Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02238516 1998-OS-26
WD 97!30051 PCTlSE99/00241
Acyclic Nucleoside Derivatives
Technical Field
This invention relates to the field of antivirals and in particular to
derivatives of
acyclic nucleosides useful against herpes and retroviral infections. The
invention
provides novel compounds, pharmaceutical compositions comprising these
compounds, methods for the treatment or prophylaxis of viral infections
employing
them, methods for their manufacture and novel intermediates.
Background to the invention
The practical utility of many acyclic nucleosides is limited by their
relatively modest
pharmacokinetics. A number of prodrug approaches have been explored in an
effort
I5 to improve the bioavailability of acycIic nucleosides in general. One of
these
approaches involves the preparation of ester derivatives, particularly
aliphatic esters,
of one or more of the hydroxy groups on the acyclic side chain.
European patent EP165 289 describes the promising antiherpes agent
9-[4-hydroxy-(2-hydroxymethyl}butyl]guanine, otherwise known as H2G. European
patent EP 186 640 discloses 6-deoxy H2G. European patent EP 343 133 discloses
that these compounds, particularly the R-(-} enantiomer, are additionally
active
against retroviral infections such as HIV. Various derivatives of H2G, such as
phosphonates, aliphatic esters (for example, the diacetate and the
dipropionate} and
ethers of the hydroxy groups on the acyclic side chain are disclosed in EP 343
133.
This patent also discloses methods far the preparation of these derivatives
comprising the condensation of the acyclic side chain to the N-9 position of a
typically 6-halogenated purine moiety or, alternatively, the imidazole ring
closure of
a pyrimidine or furazano-[3,4-d] pyrunidine moeity or the pyrimidine ring
closure of
an imidazole moiety, where the acyclic side chain is already present in the
precursor
pyrimidine or imidazole moiety, respectively. In the broadest description of
each of
SUBSTHTUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
WO 97!30051 PCTlSE97I00241
2
these methods the acyclic side chain is pre-derivatised but individual
examples also
show a one-step diacyiation of H2G with acetic or proprionic anhydride and
DMF.
Harnden, et al.,1. Med. Chem. 32, 1738 (1989) investigated a number of short
chain
aliphatic esters of the acyelic nucleoside 9-[4-hydroxy-{3-
hydroxymethyl)butyl]
guanine, otherwise known as penciclovir, and its 6-deoxy analog. Famciclovir,
a
marketed antiviral agent, is the diacetyl derivative of 6-deoxy penciclovir.
Benjamin, et al., Pharln. Res. 4 No. 2, 120 (1987) discloses short chain
aliphatic
esters of 9-[(1,3-dihydroxy-2-propoxy)-methyl]guanine, otherwise known as
ganciclovir. The dipropionate ester is disclosed to be the preferred ester.
Lake-Bakaar, et al., d15C10SeS lil Antimicrob. Agents Chemother. 33 No. i, 110-
l I2
( 1989) diacetate and dipropionate derivatives of H2G and monoacetate and
diacetate
derivatives of 6-deoxy H2G. The diacetate and dipropionate derivatives of H2G
are
reported to result in only modest improvements in bioavailability relative to
H2G.
International patent application W094/24i34, published October 27, 1994,
discloses aliphatic ester prodrugs of the 6-deoxy N-7 analog of ganciclovir,
including the di-pivaloyl, di-valeroyl, mono-valeroyl, mono-oleoyl and mono-
stearoyi esters.
International patent application W093/07163, published April 15, 1993 and
International patent application W094/22887, published October 13, 1994, both
disclose mono-ester derivatives of nucleoside analogs derived from mono-
unsaturated C18 or C20 fatty acids. U.~. Patent No. 5,216,142, issued 3une i,
1993,
also discloses long chain fatty acid mono-ester derivatives of nucleoside
analogs.
A second approach to providing prodrugs of acyclic nucleosides involves the
preparation of amino acid esters of one or more of the hydroxy groups on the
acyclic
side chain. European patent EP 99 493 discloses generally amino acid esters of
SUBSTITUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
W~ 97130051 PCTISE97/00241
3
acyclovir and European patent application EP 308 065, published March 22,
1989,
discloses the valine and isoleucine esters of acyclovir.
European patent application EP 375 329, published 3une 27, 1990, discloses
amino
acid ester derivatives of ganciclovir, including the di-valine, di-isoleucine,
di-
glycine and di-alanine ester derivatives. International patent application
W095/09855, published April 13, 1995, discloses amino acid ester derivatives
of
penCICIOVIF, including the mono-valine and di-valine ester derivatives.
io DE 19526163, published February I, 1996 and U.S. Patent no. 5,543,414
issued
August 6, 1996 , disclose achiral amino acid esters of ganciclovir.
European patent application EP 694 547, published January 3I, 1996, discloses
the
mono-L-valine ester of ganciclovir and its preparation from di-valyl-
ganciclovir.
European patent application EP 654 473, published May 24, 1995, discloses
various
bis amino acid ester derivatives of 9-[1',2'-bishydroxymethyl)-cyclopropan-
I'ylJ
methylguanine.
2o International patent application W095122330, published August 24, 1995,
discloses
aliphatic esters, amino acid esters and mixed acetate/valinate esters of the
acyclic
nucleoside 9-[3,3-dihydroxymethyl-4-hydroxy-but-i-yljguanine. This reference
discloses that bioavailability is reduced when one of the valine esters of the
trivaline
ester derivative is replaced with an acetate ester.
Brief Description of the Invention
We have found that diester derivatives of H2G bearing specific combinations of
an
amino acid ester and a fatty acid ester are able to provide significantly
improved oral
3o bioavailability relative to the parent compound (H2G). In accordance with a
first
aspect of the invention there is thus provided novel compounds of the formula
I
SUBS'I'f1"UTE SifiEl~"1' (RUL.E 26j
CA 02238516 1998-OS-26
WO 9713001 PCTlSE9710024i
4
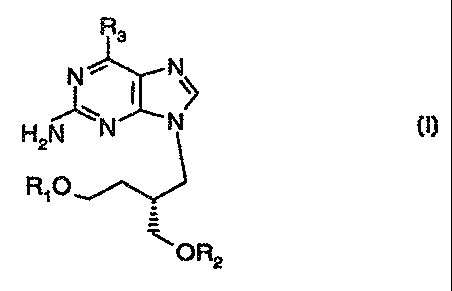
Rs
H2N~N N z
A,O
~ OR2
where a) R, is -C(U)CH(CH(CH3)~}NHS or
-C(O)CH(CH{CI=i3)CH2CH~}NHS and R2 is -C(O)C3-C2~ saturated or
monounsaturated, optionally substituted alkyl; or
b) R~ is -C(G)C3-C2; saturated or monounsaturated,
optionally substituted alkyl and R~ is -C(O)CH(CH(CH3)2)NH~ or
-C(4}CH(CI-f(CH3)CH~CH3)NH2; and
R3 is OH or H;
Ip and pharmaceutically acceptable salts thereof
The advantageous effect on oral bioavailability of the mixed fatty acid and
amino
acid esters of the invention is particularly unexpected in comparison to the
oral
bioavailability of the corresponding fatty acid esters. Based on the results
using a
I5 urinary recovery assay (Table lA) or a plasma drug assay {Table IB) of H2G
from
rats, neither the mono or di-fatty acid esters of H2G provide any improvement
in
oral bioavaitability relative to the parent compound H2G. Indeed the di-
stearate
derivative provided significantly lower bioavailability than the parent
indicating that
a stearate ester may be detrimental for improving oral bioavailability of H2G.
20 Converting one or both of the hydroxyls in certain other acyclic nucleoside
analogues to the corresponding valine or di-valine ester has been reported to
improve bioavailability. Conversion of H2G to the coresponding mono- or di-
valyl
ester derivatives produced similar improvement in bioavailability relative to
the
parent compound. Given that fatty acid derivatives of H2G are shown to be
25 detrimental for improving bioavailability, it was unexpected that a mixed
amino
acid/fatty acid diester derivative of H2G would provide improved or comparable
SU1SSTITUTE SMEET {RULE 2fi)
CA 02238516 1998-OS-26
WO 97130051 PCT/SE97100241
oral bioavailability to that of the valine diester derivative of H2G. based on
urine
recovery and plasma drug assays, respectively,
Table 1 A
Rl graug RZ group Bioavailability*
hydrogen hydrogen 8 %
hydrogen stearoyl 12
stearoyl stearoyI 1 %
valyl hydrogen 29 %
valyl valyl 36 %
valyl stearoyl 56 %
5 * see Biological Example 1 below for detaits
TABLE 1B
Rl group R2 group Bioavailability~
hydrogen hydrogen 3.8 % -
hydragen stearoyl 1.9 %
stearoyl stearoyl 4 %
valyl hydrogen 31.3 %
valyl valyl 35.61 %
valyl stearoyl 29 %
# see Biological Example 2 below for details
to The invention also provides pharmaceutical compositions comprising the
compounds of Formula I and their pharmaceutically acceptable salts in
conjunction
with a pharmaceutically acceptable carrier or diluent. Further aspects of the
invention include the compounds of Formula I and their pharmaceutically
acceptable salts fox use in therapy and the use of these compounds and salts
In the
is preparation of a medicament for the treatment or prophylaxis of viral
infection in
humans or animals.
SUBS°fiTU°fE Sl°IIrBT (RUL.E 26)
CA 02238516 1998-OS-26
i3'~ 97/30051 PCTISE97I00241
6
The compounds of the invention are potent antivirals, especially against
herpes
infections, such as those caused by Varicella zoster virus, Herpes simplex
virus
types I & 2., Epstein-Barr virus, Herpes type 6 (HHV-6) and type 8 (HHV-8).
The
compounds are particularly useful against Varicella zoster virus infections
such as
shingles in the elderly including post herpetic neuralgia or chicken pox in
the young
where the duration and severity of the disease can be reduced by several days.
Epstein Barr virus infections amenable to treatment with the compounds include
infectious mononucleosis/glandular fever which has previously not been
treatable
but which can cause many months of scholastic incapacity amongst adolescents.
The compounds of the invention are also active against certain retroviral
infections,
notably SIV, HIV-1 and HIV-2, and against infections where a transactivating
virus
is indicated.
IS Accordingly a further aspect of the invention provides a method for the
prophylaxis
or treatment of a viral infection in humans or animals comprising the
administration
of an effective amount of a compound of Formula I or its pharmaceutically
acceptable salt to the human or animal.
Advantageously group R3 is hydroxy or its tautomer =O so that the base portion
of
the compounds of the invention is the naturally occuring guanine, for instance
in the
event that the side chain is cleaved in vivo. Alternatively, R3 may be
hydrogen thus
defining the generally more soluble 6-deoxy derivative which can be oxidised
in
vivo (e.g, by xanthine oxidase) to the guanine forth.
The compound of formula I may be present in racemic form> that is a mixture of
the
2R and 2S isomers. Preferably, however, the compound of formula I has at least
7090, preferably at least 90% R form, for example greater than 95%. Most
preferably
the compound of formula I is enantiomerically pure R form.
Preferably the amino acid of group R~/R~ is derived from an L-amino acid.
SUBSTITUTE SHEET (RUL.E 26)
CA 02238516 2005-O1-18
WO 9~130Q51 PCT/SE97100241
7
Preferably the fatty acid of group R,IR~ has in total an even number of carbon
atoms, in particular, decanoyl (C,~), lauryl (C12), myristoyl (C,,~),
palmitoyI (C,6),
stearoyl (CiA) or eicosanoyl (C~~). Other useful R,/R~ groups include butyryl,
hexanoyl, octanoyl or behenoyl (C~~). Further useful R~/R~ groups include
those
derived from myristoleic, myristelaidic, palmitoleic, palmitelaidic, n6-
octadecenoic,
oleic, elaidic, gandoic, erucic or brassidic acids. Monounsaturated fatty acid
esters
typically have the double bond in the trans configuration, preferably in the
cor6, t.~-9
or t~-11 position, dependent upon their length. Preferably the R,IR~ group is
derived
from a fatty acid which comprises a C9 to C1~ saturated, or n:9
monounsaturated,
alkyl.
The saturated or unsaturated fatty acid or R,IR~ may optionally be substituted
with
up to five similar or different substituents independently selected from the
group
consisting of such as hydroxy, Ct-C~ alkyl, C~-C6 alkoxy, Cl-Cb alkoxy C~-C6
all'-5~1,
Ci-C6 alkanoyl, amino, halo, cyano, azido, oxo, mercapto and nitro, and the
like.
Most preferred compounds of the formula I are those where R~ is
-C(O)CH(CH3)~)NH~ or -C(O)CH(CHfCH3)CH~CH3)NH~ and R2 is -C(O)C9-Ci~
saturated alkyl.
The term "lower alkyl" as used herein refers to straight or branched chain
alkyl
radicals containing from 1 to 7 carbon atoms including, but not limited to,
methyl,
ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, t-butyl, n-pentyl,
1-methylbutyl, 2,2-dimethylbutyI, 2-methylpentyl, 2,?-dimethylpropyl, n-hexyl
and
the like.
The term "N-protecting group" or "N-protected" as used herein refers to those
groups intended to protect the N-terminus of an amino acid or peptide or to
protect
an amino group against undesirable reactions during synthetic procedures.
Commonly used N-protecting groups are disclosed in Greene, "Protective Groups
in
Organic Synthesis" (John Wiley & Sons. New York, 1981 ),
N-protecting groups include acyl groups such as formyl,
CA 02238516 1998-OS-26
WO 97!30051 PCTISE97100241.
acetyl, propionyl, pivalayl, t-butyiacetyl, 2-chloroacetyl, 2-bromoacetyl,
trifluoracetyl, trichloroacetyl, phthalyl, o-nitrophenoxyacetyl, a-
chiorobutyryl,
benzoyl, 4-chlorobenzoyl, 4-bromobenzoyl, 4-nitrobenzoyl, and the like;
sulfonyl
groups such as benzenesulfonyl, p-toluenesulfonyl, and the like, carbarnate
forming
groups such as benzyloxycarbonyl, p-chlorobenzyloxycarbonyl,
p-methoxybenzyloxycarbonyl, p-nitrobenzyloxycarbonyl, 2-
nitrobenzyloxycarbanyl,
p-bromobenzyioxycarbonyl, 3,4-dimethoxybenzyloxycarbonyl,
4-methoxybenzyloxycarbonyl, 2-nitro-4,5-dimethoxybenzyloxycarbonyl,
3,4,5-trimethoxybenzyioxycarbonyl, 1-(p-biphenylyl)-1-methylethoxycarbonyl,
to a"a-dimethyl-3,5-dimethoxybenzyloxycarbonyl, benzhydryloxycarbonyl,
t-butoxycarbonyl, diisopropylmethoxycarbonyl, isopropyloxyearbonyl,
ethoxycarbonyl, methoxycarbonyl, allyloxycarbonyl, 2,2,2-
trichloroethoxycarbonyl,
phenoxycarbonyl, 4-nitrophenoxycarbonyl, fluorenyl-9-methoxycarbonyl,
cyclapentyloxycarbonyl, adamantyloxycarbonyl, cyelohexyloxycarbonyl,
15 phenylthiocarbonyl, and the Like; alkyl gropus such as benzyl,
triphenylmethyl,
benzyloxymethyi and the like; and silyl groups such as trimethylsilyl and the
like.
Favoured N-protecting groups include formyl, acetyl, benzoyl, pivaloyl,
t-butylacetyl, phenylsulfonyl, benzyl, t-butoxycarbonyl (BOC) and
benzyloxycarbonyl (Cbz).
The term "activated ester derivative" as used herein refers to acid halides
such as
acid chlorides, and activated esters including, but not limited to, formic and
acetic
acid derived anhydrides, anhydrides derived from alkoxycarbonyl halides such
as
isobutyloxycarbonylchloride and the like, N-hydraxysuccinimide derived esters,
2S N-hydraxyphthalimide derived esters, N-hydroxybenzotriazole derived esters,
N-hydroxy-S-norbornene-2,3-dicarboxamide derived esters, 2,4,5-trichloraphenyl
derived esters and the like.
