the bearn splitter and is directed to a
photomuitiplier detector.
CA 02208165 1997-06-18
- 32 -
- Single S~anded Sequence of
,~ Col",'~ I,erllNucleicAcid
Fluolesoellt probe in Aqueous Env;n""~lenL
~ ~Immobilized StrandofDNA
SurfaceofOpfoalFiùro
I IyL~ndi~ed Nucleic Acid Complex
Fluorescen~ Probe in Hyd~ophoL;c Enviiunrl,ent
(In."uased Quantum Yield)
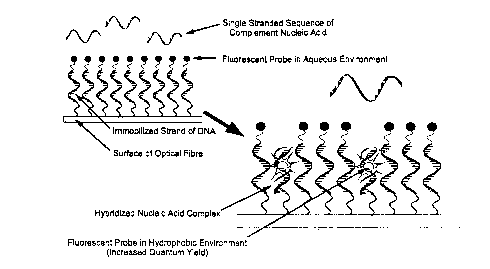
Figure 5. Illustration of the operating principles of the fiber-optic nucleic acid
biosensor. Hybridization of complement single-stranded oligonucleotide from solution
with immobilized nucleic acid probe on biosensor is followed by intercalation of the
tethered fluorescent ligand which provides transduction of the selective binding process
into a measurable analytical signal.
Fluorescence Intensity versus Temperature
100
o
,~ _
E 20 ~~~, __ _
O '--
0 10 20 30 40 50 60
T~ Jer~ture (C)
CA 02208165 1997-06-18
- 33 -
Figure 6. Fiuorescent intensity as a function of temperature for the mixed base
sequence icosanucleotide functionalized fibers. Upper Curve: response of the optical
sensor to 20pmol of linear complement icosanucleotide in the presence of 2.5 x 10 8M
ethidium bromide. Lower Curve: response of the optical sensor to 2.5 x 1 o~8 M ethidium
bromide.
Cu~ Jk ~ t6~ y Branched
Singlo Stranded Nudeic Add Sequences ~ ~i~_~
r omplem nt SHqu-nce
~r ~, Fluorescent Probe in ,~
FiuonrscentProbain /~,j~ AqueousE,.. ul.ll : ~ 7 .~/
Aqueous Envin~nment ~\,r ~ ~"~ tLow Quantum YTeld)
--(Low Quantum Yield) ~ _~, ~,, ~ ~,
Immobili~od Single~ ' Single~
Strandod of Di~iA ~ Stranded of DNA e;~
Surface of Opticai Fiber ' - Surface of Optical Fiber
Coolin~q
Cooling
Ac~o cQmplex ,~,r ~ Tripl~Stranded Nucieic
~ . .... . . Acid Complex
Hy~ hcbic Envtirmnnnooind)~ ,, FluorescencPrjobe
(Inueased Quantum Yield)
Ccolin~
E~cusionofFluore~ccntP~bc ' j~,~ Cooling
~r~m T~phx Structun3 wlth
De~e~slng Temp~ratun3 ~ Exclusion of Fluorescent Probe
2 ~from Triplex Structure with
Tripl~Sbandl3d Nuduic ~ Decneasing T
Acld Compl~x ;;;
Tnple-Stranded Nucleic
Acid Complex
7(a) 7(b)
Figure 7. (a) A Model of parallel (T~AT) triplex formation using dT1o and an optical
biosensor functionalized with immobilized dA10. dT1û:dA10 duplex is first formed upon
cooling the system below the duplex Tm followed by formation of the triple-stranded
complex with further cooling below the Tm for triplex formation. (b) The dA10 of the
optical sensor capturing the branched "V" compound 1 (see Fig. 15). Note how thefluorescent probe is excluded from the triplex as the temperature is cooled.
CA 02208165 1997-06-18
- 34 -
1 .4E-08
~,5 1.2E-08- -
1E-08-_ ---___-_ _ ----
O 8E-09~
~ 6E-09- ---
_ _ ¦ Averag~ ~ 8.8 ~l~ 0.1 - ~ l
4E-09
0 5 10 15 20
Length of Oligonuclecti~le (base units)
Figure 8. Quantity of trityl cation released during each detritylation step of the
automated phosphoramidite synthesis of dT20 onto fused silica optical fibers
functionalized by the protocols of examples 1 and 5.
.~ 800 III 757 ng ml~' cDNA
600-
r~
0 400 ~ c e
189 ng ml-' cDNA
a~ J
200- I_I II T IIT
TT T~
0 1 2 3 4 5
Time (hours)
- ssDNA on Fiber, No Ethidium Bromide Added ~ - hber Treated with C~ , ' ' DNA and Washed, No Ethidium Bromide
- ssDNA on Fiber, Treated with Ethidium Bromide and Washed ~ - dsDNA on Fiber Treated with Ethidium Bromide and Washed
- Rber Treated with dR" and washed, No Ethidium Br~mide ~i~ - Fiber Treated with dR", Ethidium Bn~mide, and washed
Figure 9(a). Response characteristics of an optical biosensor to complement and non-
complement DNA.
CA 02208165 1997-06-18
- 35 -
700- TTT
600-
e--
a~ soo -
w
400- '
300-
-
200-
C~i
'~~~ L
0 1 2
Time (hours)
ssDNA on Fiber No Ethidium Bromide Added
- ssDNA on Fiber Treated wi~h Ethidium Bromide and Washed
~- DNA/RNA Duplex on FiùerTreated with Ethidium Bromide and Washed
Figure 9(b). Response characteristics of an optical biosensor to 570 ng.ml-1 of
complement RNA.
CA 02208165 1997-06-18
- 36 -
700
600-
500
~ ~ 300 ~
r~ 200 ~
100- ~
O
0 5 lO 15 20 25 30
Time (min.)
~ Ethidium Bromide Staining ~ tion with cDNA
Figure 10. Response time of the optical sensor constructed as per the protocols in
examples 1 and 5 and effect of ethidium bromide incubation time.
700
600- /
c 500 (b) ~ (a~'/'
' 400- 7
~ ) 300 ~
3 ~ ~ / -
200-
g 100- "~ / -
. _ ~/
~y O ~
0 200 400 600 800
Concentration of cDNA (ng-ml-~)
CA 02208165 1997-06-18
- 37 -
Figure 11. Response of a DNA optical biosensor (a) after storage for one month used
without cleaning and (b) after storage for eleven months and cleaned by sonication in
ethanol for 10 minutes. Note: A 1-month-old sensor which had been cleaned by
sonication (data not shown) provided a response similar to (b).
6 0.8- Aqueous Duplexes
O ~ Immobilized Duplexes
t; 0 4-
~ /
0.2- ~ /
/,
O
50 60 70 80
- Temperature (~C)
Figure 12. Thermal denaturation profiles of aqueous dA20 + dT20 and immobilized dT20
with aqueous dA20-
-
CA 02208l65 1997-06-18
- 38 -
r2=0.988
"~, 200 ~
~ ~ /
'~ 150
G.~ 100
cn~ /
50 ~
IL / S~llsilivily = 100% per 89 pM
L.O.D. = 6 x 101~ molecules
O ;~
0 50 100 150 200 250
Concentration (pM)
Figure 13. Response of the optical sensor with immobilized nucleic acid probe for
Candida albicans to complement DNA.
0.2
0.195_
0.19
n r'
~ 0.185_ ~
? x "~
- 0.18_~ ~
a~ ~
- 0.175
0 200 400 600 800 1000
Time (s)
* - Injection of cDNA
Figure 14. Response of a reagentless biosensor as described in Example 14. The
graph measures fluorescence from the tethered dye on the terminus of the immobilized
nucleic acid as a function of time after exposure to a sample of 720 ng of cDNA.
CA 02208165 1997-06-18
- 39 -
5' ~N
HO ~
/~ ~o-rt=o
5' ~O- ~=O \o
HO~Th o O
~ O--P=O~ o--p=o 0 0--P=O
O ~Th o ~Th O ~Th
~ O O
~0--P=O ~0--1=0 ~0--P=O
O~Th O~Th o3 j~
HO HO HO
3' 3' 3'
dTIo 'IV" Compou~d (O
Figure 15. The structures of dT10 and compound 1, a branched oligonucleotide with
identical oligo(thymidine) chains linked to the 2'- and 3'-positions of a ribose branch-
point nucleoside i.e., rA2~5 d~,0 binds to dA~o to yield a triple-stranded complex
containing only T-AT (reverse Hoogsteen Watson/Crick) base triplets.
CA 02208l65 l997-06-l8
-- ~0 --
Fluorescence Intensity and ~bsorbance versus Temperature
100
~~ .~\ ~J~'
~ ' 0.8 8
8 60 ~' 0.6 ~
~' ' 'S
/ 0.4 _
c 20 ~ X ''; ~ 0.2 Z
, ~ X ~
O '' ' ------ O
0 10 20 30 40 50 60
Temperature (C)
Figure 16(a). Response (-) of the optical sensor with a 5'-end terminated recognition
sequence to 40 pmol of linear dT~o in the presence of 2.5 x 109M ethidium bromide.
Response (X) of the optical sensor to 2.5 x 108 M ethidium bromide and no dT,o.
Melting profile of the same nucleic acid system in bulk solution by measurement of
absorbance (260 nm) in 10 mM TRIS and 50mM MgCI2 at pH 7.3.
Fl2~0rescence Intensity and .4bsorbance versus Temperature
100
'~ /~\ ~'
~ '- 0.8 8
f ~ ~ ~
~ 0.6 ~
L 40 ~ , ~ 0.4
.. ~
~,i 0.2 Z
Z ~ ~ ~ ~ Y S
O ~ ~ O
0 10 20 30 40 50 60
Temperature (C)
Figure 16(b). Response (-) of the optical sensor with a 3'-end terminated recognition
sequence to 40 pmol of linear dT1o in the presence of 2 5 x 1 o-8M ethidium bromide.
Response (X) of the optical sensor to 2.5 x 10-8 M ethidium bromide and no dT10.Melting profile of the same nucleic acid system in bulk solution by measurement of
absorbance (260 nm) in 10 mM TRIS and 50mM MgCI2 at pH 7.3.
CA 02208165 1997-06-18
- 4i -
Fl1lorescence lntensify and Absorbance versus Temperature
100
C 80 / ~ \ 0.8 .
C / ~ \
/ "' ~ 0.6
~ ~
- L 40 ' ~ "' \ 0 4 ,N
.~ ''\~e ~ X ,J ~ E
E 20 ~ ,}' \ 0.2 Z
Z",'~' ~ ~ \~
O '' ' ~ l' O
0 1020 30 4050 60
Temperature (C)
Figure 16(c). Response (-) of the optical sensor with a 3'-end terminated Recognition
Sequence to 40 pmol of 1 (see Fig. 15) in the presence of 2.5 x 10-8M ethidium
bromide. Response (X) of the optical sensor to 2.5 x 1 o~8 M ethidium bromide with no 1.
Melting profile of the same nucleic acid system in bulk solution by measurement of
absorbance (260 nm) in 10 mM TRIS and 50mM MgCI2 at pH 7.3.
2 3 4 5 6 7 8 9 10
i ~ ~[ ~'~ ~ ~--
l il ilE~ ~ l ~ ~ 1 + dA~o
~ ~ ~ I ~
~ ~ ~! ~1
~ ~ ~ ,''', "~ r 5~
dT~o ~~
~ ;f~
~ ,.' '~ ~ dA,o
Figure 1 7(a). Photograph of a UV-shadowed native polyacrylamide gel containing
single strands, duplex and triple helical complexes of branched and linear controls.
CA 02208165 1997-06-18
~, --
DNA samples were loaded in 50mM MgCI~, and 30% sucrose. Lanes 4-10 are dT10,
dT10:dA1o (1:1),. dT10:dA10 (2.5:1), dT1o:dA1o(4:1), dA,0, 1 + dA10, and 1, respectively.
As can be noted the dT10:dA10 triplex (lane 7) showed a greater retardation in the
mobility relative to the corresponding duplex (lanes 5 and 6). The slowest mobility was
observed in lane 9 for 1 :dA10. Note: See Fig. 15 for the structure of 1.
- 1 2 3 4 5 6 7 8 9 10
~ 1 + dA,o
Figure 17(b). Photograph of an ethidium bromide stained native polyacrylamide gel
(same gel as Figure 2.1 4A) containing single strands, duplex and triple helicalcomplexes of branched and linear controls. DNA samples were loaded in 50mM MgCI2,
and 30% sucrose. Lanes 4-lO are dT10, dT10:dA10 (1:1),. dT1o:dA1o (2 5:1),
dT1o:dA1o(4:l)~ dA10, 1 + dA10, and 1! respectively. As can be noted the dTlo:dAlo
triplex (lane 7) showed a slight retardation in the mobility relative to the corresponding
duplex (lanes 5 and 6). The slowest mobility was observed in lane 9 for 1 :dA10. Notice
that only the duplexes and triplexes showed ethidium bromide fluorescence. Note: See
Fig. 15 for the structure of 1.
CA 02208165 1997-06-18
- 43 -
(a) \~ u, \
Incident Beam ,~ 3 c
n",,,,j",. ~ _
'~ Transmitted Beam c,
.. . ~
(b) .~ \
---- c
~". -
n",.,,"j.... \~ , _
n i ca
...... , .,., - ~ ~90~
~,
(c) ._
\ ~2~
~ ~ Reflected Beam
n,.. , ........... \,/ _
n~,, j.. , ~, -
~ , '
Figure 18. Schematic diagram illustrating the experimental concept for light scattering
investigations of a two-layer system with nFused Silica > nFilm-
CA 02208165 1997-06-18
- 44 -
(a) \ .-~ \
Incident Beam ~ ~o
n .,,"
n, ~Film~Refracted Beam
' Transmitted Beam ~~>\
~Am b.e~
(b) ~
c \
~' ~ "\~
n,. ,, . "
n, I F llm ' ~ilm / A m b ie ~~~~ ~
n...
~ ~ ~
( c ) ._
c \\
n". ~3 Film2 ~ilm I Am bien ~~ ~~~ ~
~'- "--' ' Film ¦ ~ Film I Am bienl
(d) ~
~ _ ~ c
n,.. ,,"j,. ' '-,
n,". i Fused Silica / Filn
n,,, ,. ., F ~ s ~ d S ilic a / F~
Figure 19. Schematic diagram illustrating the experimental concept for light scattering
investigations of a three-layer system with nFused Silica > nFiim > nAmbient-
CA 02208165 1997-06-18
- 45 -
(a)
Incident Beam ~,,~ ~3 u, \
n ~\, ~" \
-
n~ ~Film -\~ Refracted Beam
L~
n. ,,. --?~' Transmitted Beam
~Am ~l~ol \
(b) .~ \
n,.,, "'~\~ ~ ' \
n",~3F~lm ~l~ilm /Am~le~ Refracted Beam
n, L
'''""" ~Am !~1~0 ~,
(C ) ,_
n" ,,"~
n,"~3~Fi~m2 ~lm /Am~i~n~/~- Reflected Beam ~a ~
n,"., \/
' F i~m
Figure 20. Schematic diagram illustrating the experimental concept for light scattering
investigations of a three-layer system with nFUsed Silica < nFilm > nAmbient-
CA 02208165 1997-06-18
- 46 -
Gre-Ne Laser ~i Stepping M~r
S c rew S h aft C on tro llin g . ~ ~ P M T P owe r S u p ply and
Pivoting Arm Angle~ ~ ~gnal-Output Electronics
\\~" Hemisjpherlcai ~ ~ J,
Pivoting Goniometer Arm \~"~
PC Equipped With Interfacing ~ I fffffffffffffffffffff ¦
H ardware ,
\~ Fused Silica Wafer r,~ rl ~ I I
P h o to m u ltip lie r Tu be
~ il~ n Spacers ~ //
1~- ,' Inlet and Outlet for
~/ Analyte Solutions J /\
\~ ~ / Optical Fiber
~ _ _ Bundle
Figure 21. Schematic diagram of the instrument used for investigations of anguiarly
dependent light scatter.
CA 02208165 1997-06-18
- 47 -
P rism W afe r
(a) Interface: Fused Silica Prism /Air (d)Interface: Fused Silica Wafer/Air
50Scatter Intensity vs. Incidence Anqle Scaner Intensify vs. Incidence Angle
n",,,,".,=1.46 n,,,.,,,,=1.46
_ 40 ~ n," = 1.0003 70 - ~ n = 1.0003
E.. pe., -~ , = 42.8~ ,c 60 - '~ Experimental C, = 43.2~
--' ~ 30 - \ Calculated ~, = 43.2c .--~ 50 - ~ Calculated ~, = 43.2~
c -- 20 ~ ~ 30
~ 10 _. 20-
O . ... .... .. .
- 30 40 So 60 70 80 go 30 40 so ôO 70 80 go
Incidence Angle (deg) Incidence Angle (deg)
(b) Interface: Fused Silica Prism / Water (e) Interface: Fused Silica Wafer/ Water
Scatter Intensity vs. Incidencr~ Angle 35 Scatter Intensi~y vs. Incidence Angle
n,"",.. ,, = 1.46 - n, ,,,,.. = 1.46
~ n,,",= 1.33 _ 30 -~.... n.", = 1.33
.'~ 35 ~ '~ Expenmental ~, = 65.6~ ~'c 25 ~ Experimental ~ = 65.6~
c ~25 -~ Calculated ~ = 65.6~ C ~ 20 ~ Calculated ~. = 65.6~
c .--20 - 'J~ c 15 ~'A'''~
_ 1 5 ~ l~
-10_ ~ '' 10-
sO60 70 80 90 50 s560 65 70 7s 80 85
Incidence Angle (deg) Incidence Angle (deg)
(C)Interface: Fused Silica Prism / Cyclohexane (f) Interface: Fused Silica Wafer / Cyclohexane
Scaner Intensity vs. Incidence Angle Scaner Intensity vs. Incidence Angle
S ~, n,,,,,,,,, = 1.46 ~ 40 n,.. ,,,,,. = 1.46
n,,,.,,,,,,, = 1.427 ~ _ 35 -~ n = 1.427
'n 20 - ~ Experimental C, = 77.8~ ~ --c 80 \ Experirnental ~. = 77.9~
.--~ ~,, Calculated C, = 77.8~ ~ ~ ~ 25 - ~ Calculated ~ = 77.8~
~ 10 ~ ~ 20- ~~~
' S ''''' ' ' '''~''''''' ''
5 ........................ . . . .
ss 60 6s 70 7s 80 85 go 60 70 80 go
Incidence Angle (deg) Incidence Angle (deg)
(g) Intertace: Fused Silica Pnsm /Hybridization Buffer
80 _. S,canerlntensltyvs Incidence Angle
70 - ~ n''.'''-'1 35
o~ 60 - Expenmental ~. = 67.7~
,~ 50 - , Calculated C = 67.6~
c r'~ 40
c.~ 30
~ 20 -
10 - ~_
O ................
