Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-1_
TITLE OF THE INVENTION
DIPHENYL PYRIDYL ETHANE DERIVATIVES AS PEE IV
IIVI~BITORS
BACKGROUND OF THE INVENTION
This invention relates to compounds and pharmaceutical
compositions for the treatment of diseases by raising the level of cyclic
adenosine-3',5'-monophosphate (cAMI') through the inhibition of
phosphodiesterase IV (PDE IV).
Many hormones and neurotransmitters modulate tissue
function by elevating infra-cellular levels of adenosine 3', 5'-cyclic
monophosphate {cAMP). The cellular levels of cAMP are regulated by
mechanisms which control synthesis and breakdown. The synthesis of
cAMP is controlled by adenyl cyclase which may be directly activated
by agents such as forskolin or indirectly activated by the binding of
specific agonists to cell surface receptors which are coupled to adenyl
cyclase. The breakdown of cAMP is controlled by a family of
phosphodiesterase (PDE) isoenzymes, which also control the breakdown
of guanosine 3',5'-cyclic monophosphate (cGMP). To date, seven
members of the family have been described (PDE I-VII) the distribution
of which varies from tissue to tissue. This suggests that specific
inhibitors of PDE isoenzymes could achieve differential elevation of
cAMP in different tissues, (for reviews of PDE distribution, structure,
function and regulation, see Beavo & Reifsnyder (1990) TIPS, 11: 150-
155 and Nicholson et al ( 199I ) TIPS, ,~: 19-27].
The availability of PDE isotype selective inhibitors has
enabled the role of PDEs in a variety of cell types to be investigated. In
particular it has been established that PDE IV controls the breakdown of
cAMP in many inflammatory cells, for example, basophils (Peachell
P.T. et al., {1992) J. Immunol. 148 2503-2510) and eosinophils (Dent
G. et al., ( 1991 ) Br. J. Pharmacol. 103 1339-1346) and that inhibition
of this isotype is associated with the inhibition of cell activation.
Furthermore, elevation of cAMP in airway smooth muscle has a
spasmolytic effect. Consequently PDE IV inhibitors are currently being
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-2-
developed as potential anti-inflammatory drugs particularly for the
prophylaxis and treatment of asthma, by achieving both anti-
inflammatory and bronchodilator effects.
The application of molecular cloning to the study of PDEs
ha.s revealed that for each isotype there may be one or more isoforms.
For PDE IV, 'it is has been shown that there are four isoforms (A, B, C
and D) each coded for by a separate gene in both rodents (Swinnen 3.V.
et al., (1989) Proc. Natl. Acid. Sci. USA 86 5325-5329) said man
{Bolger G. et al., (1993) Mol. Cell Biol. 13 6558-657I).
The existence of multiple PDE IVs raises the prospect of
obtaining inhibitor$ that are selective for individual isoforms, thus
increasing the specificity of action of such inhibitors. This assumes that
the different PDE IV isoforms are functionally distinct. Indirect
evidence in support of this comes from the selective distribution of these
I5 isoforms in different tissues (Swinnen et al., 1989; Bolger et al., 1993;
Obernolte R. et al., (1993) Gene 129 239-247, ibid) and the high degree
of sequence conservation amongst isoforms of different species.
To date full length cDNAs for human PDE IVA, B and D
(Bolger et al., 1993 ibid; Obernolte et al., 1993 ibid; Mclaughlin M. et
al., (I993) J. Biol. Chem. 268 6470-6476) and rat PDE IVA, B and D
(Davis R. et al., (1989) Proc. Natl. Acid. Sci. USA 86 3604-3608;
Swinnen J.V. et al., (1991) J. Biol. Chem. 266 18370-18377), have been
reported, enabling functional recombinant enzymes to be produced by
expression of the cDNAs in an appropriate host cell. These cDNAs have
been isolated by conventional hybridisation methods. However using
this approach, only partial cDNAs for both human and rat PDE IVC
have been obtained. (Bolger et al., ibid. 1993 and Swinnen et al., ibid.
1989 and international Patent Specification No. WO 91116457.)
The design of PDE IV inhibitors for the treatment of
inflammatory diseases such as asthma, has met with limited success to
date. Many of the PDE IV inhibitors which have been synthesised have
lacked potency and/or inhibit more than one type of PDE isoenzyme in a
non-selective manner. PDE IV inhibitors that are relatively potent and
selective for PDE IV, are reported to be emetic as well. Indeed this side
CA 02239053 1998-OS-29
WO 97/22585 PCTlCA96/00838
-3-
effect has been so universal that experts have expressed their belief that
the emesis experienced upon administration of a PDE IV inhibitor, may
be mechanism based.
We have now found a novel series of tri-substituted phenyl
derivatives, members of which compared to known structurally similar
compounds are potent inhibitors of PDE IV at concentrations at which
they have little or no inhibitory action on other PDE isoenzymes. These
compounds inhibit the human recombinant PDE IV enzyme and also
elevate cAMP in isolated leukocytes. Certain compounds prevent
inflammation in the lungs induced by carrageenan, platelet-activating
factor (PAF), interleukin-5 (IL-5) or antigen challenge. These
compounds also suppress the hyperresponsiveness of airway smooth
muscle seen in inflamed lungs. Advantageously, compounds according
to the invention have good oral activity and at orally effective doses
exhibit little or none of the side-effects associated with known PDE IV
inhibitors, such as rolipram. The compounds of the invention are
therefore of use in medicine, especially in the prophylaxis and treatment
of asthma and other inflammatory conditions.
SUMMARY OF THE INVENTION
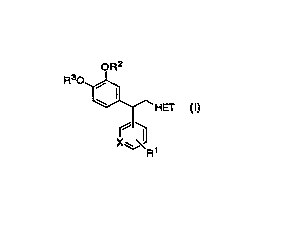
The invention encompasses novel compounds of Formula I
useful in the treatment of disease by inhibition of PDE IV, resulting in
an elevation of CAMP.
OR2
Rs0
-HEf
X~~ i
I
The invention also encompasses certain pharmaceutical
compositions and methods for treatment of diseases by inhibition of
CA 02239053 1998-OS-29
W~ 97/22585 PCT/CA96/00838
-4-
PDE IV, resulting in an elevation of cAMP, comprising the use of ,
compounds of Formula I.
DETAILED DESCRIPTION OF THE INVENTION
The invention encompasses the novel compound of Formula
I useful in the treatment of disease by inhibition of PDE IV, resulting
in an elevation of cAMP,
OR2
R
HEf
R'
I
or a pharmaceutically acceptable salt thereof wherein:
I S R I is selected from
(a) -CH=NOH,
(b) -CH(OH)-CH3,
(c) -CO-CH3,
(d) -CO-N[CH2CH2]2N-CH3;
(e) -CO-N[CH2CH2CH2CH2J, and
(f) -5-tetrazolyl;
R2 and R3 are independently selected from
(a) CI-7~y1~
(b) substituted CI-7 alkyl, wherein the substituent is. F, Cl,
Br or I,
(c) 2-phenethyl or 2-indanyl, optionally mono or di-
substituted on the benzene ring, wherein the substituents are
independently selected from the group consisting of
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
- 5 -
( 1 ) halo,
(2) C 1 _6allcoxy,
(3) Ci-6alkylthio,
(4) CN,
(5) CF3,
C 1-6~Y1~
N3~
(8) -C02H,
R4 is selected from
(a) hydrogen,
(b) C1-6alkyl,
(c) phenyl, benzyl or 2-phenethyl, optionally mono or di-
substituted on the benzene ring, wherein the substituents are
independently selected from the group consisting of
( 1 ) halo,
(2} C1-6alkoxy,
(3) Ci-6~'lthio,
(4) CN,
(5} CF3,
(6) Ci_6alkyl,
N3~
(8} -C02H,
RS and R8 are each independently selected from
(a) -CF3,
(b) Ci_6alkyl,
(c) phenyl, benzyl or 2-phenethyl, optionally mono or di-
substituted on the benzene ring, wherein the substituents are
independently selected from the group consisting of
( 1 ) halo,
. 30 (2) C1-6alkoxy,
(3) C1-6alkylthio,
. (4) CN,
(5) CF3,
(~) C1-fi~'1
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-6-
(~) N3~
(8) -CO~i,
R6 and R7 are independently selected from
(a) hydrogen, and
(b) C 1-6~Yh or
R6 and R~ may be joined to form a saturated 5, 6 or 7 membered
heterocycle, said heterocycle containing a heteroatom which is nitrogen
and optionally containing an additional hetero atom which is O or S
atom or NR4 , and optionally containing a carbonyl group;
10
HET is selected from pyridyl or imidazolyl, optionally mono or di-
substituted with substitutents selected from the group consisting of halo,
Ci-6~k3'h C1-6~oxy, C1-6alkylthio, benzyl, 2-phenethyl, NHCORS,
NR6R~~ NHS(O)ZRg~ OH, CN, or CF3, and the N-oxides thereof;