SUBSTITUTE ShtEET RULE 2B)
CA 02238516 1998-OS-26
W~ 97f300S1 JPCT/SE97fOQ24I
Preferred compounds of formula I includes
(R}-9-[2-(butyryloxymethyl)-4-(L-isoleucyloxy)butyl]guanine,
(R)-9-[2-(4-acetylbutyryloxymethyl}-4-{L-isoleucyloxy)butyl]guanine,
(R)-9-[2-(hexanoyloxymethyl)-4-(L-isoleucyloxy)butylJguanine,
{R)-9-[4-(L-isoleucyloxy)-2,-(octanoyloxymethyl)butyl]guanine,
(R)-9-[4-(L-isoleucylaxy)-2-(decanayloxymethyl)butyl]guanine,
(R)-9-[4-(L-isoleucyloxy)-2-{dodecanoyloxyrnethyl)butyl]guanine,
(R)-9-[4-(L-isoleucyloxy)-2-(tetradecanoyloxymethyl)butylJguanine,
(R)-9-[4--{L-isoleucyloxy)-2-(hexadecanoyloxymethyl)butyl]guanine,
(R)-9-[4-(L-isoleucyloxy)-2-{octadecanoyloxymethyl)butyl]guanine,
(R)-9-[2-(eicosanoyloxymethyl)-4-(L-isoieucyloxy)butyl]guanine,
(R}-9-[2-(docosanoyloxymethyl)-4-(L-isoleucyloxy)butylJguanine,
{R)-9-[4-(L-isoleucyloxy)-2-((9-tetradecenoyl)oxymethyl)butylJguanine,
{R)-9-[2-{(9-hexadecenoyl)oxymethyl)-4-{L-isoleucyloxy)butylJguanine,
(R)-9-[4-(L-isoleucyloxy)-2-((6-octadecenoyl)oxymethyl)butylJguanine,
(R)-9-[4-(L-isaleucyloxy)-2-((9-octadecenoyl)oxymethyl}-butylJguanine,
(R}-9-[2-(( 11-eicosanoyl)-oxymethyl)-4-(L-isoleucyloxy)butyl]guanine,
(R)-9-[2-(( 13-docosenoyl)-oxymethyi)-4-{L-isoleucyloxy)butyl]guanine,
(R)-2-amino-9-[2-(butyryloxymethyl)-4-(L-isoleucyloxy)butyiJpurine,
2o R}-2-amino-9-[2-(4-acetylbutylyloxymethyl}-4-{L-isoleucyloxy)butylJpurine,
(R}-2-amino-9-[2-(hexanoyloxymethyl)-4-(L-isoleucyloxy)butylJpurine,
(R)-2-amino-9-[4-(L-isoleucyloxy)-2-(octanoyloxymethyl)butyl]purine,
(R)-2-amino-9-[4-{L-isoleucyloxy)-2-(decanoytoxymethyl)butyl]purine,
(R)-2-amino-9-[4-(i.-isoleucyloxy}-2-{dodecanoyloxymethyi)butyl]purine,
(R)-2-amino-9-[4-(L-isoleucyioxy}-2-{tetradecanoyloxymethyl}butyl]purine,
(R)-2-amino-9-[4-(L-isoieucyloxy)-2-(hexadecanoyloxymethyl)butylJpurine,
(R}-2-amino-9-[4-(L-isoleucy 1 oxy)-2-(octadecanoyloxymethyl}butyl J purine,
(R)-2-amino-9-[4-(L-isoleucyloxy)-2-(eicosanoyloxymethyl)butylJpurine,
(R)-2-amino-9-[2-(eicosanoyloxymethyl}-4-(L-isoleucyloxy)butylJpurine,
(R}-2-amino-9-[2-{docosanoyloxymethyl)-4-(L-isoleucyloxy)butylJpurine,
SUBSTITUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
W~ 97/30051 PCTISE97J00241
(R)-2-amino-9-[4-(L-isoleucyloxy)-2-((9-tetradecenoyl)oxymethyl}butyl]purine,
(R)-2-amino-9-[2-{{9-hexadecenoyl)oxymethyl)-4-(L-isoleucyioxy)butyl]purine,
(R)-2-amino-9-[4-(L-isoleucyloxy)-2-((6-aetadecenoyl)oxymethyI)butyl]purine,
(R}-2-amino-9-[4-(L-isoleucyloxy)-2-((9-octadecenoyl)oxymethyl)butyl]purine,
(R)-2-amino-9-[2-(( I I-eicosanoyl)oxymethyl)-4-(L-isoleucyloxy)butyl]purine,
or
(R)-2-amino-9-[2-(( 13-docosenoyi)oxymethyl)-4-(L-isoleucyloxy)butyI]purine,
and their pharmaceutically accepable salts.
Further preferred compounds include:
10 (R}-9-[2-(butyryioxymethyl)-4-(L-valyloxy}butyl]guanine,
(R)-9-[2-(4-acetylbutyrylaxymethyl)-4-{L-valyloxy)butyl] guanine,
(R)-9-[2-(hexanoyloxymethyl)-4-(L-valyloxy)butyl]guanine,
(R)-9-[2-(octanoyloxymethyl}-4-{L-valyloxy)butyl]guanine,
(R)-9-[2-(decanoyloxymethyl)-4-(L-valyloxy)butyl]guanine,
i5 {R)-9-[2-(dodecanoyloxymethyl)-4-(L-valyloxy)butyl~guanine,
(R)-9-[2-(tetradecanoyloxymethyl-4-(L-valyloxy)butyl]guanine,
(R)-9-[2-hexadecanoyloxymethyl)-4-(L-valyloxy)butyl]guanine,
(R)-9-[2-{octadeeanoyloxymethyl}-4-(L-valyloxy)butyl~guanine,
{R)-9-[2-(eicosanoyloxymethyl)-4-(L-valyloxy)butyl]guanine,
(R)-9-[2-(eicasanoyloxymethyl}-4-(L-valyloxy)butyl]guanine,
(R)-9-[2-(docosanoyloxymethyl}-4-{L-valyloxy)butyl]guanine,
(R)-9-[2-((9-tetradecenoyl)oxymethyl}-4-(L-valyloxy}butyl]guanine,
(R)-9-[2-((9-hexadecenoyl}oxymethyl)-4.-{L-valyloxy)butyl]guanine,
(R)-9-[2-((6-octadecenoyl)oxymethyl}-4-(L-valyloxy)butyl]guanine,
(R)-9-[2-({9-octadecenoyl)oxymethyl)-4-(L-valyloxy)-butyl]guanine,
(R)-9-[2-((I I-eicosanoyl)oxymethyl)-4-(L-valyloxy)butyl]guanine,
{R)-9-[2-(( I 3-docosenoyl)oxymethyl}-4-(L-valyloxy)butyl]guanine,
(R)-2-amino-9-[2-(butyryloxymethyl)-4-(L-valyloxy)butyl]purine,
{R)-2-amino-9-[2-(4-acetylbutyryloxymethyl)-4-(L-valyloxy)butyl]purine,
(R}-2-amino-9-[2-(hexanoyloxymethyl)-4-(L-valyloxy)butyl]purine,
{R}-2-amino-9-[2-(octanoyloxymethyl}-4-(L-valyloxy)butyl]purine,
StJi~S'1'1TUT~ S>r'°IEET (RULE 26j
CA 02238516 1998-OS-26
w~ g~~~~l PCTfSE971Q0241
11
(R)-2-amino-9-[2-(decanoyloxymethyl)-4-(L-valyioxy)butyl]purine,
{R)-2-amino-9-[2-(dodecanoyloxymethyl)-4-(L-valyloxy)butyl]purine,
(R)-2-amino-9-[2-(tetradecanoyloxymethyl)-4-(L-valyloxy)butyl]purine,
{R)-2-amino-9-[2-(hexadecanoyloxymethyl}-4-(L-valyloxy)butyl]purine,
(R)-2-amino-9-[2-(octadecanoyloxymethyl)-4-(L-valyIoxy)-butyl]purine,
(R)-2-amino-9-[2-(eicosanoyloxymethyl)-4-(L-valyloxy}butyl]purine,
(R)-2-amino-9-[2-(docasanoyloxymethyl)-4-(L-valyloxy)butyl]purine,
(R}-2-amino-9-[2-((9-tetradecenoyl}oxymethyl)-4-(L-valylaxy)butyl]purine,
(R)-2-amino-9-[2-((9-hexadecenoyl)axymethyl)-4-(L-valyloxy)butyi]purine,
l0 (R)-2-amino-9-[2-((6-octadecenayl)oxymethyl)-4-{L-valyloxy)butyl]purine,
(R)-2-amino-9-[2-((9-actadecenoyi)oxymethyi}-4-(L-valyloxy)-butyl]purine,
(R)-2-amino-9-[2-((11-eicosenoyl)-oxymethyl)-4-(L-valyloxy)butyl]purine, or
{R)-2-amino-9-[2-((13-docosenayl)-oxyrnethyl}-4-(L-valyloxy)butyl]purine;
and their pharmaceutically acceptable salts.
Other preferred compounds of formula I include:
{R)-9-[4-(butyryloxy)-2-{L-valyioxymethyl)butyl]guanine,
(R)-9-[4-(4-acetylbutyryloxy)-2-(L-valyloxymethyl)butyl]guanine,
{R}-9-[4-(hexanoyloxy)-2-(L-valyloxymethyl)butyl]guanine,
(R)-9-[4-{octanoyloxy)-2-{L-valyloxymethyl)butyl]guanine,
(R}-9-[4-(decanoyloxy)-2-(L-vaiyloxymethyl)butyl]guanine,
(R)-9-[4-{dodecanoyloxy)-2-(L-valyloxymethyl)butyl]guanine,
(R)-9-[4-(tetradecanoyloxy}-2-(L-valyloxymethyl}butyl] guanine,
(R)-9-[4-hexadecanoyloxy)-2-{L-valyloxymethyl)butyl]guanine,
(R)-9-[4-{octadecanoyloxy)-2-(L-vaiyloxymethyl)butyl]guanine,
(R)-9-[4-(eicosanoyloxy)-2-{L-valyloxymethyl)butyl]guanine,
(R)-9-[4-(docosanoyioxy)-2-(L-valyloxymethyl)butyl]guanine,
(R)-9-[4.-({9-tetradecenoyl)oxy)-2-(L-valyloxymethyl)butyljguanine,
(R)-9-[4-((9-hexadecenoyl)oxy)-2-(L-valyloxymethyl)butyl]guanine,
(R}-9-[4-({6-octadecenoyl)oxy)-2-(L-valyloxymethyl)butyl]guanine,
{R)-9-[4-((9-octadecenoyl)oxy)-2-(L-valyloxymethyl)-butyl]guanine,
SUUS'fi°TU'~"~ SHEET {RULE 26)
CA 02238516 1998-OS-26
WO 97130051 PCTISE97/00241
I2
(R)-9-[4-(( 11-eicosenoyi)oxy)-Z-(L-valyloxymethyl)butyl] guanine.
(R)-9-[4-(( 13-docosenoyl)-oxy)-?-(L-valyloxymethyl}butyl]guanine,
(R)-2-amino-9-[4-(butyryloxy)-2-(L-valyloxymethyl)butyl]purine,
(R)-Z-amino-9-[4-{4-acetylbutyryloxy)-Z-(L-valyloxymethyl)butyl]purine,
(R)-2-amino-9-[4-(hexanoyloxy)-Z-(L-vaiyloxymethyl}butyl]purine,
(R)-Z-amino-9-[4-(octanoyloxy)-Z-(L-valyloxymethyl)butyl]purine,
(R}-Z-amino-9-[4-(decanoyloxy)-Z-(L-valyloxymethyl)butyl]purine,
(R)-2-amino-9-[4-(dodecanoyloxy)-Z-(L-valyloxymethyl)butyl]purine,
(R)-Z-amino-9-[4-(tetradecanoyloxy)-Z-(L-vaiyloxymethyl)butyl]purine,
i0 (R}-2-amino-9-[4-{hexadecanoyloxy)-Z-(L-valyloxymethyl)butyl]purine,
(R)-Z-amino-9-[4-(octadecanoyloxy)-Z-(L-valyloxymethyl)-butyl]purine,
{R)-2-amino-9-[4-(eicosanoyloxy)-2-(L-valyloxymethyl)butyl]purine,
(R)-Z-amino-9-[4-(docosanoyloxy)-2-(L-valyloxymethyl)butyl]purine,
(R)-2-amino-9-[4-((9-tetradecenoyl)oxy)-Z-(L-valylaxymethyl)butyl]purine,
(R}-2-amino-9-[4-((9-hexadecenoyl)oxy)-2-(L-valyIoxymethyl)butyl]purine,
(R)-Z-amino-9-[~-{(6-octadecenoyl)oxy}-2-{L-valyloxymethyl)butyl]purine,
(R)-Z-amino-9-[4-((9-octadecenoyl)oxy)-2-(L-valyloxymethyl)butyl]purine,
(R)-Z-amino-9-[4-(( I 1-eicosenoyl)oxy)-2-(L-valyloxy)butyl]purine,
(R)-Z-amino-9-[2-((13-docosenoyl}oxymethyi)-2-(L-valyloxy}butyl]purine, or
and their pharmaceutically acceptable salts.
The compounds of formula I can form salts which form an additional aspect of
the
invention. Appropriate pharmaceutically acceptable salts of the compounds of
formula I include salts of organic acids, especially carboxylic acids,
including but
not Limited to acetate, trifluoroacetate, lactate, gluconate, citrate,
tartrate, maleate,
malate, pantothenate, isethionate, adipate, alginate, aspartate, benzoate,
butyrate,
digluconate, cyclopentanate, glucoheptanate, glycerophosphate, oxalate,
heptanoate,
hexanoate, fumarate, nicotinate, palmoate, pectinate, 3-phenylpropionate,
picrate,
pivalate, proprionate, tartrate, lactobionate, pivolate, camphorate,
undecanoate and
succinate, organic sulphonic acids such as methanesulphonate,
ethanesulphonate,
2-hydroxyethane sulphonate, camphorsulphonate, 2-napthalenesulphonate,
SUBSTITUTE SWEET (RULE 26)
CA 02238516 1998-OS-26
wo 9~~soo5r ~cT~s>i;9~~oozai
13
benzenesulphonate, p-chlorabenzenesulphonate and p-toluenesulphonate; and
inorganic acids such as hydrochloride, hydrobromide, hydroiodide, sulphate,
bisulphate, hemisulphate, thiocyanate, persulphate, phosphoric and sulphanic
acids.
Hydrochloric acid salts are convenient.
The compounds of Formula 1 may be isolated as the hydrate. The compounds of
the
invention may be isolated in crystal form, preferably homogenous crystals, and
thus
an additional aspect of the invention provides the compounds of Formula I in
substantially pure crystalline form, comprising >70%, preferably >90%
homogeneous crystalline material for example >9S% homogeneous crystalline
material.
The compounds of the invention are particularly suited to oral administration,
but
may also be administered rectally, vaginally, nasally, topically,
transdermally ar
parenterally, for instance intramuscularly, intravenously or epidurally. The
compounds may be administered alone, for instance in a capsule, but will
generally
be administered in conjunction with a pharmaceutically acceptable carrier or
diluent.
The invention extends to methods for preparing a pharmaceutical composition
comprising bringing a compound of Formula I or its pharmaceutically acceptable
2o salt in conjunction or association with a pharmaceutically acceptable
carrier or
vehicle.
Oral formulations are conveniently prepared in unit dosage form, such as
capsules
or tablets, employing conventional carriers or binders such as magnesium
stearate,
chalk, starch, lactose, wax, gum or gelatin. Liposomes or synthetic or natural
polymers such as HPMC or PVP may be used to afford a sustained release
formulation. Alternatively the formulation may be presented as a nasal or eye
drop,
syrup, gel or cream comprising a solution, suspension, emulsion, oil-in-water
or
water-in-oil preparation in conventional vehicles such as water, saline,
ethanol,
3o vegetable oil or glycerine, optionally with flavourant and/or preservative
andlor
emulsifier.
SUSSTiTUTE Si-iEET (RULE 26)
CA 021238516 2005-O1-18
WO 97/30051 PCTlSE97100241
14
The compounds of the invention may be administered at a daily dose generally
in
the range 0.1 to 200 mg/kg/day, advantageously, 0.5 to 100 mg/kg/day, more
preferably 10 to SOmg/kg/day, such as 10 to 25 mQIkQ/day. A typical dosage
rate for
a normal adult will be around 50 to 500 mg, for example 300 mg, once or twice
per
day for herpes infections and 2 to 10 times this dosage for HIV infections.
As is prudent in antiviral therapy, the compounds of the invention can be
administered in combination with other antiviral agents, such as acyclovir,
valcycIovir, penciclovir, famciclovir, ganciclovir and its prodrugs,
cidofovir,
to foscarnet and the like for herpes indications and AZT, ddI, ddC, d4T, 3TC,
foscarnet, ritonavir, indinavir, saquinavir, delaviridine, Vertex VX 478,
Agouron~
AG1343 and the like for retroviral indications. '
The compounds of the invention can be prepared de novo or by esterification of
the
H2G parent compound which is prepared, for example, by the synthesis
methodology disclosed in European Patent EP 343 133,
A typical reaction scheme for the preparation of H2G is depicted overleaf:
*Trademark
CA 02238516 2005-O1-18
WO 97/30051 PCTlSE97100Z4I
CI
N~ N
I v>
i0 1 --~ HzN N N
0
0 0-
c~ o o_
0
N
2 ~N~N~ 3
NZN
HO
OH
NHZ O
N ~ N\\ hIN N\\ .
/1
H N N N 4 HZN ~N N
z
HO ~HO~
LOH SON
H2G
The condensation in step 1 is typically carried out with a base catalyst such
as
NaOH or NaaCO; in a solvent such as DMF. Step 2 involves a reduction which can
be performed with LiBH4ltetrahydrofuran in a solvent such as t-BuOH. The
5 substitution in step 3 of the clslorine with an amino group can be performed
under
pressure with ammonia. Step 4 employs adenosine deaminase which can be
conveniently immobilized on a solid support. Cooling the reaction mixture
allows
unreacted isomeric precursor to remain in solution thereby enhancing purity.
to Starting materials for compounds of the invention in which R3 is hydrogen
inay be prepared as shown in European Patent EP 186 640,
These starting materials may be
' acylated as described for H2G below, optionally after protecting the purine
2-
amino group with a conventional N-protecting group as defined above,
is especially BOC (t-Bu0-CO-), Z (Bn0-CO-) or Ph3C-.
CA 02238516 1998-OS-26
W~ 97130051 PCT/SE97/00241
16
The compounds of the invention may be prepared from I-I2G as described
below in Schemes A and B.
A. Direct acylation method
Scheme A
NHPG G
G
HO ~---~ R1
=~,QH O ~ OH
NHPG G
R* O
O WO
Deprotection
O' _ Rz* '-"Fomlula I
Scheme A depicts the preparation of compounds in which R~ is derived from
to the amino acid and R~ is derived from the fatty acid, but the converse
scheme
is applicable to compounds where Rl is derived from the fatty acid and R2 is
derived from the amino acid ester. In the variant specifically depicted in
scheme A above, G is guanine or 6-deoxyguanine, PG is an optional N-
protecting group or hydrogen, R, * is the valine or isoleucine side chain and
IS R~* is the fatty acid chain. H2G is depicted above as a starting material
but
this of course may be optionally protected at R3 or the 2 position of the
purine
with conventional N-protecting groups (not shown). The H2G (derivative)
reacts in the first step with an activated R1 a.-amino acid derivative, as
further
described below, in a solvent such as dimethylformamide or pyridine, to give
2o a monoacyiated product. The RI oc-amino acid may be suitably N-protected
with N-BOC or N-CBz or the like. Under controlled conditions, the first
acylation can be made to predominantly take place at the side chain 4-hydroxy
group on the side chain of H2G. These controlled conditions can be achieved.
for example, by manipulating the reagent concentrations or rate of addition.