40 So 60 70 80 go
Incidence Angle (deg)
CA 02208165 1997-06-18
- 48 -
Figure 22. Control experiments for the Angularly Dependent Light Scattering Technique
Using Substances of Known Refractive Index.
(a) Interface:FvsedSilicaPnsm/FeftachvelndexMdtcn~n30il,Air (b) In~er7tace:FusedSilicaPnsm/EthyleneGlycol/Ait
Scanerlnt~n~Oyvs IncidenceAngle 60 Scanerlnlensl~yvs IncirtenceAngle
40 - j n . l.S1 _ 50 _ ~ n,, , = 1.43
'n 35 - ~ EYp;nmrnl~13a~e .~35- c 40 _ ~ EYpenrnrtnlal e . 5 44.2~
j/A ~ 30 ~r~C-lcul-l-d 3 ~t e.. 43.3~ ~ .?~ ~ C~lcul~l~d e . 44.3-
c~ ~ 2 5 - ~ c~
< t S _ e O I 1~ . 79.40
tO . 0 Erp-nm~nl~10, . .7790 r
30 ~0 Sû S0 70 60 90 30 40 50 60 70 90 90
Incidence Angle (deg) Incidence Angle (deg)
Intertace:FusedSibcaW2fer/OTSMonolayer A!! d Interface:FusedSilicaWafer/OTS.Monolayer/Water
(C) 60 Scanerlnlensity~s.lnclaenceAngle ( ) 30 Sc3tterlntensityvs.1nclOenceAng/e
Experimental ~, = 43.9~ ~ Et perlmental ft . = 66.7~
?, 40- \~ .?~ 20
i-- -- 3 0 - ~ i-- r
i:~ 2 0 - ~ ~ ' ~ 1 5
O ; ~ e 10- r -
7G eo so so ss 60 65 70 2s 60 35 90
Incidence Angle (deg) Incidence Angle (deg)
Figure 23. Results of the light scattering experiments done with substrates coated with
a thin organic films.
CA 02208165 1997-06-18 .
- 49 -
Interhce: FusedSilica W~er/NucleicAddMonolayer Interfa~ FusedSilica Wafer/NucleicAudMonolayer
(a)(MesylatePr~tocol)/HyondzabonBuffer(b) (MesylateProtocol)/3:1EthyleneGlycdinWater
Sl~dter ~t~ K ~ A~ Sca5er Intensity vs. Inckbn~ Angb
400 ~, ~ ~91 o ~ Expenrner~ 64.3 ~
n , , . . .- !.4105
\ c
1 X
O .... . , .. ,,, ~ -- --
hc denc~ gb (deg) Incidence Angle (deg)
In~ce: FusedSilica ~ r/NudekAcidMonolayer
(C) (GOPS-Hydnde P,'DtOCOI) /Hybridization euffer
tern~.s. rra~a~npe
a ~47 ~
C~ ;
20- :
O . .
50 ~0 7~
en~ An~ ~ (deg)
Figure 24. Results of the light scattering experiments done with substrates coated with
covalently immobilized oligonucleotides.
Detailed Description of the Invention
Nucleic acid oligomers are covalently immobilized onto optical fibers by first
activating the surface of the optical fiber with a long chain spacer arm terminated by a
chemically protected terminus, normally a dimethoxytrityl (DMT) moiety, followed by
automated solid-phase DNA synthesis. Detection of nucleic acids or nucleic acid
analogs at the fiber surface after hybridization between immobilized nucleic acid and its
complementary nucleic acid is achieved by measuring enhanced fluorescence emission
of the fluorophore.
The optical fiber may be activated with a number of different compounds. The
method of Arnold and co-workers (Amold et al., 1989, Collect. Czech. Chem.
Commun., 54: 523) may be used for the activation of the fused silica wafers, optical
waveguides, and optical fibers whereby 25 atom-long spacer molecules terminated by a
dimethoxytrityl protected nucleoside are immobilized onto the cleaned optical fiber
CA 0220816~ 1997-06-18
- 50 -
substrate, as illustrated in Fig. 1(a). In this method, the length of the spacer between
the substrate and the first nucleoside is sufficiently long so that the environment of the
terminal nucleoside is fluid enough to permit efficient coupling with successivenucleotide monomers during automated phosphoramidite synthesis of the immobilized
nucleic acid probe. This is in accord with the report of Beaucage et al. (1992,
Tetrahedron, 48: 2223-2311) wherein it was stated that substrate linkers of lengths of at
least 25 atoms are required to achieve high (2 99.5%) synthon coupling yields. The
synthetic scheme of Arnold et al. requires inexpensive chemicals, is facile to perform,
and is done as a one pot procedure wherein product isolation and purification isobviated. Because the linker is terminated by a protected nucleoside, any reactive sites
on the support which would lead to the production of unwanted side products during
automated synthesis can be eliminated by treating the derivatized supports with acetic
anhydride prior to synthesis. Lastly, the coverage of linker on the support is easily
determined by determining the amount of trityl cation released during the first
trichloroacetic (TCA) deprotection step of the automated synthesis. This methodology
does however place limits on the types of nucleobase protecting chemistries can be
used as treatment with strong base will cleave the succinate bond between the
substrate linker and the oligonucleotide probe.
An amine-terminated solid support suitable for automated oligonucleotide
synthesis may be prepared according to the method of Brennan et al. (1993, Sensors
and Actuators B, 11: 109). A bifunctional amphiphilic support derivatization agent is
created by condensing ~-aminopropyltriethoxysilane (APTES) with 12-nitrododecanoic
acid. The resulting long chain spacer molecule is covalently immobilized onto the
surface of the optical fibers by an Sn2 reaction between the hydroxyl groups present at
the surface of the fiber and the silane moiety of the amphiphile. With the terminus of the
substrate linker in the non-reactive nitro-form, the support may then be capped using
standard methods employed during automated synthesis (acetic anhydride), or withchlorotrimethylsilane (R.T. Pon Methods in Molecular Biology, Vol.20: Protocols for
Oligonucleotides and Analogs, S. Agrawa, Ed, 1993, Humana Press, Inc. Totowa NJ.),
thereby masking other sites of reaction which may produce unwanted side products
CA 02208l6~ l997-06-l8
- 51 -
during oligonucleotide synthesis. Reduction of the terminal nitro-functionalities is then
achieved by treatment of the derivatized support with an acidic zinc solution. The
resulting amine headgroups may then be used directly for automated synthesis wherein
an ammonolysis/base resistant phosphoramidate linkage is made between the
activated support and the first nucleotide. An outline of a synthetic procedure used to
immobilize alkyl amine monolayers covalently onto fused silica substrates is depicted in
Figure t (b).
The hydrolysis resistant linkage of Maskos and Southern may also be employed
to provide waveguides functionalized with substrate linkers. Analogous to the natural
internucleotidic linkage, a phosphodiester linkage between the substrate linker and first
nucleotide is completely resistant to ammonolysis under the conditions which remove
standard base-protecting groups. This linkage is produced by derivatization of optical
fibers with the bifunctional silylating reagent 3-glycidoxypropyltrimethoxy silane via silyl-
ether bond formation with the hydroxylated waveguide surface. This yields a substrate
derivatized with short spacer molecules with terminal epoxide moieties. The length of
the spacer arm is then extended by nucleophilic attack of a polyether, such as
hexaethylene glycol (HEG), in an acid catalyzed expoxide ring-opening reaction,
yielding a stable ether linkage (U. Maskos and E.M. Southern, 1992 Nucl. Acids Res.,
20(7). 1679), as shown in Fig. 1 (c). Polyether chains provide for hydration, flexibility for
molecular motion, and improved biocompatibility in terms of minimization of non-selective binding to biological compounds. By extending the spacer molecule ensemble
to one composed of at least 25 atoms, optimal phosphoramidite synthon coupling
efficiencies are realized (Beaucage et al., 1992 Tetrahedron, 1992 48, 2223). This
support, temminated with a hydroxyl functionality, is then used directly for automated
oligonucleotide synthesis, obviating the need for tedious nucleotide functionalization of
the support.
Since polyethylene glycols are bifunctional, there exists the possibility of creating
non-reactive closed-loop structures which may significantly decrease the amount of
loading of oligonucleotides on the surface of an optical fiber, as shown in figure 1 (d). To
CA 0220816~ 1997-06-18
- 52 -
eliminate any such problem and improve upon the prior art, one terminus of the
polyether is protected with a suitable blocking group, for example, with a DMT
functionality, prior to extension of the glycidoxypropyltrimethyl silane. In the case where
a chromophoric protecting group is used (such as DMT), an additional advantage is
provided wherein facile determination of the amount of support linkers may be
determined by monitoring the absorbance of the deprotection solution (e.g. 504 nm for
DMT+). Mono-dimethoxytrityl protected polyethylene glycols may be introduced onto
the surface of fused silica waveguides by a number of methods. Waveguides first
functionalized with GOPS, as in the method of Maskos and Southern, may then be
treated with a solution of mono-dimethoxytritylated polyethylene glycol over sodium
hydride to afford linkage of the polyether to the terminal epoxide moiety of theimmobilized GOPS via a base catalyzed epoxide ring-opening reaction as shown in
figure 1(e). Mono-dimethoxytritylated polyethylene glycols (such as DMT-HEG) canalso be directly linked to the surface of fused silica waveguides by activation of the
terminal hydroxyl moiety of the polyether with methane sulfonyl chloride or ~-cyanoethyl
N,N-diisopropyl phosphityl chloride, as shown in Figs. 1 (e) and 1 (f), respectively. In the
later case, the polyether substrate linker is attached as a phosphoramidite synthon
which can be done as part of the automated oligonucleotide synthesis procedure;
thereby making the entire biosensor fabrication protocol completely automated after
cleaned waveguide pieces are introduced into the synthesis column of the automated
synthesizer.
The biorecognition element to be bound onto the terminus of the substrate linkerin configuration of the described biosensor can include immobilized nucleic acids (DNA
and RNA), modified nucleic acids, and nucleic acid analogs prepared by well-known
methods or by straight-forward extension or modification of those methods. The term
nucleic acid includes polynucleotides, oligomers, relatively short polynucleotides (up to
about 50 bases), longer polynucreotides ranging up to several hundred bases, anddoubled-stranded polynucleotides. There is no specific size limit on nucleic acids used
for immobilization in this invention. However, problems due to self-hybridization and
reduced selectivity may occur with longer nucleic acids. As used herein, the term
CA 0220816~ 1997-06-18
- 53 -
"nucleic acid analogs" inciudes modified nucleic acids. As used herein, the term"nucleotide analog" includes nucleic acids where the internucleotide phosphodiester
bond of DNA or RNA is modified to enhance bio-stability of the oligomer and "tune" the
selectivity/specificity for target molecules (Ulhmann, et al, 1990, Angew. Chem. Int. Ed.
Eng., 90: 543; Goodchild, 1990, J. Bioconjugate Chem., !: 165; Englisch et al, 1991,
Angew, Chem. Int. Ed. Eng., 30: 613). Such modifications may include and are notlimited to phosphorothioates, phosphorodithioates, phosphotriesters, phosphoramidates
or methylphosphonates.
In the present invention, nucleic acid sequences are covalently attached to the
surface of the optical fiber. In a preferred embodiment, an automated DNA synthesizer
is used to grow nucleotide oligomers onto the surface of activated optical fibers via the
well established ~-cyanoethylphosphoramidite method. Any commercially available
automated DNA synthesizer can be used. The use of an automated synthesizer to
grow nucleic acids or nucleic acid analogs on the optical fiber substrates provides many
advantages over conventional techniques of DNA immobilization. Conventionally,
nucleic acid strands are adsorbed onto a suitable support (usually nitrocellulose) with
little known about strand orientation. The use of an automated oligonucleotide
synthesizer provides full control of the oligomer sequence, strand orientation, and
packing density in association with activation of the optical fiber substrates. Control over
these parameters is critical to the development of a nucleic acid detection method
based on hybridization as the alignment of the immobilized strands with respect to the
availability of target nucleotides for hybridization and intermolecular interactions
(electrostatic and steric) between oligomers will have direct ramifications on the kinetics
and themmodynamics of hybrid formation and dissociation. The use of a gene machine,
in addition to the chemistry used to activate the surface of the optical fibers, allows for
the creation of membranes of desired density and structural order to permit rapid and
re-versible hybridization, and to coritrol refractive index.
The use of the phosphoramidite method of oligonucleotide synthesis has been
widely reviewed and has become the synthetic method of choice owing to the high
CA 0220816~ 1997-06-18
- 54 -
coupling efficiencies and robustness of the reagents, in addition to the fact that the
necessity of numerous product isolation and purification steps (which are required for
liquid phase methods) are avoided. There are two readily available types of
phosphoramidites which may be used to synthetically grow oligonucleotides, namely,
methylphosphoramidites and ~-cyanoethylphosphoramidites. The method utilizing ,B-
cyanoethyl phosphoramidites is preferable as complete deprotection of the
oligonucleotides can be done using aqueous ammonia (as opposed to thiophenol) for
the case where oligonucleotides were grown onto controlled pore glass (CPG).
Triethylamine is used to deprotect the ~-cyanoethyl protected oligonucleotides grown
onto fused silica wafers or optical fibers without liberating the oligonucleotides from the
support. An overview of the ~-cyanoethylphosphoramidite synthesis is as follows:
The first step in each cycle of solid phase automated phosphoramidite synthesis
involves the removal of the dimethoxytrityl protecting group on the immobilized
nucleotide. Detritylation is done by introducing a solution of 3% trichloroacetic acid
(TCA) in 1,2 dichloroethane (DCE) onto the synthesis column in order to yield a 5'-
hydroxyl functionality onto which the next nucleotide monomer may be coupled. TCA is
the reagent of choice for detritylation due to its rapid reaction rate so that the
oligonucleotide is only exposed to the acid for short periods of time, thereby avoiding
the acid catalyzed removal of the adenine and guanine moieties from the nucleotide
sugar groups by the process of depurination. Once the reaction has been completed,
the acid is removed by flushing the column with acetonitrile. The eluent containing the
released trityl cation is sent to a fraction collector so that the coupling efficiency of the
synthesis may be monitored by absorption spectroscopy.
Coupling is the next stage of the synthesis cycle. The contents of the synthesiscolumn are dried by alternatively washing with acetonitrile and flushing with dry argon.
This ensures that the support is anhydrous and free of nucleophiles. The desiredphosphoramidite and tetrazole are then sent into the synthesis column. Tetrazole is a
weak acid (pKa = 4.8) which is used to activate the phosphoramidite. Nucleophilic attack
by the 5'-hydroxyl group on the activated phosphoramidite moiety forms an
CA 0220816~ 1997-06-18
- 55 -
intemucleotide linkage. A ten-fold molar excess of phosphoramidite in an excess of
tetrazole is added to the synthesis column to ensure that high coupling yields are
achieved.
The next step of the synthesis is the capping step. This is done to eliminate
further growth of sequences onto which coupling did not occur. The failed sequences
are rendered unreactive by introducing acetic anhydride in the presence of
dimethylaminopyridine in order to acetylate any remaining unprotected 5'-hydroxyl
moieties.
Because the trivalent internucleotide phosphite moieties are labile to both acidic
and basic conditions, a solution of aqueous iodine is added after flushing the capping
reagents from the column. This is done in order to oxidize the trivalent internucleotide
phosphite moieties to the more stable pentavalent phosphate moieties found in
naturally occurring nucleic acids. This procedure is termed the oxidation step.
Following the oxidation step, one cycle of nucleotide addition is complete. The
process may be repeated many times until oligonucleotides of desired length and base
sequence have been constructed. After addition of the last nucleotide, a final
detritylation step is usually done in order to yield a 5'-hydroxyl group on the completed
sequence.
Triethylamine is used for the removal of ,B-cyanoethyl protecting groups on the
internucleotidic phosphotriester moieties of oligonucleotides grown onto opticalsubstrates. This procedure is known to cause quantitative loss of the phosphate
protecting groups via a ,B-elimination mechanism while not cleaving the single-stranded
nucleic acids from the optical fibers. Ammonia treatment of the immobilized
oligonucleotides is avoided by choosing an all-thymine base sequence. Thymine does
not contain a primary amine functionality which would require protection during
oligonucleotide synthesis. This approach is not limited to the use of phosphoramidite
synthons, but is compatible with all commercially available solid-phase synthesis such
CA 0220816~ 1997-06-18
- 56 -
as the H-phosphonate chemistry (Froehler, B. C., 1986, Tetrahedron Letters, 27: 5575;
Stein et al, 1990, Analytical Biochemistry, 188: 11).
Contrary to the conventional preparation of oligonucleotides by solid-phase
synthesis, post-synthesis removal of the product from the support is not desired. In
order to prevent cleavage of the oligonucleotide from the support (optical fiber) while
removing the protecting groups of the nucleobases, two modifications to the usual
synthetic protocol can be made. The approach involves the combination of a hydrolysis
resistant linkage between the oligomer and support along with the use of labile base
protecting groups. Thus, an oligomer of any sequence can be prepared and
deprotected yet remain attached to the support, available for hybridization.
The phenoxyacetyl (PAC) family of protecting groups represents a convenient
method for blocking the exocyclic amino functions of guanine, adenine and cytosine
residues (thymine or uracil require no nucleobase protection). The half-time of
deprotection with concentrated ammonium hydroxide at 20~C is 8 min, 7 min and 2 min,
respectively (Wu ét al, 1989). Under these conditions, the cyanoethyl phosphate
protecting groups are removed within seconds (Letsinger and Ogiivie, 1969), whereas
the linkage which joins the oligomer to the surface of the fused silica fiber (e.g., a
phosphodiester or phosphoramidate) is completely stable under these conditions.