15 X is selected from N, N -~ O or CH.
Within this embodiment, there is a genus of compounds
wherein
R2 is cyclopentyl, optionally mono or di or tri-substituted with
20 substituents selected from the group consisting of F, Cl, Br and I, and
R3 is methyl.
Within this genus there is a class of compounds wherein
R I is selected from
25 (a) -CH=NOH,
(b) -CH(OH)-CH3,
(c) -CO-CH3,
(d) -CO-N[CH2CH2~2N-CH3;
(e) -CO-N[CH2CHZCHZCH2], and
30 (f) -5-tetrazolyi;
R2 is cyclopentyl,
R3 is methyl,
RS and Rg are each independently selected from
(a) -CF3,
CA 02239053 1998-OS-29
w0 97/22585 PCT/CA96/00838
_'
(b) C 1 _3alkyl,
(c) phenyl, benzyl or 2-phenethyl, optionally mono or di-
substituted on the benzene ring, wherein the substituents are
independently selected from the group consisting of
( 1 ) halo,
(2) C 1 _3alkoxy,
(3) C1_3alkylthio,
(4) CN,
(5) CF3,
(6) C1_3alkyl,
(~) N3~
(8} -C02H,
R6 and R~ are independently selected from
(a) hydrogen, and
(b) C1_3alkyl, or
R~ and R~ may be joined to form a saturated 5, 6 or 7 membered
heterocycle, said heterocycle containing a heteroatom which is nitrogen
and optionally containing an additional hetero atom which is O or S
atom or NR4 , and optionally containing a carbonyl group;
HET is selected from
pyridyl or imidazolyl, optionally mono or di-substituted
with substitutents selected from the group consisting of
halo, C 1 _6alkyl, C 1 _6alkoxy, C 1 _6aikylthio, benzyl, 2-phenethyl,
NHCOR$, NR6R~~ NHS(O)2R8> OH, CN, or CF3, and the N-oxides
thereof;
X is selected from N, N -~ O or CH.
Within this class there is a sub-class of compounds wherein
R1 is selected from
(a) -CH=NOH,
(b) -CH(OH)-CH3,
(c) -CO-CH3,
(d) -CO-N[CH2CH2]~T-CH3;
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
_ g _
(e) -CO-N[CH2CH2CH2CH2], and
(f) -5-tetrazolyl;
R2 is cyclopentyl,
R3 is methyl,
RS and R8 are each independently selected from
(a) -CF3,
Cb) CI-3~y1~
R6 and R~ are independently selected from
(a) hydrogen, and
IO (b) CI_3alkyl, or
R6 and R~ may be joined to form a saturated 5, 6 or 7 membered
heterocycle, said heterocycle containing a heteroatom which is nitrogen
and optionally containing an additional hetero atom which is O or S
atom or NR4, and optionally containing a carbonyl group;
IS
HET is selected from pyridyl, optionally mono or di-substituted with
substitutents selected from the group consisting of
halo, C 1 _6alkyl, C I _6alkoxy, C I _6alkylthio, benzyl, 2-phenethyl,
NHCORS, NR6R~~ NHS(O)ZRg~ OH, CN, or CF3, and the N-oxides
20 thereof;
X is selected from CH.
As will be appreciated by those of shill in the art, Halo is
intended to include F, Cl, Br and I.
For purposes of this specif canon alkyl is defined to include
linear, branched, and cyclic structures, with C I _6alkyl including
including methyl, ethyl, propyl, 2-propyl, s- and t-butyl, butyl, pentyl,
hexyl, I,I-dimethyiethyl, cyclopropyl, cyclobutyl, cyclopentyl and
cyclohexyl. Similarly, C I _6alkoxy is intended to include alkoxy groups
of from I to 6 carbon atoms of a straight, branched, or cyclic
configuration. Examples of alkoxy groups inciude methoxy, ethoxy,
propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy, and the like.
Likewise, C I _6alkylthio is intended to include alkylthio groups of from
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-9-
1 to 6 carbon atoms of a straight, branched or cyclic configuration.
Examples of alkylthio groups include methyithio, propylthio,
isopropylthio, cycloheptylthio, etc. By way of illustration, the
propylthio group signifies -SCH2CH2CH3. C1-6tlaloalkyl means an
alkyl group in which two or more hydrogen atoms have been replaced
by halogen atoms.
Exemplifying the invention are:
(a) (R)-4-{ 2-(3-Cyclopentyloxy-4-methoxyphenyl)-2-[4-
(4-methylpiperazinocarbonyl)pheny]lethyl }pyridine,
{b) {R)-4-[2-{3-Cyclopentyloxy-4-methoxyphenyl)-2-(4-
pyrrolidinocarbonyiphenyl)ethyl]pyridine,
(c) (R)-4-[1-(3-Cyclopentyloxy-4-methoxyphenyl)-2-(4-
pyridyl) ethyl]benzaldehyde oxime,
(d) (R)-4.-{2-{3-Cyclopentyloxy-4-methoxyphenyl)-2-[4-
(tetrazol-S-yl)phenyl]ethyl }pyridine,
(e) (R)-4-{ 2-(3-Cyclopentyloxy-4-methoxyphenyl)-2-j4-
(1-hydroxyethyl) phenyl]ethyl}pyridine, and
(f) (R)-4-[2-(3-Cyclopentyloxy-4-methoxyphenyl)-2-(4-
acetylphenyl) ethyl]pyridine.
Some of the compounds described herein contain one or
more asymmetric centers and may thus give rise to diastereomers and
optical isomers. The present invention is meant to comprehend such
possible diastereomers as well as their racemic and resolved,
enantiomerically pure forms and pharmaceutically acceptable salts
thereof.
Some of the compounds described herein contain oiefinic
double bonds, and unless specified otherwise, are meant to include both
E and Z geometric isomers.
In a second embodiment, the invention encompasses
pharmaceutical compositions for treatment of disease by inhibition of
PDE IV, as disclosed herein comprising a pharmaceutically acceptable
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96100838
- 10-
carrier and a non-toxic therapeutically effective amount of compound of
formula I as described above.
Within this embodiment the invention encompasses
pharmaceutical compositions for treatment of disease by inhibition of
PDE IV, resulting in an elevation of cAMP, as disclosed herein
comprising a pharmaceutically acceptable carrier and a non-toxic
therapeutically effective amount of compound of formula I as described
above.