SUBSTITUTE SI-8EE"i' (f~IDLE 26)
CA 02238516 1998-OS-26
WO 97/30051 PCTlSE971002aI
17
especially of the acylating agent, by lowering the temperature or by ehe
choice
of solvent. The reaction can be followed by TLC to monitor the controlled
conditions.
After purification, the R1 monoacylated compounds are further acylated on the
side chain 2-CIhUH group with the appropriate activated fatty acid derivative
to give diacylated products using similar procedures as for the first
esterification step. The diester products are subsequently subjected to a
conventional deprotection treatment using for example trifluoroacetic acid,
to HCl(aq)ldioxane or hydrogenation in the presence of catalyst to give the
desired compound of Formula I. The compound may be in salt form
depending on the deprotection conditions.
The activated RIlRZ acid derivative used in the various acylations may
comprise e.g. the acid halide, acid anhydride, activated acid ester or the
acid in
the presence of coupling reagent, for example dicyclohexylcarbodiimide,
where "acid" in each case represents the corresponding Rl/Rz amino acid or
the R1/R2 fatty acid. Representative activated acid derivatives include the
acid
chloride, formic and acetic acid derived mixed anhydrides, anhydrides derived
2o from alkoxycarbonyl halides such as isobutyloxycarbonylchloride and the
like,
N-hydroxysuccinamide derived esters, N-hydroxyphthalimide derived esters,
N-hydroxy-5-norbornene-2,3-dicarboxamide derived esters,
2,4,5-trichlorophenol derived esters and the like.
SUE3ST1TUTE SHIEIeT (i~ULE 26)
CA 02238516 1998-OS-26
WO 97I300S1 FCT/SE97/00241
18
B Via protection of the side chain 4-hydroxv ~rou~:
Scheme B
G ~ G
HO~ ---~ ~--Si-O~ -------
OOH I \ OOH
i
i
G
G HO,~
-~wSi-O.
W
O
W
O R2*
O R2*
G
O O ~~
deprotection~
Formula 1
R * NHPG ~O
O R2*
wherein G, PG, R,~ and R~* are as described for scheme A.
Scheme B has been exemplified with reference to the preparation of a
compound where R1 is derived from an amino acid and RZ is derived from the
fatty acid ester, but a converse scheme will be applicable to compounds where
i0 Rz is derived from the amino acid and RI is derived from the fatty acid.
This
scheme relies on regioselective protection of the H2G side chain 4-hydroxy
group with a bulky protecting group. In scheme B above this is depicted as
t-butyldiphenylsilyI, but other regioselective protecting groups such as
trityl,
9-(9-phenyl)xanthenyl, f,l-bis(4-methylphenyl)-I'-pyrenylmethyl may also be
15 appropriate. The resulting product is acylated at the side chain
SUBST3TUTE Si°IEET (RULE 26)
CA 02238516 1998-OS-26
W~ 97130051 PC~YSE97100~41
19
2-hydroxymethyl group using analogous reagents and procedures as described
in scheme A above, but wherein the activated acid derivative is the R~ fatty
acid, for example, myristic, stearic, oleic, elaidic acid chloride and the
like.
The thus monoacylated compounds are subjected to appropriate deprotection
treatment to remove the side chain 4-hydroxy protecting group which can be
done in a highly selective manner with such reagents, depending on the
regioselective protecting group, as HF/pyridine and the like and manipulation
of the reaction conditions, viz reagent concentration, speed of addition,
temperature and solvent etc, as elaborated above. The then free side chain
l0 4-hydroxy group is acylated with the activated a-amino acid in a similar
way
as described in scheme A above.
Additional techniques for introducing the amino acid ester of RI/R~, for
instance in schemes A, B, C or D herein include the 2-axa-4-aza-cycloalkane-
1,3-dione method described in international patent application no. WO
941293 i 1.
Additional techniques for introducing the fatty acid ester of R~/R2, for
instance
in schemes A, B, C or D herein include the enzymatic route described in
Preparative Biotransformations 1.11.$ (Ed S M Roberts, J Wiley and Son,
NY> 1995) with a lipase such as SP 435 immobilized Candida antarcticus
(Novo Nordisk), porcine pancreatic lipase or Candida rugosa lipase.
Enzymatic acylation is especially convenient where it is desired to avoid N-
protection and deproteetion steps on the other acyl group or the purine 2-
amore.
An alternative route to compounds of Formula I in which R3 is hydrogen is to
6-activate the correponding guanine compound of Formula I {wherein the
amino acid ester moiety of R~/R~ is optionally protected with conventional
N-protecting groups such as BOC) with an activating group such as halo. The
thus activated 6-purine is subsequently reduced to purine, for instance with a
palladium catalyst and deprotected to the desired 6-deoxy H2G di-ester.
SUBSTITUTE SHEET (MULE 2B)
CA 02238516 1998-OS-26
V~'~ 97/3UEt51 PCT/SE97I0~241
Zo
A further aspect of the invention thus provides a method for the preparation
of the
compounds of formula I comprising
a) optionally N-protecting the purine 2 andlor 6 positions of a compound of
formula I wherein R1 and R~ are each hydrogen;
b) regioselectively acylating the compound of Formula 1 at the side chain
4-hydroxy group with either
i) an optionally N-protected valine or isoleucine group,
ii) an optionally substituted, saturated or monounsaturated C3-
to C2jCOOH derivative, or
iii) a regioselective protecting group;
c) acylating at the side chain 2-hydroxymethyl group with
i) an optionally N-protected valine or isoleucine derivative, or
ii} an optionally substituted, saturated or monounsaturated C3
C21COOH derivative;
d) replacing the regioselective protecting group at R~, if present, with
i) an optionally N-protected valine or isoleucine derivative; or
ii) an optionally substituted, saturated or monounsaturated C3-
C21COOH derivative; and
e) deprotecting the resulting compound as necessary.
Schemes A and B above employ selective acylation to stepwise add the amino
acid and
fatty acid esters, An alternative process for the preparation of the compounds
of formula I
starts with a diacylated H2G derivative, wherein both the aryl groups are the
same, and
employs selective removal of one of the acyl groups to obtain a monoacyl
intermediate
which is then acylated with the second, differing, acyl group in the same
manner as
Schemes A and B above.
Accordingly a further aspect of the invention provides a method for the
preparation of a
3o compound of the formula I, as defined above, which method comprises
A) the monodeacylation of a diacylated compound corresponding to formula I
wherein R, and R~ are both a valyl or isoleucyl ester (which is optionally
SUBSTITU'T'E SHEET (RULE 28)
CA 02238516 1998-OS-26
WO 97130051 PCT1SE97l00241
. 2t
N-protected) or are RI and R~ are both -C(=~)C3-C2~ saturated or
monounsaturated,
optionally substituted alkyl; and
B) acylating the thus liberated side chain 4-hydroxy or side chain
2-hydroxymethyl group with the corresponding valyl, isoleucyl or -C(=O)C~-C2~
saturated or monounsaturated, optionally substituted alkyl; and
C) deprotecting as necessary.
This alternative process has the advantage that the preparation of the
diacylated H2G
derivative is facile and requires little or no purification steps. Selective
removal of one
only of the acyi groups of a diacylated H2G derivative can be achieved by
manipulating
the reaction conditions, in particular the temperature, rate of reactant
addition and choice
of base.
Compounds amenable to this alternative synthesis route are thus of the
formula:
R3
N i N
H2N~N N
\oR2
where R~ and R~ are valyl or isoleucyl (which are optionally N-protected) or a
-C(=U)C3-Czi saturated or monounsaturated, optionally substituted alkyl; and
R~ is
OH or H.
2o For ease of synthesis in this alternative route, it is preferred that RI
and RZ are both
initially identical and are most preferably the same amino acid ester. Such a
di-
amino acid ester will generally be N-protected during its preparation and may
be
used directly in this condition in the selective deacylation step.
Alternatively, such
an N-protected di-aminoacylated H2G derivative may be deprotected and
optionally
reprotected, as described below. The unprotected di-aminoacyl H2G derivative
thus
comprises one of the following compounds:
SU~3STt"fUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
yV~ 97I30Q51 PCT/SE3710~Zal
22
(R)-9-j2-(L-isoicucyloxymethyl}-4--(L-isoleucyloxy)butyl]guanine,
(R}-9- j2-(L-valyIoxymethyl}-4~-(L-valyloxy)butyl]guanine,
(R)-2-amino-9-j4-(L-isoleucyloxy)-2-(L-isoleucyloxymethyl)butyl]purine, and
(R)-2-amino-9-j4-(L-valyloxy)-2-(L-valyloxymethyl)butyl]purine.
These unprotected H2G diacylated derivatives can be directly subject to
selective
deacylation of one of the acyl groups (typically the side chain 4-position
acyl)
followed by enzymatic acylation of the liberated 4-hydroxy as described above.
Alternatively, the unprotected H2G diacylated derivative can be re-protected
and
then subjected to the selective deacylation, followed in turn by conventional
acylation with the fatty acid ester, as described in Schemes A and S.
Conveniently,
such a reprotection step is done with a different N-protecting group, having
properties appropriate to the subsequent acyiation. For example, it is
convenient to
employ a lipophilic N-protecting group, such as Fmoc when preparing a di-amino
is acid H2G derivative, as the Iipophilic nature of the protecting group
assists with
separation of the acylated products. On the other hand, the lipophilic nature
of Fmoc
is of less utility when conducting an acylation with a fatty acid, and thus it
is
convenient to reprotect a diacylated H2G with an alternative N-protecting
group
such as BOC.
It will also be apparent that the preparation of the compounds of formula I
can commence
with the novel monoacylated intermediates of step b i), ii) or iii) in the
above defined first
method aspect of the invention. These compounds are thus of the formula:
R3
N i N
H2N~N N
RiO
~ ORZ
where one of RI and R~ is
i) -C(O)CIi(CH(CH~)~)NH~ or -C(O)CH(CH(CH3)CH~CH~)NH~
SU~3STiTUTE SE°~SET (f~ULE 26)
CA 02238516 1998-OS-26
WO 97/30U51 PCTlSE9710024I
23
ii) a -C(=O)C3-C~, saturated or monounsaturated, optionally substituted
alkyl, or
iii} a regioselective protecting group;
the other of R~ and R~ is hydrogen; and
S R3 is OH or H;
Useful compounds thus include:
(R)-9-[2-hydroxymethyl-4-(t-butyldiphenylsilyl)butyl]guanine,
(R)-9-[2-hydroxymethyl-4-{trityloxy}butylJguanine,
io (R}-9-[2-hydroxymethyl-4-(9-{9-phenyl)xanthenyloxy)butyl]guanine,
(R)-9-[2-hydroxymethyl-4-( i , I -bis(4-methylphenyI)-I'-
pyrenylmethyloxy)butyl]guanine,
(R)-9-[2-hydroxymethyl-4-(decanoyloxy}butyl]guanine,
(R)-9-[2-hydroxymethyl}.-4--(dodecanoyloxy)butyl]guanine,
15 (R)-9-[2-hydraxymethyl-4-(tetradecanoyloxy)butyl]guanine,
(R)-9-[2-hydroxymethyl)-4-(hexadecanoyloxy)butyl]guanine,
(R)-9-[2-hydroxymethyl--4-(octadecanoyloxy)butyl]guanine,
(R)-9-[2-hydroxymethyl)-4-(eicosanoyloxy)butyl]guanine,
(R}-9-[2-hydroxymethyl-4-(docosanoyloxy)butyl]guanine,
20 (R)-9-[4-hydroxy-2-(decanoyloxymethyl)butyl]guanine,
(R)-9-[4-hydroxy-2-(dodecanoyloxymethyl) butyl]guanine,
(R}-9-[4-hydroxy-2-(tetradecanoyloxymethyl)butyl]guanine,
(R}-9-[4-hydroxy-2-(hexadecanoyloxymethyl)butyl]guanine,
(R)-9-[4-hydroxy-2-(octadecanoyloxymethyl)butyl]guanine,
25 (R)-9-[4-hydroxy-2-(eicosanoyloxymethyl)butyl]guanine,
(R)-9-[4-hydroxy-2-(docosanoyloxymethyl)butyl]guanine,
- (R)-9-[2-hydroxymethyl-4-(L-valyloxy}butyl]guanine,
(R)-9-[2-hydroxymethyl)-4-(L-isoleucyloxy)butyl]guanine,
(R}-g-[4-hydroxy-2-{L-isoleucyloxymethyl)butyl]guanine,
30 (R)-9-[4-hydroxy-2-(L-valyloxymethyl) butyl]guanine.
(R)-2-amino-9-[2-hydroxymethyl-4-{L-valyloxy)butyljpurine,
SUBSTITUTE S9-lEET (RULE 26)
CA 02238516 1998-OS-26
W~ 97130051 PCT/SE97/00241
24
(R}-2-amino-9-[2-hydroxymethyl)-4-(L-isoIeucyloxy)butyl]purine,
{R)-2-amino-9-[4-hydroxy-2-(L-isoleucyloxymethyl)butyl]purine, and
{R)-2-amino-9-[4-hydroxy-2-(L-valyioxymethyl}butyl]purine.
Regioselectively protected, sidechain 4-hydroxy intermediates from step c} of
the
above described first method aspect of the invention are also novel compounds.
Useful compounds thus include:
(R)-9-[2-decanoyloxymethyl-4-(t-butyidiphenylsilyl)butyl]guanine,
(R)-9-[2-dodecanoyloxymethyl-4-{t-butyldiphenylsilyl)butyl]guanine,
(R}-9-[2-tetradecanoyloxymethyl-4-(t-butyldiphenylsilyl)butyl]guanine,
(R)-9-[2-hexadecanoyloxymethyl-4-(t-butyldiphenylchlorosilane)butyl]guanine,
(R)-9-[2-octadecanoyloxymethyl-4-(t-butyldiphenylsiiyl)butyl]guanine,
(R)-9-[2-eicosanoyloxymethyl-4-(t-butyldiphenylsilyl)butyl]guanine,
(R)-9-[2-docosanoyloxymethyl-4-(t-butyldiphenylsilyl)butyl]guanine,
An alternative process for the preparation of compounds of the invention of
the formula I
wherein R3 is -OH is shown in Scheme C.
SU>13S'1<t°fU'tE Si-vEE°1' (RULE 26)
CA 02238516 1998-OS-26
WO 97130051 PC~'/SE97100241
SCHEME C
OR.~
~O~X1 R402C 02R$ HO OH
8402 ~- C02R5 ---~. _-p
O R~
R~ O
OR
O R6 6
4
OC(O)R$ HO~~~ OC(O)R$
4 ~. X2/~ OC(O)Rg
lipase R~ O "~ -~-s~
O Rs ~ R~ O
cl ° R6
N N OR9
~~ N N
H N 'N HN N I
HEN '~' N
H2N N
Z ~ ' /--OH
;t ~f
R7 O _
HO HO R7 O
R O
N 6 N O R6
H N~ ~N H N~N~N O
z N z
OH
1,1. ~ ~ ~ R10
r --~ _
R7 O~ R7 O
R6 O Rfi O
OH
N N
I y
H N- 'N- N O H2N
Rio R10
1~.
O O
OH
N HP1
FORM~)../~ ~ Rii
sussT:~°u°rs s~ss~ ~~u~E 2s~
CA 02238516 1998-OS-26
'1~'Q 97!30051 PCT!~E9710024I
26
Referring to Scheme C, malonate 1 (R4 and RS are lower alkyl or benzyl or the
like) is
alkylated --by reaction with from about 0.5 to about 2.0 molar equivalents of
acetal 2 {Rg
and R~ are lower alkyl or benzyl and the Iike or R6 and R~ taken together are -
CH2CH~- or
-CH~,CH2CH~- or -CH2CH2CH~CH2-and X1 is a leaving group {for example, CI, Br
or I, or
a sulfonate such as methanesulfonate, triflate, p-toluenesulfonate,
benzenesulfonate and
the tike)) in the presence of from about 0.5 to about 2.0 molar equivalents of
a base (for
example, potassium t-butoxide or sodium ethoxide or NaH or KH and the like) in
an inert
solvent (for example, DMF or THF or dioxane or dioxolane or N-
methylpyrrolidone and
the like) at a temperature of from about -40°C to about 190°C to
provide alkyiated
malonate 3.
Reduction of 3 with from about 0.5 to about 4.0 molar equivalents of an ester
to alcohol
IS reducing agent (for example, LiBH4 or Ca(BHa)z or NaBH4 or LiAiH4 and the
like) in an
inert solvent (for example, THF or methyl t-butyl ether or t-BuOH and the
like) at a
temperature of from about -20°C to about 100°C provides diol 4.