Alternative labile protecting groups are derivatized phenoxyacetyl groups including alkyl
substituted PAC groups, more specifically t-butylphenoxyacetyl groups. The t-
butylphenoxyacetyl group can be quickly removed compared to hydrolysis of the linkage
to the spacer thereby reducing the possibility of cleavage of the immobilized sequence
from the surface. N-phenoxyacetyl deoxynucleoside 3'-cyanoethylphosphoramidites
and the analogous t-butylphenoxyacetyl phosphoramidites are commercially available.
It has been reported by Polushin and Cohen (N. N. Polushin and J. S. Cohen, Nucleic
Acids Research, 1994, 22, 54g2-5496) that the t-butylphenoxyacetyl nucleobase
protecting groups can be quantitatively be removed by treatment with ethanolamine for
10 minutes at room temperature or by treatment with a mixture of
hydrazine/ethanolamine/MeOH (1:1:5 vlvlv) for 3 minutes. Beaucage and co-workers
CA 0220816~ 1997-06-18
- 57 -
(J.H. Boal, A. Wilk, N. Harindranath, E.E. Max, T. Kempe and S.L. Beaucage, Nucleic
Acids Research, 1996, 24, 3115-3117.) also report the rapid and quantitative removal
of t-butylphenoxyacetyl protecting groups by treatment of the support-bound protected
nucleic acid with gaseous amines ({a} anhydrous ammonia gas, 10 bar, 25~C, 35 min.
or, {b} methylamine, 2.5 bar, 25~C, 2 min.)
Other possible labile protecting groups could include the "FOD" (fast
oligonucleotide deprotection available from Applied Biosystems Inc.) based on N,N-
dialkylformamidines (Vinayak et al, 1992, Nucleic Acids Research, 20: 1265-1269).
Kuijpers et al (Tetrahedron Lett., 1990, 31 6729-6732 and Nucleic Acids Res., 1993,
21, 3493-3500) have described a method of nucleobase protection using 2-(acetoxy-
methyl) benzoyl (AMB) moieties which can be removed by treatment with anhydrous
potassium carbonate in methanol for 90 minutes at room temperature. Use of
protecting groups that can be selectively removed under conditions that will not cleave
the oligomer from the support, such as the levulinyl group (removed by hydrazinetreatment) (Letsinger et al, 1968, Tetrahedron Letters, 22: 2621-2624; Hassner et al,
1975, J. Amer. Chem. Soc., 97: 1614-1615) are also contemplated by the present
invention. Even synthesis without nucleobase protecting groups is possible for nucleic
acid oligomers of up to 20 nucleobases in length using the phosphoramidite approach
(Gryaznov et al, 1991, J. Amer. Chem. Soc., 113: 5876) or H-phosphonate chemistry
(Kung et al, 1992, Tetrahedron Letters, 33: 5869). Any of these approaches
circumvents difficulties in removing nucleobase protecting groups while leaving the
oligomer attached to the support.
Free short strands of nucleic acids can also be covalently attached to the optical
fiber directly or via linker molecules. This approach allows the use of DNA or RNA
isolated from natural sources, amplified nucleic acids or their analogs, or synthetic
samples provided in the fully deprotected form. Protocols provide end-attached
oligomers of a well defined orientation. Chemically stable linkages between the support
and oligonucleotide may be employed to enhance the robustness of the biosensor.
CA 0220816~ 1997-06-18
- 58 -
Quartz (or interchangeably fused silica) optical fibers derivatized with linker
molecules terminated with either hydroxyl or amino groups can serve as substrates for
carbodiimide-mediated coupling with terminally phosphorylated single-stranded nucleic
acids. Coupling to the hydroxyl fiber produces a phosphodiester bond while coupling to
an amine fiber yields a phosphoramidate bond. Oligonucleotides can be
phosphorylated, in solution, either chemically via a modification of Ouchi's method
(Sowa et al Bull. Chem. Soc., Japan 1975, 48 2084) or enzymatically.
Covalent attachment of free short strands of single-stranded nucleic acid to theoptical fibers can be achieved by a slight modification of the method Ghosh and Musso
(Ghosh and Musso, 1987, Nucl. Acids Res. 15: 5353). Coupiing of a 5'-aminohexyl
derivatized oligomer with activated carboxyl fibers affords end-attached oligomers. This
method is known to minimize reaction at the amino groups of the DNA bases (whichwould potentially compromise the hybridization event) and affords surfaces with
excellent nucleic acid coverage. The synthesis of the 5'- or 3'-terminally modified
oligomers can be achieved readily by standard methods (Ghosh and Musso, 1987;
Beaucage and Iyer,1993).
RNA may be assembled on the support or prepared separately and linked to the
support post-synthesis. RNA monomers are commercially available, as are some 2'-O-
modified synthons. The 2'-O-methyl, allyl and 2'-deoxy-2'-fluoro RNA analogs, when
incorporated into an oligomer show increased biostability and stabilization of the
RNA/DNA duplex (Lesnik et al., 1993, Biochemistry, 32: 7832).
As used herein, the term "nucleic acid analogs" also include alpha anomers (a-
DNA), L-DNA (mirror image DNA), 2'-5' linked RNA, branched DNA/RNA or chimeras of
natural DNA or RNA and the above-modified nucleic acids. Back-bone replaced nucleic
acid analogs can also be adapted ~o use in the biosensor of the present invention.
For purposes of the present invention, the peptide nucleic acids (PNAs) (Nielsenet al, 1993, Anti-Cancer Drug Design, 8: 53; Engels et al., 1992, Angew, Chem. Int. Ed.
CA 0220816~ 1997-06-18
- 59 -
Eng., 31: 1008) and carbamate-bridged morpholino-type oligonucleotide analogs
(Burger, D.R., 1993, J. Clinical Immunoassay, 16: 224; Uhlmann, et a/, 1993, Methods
in Molecular Biology, 20, . "Protocols for Oligonucleotides and Analogs," ed. Sudhir
Agarwal, Humana Press, NJ, U.S.A., pp. 335-389) are also embraced by the term
"nucleic acid analog." Both exhibit sequence-specific binding to DNA with the resulting
duplexes being more themmally stable than the natural DNAIDNA duplex. Other back-
bone replaced nucleic acids are well-known to those skilled in the art and may also be
used in the present invention (See e.g., Uhlmann et a/ 1993, Methods in Molecular
Biology, 20, "Protocols for Oligonucleotides and Analogs," ed. Sudhir Agrawal, Humana
Press, NJ, U.S.A., pp. 335).
Optical substrates such as planar wafers and optical fibers may be used in the
present invention. A preferred embodiment utilizes optical fibers. Optical fibers are
particularly advantageous as membrane supports due to their small size, high light
transmission capability, and ability to allow total internal reflection (TIR) of light. Optical
fibers also provide a compact an rugged sensing device, and offer the ability to do
remote spectroscopic measurements (Love et a/, 1991, Biosensors with Fiberoptics,
D.L. Wise and L.B. Wingard (Eds.), Humana, NJ, pp.139-180).
There are two fundamental configurations in which alterations in fluorescence
parameters from fluorescently doped membranes on optical fibers may be monitored,
namely, extrinsic mode and intrinsic mode. Extrinsic mode configurations are those in
which the waveguide is simply used as a light pipe or conduit. End-on extrinsic mode
investigations are usually done using optical fibers. In a biosensor which uses end-on
extrinsic mode configurations, the fluorescent dyes and selective chemistry are located
on or near the distal end of the fiber. The fiber is used as a light-pipe or conduit, where
the excitation or emission radiation is simply guided from the sampling region to the
detector. Fluorescence is stimulated by coupling excitation radiation into the near end
of a fiber, and emission can be monitored by placing light sensing equipment directly
opposite the distal end of the fiber.
CA 0220816~ 1997-06-18
- 6G -
Alternatively, the detector is placed at the near end of the fiber as some of the
fluorescence may be coupled back into the fiber and totally internally reflected back to
the near end. The side-on extrinsic mode approach is typically used for investigations
carried out on planar supports, but may also be used for fibers. The immobilized single-
strand nucleic acid and fluorophore are placed along the length of the optical fiber
waveguide/wafer. The fiber is illuminated by a light source located normal to the length
of the fiber and fluorescence emission is also monitored by equipment placed normal to
the fiber. Extrinsic configurations provide the advantage that simple and inexpensive
equipment, including conventional light sources and detectors, are used (Krull et al,
1991, Fiber Optic Chemical Sensors and Biosensors, Vol. Il, O. S. Wolfbeis, Ed., CRC
Press, Boca Raton, pp. 315). However, the extrinsic sampling configuration provides
poorer sensitivity owing to the short path length and sensitivity to interferents present in
the surrounding media. In a preferred embodiment, an intrinsic mode arrangement,based on careful control of refractive index is used to monitor fluorescence emission
from the surface of optical fibers.
Fluorophores present at either the surface or just below the surface of the fiber
may be excited through the formation of a standing wave electric field which propagates
normal to the surface of the fiber upon total internal reflection of radiation in the fiber.
The process of TIR occurs when the angle of incidence, ~, at the interface between a
fiber of high refractive index, n1, and the external medium of lower refractive index, n2,
is larger than a critical angle, ~c, defined as:
sin~C = n (1)
The amplitude of the electric field of the reflecting radiation decreases exponentially as
a standing wave into the medium having the lower refractive index. This decayingradiation is referred to as an evanescent wave and can be used to excite fluorophores
located near the boundary for TIR. The propagation intensity, /, of the evanescent wave
depends on the reflection angle, ~, the wavelength of the transmitted radiation, ~, and a
Fresnel transmission factor, T:
CA 02208l6~ l997-06-l8
- 61 -
I=T(~).exp~ d ) (2)
where x represents distance nommal to the boundary for TIR, and dp is the penetration
depth which is given by (Krull et al, 1990, Talanta, 37: 801-807):
d p - (3)
4?~n 2 sin 2 (~) - n 2
The penetration depth is defined as the distance at which the intensity of the
evanescent field has decayed to 1 /e of the intensity at the reflection boundary.
Typically, the evanescent wave propagates into a thin zone beyond the surface of a
fiber with a penetration depth ranging from about 200 nm to 400 nm for visible light.
Fluorophores within the evanescent wave propagation zone are excited by that
evanescent wave to emit fluorescence. Fluorophores further from the interface with the
optical fiber will experience lower intensity of light at the excitation frequency and a
resultant concomitant decrease in intensity of emitted fluorescence.
A major limitation of the evanescent wave excitation is that less than 0.01% of all
of the excitation radiation on a fiber actually leaks beyond the fiber as an evanescent
wave, and less that 2% of the fluorescence caused by the evanescent wave is actually
recovered back into the fiber for transmission to the detector by total intemal reflection
(Love et al, 1991, Biosensors with Fiberoptics, D.L. Wise and L.B. Wingard (Eds.),
Humana, NJ, pp. 139-180). As such, the evanescent wave mode of excitation and
fluorescence signal recovery is very inefficient and not the preferred mode of operation
for optical sensor devices.
For the case where the refractive index of the immobilized layer is effectively the
same or greater than the index of refraction of the substrate for immobilization (e.g., the
silica surface of the optical element) the boundary for TIR effectively becomes the
interface between the immobilized layer and the solution. Fluorophores bound to
nucleic acid in the immobilized layer are directly exposed to the electromagnetic
radiation bound within the waveguide thereby providing a vastly improved excitation
CA 0220816F7 1997-06-18
-- 62 --
efficiency and, as a consequence, emit increased intensity fluorescence. For example,
the index of refraction of a monolayer of organic media (nmOnOIayer= 1.46 to 1.5;
Ducharme et al, 1990, J. Phys. Chem. 94: 1925) is very similar to that of fused silica or
fused silica (nqua~k = 1.46; O'Hanian, H.C. 1985, Physics, W. W Norton & Co. N Y. p.
835). Fluorophores in the immobilized layer then emit fluorescence radiation within the
waveguide itself to provide a much improved probability for transmission of the
fluorescence signal by total intemal reflection to the detector, yeilding increased
sensitivity and lower target nucleic acid detection limits.
Fluorescence is the analytical method chosen for the transduction of
hybridization events into a measurable analytical signal, since fluorescence techniques
have long been known to provide high sensitivity (comparable to radioisotopic methods)
and detailed information about structure at the molecular level (Lakowicz, 1983,Principles of Fluorescence Spectroscopy, Plenum Press, NY). Changes in the polarity,
pH, temperature, microviscosity, or orientation of molecules in the local environment of
a fluorophore may result in alteration of the electronic structure or collisional
probabilities of the fluorophore. Such environmental changes may be detected by
monitoring fluorescent signal parameters such as intensity, wavelength, lifetime, or
polarization. For example, it is not uncommon for the efficiency of fluorescenceemission (quantum yield) and fluorescence lifetime of an intercalant fluorophore to
increase by an order of magnitude or more when inserted into the rigid and hydrophobic
base stacking region of a double-stranded nucleic acids with respect to that of the
unbound dye in solution.
The present invention utilizes, and is not limited to, the fluorescence intensity
response of the bound fluorophore via monitoring in a total internal reflection
configuration along the optical fiber substrate to quantify the presence of hybridized
nucleic acids at the surface of the fiber. The fluorescence intensity is directly
proportional to the amount of target nucleic acid or nucleic acid analog initially present
in solution. It is also possible to use the time dependence of the rate of change of the
CA 0220816~ 1997-06-18
- 63 -
fluorescence intensity increase upon hybridization to determine the concentration of
target nucleic acid.
The fluorophore of the present invention can be for example ethidium bromide
(EB). The ethidium cation (3,8-diamino-6-phenyl-5-ethyl-phenanthridium) is a
fluorescent compound which strongly associates with double stranded nucleic acids by
intercalation into the base-stacking region and, in some cases, the major groove of the
double helical structure (Monaco et al., 1993, Journal of Bimolecular Structure and
Dynamics,10: 675). The ethidium cation is particularly well suited for investigations of
nucleic acid hybridization for a number of reasons. Firstly, the quantum yield of the
dye is known to increase as much as 100-fold when intercalated into the base stacking
region with respect to that of the unbourid dye in aqueous solution (Bauer et al,1989,
Proceedings of the National Academy of Science USA, 56: 7937). Secondly, the
binding affinity and the fluorescence enhancement of the dye are independent of base
composition (Cuniberti et al, 1990, Biophysical Chemistry, 38: 11). Thirdly, intercalation
of the ethidium cation is known to increase duplex stability as the two 3,8-amino
substituents hydrogen bond with the internucleotide phosphate groups on each of the
DNA strands (whereas other intercalators are known to significantly decrease duplex
stability) (Cuniberti et al, 1990, Biophysical Chemistry, 38: 11). The absorption
maximum of ethidium bromide is 510 nm, which is sufficiently close to the outputwavelength of 488 nm of an Ar+ laser which may be used to excite the fluorophore. The
dye has an emission maximum of 595 nm when bound to DNA which is a sufficiently
large Stoke's shift to make separation of the emission radiation from the excitation
radiation straight forward, and to prevent inner filter effects, by the use of a dichroic
mirror or other standard optical components (Haugland, 1992, Molecular Probes:
Handbook of Fluorescent Probes and Research Chemicals, 5th Ed:, USA: Molecular
Probes Inc.). Due to the above mentioned reasons, the use of EB has been shown to
provide a sensitive means to de~ect the presence of nucleic acid duplexes for this
application.
CA 0220816~ 1997-06-18
- 64 -
A specific example of a tethered fluorophore is illustrated in the synthetic
schemes of Figure 3a,b, and c. In this case a modified ethidium-type dye with tether,
here C13 acid moiety, is synthesized as shown (Fig. 3a). The ethidium analogue with
acid tether is attached to 5'-hexylamine functionalized oligonucleotides immobilized on
the surface of an optical fiber to generate the biosensor with the tethered fluorophore
probe. For the case where the nucleotides are grown on the support via solid phase
phosphoramidite synthesis, the 5'-hexylamine functionalization can readily be achieved
through the use of the commercially available reagent Aminolink 2~
The fluorophore or reporter group may be attached to the 5'- or 3'-end of the
oligomer by not only a hydrocarbon tether but other types of tethers such as polyether,
mixed aliphatic/aromatic, or peptidic. The tether need not be restricted to the 3'- or 5'-
ends of the oligomer but may be attached to a terminal or internal ribo-residue via the
2'-hydroxyl (Yamana et al, 1991, Tetrahedron Letters, 32: 6347). Similarly, a tether can
be attached to a terminal or internal nucleobase using pyrimidines (Pieles et al, 1990,
Nucleic Acids Research, 18: 4355) or purines (Roduit et al, 1987, Nucleosides and
Nucleotides, 6: 349). Furthermore, the internucleotidic linkage can be a site for tether
attachment (Agrawal et al, 1990, Nucleic Acids Research, 18: 5419). Obviously, any
combination of these methods could be used to site specifically incorporate multiple
reporter groups.
The choice of fluorophores which may be tethered to the oligonucleotide include
organic intercalating complexes, such as the commonly used nucleic acid stain
ethidium bromide, thiazole orange and analogs thereof as prepared by L.G. Lee et al
(1986, Cytometry 7: 508) and the YOYO, BOBO, and TOTO series of cyanine based
intercalant fluorophores which are commercially available from Molecular Probes Inc.
(Eugene, OR). Inorganic coordination complexes, such as the "molecular light switch"
Ru(phen')2 dppz PF6 developed by Jenkins et al. (1992, J. Amer. Chem. Soc. 114:
8736) may also be used as well as groove binding dyes, such as Hoechst 33258 andHoechst 33342, which are commercially available from Aldrich Chemical Co.
(Milwaukee, Wl). These fluorophores are chosen such that the fluorescent probe is
CA 0220816~ 1997-06-18
- 65 -
quenched (non-emissive) when in the presence of single-stranded nucieic acids and
provides intense luminescence when in the presence of double stranded nucleic acids.
This change in observed luminescence occurs via changes in the relative rates ofradiative and non-radiative relaxation processes of the probe when the extemal
environment changes from aqueous solution to a hydrophobic and highly structured one
in the base stacking region of double-stranded nucleic acids.
