Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02241788 2004-10-28
1
NOUVEAUX DÉRIVÉS DE N-(ARYLSULFONYL)AMINOACIDES, LEUR
PROCEDE DE PREPARATION ET COMPOSITIONS
PHARMACEUTIQUES EN CONTENANT
La présente invention a pour objet des nouveaux dérivés de
N-(arylsulfonyl)arninoacides, leur préparation, les compositions
pharmaceutiques en
contenant.
Ces composés présentent une affinité pour les récepteurs de la bradykinine
(BK). La bradykinine est un nonapeptide appartenant comme le decapeptide
kallidine à la classe des kinines et qui montre une activité physiologique
dans le
domaine cardiovasculaire et comme médiateur dans l' inflammation et la
douleur. On
distingue plusieurs récepteurs de la bradykinine : les récepteurs B1 et B~ (D.
Regoli
et al., Pharmacol. Rev., 1980, 32, 1-46). Plus précisément, les récepteurs B2
sont
les récepteurs de la bradykinine et de la kallidine: ils sont prédominants et
on les
trouve normalement dans la plupart des tissus,; les récepteurs Bl sont les
récepteurs
spécifiques de la [des-Arg9] bradykinine et de la [des-ArglO] kallidine : ils
sont
induits au cours des processus inflammatoires.
Les récepteurs de !a bradykinine ont été clonés pour différentes espèces,
notamment pour l'espèce humaine : récepteur B1 : J.G. Menke et al., ). Biol.
Chem., 1994, 269 (34) 21583-21586 ; récepteur B2 : J.F. Hess, Biochem.
Biophys.
Res. Commun., 1992, 184, 260-268.
Les revues : Drug News and Perspectives, 1994, 7 ( 10), 603-611 et Exp. Opin.
Ther. Patents, 1995, 5 (4), 331-340, font le point sur Ies antagonistes des
récepteurs
de la bradykinine. De nombreux antagonistes décrits oht des structures
peptidiques.
Comme antagonistes des récepteurs de la bradykinine, on peut citer en
particulier le
HOE-140 (F.1. Hock, Brit. J. Pharmacol. 1991, 102, 769-773) pour le récepteur
B2
et la [des-Arg9, LeuBJ bradykinine pour le récepteur B1 (M.N. Perkins et al.,
Pain.
1993, 53, 191-197). Récemment un antagoniste du récepteur B2 de structure non
peptidique le SR 173657 a été décrit dans Archiv Pharmacol., 1996, Suppl. 1,
354
{4), R6.
Selon la présente invention, on a maintenant trouvé une nouvelle famille de
composés présentant une affinité pour les récepteurs de la bradykinine, il s'
agit de
dérivés de N-(arylsulfonyl)aminoacides.
Parmi les dérivés de N-{arylsulfonyl)aminoacides, certains sont connus et
présentent différentes activités pharmacologiques. Ainsi des composés à
activité
antithrombotique sont décrits. dans les brevets ou demandes de brevet
européennes
CA 02241788 2004-10-28
2
EP 558 961, EP 236 163, EP 236 164, allemandes DD 155 954, DE 4 tl~ 468.
internationale WO 9 216 549. Dans ce domaine d'activité le NAPAP, dérivé de N-
(naphtylsulfonyl)glycine de formule
' ' i
soi r~x-cx~-co-NH- ~ ~-co N
cH2
N~AP y
~'t... ~
~o
NH
est décrit dans Pharmazie, 1987, 42 (5), 346.
De plus, des dérivés de N-tosyl-ji-alanine de formule
a
~ . SOZ NH-CH2-CH2-CO-'H-CONIb
CHz
1
20 c
dans laquelle a et b ensemble avec l' atome d' azote auquel ils sont liés
constituent un
cycle tel que pipéridine, pyrrolidine ou morpholine, sont décrits dans
Pharmazie,
1984, 39 (5), 315-317.
De même, des dérivés de N-(arylsulfonyl)proline ont été cités comme
inhibiteurs de la thrombine dans Pharmazie, 1986, 4i (4), 233-235 et
Pharmazie,
1987, 42 (2), 114-116.
Par ailleurs, la demande de brevet EP 614 911 décrit des composés de formule
Rt Ru - ~ N-Q3
30 Ari SOZ-N-C-CO NH-CH-CFi2 ~ ~ C 2
CHR'n Cp ~_Zj_Q~ _
Qz
r1 ~uRtv
CA 02241788 2004-10-28
3
dans Laquelle notamment
ArI représente un naphtyle, un phényle, un quinolyle, un isoquinolyle,
éventuellement substitués ;
- Arli représente un phényle ou un thiényle éventuellement substitués ;
- RI, RII, R'II représentent indépendamment l'un de l'autre H ou (C1-C4)alkyle
;
- ou RI ne représente rien et N est lié à ArII et éventuellement RII et R' II
forment
une double liaison ;
ou RI ou RIZ est lié à ArII et représente un (Cl-C3)alkylène ;
- RIII et RIV identiques ou différents représentent H, (C 1-C4)alkyle ou
forment
avec l' atome d' azote auquel ils sont liés un hétérocycle en (CS-C~) ;
- Zl représente un (Cl-C12)alkylène ;
Ql représente méthyle, amino, (Cl-Cç)alcoxycarbonylamino, (Cl-
C4)alkylamino, di(Cl-C4)alkylamino, pyrrolidïnyle, pipéridino, morpholino,
pipérazinyle, (Cl-C4)alkyl-4-pipérazinyle, amidino, (Cl-C4)alkylamidino,
guanidino, (Cl-Cq,)alkylguanidino, pyridyle, imidazolyle, pyrimidinyle,
indolyle,
hydroxy, (Cl-Cq.)alcoxy, (C2-Cg)alcoxycarbonyle, amino(Cl-C4)alkyl-N-(Cl-
C4)alkylamino, carbamoyle ou phényle éventuellement substitué ;
- Q2 représente H ou (Cl-Cç)alkyle ;
- Q3 représente H ou (Cl-Cq,)alkyle ou Ql et Q3 sont liés pour former un
hétérocycle et représentent ensemble (C2-Cg)alkylène tandis que Zl ne
représente
rien, sous forme d'énantiomères purs ou de leurs mélanges en proportions
quelconques ;
ainsi que leurs sels avec des acides.
Ces composés ont une affinité pour les récepteurs biologiques du neuropeptide
Y.
Selon la présente invention, on a maintenant trouvé des nouveaux composés
présentant, de façon inattendue, une affinité pour les récepteurs de la
bradykinine.
La présente invention a pour objet les composés de formule
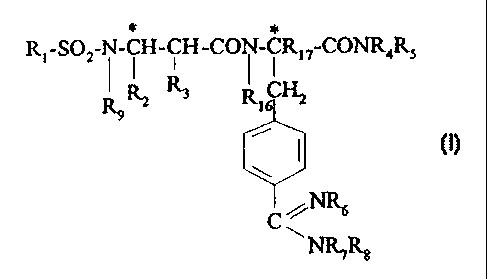
* *
Rt-SOZ ~ -ÇH-ÇH-CO ~ -ÇRyCONR4R5
RsRz R.' RIC
I (I)
C
CA 02241788 2006-O1-20
4
dans laquelle:
- R1 représente un phényle, un naphtyle, un tétrahydronaphtyle, un quinolyle,
ou un isoquinolyle, lesdits cycles étant non substitués ou substitués une ou
plusieurs fois par R10;
- R2 représente un phényle non substitué ou substitué une ou plusieurs fois
par R11, un phényl(C1-C4)alkyle non substitué ou substitué une ou plusieurs
fois sur le phényle par R11, un naphtyle non substitué ou substitué une ou
plusieurs fois par R11, ou un cyclohexyle non substitué ou substitué une ou
plusieurs fois par R11;
- ou R2 et R9 sont liés ensemble et constituent un (C3-CS)alkylène non
substitué
ou substitué par R12 ou un (C2-C4)alkylène interrompu par un atorne d' oxygène
ou un atome de soufre et non substitué ou substitué par R12 ;
- ou RZ et R9 ensemble avec l' atome de carbone et l' atome d' azote auxquels
ils
sont liés constituent la tétrahydroisoquinoline non substituée ou substituée
une ou
plusieurs fois par un halogène, un hydroxy, un (C1-Cq,)alkyle, un (C1-
Cç)alcoxy, ou un benzyloxy ;
- R3 représente l'hydrogène ou un hydroxy ;
- R4 et RS représentent chacun,indépendamment l'hydrogène ou un (Cl-C4)alkyle;
- ou R4 et R5 ensemble avec l'atome d'azote auquel ils sont liés constituent
un
radical hétérocyclique choisi parmi pyrrolidin-1-yle, pipérid-1-yle,
perhydroazépin-1-yle, morpholin-4-yle, 4-oxopipérid-1-yle, dihydropyrro-1-yle
et dihydroimidazol-2-yle, lesdites radicaux hétérocycles étant non substitués
ou substitués une ou plusieurs fois par R13;
- Rg représente l'hydrogène ou optionnellement représénte un benzyle non
substitué ou substitué sur le phényle une ou plusieurs fois par R13 ou un
groupe ZR14 lorsque R7 est l'hydrogène;
- R7 représente l'hydrogène ou un (C1-Cçalkyle;
- Rg représente l'hydrogène; un benzyle non substitué ou substitué sur le
phényle une ou plusieurs fois par R13; ou un groupe ZR14;
CA 02241788 2005-08-22
4a
- ou R7 et Rg ensemble avec l'atome d'azote auquel ils sont liés constituent
un
radical hétérocyclique choisi parmi pyrrolidin-1-yle, pipérid-1-yle
perhydroazépin-1-yle, morpholin-4-yle, tétrahydropyrimidin-2-yle, pipérazin-1-
yle et piperazin-1-yle substitué en position 4 par un (C1-C4)alkyle ou un
benzyle;
- ou, lorsque R7 est l'hydrogène, Rg et Rg sont optionneilement liés ensemble
pour former un (C2-C4alkylène non substitué ou substitué une ou plusieurs
fois par un (C1-Cq,)alkyle;
- R9 représente l'hydrogène, un (C1-C4)allcyle ou un phényl(C1-C4)alkyle non
substitué ou substitué sur le phényle une ou plusieurs fois par R11 ;
- R 1p représente un halogèné, un (C 1-C4)alkyle, un (C 1-C4)alcoxy, un
hydroxy,
un amino, un (C1-C4)alkylarnino ou un di(C1-Cq.)alkylamino ;
CA 02241788 1998-07-07
WO 97/25315 5 PCT/FR97100026
R11 -représente un halogène, un (CI-C4)alkyle, un trifluorométhyle, un
phényle,
un hydroxy, un (C 1-C4)alcoxy ou un benzyloxy ;
ou R11 est en position ortho du phényle représentant R2 et forme avec R3 un
groupe méthylène ou un groupe éthylène ;
- ou R11 est en position ortho du phényle représentant R2 et forme avec R9 un
groupe méthylène ou un groupe éthylène ;
- R12 représente un halogène, un (Cl-C4)alkyle, un hydroxy, un (Cl-C4)alcoxy,
un benzyloxy, un oxo, un phényle, un acétyloxy ou un trifluoroacétyloxy ;
- R13 représente un (C1-C4)alkyle, un halogène ou un hydroxy ;
- R14 représente un méthyle, un amino, un (C1-C4)alkylamino, un di(C1-C4)
alkylamino, un tri(Cl-C4)alkylammonium, un amidino, un (C1-Cç)alkylamidino,
un guanidino, un (C 1-C4)alkylguanidino, un hydroxy, un (C 1-C4)alcoxy, un
(C1-C4)alcoxycarbonyle, un groupe -AIkN(R15)Alk'N(R' 15)2~ ou un radical
hétérocyclique choisi parmi pyrrolidin-1-yle, pipéridin-1-yle, perhydroazépin-
1-
yle, pyridyle, imidazolyle, dihydroimidazolyle, imidazolidinyle, pyrimidinyle,
et
indolyle ;
- R 15 et R' 15 représentent indépendamment l' un de l' autre l' hydrogène ou
un (C 1-
C4)alkyle ;
R 16 représente l' hydrogène ou un méthyle, ou R 16 forme avec R9 un groupe
méthylène ;
- R 1 ~ représente l' hydrogène ou un méthyle ;
Alk et Alk' représentent indépendamment l'un de l'autre un (C1-CrI)alkylène ;
- Z représente un (C2-C 12)alkylène ou un (C 1-C6)alkylène interrompu ou
substitué
par un (C5-C~)cycloalkyle ou par un phényle ;
- C* représente un atome de carbone asymétrique ;
ainsi que leurs sels avec des acides minéraux ou organiques.