Enzymatic esterification
of 4 by reaction with from about 1.0 to about 20.0 molar equivalents of a
vinyl ester 5 (Rg
is C3-C21 saturated or monounsaturated, optionally substituted alkyl) in the
presence of a
lipase (for example, Lipase PS-30 or Lipase PPL or Lipase CCL and the like) or
a
phospholipase (for example phospholipase D and the like) provides the desired
stereoisomer of ester 6. This reaction can be carried out in the absence of
solvent or in the
presence of an inert solvent (far example, methyl t-butyl ether or toluene or
hexane and the
Iike). The reaction is carried out at a temperature of from about -20°C
to about 80°C.
zs
The alcohol substituent of 6 is converted to a leaving group (for example, a
halogen or a
sulfonate) by reaction with a halogenating agent (for example NBSIP(Ph)3 or
NCS/P{Ph)3
or POCI3 or NCS/P(Ph)3JNa1 in acetone and like) in an inert solvent (for
example,
methylene chloride or toluene or ethylacetate and the like) or by reaction
with from about
0.8 molar equivalents to about 2.0 molar equivalents of a sulfonyl halide {for
example,
benzenesulfonylchloride, toluenesuifonylchloride or methane sulfonylchloride
and the
SU~STITUTE SHEET (RULE ~E)
CA 02238516 1998-OS-26
WO 97!30051 PCT/SE97100241
z~
like) in the presence of from about I .0 to about 4.0 molar equivalents of a
base (fox
example, triethylamine or potassium carbonate or pyridine or
dimethylaminopyridine or
ethyldiisopropylamine and the like) in an inert solvent (for example methylene
chloride or
toluene or ethylacetate or pyridine or methyl t-butyl ether and the like) at a
temperature of
from about -25°C to about I00°C to provide ester 7. (X2 is a
halogen or sulfonate leaving
group).
Reaction of 7 with from about 0.9 to about 2.0 molar equivalents of 2-amino-4-
chioropurine 8 in the presence of from about 1.0 to about 6.0 molar
equivalents of a base
l0 (for example, potassium carbonate or NaH or KH or NaOH or KOH or lithium
diisopropylamide and the like) in an inert solvent (for example, DMF or THF or
acetonitrile or N-methylpyrrolidone or ethanol and the tike) at a temperature
of from about
-25 -°C to about 140°C provides substihxted purine 9.
I5 Alternatively Mitsunobu coupling (for example P(Ph)3ldiethyl
azidocarboxylate) of
alcohol 6 with 2- _ -amino-4-chloropurine 8 provides 9.
Reaction of 9_ with from about 2.0 to about 20 molar equivalents of an alcohol
R90H {R9
is an alcohol protecting group such as benzyl and the like) in the presence of
from about
20 1.0 to about 6.0 molar equivalents of a base (for example, potassium t-
butoxide or
potassium carbonate or NaH or KH or lithium diisopropylamide and the Like) in
an inert
solvent (for example, THF or DMF and the like) at a temperature of from about -
25°C to
about 150°C provides alcohol 10.
25 Removal of the alcohol protecting group R9 of I0 (for example, by catalytic
hydrogenation
in an inert solvent such as ethanol or benzyl alcohol or methanol or THF and
the like in the
presence of an hydrogenation catalyst such as Pd/C or Pd{OH)2 and the like)
provides
substitued guanine 11.
30 Esterification of 11 by reaction with a) from about 0.8 to about 2.0 molar
equivalents of
RJOCOOH and a coupling agent (for example DCC/DMAP) and the like in an inert
solvent {for example THF or DMF and the like) or b) from about 0.8 to about
2.0 molar
Sl95STITUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
PCTISE97100241
WO 9?130051 '
28
equivalents of an activated derivative of RIpCOOH (for example, the acid
chloride or
N-hydroxysuccinimide ester or R~aC(O)OC(O)R!~ and the like) in the presence of
from
about 0 to about 3.0 molar equivalents of a base (for example, pyridine or
triethylatnine or
ethyldiisopropylamine or DFU or potassium carbonate and the like} in an inert
solvent (for
example, methylene chloride or THF or pyridine or acetonitrile or DMF and the
Iike) at a
temperature of from about -25°C -to about 100°C provides ester
I2.
The acetal substituent of 12 is deprotected and the resulting aldehyde is
reduced by first
reacting 12 with from about 0.1 to about 10.0 molar equivalents of an acid
(for example,
i0 triflic acid or HCl or acetic acid or sulfuric acid and the like} in an
inert solvent (for
example, THFIH20 or methylene chloridelH20 or ethylacetatelH2O or ethanollH~O
or
methanollHzO and the Like) at a temperature of from about -25 °C to
about 100°C. To the
crude reaction mixture is added from about 0.1 to about I0.0 molar equivalents
of a base
(for example, sodium bicarbonate or potassium carbonate or triethylamine or
pyridine or
KOH and the like), additional inert solvent (for example, THF and or methylene
chloride
or ethylacetate or methyl t-butyl ether or isopropoanol and the like) and from
about 0.3 to
about 5.0 molar equivalents of an aldehyde reducing agent (for example, sodium
borohydride or RaNi/H2 and the like) at a temperature of from about -25
°C to about
100°C ato provide alcohol 13.
Reaction of 13 with from about 0.8 to about 3.0 molar equivalents of N-
protected amino
acid PINHCH(R1 I)COOH or an activated derivative thereof (PI is an N-
protecting group
and R11 is isopropyl or isobutyl) in an inert solvent {for example, THF or
dioxane or
dioxolane or DMF or methylene chloride and the like) at a temperature of from
about 2S°C
to about 100°C provides alcohol 14. N-deproteetion of 14 provides the
compound of the
invention of formula lC wherein R3 is -OH.
Alternatively compound I3 can be reacted with the symmetrical anhydride
derived from
P,NHCH{Ril)COOH (i.e.PiNHCH{Rt1)C(O)O-C(O)CH{Rsi)NHP1) to provide I wherein
Rs is OH.
SUBSTITUTE St3EET (RULE 26)
CA 02238516 1998-OS-26
'S~Ct 97130051 PCTlSE97100241
29
Another alternative process for the preparation of compounds wherein R3 is -
OFi is shown
in Scheme D.
SCHEME D
OR12
~X1 R402 0285 HO OH
R402C~ C02R~ --b
OR12 OR12
1Z
OC(O)R8 HO~~ OC{O)R$ X !~~ OC(O)R
2
tipa~ ~ ig
1~
O R12
CI ORi2
Cl
N N N
N
H N N HN N ---y'
z ~ HzN N
~ HzN
~OC(O)R$
HO HO
OR12 N
R
N ~ \~ N O 12
N HZN N
HZN~ N ~
~oH
'~ ?.~ R10
-.- _
OR12
O X12
OH
N N
O HzN~ O
HzN N
R10
R10
2~----s
_ O O
OH NHP1
FORMULA f R11
SUBSTiTU'I°E Si~iEE't' (RULE 26)
CA 02238516 1998-OS-26
WO 9713001 T'CTISE97/002~i
Malonate I (Ra and RS axe Iower alkyl or benzyl and the like) is alkylated
with from about
0.5 to about 2.0 molar equivalents of ether I5 wherein X~ is a leaving group
(for example
Cl, Br or I, or a sulfonate such as methane sulfonate, triflate, p-
toluenesulfonate,
benzenesuifonate and the like) and R~~ is -CH(Ph)2, -C(Ph)3 or -Silt-Bu}{Me)2
and the like
(Ph = phenyl) in the presence of from about 0.5 to about 2.0 molar equivalents
of a base
(for example potassium t-butoxide or sodium ethoxide or NaH or KH and the
like) in an
inert solvent (for example DMF or THF or dioxane or dioxolane or N-methyl
pyrrolidinone and the like) at a temperature of from about -40°C to
about 190°C to provide
alkylated malonate 16.
Reduction of 16 with from about 0.5 to about 4.0 molar equivalents of an ester
to alcohol
reducing agent (for example L18H4 or Ca(BH4)~ ar NaBH4 or LaAlH4 and the like)
in an
inert solvent {for example THF or methyl t-butyl ether or ethanol or t-butanoi
and the like)
at a temperature of from about -20°C to about 100°C provides
diol 17. Enzymatic
esterification of 17 by reaction with from about 1.0 to about 20.0 molar
equivalents of a
vinyl ester 5 (R$ is C3-C~i saturated ar monounsaturated, optionally
substituted alkyl) in
the presence of a lipase (for example, Lipase PS-30 or Lipase PPL or Lipase
CCL and the
like) or a phospholipase (for example phospholipase D and the Iike) provides
the desired
stereoisomer of ester 18. The reaction can be carried out in the absence of
solvent or in the
presence of an inert solvent (for example methyl t-butyl ether or toluene or
hexane or the
like). The reaction is carried out at a temperature of from about -20°C
to about 80°C.
The alcohol substituent of 18 is converted to a leaving group (for example a
halogen or
sulfonate) by reaction with a halogenating agent (for example NBSIP(Ph)3 or
NCSIP(Ph)3
or POCl3 or NCS/P{Ph)3lNal in acetone and the Like) in an inert solvent (for
example
methylene chloride or toluene or ethylacetate and the like) or by reaction
with from about
0.8 molar equivalents to about 2.0 molar equivalents of a sulfonyl halide (for
example
benzenesulfonylchloride, toluenesulfonylchloride or methane sulfonylchloride
and the
like) in the presence of from about 1.0 to about 4.0 molar equivalents of a
base (for
example triethylamine or potassium carbonate or pyridine or methyl t-butyl
ether and the
like} at a temperature -of from about -25°C to about 100°C to
provide ester 19. (Xa is a
halogen or sulfonate leaving group).
SUBSTITUTE SHEET (RULE 2B)
CA 02238516 1998-OS-26
wo 9m300st PCTISE9~I002~1
31
Reaction of i9 with from about 0.9 to about 2.0 molar equivalents of 2-amino-4-
chloropurine 8 in the presence of from about 1.0 to about 6.0 molar
equivalents of a base
(for example potassium carbonate or NaH or KH or NaOH or KOH or lithium
diisopropylamide and the like) in an inert solvent (for example DMF or THF or
acetonitrile or N-methylpyrrolidone or ethanol and the like) at a temperature
of from about
-25°C ._to about I40°C provides substituted purine 20.
Alternatively, Mitsunobu coupling (for example, P(PH)3ldiethyl
azidocarboxylate) of
_ -alcohol 18 with 2-amino-4-chloropurine 8 provides 20.
Reaction of 20 with from about 2.0 to about 20.0 molar equivalents of an
alcohol R90H
{R9 is an alcohol protecting group such as benzyl and the Like) in the
presence of from
about 1.0 to about 5.0 molar equivalents of a base (for example, potassium t-
butoxide or
I5 potassium carbonate or NaH or KH or lithium diisopropylamide and the like
in an inert
solvent {for example, THF or DMF and the like} at a temperature of from about -
25°C to
about I50°C provides alcohol 21.
Removal of the alcohol protecting group R9 of 2I (for example by catalytic
hydrogenation
2o in an inert solvent such as ethanol or benzyl alcohol or methanol or THF
and the like in the
presence of an hydrogenation catalyst such as PdIC or Pd(OH)2 and the like)
provides
substituted guanine 22.
The ether substitutent of 23 is deprotected by reaction with a) a reducing
agent (for
25 example, HC02H and PdIC and the Like} wherein Rig is -CH(Ph)2 or -C(Ph)3,
or b} a
desilylating agent (for example Bu4NF and the like} wherein RI2 is -Silt-
Bu}(Me)Z and the
like to provide 13.
3Q
Alcohol I3 can be converted to I as outlined in scheme C.
An additional alternative involves enzymatic esterification of alcohol 4 or 17
with the
vinyl ester CH2=CH-OC(O)R,o (i.e. R$ = RIO in Schemes C and D) to directly
incorporate
SU~STtTUT~ SH~~T RULE 26)
CA 02238516 1998-OS-26
wo 9m3oosl ~cT~sE9~~ooz4~
sz
into 6 or 1~8 the desired carboxylic acid ester of the final product I. This
allows the
elimination of the ester hydrolysis and reesterification involved in going
from 9 to 12 or
from 20 to 23.
The processes of Schemes C and D are characterized by the fact that each of ,
the hydroxyl groups of the acyclic side chain is differentiated by the use of
different hydroxy protecting groups or precursor groups. This allows the
selective acylation of each of the hydroxy groups with either an amino acid or
a fatty acid group.
Schemes C and D have been illustrated and described with reference to
embodiments of the invention wherein R, is derived from an amino acid and
R2 is derived from a fatty acid. However, it will be apparent that respective
converse schemes will apply to compounds where Ri is derived from a fatty
acid and R~ is derived from an amino acid.
Detailed DescriQtion of the Invention
The invention will now be illustrated by way of example only with reference to
the
following non-limiting Examples, comparative examples and the accompanying
Figures, in which:
Figure 1 depicts plasma H2G levels as a function of time in eynamalgus
monkeys administered with a compound of the invention or with an
alternative prodrug derivative of H2G, as further explained in Biological
Example 3; and
Figure 2 depicts survival as a function of time for Herpes simplex infected
mice administered with various doses of a compound of the invention or a
prior art antiviral, as further explained in Biological Example 4.
SU~ST1TUTE SHEET (RUl.IE 2~)
CA 02238516 1998-OS-26
WD 97/30051 . PCTISE97100241
33
EXAMPLE 1
(R)-9-f 2-(Stearo~oxvmethyl~4-(L-valyloxy)butyll Guanine
This example illustrates the application of preparation scheme A.
a) (R)-9-[4-{N-tert-Butoxycarbonyl-L-valyloxy)-2-{hydroxymethyl)
butyl]guanine.
H2G (5 g, 19.7 mmol) was dissolved in DMF (300 ml) under heating and was
cooled to room temperature before addition of N-t-Boc-L-valine {5.58 g, 25.7
to mmoI), DMAP (0.314 g, 2.57 mmol) and DCC (6.52 g, 31.6 mmol). The mixture
was stirred at room temperature for 24 h and was then filtered. The product
was
ehromatographed on silica gel and eluted with CH2CIz/MeOH to give 2.4 g of the
desired intermediate product.
I5 tI~-NMR (250 MHz, DMSO-db): 8 0.95 (d, 6H)> 1.47 (s, 9H), 1.5-1.8 (m, 2H),
1.96-
2.20 (m, 2H), 3.40 (m, 2H), 3.91 (t, 1H), 4.05 (m, 2H), 4.21 (t, 2H), 4.89 (t,
1H), 6.6
(br s, 2H), 7.27 (d, 1H)> 7.75 (s, 1H), 10.7 (br s, 1H).
b) {R}-9-j4-(N-tert-Butoxycarbonyl-L-valyioxy)-2-{stearoyloxymethyl)
zo butyl]guanine
The product from step a} ( 185 mg, 0.41 mmol) was dissolved in pyridine (5
ml), the
solution was cooled in an ice bath and stearoyl chloride ( 179 ~.I, 0.531
mmol) was
added. The solution was kept in the ice bath for 2 h, then at room temperature
for 1
h. It was then evaporated and chromatographed on silica gel. it was eluted
with
25 dichloromethanelmethanol to give 143 mg of the desired intermediate
product.
e) (R}-9-j2-(Stearoyloxymethyl)-4-(L-valyloxy)butyl]guanine.
The product from step b) (138 mg, 0.192 mmol) was cooled in an ice bath and
trifluoroacetic acid (5 ml) was added. The solution was kept in the ice bath
for 45
30 minutes and was then evaporated to give an oil. 'Water (0.5 to 1 ml) was
added and
evaporated twice. The residue was once more dissolved in water (5 ml ),
filtered and
freeze-dried to give 148 mg of the desired product as the bistrifluoracetate
salt.
SUBSTITUTE St-iEE3' (RULE 26)
CA 02238516 1998-OS-26
WO 9'7130051 PCT/SE97I00241
34
1H NMR (250 MHz, DMSC?-d6): 0.97 (t, 3H), I .05 (dd, 6H), I.34 (br s, 28 H),
1.59
(m, 2H), 1.80 (m, 2H)> 2.25 (m, 1H), 2.36 (t, 2H), 2.50 (m, IH), 3.98-4.18 (m,
5H),
4.35 (t, 2H), 6.6 (br s, 2H), 8.0 (br s, IH), 8.4 (br s, 3H), I0.9 (br s, 1H).
S
EXAMPLE 2
(R)-9-C2-(Myristo~lox~methyl)-4-(L-valyloxy}hutyllguanine
The titled compound was obtained as the bistrifluoracetate salt in a manner
analogous to Example I using myristayl chloride instead of stearoyl chloride
in
step b).
EH NMR (250 MHz, DMSO-d6): b 0.97 (t, 3H), 1.05 (dd, 6H), 1.34 (br s, 20H),
1.57
{m, 2H), 1.78 (m, 2H), 2.24 (m, 1H), 2.35 (t, 2H}, 2.51 (m, 1H), 3.97-4.20 (m,
5H),
4.36 (t, 2H), 6.8 (br s, 2H}, 8.2 (br s, 1H), 8.5 (br s, 3H), 11.1 (br s, lI-
I}.
EXAMPLE 3
(R)-9-f2-(Oieoylox~methyI)-4-(L-val,~loxy)butyll guanine
The titled compound was obtained as the bistrifluoroacetyl salt in a manner
analogous to Example 1 using oleoyl chloride instead of stearoyl chloride in
step b).
IH NMR (250 MHz, DMSO-d6): 0.96 {t, 3I-I}, 1.05 (dd, 6H), I.35 (br s, 20H),
1.59
(m, 2H), 1.76 (m, 2H), 2.09 (m, 4H), 2.24 (m, 1H), 2.35 (t, 2H), 2.50 (m, 1H),
3.97-
4.17 (m, 5H), 4.35 (t, 2H), 5.43 (t> 2H), 6.7 (br s, 2H), 8.0 (br s, IH), 8.5
(br s, 3H),
11.1 (br s, 1H).