Other examples of classes of fluorophores which can be used in the present
invention include acridine dyes, phenanthrides, phenazines, phenothiazines, quinolines,
alfatoxin, polycyclic hydrocarbons, oxirane derivatives, actinomyces, anthracyclinones,
thiaxanthenones, anthramycin, mitomycin, platinum complexes, polyintercalators,
norphilin-A, fluorenes and fluorenones, furocoumarins, benzodipyrones and monostral
fast blue. Preferred dyes are also those that provide large Stoke's shifts, can be
excited at long wavelengths and have large differences in fluorescence lifetime,quantum efficiency, and/or wavelength of excitation and emission when in solution as
compared to when bound to hybridized nucleic acids.
Light emitted from fluorophores (after direct excitation) at the surface of the fiber
is preferentially coupled back into the fiber and can be monitored by a photomultiplier
tube (PMT) or any other suitable light detection equipment. Increasing the length of
coated fiber results in a greater optical path length and better sensitivity (Krull et al,
1991, Fiber Optic Chemical Sensors and Biosensors, Vol. Il, O. S. Wolfbeis, Ed., CRC
Press, Boca Raton, pp. 315). Direct excitation of fluorophores in an immobilized layer
extending from the biosensor results in improved signal to noise ratio as interferences
from background fluorescence in the bulk environment are avoided.
One instrument used for fluorescence intensity measurements is based on a
fluorescence microscope as desc-ribed elsewhere (Brennan et al, 1990, Anal, Chim.
Acta., 237: 253) and shown in Fig. 4(a). An instrument as shown in Fig 4(b) may also
be used in which the output from a suitable light source, for example an argon ion laser,
is directed into an optical fiber via a lens with a numerical aperture which is equal to or
CA 0220816~ 1997-06-18
- 66 -
greater than the numerical aperture of the nucleic acid functionalized waveguide when
in the hybridization buffer solution used for analyte detection. The excitation radiation
may be coupied into a delivery fiber via a twisted optical fiber waveguide assembly such
that all modes carried by the first fiber into which the excitation radiation was first
coupled would be delivered to the second fiber to provide optimal excitation of
fluorophores associated with the biosensor. The excitation radiation may be totally
internally reflected along the length of the delivery fiber to a sensing fiber functionalized
with immobilized oligonucleotide and fluorophore. Coupling of the radiation between
fibers may be achieved by abutting the distal terminus of the delivery fiber to the
proximal terminus of the sensing fiber in a suitable non-fluorescent fiber coupler. The
terminus of the coupler is preferentially designed as a compression-fit end which
provides a solution-tight seal to prevent contaminants from diffusing int~ the fiber
coupler and causing drift in the analytical signal. The sensing fiber is situated within in
a small volume, stop-flow, hybridization chamber made of a suitable inert material with
good thermal conductivity (e.g. stainless steel or titanium). The temperature of the
hybridization may be controlled by use of a suitable thermoelectric housing to provide
rapid thermostating to the desired temperature and computer control. The temperature
of the solutions in the hybridization cell may be accurately determined ( ~ 0.2 ~C) by use
of a glass encapsulated thermistor incorporated into the hybridization cell. Solutions
delivery to the hybridization cell and sensing fiber may be done by use of a computer
controlled pump (e.g. peristaltic pump) where all solutions originate from a computer
controlled autosampler. Fluorescence emission from fluorophores associated with
immobilized nucleic acid complexes was totally internally reflected within the sensing
fiber. The portion of the light coupled back into the delivery fiber was directed towards
an interference filter with the appropriate bandpass window for the emission of the
fluorophore used with the optical sensor. Fluorescence radiation traversing the
interference filter then enters a photomultiplier tube to provide a quantitative measure of
the fluorescence intensity. In alternative embodiments, the radiation source can be a
frequency doubled laser, a semiconductor laser, bright lamp or LED. Coupling into the
waveguide can be accomplished with fiber couplers, and the detector can be an
avalanche diode rather than a PMT.
CA 0220816~ 1997-06-18
- 67 -
In one embodiment of the invention the biosensor operates as follows. The
optical fiber with attached fluorescently labeled single-stranded nucleic acid is placed in
a flow through cell and immersed in hybridization buffer solution. When single-stranded
nucleic acids or nucleic acid analogs which are complementary to the immobilizedstrands are introduced to the flow cell, hybridization occurs followed by intercalation (or
other suitable ligand binding motif) and enhanced fluorescence emission of the
attached fluorescent probe, as illustrated in figure 5. Fluorescence intensity is
monitored in a total intemal reflection configuration wherein the optical fiber and organic
coating form a waveguide to provide excitation to the surface immobilized nucleic acid
and fluorescent probe, as well as to collect fluorescence emission. By monitoring the
fluorescence intensity from the fiber, a rrieasure of the amount of target nucleic acid in
solution can be determined.
Regeneration of the biosensor can be achieved by thermal methods such as by
elevating the temperature within the flow-through hybridization cell or by chaotropic
methods in which solutions of highly polarized salts alter the hydrogen bonding
structure of the solution to affect denaturation of the hybridized complex. In either case,
the complex stability in the system is reduced to the point where hybridization is not
energetically favorable and the complement strands are dissociated from the covalently
immobilized oligomers and may be flushed out of the flow cell. Regeneration methods
as described herein can be employed to recycle biosensors.
Formation of multi-stranded nucleic acids (i.e. nucleic acid complexes composed
of 3 or more strands), such as triplex nucleic acids, may be determined from thetemperature dependence of the fluorescent signal. Normally, the fluorescence
efficiency of a fluorophore increases with decreasing temperature owing to the reduced
collisional deactivation as a consequence of the reduced kinetic energy of the
molecules surrounding the fluorophore. Fluorescence efficiencies with negative
-temperature coefficients are readily observed for fluorophores in solution and well as for
fluorophores intercalated into nucleic acids, as illustrated in figure 6. When multi-strand
CA 0220816~ 1997-06-18
- 68 -
formation occurs, (e.g. binding of a third strand in the major groove of a double-helical
nucleic acid) exclusion of the bound ligand often follows as the partition coefficient for
the fluorophore in the multi-stranded nucleic acid is often much reduced with respect to
that of the same fluorophore in double-stranded nucieic acid. The ligand exclusion
process will also show a temperature dependence where reduced ligand binding is
observed as the temperature of the system is decreased. As such, a positive
temperature coefficient of fluorescence intensity would be observed for fluorophores
associated with multi-stranded nucleic acids as increasing amounts of fluorophore
become excluded from the highly-structured environment within the nucleic acid
complex into bulk solution where the probability for collisional quenching of
fluorescence is far greater. A net positive temperature coefficient of fluorescence
iritensity would then be observed for a fluorescent nucleic acid binding ligand in a multi-
stranded nucleic acid. The temperature at which multi-strand formation occurs could
also be assayed from the maxima in a fluorescence intensity versus temperature plot
where the temperature coefficient changes from negative (for the dye bound in double-
stranded nucleic acid) to positive (for the dye being excluded from the multi-stranded
nucleic acid complex). This process is illustrated in Figs. 7(a & b) for triplex formation
on the sensor surface with linear and branched nucleic acids.
The biosensor of the present invention may then provide for rapid clinical testing
for viruses (e.g., HIV, T cell Iymphotropic virus 1 and 2, hepatitis B and C), and
pathogenic bacteria (e.g. E. coli., Salmonella, Listeria, Chlamydia ssp., Trichomonas
vaginalis, Gradenerella vaginitis) as well as other microorganisms. Detection of genetic
disorders (e.g., cystic fibrosis and sickle-cell anemia) and diseases such as cancer is
also contemplated by the method and apparatus of the present invention as well as
potential therapeutics to treat such diseases (e.g. branched antisense nucleic acids
which inhibit expression of targeted nucleic acid sequences via triplex formation with
that particular sequence, effectively shielding the genetic information from being read
by transcription enzymes).
CA 0220816~ 1997-06-18
- 69 -
Examples
Example 1.
Pl~"ardlion of Fused silica Optical Fibers Derivdli~l with Long Chain Aliphatic
er M~le~ -les Terminated with a 5'-0-dimethoxytrity1-2'-deoxythymidine
Nucleoside.
Plastic-clad silica optical fibers with a diameter of 400 ,um were purchased from 3M
Specialty Optical Fiber (North York, ON, Canada). The cladding on the fibers wasmechanically removed, and the fibers were cut to lengths of about 1 cm. One face on
each fiber was polished by suspending the fiber over (and placing the end face of the
fiber in contact with) the rotating plate of a Thermolyne type 37600 speed controlled
mixer (Sybron Corporation, Dubuque, IO, USA) onto which 1200 grade emery paper
was immobilized. All fused silica optical fibers were cleaned using a Harrick PDG-32G
plasma cleaner (Harrick Scientific Corporation, Ossining, NY, USA) before activation
with aminopropyltriethoxy silane (APTES).
The fibers were then washed with a 1:1 acetone/methanol mixture and stored in a
vacuum desiccator. The optical fibers were plasma cleaned for 5 minutes at low power
(40 W) and were placed in a solution of 1 :200 (v/v) aminopropyltriethoxy silane(APTES) in d~ toluene. This was done under a nitrogen atmosphere using glasswarewhich was oven dried and previously treated with octadecyltrichlorosilane. The structure
of the APTES coatings on fused silica substrates has previously been investigated by
Vandenberg etal. (1991, J. Colloid and Interface Sci., 147: 103). The method of Arnold
and co-workers (1989, Collect. Czech. Chem. Commun., 54: 523) was used to
synthesize an aliphatic spacer arrn terminated with 5'-O-dimethoxytrityi-2'-
deoxythymidine. In this method 1,10-decanediol was condensed with succinic
anhydride to form 1,10 decanediol bis-succinate, as illustrated in Figure 1(a). The bis-
succinate was reacted with N-hydroxysuccinimide and 5'-O-dimethoxytrityl-
2'deoxythimidine in the presence of N, N'-dicyclohexyl-carbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) to yield a nucleoside functionalized spacer molecule.
CA 0220816~ 1997-06-18
- 70 -
The spacer was then attached to the surface of the APTES treated optical fiber via
amide formation.
Example 2.
Pl ~,aralion of Fused silica Optical Fibers Derivati~.l with
Glycidoxypropyltrimethoxysilane Extended with Mono-Dimethoxytritylated
Hexaethylene Glycol Substrate Linker Molecules.
In order to grow oligonucleotides onto the surface of silica substrates (such asfused silica) by automated solid phase synthesis, the surface is functionalized with
spacer molecules of at least 25 A in length which had either an amine or a hydroxyl
functionality at the terminus of the spacer molecule. A chemically resistant, non-
hydrolyzable spacer molecule is employed. The method used was a modification of that
reported by U. Maskos and E.M. Southern suPra wherein the silica surface was treated
with glycidoxypropyltrimethoxysilane (GOPS), followed by extension via treatment with
hexaethylene glycol (HEG) under acidic conditions. For the purpose of creating
biosensors with higher sensitivity and lower detection limits, this method is
advantageous over the use of hydrocarbon tethers. The water soluble HEG linker will
provide a more fluid environment (which should not self-assembie) so as to improve the
ability of the immobilized DNA strands to hybridize with complementary material in
solution (in terms of energetics and kinetics). The hydrophilicity of the linker will also
facilitate the removal of adsorbed contaminants (e.g. proteins, organics) which may
occlude the surface and contribute to drift in the fluorescence intensity. However, as
HEG is bifunctional, there exists the possibility of creating non-reactive closed-loop
structures which may significantly decrease the loading of oligonucleotides on the
surface of the fibers. In order to eliminate this problem, one terminus of the HEG is
protected with a dimethoxytrityl functionality prior to extension with GOPS. This strategy
permits facile determination of the amount of support linkers bound to the silica surface.
Removal of the trityl protecting groups by treatment with acid yields the highly colored
trityl cation, which can be quantitatively measured by monitoring A(504 nm) of the
deprotection solution. Knowing there is one trityl group released per linker molecule
attached to the surface, the loading of HEG can easily be determined. Immobilization of
CA 02208l6~ l997-06-l8
- 71 -
a protected linker molecule provides the additional advantage that the hydroxyl groups
produced after the attachment of the HEG to the epoxide moiety and all other surface
silanols can be capped to prevent unwanted oligonucleotide growth at these sites. The
presence of side product oligonucleotides, which are prematurely terminated due to the
lack of a suitable support molecule, may decrease the sensitivity and selectivity of the
sensor. The additional charge imparted from the anionic backbone of a side product
strand may inhibit hybridization between the analyte strands and neighboring probe
sequences. See: R.T. Pon Methods in Molecular Bioloqy. Vol.20: Protocols for
Oligonucleotides and Analogs, S. Agrawa, Ed, 1993, Humana Press, Inc. Totowa NJ. In
conjunction with the use of non-hydrolyzable spacer molecules, t-butylphenoxyacetyl
protected phosphoramidite synthons were employed. This labile protecting group can
be quickly removed (i5 min ~ 55~C or 120 min. at room temperature as compared to12-16 hours ~ 55~C using 27% aqueous ammonia) thereby reducing the possibility of
cleavage of the immobilized sequences by hydrolysis of the silyl ether bonds which
ultimately anchor the strands to the fiber surface.
i) Cleaninq of Silica Substrates Prior to Functionalization with GOPS:
The buffer coating was mechanically stripped from the pre-cut optical fiber pieces (400
~lm diameter x 44 mm) and the cladding was dissolved by treatment with acetone. The
fused silica substrates, i.e., optical fibers or wafers, were added to a 1:1:5 (v/v) solution
of 30% ammonium hydroxide/30% hydrogen peroxide/water and the mixture was stirred
at 80~C for five minutes. The substrates were then removed and treated with a solution
of 1:1:5 (v/v) conc. HCI/30% hydrogen peroxide/water and the mixture stirred at 80~C
for five minutes. The substrates were then sequentially washed with methanol,
chloroform and diethyl ether, respectively, and dried in-vacuo.
ii) Synthesis and Purification of mono-dimethoxytritylated hexaethvlene qlycol (DMT-
HEG):
Dimethoxvtrityl chloride (7.19) was dissolved in 10 ml of dry pyridine and addeddropwise to a stirred solution of hexaethylene glycol (5.65 ml) in 5 ml of dry pyridine
under an argon atmosphere. Stirring was continued for 16 hours after which time the
CA 0220816~ 1997-06-18
- 72 -
reaction mixture was combined with 50 ml of dichloromethane. The dichloromethanesolution mixture was twice shaken with 900 ml portions of 5% aqueous sodium
bicarbonate and then with three 900 ml portions of water in order to remove unreacted
HEG, pyridine, and pyridinium salts. The product was purified by liquid chromatography
using silica gel and a solvent system of 0.1% triethylamine in 1:1 dichloromethane /
diethyl ether. The identity of the product was confirmed by proton NMR spectroscopy
(200 MHz).
iii) Functionalization of Fused Silica Substrates with 3-
GlycidoxvProPyltrimethoxvsilane (GOPS):
The cleaned fused silica substrates were suspended in a stirred solution
composed of 40 ml xylene, 12 ml GOPS, and a trace of Hunig's base at 80~C overnight.
The fibers were then sequentially washed with methanol, chloroform, ether, and then
dried in-vacuo.
iv) Linkaqe of DMT-HEG to GOPS Functionalized Silica Substrates:
The GOPS functionalized fibers were suspended in a stirred solution of 1:4:8
(v/v) DMT-HEG / diethyl ether / toluene containing a catalytic amount of sodium hydride
under an argon atmosphere. The reaction mixture was stirred for 14 days after which
time the fibers were removed and washed sequentially with methanol, chloroform,
ether, and then dried in vacuo.
v) CaPPinq of Unreacted Silanol and Hydroxyl Functionalities with Chlorotrimethyl-
silane:
The fused silica fibers functionalized with DMT-HEG were suspended in a
solution of 1:10 (v/v) chlorotrimethylsilane / pyridine for 16 hours under an argon
atmosphere at room temperature.
-
Example 3.
CA 0220816~ 1997-06-18
- 73 -
Pl ~,~.ardlion of Fused silica Optical Fibers Derivdli~ with Mono-
Dimethoxytritylated Hexaethylene Glycol Su~,stlate Linker Molecules via MesylateActivation.
The details of the preparation of the fused silica substrates and DMT-HEG
synthesis are provided in example 2(i and ii). DMT-HEG (0.5g) was suspended in 50 ml
anhydrous pyridine. The solution was maintained under an anhydrous argon
atmosphere and stirred. while 1.2 equivalents of methanesulfonyl chloride was added
dropwise. The reaction allowed to proceed for 60 minutes at room temperature with
stirring. The cleaned fused silica and silicon substrates were introduced into the
solution containing the mesylated DMT-HEG and the substrate functionalization
reaction allowed to proceed for 4 days with stirring at 40~C under an argon atmosphere.
Following the 4 day incubation period, the solution was decanted away from the
functionalized substrates which were then washed with copious amounts of
dichloromethane. Washings were continued until no discernible absorbance at 504nm
was observed from the wash solution made acidic by treatment with trichloroacetic acid.
The functionalized substrates were then capped as per the methods of example 2(v)
and stored in-vacuo and over P2O5 until needed.
Example 4.
Preparation of Fused silica Optical Fibers Derivdli~t:J with Mono-
Dimethoxytritylated Hexaethylene Glycol Phosphoramidite Substrate Linker
Molecules via Standard ~-Cyanoethylphosphoramidite Coupling on an Automated
Synthesizer.
The details for the preparation of the fused silica substrates and DMT-HEG are
provided in example 2(i) and 2(ii), respectively. DMT-HEG (0.5g) was suspended in a
solution consisting of 12 ml of anhydrous THF and 4 ml of anhydrous N,N-diisopropyl
ethylamine. The solution was maintained under an anhydrous argon atmosphere and
stirred at all times. 1.1 equivalents of 2-cyanoethyl-N,N-diisoproylamino-
phosphochloridite was added dropwise to the DMT-HEG solution and the reaction
allowed to proceed for 90 minutes at room temperature. TLC analysis (1:1 CH2CI2 /
diethyl ether) indicated quantitative formation of the DMT-HEG phosphoramidite
CA 0220816~ 1997-06-18
- 74 -
synthon (Rf = 0.7). The reaction product was thrice extracted into ethyl acetate from a
5% sodium bicarbonate solution. The organic phase was separated from the aqueouslayer, dried over NaSO4, filtered and the solvent removed under reduced pressure. The
product was then stored dry and at -20~C under an anhydrous argon atmosphere until
required. Functionalization of fused silica substrates was then done as part of a
standard coupling cycle using the automated solid phase DNA synthesizer and a 0.1 M
solution of the DMT-HEG phosphoramidite in anhydrous THF. The methods for
automated oligonucleotide synthesis are detailed in example 5. The capping procedure
detailed in example 2(v) was then done in order to block any undesired reactive sites on
the substrates.