Les sels sont en général préparés avec des acides pharmaceutiquement
acceptables mais les sels d' autres acides utiles pour la purification ou l'
isolement des
composés de formule (I) font également partie de l'invention. Les sels des
composés
de formule (I) pharmaceutiquement acceptables sont par exemple le
chlorhydrate, le
bromhydrate, le sulfate, le méthanesulfonate, le benzènesulfonate, le
naphtalènesulfonate, le maléate, le fumarate, le citrate, l'acétate, le
gluconate, le
dobésilate, ou le sultosilate.
Les composés de formule (I) comprennent 2 atomes de carbone asymétriques
(ou éventuellement plus) et les 4 (ou éventuellement plus) énantiomères purs
ainsi
que leur mélange en proportions quelconques sont objets de l'invention.
CA 02241788 1998-07-07
WO 97/25315 6 PCT/FR97/00026
Par halogène, on entend chlore, fluor, brome, iode ; le chlore et le fluor
étant
préférés.
Par alkyle, alkylène, ou respectivement alcoxy, on entend un radical alk;yle,
un
radical alkylène ou respectivement un radical alcoxy linéaire ou ramifié.
On préfère les composés de formule (I) dans laquelle
dans laquelle
RI représente un phényle, un naphtyle, un tétrahydronaphtyle, un quinolyle, un
isoquinolyle, lesdits cycles étant non substitués ou substitués une ou
plusieurs foïs
P~ RIO ;
- R2 représente un phényle non substitué ou substitué une ou plusieurs fois
par
RI1, un phényl(Cl-C4)alkyle non substitué ou substitué une ou plusieurs fois
sur
le phényle par R l I ou un naphtyle non substitué ou substitué une ou
plusieurs
fois par R I l ;
- ou R2 et R9 sont liés ensemble et constituent un (C3-C5)alkylène non
substitué
ou substitué par R I2 ou un (C2-C4)alkylène interrompu par un atome d' oxygêne
ou un atome de soufre et non substitué ou substitué par R I2 ;
- ou R2 et R9 ensemble avec l' atome de carbone et l' atome d' azote auxquels
ils
sont liés constituent la tétrahydroisoquinoline non substituée ou substituée
une ou
plusieurs fois par un halogène, un hydroxy, un (C 1-C4)alkyle, un (C 1-Cq.
)alcoxy
ou un benzyloxy ;
- R3 représente l' hydrogène ou un hydroxy ;
R4 et R5 représentent chacun indépendamment l' hydrogène ou un {C 1-C4)alkyle;
- ou R4 et R5 ensemble avec l' atome d' azote auquel ils sont liés constituent
un
radical hétérocyclique choisi parmi pyrrolidin-1-yle, pipériè~-1-yle,
perhydroazépin-1-yle, morphoün-4-yle, 4-oxopipérid-1-yle, lesdits radicaux
hétérocycliques étant non substitués ou substitués par R 13 ;
- R6 représente l'hydrogène, R6 peut également représenter Rg lorsque R~ est
l'hydrogène ;
- R~ représente l' hydrogène ou un (C 1-C4)alkyle ;
- Rg représente l'hydrogène; un benzyle non substitué ou substitué sur le
phënyle
une ou plusieurs fois par R13 ; ou un groupe ZR14 ;
- ou R~ et Rg ensemble avec l'atome d'azote auquel ils sont liés constituent
un
radical hétérocyclique choisi parmi : pyrrolidin-1-yle, pipérid-1-yle,
perhydroazépin-1-yle, morpholin-4-yle, pipérazin-I-yle ou piperazin-1-yle
substitué en position 4 par un {CI-C4)alkyle ou un benzyle ;
CA 02241788 1998-07-07
WO 97/25315 7 PCT/FR97/00026
- ou, lôrsque R~ est l' hydrogène, R6 et Rg sont liés ensemble pour former un
(C2-
C4)alkylène non substitué ou substitué une ou plusieurs fois par un (C1-
C4)alkyle;
R9 représente l'hydrogène, un (C1-C4)alkyle ou un phényl(C1-C4)alkyle non
substitué ou substitué sur le phényle une ou plusieurs fois par R11 ;
- R10 représente un halogène, un (C1-C4)alkyle, un (C1-C4)alcoxy, un hydroxy,
un amino, un (C1-C4)alkylamino ou un di(C1-C4)alkylamino ;
- R11 représente un halogène, un {C1-Cq)alkyle, un hydroxy, un (Cl-C4)alcoxy
ou un benzyloxy ;
- R12 représente un halogène, un (Cl-C4)alkyle, un hydroxy, un (C1-C4)alcoxy,
un benzyloxy, un oxo ou un phényle ;
R13 représente un (C1-C4)alkyle, un halogène ou un hydroxy ;
Rlq. représente un méthyle, un amino, un (C1-C4)alkylamino, un di(Cl-C~)
alkylamino, un tri(C1-C4)alkylammonium, un amidino, un {C1-C4)alkylamidino,
un guanidino, un (C1-C4)alkylguanidino, un hydroxy, un (C1-C4)alcoxy, un
(Cl-C4)alcoxycarbonyle, un groupe -AIkN(R15)Alk'N(R' 15)2> ou un radical
hétérocyclique choisi parmi pyrrolidin-1-yle, pipéridin-1-yle, perhydroazépin-
1-
yle, pyridyle, imidazolyle, dihydroimidazolyle, imidazolidinyle, pyrimidinyle,
et
indolyle ;
- R 15 et R' 15 représentent indépendamment l' un de l' autre l' hydrogène ou
un {C 1-
C4)alkyle ;
- R16 représente l'hydrogène ;
- R 1 ~ représente l' hydrogène ;
- Alk et Alk' représentent indépendamment l'un de l'autre un (C1-C4)alkylène ;
- Z représente un (C2-C 12)alkylène ou un (C 1-C6)alkylène interrompu ou
substitué
par un (CS-C~)cycloalkyle ou par un phényle ;
ainsi que leurs sels avec des acides minéraux ou organiques.
Certaines valeurs des substituants sont préférées. Ainsi on préfère les
composés
de formule (I) répondant à au moins l'une des conditions suivantes
a -R1 représente un naphtyle, un quinolyle ou un trichlorophényle; RZ, R3, R4,
R~,
R6, R~, RA, Rg, R,6 et R,~ étant tels que définis ci-dessus pour la formule I;
b -R2 représente un phényle non substitué ou substitué par R11 ; R,, R3, R4,
R5, R~,.
R~, RR, R9, R,6 et R~~ étant tels que définis ci-dessus pour la formule I;
c -NR4R5 représente un groupe pyrrolidin-1-yle; RE, R2, R3, R6, R~, Rg, R9,
Ris et
R,~ étant tels que définis ci-dessus pour ia formule I;
CA 02241788 2004-10-28
ô
d -C(NR6)NR~Rg représente un 4,5-dihydroimidazol-2-yle ; R,, R2, R3, R4, Rs,
R9,
R~6 et R17 étant tels que définis ci-dessus pour !a formule I;
e- R3 = R9 = R16 = R1~ = H; Rl, R2, R4, RS. Rb, R~ et Rg étant tels que
définis
ci-dessus pour la formule I,
ainsi que leurs sels avec des acides minéraux ou organiques.
Selon l'invention on préfère les composés de formule
R,-sot-rrH-çH-cH2-corrl-~ çH-coN~~
"2a
(Ia)
C~ ~
y
dans laquelle
- R2a représente un phényle non substitué ou substitué en position meta ou
para par
R I I ; un napht- I-yle ou un napht-2-yle ;
- RI, R6, R~, Rg et R11 sont tels que définis ci-dessus pour (I) ;
ainsi que leurs sels avec des acides minéraux ou organiques.
Tout particulièrement, on préfere les composés de formule
Rlà SOZ-NH-ÇH-CHz-CONH-CH-CON
~.I
CHz
(ha)
N
N
H
dans Iaquehe
- Rla représente un napht-I-yle, un napht-2-yle, un 2, 4, 6-trichlorophényle
ou un
quinolin-2-yle ;
- R2a est tel que défini ci-dessus pour (Ia) ;
ainsi que leurs sels avec des acides minéraux ou organiques.