EXAMPLE 4
f R)-9-12-(Butyr~o ~meth~L-4-(L-valyloxy~butyl l guanine
a) (R)-9-[4-(N-tert-Butoxycarbonyl-L-valyloxy)-2-(butyryloxymethyl)
butyl]guanine
DCC (110 mg, 0.53 mmol) was dissolved in dichloromethane ( 10 ml) and butyric
acid (82 mg, 0.93 mmol) was added. AFter 4 hours at room temperature the
mixture
SUBSTfTUTE SE°3~~T (RUL.1; 26)
CA 02238516 1998-OS-26
WO 97I300SI, I'CTlSE97100241
3S
was filtered and the filtrate was evaporated. The residue was dissolved in
pyridine
(5 ml) and (R)-9-[4-(N-tert-Butoxycarbonyl-L-valyloxy)-2-hydroxymethylbutylj
guanine (200 mg, 0.44 mmol) (Example I, step a} was added. The mixture was
stirred for 120 hours at room temperature. According to TLC the reaction was
incomplete and more anhydride was made using the procedure above. This
anhydride was added and the mixture was stirred for an additional 20 hours.
The
reaction mixture was evaporated and chromatographed first an silica gel and
then on
aluminium oxide, in both cases eluted with dichloromethanelmethanol to give
79 mg of the intermediate product.
b) (R)-9-[2-(Butyryloxyrnethyl)-4-(L-vaIyloxy}butyl]guanine
The intermediate product of step a was deprotected in a manner analogous to
Example 1, step 3 to give 84 mg of the desired product as the
bistrifIuoracetate salt.
1H NMR {250 MHz, D20): & 0.88 (t, 3H)> 1.06 (dd, 6H)> 1.53 {m, 2H), 1.93 (q>
2H},
2.25 (t, 2H), 2.36 (m, 1H), 2.60 {m, 1H), 4.06 (d, IH), 4.14-4.30 (m, 2H),
4.43
(m, 4H}, 8.99 (br s, IH}.
EXAMPLE 5
2D (R)-9-12-(Decan~Ioxymethyl~-4-(L-valyloxy)butyll ug arsine
The titled compound was obtained as the bistrifiuoroacetate salt in a manner
analogous to Example 1 using decanoyl chloride instead of stearoyl chloride in
step b.
zs
iH NMR {250 MHz, D20): 8 0.90 (m, 3H), I.OI (d, 6H), I.28 (br s, 12H), I .5
(m,
ZH), 1.8 (m, 2H), 2.3 (m, 3H), 2.5 (m, IH), 4.0-4.4 (m, 7H), 8.I (br s, IH}.
suBS~r~~ru're sc~~~T (RUSE 2~)
CA 02238516 1998-OS-26
fi'~ 97130051 PCTISE97IOOZ41
36
EXAMPLE 6
(R)-9-f ~-Docosanoyloxymethvl-4-(L-valylox~butyll~uanine
The titled compound was obtained as the bistrifluoroacetate salt in a manner '
analogous to Example 1 but using in step b the DMAPIDCC conditions of Example
I, step a) in conjunction with docosanoic acid in place of the N-t-Boc-L-
valine and
a mixture of DMF and dichloromethane as solvent.
1H NMR (250 MHz, DMSO-d6): 8 0.97 {t, 3H), 1.05 (dd, 6H), 1.34 (br s, 36 H),
io 1.58 (m, 2H)> 1.77 {m, 2H), 2.24 (m, 1H}, 2.35 {t, 2H), 2.50 (m, 1H), 3.97-
4.17 (m,
5H), 4.35 (t, ZH), 6.7 (br s, ZH), 8.1 (br s, 1H), 8.4 {br s, 3H), 11.0 (br s,
1H).
EXAMPLE 7
R-9-l4-(L-Isoleucyloxy)-~-(5tearoyloxymeth~l)butyllQuanine
This example illustrates the application of preparative scheme B.
a) ( R)-9-[2-hydroxymethyl 4-(t-butyldiphenylsilyloxy) butyl]guanine
HZG (2g, 8 mmole) was coevaporated with dry DMF two times and was then-
suspended in dry DMF (I20 ml) and pyridine (I ml). To the suspension was
added dropwise t-butyldiphenylchlorosilane (2.1 ml, 8.2 mmole) in
dichloromethane (20 ml) at 0 °C over a period of 30 min. The reaction
mixture became a clear solution at the completion of the dropwise addition.
The reaction continued at 0 °C for two hours and was then kept at
4 °C
overnight. Methanol (5 mI) was added to the reaction. After 20 min at room
temperature, the reaction mixture was evaporated to a small volume, poured
into aqueous sodium hydrogen carbonate solution and extracted with
dichloromethane two times. The organic phase was dried over sodium
3o sulphate and evaporated in vacuo. The product was isolated by silica gel
column chromatography using a methanol/dichloromethane system with a
stepwise increasing MeOH concentration . The product was eluted with 7%
MeOH in CH~Ch to yield 1.89 g.
SUBSTITUTE SH~ST (RU?l.E 26)
CA 02238516 2005-O1-18
WO 97130051 PCT/SE97100241
37
b) (R)-9-[2-(Stearoyloxymethyl)-4-(t-butyldiphenylsilyloxy)butyl]guanine
(R)-9-[2-Hydroxymethyl 4-(t-butyldiphenylsilyloxy)butyl]guanine (?.3Ig,
~ mmole) was coevaporated with dry pyridine twice and dissolved in pyridine
(20 ml). To the solution was slowly added dropwise stearoyl chloride (1.86
mI, 5.~ mmole, technical grade) in dichloromethane (2 ml) at -5 °C. The
reaction was kept at the same temperature for 1 hr and then at ~ °C for
2 hr.
The reaction was monitored by TLC. Additional stearoyI chloride (0.29 ml) at
- 5° C was added due to incompletion of reaction. After 30 min at S
°C, .
methanol (3 ml) was added and the reaction mixture stirred for 20 min. It was
then poured into aqueous sodium hydrogen carbonate solution, and extracted
with dichloromethane. The organic phase was dried and the product purified
by silica ge column chromatography with stepwise~increa5ing MeOH, eluting
with 3.5 % MeOH in CH~Cl2, (Yield 2.7 g).
i5
c) (R)-9-[(4-Hydroxy-2-(stearoyloxymethyl)butyl]guanine
(R}-9-[2-(Stearoyloxymethyl)-4-(t-butyldiphenylsilyloxy)butyl]guanine (2.7 g,
3.56 mmole) was dissolved in dry THF (30 ml) and hydrogen fluoride-pyridine
(1.5 ml) added to the solution. The reaction was kept at 4°C overnight
and
monitored by TLC. The reaction reached about 80 ~Io conversion. Additional HF-
pyridine was added (0.75 ml). After 4 hr, TLC showed that the starting
material had
disappeared. The reaction mixture was concentrated in vacuo without raising
the
temperature and more pyridine (5 ml ) was added and evaporated again. The
product
was isolated by silica gel column chromatography. (Yield 1.26 g).
dj (R)-9- [4-(N-BOC-L-isoleucyloxy)-2-(stearoyloxymethyl)butyl]guanine
(R}-9-[4-Hydroxy-2-(stearoyloxymethyl)butyl] guanine ( 135 mg, 0.26 mmole) and
N-BOC-L-isoleucine (180 mg, 0.78 mmole) were coevaporated with dry DMF twice
and dissolved in the same solvent (3.5 ml). To the solution was added
I,3-dicyclohexylcarbodiimide (160 mg, 0.78 mmole) and 4-dimethylaminopyridine
{4.8 mg, 0.039 mmoIe). After reaction for 18 hours, the reaction mixture was
filtered through Celite and worked up in a conventional manner. The product
was
* Trademark
CA 02238516 1998-OS-26
WO 97/30051 PCTlSE97/00241
38
isolated by silica gel column chromatography, eluting at 5 % MeOH in CH2C12.
(Yield 160 mg}
e) (R)-9-j4-{L-Isoleucyloxy}-2-{stearoyloxymethyl)-butyl]guanine
(R}-9-[4-{ N-BOC-L-isoleucyloxy)-2-(stearoyloxymethyl)butyl]guanine (I50 mg, >
0.205 mmoie) from step d) was treated with trifluoroacetic acid (3 mI) at
0°C for 20
min. The solution was evaporated in vacuo. The residue was coevaparated with
toluene twice and kept under vacuum for several hours. The residue was
dissolved
in MeOH (2 ml) and evaporated to give the trifluoracetate salt as a glass-like
~o product (Yield I91 mg).
H1 NMR (DMSO-d6 + D20): 8 8.35 (s>1H, base), 4.21 (t, 2H, H-4), 4.10 (d, 2H)
3.96 (d, 2H), 3.90 (d, 1 H, isoleucine), 2.48 (m, l I-i, H-2), 2.I5 (2I1,
stearoyl), I .85
(m, 1H, isoleucine), I.68 (m, 2H)> 1.48 (m, 4H), 1.68 (m, 28H), 0.81 (m, 9H).
EXAMPLE 8
(R) ~9-j2-(Decanoyloxymethvl)-4-( L-isoleucyloxy)butyllauanine
The title compound was obtained as the bistrifluoroacetyl salt in a manner
analogous to Example 7 using decanoyl chloride instead of stearoyl chloride in
step
b).
'HNMR (DMSO-d6): b 1 I.1 {s, 1H, NH), 8.35 (s, br, 3H), 8.28 { s, IH, base),
6.75
(s, 2H, NHZ ), 4.23 (t, 2H), 4..07 {d, 2H), 4.05 (m, 3H), 2.4 (m, IH), 2.2I
(t, 2H),
1.83 (m, IH), 1.66 (m, 2H), 1.45 (m, 2H), 1.39 (m, 2H), 1.22 (s, 12H ), 0.84
(m, 9H).
SLIB~TITUT~ rl~9~E'1' (RULE 2~)
CA 02238516 1998-OS-26
WQ 97130051 1'CTl~E9710024I
39
EXAMPLE 9
(R) 9 (4 (L Isoleucyloxy)-2-(myristoyloxymethyl)butyll~uanine
The title compound was obtained as the bistrifluoroacetyl salt in a manner
analogous to Example I using N-BOC-L-isoleucine instead of N-BOC-valine in
step
a) and myristoyl chloride in step b).
~H-NMR (DMSO-d6}: ~ 10.99(s, IH), 8.34 (br s, 3H) 8.15 (s, 1H ), 6.67 ( br s,
2H),
4.23 (t, 2H), 4.05 (d, 2H), 3.97 (m, 3H), 2.48 (m, IH), 2.20 {t, 2H), i.85 (m,
IH),
to 1.65 (m, 2H), 1.4I (m, 4H}> I.23 {s, 20H), 0.85 (m> 9H).
EXAMPLE 10
~R) 9 12 (4 Acetylbutyryloxymethyl-~4-(L-valyloxy)butyll~uanine
The titled compound was obtained as the bistrifluoroacetate salt in a manner
analogous to Example 1 but using in step b) the DCClDMAP conditions of Example
1, step a) in conjunction with 4-acetylbutyric acid instead of N-t-Boc-L-
valine.
'H-NMR (250 MHz, DMSO-d6}: 8 I.05 (dd, 6H), I.77 (m, 4H), 2.19 (s, 3H), 2.24
(m, IH), 2.36 (t, 2H), 2.44-2.60 (m, 3H), 3.95-4.20 {m, 5H), 4.36 (m, 2H), 6.8
(br s,
2H), 8.3 (br s, 1H), 8.5 (br s, 3H), I I.1 (br s, IH).
EXAMPLE 1 I
R 9 2 Dodecanoylvxvmeth ~'~1-4-(L-valylaxy)butyll u~anine
The titled compound was obtained as the bistriflouroacetate salt in a manner
. analogous to Example I using dodecanoyl chloride instead of stearoyl
chloride in
step b).
$uBS~rt~u~rE st~~~T (~u~.~ zs~
CA 02238516 1998-OS-26
'W~ 97130051 PCTISE97/00241
' 40
EXAMPLE I2
(R)-9-12-Palmitoyloxvmethyl-4-(L-valvloxy)butyll~uanine
The titled compound was obtained as the bistriflouroacetate salt in a manner
analogous to Example 1 using palmitoyl chloride instead of stearoyl chloride
in step ,
b).
1H-NMR( 250 MHz, DMSO-db): fi 0.97 (t, 3H), 1.05 (m, 6H), 1.35 (br s, 24H),
1.58
{m, 2H), 1.78 (m, 2H), 2.25 (m, 1H), 2.35 (t, ZH}, 2.51 (m, 1H), 3.97-4.18 (m,
SH),
Io 4.35 (t, 2H), 6.7 (br s, 2H), 8.1 (br s, 1H), 8.5 (br s, 3H), 11.0 (br s,
1H).
EXAMPLE i 3
(R)- 2-Ammo-9- 2-stearo lox meth 1-4-(L-val loxy)but 1) urine
Y. y Y y Y L -
This example shows the deoxygenation of group R~.
a) (R)-2-Amino-9-(2-stearoyloxymethyl-4-(N-tert-butoxycarbonyl-L-
valyloxy)butyl)-6-chloropurine:
To a solution of (R}-9-(2-stearoyloxymethyl-4-(N-tert-butoxycarbonyl-L-
valyloxy)butyl)guanine from step 2 of Example 1 (646 mg, 0.9 mrnole) in
aeetonitrile were added tetramethylammonium chloride (427 mg, 2.7 mmole), N,N-
diethylaniline (0.716 ml, 4.5 mmole) and phosphorous oxychloride (0.417 ml,
4.5mmole). The reaction was kept under reflex and the progression monitored by
TLC. After 3 hours the reaction mixture was evaporated in vacuo and the
residue
was dissolved in dichloromethane, then poured into cold sodium hydrogen
carbonate aqueous solution. The organic phase was evaporated and purified by
silica
gel column chromatography. Yield: 251 mg. '
3o Ht-NMR (CDCL3): b 7.76 (1H, H-8), 5.43 (br,2H, NHZ), 4.45-4.00 (m, 7H),
2.53
(m, 1H), 2.28 (t 2H), 2.12 (m, 1H}, 1.75 {m, 2H), 1.59 (m, 2H), 1.43 (9H),
1.25 (m,
28H), 0.96 (d, 3H), 0.87 (m, 6H).
~UBSTfTU't°E EMEET (MULE 26j
CA 02238516 1998-OS-26
W~ 97/3(1051 PCTl~E971OU24I
41
b) (R)- 2-Amino-9-(2-stearoyloxmethyl-4-(N-tert-butoxycarbonyl-L-
vaiyloxy)butyl)purine:
To the solution of (R)-2-amino-9-(2-stearoyloxymethyl-4-(N-tert-butoxycarbonyl-
L-valyloxy)butyl)-6-chloropurine (240 mg, 0.33 mmole) in methanollethyi
acetate
(6 ml, 3:1 VIV) were added ammonium formate (105 mg, I.65 mmole) and 10%
palladium on carbon ( 15 mg). The reaction was kept under refiux for 1 hour
and
recharged with ammonium formate {70 mg). After one hour more the TLC showed
completion of the reaction and the mixture was filtered through Celite and
washed
extensively with ethanol. The filtrate was evaporated and purified by silica
gel
column. Yield: i 93 mg.
H~-NMR (CDCL3): 68.69 {s,lH, H-6), 7.74 (s, 1H, H-8), 5.18 (br, s, 2H, NHS),
4.45-4.01 (m, 7H), 2.55 (m, 1H), 2.28 {t, 2H), 2.i0 {m, 1H), 1.75 (m, ZH),
1.60 (m,
2H), 1.43 (s, 9H), 1.25 (s, 28H), 0.96 (d, 3H), 0.87 (m, 6H).
c) (R)-2-Amino-9-{2-stearoyloxymethyl-4-(L-valyloxy)butyl)purine:
(R)-2-Amino-9-(2-Stearoyloxmethyl-4-(N-tent-butoxycarbonyl-L-
vaiyloxy)butyl)purine (180 mg, 0.26 mmole) was treated with trifluoroaeetic
acid
(5m1) at 0°C for 40 min. It was then evaporated in vacuo and
coevaporated
successively with toluene and methanol. The residue was freeze-dried overnight
to
give 195 mg of the desired product.
'H-NMR (DMSO-d6): ~ 8.78 (s, 1H, H-6), 8.32 (br, 3H), 8.29 (s, 1H, H-8), 4.27
(t, 2H), 4.13 (d, 2H), 3.98 (t, 2H, 2H), 3.89 (m, 1H), 2.47 (m, 1H), 2.18 (m>
3H),
1.43 (m, 2H), I.23 (28H), 0.93 (m, 6H), 0.85 (t, 3H).
suesTrruT~ sHesr t~u~.E a~>
CA 02238516 1998-OS-26
WO 97130051 PCT/SE97I00241
42
EXAMPLE 14
Alternative~reparation of (R)-9-t4-Hydroxy-2-(stearoyloxymethyl?butyl]guanine
a) Preparation of ethyl 4,4-diethoxy-2-ethaxycarbonyl-butyrate
Et02C.~.CO~Et
~~OEt
OEt
Potassium tort-butoxide ( 141.8g, i .11 equiv.) was dissolved in dry DMF ( 1
L).
Diethyl malonate (266 mL, 1.54 equiv.) was added over 5 minutes.
Bramoacetaldehyde diethylacetal ( 172 mL, 1.14 mole) was added over 5 minutes.
The mixture was heated to 120° C (internal temperature), and stirred at
120° C far S
hours. The mixture was allowed to cool to room temperature, poured into water
(5
L}, and extracted with methyl tert-butyl ether (MTBE, 3 x 600 mL). The organic
solution was dried over MgSOd, filtered, concentrated, and distilled (0.5 mm,
95-
140° C) to yield the desired diester {244 g, 78%} as a colorless oil.