For purposes of this specification a compound is said to
selectively inhibit PDE IV in preference to other PDE's if the ratio of
the IC50 concentration for aiI other PDE inhibition to PDE IV
inhibition is 100 or greater.
The pharmaceutical compositions of the present invention
comprise a compound of Formula I as an active ingredient or a
15 pharmaceutically acceptable salt, thereof, and may also contain a
pharmaceutically acceptable carrier and optionally other therapeutic
ingredients. The term "pharmaceutically acceptable salts°' refers to
salts
prepared from pharmaceutically acceptable non-toxic bases including
inorganic bases and organic bases. Salts derived from inorganic bases
20 include aluminum, ammonium, calcium, copper, ferric, ferrous,
lithium, magnesium, manganic salts, manganous, potassium, sodium,
zinc, and the like. Particularly preferred are the ammonium, calcium,
magnesium, potassium, and sodium salts. Salts derived from
pharmaceutically acceptable organic non-toxic bases include salts of
25 primary, secondary, and tertiary amines, substituted amines including
naturally occurring substituted amines, cyclic amines, and basic ion
exchange resins, such as arginine, betaine, caffeine, choline, N,N=
dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-
dimethylaminoethanol, ethanolamine, ethylenediamine, N-
30 ethylmorpholane, N-ethylpiperidine, glucamine, glucosamine, histidine,
hydrabamine, isopropylamine, lysine, methylglucamine, morpholine,
piperazine, piperidine, polyamine resins, procaine, purines,
theobromine, triethylamine, trimethylamine, tripropylamine,
trornethamine, and the like.
CA 02239053 1998-OS-29
WO 97/22585 PCTlCA96/00838
-il-
It will be understood that in the discussion of methods of
treatment which follows, references to the compounds of Formula I are
meant to also include the pharmaceutically acceptable salts.
Compounds according to the invention are selective and potent
inhibitors of PDE IV. The ability of the compounds to act in this way
may be simply determined by the tests described in the Examples
hereinafter.
The compounds according to the invention are thus of
particular use in the prophylaxis and treatment of human diseases where
an unwanted inflammatory response or muscular spasm (for example
bladder or alimentary smooth muscle spasm) is present and where the
elevation of cAMP levels may be expected to prevent or alleviate the
inflammation and relax muscle.
Particular uses to which the compounds of the invention
I S may be put include the prophylaxis and treatment of asthma, especially
inflamed lung associated with asthma, cystic fibrosis, or in the treatment
of inflammatory airway disease, chronic bronchitis, eosinophilic
granuloma, psoriasis and other benign and malignant proliferative skin
diseases, endotoxic shock, septic shock, ulcerative colitis, Crohn's
disease, reperfusion injury of the myocardium and brain, inflammatory
arthritis, chronic glomerulonephritis, atopic dermatitis, urticaria, adult
respiratory distress syndrome, diabetes insipidus, allergic rhinitis,
allergic conjunctivitis, vernal conjunctivitis, arterial restenosis and
artherosclerosis.
Compounds of the invention also suppress neurogenic
inflammation through elevation of CAMP in sensory neurones. They
are, therefore, analgesic, anti-tussive and anti-hyperalgesic in
inflammatory diseases associated with irritation and pain.
Compounds according to the invention may also elevate cAMP in
lymphocytes and thereby suppress unwanted lymphocyte activation in
immune-based diseases such as rheumatoid arthritis, ankylosing
spondylitis, transplant rejection and graft versus host disease.
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
- 12-
Compounds according to the invention have also been found
to reduce gastric acid secretion and therefore can be used to treat
conditions associated with hypersecretion of gastric acid.
Compounds of the invention suppress cytokine synthesis by
inflammatory cells in response to immune or infectious stimulation.
They are, therefore, useful in the treatment of bacterial, fungal or viral
induced sepsis and septic shock in which cytokines such as tumour
necrosis factor (TNF) are key mediators. Also compounds of the
invention suppress inflammation and pyrexia due to cytokanes and are,
therefore, useful in the treatment of inflammation and cytokine-
mediated chronic tissue degeneration which occurs in diseases such as
rheumatoid or osteo-arthritis.
Over-production of cytokines such as TNF in bacterial,
fungal or viral infections or in diseases such as cancer, Ieads to cachexia
and muscle wasting. Compounds of the invention ameliorate these
symptoms with a consequent enhancement of quality of life.
Compounds of the invention also elevate cAMP in certain
areas of the brain and thereby counteract depression and memory
impairment.
Compounds of the invention suppress cell proliferation in
certain tumour cells and can be used, therefore, to prevent tumour
growth and invasion of normal tissues.
For the prophylaxis or treatment of disease the compounds
according to the invention may be administered as pharmaceutical
compositions, and according to a further aspect of the invention we
provide a pharmaceutical composition which comprises a compound of
formula ( 1 ) together with one or more pharmaceutically acceptable
carriers, excipients or diluents.
For the treatment of any of these, compounds of formula I
may be administered orally, topically, parenteraily, by inhalation spray
or rectally in dosage unit formulations containing conventional non-
toxic pharmaceutically acceptable carriers, adjuvants and vehicles. The
term parenteral as used herein includes subcutaneous injections,
intravenous, intramuscular, intrasternal injection or infusion techniques.
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-13-
In addition to the treatment of warm-blooded animals such as mice, rats,
horses, cattle sheep, dogs, cats, etc., the compound of the invention is
effective in the treatment of humans.
The pharmaceutical compositions containing the active
ingredient may be in a form suitable for oral use, for example, as
tablets, troches, lozenges, aqueous or oily suspensions, dispersible
powders or granules, emulsions, hard or soft capsules, or syrups or
elixirs. Compositions intended for oral use may be prepared according
to any method known to the art for the manufacture of pharmaceutical
compositions and such compositions may contain one or more agents
selected from the group consisting of sweetening agents, flavoring
agents, coloring agents and preserving agents in order to provide
pharmaceutically elegant and palatable preparations. Tablets contain the
active ingredient in admixture with non-toxic pharmaceutically
acceptable excipients which are suitable for the manufacture of tablets.
These excipients may be for example, inert diluents, such as calcium
carbonate, sodium carbonate, lactose, calcium phosphate or sodium
phosphate; granulating and disintegrating agents, for example, corn
starch, or alginic acid; binding agents, for example starch, gelatin or
acacia, and lubricating agents, for example, magnesium stearate, stearic
acid or talc. The tablets may be uncoated or they may be coated by
known techniques to delay disintegration and absorption in the
gastrointestinal tract and thereby provide a sustained action over a
longer period. For example, a time delay material such as glyceryl
monostearate or glyceryl distearate may be employed. They may also
be coated by the technique described in the U.S. Patent 4,256,108;
4,166,452; and 4,265,874 to form osmotic therapeutic tablets for
control-release.-
Formulations for oral use may also be presented as hard
gelatin capsules wherein the active ingredient is mixed with an inert
solid diluent, for example, calcium carbonate, calcium phosphate or
kaolin, or as soft gelatin capsules wherein the active ingredients is mixed
with water or an oil medium, for example peanut oil, liquid paraffin, or
olive oiL
CA 02239053 1998-OS-29
w0 97/22585 PCT/CA96/00838
- 14-
Aqueous suspensions contain the active material in
admixture with excipients suitable for the manufacture of aqueous
suspensions. Such excipients are suspending agents, for example sodium
carboxymethyl-cellulose, methylcellulose, hydroxy-
S propylmethycellulose, sodium alginate, polyvinyl-pyrrolidone, gum
tragacanth and gum acacia; dispersing or wetting agents may be a
naturally-occurring phosphatide, for example lecithin, or condensation
products of an alkylene oxide with fatty acids, for example
polyoxyethylene stearate, or condensation products of ethylene oxide
with long chain aliphatic alcohols, for example heptadecaethylene-
oxycetanol, or condensation products of ethylene oxide with partial
esters derived from fatty acids and a hexitol such as polyoxyethylene
sorbitol monooleate, or condensation products of ethylene oxide with
partial esters derived from fatty acids and hexitol anhydrides, for
example polyethylene sorbitan monooleate. The aqueous suspensions
may also contain one or more preservatives, for example ethyl, or n-
propyl, p-hydroxybenzoate, one or more coloring agents, one or more
flavoring agents, and one or more sweetening agents, such as sucrose,
saccharin or aspartame.