Example 5.
Synthesis of Oligonucleotides onto Substrate Linker Functionalized Fused Silica
Waveguides.
All DNA synthesis was done by the well established ~-
cyanoethylphosphoramidite method with an either an Applied Biosystems 381A or 391
EP DNA Synthesizer using susbtrate linker functionalized controlled-pore glass beads,
fused silica optical fibers, planar fused silica wafers, or planar silicon wafers.
Automated solid-phase DNA synthesis is well known and is described in detail
elsewhere (Beaucage et al., 1992, Tetrahedron Letters, 48: 2223-2311;
Oliqonucleotides and Analogues: A Practical ApProach, F. Eckstein, Ed. Oxford
University Press, NY, 1991). The substrate linker functionalized optical fibers were
placed into an emptied Applied Biosystems (ABI) Oligonucleotide Purification Cartridge
column (OPC-column) or 10 ,umol scale synthesis column with the dead volume being
taken up by inert polyethylene pieces. The end filter papers were replaced (ABI) and
the column ends were crimped closed using aluminum seals. Synthesis of oligomersonto the optical fibers was carried out at the 0.2 ~Lmol. scale with a pulsed-delivery cycle
in the "trityl off" mode. The ~-cyanoethylphosphoramidite cycle was used as supplied by
ABI with the exception of extended nucleoside coupling times (2-10 min.) and increased
solution delivery times to accommodate the larger synthesis columns. With the
exception of thymidine synthons, t-Butylphenoxyacetyl protected phosphoramidite
CA 0220816~ 1997-06-18
- 75 -
synthons were used in conjunction with a t-butylphenoxyacetic anhydride capping
solution as suppiied by Millipore Inc.
In the case where polythymidilic acid oligonucleotides were grown on the opticalfibers, deprotection of the phosphate blocking groups from the immobilized oligomer
was achieved by standing the fibers in a solution of 2:3 triethylamine/acetonitrile at
room temperature for 1.5 hours. This procedure caused the loss of the phosphate
blocking group via ,13-elimination while not cleaving the single-stranded DNA (ssDNA)
from the optical fibers. In the case where oligonucleotides containing bases other than
thymine were grown, the following protocol was used for phosphate and nucleobasedeprotection. A 30% ammonium hydroxide solution was drawn up into the synthesis
column containing the optical fibers functionalized with immobilized oligonucleotides
using a syringe and a male-to-male luer adapter. The fibers submerged in ammoniawere allowed to stand for two hours at room temperature after which time the ammonia
solution was expelled from the column and the contents of the column were washedfive times with 5 ml portions of sterile water. The deprotection solutions and washings
were collected and concentrated to a total volume of 1 ml. A260nm of the concentrated
deprotection solution was measured in order to determine the quantity of DNA liberated
from the fused silica substrates. Based on the results of the trityl cation assay and
A260nm of the deprotection solution, it was found that -20% of the oligomers remained
attached to the surface following the ammonia deprotection procedure. In the case
where oligonucleotides of mixed base sequence were grown onto optical fibers, the
oligonucleotide sequence was (5'-TAG GTG AGA CAT ATC ACA GA-3' ), which is a
nucleic acid probe for the E03 forward sequence of the Candida albicans genome.
Fibers coated in ssDNA were either stored dry or kept in a solution of 1:1
ethanol/water. All fibers were cleaned prior to use by sonication in a solution of 1:1
ethanol/water for 5 minutes in order to remove any fluorescent contaminants adsorbed
to the surface of the fibers.
Example 6.
CA 0220816~ 1997-06-18
- 76 -
Biosensor Characterization by Trityl Cation Assay.
All oligonucleotide syntheses were evaluated by spectroscopic quantitation of
trityl cation released during the trichloroacetic acid treatment steps of the automated
synthesis. The collected fractions of trityl cation were diluted with 2.0 ml of 5% TCA in
1 ,2-dichloroethane.immediately prior to making absorbance measurements. Absorption
at 504 nm was measured in order to detemmine the concentration and the total number
of trityl cation moieties released during each TCA deprotection step of the synthesis. In
this way, the total number of oligomers successfully grown onto the solid supports was
determined.
As there exists no discernible decrease in the amount of trityl cation released
during successive deprotection steps, it may be safely assumed that the couplingefficiency of 99.5% or better suggested by the manufacturer of the automated
synthesizer (ABI) was achieved. The results of a trityl cation assay for a synthesis of
dT20 onto optical fibers by the methods given in Examples 1 and 5 are shown in Fig. 8.
Example 7.
Generation of Complementary and Non-complementary Nucleic Acids.
Synthesis of dA20 and rA20 was done using a conventional LCAA-CPG support
with the ,~-cyanoethylphosphoramidite cycle supplied by ABI. A nonadecamer of
random base composition (dR19) was also prepared by simultaneously introducing all
four phosphoramidite reagents to the column at each coupling step. Standard
deprotection with aqueous ammonia (29%, 1.5 ml, 24 h) was used to liberate the
oligomers from the solid support and remove the base protecting groups. For the case
of the rA20, deprotection of the phosphate blocking groups, base protecting groups and
cleavage from the CPG support was done by treating the oligomers with 1.5 ml of a
solution consisting of 4 parts aqueous ammonia and 1 part ethanol for 4& hours at room
temperature. The aqueous solutiori containing the. oligonucleotides was then collected,
evaporated to dryness, and the residue treated with 300 ~l of an anhydrous solution of
1 M tetra-N-butyl ammonium fluoride in THF overnight at room temperature. After the
incubation time, the reaction was quenched by adding 1 ml of water to the reaction
CA 0220816~ 1997-06-18
- 77 -
mixture. Crude oligomer was purified by polyacrylamide gel electrophoresis and reverse
phase liquid chromatography or size exciusion chromatography.
Example 8.
Detection and Quantification of Complement DNA (cDNA), Complement RNA
(cRNA) and Non-comFle.,.E..t Nucleic Acid by the Optical Sensor Fabricated by
the Ploc~.~ures of Examples 1 and 5.
An optical fiber functionalized with polythymidilic acid icosanucleotide was
selected at random from the batch of fibers (ca. 25) created in example 1 and was
positioned under the objective of the microscope, as illustrated in Fig. 4(a). In this
orientation the incident laser radiation entered the proximal terminus and was totally
intemally reflected throughout the fiber. The majority of the fiber was submerged in a
hybridization buffer solution consisting of 1.0 M NaCI and 50 mM sodium phosphate
(pH 7.4) in sterile water contained within a 4 ml plastic cuvette. Hybridization buffer was
passed through an acrodisc~ filter immediately prior to introduction to the cuvette.
Emitted fluorescent radiation, from the stimulated fluorescent molecules associated with
the double-stranded nucleic acids, was directed back towards the microscope by total
intemal reflection. The emission from the fluorescent molecules was separated from the
excitation radiation by a dichroic mirror and directed to a photomultiplier tube. The
photomultiplier tube provided measurements of the intensity of fluorescence emission.
Fluorescence intensity values are reported with the system at 25~C to avoid
inconsistencies cause by the temperature dependence on fluorescence quantum
efficiency and as relative quantities, thereby obviating the need to control experimental
parameters such as laser intensity, optical alignment and photomultiplier tube (PMT)
gain which are beyond accurate control from day to day.
In order to affect hybridization with the immobilized nucleic acid strands, dA20ssDNA was added to the plastic cuvette containing the suspended fiber in fresh
hybridization buffer at a temperature of 85~C. This temperature was chosen as it is
sufficiently greater than the 60~C duplex melting temperature (Tm~ the temperature at
which half of all the duplexes present are dissociated) and is well below the boiling point
CA 0220816~ 1997-06-18
- 78 -
of the buffer. Incubation at temperatures below Tm has been shown to cause
incomplete hybridization wherein only a fraction of the bases on each strand interact to
form partially hybridized complexes (Rubin et al, 1989, Nucleic Acid and Monoclonal
Antibody Probes, B. Swaminathan and G. Prakash, Eds., Marcel Dekker, Inc., NY, pp.
185-219). Though covalent immobilization of ssDNA removes one degree of freedom
from the oligomer, hybridization at temperatures initially above the duplex Tm ensures
the formation of duplexes with the greatest possible extent of overlap. In all cases, no
appreciable intensity change from that of the baseline was observed after the 90 minute
incubation period. The solution was allowed to stand and cool to room temperature
(25~C) between 30 and 90 minutes after which the fiber was flushed with 60 ml ofhybridization buffer (25~C) to remove the excess strands.
Intercalation of the fluorophore into the dsDNA was achieved by injecting 10 ~l of
a 1 mg-ml ' aqueous solution of ethidium bromide (EB) into the cuvette and allowing the
solution to stand for 15 minutes followed by washing the fiber by flushing the cuvette
with 60 ml of fresh hybridization buffer (25~C)
The response of the fiber optic DNA biosensor to EB and cDNA is shown in
Figure 9. As a control experiment, 10 ,~L of a 1 mgml~1 aqueous solution of EB was
added to the cuvette in which the fiber functionalized with ssDNA was suspended. After
15 minutes, 60 ml of fresh hybridization buffer (25~C) was flushed through the cuvette
in order to remove any non-specifically bound ethidium cation and no discernibleincrease in fluorescence intensity from the fiber was observed. A 104 + 15% increase
in the fluorescence intensity was observed from the fiber which was exposed to 189
ng.ml-1 of cDNA and stained with EB relative to the baseiine value for the cleaned
sensor with only ssDNA on the waveguide surface. The fiber was regenerated by
flushing the cuvette and optical sensor 30 ml of hot (85 ~C) buffer solution over a period
of c.a. 30 seconds and the system allowed to stand for five minutes. After the five
minute wait, an additional 30 ml of hot buffer was flushed through the cuvette to wash
away the dissociated cDNA strands. This procedure is known to melt DNA duplexes as
CA 0220816~ 1997-06-18
- 79 -
the buffer temperature was well above the Tm of the dsDNA. The fluorescence intensity
returned, within experimental uncertainty, to the initial intensity observed at the
beginning of the experiment. The same hybridization experiment was repeated where
the optical sensor was exposed to 757 ng.ml-1 of cDNA during the hybridization
procedure. This yielded an increase of 720 + 20% in the observed fluorescence
intensity from the sensor with respect to the baseline value. In contrast, a similar
concentration of non-complementary sequences (a nonadecamer of random base
sequence) gave essentially no response, as shown in Fig. 9(a).
A 3.8 ng-~ solution of rA20(45o~Ll) was introduced into a cuvette containing hothybridization buffer and the sensor to provide a 570 ng ml~1 solution of cRNA. The
same hybridization and staining procedure as used for cDNA was followed. The
response profile for this hybridization procedure is shown in Fig. 9(b). A comparison of
response of the biosensor with immobilized dT20 to cDNA and cRNA agree to withinexperimental error.
Example 9.
Effective of Ethidium Bromide (EB) Staining Time and Concentration
The staining time of the sensor with EB, was changed after each hybridization
with cDNA. For each determination, injections of 30,u1 of 56.8~lg-ml~1 solution of
aqueous dA20 were made and .the hot hybridization buffer in the cuvette, which
contained the cDNA strands, was allowed to cool to room temperature over a time of 30
minutes. A 1 mg-ml~1 solution of EB in water (I0~11) was added to the cuvette after each
hybridization to provide an EB concentration of 8.4 X 10-3 M. A staining time of 20 min.
with 8.4 X 10-3 M EB was required to generate 299% of the full signal, as shown in Fig.
1 O~a).
To study the effect of EB concentration during dsDNA staining, all hybridizationparameters were the same as those used to study staining time and a staining time of
20 min. was used. Staining with EB solutions of concentrations of 8.5 X 10-3M or
CA 0220816~ 1997-06-18
- 80 -
greater were required to generate 299% of the full staining in 20 min., as shown in Fig.
1 O(b).
Examp/e 10.
Long Term and Thermal Stability of the Nucleic Acid Sensor.
The robustness of the optical sensors, and DNA as a biorecognition element,
was made evident by the maintenance of activity after long term storage and vigorous
cleaning conditions. Fibers that were stored for over 1 year in vacuo, in 1:1
ethanol/water solutions, absolute ethanol, or dry at -20~C provided identical response
characteristics to freshly prepared fibers. Adsorbed fluorescent contaminants which
were accumulated through long temm storage were completely removed (as confirmedthrough fluorescence microscopy) by sonicating the fibers in a solution of 1:1
ethanol/water with full maintenance of activity and sensitivity. Figure 11 (a) shows the
response of a 1 month old fiber (stored in vacuo) used with no cleaning of the surface
and (b) an 11 month old fiber (stored dry at -20~C) which had been cleaned by
sonication in ethanol solution. It should be noted that the sensitivity of the cleaned 11
month old fiber is identical to that of 1 month old fibers cleaned by the same procedure
(data not shown). Cleaning of the sensor by sonication prior to use has consistently
been observed to increase the sensitivity of the device by a factor of c.a. 2.5. The
sensors have provided femtomolar detection limits and a response which is linear with
the concentration of cDNA (M.W. = 6199 g mol~'). The regression lines shown in figure
11 show good fits to the data points with r2 values of 0.965 and 0.968 for the 1 month
and 11 month-old fibers, respectively. From this data, the sensitivity of the optical
sensor (11 month-old, fabricated by the protocols of examples 1 and 5) was determined
to be an increase in fluorescence intensity of 203% per 100 ng ml-' of cDNA with a
measured limit of detection of 86 ng ml-'. Maintenance of calibration has been
observed for all experiments done thus far in which as many as 5 regenerations have
been done over durations of up to 12 hours.
The ability to clean and sterilize a bioprobe or biosensor device so that it may be
usable in an on-line configuration is a significant advantage. As the specific binding
properties of nucleic acids are based on secondary structure, the use of nucleic acids in
CA 02208l6~ l997-06-l8
- 81 -
biosensor fabrication leads to devices which are not only stable to prolonged storage,
but also to harsh washing conditions and sterilization. A summary of the effects of
cleaning by sonication in absolute ethanol (15 minutes) and autoclaving (120 ~C for 20
minutes at 4 atmospheres pressure in sterile water) on the response of the sensors to
(-400 ng mL~1) is shown in the Table which follows. Both sonication in ethanol and
autoclaving are observed to improve the response of the sensor, most likely through the
removal of contaminants on the surface of the sensor (stored dry for 11 months, or
stored in ethanol).
Table 1. Effect of storage conditions, cleaning, and sterilization on sensor response to
400 ng ml~~ cDNA.
StorageConditions Cleaning/Sterilization Relative Fluorescence
Conditions Intensity Increase (%)
1 :1 Ethanol/Water (25~C) - 333+20
95% Ethanol (25~C) - 395+20
Dry (-20~C) - 341~20
1:1 Ethanol/Water(25~C) Autoclave 430+20
1:1 Ethanol/Water(25~C) Sonication 453+20
Example 11.
Thermal Denaturation Studies of the cDNA: Immobilized DNA Complex on the
Sensors Create~l from the Protocols of Examples 1 and 5 and Comparison to that
of the Same Oligonucleotide Complex in Solution.
i) Thermal Denaturation InvestiqaVons of Aqueous dT2n with Aqueous dA~n.
Equimolar amounts of each oligomer in hybridization buffer (1 M NaCI, 1 OmM
PO4, pH = 7.0) were mixed so that the final concentration was approximately 1 ~JM in
each strand. Prior to thermal melt studies the oligonucleotide mixture was heated
CA 0220816~ 1997-06-18
- 82 -
bnefly to 80~C and slowly cooled to 20~C in order to hybridize all of the strands. The
samples were held at the low temperature limit for 15 minutes prior to initiating melt
studies, to allow for thermal equilibration. The temperature was then ramped at 0.5~C
intervals at a rate of 0.5~C/min. while the absorbance was recorded at 260 nm.
ii) Melt-Cun/e Investiaations of Immobilized dT~n with cDNA.
Sequences of dT20 were immobilized onto planar fused silica wafers (5 mm x
10mm x 1mm) according to the protocols in examples 1 and 5. The immobilized dT20was hybridized with complementary dA20 sequences by immersing the wafer in a 56.8
ng ml~' solution of dA20 at 85~C and allowing the immersed wafer to cool to roomtemperature (25~C). The wafer was then removed from the cDNA solution and washedwith room temperature hybridization buffer solution. The wafer was then suspended in
a quartz cuvette that was placed in the temperature controlled cuvette housing of the
UV-vis spectrometer. The placement of the wafer was adjusted so that it rested in the
path of the light beam. The dead volume beneath the wafer was taken up by inert
packing material. Absorption spectra were collected at approximately 2~C increments
of temperature in the range from 29~C to 76~C. The temperature in the cuvette was set
by programming an extemal circulating bath to a specific temperature and the
temperature of the buffer solution surrounding the fused silica wafer was quantitatively
measured using a silanized glass encapsulated thermistor. Measurements of
absorption at each temperature were done by integrating 100 spectra in the wavelength
range between 220 nm and 320 nm.
iii) HyPochromicitv and Melt-Curve Thermodvnamics.
The transition between an ordered duplex state and the disordered denatured
state for systems of complementary nucleotides can be monitored and analyzed by UV-
Visible absorbance spectroscopy to determine the duplex melting temperature (Tm)~
The extent of hybridization (i.e. the number of base pairs formed per duplex) was
CA 0220816~ 1997-06-18
- ~3 -
determined by a comparison of melt profiles for the immobilized oligonucleotides to
similar reported values and dA20 + dT20 in solution.
The fraction of single strands present in the system at any temperature (fss(T))may be determined through the use of the following equation:
55 A5s (T) - Ads (T)
where A(T), Ass(T), and Ads(T) are the absorbances of the experimentally obtained
melting curve, the upper baseline (single stranded oligomers), and the lower baseline
(double stranded oligomers) respectively at temperature T(Nelson, J.W.; Martin, F.H.;
Tinoco Jr., I. Biopolymers 1981, 20, 2509-2531.). By plotting fss against temperature,
the duplex melting temperature can be obtained by determining the temperature atwhich fss~ 0 5
iv) Melt-Curve Studies of Support Bound Du~lex DNA and Aqueous Phase DNA.