'H NMR {CDC13) b 1.19 (t, 6H), 1.28 {t, 6H), 2.22 (dd, 2H}, 3.49 (m, 2H), 3.51
(t,
1H), 3.65 (m, 2H) 4.20 (qd, 4H}, 4.54 (t, 1H}.
b) Preparation of 4,4-diethoxy-2-(hydroxymethyl)-butanal
HO~''~OH
~OEt
t?Et
LiBH4 (purchased solution, 2M in THF, 22.5 rnL) and the product of Example 14
step a) (5 g in I 5 mL of THF, 18.1 mmol) were combined and warmed to
60° C and
stirred at 60° C for 4 hours. The reaction mixture was allowed to cool
to room
temperature and the reaction vessel was placed in a cool water bath. Then
triethanolamine (5.97 mL, 1 equiv.) was added at such a rate that the
temperature of
the reaction mixture was maintained between 20-25 °C. Brine (17.5 rnL)
was
added at a rate such that gas evolution was controlled and the mixture was
stirred fox
45 minutes at room temperature. The layers were separated, the organic layer
was
SU~ST1TUTE Si-11~ET (RUL.E 26)
CA 02238516 1998-OS-26
WO 97/30D51 PCTISE9710U241
43
washed with brine (2 x I5 znL). The combined brine washes were extracted with
MTBE (methyl tert-butyl ether, 3 x 20 mL}. The combined organic extracts were
evaporated and the residue was dissolved in MTBE (50 mL) and washed with brine
(25 mL). The brine layer was back-extracted with MTBE (3 x 25 mL). The,
combined organic extracts were dried over Na2S04, filtered, and concentrated
to
yield the desired diol (3.368, I5.5 mmol, 97%) as a colorless oil.
'H NMR (CDC13) ~ 1.22 (t, 6H}, 1.73 (dd, 2H), 1.92 (m, IH), 2.67 (bs, 2H),
3.52
(m, 2H), 3.69 (m, 2H), 3.72 (m, 4H), 4.62 (t, IH}.
c} Preparation of (2R}-2-acetoxymethyl-4,4-diethoxy-butanol
HO~OAc
Et0
O~t
into a 10 ml 1 neck round bottom flask was charged the product of Example I4
step
b) (3.84 g, 20 mmol), followed by addition of vinyl acetate (2.6 g, 30 mmol)
and
finally Lipase PS 30 (69 mg, purchased from (Amano, Lombard, Illinois}. The
mixture was allowed to stir at ambient temperature far I6 hours. Progress of
the
reaction was closely monitored by TLC (2/1 hexane - EtOAc; stained wzth
Ce2(S04)3 and charred on hot plate; r.f. of diol is O.I, monoacetate is 03,
his acetate
is 0.75). The reaction mixture was diluted with CH2C12 and filtered through a
5
micron filter. The filter was washed with additional CH~Cl2.
The filtrate was then concentrated in vacuo to afford the desired product.
d) Preparation of (2S}-2-acetoxymethyl-4,4-diethoxybutyl toluenesulfonate
TsO~~OAc
Et0
OEt
SUBSTITUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
WO 97!30051 pCTJSlE97100241
44
Into a 100 tnL 1-neck round bottom flask, equipped with a magnetic stir bar
and
septum under N2 was charged the crude product of Example I4 step c) (4.62 g,
19
mmol), dry CHZCIZ (20 mL) and Et~N (5.62 mL, 40 mmol). To this solution was ,
added tosyl chloride (4.76 g, 25 rnmol). The resulting mixture was stirred at
ambient temperature for 4 hours. Charged Ii~O {0.27 g, I5 mmol) and stirred ,
vigorously for 4 hours. The reaction mixture was diluted with 8fl mL EtOAc and
50
mL H20 and the aqueous layer was separated. To the organic layer was added 75
ml of a 5 % aq, solution of KH~P04. After mixing and separation of the layers,
the
aqueous layer was removed. The organic layer was washed with 50 mL of
saturated
i0 NaHC03 solution, dried over Na~S04, filtered and concentrated in vacuo to a
constant weight of 7.40 g of the desired product.
'H NMIZ (CDCl3) 8 i.I7 (t, 6H); 1.62 (m, 2H); 1.94 (s, 3H); 2.19 {m, 1H); 2.45
(s,
3H); 3.42 {m, 2H); 3.6 (m, 2H); 4.03 (m, 4H}; 4.51 (t, IH); 7.36 (d, 2H}; 7.79
(d,
is 2H).
e) Preparation of
C1
NCI
H2N~N,~_.N
~OAc
Et0 -
OEt
20 Into a 50 mL 1 neck round bottom flask was charged the product of Example
14 step
d) (3.88 g, IO mmol), anhydrous DMF (20 mL), 2-amino-4-chloro-purine (2.125 g,
12.5 mmol) and KZC03 (4.83 g). The resulting suspension was stirred at 40
°C
under a N~ blanket for 20 hours. The mixture was concentrated to remove most
of '
the DMF on a rotary evaporator. The residue was diluted with EtOAc {50 mL) and
25 HBO (50 mL). The reaction mixture was transferred to a separatory funnel,
shaken
and the aqueous layer was separated. The aqueous layer was extracted with
EtOAc
(25 mL). The organic layers were combined and washed with 5 % KH2PO4 (?5
SUBSTITUTE : HEET (RULE ~6)
CA 02238516 1998-OS-26
WO 97130051 PC"ft"lSE97I00Z41
mL). The organic layer was separated and washed with H~0 (75 mL), brine (75
mL), dried over hIazS04, filtered and concentrated in vacuo to afford 3.95 g
of crude
product. The crude product was scurried with 40 mL of methyl-t-butyl ether.
This
mixture was stirred overnight at 4°C and the mixture was filtered. The
filtrate was
5 coneentraxed to afford 3.35 g of the product as an oil (containing 2.6 g of
the desired
product based upon HPLC analysis).
300 MHz 1H NMR (CDCI3) b I .19 (m, 6H); I .69 (2H); 1.79 {s, I H); 2.03 (s,
3H);
2.52 (m, 1H); 3.48 (m> 2H); 3.62 (m, 2H); 4.04 (m, 2H); 4.16 (m, 2H); 4.61
(t,lH);
l0 5.12 (bs, 2H); 7.81 (s, 1 H).
f) Preparation of
Bn
I
. H2N~N ,. N
OH
EtO~
OE!
z5 (Bn-benzyl)
Into a S00 mL 1 neck round bottom flask was charged benzyl alcohol ( 136 mL),
cooled to 0 °C, followed by portionwise addition of KO-t-Bu (36 g, 321
mmol).
The temperature was allowed to warm to 40°C, and the mixture was
stirred 20
minutes. To this mixture was added at 0 °C the crude product of Example
14 step e)
20 (24.7 g, 64.2 mmol) dissolved in 25 mL anhydrous THF and benzyl alcohol (30
mL). The temperature was allowed to slowly warm to 8 °C over 2 hours.
The
reaction mixture was poured into 500 mL ice and was extracted with 500 mL
MTBE. The organic layer was washed with 250 mL of brine, dried over Na2SO4,
- filtered and concentrated in vacuo to afford 193 g of a benzyl alcohol
solution of the
25 desired product. HPLC analysis indicated that the solution contained 25.96
g of the
desired product.
SU~STflf'UT~ SHS~'~' (MULE 26)
CA 02238516 1998-OS-26
'W~ 97130051 PCT/SE97/002~t1
46
300 MHz 1H NMR (CDC13) F 1.22 (m,6H); 1.55 (2H); 2.18 (m, 1H); 3.15 (m, 1H);
3.40 (m, 1H); 3.5I (m, 2H); 3.70 (m, 2H); 4.25 (m, 2H); 4.63 (t,lH); 4.90 (bs,
2H);
5.25 (m, 1H); 5.58 (s, 2H); 7.35 (m, 3H); 7.SI (m, 2H); 7.72 (s, 1H).
MS = (M + H)+ = 416 (CI).
,
g) Preparation of
OH
N
~~ N
Hz
OH
EtO
oEt
Into a 100 mL i neck round bottom flask was charged the crude product of
Example
14 step f) (9.65 g of the benzyl alcohol solution, containing 1.30 g, 3.13
mmol of the
i0 product of Example 14, step f) dissolved in absolute EtOH (20 mL). To this
was
added 0.45 g of 10 % PdJC slurried in 5 mL absolute EtOH. The reaction flask
was
evacuated and charged with H2 three times with a balloon of H2. The reaction
flask
was pressurized with 1 atm. HZ and the reaction mixture was stirred overnight.
The
reaction mixture was filtered through a pad of diatomaceous earth to remove
Pd/ C.
The volatiles were removed in vacuo. The residue was mixed with 25 mL of
isopropyl acetate and then concentrated in vacuo. The residue was diluted with
EtOAc ( I O mL), seeded with the desired product, heated to reflex and then
CH3CN
(2 mL) and MTBE (35 ml) were added. The mixture was stirred for 30 minutes.
The precipitate was filtered and dried to a constant weight of 600 mg of the
desired
za product.
300 MHz tH NMR (d6-DMSO) 8 1.16 (m,6H); 1.45 ( m, 1H); 1.61 ( m, 1H); 2.16
(m, IH); 3.45 (rn, 2H); 3.40 (m, 1H); 3.62 (m, 2H); 4.02 {m,2 H); 4.53 {t,
1H); 4.85
(t, IH); 6.55 (bs. 1H); 7.75 (s, 1H). MS = (M + H)+ = 416 (CI). .
SUBST6TUTE SHEET (f;UL~ 26}
CA 02238516 1998-OS-26
VE~U 97/30051 PCTISE97/00241
47
h) Preparation of
ot~
. ~2 N
o~ ~CNf2~16CH3
EtO~
oEt
Into a 2S mL I neck round bottom flask was charged the product of Example 14
step
g) (0.650 g, 2.0 mmol), pyridine (4 zn.L.} and CHZCIZ {2 mL), DMAP ( l Omg).
The
S mixture was cooled to -5 °C and stearoyl chloride (790 mg, 2.6 moral)
dissolved in
CHaCl2 (0.5 mL) was added over S minutes. The resulting mixture was stirred 16
hours at -S °C. Absolute EtOH (0.138 g, 3.0 mmol) was added and the
mixture was
stirred an additional 1 hour. The reaction mixture was concentrated in vacuo.
Toluene (30 mL) was added to the residue and then the mixture was concentrated
in
to vacuo. Again, toluene (30 n~I,) was added to the residue and then the
mixture was
concentrated in vacuo. To the residue was added I % KHZP04 (25 mL} and this
mixture was extracted with CH~Clz (60 mL). The organic layer was separated and
was dried over Na~S04, filtered and concentrated in vacuo to a constant weight
of
1.65 g. The crude product was chromatographed on 40 g of Si02, eluting with
95/5
15 CH2CI2 - EtOH, affording 367 mg of the desired product.
300 MHz 1H NMR (CDCI3) 8 0.89 (t, 3H); 1.26 (m, 30 H); 1.65 (m,3 H); 2.32
(m, 1H}; 3.45 (m, 1 H); 3.60 {m, 2H); 4.08 {m, 2H); 4.60 {m, 1 H}; 6.0 (bs,
2H};
7.53 (s, 1 H).
i) Preparation of
o~
N~N
H2N~N~N O
~O~ (CHI 1 sCEi3
' HO --~
Into a 25 mL 1 neck round bottom flask was charged the product of Example 14,
step h) (0.234 g, 0.394 rximol) dissolved in THF (1.7 mL). To this solution
was
SUBSTITUTE aHEET (F~UL.E 26)
CA 02238516 1998-OS-26
WD 97I30U~1 ~'CTISE97100241
48
added triflic acid (0.108 g) in H2O 180 mg. The mixture was stirred overnight
at
room temperature. To the reaction mixture was added saturated NaHCO~ solution
(10 mL), THF (5mL), CH2Cl~ (2 mL) and NaBH4 (0.10 g). This mixture was
stirred for 30 minutes. To the reaction mixture was added a 5 % solution of
KH~P04 {30 mL). This mixture was extracted with 2 x 15 ml of CHzCl2. The _
organic layers were combined and dried over Na2S04, filtered and concentrated
in
vacuo to a constant weight of 207 mg. This material was recrystallized from
EtOAc (8 mL) and CH3CN (0.5 mL) affording 173 mg of the desired product.
300 MHz ~H NMR (d6-DMSO) 8 0.82 (t, 3H); 1.19 (m, 30H); 1.41 (m, 4H); 2.19
{t, 2H); 2.32 (m, iH); 3.40 {m, 2H}; 3.9 (m, 4H); 4.49 (m, 1H}; 6.4 (bs, 2H);
7.61
(m, 1.5H); 9.55 (m, 0.5H).
EXAMPLE 15
Alternative preparation of (Rl-9-f4-~N-tert-but~loxycarbonyl-L-valyloxv)-2-
(stearoyloxymeth rl but 1 uanine
{R)-9-[2-(Stearoyloxymethyl)-4-(t-butyldiphenylsilyloxy)butyl]guanine (45g)
arid
THF (950 ml) were combined in a ZL flask. Then Boc-L-valine (3.22 g, 0.25 eq)
was added, followed by tetrabutylammonium fluoride ( i M in THF, 89.05 mL)
over
10 minutes. The clear reaction mixture was stirred at roam temperature for 2
hours
and 50 minutes with monitoring of the reaction progress by TLC (90110
CH2Cl2/MeOH).
To the reaction mixture was added Boc-L-valine (35.43 g, 2.75 eq), DCC (36.67
g,
2.75 eq} and dimethylaminopyridine ( 1. i g, 0.15 eq) in THF (25 ml). The
reaction
mixture was stirred at room temperature for 24 hours. DCI3 was filtered off
and
washed with CIi?C12. The filtrate was concentrated, and the residue was taken
up in
2 litres of CH2CL2 and washed with 2L of i/2 saturated sodium bicarbonate and
brine
solutions. On drying and evaporation, approximately 100 g of cnzde product was
obtained. The material was purified by silica chromatography (6000 ml of
silica)
SUUSTITt3'f~ SHEET (RULE 26)
CA 02238516 2005-O1-18
WO 97/30051 PCT/SE97100241
49
using 3% MeOH/CH~CI~ to 5~7o MeOH/CH~CI~ to obtain 38.22 mg of the desired
product.
EXAMPLE 16
Alternative preparation of (R)-9-f 2-(stearovloxvmethvll-4-(L-valyloxv)
butyll guanine
a) (R)-9-[2-Hydroxymethyl)-4-(t-butyldiphenylsilyloxymethyl)butyl]guanine
H2G (450.0 g, 1.78 mol) and N,N dimethylformamide (6.4 kg) were charged into a
Bucchi evaporator and the mixture warmed to dissolve the solid. The solution
was
concentrated to dryness under vauum at na more than 90°C. The resulting
powder
was transferred to a 22 litre flask with stirrer, addition funnel and and
temperature
probe. N,N-dimethylformamide ( 1 ~7 kg) was added followed by pyridine (3.53
kg).
The resulting suspension was cooled to -10°C under nitrogen and stirred
at -5 ~5°C
as t-butylchlorodiphenylsilane (684 g, 2.49 moI) was added dropwise. The
resulting
mixture was stirred at -5 ~5°C until the reaction was complete (as
monitored by
TLC (i0:1 methylene chloride/methanol) and HPLC (4.6 x 250 mm Zorbax RxC8
(5 micron); 60:40 acetonitrile-aq. NHaO?.C (0.05 M) at 1.5 ml/min; UV
detection
at 254 nm)). Water ( 16 kg) was added and the mixture was stirred for 30
minutes to
precipitate the product, then the mixture was cooled to 0°C for 30
minutes. The
solid was isolated by filtration and the product cake was washed with cold
water and
sucked dry with air to provide the crude product as an off-white solid. The
crude
solid was taken up in pydridine (3 kg) and concentrated under vacuum at
60°C to
remove water. The dry solid residue was slurried with methanol (10 kg) at
60°C for
1-2 hours and filtered while hot. The filtrate was concentrated under vacuum
and the
solid residue was refluxed with isopropyl acetate (7 kg) for 30 minutes. The
mixture
was cooled to 20°C and filtered. The filter cake was dried under vacuum
at 50°C to
provide the title compound as a white solid (555 g),
b) (R)-9-[2-(Stearoyloxymethyl)-4-(t-butyldiphenylsilyloxy)butyl]guanine
The product of Example 16, step a) (555 g, 1.113 mot) was charged to a 50
litre
Buchi evaporator. Pyridine (2.7 kg) was added dropwise to dissolve the solid
and
*Trademark
CA 02238516 2005-O1-18
WO 97130051 PCT15E97100241
the mixture was distilled to dryness under vacuum at 60°C. The residue
was taken
up in fresh pyridine (2.7 kg) and transferred to a 22 litre flask with
stirrer. addition
funnel and temperature probe. The solution was cooled to -5°C under
nitrogen. A
solution of stearoyl chloride (440 g, 1.45 mol) in methylene chloride ( 1.5
kg) was
5 added so as to maintain a temperature below 0°C. 4-(Ivl,N-
dimethylamino)pyridine
( 15 g, 0. 12 mol) was added and the mixture was stirred at -~ - 0°C
for 2-4 hours
until conversion was complete (as monitored by TLC (10:1 methylene
chIoride/methanol) and HPLC (4.6 x 250 mm Zorbax RxC8 (5 micron); 60:40
acetonitrile-aq. NH40Ae (0.05 M) at 1.5 ml/min; UV detection at 254 nm)). At
the
10 end of the reaction, acetonitrile (8.7 kg) was added and the mixture was
stirred for
not less than 15 minutes to precipitate the product. The slurry was cooled to
0°C for
2 hours and the solid isolated by filtration and the filter cake washed with
acetonitrile (2 kg). The desired product was obtained as a white solid (775
g).
~5 c) (R)-9-[4-Hydroxy-2-(stearoyloxymethyl)butylJguanine
A solution of the product of Example 16, step b) (765 g, 0.29 mol) in
tetrahydrofuran (10 kg) was prepared in a reactor. A solution of
tetra(n-butyl)ammonium fluoride in tetrahydrofuran ( 1.7 kg of 1 M solution, i
.7
mol) was added and the resulting clear solution was stirred at 20 ~ 5°C
for 4 hours.