CA 02241788 1998-07-07
WO 97/25315 9 PCTIFR97/00026
Plus particulièrement, on préfere les composés de formule (I' a) dans laquelle
R2a est un phényle non substitué ou substitué en position méta ou en position
para
par R11.
De manière toute particulière, on préfère les composés de formule {I), {Ia) ou
(I'a) présentant une isomérie (R,R) sur les atomes de carbone marqués C*.
Dans la description et dans les revendications on utilise les abréviations
suivantes:
Me : méthyle
Et : éthyle
iPr : isopropyle
nBuOH : n-butanol
iPrOH : isopropanol
EtOH : éthanol
MeOH : méthanol
Et20 : éther : éther diéthylique
DMF : diméthylformamide
DCM : dichlorométhane
THF : tétrahydrofurane
AcOH : acide acétique
2fl AcOEt : acétate d' éthyle
DIPEA : düsopropyléthylalnine
DMAP : 4-diméthylaminopyridine
DCC : 1,3-dicyclohexylcarbodümide
DCU : dicyclohexylurée
NSuOH : N-hydroxysuccinimide
NSu : succinimido
NBS : N-bromosuccinimide
Fmoc : Fluorénylméthoxycarbonyle
Boc : sert-butoxycarbonyle
(Boc}20 : Bicarbonate de ditertiobutyle
NEt3 : triéthylamine
Bn : benzyle
Pd/C : palladium sur charbon
Sephadex~ LH 20 : commercialisée par PHARMACIA
Sephadex~ G 25 : commercialisée par PHARMACIA
Alcalase~ : subtilisine Carlsberg, commercialisée par NOVO (Danemark)
CA 02241788 1998-07-07
WO 97/25315 10 PCT/FR97/00026
Pénicilline Amidase : Pénicilline amidohydrolase, commercialisée par Sigma
BOP : hexafluorophosphate de benzotriazol-1-yloxytris(diméthylamino)
phosphonium
K2C03 : carbonate de potassium
K2S04 : sulfate de potassium
KHS04 : hydrogénosulfate de potassium
KHS04/K2S04 : solution de 16,66 g de KHS04 et 32,32 g de K2S04 dans 1 1
d'eau
NaCI : chlorure de sodium
Na2S04 : sulfate de sodium
MgS04 : sulfate de magnésium
NaOH : soude
NH40H : ammoniaque
HCI . acide chlorhydrique
TFA : acide trifluoroacétique
éther chlorhydrique : solution saturée de gaz chlorhydrique dans l'éther
diéthylique
mPa. s : milli Pascal/seconde
F : point de fusion
TA : température ambiante
RMN : résonance magnétique nucléaire
DMSO : diméthyl sulfoxyde
8 : déplacement chimique
s : singulet ; se : singulet élargi ; sd : singulet dédoublé ; d : doublet ;
dd : doublet dédoublé ; t : triplet ; te : triplet élargi ; qd : quadruplet ;
qt : quintuplet ; mt : multiplet ; m : massif
La présente invention a également pour objet le procédé de préparation des
composés de formule (I) et leurs sels. Ce procédé, appelé procédé 1, est
caractérisé
en ce que
a 1 } on traite un composé de formule
CA 02241788 1998-07-07
WO 97/25315 1 1 PCTIFR97/00026
R 1-SOZ-N-CH-CH-CON-CRI ~ -CONR4R5
i
~Rz ~ R16 ~ (II)
_' l
ï
CN
dans laquelle R1, R2, R3, R4, R5, R9, R16, R17 et C* ont les définitions
données
ci-dessus pour (I), sous forme d'un énantiomère pur ou d'un mélange d'isomères
en
proportions quelconques, avec un alcool de formule R-OH dans laquelle R
représente un (C 1-C4)alkyle, en milieu acide, pour former un imidate
intermédiaire
sur lequel on fait réagir une amine de formule HNR7Rg (III) ou une diamine de
formule H2NR6R8NH2 (IV) dans lesquelles R6, R7, Rg ont les définitions données
ci-dessus pour (I) ;
b1) on isole le composé de formule (I) ainsi obtenu sous forme de base ou de
sel,
cl) le cas échéant, on prépare un autre sel du composé de formule (I).
De nombreux procédés de synthèse des amidines sont décrits dans l' ouvrage
"The chemistry of an~idines and imidates", D.G. Neilson, Ed. Saul Pat~i, Wiley
ans
Sons, 1975, 389-394. La préparation de certaines amidines est décrite
précisément
dans la demande de brevet EP 614 911 A.
La formation de l' imidate est effectuée de préférence en milieu acide fort,
tandis
que l'imidoester sous forme de base libre ou sous forme de sel, est mis à
réagir avec
l'amine (III) ou la diamine (IV) dans un solvant polaire inerte par exemple un
alcool, à une température comprise entre 0°C et la température de
reflux du solvant.
On fait réagir sur l' imidate intermédiaire une amine dont la formule dépend
de
celle du composé (I) que l'on veut obtenir. Pour préparer un composé de
formule (I)
dans laquelle R6 = H, on fait agir une amine HNR~RB; pour préparer un composé
de
formule (I) dans laquelle R6 = Rg et R~ = H, on fait réagir deux moles d'une
amine
de formule H~NRs par mole d'imidate; pour préparer un composé de formule (I)
dans laquelle R; est l'hydrogène et R6 et Rg sont liés ensemble pour former un
(C~-
C4)alkylène non substitué ou substitué une ou plusieurs fois par un alkyle, on
fait
agir une diamine de formule H,NR~RANH~.
La plupart des amines (III) et des diamines (IV) sont connues et les produits
nouveaux peuvent être préparés en appliquant des principes et méthodes bien
connus
de l'homme du métier. Par exemple, pour les dérivés dans lesquels R14 est un
CA 02241788 1998-07-07
WO 97/25315 12 PCTIFR97/00026
imidazolyle, on se réfèrera au brevet US 3,881,016 et à la publication Synth.
Communie. 1987, 17 (21), 223-227.
Les composés de formule (I) dans laquelle R14 représente NH2 ou alkylamino
peuvent être préparés par hydrolyse des composés de formule (I) dans laquelle
R14
contient un groupe t-butoxy-carbonylamino, lui-même obtenu selon Synth.
Commun. 1990, 20 ( 16), 2559-2564.
On peut préparer les composés de formule (I) dans laquelle R14 représente un
groupe guanidino, substitué ou non, par action sur le composé de formule (I)
dans
laquelle R14 = NH2, d'un composé de formule
HZN-C-Y
NT
dans lequel T représente H ou (C 1-C4)alkyle et Y représente un nucléofuge,
tel que
S03H, par exemple , l'acide aminoiminométhane sulfonique (dans les conditions
décrites dans Tetrahedron Letters, 1988, 3183-3186) ou l'acide N-
méthylaminoiminométhane sulfonique (obtenu selon le procédé décrit dans J.
Org.
Chem. , 1986, 51 ( 10), 1882).
L'action d'une amine (III) de formule H2NRg en excès sur l'imidate résultant
de la réaction de ROH sur un composé de formule (II) conduit à la formation
d'un
mélange de 2 composés de formule (I) : pour l'un R6 = R7 = H et pour l'autre
R6
= R8 et R7 = H.
Les composés de formule (I) dans laquelle R6 et Rg ensemble forment un (C2-
C4)alkylène non substitué ou substitué une ou plusieurs fois par un (C1-
C4)alkyle
peuvent être préparés de façon connue en soi, par action d'une diamine
H2N-R6Rg-NH2 sur l' imidoester ou éventuellement par action de la même diamine
dont l'une des fonctions est protégée par un groupement labile (tel que Boc ou
Fmoc
par exemple) qui sera éliminé avant cyclisation.
On peut préparer les composés de formule (I) dans laquelle R 14 représente un
dihydroimidazole par action d'un alcool en milieu acide sur un composé de
formule:
35
CA 02241788 1998-07-07
WO 97125315 13 PCT/FR97/00026
R 1-SOZ-N-CH-CH-CON-CRi ~-CONR4R5
C ~ i
~ Rz ~ R16 Hz (II')
~C~~
NH NH
Z
CN
pour former un imidate intermédiaire, que l'on fait réagir sur une amine
appropriée selon les méthodes habituelles.
Les nitriles de formule (II) sont préparés en utilisant les méthodes
classiques de
la chimie des peptides, par exemple celles décrites dans The Peptides Ed. E.
Gross
et J. Meienhofer, Academic Press, 1979, l, 65-104. Des méthodes connues
permettent d'effectuer des couplages peptidiques sans racémisation des atomes
de
carbone de chaque aminoacide constitutif ; de plus, les (3-alanines ~i-
substituées pour
lesquelles le carbone chiral n'est pas adjacent au groupe carboxyle sont
réputées ne
pas subir de racémisation {Ann. Rev. Biochem., 1986, 55, 855-878). Par
ailleurs, la
demande de brevet EP 236 163 décrit des procédés permettant de conserver la
chiralité de chaque aminoacide.
En général les réactions de couplage entre les 2 aminoacides ont lieu à des
températures comprises entre 0°C et 40°C dans un solvant inerte
tel que le
dichlorométhane, l'acétonitrile, le tétrahydrofurane ou le diméthylformamide,
en
présence d'un agent de couplage et d'au moins un équivalent d'une amine
tertiaire
telle que la triéthylamine, la N-éthylmorpholine ou la düsopropyléthylamine.
Ainsi la prëparation d'un nitrile de formule (II) peut être réalisée selon
l'une des
voies de synthèse ci-après.