Oily suspensions may be formulated by suspending the
active ingredient in a vegetable oil, for example arachis oil, olive oil,
sesame oil or coconut oil, or in mineral oil such as liquid paraffin. The
oily suspensions may contain a thickening agent, for example beeswax,
hard paraffin or cetyl alcohol. Sweetening agents such as those set forth
above, and flavoring agents may be added to provide a palatable oral
preparation. These compositions may be preserved by the addition of
an anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation
of an aqueous suspension by the addition of water provide the active
ingredient in admixture with a dispersing or wetting agent, suspending
agent and one or more preservatives. Suitable dispersing or wetting
agents and suspending agents are exemplified by those already
mentioned above. Additional excipients, for example sweetening,
flavoring and coloring agents, may also be present.
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
- 15-
The pharmaceutical compositions of the invention may also
be in the form of an oil-in-water emulsions. The oily phase may be a
vegetable oil, for example olive oil or arachis oil, or a mineral oil, for
example liquid paraffin or mixtures of these. Suitable emulsifying
agents may be naturally-occurring phosphatides, for example soy bean,
lecithin, and esters or partial esters derived from fatty acids and hexitol
anhydrides, for example sorbitan monooleate, and condensation
products of the said partial esters with ethylene oxide, for example
polyoxyethylene sorbitan monooleate. The emulsions may also contain
sweetening and flavouring agents.
Syrups and elixirs may be formulated with sweetening
agents, for example glycerol, propylene glycol, sorbitoi or sucrose.
Such formulations may also contain a demulcent, a preservative and
flavoring and coloring agents. The pharmaceutical compositions may be
in the form of a sterile injectable aqueous or oleagenous suspension.
This suspension may be formulated according to the known art using
those suitable dispersing or wetting agents and suspending agents which
have been mentioned above. The sterile injectable preparation may also
be a sterile injectable solution or suspension in a non-toxic parenterally-
acceptable diluent or solvent, for example as a solution in 1,3-butane
diol. Among the acceptable vehicles and solvents that may be employed
are water, Ringer's solution and isotonic sodium chloride solution. In
addition, sterile, fixed oils are conventionally employed as a solvent or
suspending medium. For this purpose any bland fixed oil may be
employed including synthetic mono- or diglycerides. In addition, fatty
acids such as oleic acid find use in the preparation of injectables.
Compounds of formula i may also be administered in the
form of a suppositories for rectal administration of the drug. These
compositions can be prepared by mixing the drug with a suitable non-
irritating excipient which is solid at ordinary temperatures but liquid at
the rectal temperature and will therefore melt in the rectum to release
the drug. Such materials are cocoa butter and polyethylene glycols.
For topical use, creams, ointments, jellies, solutions or
suspensions, etc., containing the compound of Formula I are employed.
CA 02239053 2003-06-20
WO 97/Z2585 PCT/CA96/90838
~, ~1 r
(For purposes of this application, topical application shall include mouth
washes and gargles.)
Dosage levels of the order of from about 0.01 mg to about
140 mglkg of body weight per day are useful in the treatment of the
above-indicated conditions, or alternatively about 0.5 mg to about 7 g
per patient per day» For example, inflammation may be effectively
treated by the administration of from about 0.01 to 50 mg of the
compound per kilogram of body weight per day, or alternatively about
0.5 mg to about 3.5 g per patient per day.
The amount of active ingredient that may be combined with
the carrier materials to produce a single dosage form will vary
depending upon the host treated and the particular mode of
administration. For example, a formulation intended for the oral
administration of humans may contain from 0.5 mg to 5 g of active
agent compounded with an appropriate and convenient amount of
carrier material which may vary from about 5 to about 95 percent of
the total composition. Dosage unit forms will generally contain between
from about 1 mg to about 5(10 mg of an active ingredient, typically 25
mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, fi00 mg, 800
mg, or 1000 mg.
It will be understood, however, that the specific dose level
for any particular patient will depend upon a variety of factors
including the age, body weight, general. health, sex, diet, tine of
administration, route of administration, rate of excretion, drug
combination and the severity of the particular disease undergoing
therapy.
~~Q~
The compounds of the present invention can be prepared
according to WO 94!14742, published on '~ .luly 1994, or according to
WO 95/1738b, published on 29 June 1995, ~r by tn~
methods described below. Iii waJ.~. be apparent tca
one skilled in the art that sim~.lar methodology could be
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
used to prepare the enantiomers or the racemates of the illustrated
compounds.
CA 02239053 1998-05-29
WO 97/22585 PCT/CA96/00838
-18-
CHEMISTRY
Scheme 1
Carboxylic acid derivatives were prepared by the method
presented in Scheme 1. The diastereoselective addition of Grignard
reagents derived from suitable bromoaryl-1,3-dioxolanes to acylsultam
Michael acceptor II afforded triarylpropanoylsultam intermediates.
Removal of the chiral auxiliary, subsequent decarboxylation and
aldehyde deprotection were then performed in a one-pot fashion by
successive treatment with a suitable lithium thiolate followed by a
saponification with aqueous hydroxide and then by an aqueous acidic
treatment. In this way, arenecarboxaIdehyde intermediates 1 were
obtained. Oxidation of those by sodium chlorite gave access to
arenecarboxylic acid intermediates 2. Carboxylic amides were prepared
by treating acids 2 with amines in the presence of diethyl
cyanophosphonate and triethylamine.
Scheme 2
Reaction of hydroxyiamine hydrochloride with
arenecarboxaldehyde intermediates Z gave access to oxime derivatives
(Scheme 2). Dehydration of these oximes with trichloromethyl
chloroformate ( or other suitable dehydrating agent such as acetic
anhydride, ethyl orthgoformate/H+, CC13COCl/Et3N, etc.) yielded the
corresponding aren~nitriles which were transformed into 5-tetrazolyl
derivatives following reaction with a suitable azide.
Scheme 3
Addition of methylmagnesium bromide to
arenecarboxaldehyde intermediates 1 gave the corresponding secondary
alcohols which were oxidized with pyridinium chlorochromate to afford
the acetyl derivatives .
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-19-
Scheme 1
O R2 O
R 3° / HET ~ ~~Br
W ~ / N
O
2
B
OR2
R3° / HET
1 ) PrSH / n-BuLi
2) NaOH
3) HCI
~z
°J
OR2 OR2
3 3
R O ' , NaCl02 _ R O \
~HET HET
C H O ~ C02H
OR2
HNRIRp / (Et0)2POCN / Et3N R3° /
HET
CONR~R2
HNRiR2 = N-methyl piperazine or pyrrolidine
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-20-
Scheme 2
OR2 OR2
R30 , R30
HEf
H2NOH~HCI _ HET CI3C02CC1
'CHO ~\CH=NOH
OR2 OR2
R30 / R30
HET B~3SnN3 ~ ' HEl'
\CN
N.N.N
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-2I -
scheme 3
o R2 o R2
R3o ~ R3o
CH3MgBr~
HET HET
CHO CH(OH)-CH3
O R2
R30
PCC~,
HET
CO-CH3
CA 02239053 1998-OS-29
WO 97!22585 PCT/CA96/00838
-22-
Representative compounds
Table I illustrates the compounds of the instant invention:
Table 1
" 'o
CH30
v
~N
R1
Scheme
C O- V -CH3
C O-NJ
CH=NOH
5 tetrazolyl
5 CH(OH}-CH3
6 CO-CHI
CA 02239053 1998-05-29
WO 97/22585 PCT/CA96/00838
-23-
Table 2. In Vitro Potency of
PDE N inhibitors.