The purpose of the thermal denaturation studies was to examine whether linkage
of an oligonucleotide to a solid support through a terminal nucleotide phosphate would
cause the loss of degrees of freedom with respect to the avaiiability of each nucleotide
to partake in formation of the double stranded structure. Melt profiles for the thermal
denaturation of dsDNA immobilized on the surface of a fused silica wafer and dsDNA in
solution were obtained, and the results of these investigations are summarized in Fig.
12. The duplex melting temperature of the immobilized strands with aqueous phasecomplement strands was 62.4 + 0.3~C. The Tm value for the aqueous phase dA20 +
dT20 duplex was determined to be 60.5~C using the software supplied by Varian.
Kibler-Herzog et. al. (Kibler-Herzog, L.; Zon, G.; Whittier, G.; Mizan, S.; Wilson, W.D.
Anti-CancerDrug Design 1993, 8, 65-79.) have reported the melting temperature of a
dA19 + dT19 duplex in 1.02 M NaCI to be 61 .1~C. This suggests that for the immobilized
oligomers investigated in this workl the extent of hybridization was complete with base
pairing of 20 bases per strand. The small differences in the three Tm values may be
accounted for by the fact that each of these experiments was done on a differentinstrument at different times, and the salt concentration used in this work was slightly
CA 0220816~ 1997-06-18
- &~ -
lower than that used by Kibler-Herzog et al. As the duplex stability in low ionic strength
buffers is less than that in high ionic strength buffers, it would be expected that the
melting temperature of the immobilized dT20 + dA20 duplex would be higher in the buffer
of lower stringency (Puglisi, J.D.; Tinoco Jr., I.; Methods in Enzymology, 1989, 180,
304-325.). In addition to this, a greater value of Tm for the immobilized duplex over the
aqueous phase duplexes should not be considered unusual as only one of the strands
will experience a significant gain in entropy upon melting of the immobilized duplex.
These factors lead to the conclusion that, within experimental uncertainty, the
immobilized dT20 + dA20 duplexes were more stable, if not as stable, as the aqueous
phase dA20 + dT20 and dA19 + dT~g duplexes. This also suggests that no hindrance of
duplex formation is observed with respect to the availability of the bases for
hybridization. This result is in accord with the investigations of Wolf et al. (Wolf, S.F.;
Haines, L.; Fisch, J.; Kremsky, J.N.; Dougherty, J.P.; Jacobs, K. Nuclelc Acids
Research 1987, 15, 2911-2926.), in which oligonucleotides bound to solid supports via
a long chain aliphatic tether at the strand termini (3'-end) were not observed to be
hindered with respect to hybridization efficiency
Example 12.
Detection of cDNA with and Optical Sensor functionalized with an
Oligonucleotide Probe Sequence for Candida albicans.
Optical Sensors were created by the protocols in examples 2 and 5 where the
oligonucleotide sequence (5'-TAG GTG AGA CAT ATC ACA GA-3' ), which is a nucleicacid probe for the E03 forward sequence of the Candida albicans genome, was
assembled onto the substrate linker functionalized fibers. Hybridization and staining
protocols as reported in example 9 were followed. The response of the sensor to cDNA
(20 nucleotides in length) is shown in Fig. 13. Linear calibration (r2=0.988), good
sensitivity (100% fluorescence intensity increase per 89 pM increase in concentration in
the 4ml of solution surrounding the optical sensor) and low detection limits (6 x 101~
molecules) were observed for the device.
. CA 02208165 1997-06-18
J ~
Example 13.
Fabrication of optical sensors with immobilized Polythymidilic Acid
Icosan~ otides functionalized at the 5'-terminus with N5-Tethered 3,8-Diamino-
6-phenylphenanthridium Cation.
i) Synthesis of methyl-(12-hydroxy)dodecanoate.
12-hydroxydodecanoic acid (59) was dissolved in 100 ml of dry methanol to
which was added a solution of p-toluenesulfonic acid (88 mg) in 5 ml of methanoldropwise over a 15 minute time-span. The solution was refluxed for 16 hours after
which time the solvent was removed under reduced pressure. The product was then
twice extracted into chloroform from a 5% aqueous solution of sodium bicarbonate.
The organic phase was recovered, dried over NaSO4, and the solvent removed underreduced pressure.
ii) Tosvlafion of methyl-(12-hvdroxy)dodecanoate
Methyl-(12-hydroxy)dodecanoate (1.6 g, 7 mmol) was placed in and oven dried
flask cooled under anhydrous argon and treated with 3 ml of a solution of p-
toluenesulfonyl chloride (1 eq., 7 mmol, 1.31 g) in dry pyridine. The solution was stirred
at 25~C under an inert atmosphere for 16 hours. The solvent was then removed under
reduced pressure and the tosylated product was stored dry at -20~C until needed.
iii) N-A/kylation of 3.8-dinitro-6-Phenyl-Pnenanthridine wifh the tosvlate of methvl-(12-
hydroxy)dodecanoate.
3,8-Dinitro-6-phenyl-phenanthridine (3.5 mmol, 1.2g) was combined with the
tosylated methyl-(12-hydroxy)dodecanoate (7 mmol, 2.7 g) in dry nitrobenzene and the
solution stirred for 6 hours at 160~C under an argon atmosphere. The alkylated
quaternary ammonium salt was precipitated from the mother liquor by addition of diethyl
ether and collected by filtration. The product was further purified by silica gel column
chromatography (25% methanol in chioroform) and recovered as a dark purple solid.
CA 0220816~ 1997-06-18
- ~6 -
ivJ Reduction of 3.8-dinitro-5-methvldodecanoate-6-Phenyl-Phenanthridium Chloride
3,8-Dinitro-5-methyldodecanoate-6-phenyl-phenanthridium chloride (1.3mmol,
0.729) was dissolved in 10 ml of THF and stirred over NiCI2-6H2O (10.68g) and
powdered Al (0.81g). Water (0.3 ml) was then added to initiate the formation of the
black Ni / Al catalyst and the reaction allowed to proceed for 15 minutes. The solution
containing the reduced product was recovered by filtration, followed by removal of the
solvent under reduced pressure. The product was purified by silica gel column
chromatography (25% methanol in chloroforrn) and recovered as a dark purple solid
(4%, 0.03g)-
vJ Tritylation of 3.8-diamino-5-methyldodecanoate-6-phenYl-phenanthridium Chloride
3,8-diamino-5-methyldodecanoate-6-phenyl-phenanthridium Chloride (0.039, 62
mol) was dissolved in dry pyridine and treated with dimethoxytrityl chloride (3 eq, 68
mg) suspended in dry pyridine (4ml). The reaction was allowed to proceed for 16 hours
at 25~C with stirring under an inert atmosphere. The solvent was then removed under
resuced pressure and the product purified (58%, 36,umol) by reverse phase HPLC
(isocratic elution with 1:1 methanol / water).
vi) DeProtection of the methyl-ester protectina qroup on 3.8-Bis(dimethoxytritylamino)-5-
methvldodecanoate-6-phenyl-~henanthridjum Chloride
3,8-Bis(dimethoxytritylamino)-5-methyldodecanoate-6-phenyl-phenanthridium
chloride (36 ,umol) was suspended in 80ml of a solution of 1:3 water / methanol. The
solution was degassed and treated with KOH (4 eq., 160 ,umol) for 16 hours with stirring
at 25~C. The reaction was quenched and the pH neutralized by treatment with HCI (1
eq, 1 5,ul of conc.) .
vil) SYnthesis of 5'-aminohexYl-dT~n Functionalized OPtical Sensors.
DMT-HEG-GOPS functionalized optical fibers (prepared according to the method
of Examples 2) were functionalized with polythymidilic acid icosanucleotide (according
to the method of example 5) temminated an N-trifluoroacetamide protected aminohexyl
moiety at the 5'-end by use of the commercially available Aminolink 2~ phosphoramidite
CA 0220816~ 1997-06-18
- 87 -
synthon from ABI. Deprotection of the phosphate blocking groups from the immobilized
oligomers was achieved by standing the fibers in a solution of 2:3 (v/v)
triethylamine/acetonitrile at room temperature for 1.5 hours. Removal of the
trifluoroacetamide protecting group on the aminohexyl functionality located at the 5'-end
of the immobilized strands was done by exposing the fibers to a 103 M solution of
sodium borohydride in absolute ethanol for 1 hour at room temperature. The fibers
were then washed once in a solution of 10-3 M HCI followed by washing with copious
amounts of sterile water.
viii) Attachment of the Tritvl-Protected Tethered Ethidium Analoque to the Aminohexyl
Functionalked Optical Fibers:
~ The fully deprotected fused silica optical fibers functionalized with 5'-aminohexyl
polythymidilic acid icosanucleotides were immersed in a solution containing 5mg of the
DMT-protected tethered ethidium analogue, 40,u1 of 1-methylimidazole, and 1.91 9 of 1-
ethyl-3-(3-dimethylaminopropyl)carbodiimide in 50 ml of water. After a 7 day incubation
period at room temperature, the fibers were washed five times each with 50 ml portions
of water, ethanol, and dichloromethane, respectively. The proportion dye-functionalized
oligonucleotides was determined by measurement of the amount of dimethoxytrityl
released from each detritylation step during automated synthesis and that from the
deprotection procedure used to restore the primary amine moieties on the dye. From
these assays it was determined that 63% of the immobilized oligonucleotides werefunctionalized with tethered dye.
ix) Characterization of the Fluorescence Response of the F~eaqentless Sensors with
Tethered Fluorophore:
The response of the reagentless sensor to 720 ng of complement DNA is shown
in Fig. 14. Hybridization was done at 40~C in a buffer consisting of 1 M NaCI and 50
mM phosphate (pH 7.0). It should ~e noted that this sensor has a significantly improved
response time over the sensors without tethered dye. 99% of the full analytical signal
was reached in c.a. 6 minutes after injection of the complementary strands for the
CA 0220816~ 1997-06-18
- 88 -
reagentless system while 45 minutes was required for full signal generation by the
sensors without tethered dye (see figure 1 0{a}).
Example 14.
Detection of TAT Triple-Helical DNA Using a Fiber Optic Biosensor
i) Backqro~Jnd
One important avenue not yet explored by the fiber-optic nucleic acid biosensor
community is the investigation of triple-stranded oligonucleotide formation. Typically, a
number of spectroscopic techniques (CD, NMR, UV and fluorescence spectroscopy) in
addition to gel mobility shift assays need to be implemented in order to study the
formation of triple-helical nucleic acids. However, each of these methods have
problems in terms of either the amount of material that is required for analysis (NMR,
CD, and gel mobility assays), or that they are limited to investigations of only certain
triplex systems (e.g. only TAT triplexes can be monitored by UV absorption
spectroscopy at 284nm).
Various groups have developed methods for triplex detection ({i} Geselowitx,
D.A.; Neumann, R.D. Bioconjugate Chem., 1995, 6, 502. {ii} Bates, P.J., Dosanjh, H.S.;
Kumar, S.; Jenkins, T.C.; Laughton, G.A.; Neidle, S. Nucleic Acids Res., 1995, 23,
3627.). The use of nucleic acid bindin~ ligands to identify DNA structures and
morphology is one such method. Many ligands are known to interact in a noncovalent
manner with the target oligonucleotide. Binding modes can be characterized as: (i)
intercalation of the ligand, in which typically a planar aromatic moiety slides between
the DNA bases - stabilized by ~ stacking and dipole interactions, or (ii) minor or major
groove interaction which is stabilized by hydrogen bonding, hydrophobic and/or
electrostatic interactions (Long, E.C.; Barton, J.K. Acc. Chem. Res. 1990, 23, 273.).
Ethidium bromide binds to both duplexes and triplexes via an intercalative mode
(Waring, M.J. Biochim. Biophys. Acta, 1966, 114, 234.), and this has been studied
extensively by fluorescence methods. The fluorescence quantum efficiency of the
ethidium cation increases when intercalated into duplexes (LePecq, J.B.; Paoletti, C. J.
Mol. Biol., 1967, 27, 87.) and triplexes (Mergny, J.L.; Collier, D.; Rougée, M.; Montenay-
CA 0220816~ 1997-06-18
- 89 -
Garestier, T.; Hélèn, C. Nucleic Acids Research, 1991, 19, 1521., Scaria, P.V.; Shafer,
R.H. J. Biol. Chem., 1991, 266, 5417), however, it has been shown that there is a
marked difference in the binding efficiency and hence fluorescence intensity between
the two types of complexes. LePecq and Paoletti were the first to observe that the
fluorescence enhancement of ethidium during interaction with the duplex (poly rA) (poly
rU) was significantly greater than for binding to the triplex (poly rA) (poly rU)2 (LePecq,
J.B.; Paoletti, C. C. R. Acad. Sc. Paris 1965, 260, 7033.). More recent studies have
confirmed that the fluorescence intensity of intercalated ethidium bromide is greater for
duplexes than triplexes of ribonucleic acid, and that a temperature dependence exists
for deoxyribonucleic acids (Mergny, J.L.; Collier, D.; Rougée, M.; Montenay-Garestier,
T.; Hélène, C. Nucleic Acids Res., 1991, 19, 1521., Scaria, P.V.; Shafer, R.H. J. Biol.
Chem., 1991, 266, 5417., Fang, Y.; Bai, C.L.; Zhang, P.C.; Cao, E.H.; Tang, Y.Q.Science In China (Ser. B), 1994, 37, 1306.). The results of molecular modeling studies
suggest that a reduced binding affinity of ethidium for triplexes (relative to duplexes)
exists due to the energetic cost of destacking base triplets as compared to successive
base pairs (Sun, J.S.; Lavery, R.; Chomilier, J.; Zakrzewska, K.; Montenay-Garestier,
T.; Hélène, C. J. Biomol. Struct. Dynam., 1991, 9, 425.). This is partially offset by the
quantum efficiency of ethidium bromide in triplex DNA which is greater than that for
duplex DNA. Short homopolymeric TsAT triplexes have been the subject of seminal
fluorescence studies. Letsinger et al. (Salunkhe, M., Wu, T. & Letsinger, R.L. (1992) J.
Am. Chem. Soc. 114, 8768-8772.) have shown that for parallel T~AT triplexes, thefluorescence intensity of ethidium cation decreases dramatically in comparison to
fluorescence intensity of the ligand bound to AT duplexes. Independent confirmation of
decreased fluorescence intensity for ethidium bound to parallel T~AT triplexes
(2XdT1o:dA1o) relative to duplexes (dT1o:dA10) has appeared (Fang, Y.; Bai, C.L.; Zhang,
P.C.; Cao, E.H.; Tang, Y.Q. Science In China (Ser. B), 1994, 37, 1306.).
We chose to investigate both parallel and antiparallel TAT triplexes as these
sequences have been well documented in the literature (Plum, G.E.; Pilch, D.S.;
Singleton, S.F.; Breslauer, K.J. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 319, and
references therein). Branched nucleic acids as described by Damha et al. (Hudson,
CA 0220816~ 1997-06-18
-- 90 --
R.H.E.; Damha, M.J. Nucleic Acids Res. Symp. Ser. 1993, 29, 97., R. Hudson, A.
Uddin, and M. Damha J. Am Chem. Soc., 1995, 117, 12470.) were used in this studyas the unique architecture of bNAs has been utilized to stabilize reversed-Hoogsteen
and Hoogsteen TAT triplexes. Investigations were also done to deterrnine the best
oligonucleotide orientation on the support for the detection of TAT triplexes. The
motivation behind this research endeavor was then to create a rapid, reliable,
reproducible assay for the detection of triple-helical nucleic acid formation. The
development of a triple-helical assay, is an extension of work initiated for the detection
of nucleic hybridization (Watson-Crick motif) using fiber optic total internal reflection
fluorecence (TIRF) sensors functionalized with single-stranded deoxyribonucleic acid
(ssDNA) probes.
ii) Svnthesis of Oliqonucleotides on OPtical Fibers.
Optical fibers were activated by protocols given in example 2 and polyadenilic
decanucleotides were assembled onto the substrate linker molecules on the fiber
surface as per the methods given in example 5. Two batches of fiber were created, the
first using commercially available N6-phenoxyacetyl-5'-O-DMT-2'deoxyadenosine-3'-O-
[(~-cyanoethyl)N,N-diisopropyl]phosphoramidite from Millipore Inc. to assemble
decanucleotides with the 5'-temminus oriented away from the fiber surface. N6-
phenoxyacetyl-3'-O-DMT-2'deoxyadenosine-5'-O-[(~-cyanoethyl)N ,N-diisopropyl]
phosphoramidite was prepared via standard protocols and used to grow oligonucleotide
on the functionalized fibers in a reversed (fiber~substrate linker~5'-dA10-3')
orie,)tatiG".
iii) Svnthesis of Branched Oliqonucleotides.
The "V" branched sequence 1 (Figure 15) was synthesized on an Applied
Biosystems 381 A instrument using a 1 !lmol scale synthesis cycle and ,B-
cyanoethylphosphoramidite chemistry. Purification, desalting, and analysis of the
branched oligonucleotide 1 by polyacrylamide gel electrophoresis was accomplished by
our detailed protocols (Damha, M.J., Ganeshan, K., Hudson, R.H.E., Zabarylo, S.V.,
(1992) Nucleic Acids Res 20, 6565-6573; Damha, M.J.; Ogilvie, K.K. In Methods in
CA 0220816~ 1997-06-18
-- 91 --
Molecular Biology, Vol.20: Protocols for Oligonucleotides and Analogs; Agrawal, S.,
Ed.; Humana Press, Inc.: Totowa, NJ, 1993, pp 81-1 14). Typical yields of this
branched oligomer were 5-15 A260 units (15-25%). The complement dA~o for thermaldenaturation studies was obtained from Dalton Laboratories (Toronto, Canada).
iv) Thermal Denaturation Profiles.