35
CA 02241788 2004-10-28
14
SCHÉMA 1 : Voie 1
RISOzN-ÇH-ÇH-COzH + HNRIS- 'R~~-CONR4R5
~Rz ~ ~z
(VI)
CN
couplage
'
SCHÉMA 2 : Voie 2
*
Boc-N-ÇH ÇH-COzH + HNRts ~RI~ CONRaRs
(~ ~ '~ (~)
.~
'-'-' Boc- ~ - i,H- ~ H-CONR16-' RyCONR4R5
_ g~~ ~ C~ (~)
CN
TFA
-= , H- ~ - i H-ÇH-CONR~s-ÇR~~-CONR4Rs
g~Rz ~ ~z
(I7~
2) R1S ~1 {~~ CIV
CA 02241788 1998-07-07
WO 97/25315 15 PCTIFR97/00026
Pour préparer un composé de formule (V), utile dans la voie 1, le radical R,SO
est introduit de façon classique par action d'un halogénure de sulfonyle de
formule
R~SOZHaI dans laquelle R, est tel que défini ci-dessus pour (I) et Hal
représente un
halogène, de préférence le chlore, sur un composé de formule
H-N-CH-CH-COOR'
i
~RZ
dans laquelle R2, R~, R9 ont les significations données ci-dessus pour (/j et
R'
représente l'hydrogène ou un (C,-C4)alkyle
On peut également préparer un composé de formule (V) dans laquelle R3 est
l' hydrogène en 2 étapes : tout d' abord on prépare un composé de formule
Y-N- ~ H-COOR' (V bis)
~R2
dis laquelle Y représente R1S02 ou un groupe protecteur tel que Boc ou Fmoc,
puis on allonge la chaîne carbonée d'un atome en utilisant des méthodes
connues,
puis si nécessaire on élimine le groupe protecteur et on fait réagir un
halogénure de
sulfonyle de formule R1 S02Ha1 et enfin on élimine le groupe R' , si celui-ci
est
différent de H.
Ce mode opératoire est approprié par exemple lorsque R2 et R9 sont liés
ensemble et constituent un (C3-C5)alkylène non substitué ou substitué par R12
ou
un C2-Cq alkylène interrompu par un atome d' oxygène ou un atome de soufre non
substitué ou substitué une ou plusieurs fois par R 12.
Les halogénures de sulfonyle sont connus ou préparés par des méthodes
connues.
Les composés de formule (VI) sont préparés par action d'une amine HNR4R5
sur un aminoacide de formule
NHR~ 6-ÇR 1 oCOOH
CI H2
i
CN
dans lequel l' amine est protégée, par exemple par un groupe Boc ou Fmoc,
suivie de
l'élimination du groupe protecteur.
CA 02241788 2005-08-22
16
Dans la séquence réactionnelle de la voie 2, l' amine est déprotégée en milieu
acide (TFA) et le radical R~SOz est introduit de façon classique par action
d'un
halogénure de sulfonyle de formule : R,S02Ha1 dans laquelle Ri est tel que
défini ci-
dessus pour (I) et Hal représente un halogène, de préférence le chlore.
Dans la voie 1 comme dans la voie 2, l'introduction du groupe RiSOZ est
effectuée en présence d'une base, éventuellement en milieu biphasique, en
présence
d'un catalyseur de transfert de phase.
Un substituant Rg peut être introduit sur un composé de formule (II] dans
laquelle Rg = H par. des méthodes connues, par exemple par action d' un
halogénure
de formule R9Hal, dans laquelle Hal représente un atome d' halogène par
exemple le
chlore.
Les composés de formule (II) dans laquelle Rg et R16 ensemble forment un
groupe méthylène sont préparés par action du paraformaldehyde sur des composés
de formule (II) dans laquelle Rg = R16 = H.
Les composés de formule (II) sont nouveaux et constituent un aspect ultérieur
de
la présente invention.
Certains composés de formule (V), dérivés de toluènesulfonamide ou de
phénylsulfonamide ont été décrits dans les publications suivantes
Tetrahedron, 1993, 49 (48), 11329-11340 ;
Centralblatt, 1929, II, 1398 ;
J. Org. Chem., 1978, 43 (23), 4.4.38-4441 ;
J. Org. Chem., 1966, 31 (7), 2385-2386.
Certains autres composés de formule:
R1S02 N-ÇH-ÇH-C02H
RgR2 R3
dans laquelle R1 représente un phényle, un naphtyle, un tétrahydronaphtyle, un
quinolyle ou un isoquinolyle, lesdits cycles étant non substitués ou
substitués
une ou plusieurs fois par R10 tel que défini pour (I), à condition que R1 ne
représente pas p-toylyle; et
R2, Rg, Rg et C* sont tels que définis pour (I),
étant entendu que
CA 02241788 2004-10-28
16a
- lorsque R1 représente phënyle et R3 représente l'hydrogène, alors R2 et Rg
ensemble avec les atomes de carbone et d'azote auxquels ils sont liés, ne
représentent ni pyrrolidinyle, ni 6,7-diméthoxy-1,2,3,4-
tétrahydroisoquinolyle,
sont nouveaux et font partie de l'invention.
Selon un mode de réalisation préféré de l'invention, dans cette formule V,
R1 représente naphtyle, quinolyle ou trichlorophényle et R3 représente un
atome d'hydrogène.
CA 02241788 1998-07-07
WO 97/25315 l 7 PCTIFR97/00026
Pour préparer un composé de formule (I) dans laquelle Rg et R11 ensemble
forment un groupe méthylène ou éthylène, on prépare d' abord un béta-
aminoacide
de formule (XIII)
H- i ~H~H-COOH
(XIII)
dans laquelle Rg et R11 forment un groupe méthylène ou éthylène en utilisant
des méthodes connues, par exemple celle décrite dans J. Org. Chem., 1987, 52,
616-622.
Pour préparer un béta-aminoacide de formule (XIII) dans laquelle R3 et RI1
forment ensemble un groupe méthylène ou éthylène, on utilise des méthodes
connues. Ainsi on prépare l'ester méthylique de l'acide 1-aminoindane-2-
carboxylique en 3 étapes à partir de l'indan-1-one : selon J. Med. Chem.,
1970,
650, l' action du carbonate de méthyle en présence d' hydrure de sodium permet
d'obtenir l'ester méthylique de l'acide (1-oxo)indane-2-carboxylique, puis,
selon J.
Heterocycl. Chem. , 1974, 11, 982, on prépare l' ester méthylique de l' acide
1
hydroxyiminoindan-2-carboxylique, enfin l' hydroxylamine est réduite, en amine
en
présence d'un catalyseur.
L'acide 1-amino-1,2,3,4-tétrahydronaphtalène-2-carboxylique peut être préparé
en suivant la méthode décrite dans J. Chromatogr. , A 1994, 676, 297-302.
Alternativement selon un autre procédé, lorsque le groupement amidine
C(=NR6)NR7Rg ne comprend pas de fonction susceptible de réagir dans une étape
ultérieure dans les conditions du couplage peptidique, on peut préparer un
dérivé
d'aminoacide comportant le groupement amidine à partir du dérivé correspondant
comportant un groupe cyano, et effectuer ensuite les couplages nécessaires
pour
obtenir un composé selon l'invention.
Ainsi selon un aspect ultérieur, la présente invention a pour objet un autre
procédé pour la préparation d'un composé de formule (I), appelé procédé ~,
caractérisé en ce que
a2) on traite un composé de formule
CA 02241788 1998-07-07
WO 97125315 I g PCT/FR97/00026
XNR~~-CR1~-CONR4R5
ÇH2
(VI bis)
i y
,,
CN
dans laquelle X représente l'hydrogène ou un groupe Boc et R4, RS et C* sont
tels
que définis pour (I), sous forme d'un énantiomère pur ou d'un mélange
d'isomères
en proportions quelconques, avec un alcool de formule R-OH dans laquelle R
représente un (C 1-C4)alkyle, en milieu acide, pour former un imidate
intermédiaire
sur lequel on fait réagir une amine de formule HNR~Rg (III) ou une diamine de
formule H2NR6RgNH2 (IV) dans lesquelles R6, R~, Rg ont les définitions données
ci-dessus pour (I)
b2) on effectue le couplage du composé ainsi obtenu de formule
HNR~ 6-CRl ~-CONR4R5
ÇH2
1
ò ~ NR6
C'
~rRa
- soit avec un composê de formule
Pr-N-CH-CH-C02H
(XII)
~R2
dans laquelle R2, R3 et R9 sont tels que définis pour (I) et Pr représente un
groupement protecteur, par exemple Boc ou Fmoc puis, après déprotecaion de
l'amine en milieu acide, on fait agir un halogènure de sulfonyle de formule
R I S02Ha1 dans laquelle R 1 est tel que défini pour (I) et Hal représente un
halogêne,
par exemple le chlore ;
- soit avec un composé de formule
Rt SOz-N-ÇH-ÇH-C02H
!,
R9~ ~ (V)
dans laquelle R1, R2, R3 et R9 sont tels que définis pour (I)
CA 02241788 2004-10-28
19
c2) on isole le composé de formule (I) ainsi obtenu sous forme de base ou de
sel;
d2) le cas échéant, on prépare un autre sel du composé de formule (I).
Les composés de formule (VI) et (XI) sous forme optiquement pure peuvent être
obtenus à partir d'un ester, par exemple, l'ester éthylique de la 4-
cyanophénylalanine racémique selon le schéma réactionnel décrit ci-agrès
SCHÉMA 3
HCI, HzN-CH-COi Et
CH2 ~ ~ CN
(Boc)20INEt3
BocNH-G'~i-C02Et
C
~ Alcaiase ~
CD)
' Boc-NH-CH-C02Et
(D) S CH2 ~ ~ CN
NaOH
(I,)
BocNH-CH COzH ~D)
BocNH-CH-C02H
CHz CN
~ -CN
CA 02241788 2004-10-28
NSuOHlDCC NSuOH/DCG
(L) (D)
BocNH-CH-C02NSu BocNH-CH-COzNSu
CHz ~ ~ CN (D) ~ CHZ ~ I CN
bars bars
CL) CD)
BocNH-CH-CO NRaRs BocNH-CH-CO-NR4Rs
CN
(L) g C~ ~ ~ CN (D} 8 ~ ~ I
TFA TFA
Cl-.) CD)
TFA, H2N-CH-CONRaRs TF~ ~N-CH-CONR4Rs
C~ ~ ~ CN
(L) (~ ~ ~ CD) (~
1} HCI, ROH 1} HCI, ROH
2) (~) 2} HI'(~)
ou ou
~z~sRs~ Hz~6ReNHz
(L) (D)
2HC1, HzN-CH-CONRaRs 2HC1, HZN-CH-CONRaRs
%NR6 %NR6
L'ester éthylique de la 4-cyanophénylalanine racémique est décrit dans la
demande de brevet EP 614 911 A. La protection de la fonction amine de ce
composé est réalisée de façon classique, par action de (Boc)20 en présence de
triéthylamine. L'action d'une enzyme, l'Alcalase~, sur le composé 4 ainsi
obtenu
CA 02241788 1998-07-07
WO 97/25315 2 I PCT/FR97/00026
permet d' hydrolyser sélectivement la fonction ester de l' aminoacide de
configuration
(L) et d' isoler ainsi chacun des composés 5 et 6 sous forme optiquement pure
(Synthesis, 1983, 1041-1043).