ICsfl {nM)
E7C GST-Met 248 PDE iVa
1 0.8
2 1
3 3
4 2
2
6 3
Assays for Determining Biological Activity
5 Establishment of CHO-Kl cell lines stable expressing
PDE IVa enzyme
CHO-K1 cells stably expressing the prostacyclin receptor and grown
under 6418 selection as described previously (Y. Boie, et al, J. Biol.
Chem.: 269, 12173-12178, 1994) were plated at a density of 1.75 x 106
cells/I75cm2 in a T-175 flask (Gibco, Burlington, VT) containing alpha
MEM media; I0% heat inactivated fetal bovine serum {FBS); 1% (v/v)
penicillin/streptomycin; 25 mM Hepes, pH 7.4; and 500 mg/ml G4I8
(complete media). The cells were placed in an incubator for 24 hr at
37°C and 5% C02. The cells were then washed with warmed sterile
phosphate buffered saline (PBS) and incubated with 2mg/ml DNA, and 9
mg/ml lipofectamine reagent in Opti-MEM for 7 hr. At 37°C and 5%
C02. The incubation solution was diluted 1:2 with Opti-MEM
containing 20% FBS and incubated overnight. Following the overnight
incubation, the media was replaced by complete media containing 500
mg/ml hygromycin B. Colonies were identified and grown in T-17~
flasks for further characterization.
Measurement of whole-cell cAMP content
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-24-
CHO-K1 cells were plated at a density of 106 cells/175 cm2 containing
complete media with 500 mg/ml hygromycin. The flasks were
maintained in an incubator at 37°C with 5.0% C02 for 72 hr. The
media was changed and the cells were allowed to grow overnight. The
cells were washed and dissociated from the plate with PB S containing
0.5 mM EDTA. Cellular CAMP content was measured by centrifuging
the cell suspension at 150 g x 10 min. And resuspending the cells in a
Hanks buffered salt solution at a density of 0.2 x 106 cells/ml. The cells
were preincubated at room temperature for 15 min. and then incubated
with 10 mM prostaglandin I2 (PGI2) and the indicated compound for an
additional 10 min. Basal CAMP levels were determined by incubating
the cells in 0. I % DMSO. The incubations were terminated by the
addition of HCl (0.1 N final) and the cells measured for cAMP as
described below.
- 15
Determinations of whole-cell cAMP content were performed by
incubating I00 ml reconstituted rabbit anti-succinyl CAMP serum with
I00 ml of the whole-cell reaction or known CAMP standard and 30
pmol of 125I-CAMP THE in a ScintiStripTM well (300 ml final
volume) at room temperature for 18 h. Total cpm (Bo) was determined
in the absence of sample of cAMP standard. The reaction mixture was
then aspirated out of the well, and the individual wells were counted in a
Beckman LS 6000SC with the window open from 10-999 for 1 min.
The data were expressed as °!oB/Bo = [(standard or sample cpm -
non-
specific cpm) / (Bo cpm - non-specific cpm)~ x 100. Non-specifac cpm
were determined by incubating only the 125I-cAMP THE with assay
buffer (50 nM acetate; pH 5.8) in the ScintiStripTM well. AlI
determinations were performed in triplicate.
Phosphodiesterase Scintillation Proximity Assay
CHO-K1 cells were lysed by sonication for IO secs at a power setting of
50% (Braunsonic Model 2000) in an ice cold solution containing 50
mM Tris, pH 7.5; 1mM EDTA; and 200 mM b-mercaptoethanol.
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-25-
The soluble and particulate fractions of the cell were obtained by
centrifuging the sonicate for 90 min. at 100,000 x g at 4°C. PDE
activity was measured in a solution containing 50 mM Tris, pH 7.5;
lOmM MgCl2; 1 mM EDTA; and 100 nM (or indicated) 3H-cAMP
(100 ml final volume) in the presence of varying concentrations of
inhibitor. The reaction mixture containing enzyme was incubated
for 10 min. at 30°C in 96-well View Plates (Packard), and
terminated by the addition of 50 ml Phosphodiesterase Scintillation
Proximity Assay (SPA) Beads (Amersham) containing 18 mM
ZnS04. The amount of 3H-cAMP hydrolysis was determined by
counting the plates in a Wallac 1450 mBeta LSC counter.
The Elevation of CAMP in Leukocytes
The effect of compounds of the invention on intracellular cAMP
was investigated using human neutrophils or guinea pig eosinophils.
Human neutrophils were separated from peripheral blood,
incubated with dihydrocytochalasiri B and the test compound for 10
min and then stimulated with FMLP. Guinea pig eosinophils were
harvested by peritoneal lavage of animals previously treated with
infra-peritoneal injections of human serum. Eosinophils were
separated from the peritoneal exudate and incubated with
isoprenaline and test compound. With both cell types, suspensions
were centrifuged at the end of the incubation, the cell pellets were
resuspended in buffer and boiled for 10 min prior to measurement
of cAMP by specific radioimmunoassay (DuPont).
The most potent compounds according to the Examples induced a
concentration -dependent elevation of cAMP in neutrophils and/or
eosinophils at concentrations of 0.1 nM to 1 mM.
Suppression of Leukoc3~te Function
Compounds of the invention were investigated for their effects on
superoxide generation, chemotaxis and adhesion of neutrophils and
eosinophils. Isolated leukocytes were incubated with dihydrocyto-
CA 02239053 1998-05-29
WO 97122585 PCT/CA96/00838
-26-
chalasin B for superoxide generation only and test compound prior
to stimulation with FMLP. The most potent compounds of the
Examples caused a concentration-dependent inhibition of
superoxide generation, chemotaxis and adhesion at concentrations
of 0. I nM to I mM.
Lipopolysaccharide (LPS)-induced synthesis of tumour necrosis
factor {TNF) by human peripheral blood monocytes (PBM) is
inhibited by compounds of the Examples at concentrations of
IO O.OlnM to lOmM.
Relaxation of Constricted Airwav Smooth Muscle in vitro
The effects of compounds of the invention on guinea-pig isolated
tracheal smooth muscle were investigated. Isolated tracheal rings
I S were suspended in organ baths and immersed in oxygenated Krebs'
solution. The smooth muscle was contracted with sub-maximal
concentrations of histamine or carbachol prior to the addition of
increasing concentrations of test compound to the organ baths. The
most potent compounds of the Examples caused a concentration-
20 dependent reversal of both histamine and carbachol-induced
contractions at concentrations of InM to 100mM. The compounds
were generally more potent in reversing histamine-induced tone
than carbachol-induced tone.
25 Effects on Cardiac Muscle in vitro
Compounds of the invention have been tested for their effects on
isolated cardiac muscle. Right atrial and papillary muscles were
dissected out from the hearts of guinea pigs and suspended in organ
baths for measuring the rate {chronotropic) of spontaneously
30 beating atria and force (isotropic) of the electrically stimulated
papillary muscle. In these preparations, selective PDE IV
inhibitors such as rolipram do not have any direct effects whereas
selective PDE III inhibitors such as mi.lrinone have positive
chronotropic and isotropic effects. The non-specific PDE inhibitor
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-27-
theophylline, which is used in asthma as a bronchodilator, also
causes significant cardiovascular changes such as tachycardia.