Absorbance versus temperature profiles of the nucleic acid complexes were
measured at 260 nm using a Varian Cary I UV-VIS spectrophotometer equipped with a
variable temperature cell holder controlled by an external variable temperature
circulating bath. Data were collected with the spectrophotometer set on dual beam
optical mode to reduce optical drift. The data were collected at 260 nm at 0.5 ~C
intervals with an equilibration time of 60s for each measurement point. Absorption
coefficients of the branched molecules were assumed to be similar to their
corresponding linear sequences and were calculated from those of mononucleotidesand dinucleotides according to the nearest-neighbor approximation (Puglisi, J.D.;
Tinoco, I., Jr. In Methods in Enzymology; Dahlberg, J.E., Abelson, J.N., Eds.; Academic
Press, Inc.: San Diego, 1989; Vol. 180, 304.). Samples for thermal denaturation
analysis were prepared by mixing the pyrimidine containing strand with the target
(2mM), Iyophilizing the solution to dryness, and dissolving the oligomers in 10 mM Tris,
50 mM MgCI2, pH 7.3 adjusted with HCI. The mixtures were then transferred to Hellma
QS-1.000-104 cells. Oligonucleotide solutions were heated to 80~C for 15 min and then
slowly cooled to room temperature prior to melting experiments. Normalized plots were
constructed according to the method of Kibler-Herzog et al. (Kibler-Herzog, L.; Zon, G.;
Whittierm, G.; Shaikh, M.; Wilson, W.D. Anti-Cancer Drug Des. 1993, 8, 65.) based on
{(At - Ao)/(Af - Ao)} where Ao is the initial absorbance, Af is the final absorbance and At
is the absorbance at any temperature. All complexes showed sharp melting transitions.
The melting temperature (Tm) was determined from the first derivative of each thermal
curve. A precision in Tm values, determined from variance in repeated experiments, of
+0.5 ~C or better was obtained for all of the denaturation profiles investigated.
CA 0220816~ 1997-06-18
- 92 -
v) Instrument Setu~ and Fluorescent Measurements
The laser radiation exiting the immersion lens of the fluorescence microscope
(as described in Example 9) was coupled into a delivery fiber of similar numerical
aperture (0.48) aligned beneath the objective, as illustrated in Fig. 4(c). The light was
totally internally reflected along the delivery fiber to a sensing fiber functionalized with
immobilized oligonucleotide. Coupling of the radiation between fibers was achieved by
abutting the distal terminus of the delivery fiber to the proximal terminus of the sensing
fiber. A loss in optical transmission of no greater than 2% was observed for thecoupled system. The temmini of the teflon fiber coupler were designed as compression-
fit ends which provided a solution-tight seal that prevented contaminants from diffusing
into the fiber coupler and causing drift in the analytical signal. The sensing fiber was
placed in a small volume, stop-flow, stainless steel hybridization chamber (1.5mm i.d. x
48mm) which provided a solution volume of 79 ,ul exposed to the sensing fiber. The
temperature of the hybridization cell was controlled by placing the cell in a thermostated
housing in which glycol solutions from external variable temperature circulating baths
were made to flow. The temperature of the solutions in the hybridization cell were
accurately determined (+ 0.2~C) by use of a glass encapsulated thermistor incorporated
into the hybridization cell and in contact with the solution at the exit of the hybridization
chamber. Solutions containing hybridization buffer, ethidium bromide, and
complementary nucleic acid sequences were delivered to the hybridization cell and
sensing fiber by use of a peristaltic pump. Fluorescence emission from ethidium
bromide that was intercalated into immobilized nucleic acid complexes was totally
internally reflected within the sensing fiber. The portion of the light coupled back into
the delivery fiber was directed towards the microscope objective where it was collimated
and directed to the dichroic mirror. The fluorescence radiation was of longer
wavelength (~ma~ =595 nm) than the dichroic cut-off, and was transmitted through the
mirror and directed towards a photomultiplier tube, where the fluorescence intensity
could be quantitatively measured. Drift caused by variations in the efficiency of optical
coupling, laser intensity and photomultiplier gain were obviated by normalization of all
signals to that of a standard solution of ethidium bromide at 25~C prior to and at the
completion of each analysis.
CA 0220816~ 1997-06-18
- 93 -
vi) PAGE Mobility RetardaVon Assay.
The solutions of oligonucleotides were Iyophilized to dryness, incubated in 10~1L
of 30% sucrose with 50 mM MgCI2 at 75 ~C for 15 min., and then cooled to room
temperature slowly. After a 4 day incubation at 4 ~C, the samples were loaded onto the
gel. The running buffer contained 90mM tris-borate buffer (pH 8.0). The non-denaturing
15% polyacrylamide gels contained 90mM tris-borate (pH 8.0) and 50mM MgCI2. The
native gels were run at 12.5mA for 12h. Following electrophoresis, the gels werecovered with Saran Wrap~ and photographed with a Polaroid MP4 Land Camera over afluorescent TLC plate (Merck, distributed by EM Science, Gibbstown, NJ) illuminated by
a UV lamp (Mineralight lamp, Model UVG-54, San Gabriel, CA). Instant Sheet Film
(#52, medium contrast, lSO 400/ 21~C) was used and the exposure (f4.5, 1.5s) made
through a Kodak Wratten gelatin filter (#58). The gels were subsequently stained for 5
min in a 5,ug/ml solution of ethidium bromide and destained in distilled water for 30s.
The gels were then covered with Saran Wrap~, illuminated by a UV lamp and
photographed (f4.5, 2s) through a Hoya orange filter over a white background.
vii) Parallel and AntiParallel TA T TriPlex Considerations.
In the formation of the intermolecular triplex 2 x dT10 / dAlo triplex, the third dT10
strand interacts by means of Hoogsteen hydrogen bonds with the dA,o strand in target
Watson-Crick duplex, and is oriented parallel to it. In melting experiments (Mg+2 buffer),
the triplex 2 x dT10/dA10 has two clearly resolved transitions, one for dissociation of the
third strand from the duplex, i.e., dT1o~dA1o/dT10 ~ dT10 + dA10/dT10 (Tm 18~C), and
one for dissociation of the duplex into its component strands, i.e., dA10 / dT10 ~ dA10 +
dT,o (Tm 32~C). Thus for this complex, association of the third (dT10) strand with the
duplex (dA10/dT10) is thermodynamically weakerthan duplex formation itself.
Branched oligonucleotides are useful probes for stabilizing triplex DNA (R.
Hudson, A. Uddin, and M. Damha J. Am. Chem. Soc., 1995, 117,12470.). The
branched oligomer 1 (Fig. 15) for instance, binds to dA10 to give a novel TAT triple-
stranded complex in which both dT10 strands are antiparallel to the purine (dA10) strand.
CA 0220816~ 1997-06-18
- 94 -
Although this motif had been observed for TAT bases in complexes dominated by
pur-pur/py bonding (e.g. G-GC, A-AT) ({i} Moser, H.E.; Dervan, P.B. Science, 1987,
238, 645. {ii} Strobel, S.A.; Doucettestamm, L.A.; Riba, L.; Housman, D.E.; Dervan,
P.B. Science, 1991, 254,1639 {iii} Hoogsteen, K. Acta Crysfallogr. 1959, 12, 822-823;
(b) Felsenfeld, G.; Davies, D.R.; Rich, A. J. Am. Chem. Soc. 1957, 79, 2023-2024; {iv}
R. Hudson, A. Uddin, and M. Damha J. Am. Chem. Soc., 1995, 117, 12470.), it had not
been observed previously for dT~/dAn complexes. The formation of this triplex was
induced by linkage of two dT10 strands through their 5' ends via coupling to
riboadenosine at the neighboring 2' and 3' oxygen atoms (Fig.15). This arrangement
c~uses the initial direction of the two dT10 strands to be parallel, and forces the
formation of a triplex in which the third dT,o strand runs antiparallel to the dA,o strand,
and is bound to it via reversed-Hoogsteen interactions. Thermal denaturation profiles of
a mixture of 1 and dA10 (1: 1) in Mg+2 buffer, show a single transition from bound to
unbound complex, consistent with its formation involving one rather than two
bimolecular reaction steps, i.e., 1 + dA~o ~ triplex 1/dA1o (Tm 35~C).
viiiJ Triplex Studies Usinq Derivatized Opt/cal Fibers with Normal ~Fiber-3'-dAI0-5')
Oliqonucleotide Orientation.
Characterization of the triple-helical complexes via thermal denaturation studies
were done. The dA10 was grown in the conventional 3'-to-5' direction from the fiber
surface. Solutions of ethidium bromide, ethidium bromide with dT10, or ethidium
bromide with 1 were heated in the hybridization chamber containing the decaadenylic
acid functionalized optical fibers. Upon slow cooling, fluorescent measurements were
taken at various temperatures. Fig. 16(a), illustrates that as the dT10:dA10 duplex was
forrned by lowering the temperature, there was an increase in the fluorescence intensity
corresponding to ethidium bromide intercalation and quantum yield enhancement of the
ligand in this complex. After further lowering of the temperature, we observed adecrease in the fluorescence intensity with decreasing temperature, indicative of
exclusion of the ligand as a result of triplex formation (2 x dT10:dA10). This process is
illustrated in Fig. 7(a). In order to verify that triplex formation was alone responsible for
the exclusion of the ethidium cation and hence the decrease in fluorescence intensity
CA 0220816~ 1997-06-18
- 95 -
from the fiber, a control experiment was done using optical fibers functionalized with a
twenty-nucleotide probe sequence of mixed base composition incapable of forming
triplex structures. The hybridization experiment was done using the same methodology
as for the decaadenylic acid functionalized fibers with the exception of the hybridization
buffer (1M NaCI, 50mM P04, pH 7.0). Having this nucleic acid sequence and buffercomposition, only double-stranded complexes could form between the immobilized
probe sequence and the complementary sequence. As can be seen in Fig. 6, a
fluorescence intensity with a negative temperature coefficient was observed for the
duplex system over the temperature range studied. The denaturation temperature for
this system of nucleic acids and hybridization buffer was determined to be 73~C by UV-
visible thermal denaturation studies. Only double-stranded complexes existed over the
temperature range investigated, as indicated by the enhanced fluorescence intensity for
the experiment using the ethidium bromide and complementary oligonucleotide. Thecontrol experiment with ethidium bromide and no complementary oligonucleotide
showed no such dramatic increase in intensity.
Upon exposure of the optical sensor to the reversed-Hoogsteen forming 1, no
significant increase in fluorescence intensity over that of the ethidium bromide alone in
solution was observed. The geometrical constraints of compound 1 are such that, if a
complex is formed with the immobilized dA,0 strand in this particular (fiber-3'~5')
orientation, the branch-point riboadenosine should be oriented toward the fiber surface
thus presenting a steric barrier to triplex forrnation. In order to test whether steric
interference surrounding the branch-point prevented triple-helical formation, an optical
fiber having a dA,o strand in the opposite orientation from the surface (i.e. fiber-5'~ 3')
was synthesized.
ix) TriPlex Studies Usinq Derivatized OPtical Fibers with Reversed (Fiber-5'-dAl0-3')
Oliqonucleotide Orientation.
From Figure 16(b), the fluorescent intensity versus temperature profile indicated
that with dT,o there was an initial increase in fluorescence indicative of duplex
formation. This was followed by a decrease in the intensity which was indicative that
CA 0220816~ 1997-06-18
- 96 -
triplex formation had occurred. Upon treatment of the optical sensor with 1, a decrease
in fluorescence intensity for the reverse Hoogsteen complex at temperatures below the
Tm t35~C) was observed Fig. 1 6(c), which is consistent with triplex formation. From the
data of Scaria and Shafer (Scaria, P.V.; Shafer, R.H. J. Biol. Chem., 1991, Z66, 5417),
it can be inferred that a temperature below 25~C is required for the ethidium cation
CA 0220816~ 1997-06-18
- 97 -
reverse-Hoogsteen paired TAT triplexes, and that of their component strands), was
studied at 4~C in a buffer containing magnesium. Following eiectrophoresis, the gels
were visualized by UV shadowing, and by staining with ethidium bromide, as shown in
Figs. 17(a and b), respectively. The Hoogsteen triplex migrated more slowly than the
duplex due to the presence of the third dT10 strand. The reversed-Hoogsteen triplex
has the slowest mobility of all, and is characteristic of branched nucleic acid structures
[ref Hudson and Damha, JACS 1993; Wallace, J.C.; Edmons, M. PNAS vol. 80, 950-
954, 1983). Association of 1 and dA,o was quantitative as evidenced by the complete
disappearance of compound 1 and dA1o, when mixed in equimolar amounts. The
stoichiometry of interaction between dT10 and dA10 for the duplex and Hoogsteen triplex
was also confirmed by studies at different concentrations of the two oligonucleotides.
When the gel shown in Fig. 17(a) was stained with ethidium bromide and illuminated by
a UV lamp, fluorescence was observed only in the bands corresponding to the
complexes (not single strands). This is consistent with the well-known intercalation
mechanism of ethidium bromide (Lim, C.S. BioTechniques, 1994, 17, 626.). As
previously suggested by the biosensor studies, the 1/dA~0 reverse Hoogsteen triplex
gave the lowest fluorescence intensity, which could be related to the limited ability of
ethidium to bind to this complex.
Example 15.
Optical Sensors Which Function by the Intrinsic Mode of Operation.
Background
Angularly dependent light scattering experiments were done to determine the
refractive index of oligonucleotide monolayers covalently immobilized onto fused silica
substrates. With knowledge of the refractive index of the immobilized oligonucleotide
filrn, the mode of operation of the devices, namely, intrinsic or evanescent total internal
reflection fluorescence, can be elucidated. The concept of the experiments done is
based on classical optical theory with respect to how alterations in the direction of a ray
or collimated beam of light traversing an interface between two dielectric materials may
CA 0220816~ 1997-06-18
- 98 -
be predicted based on the difference in the refractive index of the two materials, and
vice versa. In particular, Snell's law of refraction states that for a ray of light traveling in
a plane normal to that defined by the interface between two materials of refractive index
n1 and n2, the angular trajectory of the transmitted ray, ~ t, from the normal to the
interfacial plane will differ from that of the incident ray, ~ j, by a quantity dependent on
the difference in refractive index of the two materials. This can be solved for
mathematically using the following equation (Ohanian, H.C.; Physics, W.W. Norton and
Company, New York (1985) p. 837):
n1-sin(~j) = n2-sin(~t) (4)
Fig. 18(a) illustrates this concept for the case where the upper medium is fusedsilica and the lower medium is the ambient, as characterized by nFused Silica and nAmbjent,
respectively, where nFUsed Silica > nAmbient As shown in Figs. 18(a) and 18(b), as the
angle of incidence is increased, the angle of the refracted beam will be deflected by
increasing amounts toward the interface, where at all times ~ j < ~ t. This trend is
continued to the point where the refracted beam is directed into the interfacial plane
(i.e. ~ t = 90~). The angle of incidence for which this occurs is known as the critical
angle, ~c, and can be calculated from the following relation:
-I nAmbi~nt ~ (5)
nFuscd Siiica
For the case where ~ j > ~c, the incident ray undergoes total internal reflection (TIR) at
the interface. The angle of the reflected beam with respect to the normal to theinterface is then equal to that of the angle of incidence for all B, 2 ~c, as illustrated in Fig.
1 8(c).
- If a detector for optical radiation was placed directly beneath the intersection
point of the light ray with the interface and intensity was recorded as a function of
incidence angle, a continuously decreasing intensity with increasing incidence angle
would be observed. This observation is the result of the refracted beam being
CA 0220816~ 1997-06-18
_ 99 _
increasingly deflected out of the optical axis of the detector. An illustration depicting the
trends in detector response is included on the right hand side of each ray diagram in
Figs. 18-20. As the refracted beam closely approaches the interface between the two
dielectric materials, a local maxima in the detector response would be observed as a
result of the beam being scattered by surface roughness and other imperfections at the
interface. This would continue until ~ c, after which point the incident beam would
undergo TIR and hence provide negligible amounts of signal to the detector, with the
exception of that which leaks out via scatter from imperfections at the interface and
within the waveguiding media. As such, the critical angle for TIR can be determined
directly from this point on the plot of scatter intensity versus incidence angle (Fig.
18(c)). For the case where the refractive index of one of the media is known, the
refractive index of the other can be solved using equation (5).
A three-layer model must be considered for the case where a thin film of organicmaterial is placed at the interface, as shown in Figs. 19 and 20. Each medium type is
herein characterized by the refractive index of the material, as given by nFused Silica, nFilm,
and nAmbjent, respectively, for the fused silica, organic film, and ambient. The interaction
of a light ray at each interface must be considered independently. An incident ray in the
fused silica medium at an angle ~j relative to the interfacial normal will be refracted to a
differing angle after traversing each interface. The propagation angle of the ray will
then be ~tF~ and ~t~ in the organic film and ambient media, respectively, relative to
the interfacial normal.
For the case where nFUsed Silica > nFilm > nAmbient, the reciprocal trend will be
observed with respect to the propagation direction of the refracted rays, where ~ j <
~tF~< ~t~, as shown in Fig. 19. As B, is increased, a local maxima in the detector
response will be observed as ~t passes through the critical angle for TIR at the film-
ambient interface. This is illustrated in Fig. 19 (b and c). A second local maxima will berevealed as ~j passes through the critical angle for TIR at the fused silica-film interface,
as shown in Fig. 19(d). Given nFUsed Silica and nAmbjent, the refractive index of the organic
CA 0220816~ 1997-06-18
- 100 -
film can be directly detemmined from analysis of traces of scatter intensity versus ~ j.
The critical angle for the TIR at the fused silica-film interface can be directly obtained
from the point where the second local maxima intersects the baseline scatter intensity,
as described previously for the two-layer model and shown in Fig 1 9(d). By substituting
~CF"",~5jl; /r", and nFused Silica into equation 5, nFjlm can be solved for directly. Verification of
this result can be acquired by substituting the calculated value of nFjlm and nAmbjent, and
into equation 5 to detemmine the value of ~CFih~ Using the value of ~ j from the point
where the first local maxima intersects the baseline scatter intensity, nFused Silica, nFilm
and equation 4, a second method for calculating ~CFihl~i is provided. The goodness of
agreement between the two values of ~CFihl~i would indicate the validity of the
calculated value of nhlm.