Le composé de configuration (L) est isolé sous forme acide : (L) 6. Le composé
de configuration (D) est isolé sous forme d'un ester éthylique : (D) 5 ; ce
dernier est
hydrolysé par la soude pour obtenir la forme acide : (D) 6. Chacun des 2
composés
de formule 6 est ensuite traité par la N-hydroxysuccinimide dans un solvant
inerte
tel que le DMF ou le dioxane, en présence d'un agent de couplage tel que le
DCC
pour obtenir un composé de formule 7. L'action d'une amine HNR4R5, suivie de
l' action de l' acide trifluoroacétique permet de préparer les composés de
formules (L)
(VI) et (D) (VI) ; on effectue ensuite les réactions classiques décrites ci-
dessus pour
obtenir les amidines souhaitées de formule (L) (XI) et (D) (XI).
Les béta-aminoacides de formule XIII
H- i -ÇH- i H-COOH (XIII)
ou les esters aliphatiques correspondants sont connus ou peuvent être préparés
par différentes méthodes par exemple selon J. Am. Chem. , 1936, 58, 299. Un
mode
de préparation particulier des beta-aminoacides consiste à effectuer un
allongement
de chaîne à partir d'un alpha-aminoacide selon Tetrahedron, 1994, 50, 9457-
9470
ou selon J. Chem. Soc. , Perkin Transact. II, 1977, 370.
Plus précisément, la préparation de certains beta-aminoacides de formule (XIV)
HAN-CH{R2)-CH2-COOH est décrite dans les publications ou brevets cités ci-
après.
30
CA 02241788 2004-10-28
22
R2 Référence
(R, S) : commercial
(R) : Ber. 1910, 43, 2020
\ / (S) J. Org. Chem., 1991, 56, 5883
J. Chem. Soc. Chem. Commun., 1993, 1153
Ct
Ct Heterocycles, 1989, 28, 1015
Cl ~ l Bull. Soc. Chim. Fr., 1987, 1079
Heterocycles, 1978, 1277-1285
OMe
/ C~~- Tetrahedron, 1987, 43, 3509-3517
EP-355819
Tetrahedron Lett. , 1988, 29, 6465
Heterocycles, 1989, 28, 1015
O n
J. Agric. Food Chem., 19?7, 25, 965
OH
Tetrehedron, 1994, 50 (31), 9457-9470
Tétrahedron Lett. , 1990, 3 I , 5153-5156
Les composés (I) dans lesquels R2 et R9 sont liés ensemble et constituent un
C3-CS alkylène non substitué ou substitué par R 12 sont préparés à partir de
composés de formule (XIII) connus ou préparés par des méthodes connus.
CA 02241788 1998-07-07
WO 97/25315 23 PCT/FR97/00026
Pour obtenir des composés optiquement purs de formule (XIII) ou (XIV), on
peut par exemple utiliser un ester de menthol selon la technique décrite dans
Tetrahedron Lett., 1988, 29, 6465-6466 ; on peut également utiliser un sel de
quinine ou de quinidine selon Chem. Ber., 1910, 43, 2020, ou bien le dérivé
phénylacétamido du composé de formule (XIV) sous forme racémique peut être
utilisé pour une résolution enzymatique avec une pënicilline amylase (Synlett,
1993,
339). On peut également mettre en oeuvre des synthèses énantiosélectives
Tetrahedron, 1994, 50, 9517 ; Aldrichimica, Acta, 1994, 27 ( 1 ), 3.
Pour préparer un alpha-hydroxy-beta-aminoacide, on peut utiliser la méthode
décrite dans Bull. Soc. Chim., France, 1940, 7, 593-603.
L' affinité des composés selon l' invention pour les récepteurs B 1 de la
bradykinine a été mesurée sur des suspensions de membranes de cellules MRCS en
utilisant une technique proche de celle décrite par K.H. Schneck et al., dans
Eur. J.
Pharmacol. , 1994, 266, 227-282. Dans ce test, l' affinité de [des-Arg9]
bradykinine
est comprise entre 10-6M et 10-7M, celle de la [des-Arg 10]kallidine est 2.10-
9M ;
et les composés de l' invention présentent une affinité allant jusqu' à 10-1
OM.
L'affinité des composés selon l'invention pour les récepteurs B2 de la
bradykinine a été mesurée sur des suspensions de membranes de cellules MRCS
selon une technique proche de celle décrite par D.G. Sawutz et al., dans Eur.
J.
Pharmacol. , 1992, 227, 309-315. Dans ce test, l' affinité de la bradykinine
est
voisine de 10-9M et celle des composés de l' invention varie autour de 10-6M
ou
10-7M.
La toxicité des composés selon l' invention est compatible avec leur
utilisation
thérapeutique.
Les composés selon l'invention pourront être utiles pour le traitement ou la
prévention de nombreuses pathologies, en particulier les pathologies de
l'inflammation, les maladies inflammatoires persistantes ou chroniques (Drug
News
and Perspectives, 1994, 10(7), 603-611). A titre d'exemple, on peut citer
l'inflammation neurogénique, la douleur (Brit. J. Pharmacol., 1993, 110, /93
198}, le choc septique, l' asthme, l' arthrite rhumatoïde, les maladies
inflammatoires
des articulations, les brûlures {Pain, 1993, 53, 191-197), les plaies, les
maladies des
voies respiratoires, par exemple les rhinites d'origine virale ou allergique,
le
syndrome de réponse inflammatoire systématique, l'oedème (Brit. J. Pharmacol.,
1995, 114, 1005-/013), l'angiogénèse (Brit. J. Pharmacol., 1993, 109, 14-17),
le
diabète infectieux de type I (Abst. l4th Intern. Symp. on Kinins, C49, Denver
Colorado, 10-15 Sept. 1995) l'hypertrophie ventriculaire associée au diabète,
la
CA 02241788 1998-07-07
WO 97J25315 24 PCT/FR97J00026
fibrose pulmonaire et la sclérose progressive systémique, l'entérocolite, et
plus
généralement toutes les pathologies bradykinine-dépendantes.
Les composés selon l'invention sont généralement administrés en unité de
dosage.
L' invention concerne aussi les compositions pharmaceutiques comprenant
comme principe actif l'un des énantiomères des composés de formule (I), l'un
de
leur mélange ou les sels de ceux-ci avec un acide pharmaceutiquement
acceptable,
ainsi qu'un excipient convenable pour une administration par voie orale,
injectable,
topique ou transdermique. Les doses journalières sont fonction de la
pathologie à
traiter et du malade.
La présente invention a également pour objet des compositions pharmaceutiques
contenant une dose efficace d' un composé selon l' invention ou d' un sel
pharmaceutiquement acceptable et des excipients convenables.
Lesdits excipients sont choisis selon la forme pharmaceutique et le mode
d' administration souhaité.
Dans les compositions pharmaceutiques de la présente invention pour
l' administration orale, sublinguale, sous-cutanée, intramusculaire,
intraveineuse,
topique, intratrachéale, intranasale, transdermique ou rectale, les principes
actifs de
formule (I) ci-dessus, ou leurs sels éventuels, peuvent être administrés sous
formes
unitaires d' administration, en mélange avec des supports pharmaceutiques
classiques, aux animaux et aux êtres humains pour la prophylaxie ou le
traitement
des troubles ou des maladies ci-dessus. Les formes unitaires d'administration
appropriées comprennent les formes par voie orale telles que les comprimés,
les
gélules, les poudres, les granules ou les solutions ou suspensions orales, les
formes
d'administration sublinguale, buccale, intratrachéale, intranasale, !es formes
d' administration sous-cutanée, intramusculaire ou intraveineuse et les formes
d'administrations rectale. Pour l'application topique, on peut utiliser les
composés
selon l'invention dans des crèmes, pommades, gels ou lotions.
Afin d'obtenir l'effet prophylactique ou thérapeutique désiré, la dose de
principe actif peut varier entre 0,01 et 50 mg par kg de poids corporel et par
jour.
Chaque dose unitaire peut contenir de 0,5 à 1000 mg de préférence de :a à 500
mg, d'ingrédients actifs en combinaison avec un support pharmaceutique. Cette
dose
unitaire peut être administrée 1 à 5 fois par jour de façon à administrer un
dosage
journalier de 0,5 à 5000 mg, de préférence de 1 à 2500 mg.
Lorsqu' on prépare une composition solide sous forme de comprimés, on
mélange l'ingrédient actif principal avec un véhicule pharmaceutique tel que
la
CA 02241788 1998-07-07
WO 97125315 25 PCT/FR97/00026
gélatine, l'amidon, le lactose, le stéarate de magnésium, le talc, la gomme
arabique
ou analogues. On peut enrober les comprimés de saccharose, d'un dérivé
cellulosique, ou d'autres matières appropriées ou encore on peut les traiter
de teile
sorte qu' ils aient une activité prolongée ou retardée et qu' ils libèrent d'
une façon
continue une quantité prédéterminée de principe actif.
On obtient une préparation en gélules en mélangeant l'ingrédient actif avec un
diluant tel qu'un glycol ou un ester de glycol et en versant le mélange obtenu
dans
des gélules molles ou dures.
Une préparation sous forme de sirop ou d'élixir ou pour l'administration sous
forme de gouttes peut contenir l'ingrédient actif conjointement avec un
édulcorant,
acalorique de préférence, du méthylparaben et du propylparaben comme
antiseptique, ainsi qu'un agent donnant du got~t et un colorant approprié.
Les poudres ou les granules dispersibles dans l' eau peuvent contenir l'
ingrédient
actif en mélange avec des agents de dispersion, des agents mouillants ou des
agents
de mise en suspension, comme la polyvinylpyrrolidone, de même qu'avec des
édulcorants ou des correcteurs du goût.
Pour une administration rectale, on recourt à des suppositoires qui sont
préparés
avec des liants fondant à la température rectale, par exemple du beurre de
cacao ou
des polyéthylèneglycols.
Pour une administration parentérale, on utilise des suspensions aqueuses, des
solutions salines isotoniques ou des solutions stériles et injectables qui
contiennent
des agents de dispersion et/ou des agents mouillants pharmacologiquement
compatibles, par exemple le propylèneglycol ou Ie butylèneglycol.
Pour une administration locale on peut utiliser des crèmes, des pommades, des
lotions ou des gels par exemple.
Pour l' administration transdermique, on peut utiliser des patches sous forme
multilaminée ou réservoir dans lequel le principe actif peut être en solution.