Selective PDE IV inhibitors have advantage over theophylline,
therefore, through reduced cardiovascular side effects. The most
potent and selective compounds of the Examples had no direct
effects on the atrial and papillary muscles in vitro at concentrations
up to lOmM but in combination with PDE III inhibitors, these
inhibitors showed an enhancement of chronotropic and inotropic
activity, typical of selective type IV inhibitors.
Anti-inflammato~y Actiyity in vivo
Interleukin-S {IL-5)-induced pleural eosinophilia in the rat (Lisle,
et al, 1993, Br.J. Pharmacol. ~ 230p) is inhibited by compounds
of the Examples given orally at doses of 0.0001 to lO.Omg/kg. The
most potent compounds cause a dose-dependent reduction in
migrating-oos~ophiis-~D~s ofd-003-to-0:03trigfkg -p.o. -
Compounds of the invention also reduce the inflammatory
responses induced in rats by platelet activating factor (PAF).
Anti-allergic Activity in vivo
Compounds of the invention have been tested for effects on an IgE-
mediated allergic pulmonary inflammation induced by inhalation of
antigen by sensitised guinea pigs. Guinea pigs were initially
sensitised to ovalbumin under mild cyclophosphamide-induced
immunosuppression, by intraperitoneal injection of antigen in
combinations with aluminium hydroxide and pertussis vaccine.
Booster doses of antigen were given two and four weeks later and
at six weeks, animals were challenged with aerosolised ovalbumin
whilst under cover of an intraperitoneally administered anti-
histamine agent {mepyramine). After a further 48h, bronchial
alveolar lavages (BAL) were performed and the numbers of
eosinophils and other leukocytes in the BAL fluids were counted.
The lungs were also removed for histological examination for
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
- 28 -
inflammatory damage. Administration of compounds of the
Examples (0.001-lOmglkg i.p. or p.o.), up to three times during
the 48h following antigen challenge, lead to a significant reduction
in the eosinophilia and the accumulation of other inflammatory
Leukocytes. There was also less inflammatory damage in the lungs
of animals treated with compounds of the Examples.
Effects on Pulmonary Dynamics
Compounds of the invention (0.001-l0mg/kg by oral or other route
of aministration) reduce the allergic bronchoconstruction caused by
antigen in sensitized guinea pigs.
Compounds of the invention have been tested for their effects on
ozone-induced hyperreactivity of the airways of guinea pigs.
Following the inhalation of ozone, guinea pigs become very much
more sensitive to the bronchoconstrictor effects of inhaled
histamine than naive animals (Yeadon .~t al. 1992, P. ulmonary
~Pharm., ~, 39). There is a pronounced shift to the left (10-30 fold)
of the dose response curve to histamine and a highly significant
increase in the maximum increase in pulmonary resistance.
Compounds of the Examples administered lh prior to ozone by the
intraperitoneal or oral (0.001-lOmg/kg) route caused a dose-
dependent inhibition of ozone-induced hyperreactivity.
Adverse Effect
Compounds of the invention are free from adverse effects
following repeated overdosage to rats or dogs. For example, over
administration of 125mgJkg/day of active compounds of the
Examples to rats for 30 days is not associated with adverse toxicity.
SPA based PDE activity assa~protocol
CA 02239053 1998-OS-29
WO 97/22585 PCTlCA96/00838
-29-
Compounds which inhibit the hydrolysis of cAMP to AMP by the
type-IV cAMP-specific phosphodiesterases were screened in 96-
well plate format as follows:
In a 96 well-plate at 30 oC was added the test compound (dissolved
in 2 ul DMSO), 188 ml of substrate buffer containing [2,8-3H]
adenosine 3',5'-cyclic phosphate (CAMP, 100 nM to 50 ~t.M), 10
mM MgCl2, 1 mM EDTA, 50 mM Tris, pH 7.5. The reaction was
initiated by the addition of 10 ml of human recombinant PDE-IV
(the amount was controlled so that ~ 10% product was formed in
10 min. at 30 °C). The reaction was stopped after 10 min. by the
addition of 1 mg of PDE-SPA beads (Amersham). The product
AMP generated was quantified on a Microbeta 96-well plate
counter. The signal in the absence of enzyme was defined as the
background. 100% activity was defined as the signal detected in the
presence of enzyme and DMSO with the background subtracted.
Percentage of inhibition was calculated accordingly. IC50 value
was approximated with a non-linear regression fit of the standard
4-parameter/multiple binding sites equation from a ten point
titration.
IC50 values shown in Table 2 were determined with 100 nM cAMP
using the purified GST fusion protein of the human recombinant
phosphodiesterase IVa (met-248) produced from a baculovirus/Sf 9
expression system.
The most potent compounds of the invention are 20-30 times less
active than rolipram in inducing behavioural changes, sedation or
emesis in rats, ferrets or dogs.
The invention will now be illustrated by the following non-
limiting examples in which, unless stated otherwise:
CA 02239053 1998-OS-29
WO 97/22585 PC'1'/CA96/00838
-30-
(i) all operations were carned out at room or ambient
temperature, that is, at a temperature in the range 18-25°C;
evaporation
of solvent was carried out using a rotary evaporator under reduced
pressure (600-4000 pascals: 4.5-30 mm. Hg) with a bath temperature of
up to 60°C; the course of reactions was followed by thin layer
chromatography (TLC) and reaction times are given for illustration
only; melting points are uncorrected and 'd' indicates decomposition; the
melting points given are those obtained for the materials prepared as
described; polymorphism may result in isolation of materials with
different melting points in some preparations; the structure and purity
of all final products were assured by at least one of the following
techniques: TLC, mass spectrometry, nuclear magnetic resonance
(NMR) spectrometry or microanalytical data; yields are given for
illustration only; when given, NMR data is in the form of delta (d)
values for major diagnostic protons, given in parts per million (ppm)
relative to tetramethylsilane (TMS) as internal standard, determined at
300 MHz or 400 MHz using the indicated solvent; conventional
abbreviations used for signal shape are: s. singlet; d. doublet; t. triplet;
m. multiplet; br. broad; etc.: in addition "Ar" signifies an aromatic
signal; chemical symbols have their usual meanings; the following
abbreviations have also been used v (volume), w (weight), b.p. (boiling
point), m.p. (melting point), L (liter(s)), mL (milliliters), g (gram(s)),
mg (milligrams(s)), moI (moles), mmol (millimoles), eq (equivalent(s)).
The following abbreviations have the indicated meanings:
Ac _ acetyl
Bn - benzyl
cAMP cyclic adenosine-3,5-monophosphate
DBU - 1,8-diazabicyclo[5.4.0]under-7-ene
DIBAL - diisobutylaluminum hydride
DMAP - 4-(dimethylamino)pyridine
DMF - N,N-dimethylformamide
Et3N - triethylamine
GST glutathione transferase
CA 02239053 1998-OS-29
WO 97!22585 PCT/CA96/00838
-31 -
LDA - lithium diisopropylamide
m-CPBA - metachloroperbenzoic acid
MMPP - monoperoxyphtalic acid
MPPM - monoperoxyphthalic acid, magnesium salt
6H20
Ms - methanesulfonyl = mesyl = S02Me
Ms0 - methanesulfonate = mesylate
NSAm - non-steroidal anti-inflammatory drug
OXONE~ = 2KHS05KHS04K2S04
PCC - pyridinium chlorochromate
PDC - pyridinium dichromate
PDE phosphodiesterase
Ph - phenyl
Phe - benzenediyl
Pye - pyridinediyl
r.t. - room temperature
rac. - racemic
SAM - aminosulfonyl or sulfonamide or
S42~H2
SPA scientillation proximity assay
TBAF - tetra-n-butylammonium fluoride
Th - 2- or 3-thienyl
TFAA - trifluoroacetic acid anhydride
- tetrahydrofuran
Thi - thiophenediyl
TLC - thin layer chromatography
TMS-CN - trimethylsilyl cyanide
Tz - 1H (or 2H)-tetrazol-5-yl
C3H5 - ally!