20 Water (32 kg) was added and the resulting slurry was stirred for 1 hour and
then
cooled to 0°C for 30 minutes. The precipitate was isolated by
filtration and the filter
cake was washed successively with water ( 10 kg) and acetonitrile (5 kg).
After
drying under vacuum at 25°C, 702 g of crude product was obtained. The
crude
product was dissolved in refiuxing THF (4.2 kg) and water ( 160 g), then
cooled to
25 40°C and treated with methylene chloride ( 14.5 kg). The mixture was
allowed to
cool to 25~5°C for l hour, then it was cooled to ~~5°C for 1
hour to complete
precipitation. The slightly off-white powder was isolated by filtration and
dried
under vacuum at 40°C to yield the desired product (416 g).
30 d) (R)-9-[4-(N-Cbz-L-valyloxy)-2-(stearoyloxymethyl)butyl]guanine
A solution of N-Cbz-L-valine ( 169 g, 0.67 mol) in dry THF (750 ml) was
prepared
in a 2 litre flask with mechanical stirrer, thermometer and addition funnel. A
* Trademark
CA 02238516 1998-OS-26
WO 971300~f PCT/SE97/0024I
51
solution of dicyclohexylcarbodiimide (69.3 g, 0.34 mol) in THF (250 ml) was
added
over 5 minutes and the resulting slurry was stirred at 20+_5°C for 2
hours. The slurry
was filtered and the filter cake was washed with THF (300 ml). The filtrate
and
wash were charged to a 3 litre flask with stirrer and thermometer. The product
of
Example 16, step c) (116 g, 0.22 mol} was added as a solid, with a rinse of
THF
{250 mI). 4-(N,N-dimethylamino)pyridine (2.73 g, 0.022 mol) was added and the
white slurry stirred at 20~S°C. Within IS minutes, the solids were all
dissolved and
the reaction was complete within 1 hour (as determined by HPLC: 4.G x 250 mm
Zorbax RxC8 column; 85: I S acetonitrile- 0.2 % aq. HCI04 at 1 mI/min.; UV
1o detection at 254 nm; starting material elutes at 4.1 min. and product
elutes at 5.9
min.). The reaction was quenched by addition of water (5 ml) and the solution
was
concentrated under vacuum to leave a light yellow semisolid. This was taken up
in
methanol ( 1.5 litres} and warmed to reflux for 30 minutes. The solution was
cooled
to 25°C and the precipitate was removed by filtration. The filtrate was
concentrated
i5 under vacuum to leave a viscous, pale yellow oil. Acetonitrile, ( I L) was
added and
the resulting white suspension was stirred at 20 ~°C for 90 minutes.
The crude
solid product was isolated by filtration, washed with acetonitrile (2 x 100
ml} and
air-dried overnight to provide the desired product as a waxy, sticky solid (
122 g}.
This was further purified by crystallization from ethyl acetate (S00 ml) and
drying
2E~ under vacuum at 30°C to provide the desired product as a white,
waxy solid ( 104 g}.
e) (R)-9-[4-(L-valyloxy)-2-(stearoyloxymethyl)butyl]guanine
A solution of the product of~Example 16, step d}, (77 g) in warm {40°C)
ethanol
(2.3 L) was charged to an hydrogenation reactor with 5 % Pd-C ( 15.4 g). The
25 mixture was agitated at 40°C under 40 psi hydrogen for 4 hours,
evacuated and
hydrogenated for an additional 4-10 hours. The catalyst was removed by
filtration
and the filtrate was concentrated under vacuum to provide a white solid. This
was
stirred with ethanol (38S mI) at 25°C for 1 hour, then cooled to
0°C and filtered.
The filter cake was dried with air, then under vacuum at 35°C to yield
the title
30 compound as a white powder (4G g).
SUBSTITUTE SHEET (RULE Z6)
CA 02238516 1998-OS-26
WO 97!30051 PCTlSE97l00241
52
EXAMPLE I7
R)-9-C2-(L-Valyloxymethyl)-4-(stearovloxylbutyll canine
a) (R)-9-[2-Hydroxymethyl-4-(stearoyIoxy)butyl]guanine.
H2G (506 mg; 2.0 mmol) was dissolved in dry N>N-dimethylformamide (40 ml}
with pyridine (400 mg; 5.06 mmol) and 4-dimethylaminopyridine {60 mg; 0.49
mmol). Stearoyl chloride (1500 mg; 4.95 mmol) was added and the mixture kept
overnight at room temperature. Most of the solvent was evaporated in vacuo,
the
residue stirred with 70 ml ethyl acetate and 70 ml water, and the solid
filtered off,
~,vashed with ethyl acetate and water and dried to yield 680 mg of crude
product.
Column chromatography on silica gel (chloroform:methanol 15:1 ) gave pure
title
compound as a white solid.
IH NMR (DMSO-d6} 8: 0.86 (t, 3H); 1.25 (s, 28H); 1.51 (qui, 2H); 1.62 (m, 2H);
2.06 (m, 1H}; 2.23 (t, 2H}; 3.34 (d, 2H); 3.96 (ABX, 2H); 4.07 (dd, 2H); 6.30
(br s,
2H); 7.62 (s, 1H); 10.45 (s, 1H).
13C NMR (DMSO-d6) 8: 13,8 (C18); 22.0 (C17); 24.4 (C3); 27.7 (C3'); 28.4-28.8
(C4-6, C15); 28.9 (C7-14); 31.2 (C16); 33.5 (C2); 38.0 (C2'); 44.0 (Cl');
60.6/61.8
(C4', C2"); 116.5 (guaCS); 137.7 (guaC7); 151.4 (guaC4); 153.5 (guaC2); 156.7
(guaC6); 172.7 (COO}.
b) (R)-9-[2-(N-Boc-L-valyloxymethyl)-4-(stearoyloxy)butyl]guanine.
A mixture of N-Boc-L-valine (528 mg; 2.1 mmoi) and N,N'-dicyelohexyl
carbodiimide (250 mg; 1.21 mg) in dichloromethane (20 mi) was stirred over
night
at room temperature, dicyclohexylurea filtered off and extracted with a small
volume of dichloromethane, and the filtrate evaporated in vacuo to a small
volume.
(R}-9-[2-Hydroxymethyl-4-(stearoyloxy)butyl]guanine (340 mg; 0.654 mmol), 4-
dimethylaminopyridine (25 mg; 0.205 mmol), and dry N,N-dimethylformamide (15
3o ml) were added and the mixture was stirred for 4h at 50oC under N2. The
solvent
was evaporated in vacuo to a small volume. Column chromatography on silica
gel,
suBS°r~TU-r~ sH~ET (~tuLS 2sy
CA 02238516 1998-OS-26
WO 97130051 PCTISE9710024I
53
then on aluminum oxide (ethyl acetate:methanol: water 15:2:1 as eluent} gave
185
mg (39%v) pure title compound as a white solid.
IH NMR (CHC13) 8: 0.85-1.0 (m, 9H) 18-CH3, CH(CH3)2; 1.25 (s, 28H) 4-17-
CH2; i .44 (s, 9H) t-Bu; 1.60 (qui, 2H) 3-CH2; 1.74 {qua, 2H) 3'-CH2; 2.14 (m,
1H)
2'-CH; 2.29 (t, 2H) 2-CH2; 2.41 (m,lH) CH(CH3)2; 4.I-4.3 {m, 6H) C1'-CH2, C2"-
CH2, C4-CH2; 5.4 {d, 1H) aCH; 6.6 (br s, 2H) guaNH2; 7.73 (s> 1H) guaHB; 12.4
(br s}.
l0 13C NMR (CHCl3) 8: 13>9 (C18); 17,5/18.9 (2 VaI CH3); 22.4 (C17); 24.7
(C3);
28.1 (C3'); 28.9-29.3 {C4-6, Ci5); 29.4 (C7-14); 30.7 (Val ~3C); 3 i.7 (C16);
34.0
(C2); 35.9 (C2'); 43.9 (CI'}; 58.7 (Val aC}; 61.4/63.6 (C4', C2"); 79.9
(CMe3);
116.4 (guaCS); 137.9 (guaC7); I5I.7 (guaC4); 153.7 (guaC2); 155.7 (CONH);
158.8 (guaC6); 172.1 (CHCOO}; 173.5 (CH2COO).
F5
c) (R)-9-j2-(L-Valyloxymethyl}-4-(stearoyloxy)butyl]guanine.
Chilled trifluoroacetic acid (2.0 g} was added to {~-9- j2-(N-Boc-L-
valyloxymethyl}-4-(stearoyloxy)butyl]guanine {I80 mg; 0.25 mmol) and the
solution kept at room temperature for Ih, evaporated to a small volume, and
20 lyophilized repeatedly with dioxane until a white amorphous powder was
obtained.
The yield of title compound, obtained as the trifluoracetate salt, was
quantitative.
tH NMR (DMSO-d6) b: 0.87 (t, 3H) 18-CH3, 0.98 (dd, 6H) CH(CH3)2; 1.25
(s, 28H) 4-17-CHz; i.50 {qui, 2H) 3-CH2; 1.68 (qua, 2H) 3'-CH2; 2.I9 (m, 1H)
2'-
2~ CH; 2.26 (t, 2H) 2-CH2; 2.40 (m,lH) CH(CHg}2; 3.9-4.25 (m, 7H) CI'-CH2, C2"-
CH2, C4-CH2, aCH; 6.5 (br s, 2H) guaNH2; 7.79 {s, 1H) guaHB; 8.37 (br s, 3H)
NHg'~; 10.73 (br s, 1H) guaNH.
t3C NMR (DMSO-d6) 8: 14.2 {C18); 17.9118.3 {2 Val CH3); 22.3 (C17); 24.6 (C3);
34 27.7 (C3'); 28.7-29.1 (C4-6, C15); 29.2 (C7-14); 29.5 {Val j3C); 31.5
(C16); 33.7
SUBSTITUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
'V4~0 97130051 PCT/SE97100241
54
(C2}; 35.0 (C2'); 44.1 (Cl'); 57.6 {Val aC); 61.6/65.2 (C4', CZ"}; I 16.1
(guaCS}; I 16.3 (qua, J 290Hz, CF~);137.9 (guaC7}; I S I .5 (guaC4); 154.0
(guaC2);
I56.7 {guaC6);158.3 (qua, J lSHz, CF3COO) 169.1 (CHCOO); 173.1 (CH6C00).
s EXAMPLE 18
Alternative preparation of (R)-9-f2-h~ dr roxymethyl-4-(stearovlox~yll~uanine
H2G {7.60 g, 30 mmol) was heated to solution in dry DMF (200 ml). The solution
was filtered to remove solid impurities, cooled to 20° C (H2G
cystallized) and
1o stirred at that temperature during addition of pyridine {9.0 g, I 14 mmol},
4-
dimethylaminopyridine (0,46 g, 3.75 mmol) and then, slowly, stearoyl chloride
(20.0
g, 66 mmol). Stirring was continued at room temperature overnight. Most of the
solvent was then evaporated off in vacuo, the residue stirred with 200 ml
ethyl
acetate and 200 ml water and the solid filtered off, washed with ethyl acetate
and
15 water and dried to yield crude product. As an alternative to
recrystallization, the
crude product was briefly heated to almost boiling with 100 mI of ethyl
acetate:
methanol: water ( 15:2:1 ) and the suspension slowly cooled to 30° C
and filtered to
leave most of the 2" isomer in solution (the 2" isomer would crystallize at
lower
temperature). The extraction procedure was repeated once more to yield, after
2o drying in vacuo, 6.57 g {42%) of almost isomer free product.
EXAMPLE 19
Preparation of crystalline (R)-9-f2-stearo~loxymethyl)-4-(L-
valyloxy)butyllguanine
2s The product of Example 16, step c) (20.07 g, 32.5 mmol) was dissolved in
absolute
ethanol (400 ml) with heating, filtered, and further diluted with ethanol (1
I7.S ml).
To this solution was added water {HPLC grade, 103.5 ml), and the mixture was
allowed to cool to 35-40°C. After the mixture was cooled, water (HPLC
grade,
931.5 mI) was added at a constant rate over 16 hours with efficient stirring.
After all
3o the water was added, stirring was continued for 4 hours at room
temperature. The
resulting precipitate was filtered through paper and dried under vacuum at
room
SUIE~STJTUTE SI~~ET (RULE 26)
CA 02238516 1998-OS-26
WO 97/30051 PCTISE97100241
temperature to obtain the title compound as a white, free flowing crystalline
powder
(I9.43 g, 97%), m pt I69-170 °C.
EXAMPLE 20
S 9-R-(4-Hydroxy-2-(L-valyloxvmethyl)bu~l ) guanine
a} To a solution of 9-12-(4-(tert-butyldiphenylsilyloxy)-2-
(hydroxymethyl)butyl)guanine (695 mg, 1.5 mmole) in DMF (30 ml) were
added N-Boc-L-saline (488 mg, 2.25 mmole), 4-dimethylamino pyridine (30
to mg, 0.25 mmoie) and DCC (556 mg, 2.7 mmole). After 16 hr, the reaction
was recharged with N-Boc-L-valine (244 mg) and DCC (278 mg), and was
kept for an additional 5 hours. The reaction mixture was filtered through
Celite and poured into sodium hydrogen carbonate aqueous solution, and
then it was extracted with dichloromethane. The organic phase was
1S evaporated and purified by silica gel column chromatography, giving 950 mg
the N-protected monoamino acyl intermediate.
b) The above intermediate (520 mg, 0.78 mmoie} was dissolved in THF
I5 mI}. To the solution was added hydrogen fluoride in pyridine (70 % l
zo 30 %, 0.34 ml). After two days, the solution was evaporated and
coevaporated with toluene. Purification by silica gel column chromatography
gave 311 mg of the protected monoamino acyl compound.
~H-NMR (DMSO-d6): & I0.41(s, IH), 7.59 (IH), 6.26 (br s, 2H}, 4.32 (t,
2S IH), 3.95 (m, 5H}, 3.46 (m, 2H), 2.4I (m, 1H}, 2.06 (m, 1H), I.45 (m, 2H)>
I.39 (s, 9H), 0.90 (d, 6H}.
' c) The product of step b) (95 mg, 0.2i mmoie) was treated with a
mixture of trifluoroacetic acid (4 ml) and dichloromethane (6 ml) far 1 hr.
3Q The solution was evaporated and freeze-dried, give I25 mg of the
unprotected monoaminoacyi product.
SUBSTITUTE SHEET (i3ULE 2~)
CA 02238516 1998-OS-26
WO 97/30051 PCT/SE97100241
56
'H-NMR (D20): b 8.88 (s, IH), 4.32 (m, 4H), 3.96 (d, 1H), 3.68 (m, 2H),
2.63 (m, 1H), 2.22 (m, 1H), 1,73 (m, 2H}, I.00 (m, 6H).
EXAMPLE 21
(R)-9-(2-Hydroxvmethvl-4-lT_ isoleucyloxy)butvI) ug arsine
a) To a solution of (R)-9-(2-hydroxymethyl-4-hydroxybutyl)guanine
(2.53 g, 10 mmole) in DMF {2S0 ml) were added N-Boc-L-isoleucine(2.77 g,
12 mmole), 4-dimethylaminopyridine (61 mg, 0.6 mmole) and DCC {3.7 g,
18 mrnole). After reaction for 16 hr at 0°C, N-Boc-L-isoleucine ( i .3
g) and
DCC ( 1.8 g) were recharged, and the reaction was kept overnight at room
temperature. The reaction mixture was filtered through Ceiite and the filtrate
was evaporated and purified by silica gel column chromatography, giving
I .25 g of the N-protected monoaznino acyl intermediate.
'H-NMR (DMSO-d6): 8 10.56 (s, 1H), 7.62 (s, 1H), 6.43 (s, 2H), 4.75
{t, IH), 4.15 - 3.80 (m, 5 H), 3.25 (m, 2H} 2.05 (m, 1H), 1.80-I-05 (m, 14H),
0.88 (m, 6H).
b) The intermediate from step a) ( 100 mg, 0.21 mmole} was treated with
trifluoroacetic acid (3 m) and fox 30 min at 0°C. The solution was
evaporated
and freeaze-dried, give the titled unprotected mono-aminoacyl product in
quantitative yield.
z5 1H-NMR (DMSO-d6 + D20): 8 8.72 {s, 1H), 4.15 (rn, 4F1), 3.90 (d, 1H), 3.42
(m, 2H), 2.09 {m, 1H), 1.83 (m, 1H), 1.61 (m, 2H), 1.I5 (m, H), 0.77 (d, 3H),
0.71
(t, 3H).
SUBSTfTU'fE SMEIE'T' {RULE 2~)
CA 02238516 1998-OS-26
WO 97130051 PCTISE97I00241
57
ExAMPLE 22
(R) 9-f2-Hydroxvmethyl-4-( L-valyloxv)butyll~uanine
The product of Example 1, step a) was deprotected with trifiuoroaacetic acid
in the
same manner as Example I > step c)
1H-NMR (250 MHz, DMSO-db}: $ I.04 (dd, 6H), I.55-I.88 (m, 2H), 2.2I (m, 2H},
3.48 (m, 2H), 4.00 (m, 1H), 4.13 (m, 2H), 4.34 {t, 2H), 6.9 (br s, 2H), 8.2I
(s, 1H),
1o 8.S (br s, 3H), 1 I.I (br s, 1H).
EXAMPLE 23
(R) 9 h-(L-Valytoxymethyt)-4-(valyloxy)butyll~uanine
t5 a) (R)-9-[4-(N-Boc-~.-valyloxy)-2-(N-Boc-L-valyloxymethyl)butyl]guanine
25
Application of the technique described in Example 1, step a), but using 2.7
eqs, 0.28
eqs, and 3.2 eqs of N-Boc-~.-valine, DMAP, and DCC, respectively, resulted in
the
tine compound.