Le principe actif peut être formulé également sous forme de microcapsules,
éventuellement avec un ou plusieurs supports ou additifs.
Les compositions de la présente invention peuvent contenir, à côté des
produits
de formule (I) ci-dessus ou d'un des sels pharmaceutiquement acceptables,
d'autres
principes actifs qui peuvent être utiles dans le traitement des troubles ou
maladies
indiquées ci-dessus.
Les Préparations et les Exemples suivants illustrent l' invention. Sauf
indication
contraire les composés sont obtenus sous forme d'un mélange de
diastéréoisomères.
CA 02241788 1998-07-07
WO 97/25315 26 PCT/FR97/00026
Les spectres de résonance magnétique nucléaire (RMN) sont enregistrés à 200
MHz dans le DMSO deutérié contenant éventuellement du TFA, en utilisant le
tétraméthylsilane comme référence. Les déplacements chimiques sont indiqués en
ppm.
Les spectres de masse indiquent la valeur MH+
PRÉPARATIONS
Préparation 1.1
1-[2-Amino-3-(4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate.
On mélange 4,06 g d' acide 2-[N-(Boc)amino]-3-(4-cyanophényl)propionique
avec 1,15 ml de pyrrolidine et 7,5 g de BOP dans 20 ml de DMF ; on laisse sous
agitation 2 heures à TA en maintenant à pH = 7 par addition de NEt3. Après
évaporation à sec, on chromatographie sur silice en éluant par le mélange
chloroforme/méthanol (9/0,5 ; v/v). Après évaporation des solvants, le solide
obtenu est traité par 20 ml de TFA dans 20 ml de DCM pendant 30 minutes à TA.
On évapore les solvants, reprend à l'éther puis sèche pour obtenir 3,95 g du
produit
attendu.
Préparation 1.2
1-[2-Amino-3-(4-(3,4-dihydroimidazol-2-yl)phényl)propionyl]pyrrolidine,
dichlorhydrate.
On dissout 18 g de 1-[2-((N-Boe)amino)-3-(4-
cyanophényl)propionylJpyrrolidine dans 90 ml de DCM, ajoute 90 ml de TFA et
laisse sous agitation 40 minutes à TA. Après évaporation à sec, le résidu est
repris
par DCM puis évaporé (2 fois). On reprend ensuite par 300 ml d'ëthanol anhydre
saturé en HCI à 0°C et on laisse 18 heures à +4°C. Le milieu est
évaporé à sec,
repris par de l'éthanol, évaporé à nouveau puis repris par du DCM et évaporé
(2
fois). On obtient 23 g d'imidate intermédiaire. Le produit obtenu est dissous
dans 1
litre d'éthanol anhydre et l'on ajoute en 30 minutes 4,41 g d'éthylènediamine
dans
50 ml d'éthanol anhydre. Après 48 heures sous agitation à TA, on concentre le
milieu puis on acidifie à pH = 2 par addition d'une solution saturée d'HCl
dans du
méthanol. On essore, évapore à sec, puis on reprend par de l'éther, essore et
sèche
pour obtenir 17,4 g du produit attendu.
RMN (DMSO + TFA) : 1,50-1,80 : m : 4H ; 2,50-3,60 : m : 6H ; 4,00 : s
4H
4,35:se:1H;7,50:d:2H;8,15:d:2H;8,60:se:3H;11,10:s:2H.
Préparation I.3
CA 02241788 1998-07-07
WO 97/25315 27 PCT/FR97/00026
1-[2-Amino-3-(4-(N'-[3-(diméthylamino)propyl]amidino)
phényl)propionyl]pyrrolidine, tris-trifluoroacétate.
A) 1-[2-Amino-3-(4-(N~-[3-(diméthylamino)propyl]amidino}
phényl)propionyl]pyrrolidine, trichlorhydrate.
On dissout 809 mg de 1-[2-amino-3-(4-cyanophényl)propionyl]pyrrolidine
trifluoroacétate dans 50 ml de méthanol saturé en acide chlorhydrique à
0°C: et on
laisse une nuit au réfrigérateur. Après évaporation à sec, l'imidate formé est
repris
par du toluène, puis le milieu est évaporé et le résidu est séché sous vide
sur de la
potasse. Le produit obtenu est dissous dans 75 ml de méthanol anhydre et on
ajoute
lentement 570 p1 de N-(diméthyl)propane-1,3-diamine en solution dans 15 ml de
méthanol anhydre. Après 3 heures sous agitation à TA, on évapore à sec puis le
résidu est dissous dans 30 ml de méthanol en ajoutant 5 ml d'HC1 4N dans du
dioxane et on évapore à sec.
B) 1-[2-(N-Boc)amino-3-(4-(N1-[3-(diméthylamino)propyl]amidino)
phényl)propionyl] pyrrolidine, dichlorhydrate.
Le produit brut obtenu à l'étape précédente est dissous dans 10 ml de dioxane
et
10 ml d'eau. On ajoute de la triéthylamine pour atteindre pH = 8,3 puis 600 mg
de
(Boc)20 et on laisse sous agitation à TA pendant 15 heures. Le milieu est
dilué par
de l'eau, lavé 2 fois à l'éther, acidifié à pH = 1,5 par addition d'HCl 1 N
puis lavé
au DCM. On porte à pH = 7 par addition de soude 1 N et on évapore à sec. Le
résidu est repris dans du DCM, on filtre l'insoluble puis on purifie par
chromatographie sur silice en éluant avec le mélange chloroforme/méthanol
(85/15 à
80120 ; v/v). On obtient 560 mg du produit attendu.
C) 1-[2-Amino-3-(4-(N1-[3-(diméthylamino)propyl]amidino)
phényl)propionyl]pyrrolidine, tris-trifluoroacétate.
On mélange 550 mg du composé de l'étape B avec 15 ml de DCM et 15 ml de
TFA et on laisse 45 minutes sous agitation à TA. Après évaporation à sec, le
résidu
est dissous dans l'isopropanol, on évapore à nouveau à sec, reprend 2 fois par
de
l'éther. On décante puis sèche sous vide en présence de potasse. On obtient
,80 mg
du composé attendu.
Préparation 1.4
1-[2-Amino-3-(4-c yanophényl)propionyl]-2, 5-diméthyl-2 , 5-dihydropyrrole ,
trifluoroacétate.
A) 1-[2-(N-Boc)amino-3-(4-cyanophényl)propionyl]-2,5-diméthyl-2,5-dihydro-
pyrrole.
CA 02241788 1998-07-07
WO 97125315 2g PCTIFR97/00026
On mélange 2,1 g d'acide 2-[(N-Boc)amino]-3-{4-cyanophényl)propionique
avec 0,715 g de 2,5-diméthyl-2,5-dihydropyrrole dans 10 ml de DMF, on ajoute
3,9
g de BOP et on ajuste à pH = 7 par addition de NEt3. Après 18 heures sous
agitation à TA, on évapore à sec, puis on reprend par AcOEt, lave par une
solution
de NaHC03, par KHS04/K2S04, par une solution saturée de NaCI. On sèche sur
Na2S04 puis évapore. On reprend par un mélange étherlhexane puis sèche sur
Na2S04. On obtient 1,4 g du composé attendu.
RMN : (DMSO + TFA) 1,15-1,25 : m : 6H ; 1,30 : s : 9H ; 2,9-3,1 : rr~ : 2H;
4,3-4,6 : m : 1H ; 4,6-4,75 : m : 1H ; 4,9-5,1 : m : 1H ; 5,75-5,9 : m : 2H ;
7,5
d:2H;7,9:d:2H.
B) 1-[2-Amino-3-{4-cyanophényl)propionyl]-2,5-diméthyl-2,5-dihydropyrrole,
trifluoroacétate.
On dissout le composé de l'étape précédente dans 5 ml de DCM, on ajoute 5 ml
de TFA puis on laisse sous agitation 40 minutes à TA. On évapore à sec,
reprend
par DCM et évapore (3 fois) puis on reprend par un mélange éther/hexar~e. On
essore puis sèche sur Na2S04 pour obtenir 1,5 g du produit attendu.
En procédant comme dans la Préparation 1.4, étape A ci-dessus, on prépare les
composés décrits dans le tableau ci-après
TABLEAU 1
BocHN-CH-CO-NR4R5
CH2
i
CN
35
CA 02241788 1998-07-07
WO 97/25315 ~9 PCT/FR97/00026
Prparation -NR4R5 RMN {DMSO + TFA)
1.5 /~ 1,20 : s : 9H ; 2,75-2,95 :
mt : 2H ;
-N O
~ 3,30-3,60 : m : SH ; 4,60 :
mt : 1H ;
7,40 : d : 2H ; 7,70 : d :
2H
1.6 Me 0,8-1,2 : m : 6H ; 1,3 : s
: 9H ;
-N-iPr 2,7-3 : m : SH ; 4-4,8 : m
: 2H ;
7,S:d:2H;7,8:d:2H
1.7 Me 0,8-1,1 : m : 6H ; 1,15 : s
: 9H ;
-N 1,5-2,1 : m : 4H ; 2,7-2,9
: m : 2H ;
3,6-4 : m : 2H ; 4,1-4,4 :
m : 1H ;
Me 7,4:d:2H;7,7:d:2H
I S Préparation 1. 8
1-[2-N-méthylamino-3-{4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate.
A) Acide 2-(N-Boc-N-méthyl)amino-3-(4-cyanophényl)propionique.
On dissout 1,16 g d'acide 2-[N-Boc-amino]-3-(4-cyanophényl)propioniquc: dans
ml de THF, on ajoute à 0°C 2 ml d'iodure de méthyle puis, par petites
portions,
20 360 mg d'hydrure de sodium à 80 % dans l'huile. On laisse sous agitation
une nuit à
TA. Le milieu réactionnel est dilué par AcOEt puis on ajoute de l'eau et amène
à
pH = 2,5 par HCl IN. La phase organique est décantée puis lavée par de l'eau,
une
solution saturée de NaCI, puis séchée sur Na2S04 et évaporée. Le résidu est
repris
par un mélange Et20-hexane (1/1 ; v/v). La poudre formée est filtrée et séchée
pour
donner l, I 1 g du composé attendu.
RMN {DMSO + TFA) : 1,20 : sd : 9H ; 2,55 : sd : 3H ; 2,90-3,25 : m : 2H ;
4,50-4,80 : m : 1H ; 7,30-7,70 : m : 4H.