For the case where nFUsed Silica < nFilm > nAmbient, an estimate of the value for nFjlm
may be attained provided the values of nFUsed Silica, /7Ambient are known. A direct
determination of nFjlm cannot be achieved in this case as TIR will not occur at the
interface between the fused silica and organic film for light incident in the fused silica,
yielding no mechanism for the detemmination of ~tF~ . The ray diagrams and detector
response trends for this scenario are shown in Fig. 20. A slight underestimate for the
value of nhlm may be had by assuming that ~CFihl~ is equal to the value of ~ j at the
termination point of the local maxima from the plot of scatter intensity versus incidence
angle. This overestimate ~f ~cr and nAmbjent can be used in equation 5 to provide
an underestimate of the value of nF,Im. This value of nFOm along with those for nFused Silica
and ~j can then be substituted into equation 4 to provide an underestimate of ~cfir ,~j~ .
This underestimate of ~cFh and nAmbjent can again be used in equation 5 to provide
an overestimate of nFjlm. An average of these two values should provide a good
estimate of the true value of nFjlm to within the uncertainty limits set by the low and high
value extremes.
Materials and Methods
CA 0220816~ 1997-06-18
- 101 -
Planar Suprasil~ fused silica wafers (Heraeus Amersil, Duluth, GA, USA) with
dimensions of 10 x 5 x 1mm, a refractive index of 1.46008 and a surface flatness of 10
waves/inch, were functionalized with substrate linker molecules by the methods of
examples 2 and 3. Similarly, silicon wafers (Heraeus Amersil, Duluth, GA, USA) with
dimensions of 10 x 5 x 1 mm were functionalized with substrate linker molecules by the
methods of example 3. Polythymidilic acid icosanucleotides were then assembled onto
the functionalized wafers by automated solid-phase oligonucleotide synthesis, as per
the methods provided in example 5. All water used in the light scattering experiments
was obtained from a Milli-Q 5 stage cartridge purification system (Millipore Corp.,
Mississ~llga, ON, Canada) and had a specific resistance not less than 18 MQ cm. The
hybridization buffer was the same as that used for hybridization experiments on optical
fibers and described in example 8. The refractive index of the hybridization buffer was
determined by use of an Bausch & Lomb Abbe-3L Refractometer (Fisher Scientific,
Nepean, ON, CA) to within the reported accuracy of 0.0001. Octadecyltrichlorosilane
(OTS), ethylene glycol, hexadecane, carbon tetrachloride, chloroform and cyclohexane
were of analytical grade or better from Aldrich Chemical Co. (St. Louis, MO, USA) and
used as received unless stated otherwise.
OTS functionalization of fused silica wafers
Fused silica wafers were cleaned by treatment with solutions of NH40H / H2O /
H2O2 and HCI / H2O / H2O2 respectively, as per the method detailed in example 2 (i).
Prior to use, carbontetrachloride and chloroform were dried by reflux over P2O5 under
an argon atmosphere followed by distillation under the same conditions.
Functionalization of the substrates with OTS monolayers was then done as per themethods of von Tscharner and McConnell (von Tscharner, V. and McConnell, H.M.,
Biophys. J., 36 (1981) 421) and as described in the following. The cleaned substrates
were treated with a solution of 80% hexadecane, 12% carbon tetrachloride, 8%
chloroform and 0.1% OTS (v/v) for 15 minutes at 25~C with stirring under an anhydrous
argon atmosphere. The reaction mixture was then decanted and the functionalized
fused silica wafers were then washed thrice with distilled chloroform and stored in-
vacuo and over P2Os until required.
CA 02208l6~ l997-06-l8
- 102 -
Instrume,~taliGn used for Liqht Scatterinq ExPeriments
Wafers were placed in a custom-built stop-flow cell, beneath a Harrick EA 7X89
fused silica hemispherical prism with a radius of 8mm (Harrick Scientific Corp.,Ossi"ylol1, NY, USA), as illustrated in Fig. 21. Optical contact between the fused silica
hemispherical prism and fused silica wafer functionalized with an oligonucleotide
monolayer was made by applying a thin film of fluorescence free Zeiss Immersionsoel
518C refractive index matching oil (n = 1.515, Carl Zeiss Canada Ltd., Don Mills, ON,
CA) at the interface between the two. The other face of the wafer was exposed to a
solution compartment with dimensions of 9 x 2 x 1 mm (I x w x h). The flow cell was
mounted at the vertex of a modified goniometer element obtained from a type 43702-
200E Thin Film Ellipsometer (Rudolph Research Corporation, Flanders, NJ, USA) with
an angular accuracy and precision of 0.005~. 543 nm optical radiation from a Gre-NeTM
Laser (Melles Griot, Carlsbad, CA, USA, 1mW output power, 1.5 mm beam diameter,
0.01mrad beam divergence) mounted on one arm of the goniometer was passed
through the hemispherical prism and impinged on the planar fused silica wafer. The
hemispherical prism guaranteed that alterations in the incidence angle owing to
refraction at the air- prism interface were eliminated as the beam invariably entered the
prism norrnal to the prism / air interface. A M062-FC03 Slo-Syn stepping motor
(Superior Electric Co., Bristol, CT, USA, 200 steps per revolution) was coupled via a set
of gears to the screw shaft of the goniometer used to drive the pivoting mechanism of
the goniometer arms. The gear ratio used provided the motor with a 7x mechanicaladvantage so as to reduce the load on the motor and prevent it from slipping. TTL
signals from a standard PC parallel interface were used to operate the advance
mechanism of the stepper motor so as to offer accurate control of the angle of the
goniometer arms and incidence of the laser beam. One end of a fiber-optic bundle(Oriel Corp, Stratford, CT, USA, model no. 77533) was mounted in the base of the flow
cell ca. 1 mm from the exposed face of the fused silica wafer. The other terminus of the
fiber bundle was directed to a 630nm long-pass colloidially-colored glass filter (Schott
Glass Technologies, Duryea, PA, USA) placed before the window of an R-928
photomultiplier tube (Hamamatsu Corp., Bridgewater, N.J., USA) operated using a DZ-
CA 02208l6~ l997-06-l8
- 103 -
112 Photoelectric Indicator (Rudolph Research, Flanders, N.J., USA). The long-pass
filter provided for attenuation of the light intensity transmitted by the fiber bundle by a
factor of 105. This was done in order to prevent an overload condition in the PMT form
occurring, guarantee the linearity of response and preserve the useful lifetime of the
detector. The current from the PMT was converted to an analog voltage output (0 - 5
VDC) from the signal processing electronics contained within the Photoelectric Indicator
and passed to a 12 bit analog to digital converter (Metra-Byte, Taunton, MA, USA) for
data acquisition on a PC computer using software created in-house to acquire plots of
intensity versus incidence angle.
Results and Discussion
In order to test the validity of the light scattering approach for refractive index
determination, samples of known refractive index were introduced into the flow cell
beneath the hemispherical prism and analyzed by ramping the incidence angle of the
laser mounted on the goniometer arm from low to high incidence angles while recording
the observed scatter intensity. The results of experiments done using samples of air (n
= 1.0003), water tn = 1.33), and cyclohexane (n = 1.4266) are shown in Fig. 22(a -c),
respectively and summarized in Table 2. Knowing the refractive index value of the
prism material (fused silica, n = 1.46) and the analyte, equation 5 was used to
determine the critical angle values for TIR in each system. Good correlation with the
predicted values was observed in all three cases with no more than 1% error between
the experimental and theoretical values for ~c. An unmodified fused silica wafer was
then coupled to the base of the prism and the same three control samples were again
analyzed. Identical results within the resolution of the experimental technique were
observed between the theoretical and observed values of critical angle for analyses
done with and without the fused silica wafer. This indicated that no additional
modifications to the instrument or correction factors would need be applied as a result
of moving the intersection point of the laser beam 1mm below the base of the prism.
Hybridization buffer (n = 1.35) was also analyzed by the light scattering method (Fig.
22(g)) and provided good agreement with the calculated value for ~c based on therefractive index of the buffer as determined using a standard Abbe refractometer.
CA 02208l6~ l997-06-l8
- 104 -
Table 2 - Summary of the Results from the Angularly Dependent Light Scattering
Experiments and Correlation with Controls of Known Refractive Index.
InterfaceType nAmbjentExperimental ~c Calculated ~c % Error
Fused SilicaPrism -Air 1.0003 42.8~ 43.2~ 0.9Fused Silica Prism - Water 1.33 65.6~ 65.6~ 0.0Fused SilicaPrism - 1.427 77.8~ 77.8~ 0.0-Cyclohexane
Fused Silica Prism - 1.35 67.7~ 67.6~ 0.1Hybridization Buffer
Fused Silica Wafer - Air 1.0003 43.2~ 43.2~ 0.0Fused Silica Wafer - Water 1.33 65.6~ 65.6~ 0.0Fused Silica Wafer- 1.427 77.9~ 77.8~ 0.1Cyclohexane
Experiments were then done using films of organic media of known refractive
index in order to test the validity of the technique as applied to the previously described
the three-layer model. Thin films (10 - 50 ,um) of refractive index matching oil and
ethylene glycol were applied to the exposed surface of the hemispherical prism and
analysis was then done where air was used as the ambient in both cases. The results
of the light scattering experiments for these samples are shown in Figs. 23a and 23b,
respectively. As can be seen in figure 23a, the experimentally determined value of
= 41.7~ for the oil-air interface, based on a value of ~ j of 43.5~, and that
C~
predicted from theory, well agree after taking into account refraction of the beam upon
traversing the fused silica - oil interface. However, for this particular example, the two-
step approximation method for detemlination of nFjlm cannot be used in this example.
The first assumption that ~ t used in this treatment leads to a first estimate of nFi,m
which is iower than that of nfused silica This would lead to the result that the transmitted
beam in the oil would be refracted away from the normal as opposed to towards the
nommal, as would be the case for nFilm > nFused Silica. This causes the next approximation
of nFjlm to be a more exaggerated underestimate of the true value. As such, films of
refractive index slightly greater than that of the fused silica substrate cannot be solved
for.
CA 02208l6~ l997-06-l8
- 105 -
The results for the light scattering experiment using an ethylene glycol film
provided very good agreement between the values of ~c at each interface with respect
to that calculated from theory. Of significance in this plot of scatter intensity versus
incidence angle is the appearance of two distinct maxima. The observation of the two
maxima concurs with that proposed for the three-layer model (Fig. 19) for the case
where nFUSed Silica > nFilm > t7Ambient As such, information with regard to whether the
refractive index of the organic film is greater than or less than that of the substrate
material can be obtained by quick inspection.
A monolayer film of OTS was covalently attached to the surface of a fused silicawafer by a method previously shown to provide dense surface pâcking and a theoretical
refractive index in the range of 1.4 -1.6. ({a}Ducharme, D. et al., J. Phys. Chem, 94
(1990) 1925. {b} von Tscharner, V. and McConnell, H.M.; Biophys J., 36 (1981) 421).
The results of the light scattering experiments are shown in Fig 23(c) and 23(d),
respectively, for OTS functionalized fused silica wafers exposed to air and water as the
ambient. Using the following rearrangement of equation 5:
nAmbi~nr (6)
CFilm / ,~m/~ic~
the value of the refractive index for the OTS monolayer could be solved for. Values of
1.44 and 1.45 for the refractive index of the monolayer were determined from theanalyses using air and water as the ambient, respectively. Given that the refractive
index values determined for the OTS monolayer differed by ~ 1 % with that of the fused
silica substrate, the same limitation as observed for the refractive index matching oil
layer applies herein. As such, it can only be assumed that the refractive index of the
OTS overlayer is only slightly greater than that of the fused silica. This is reinforced by
the fact that of only one local maxima in the plot of scatter intensity versus incidence
angle was observed. If the film refractive index was indeed less than that of the fused
silica substrate then two local maximas should have been observed, in accord with the
three-layer model concept and as clearly demonstrated by the experiment using
ethylene glycol for the film (Fig. 23(b)).
CA 02208l6~ l997-06-l8
- 106 -
Samples of fused silica wafer functionalized with substrate linker molecules by
the methods of example 2 and 3 onto which polythymidilic acid icosanucleotides were
assembled by the method of example 5 were analyzed by the angularly dependent light
scattering technique. The results of the analysis are shown in Fig. 24(a) and 24(b) for
samples prepared by the mesylate activation scheme as detailed in example 3. Theresults for samples prepared by the GOPS-HEG protocol given in example 2 are shown
in Fig. 24(c). For the samples prepared by mesylate activation for which water and 3:1
ethylene glycol in water solution was used as the ambient, a value of 1.57 was
detem~ined in both cases for the underestimate of nfjlm, based on the assumption that
~t~ = ~. Subsequent to the recalculation of ~t~ based on the underestimated value of
n~,m, overestimates for n~lm of 1.67 and 1.68, respectively, were determined from the
cases where water and 3:1 ethylene glycol in water were used as the ambient. This
provided an average value for nFjlm of 1.62 + 0.05. Similarly, analysis of the fused silica
wafer functionalized with polythymidilic acid icosanucleotide on GOPS-HEG substrate
linkers (example 2) yielded an average value for nFjlm of 1.48 + 0.01. The fact that
estimates of nFilm for both types of nucleic acid - substrate linker overcoating could be
solved for strongly reinforces the fact that these overlayers on the fused silica
substrates indeed posses a larger value of refractive index than that of the substrates
onto which they are immobilized.
In addition to the light scattering experiments, ellipsometry was done in order to
provide secondary confimmation of the experimentally determined values of the
refractive index for the oligonucleotide monolayers. Ellipsometry was done on samples
of silicon wafer functionalized with substrate linker molecules by the methods of
example 3 onto which molecules of polythymidilic acid icosanucleotide were assembled
by automated solid-phase oligonucleotide synthesis as detailed in example 5. Silicon
wafers were necessarily used as the substrate material for these experiments as the
fused silica substrates used for the light scattering experiments provide little reflection
of the laser beam incident at an angle 70~ in the ambient. The surface of the silicon
wafers was made similar to that of fused silica via the cleaning procedure used prior to
CA 02208l6~ l997-06-l8
- 107 -
functionalization of the substrate. This cleaning procedure is known to provided a layer
of oxidized silicon at the surface of the silicon wafers (Kem, W. and Puotinen, D.A.;
RCA Review, 31 (1970) 187-206). As such, silanol moieties then present at the
oxidized silicon-ambient interface provide attachment points for the substrate linker
molecules.
McCracken (F.L. McCracken, NBS Technical Note 479, Washington DC (1969))
has developed software capable of providing values of thickness and refractive index
from ellipsometric measurements of thin films using the exact Drude equations for
ellipsometry. The Film 85 software provided with the AutoEL-II null reflection
ellipsometer (Rudolph Research Corp., Flanders, NJ, USA) was based on that originally
developed by McCracken and used for the analysis of ellipsometric data from the
experiments described herein. Ellipsometric analysis of the cleaned substrate revealed
the formation of a 20A thick layer of oxidized silicon on the surface of the wafers. Three
silicon wafers functionalized with substrate linker and oligonucleotide where then
analyzed. Ten different locations on the wafer surfaces were chosen at random and
the results of the ellipsometric analysis are summarized below in Table 3.
Table 3 - Results of Ellipsometric Analysis of Oxidized Silicon Substrates
Functionalized with Substrate Linker Molecules by the Methods of Example 3 and
Polythymidilic Acid Icosanucleotide by the Methods of Example 5.
Film Thickness (A) Film Refractive Index Corrected Refractive
SampleEstimated Using the Estimated Using the Index for a Film of
NumberIterative CalculationIterative Calculation 100A Thickness.
Method of Program 12. Method of Program 12.
84 2.0 C ~.76
2 03 .7~ -.8-
3 13 ~ 3~"
C~ .~C" . ~-
' 7 2. 5 .j9
6 7" 2. 39 .58
7 7~ 2.091 .59
8 82 2. 29 ~.7ei
9 8 2. 19 .72
7' 2. 32 .56
CA 0220816~ 1997-06-18
- 108 -
¦¦ AverageValue ~ ~ ¦ 1.6+0.1
As can be seen by inspection of the data shown in Table 3, determination of thickness
and refractive index concurrently via the iterative process provides a large degree of
variation. This is based largely on the fact that the covalently immobilized nucleic acid
membrane system is not ideal for ellipsometric analysis in that it violates many of the
assumptions of the Drude equations. Of particular significance is the fact that a
-
densely packed oligonucleotide film with the nucleic acid strands oriented perpendicular
to the air-film boundary would be uniaxially anisotropic. This would cause alterations in
the speed of the p- and s-polarized components of the light beams upon passage
through the oligonucleotide film. This effect has been known to produce relative errors
in thickness of up to10% (R.M.A. Azzam and N.M. Bashara, ElliPsometrv and Polarized
Liaht, North Holland Publishing Company, New York (1977)).
A better estimate of the refractive index of the immobilized nucleic acid films may
be achieved by application of Maxwell-Garnet theory (R.M.A. Azam and N.M. Bashara,
ElliPsometrv and Polarized Light. North Holland Publishing Company, New York (1977),
p. 359). The concept of Maxwell-Garnet theory, as applied herein, is based on the
notion that a partially formed monolayer film of coverage ~ is optically equivalent to a
fully formed monolayer film of refractive index (nFI~m) and relative thickness (T~) such
that the observed film thickness ( T) is related to that of the fully formed film by:
T = ~3 Tf (7)
Likewise, the same scaling factor, ~3, can be applied to the refractive index value given
from ellipsometric analysis so that values more representative of that for the actual
immobilized layers can be obtained. The results after applying this correction are given
in Table 3 and provided an average value of 1.6 + 0.1 for nFjlm.
The good correlation between the result of the light scattering experiments and
ellipsometry provides unequivocal evidence that monolayers of oligonucleotides can be
assembled onto substrate linker functionalized fused silica substrates of higherrefractive index than that of the substrate material. Oligonucleotides assembled onto
CA 02208l6~ l997-06-l8
- 109 -
fused silica wafers functionalized with substrate linker molecules via the mesylate
activation scheme, as outlined in example 3 were observed to provide immobilizednucleic acid monolayers with a refractive index of 1.62 + 0.05 by light scattering
investigations. This correlated well to the refractive index value of 1.6 + 0.1 obtained by
ellipsometric investigations. Light scattering investigation of oligonucleotidesassembled onto fused silica wafers functionalized with substrate linker molecules via
the methods of example 2, revealed a nucleic acid film refractive index of 1.48 + 0.01,
which also is higher than that of the fused silica substrates onto which they are
covalently attached. As such, optical sensors created by the methods reported herein
will then function by and provide the signal throughput advantages associated with the
intrinsic TIRF motif described previously.