1H NMR (250 MHz, CHC13) b: 0.95 (m, I2H}, 1.42 {br s, 18H}, 1.8 (m, 2H), 2.14
(m, 2H), 2.47 lm, IH), 4.0-4.4 (m, 8H}, 6.5 (br s, 2H), 7.67 (s> IH).
b) (R)-9-[4-(L-Vaiyloxy)-2-(L-valyloxymethyl)butyl~guanine
The titled compound was obtained as the tris-trifluoroacetate salt from the
intermediate of Example 20 step a) by deprotection in a manner analogous to
Example 1 step c).
~H NMR {250 MHz, D20) s: 1.0 (m> 12H), I.89 (m, 2H)> 2.29 (m, 2H}, 2.62 (m,
IH), 4.02 (dd, 2H), 4.38 (m, 6H), 4.89 (br s, ca. IOH), 8.98 (s, 1H).
EXAMPLE 24
{R) 9-(4-hydroxv-2-(stearoyloxymethyl)butyll~uanine
The titled compound is prepared according to steps a) to c) of Example 7.
SUBSTITUTE SHEET (RULE 2B)
CA 02238516 1998-OS-26
W~ 97!30051 PCTISE97I00241
58
tH NMR {250 MHz, DMSO-d6 ): $ 10.52 (s, iH), 7.62 (s, 1H}, 6.39 (s, 2H}, 4.50
(t,
1H), 3.93 (m, 4H), 3.42 (m, 2H), 2.45 (rn, 11-i), 2.23 (t, 2H), 1.48 (m, 4H),
1.22 (s,
28H}, 0.89 (t, 3H} ,
EXAMPLE 25 -
fR) 9 t2-H~droxvmethyl-4-(stearoyloxy)butyll~uanine.
The titled compound is prepared by the procedure of Example 17, step a)
to 1T-~ NMR (DMSO-d6) b: 0.86 (t, 3H); 1.25 (s, 28H); 1.5 i (qui, 2H); 1.62
(m, 2H);
2.D6 (m, 1H); 2.23 (t, 2H); 3.34 (d, 2H); 3.96 (ABX, 2H); 4.07 {dd, 2H); 6.30
(br s. ZH); 7.62 (s, 1H); ID.45 (s, 1H).
1s EXAMPLE 26
Alternative re aration of R -9- 2-stearo lox meth 1 -4- L-valvlox but I uanine
a) (R)-9-[4-N-benzyloxycarbonyl-L-valyloxy)-2-{hydroxymethyl)-
butyl]guanine
2o Dry H2G (252 mg, lmmol), 4-dimethylaminopyridine (122 mg, 1 mmol) and N-
Cbz-L-valine p-nitrophenyl ester (408 mg, 1. l mmol) were dissolved in dry
dimethyl fonnamide (16 ml). After stirring at 23°C for 30 hours, the
organic solvent
wa,s removed and the residue carefully chromatographed (silica, 2%-7%
methanollmethylene chloride) to afford the desired product as a white solid {
I5 I
25 mg, 31 %).
b) (R)-9-[4-N-benzyloxycarbonyl-L-valyloxy)-2-(stearoyloxymethyl)-
butyl]guanine
A solution of stearoyl chloride (394 mg, I.3 mmol) in dry methylene chloride
(2 ml)
3o was added slowly dropwise under nitrogen to a solution of the product of
step a)
(243 mg, 1 mmol} and 4-dimethylaminopyridine (20 mg) in dry pyridine (5 mI) at
-5°C. The reaction mixture was stirred at that temperature for 12
hours. Methanol (5
SUBSTtTUTE SHEET (RULE 2S)
CA 02238516 1998-OS-26
W~ 97l3U05I PCTlSE97/U0241
59
mI) was added and the reaction stirred for i hour. After removal of the
solvent, the
residue was triturated with acetonitrile and chromatographed (silica, 0-5%
methanollmethylene chloride) to afford the desired product (542 mg, 72%).
' 5 c) (R)-9-[2-stearayloxymethyl)-4-(L-valyloxy)butyl]guanine
The product of step b) (490 mg, lmmol) was dissolved in methanol (30 ml) and
5%
PdIC (100 mg) added. A balloon filled with hydrogen was placed on top of the
reaction vessel. After 6 hours at 23°C, TLC showed the absence of
starting material.
The reaction mixture was filtered through a 0.45 micron nylon membrane to
remove
to the catalyst and the solvent was removed to afford the desired product as a
white
solid (350 mg, 99%) which was identical (spectral and analytical data) to
Example
16.
EXAMPLE 27
15 Alternative re aration of R -9 - 4-h drox -2- -val lox meth 1 but 1 uanine
(R)-9-(4-(L-valyloxy)-2-(L-valyloxymethyl) butyl)guanine from Example 23 step
b)
( 100 mg, 0,126 mmole) was dissolved in 0.1 N NaOH aqueous solution (6.3 ml,
0.63 mmole) at roam temperature. At intervals, an aliquot was taken and
20 neutralized with 0.5 N trifluoroacetic acid. The aliquots were evaporated
and
analyzed by HPLC to monitor the progress of the reaction. After 4 hours, 0.5 N
trifluoroacetic acid solution (1.26 ml, 0.63 mmoie) was added to the solution
and
the reaction mixture was evaporated. The desired product was purified by HPLC,
('YMC, 50 x 4.6 rnm, gradient 0.1 % TFA + 0-50% 0.1 % TFA in acetonitrile, in
20
25 minutes, U'V detection at 254 nm. Yield: 13.6 %
'H-NMR (Dz0): 8 8.81 (s, 1H), 4.36 (m, 4H), 4.01 {d, 1H), 3.74 {m, 2H), 2.64
(m,
1H), 2.25 (m, 1H), 1.73 (m, 2H), i.03 (dd, 6H).
su~swTUT~ s~E~°r (~uLE 2~)
CA 02238516 1998-OS-26
WO 97/3fl05I PCTISE97/Ofl241
EXAMPLE 28
Alternative-preparation of (Rl-9-(2-hydroxymethvl-4-(L-valyloxylbut~, u~,
anine
HPLC separation of the reaction solution from Example 27 gave the titled
5 compound in 29.2% yield. ,
~H-NMR (DMSO-db): 8 8.38 (s, 3H), 8.26 (s, IH), 6.83 ( br s, 2H), 4.23 (m,
2H),
4.06 (m, 2H), 3.91 (m, IH), 3.40 (m, 2H), 2.i9 (m, 2H), 1.8 -I.40 (m, 2H),
0.95 (dd,
6H).
EXAMPLE 29
R -9- 2-stearovlox meth I -4- L-val lox but I uanine monoh drochloride
The product of Example 16, step d) (360 mg, 0.479 mmol) was dissolved in a
i5 mixture of methanol (10 mi) and ethyl acetate (10 ml). To the solution was
added
l001o PdJC (100 mg) and IN HCl (520 microiitres). The reaction mixture was
stirred
at room temperature for 2 hours under 1 atm. H~. The reaction mixture was
filtered
and the solvent evaporated from the. filtrate to provide the desired product
as a
crystalline solid (300 mg).
2fl
FORMULATION EXAMPLE A
Tablet formulation
The following ingredients are screened through a O.IS mm sieve and dry-mixed
25 10 g (R)-9-[2-(stearoyloxymethyl)-4-(L-vaiyloxy)butyl]guanine
40 g lactose
49 g crystalline cellulose
1 g magnesium stearate
A tabletting machine is used to compress the mixture to tablets containing 250
mg
30 of active ingredient.
FORMULATION EXAMPLE B
SUBSTITt,IT3E SHEET (RtIE.E 25)
. ~ CA 02238516 2005-O1-18
~'O 97!30051 PCTlSE97100241
61
Enteric coated tablet
The tablets of Formulation Example A are spray coated in a tablet coater with
a
solution comprising
S 120 g ethyl cellulose
30 g propylene glycol
IOg sorbitan monooleate
. ad I 000 ml aq. dist.
1o FORMULATION EXAMPLE C
Controlled release formulation
~0 g (R)-9-[2-(stearoyIoxymethyl)-4-(L-valyloxy)butyl]guanine
*
I2 g hydroxypropylmethylcellulose (Methocell K15) '
IS 4.5 g lactose
are dry-mixed and granulated with an aqueous paste of povidone. Magnesium
stearate (0.5 g) is added and the mixture compressed in a tabletting machine
to 13
mm diameter tablets containing 500 mg active agent.
2o FORMULATION EXAMPLE D
Soft capsules
250 g (R)-9-[2-(stearoyloxymethyl)-4-(L-valyloxy)butyl]guanine
100 g lecithin
25 100 g arachis oil
The compound of the invention is dispersed in the lecithin and arachis oil and
filled
into soft gelatin capsules.
*Trademark
CA 02238516 1998-OS-26
WO 97I300S1
62
PCTISE97I00241
BIOLOGY EXAMPLE 1
Bioavailability testing in rats
The bioavailability of compounds of the invention were compared to the parent
compound H2G and other H2G derivatives in a rat model. Compounds of the
invention and comparative compounds were administered, per oral (by catheter
into
the stomach), to multiples of three individually weighed animals to give 0.1
mmolll:g of the dissolved prodrug in an aqueous (Example 4, 5, Comparative
example 1 - 3, 5, 8), peanut oil (Comparative examples 4, 9,10) or propylene
glycol
1U {Example I - 3, 6 - 12, 17, Comparative example 6, 7) vehicle dependent on
the
solubility of the test compound ingredient. The animals were fasted from 5
hours
before to approximately 17 hours after administration and were maintained in
metabolic cages. Urine was collected for the 24 hours following administration
and
frozen until analysis. H2G was analysed in the urine using the HPLC/UV assay
of
Stable & C~?berg, Antimicrob Agents Chemother. 36 No 2, 339-342 ( 1992),
modified
as follows: samples upon thawing are diluted 1:100 in aq dist H20 and filtered
through an amicon filter with centrifugation at 3000 rpm for 10 minutes.
Duplicate
30 lt,l samples are chromatographed on an HPLC column; Zorbax SB-C18; 75 x 4.6
mm; 3.5 micron; Mobile phase 0.05M NHaPO4, 3 - 4 % methanol, pH 3.3 - 3.5; 0.5
2o ml/min; 254 nm, retention time for H2G at MeOH 4% and pH 3.33, --I2.5 min.
Bioavailability is calculated as the measured H2G recovery from each animal
averaged over at least three animals and expressed as a percentage of the
averaged
24 hour urinary H2G recovery from a group of 4 individually weighed rats
respectively injected i.v.juguiaris with O.x mmol/kg H2G in a FZinger~s buffer
vehicle and analysed as above.
Comparative example 1 (H2G) was from the same batch as used for preparation of
Examples i to 12. The preparation of Comparative example 2 (monoVal-H2G) and
SUHST1TUTE SHEET {RULE 26)
CA 02238516 1998-OS-26
WO 97/30051 , PCTlSE97/0024i
63
3 (diVal-H2G) are shown in Examples 21 and 23. Comparative example 4
{distearoyl H2G) was prepared by di-esterification of unprotected H2G in
comparable esterification conditions to step 2 of Example 1. Comparative
examples 5 & 8 (VailAc H2G) were prepared analogously to Example 4 using
acetic
anhydride with relevant monovaline H2G. Comparative example 6 (Alalstearoyl
H2G) was prepared analogously to Example 6 using 1~1-t-Boc-L-alanine in step
4.
Comparative example 7 (Gly/decanoyl) was prepared analogously to Example 5 but
using the step I intermediate made with N-t-Boc-L-gl~ycine. The preparation of
Comparative examples 9 and 10 is shown in Examples 24 and 25 respectively. The
to results appear on Table 2 overleaf:
SUBSTITUTE S~°IEE°T {RULE 2~)
CA 02238516 1998-OS-26
vvo 9'r3oosz rcmsE9~~ooz4a
64
TABLE 2
R, I R' .._ ~ Bi
Comparative example hydrogen hydrogen 8 %
I
Comparative example valyl hydrogen 29 % ,
2
Comparative example valyl valyl 36 070
3
Example I valyl stearoyl 56 %
Comparative example stearoyl stearoyl 1 %
4
Example 2 valyl myristoyl 57 %
Example 3 valyl oleoyl 51 %
Example 4 valyl butyryl 45 %a
Comparative example valyl acetyl 11
S
Example 5 , valyl decanoyl 48 %
Example 6 valyl docosanoyl 48 %
Example 7 isoleucyl stearoyl 53 %
Example 8 isoleucyl decanoyl 57 %
Example 9 isoleucyl myristoyl 49 %
Example 10 valyl 4-acetylbutyryl52 %
Example 11 valyl dodecanoyl 46 %
Example 12 valyl palrnitoyi 58 %
Example 17 stearoyl valyl S2 %
Comparative example alanyl stearoyl 23
6
Comparative example glycyl decanoyl 2S %p
7
Comparative Example acetyl valyl 7 %
8
Comparative Example hydrogen stearoyl i 2% ,
9
Comparative Example stearo 1 h dro en 7%
IO
Comparison of the bioavailabilities of the compounds of the invention with the
comparative examples indicates that the particular combination of the fatty
acids at
R~IR~ with the amino acids at R,IR2 produces bioavailabilities significantly
greater
SU~STtTUTIr SHSET (IE~ULE 26)
CA 02238516 1998-OS-26
Wi7 97!30051 PCTlSE97100Z41
than the corresponding diamino acid ester or difatty acid ester. For example,
in this
model, the compound of Example 1 displays 55 °yo better bioavailability
than the
corresponding divaline ester of Comparative example 3. The compound of Example
4 displays 25 °lo better availability than the corresponding divaline
ester.
5
It is also apparent, for instance from Comparative examples 5, 6 and 7 that
only the
specified fatty acids of this invention in combination with the specified
amino acids
produce these dramatic and unexpected increases in pharmacokinetic parameters.
1o BIOLOGY EXAMPLE 2
Plasma concentrations in rats
A plasma concentration assay was done in male Sprague Dawley derived rats. The
animals were fasted overnight prior to dosing but were permitted free access
to
15 water. Each of the compounds evaluated was prepared as a
solution/suspension in
propylene glycol at a concentration corresponding to 10 mg H2G !ml and shaken
at
room temperature for eight hours. Groups of rats (at least 4 rats in each
group)
received a 10 mg/kg ( 1 mllkg) oral dose of each of the compounds; the dose
was
administered by gavage. At selected time points after dosing (0.25, 0.5, l,
1.5, 2, 4,
20 6, 9, 12, 15, and 24 hours after dosing), heparinized blood samples (0.4
ml/sampie)
were obtained from a tail vein of each animal. The blood samples were
immediately
chilled in an ice bath. Within two hours of collection, the plasma was
separated
from the red cells by centrifugation and frozen till analysis. The components
of
interest were separated from the plasma proteins using acetonitrile
precipitation.
25 Following lyophilisation, and reconstitution, the plasma concentrations
were
determined by reverse phase HPLC with fluorescence detection. The oral uptake
of
H2G and other test compounds was determined by comparison of the H2G area
under the curve derived from the oral dose compared to that obtained from a 10
mg/kg intravenous dose of H2G, administered to a separate group of rats. The
3o results are depicted in Table 1B above.
BIOLOGY EXAMPLE 3
SU~STfrU'fE SHEE°f (RULE 2fs)
CA 02238516 1998-OS-26
WO 9'1130051 , PC'~/SE97100241
66
Bioavailability in monkey5.
The compounds of Example 1 and Comparative example 3 (see Biology Example 1
above) were administered p.o. by gavage to cynornoigus monkeys. The solutions
comprised: '
Example 1 150 mg dissolved in 6.0 ml propylene glycol, corresponding
to 25 mglkg or 0.0295 mmollkg.
Comparative 164 mg dissolved in 7.0 ml water, corresponding to 23.4
Example 3 mg/kg or 0.0295 rnmollkg.
Blood samples were taken at 30 min, 1, 2, 3, 4, 6, IO and 24 hours. Plasma was
separated by centrifugation at 2500 rpm and the samples were inactivated at
54°C
for 20 minutes before being frozen pending analysis. Plasma H2G levels were
monitored by the HPLClUV assay of Example 30 above.
Figure I depicts the plasma H2G recovery as a function of time. Although it is
not
possible to draw statistically significant conclusions from single animal
trials, it
appears that the animal receiving the compound of the invention experienced a
somewhat more rapid and somewhat greater exposure to H2G than the animal which
received an alternative prodrug of H2G.
BIOLOGY EXAMPLE 4
Antiviral activity
Herpes simplex virus-1 (HSV-1)- infected mouse serves as an animal model to
determine the efficacy of antiviral agents in vivo. Mice inoculated
intraperitoneally
with HSV-1 at 1000 times the LDSO were administered either with a formulation
.
comprising the currently marketed anti-herpes agent aeyclovir (21 and 83 mg/kg
in a
2%a propylene glycol in sterile water vehicle, three times daily, p.o.) or the
compound of Example 29 (21 and 83 mg/kg in a 2~o propylene glycol in sterile
SUBSTITUTE SHEET (RULE 26)
CA 02238516 1998-OS-26
i'VO 97130051 PCTlsE97l00241
67
water vehicle, three times daily, p.o.) for 5 consecutive days beginning 5
hours after
inoculation. The animals were assessed daily for deaths. The results are
displayed in
Figure 2 which charts the survival rate against time. In the legend, the
compound of
the invention is denoted Ex.29 and acyclovir is denoted ACV. The percentage of
' 5 mice surviving the HIV-1 infection was significantly greater following a
given dose
of the compound of the invention relative to an equivalent dose of acyclovir.
The foregoing is merely illustrative of the invention and is not intended to
limit the
invention to the disclosures made herein. Variations and changes which are
obvious
tp to one skilled in the art are intended to be within the scope and nature of
the
invention as defined in the appended claims.
SUSSTiTUTE SrtSET (F~U~E 26) '