B) 1-[2-(N-Boc-N-méthyl)amino-3-(4-cyanophényl)propionyl]pyrrolidine.
On mélange sous agitation I,10 g du composé de l'étape précédente, 0,35 ml de
pyrrolidine et 1,78 g de BOP dans 15 ml de DMF, on ajuste à pH = 6 par
addition
de DIPEA. Après 2 heures et demie sous agitation, on extrait par AcOEt puis on
lave la phase organique par NaOH 0,25 N, HC1 0,25 N, H20 puis une solution
saturée de NaCI. La cire épaisse formée prend en masse après quelques jours à
+4°C. On obtient 1,25 g du composé attendu.
RMN (DMSO + TFA) : 1,10 : sd : 9H ; 1,60-I,85 : m : 4H ; 2,60 : sd : 3H ;
2,75-3,40 : m : 6H ; 4,80-5,10 : m : 1H ; 7,25-7,7U : m : 4H.
CA 02241788 1998-07-07
WO 97125315 30 PCT/FR97100026
C) 1-[2-N-méthylamino-3-(4-cyanophényl)propionyl]pyrrolidine,
trifluoroacétate.
0,72 g du composé de l'étape précédente sont placés dans 12 ml de TFA et 12
ml de DCM. Après 40 minutes sous agitation à TA, le milieu réactionnel est
concentré sous vide puis évaporé avec du DCM. On obtient 0,75 g du composé
attendu sous forme d'une cire épaisse.
Préparation 1.9
N,N-Diéthyl-[2-amino-3-(4-{3,4-dihydroimidazol-2-yl)phényl}]propionamide,
dichlorhydrate .
On prépare le N-N-diéthyl-[2-(N-Boc)amino-3-(4-cyanophényl)]propionamide
en procédant selon le mode opératoire décrit à la Préparation 1.4, étape A. Le
produit attendu est ensuite obtenu en suivant le mode opératoire décrit à la
Préparation 1.2.
Préparation 1.10
Ester ëthylique de l'acide 2-(N-Boc)amino-3-(4-cyanophényl)propionique.
26 g de Boc20 en solution dans 100 ml de DCM sont ajoutés progressivement à
une solution de 25,5 g du chlorhydrate d'ester éthylique de l'acide 2-amino-3-
{4-
cyanophényl) propionique et 13,9 ml de NEt3 dans 400 ml de DCM. Après 6 heures
d' agitation à TA le milieu réactionnel est lavé par une solution KHS04/K2SO4,
par
une solution saturée de NaHC03, par une solution saturée de NaCI. Après
séchage
sur Na2S04 et évaporation du DCM, le résidu est trituré dans l'heptane pour
donner
29 g de poudre blanche.
Préparation 1.11
Ester éthylique de l'acide (R) 2-(N-Boc)amino-3-(4-cyanophényl)propionique
composé B et acide (S) 2-(N-Boc)amino-3-(4-cyanophényl)propionique : composë
A.
Un mélange de 24 g du produit obtenu ci-dessus et 8,4 g de NaHC03 dans 900
ml d' AcOEt et 500 ml H20 est traité par 2 ml d' Alcalase~ pendant 24 heures à
TA .
Les 2 phases sont décantées ; la phase AcOEt est relavée par 100 ml d' un
solution à
10 % de NaHC03 qui est jointe à la première phase aqueuse et la phase aqueuse
est
relavée par 100 ml d'AcOEt jointes à la première phase AcOEt. La phase AcOEt
ainsi obtenue est séchée sur Na2S04 puis évaporée à sec ; on obtient 12,45 g
de
composé B
aD = +8,8° (c = 1 ; MeOH)
CA 02241788 1998-07-07
WO 97/25315 31 PCTIFR97/00026
La phase aqueuse est reprise avec de l'AcOEt et on amène à pH = 2,5 par HCI
6N. AcOEt est décanté, relavé par une solution KHS04/K2S04, par une solution
saturée NaCI, séché sur Na2S04 et évaporé à sec, on obtient 10,1 g du composé
A.
a.D = +8,8° (c = 1 ; MeOH)
Préparation 1.12
I-[2-Amino-3-(4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate, isomère
(S).
A) Ester de 2,5-dioxopyrroüdin-1-yle de l'acide 2-(N-Boc)amino-3-(4-
cy~ophényl) propionique, isomère (S).
On dissout dans 100 ml de dioxane 9,85 g d'acide 2-(N-Boc)amino-3-(4-
cyanophényl)propionique, isomère (S) et 4,14 g de NSuOH et on ajoute lentement
8,5 g de DCC en solution dans 30 ml de dioxane à TA puis on laisse sous
agitation
8 heures à TA. On filtre le DCU et le lave à l'acétone. Après évaporation à
sec du
filtrat, le résidu est dissous dans l'acétone puis on abandonne une nuit à TA.
Le
DCU restant qui a précipité est éliminé puis le filtrat est évaporé à sec,
trituré dans
Et20 puis essoré, lavé par Et20 et séché. On obtient 11,5 g du composë
attendu,
aD = -29,7° (c = 1, MeOH)
B) 1-[2-(N-Boc)amino-3-(4-cyanophényl)propionyl]pyrrolidine, isomère (S).
On place 11 g du composé de l'étape précédente dans 150 ml d'acétonitrile et
20 ml de DMF et on ajoute en IO minutes 2,5 ml de pyrrolidine dans 30 ml
d'acétonitrile. On laisse sous agitation 2 heures à TA puis on abandonne une
nuit à
TA. On évapore à sec, reprend par AcU~t puis on ajoute un tampon
~504/K2S04. On extrait par AcOEt puis lave par une solution saturée de
NaHC03 puis une solution saturée de NaCI ; on sèche sur Na2S04 puis on
c:vapore
à sec. Le résidu est trituré dans Et20, essoré, lavé par Et20 et séché. On
obtient
6,95 g du composé attendu,
a. D = + 26 ° {c = I , MeOH)
C) 1-[2-Amino-3-(4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate,
isomère
(S).
On dissout 6,75 g du composé de l'étape précédente dans 50 ml de DCM et on
filtre l'insoluble. On ajoute 50 ml de TFA et on laisse sous agitation pendant
45
minutes. On évapore à sec, redissout dans l'isopropanol, évapore à sec à
nouveau
puis on triture dans Et20, essore, lave par Et2O et sèche sur Na2S04. On
obtient
6, I3 g du composé attendu, F = 193-196°C.
CA 02241788 1998-07-07
WO 97/25315 32 PCTIFR97100026
aD = +51 ° {c = 1 ; MeOH)
Préparation 1.13
1-[2-Amino-3-(4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate, isomère
(R).
A) Acide (R) 2-(N-Boc)amino-3-(4-cyanophényl)propionique.
A 12,18 g du composé B de la Préparation 1.11, en solution dans 180 ml de
MeOH, on ajoute 43 ml de solution NaOH 1N et agite 1 heure à TA. Puis on
additionne 43 ml de solution HCl 1N et évapore 150 ml de méthanol puis on
reprend dans de l'AcOEt, et on lave par de l'eau puis par une solution saturée
de
NaCI. On obtient 11 g de composé attendu. Après cristallisation par un mélange
Et2Olheptane.
aD = -9,5° {c = 1 ; MeOH)
B) Ester de 2,5-dioxopyrrolidin-1-yle de l'acide (R) 2-(N-Boc)amino-3-{4-
cyanophényl)propionique.
A 10 g de l'acide obtenu ci-dessus dissous dans 10 ml de dioxane, on ajoute
4,2
g de NSuOH puis en 20 minutes on ajoute 8,62 g de DCC dissous dans 30 ml de
dioxane. Après 1 nuit d'agitation à TA la DCU formée est filtrée, lavée par du
~oxane. Le filtrat est évaporé à sec et le résidu trituré dans de l'éther pour
donner
un solide qui est filtré et séché. On obtient 12,09 g du composé attendu.
a,D = +27,1 ° (c = 1 ; MeOH)
C) 1-[2-(N-Boc)amino-3-(4-cyanophényl)propionyl]pyrrolidine, isomère (R).
A 11,6 g du composé obtenu à l'étape ci-dessus dissous dans 1:i0 ml
d'acétonitrile plus 20 ml de DMF on ajoute en 10 minutes 2,6 ml de pyrrolidine
dissoute dans 20 ml d' acétonitrile. Après 1 nuit d' agitation à TA, on
élimine un peu
d' insoluble et concentre ie filtrat sous vide. Le résidu est repris dans de
l' AcOEt et
on lave par une solution KHS04/K2S04, une solution saturée de NaHCO_;, une
solution saturée par NaCI ; après séchage sur Na2S04 l'AcOEt est évaporé sous
vide et le résidu trituré dans l'éther, on obtient 9,3 g du composé attendu
sous forme
d'un solide blanc.
a25 - -29,2° (c = H ; MeOH)
D
CA 02241788 1998-07-07
WO 97/25315 33 PCT/FR97/00026
D) 1-[2-Amino-3-(4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate,
isomère
(R).
8,7 g de produit obtenu à l'étape précédente sont agités 35 minutes dans un
mélange de 50 ml de DCM et 50 ml de TFA. Après évaporation à sec, on reprend
le
résidu dans de l'isopropanol et on réévapore à sec, on obtient 8,67 g du
composé
attendu sous forme solide.
aD = -46° (c = 1 ; MeOH)
Préparation 1.14
1-[2-Amino-2-méthyl-3-(4-cyanophényl)propionyl]pyrrolidine, trifluoroacétate.
A)
Un mélange de 7 g d'ester mêthylique de la (D,L) alanine, 14 ml de NEt.3, 4,2
g de MgS04, 3 H20 et 5,1 ml de benzaldéhyde dans 100 ml de dichlorométhane est
agité 18 heures à TA. L'insoluble est filtré, le filtrat concentré sous vide
et le résidu
repris dans un mélange éther-eau ; l'éther est décanté, relavé à l'eau puis
par une
solution saturée de NaCI, sèché sur Na2S04 et évaporé. On obtient 8,3 g du
composé attendu sous forme d'huile.
B)
A 2,29 g du produit de l'étape précédente dissous dans 60 ml de THF on ajoute
à -70°C 13,2 ml de bis(triméthylsilyl)amidure de lithium (1M dans le
THF) en 20
minutes. Après 30 minutes on ajoute en 15 minutes 2,35 g de 4-
bromométhylbenzonitrile dissous dans 15 ml de THF, puis on laisse remonter
doucement la température. Après 3 heures et demie le milieu réactionnel est
repris
dans AcOEt et on lave par de l'eau, par une solution saturée de NaCI. Après
séchage et évaporation d'AcOEt, on obtient 3,6 g du composé attendu sous forme
d'huile.