Alkvl Group Abbreviations
Me - methyl
Et - ethyl
n-Pr - normal propyl
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-32-
i-Pr - isopropyl
n-Bu - normal butyl
i-Bu - isobutyl
s-Bu - secondary butyl
t-Bu - tertiary butyl
c-Pr ~ - cyclopropyl
c-Bu - cyclobutyl
c-Pen - cyclopentyl
c-Hex - cyclohexyl
EXAMPLE 1
(R~-4-12-(3-Cyclopentyloxy-4-methoxyphen' 1~-2-[4-t4-
n~ethXlpiperazinocarbon~phen l~thvl }pyridine
A solution of carboxylic acid 2 {100 mg, 0.24 mmol), 1-
methyipiperazine (0.03 mL, 0.26 mmol), diethyl cyanophosphonate
(0.05 mL, 0.31 mmol) and triethylamine (0.05 mL, 0.34 mmol) in DMF
(2 mL) was stirred at room temperature for 16 hours before EtOAc was
added. The organic phase was washed with water and brine, dried
(MgS04) and evaporated. The residue was purified by fiash-
chromatography {silica gel, CH2CI~MeOH 90:10) to afford the title
compound as a white solid (60 mg, 50%). 1H NMR (400 MHz, CDCl3):
d 1.55 {m, 2H), 1.75 (m, 6H), 2.30 (s, 3H), 2.30 (m, 2H), 2.45 {m, 2H),
3.30 (d, 2H), 3.40 (m, 2H), 3.75 (m, 2H), 3.80 (s, 3H), 4.15 (t, 1H),
4.60 {m, 1H), 6.60 (s, 1H), 6.65 (d, 1H), 6.75 (d, 1H), 6.90 {d, 2H),
7.20 {d, 2H), 7.30 (d, 2H), 8.40 (d, 2H).
EXAMPLE 2
LR)-4-f 2-(3-Cvciopentyloxy-4-methoxyphenvl)-2-t4-
p~ olidinocarbonvlphenvl)ethv~vridine
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-33-
Following the procedure described in Example 1 but
substituting pyrrolidine for i-methylpiperazine, the title product was
obtained as an off white solid. IH NMR {400 MHz, CDC13): d I.55 (m,
2H), 1.80 (m, 8H), I.95 (m, 2H), 3.30 (d, 2H), 3.40 (t, 2H), 3.60 (t,
2H), 3.80 (s, 3H), 4.15 {t, 1 H), 4.60 (m, 1 H), 6.60 (s, 1 H), 6.65 (d, 1 H),
6.75 (d, IH), 6.90 (d, 2H), 7.20 (d, 2H), 7.45 (d, 2H), 8.40 {d, 2H).
lR)-4-f 1-(3-CyclopentYloxv-4-methoxvphenvl)-2-l4-pvrid~)
ethy~benzaldehvde oxime
Hydroxylamine hydrochloride { 100 mg, I .4 mmol) was
added to a solution of aidehyde 1 (406 mg, 1.0 mmol) in pyridine {4
mL) and the reaction mixture was stirred at room temperature for 18
hours. The volatiles were then evaporated and EtOAc was added to the
residue. The organic phase was washed with 5% aqueous NaHC03,
dried (MgS04) and evaporated. The residue was purified by flash-
chromatography (silica gel, CH2C12/MeOH 95:5) to afford the title
compound as a white solid (306 mg, 89%). 1 H NMR (400 MHz,
CDCl3): d 1.55 {m, 2H), 1.80 (m, 6H), 3.30 (d, 2H), 3.80 {s, 3H), 4.15
(t, 1 H), 4.65 (m, I H), 6.60 (s, 1 H), 6.65 (d, 1 H), 6.75 {d, 1 H), 6.90 (d,
2H), 7.20 (d, 2H), 7.45 (d, 2H), 7.60 (s, 1H), 8.10 {s, IH), 8.40 (d, 2H).
EXAMPLE 4
fRl-4-12-(3-Cvclopent~y-4-methoxyphenyl)-2-f4-(tetrazol-5-
hen
ethyl }pyridine
Steg i:
{R)-4-[ 1-(3-Cyclopentyloxy-4-methoxyphenyl)-2-{4-
pyridyl)ethyl~benzenenitrile
CA 02239053 1998-OS-29
WO 97!22585 PCT/CA96/00838
- 34 -
Trichloromethyl chloroformate (0.18 mL, 1.5 mmol) was
added to a 0°C solution of (R)-4-j 1-(3-cyclopentyloxy-4-
methoxyphenyl)-2-(4-pyridyl)ethyl]benzaldehyde oxime from Example
3 (425 mg, 1.0 mmol) in acetonitrile (4 mL). The reaction mixture was
stirred at room temperature for 1 hour before water was added. The
mixture was then extracted with EtOAc. The organic phase was washed
with 5% aqueous NaHC03, water and brine, dried (MgS04) and
evaporated to afford the title compound as an off white solid (350 mg,
86%).
IO
Step 2:
(R}-4-{ 2-(3-Cyclopentyloxy-4-methoxyphenyl)-2- j4-(tetrazol-5-
yI)phenyl]
ethyl }pyridine
A solution of (R)-4-jl-(3-cyclopentyloxy-4-
methoxyphenyl)-2-(4-pyridyl)ethyl]benzene-nitrile from Step I (350
mg, 0.88 mmol) and tributyltin azide (1.17 g, 3.5 mmol) in 1,2-
dichlorobenzene (4 mL) was stirred at 150°C for 2 hours. The reaction
mixture was then cooled to room temperature and acetic acid (0.5 rnL)
was added. After 15 min, aqueous 1 N NaOH was added and the aqueous
phase was washed 3 times with ether. The aqueous phase was then
treated with 25% aqueous NH40Ac and 6 N HCI to pH 7 before it was
extracted twice with CH2CI2. The organic phase was then dried
(MgS04) and evaporated to afford the title compound as a yellow solid
(341 mg, 88%). 1H NMR (400 MHz, CDCI3): d 1.55 (m, 2H), 1.80 (m,
6H), 3.30 (m, iH), 3.45 (m, 1H), 3.80 (s, 3H), 4.20 (m, 1H), 4.70 (m,
1H), 6.70 (s, 1H), 6.80 (m, 2H), 7.00 (d, 2H}, 7.20 (d, 2H), 7.90 (d,
2H), 8.40 (d, 2H).
EXAMPLE 5
(R)-4-f 2-(3-C~pent~v-4-methoxv henvl)-2-f4-(1-hvdroxyeth
phen l~vl }pyridine
CA 02239053 1998-OS-29
WO 97!22585 PCT/CA96/00838
-35-
Methylmagnesium bromide ( I .4 M solution in toluene-THF;
1.1 mL, 1.5 mmol) was added dropwise to a -78°C solution of aldehyde
1 {509 mg, I.3 mmol) in THF (4 mL) and the mixture was allowed to
warm to room temperature. After 6 hours, 5% aqueous NaHC03 was
added and the mixture was extracted twice with EtOAc. The organic
phase was washed with brine, dried (MgS04) and evaporated. The
residue was purified by flash-chromatography (silica gel, hexane/EtOAc
25:75) to afford the title compound as a colorless gum (275 mg, 52%).
IH NMR (400 MHz, CDCl3): d 1.45 (d, 3H), 1.55 (m, 2H), 1.75 (m,
6H), 2.40 (s, 1 H), 3.30 (m, 2H), 3.80 (s, 3H), 4.15 (t, 1 H), 4.65 (m,
1H), 4.85 (m, 1H), 6.60 (s, IH), 6.65 (d, IH), 6.70 (d, 1H), 6.90 (d,
2H), 7.15 (d, 2H), 7.25 (d, 2H), 8.30 (d, 2H).
EXAMPLE 6
(Rl-4-f2-(3-C~pentvloxy-4-methoxvphenvll-2-(4-acet~phen~l
eth~~p~ridine
A solution of (R)-4-{ 2-(3-cyclopentyloxy-4-
methoxyphenyl)-2-[4-(I-hydroxyethyl)-phenyl]ethyl}pyridine from
Example 5 {205 mg, 0.49 mmol) in CH2Cl2 (4 mL) was added to a
suspension of pyridinium chlorochromate (272 mg, I.3 mmol) and
molecular sieve in CH2Cl2 (3 mL). The reaction mixture was stirred at
room temperature for 1 hour before the volatiles were evaporated. The
residue was then triturated in EtOAc and filtered over celite.
Evaporation of the filtrate afforded a residue which was purified by
flash-chromatography (silica gel, hexane/EtOAc 25:75) to afford the
title compound as a white foam (39 mg, I9%). 1H NMR (400 MHz,
CDCl3): d 1.55 (m, 2H), 1.75 (m, 6H), 2.55 (s, 3H), 3.30 (d, 2H), 3.80
(s, 3H), 4.20 (t, I H), 4.65 (m, 1 H), 6.60 (s, 1 H), 6.65 (d, 1 H), 6.75 {d,
1H), 6.90 (d, 2H), 7.25 (d, 2H), 7.85 (d, 2H), 8.40 (d, 2H).
PREPARATION OF INTERMEDIATES
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/0~838
-36-
INTERMEDIATE 1:
~R)-4-f 1-(3-Cvclopentvloxy-4-methoxyphenyl)-2-(4-pvrid~l)
etlz"y_~Ilbenzaldehvde.
" 'o
CH30
v
~ ~N
O ~H
Step 1: 2-(4-Brornophenyl)-1,3-dioxolane
A solution of 4-bromobenzaldehyde (66.5 g, 359 mmol),
ethylene glycol (22 mL, 395 mmoi) and p-toluenesulfonic acid
monohydrate ( 120 mg) in benzene (400 mL) was refluxed for 3 hours
with concomitant Dean-Stark water trapping. The organic phase was
then washed successively with 5% aqueous NaHC03, water and brine,
dried (MgS04) and evaporated. The residue was distilled under reduced
pressure (bp: 112°C/0.4 mmHg) to afford the title compound as a
colorless liquid (79.8 g, 97%).
Step 2: (1R, SS~-N { (3R)-3-(3-Cyclopentyloxy-4.-methoxyphenyl)-3-[4-
(1,3-dioxolan-2-yl)phenyl]-2-(4-pyridyl)propanoyl~-10,10-dimethyl-3-
thia-4-azatricyclo[5.2.1.01 ~5]decane-3,3-dioxide
A solution of 2-(4-bromophenyl)-1,3-dioxolane from Step
1 (55.3 g, 241 mmol) in THF (40 mL) was added to magnesium
turnings (6.16 g, 254 mmol) in THF (140 mL) containing 1,2-
dibromoethane (0.1 mL) at a sufficient rate to maintain a gentle reflux.
The mixture was stirred at room temperature for 2 hours before the
solution was added dropwise to a 0°C solution of acyl sultam II
CA 02239053 1998-OS-29
WO 97/22585 PCT/CA96/00838
-37-
{Celltech, World Patent Application 95/17386, 29 June 1995) (43.2 g,
80.5 mmol) in THF (700 mL). The reaction mixture was stirred at
room temperature for I6 hours before 10% aqueous NH4Cl was added.
The mixture was extracted with EtOAc and the organic phase was
washed with water and brine, dried (MgS04) and evaporated. The
residue was recrystallized twice in EtOH to afford the title compound as
an off white solid (25.6 g, 46%).
Step 3:(R)-4-[1-(3-Cyclopentyloxy-4-methoxyphenyl}-2-(4-
pyridyl}ethyl]benzaldehyde
n-Butyllithium (2.4 M solution in hexane; 10.6 mL, 25
mmol} was added dropwise to a 0°C solution of propanethiol (4.6 mL,
51 mmol) in THF (I50 mL}. After 15 minutes, (1R, SSA-N { (3R)-3-(3-
Cyclopentyloxy-4-methoxyphenyl)-3-[4-(1,3-dioxolan-2-yl)phenyl]-2-
(4-pyridyl)propanoyl }-I0,10-dimethyl-3-thia-4-
azatricyclo[5.2.1.01~5]decane-3,3-dioxide from Step 2 (9.7 g, 14.1
mmol) was added as a solid and the reaction mixture was stirred at
room temperature for 3 days. The volatiles were then evaporated and
the resulting residue was dissolved in EtOH (80 mL) and water (40
mL). Sodium hydroxide (4.5 g, 113 mmol) was added and the mixture
heated to reflux for 2 hours. The reaction mixture was cooled to room
temperature and treated with HCl 6 N to pH 4.5 then heated to reflux
for 1 hour. The reaction mixture was cooled to room temperature and
treated with NaOH I N to pH 14 then extracted twice with EtOAc. The
organic phase was washed with brine, dried (MgS04) and evaporated.
The residue was purified by flash-chromatography (silica gel, ether) to
afford the title compound as a white foam (2.3 g, 41%). 1H NMR (400
MHz, CI~C13): d 1.55 (m, 2H), 3.75 (m, 6H), 3.30 (m, 2H}, 3.80 (s,
3H), 4.20 (t, 1H), 4.60 (m, 1H), 6.60 (s, 1H), 6.65 {d, 1H), 6.75 (d, 1H),
6.90 (d, 2H), 7.35 (d, 2H), 7.75 (d, 2H), 8.40 (d, 2H), 9.95 (s, 1H).
Intermediate 2:
CA 02239053 1998-05-29
WO 97/22585 PCT/CA96/00838
-38-
C
(T2)-4-jI-f3-Cvclopentvloxy-4-methox henyl)-2-(4-
p rid l~3rilbenzoic acid
" 'o
H30
j ~n
OOH
A solution of NaC102 (149 mg, 1.64 mmol) and
NaH2P04~H20 (227 mg, I.64 mmoi) in water (I mL) was added to a
solution of aldehyde I (508 mg, 1.27 mmol) and 2-methyl-2-butene
(0.94 mL, 8.9 mmol) in t-BuOH (6 mL). The reaction mixture was
IO stirred at room temperature for I9 hours before 25% aqueous NH40Ac
was added. The mixture was then extracted twice with EtOAc. The
organic phase was washed with brine, dried (MgS04) and evaporated to
afford the title compound as a white solid (540 mg}. I H NMR (400
MHz, ClDCl3}: d I.55 (m, 2H), I.75 (m, 6H), 3.35 (m, 2H), 3.80 (s,
3H), 4.20 (t, IH}, 4.60 (m, IH), 6.60 (s, IH), 6.65 (d, IH), 6.75 (d, IH),
7.00 (d, 2H), 7.25 (d, 2H}, 8.00 (d, 2H), 8.45 (d, 2H).