C)
Le composé de l' étape ci-dessus est repris dans 60 ml d' éther plus 60 ml ci'
HCl
1N et on agite 18 heures à TA. La phase aqueuse est décantée, mise au contact
d'AcOEt et on amène à pH = 10 par NaOH lON ; AcOEt est décanté, relavé par
H20, une solution NaCI saturée, séché, évaporé. On obtient 2,20 g du composé
attendu sous forme d'huile.
D)
A 2,18 g du composé de l'étape ci-dessus dissous dans 10 ml de dioxane on
ajoute à 10°C en 10 minutes 2,42 g de Boc20 dissous dans 10 ml de
dioxane ; puis
on agite 1 nuit à TA puis 3 heures à 40°C. Le milieu réactionnel est
repris dans
CA 02241788 1998-07-07
WO 97/25315 34 PCT/FR97/OOU26
AcOEt que l'on lave à l'eau, puis par une solution saturée de NaCI, que l'on
sèche
puis évapore pour donner une huile utilisée directement dans l'étape suivante.
E)
Le produit de l'étape précédente est dissous dans 10 ml de méthanol et on
ajoute
2,4 ml de solution KOH 8,36 N et agite 1 nuit ; on rajoute 2,4 ml de la
solution
KOH et porte à reflux 1 heure. Le milieu réactionnel est refroidi, repris par
un
mélange eau-éther ; la phase aqueuse est décantée, reprise avec de l'AcOEt et
on
amène à pH = 2 par HCl 1N. AcOEt est décanté, relavé par H20, par une solution
NaCI saturée, séché au Na2S04 puis évaporé. On obtient 0,99 g du composé
attendu après chromatographie sur silice en éluant par chloroforme/MeOH/'AcOH
(94/6/0,2 ; v/v/v).
F)
A 0,98 g du composé de l'étape précédente dans 10 ml de DMF on ajoute 0,30
ml de pyrrolidine, 1,55 g de BOP et de la DIPEA pour obtenir un pH de 6-7.
Après
2 heures et demie d'agitation le milieu réactionnel est repris par AcOEt et on
lave
par H20, NaOH 0,2N, HCl 0,2N, H20, une solution NaCI saturée. Après séchage
et évaporation on obtient 1,13 g du composé attendu, F = 84-87°C.
RMN (DMSO + TFA) : 1,20 : s : 3H ; 1,45 : s : 9H ; 1,60-1,90 : m : 4H ;
3,00-3,50 : m : 6H ; 7,30 : d : 2H ; 7,80 : d : 2H;
G)
1,12 g du composé de l'étape précédente sont agités 1 heure dans un mélange de
10 ml de DCM et 12 ml de TFA. Après évaporation et séchage on obtient 1,15 g
du
composé attendu sous forme de solide.
Prëparation 2.1
Acide 3-amino-3-(napht-2-yl)propionique, chlorhydrate.
On utilise la méthode décrite dans J. Am. Chem. Soc., 1936, 58, 299. On
mélange 15,5 g de 2-formylnaphtalène, 10,4 g d'acide malonique et 15,4 g
d'acétate
d'ammonium dans 150 ml d'éthanol et on chauffe à reflux pendant 6 heures.
Après
refroidissement à TA, on essore, lave par EtOH et sèche. Le produit obtenu est
dissous dans une quantité suffisante d'HCl 2N puis on filtre l'insoluble. La
solution
acide est concentrée par évaporation sous vide et le solide ainsi formé est
recristallisë dans 20 ml de mélange AcOH/H20 (1/1 ; vlv). On obtient 3,8 g du
produit attendu.
Selon le mode opératoire décrit ci-dessus, on prépare les composés du
TABLEAU 2 ci-après.
CA 02241788 2004-10-28
TABLEAU 2
H2N-ÇH-CHZ-COOH, HCl
Rz
Préparations R2
2.2
CI
2.3
10 \ ~ OBn
2.4 OHn
\ /
2.5 C1
CI
Préparation 2.6
20 Acide 3-(N-Boc)amino-3-(3,4-dichlorophényl)propionique.
A partir du composé de la Préparation 2.5, on prépare le béta-aminoacide N-
protégé de la façon suivante.
On dissout I0 g d'acide 3-amino-3-(3,4-dichlorophényl)propionique dans 100
ml d'eau et on ajoute 9 ml de triéthylamine, puis, progressivement, 9,3 g de
(Boc)20 dans 100 ml de dioxane. Après une nuit à TA sous agitation, on
concentre
le milieu puis on reprend par de l'eau en ajustant à pH = 9 par addition de
soude 1
N. On lave 2 fois à l'éther, reprend la phase aqueuse et acidifie à pH = 3 par
addition d'HCl 1 N, on extrait par AcOEt et concentre pour obtenir 3,9 g du
composé attendu, F = 128°C.
RMN (DMSO)
1,25 ppm : s : 9H ; 2,50-2,65 ppm : mt : 2H ; 4,75 ppm : q : IH ;
30 7,25 ppm : d : 1H ; 7,20-7,40 ppm : m : 3H
Préparation 2.7
CA 02241788 1998-07-07
WO 97/25315 36 PCT/FR97/00026
Chlorhydrate de l'acide 3-amino-3-(3-isopropyloxyphényl)propionique.
A) {3-isopropyloxyl)benzaldehyde.
A 12,2 g de 3-hydroxybenzaldehyde dans 100 ml de DMSO on ajoute 3,4 g de
K2C03 et 1,5 g de chlorure de benzyltriéthylammonium puis on ajoute en 30
minutes 10 ml d' iodure d' isopropyle en solution dans 20 ml de DMSO et on
laisse
une nuit sous agitation à TA. On verse sur 400 ml d'eau puis on extrait par
AcOEt,
lave par une solution saturée de NaCI puis sèche sur Na2S04 et évapore à sec.
L'huile obtenue (14,4 g) est utilisé telle quelle à l'étape suivante.
B) Chlorhydrate de l' acide 3-amino-3-(3-isopropyloxyphényl)propionique.
On chauffe une nuit à 80°C un mélange contenant 14 g de l'huile
obtenue à
l'étape précédente, 150 ml de méthoxyéthanol, 8,95 g d'acide malonique et 13,2
g
d'acétate d'ammonium. Après refrodissement, on évapore à sec puis on dissout
l' huile formée dans l' éthanol et 50 ml d' HCl 2N . On procède par
évaporation
fractionnée pour obtenir le composé attendu qui est essoré puis lavé par
AcOEt. On
obtient 4,56 g.
RMN {DMSO) : 1,25 : d : 6H ; 4,65 : heptuplet : 1H ; 7,20-7,55 : mt : 4H ;
9,95 : s : 1H.
En suivant le mode opératoire décrit dans la Préparation ci-dessus, on prépare
les composés décrits dans le tableau ci-dessous.
TABLEAU 3
H2N-ÇH-CH2-COOH, HC1
Prparation R2 RMN
2.8 -- (DMSO)
2,85-3,15 : mt : 2H ; 3,85
: s : :3H ;
OMe 4,60 : t : 1H ; 6,95-7,45 :
mt : 4H
2.9 -
(DMSO + TFA)
OM
\ ~ 2,50-3,00 : mt : 2H ; 3,70
e : s : :3H ; '
4,50: t: 1H;6,90:d:2H;7,40:d:
2H
2.10 -' (DMSO + TFA)
\ / 2,80-3,10 : mt : 2H ; 4,60
: t : 1H ;
Cl 7,30-7,60 : m : 4H
CA 02241788 1998-07-07
WO 97/25315 37 PCT/FR97/00026
2.11 - (DMSO + TFA)
2,90-3>20 : mt : 2H ; 4,75
: t : lH ;
CF3 7,00-8,05 : mt : 4H
2.12 (DMSO + TFA)
3,00-3,20 : mt : 2H ; 5,40-5,60
: mt
1H ;
7,50-8,30 : m : 7H
2.13 F (DMSO + TFA)
2,75-3,10 : mt : 2H ; 5,60
: t : :lH ;
~ ~
7,00-7,45 : m : 4H
2.14 (DMSO + TFA)
0,90-1,75 : m : 11H ; 2,40-2,70
; mt
2H ;
3,15-3,30 : m : 1H
1
5
2.15 - (DMSO + TFA)
2,25 : s : 3H ; 2,80-3,05 :
mt : 'ZH ;
Me 4,55 : t : 1H ;7,10-7,30 :
m : 4H
Préparation 2.16
Acide 3-(N-Boc)amino-3-biphényl-4-ylpropionique.
A) Chlorhydrate de l'acide 3-amino-3-biphényl-4-yl-propionique.
On mélange 18,2 g de 4-phénylbenzaldehyde, 10,4 g d'acide malonique et 15,4
g d'acétate d'ammonium dans 150 ml de méthoxyéthanol. Après une nuit de
chauffage à 80°C, on laisse refroidir puis le produit formé est lavé à
l'éthanol, à
l'éther, puis séché. Après un nouveau lavage à l'eau, le produit est
recristallisé dans
un mélange mëthanol-eau avec un peu d'HCI. On obtient un mélange du produit
attendu et de chlorhydrate de l'ester méthylique de l'acide 3-amino-3-biphényl-
4-yl-
propionique, utilisé tel quel à l'étape suivante.
B) Acide 3-(N-Boc)amino-3-biphényl-4-ylpropionique.
On place le produit de l'étape précédente dans 200 ml de dioxane, on ajoute 55
ml de NaOH 2N et on laisse sous agitation à TA pendant 1 heure 40 minutes. On
ajoute 13 g de Boc20 et maintient l'agitation pendant une nuit. On filtre
l'insoluble,
dilue par 200 ml d'eau, lave le mélange par Et20 puis acidifie à pH = 2,5 par
addition d'HCl 2N en présence de 100 ml d'AcOEt. On extrait par AcOEt, lave
par
CA 02241788 1998-07-07
WO 97!25315 38 PCTIFR97/00026
KHS04/K2S04, par une solution saturëe de NaCI puis on sèche sur Na2S04 et
évapore à sec pour obtenir 11,03 g du composé attendu.
RMN (DMSO) : 1;40 : s : 9H ; 2,60-2,85 : mt : 2H ; 5,00 : qd : 1H ; 7,35-
7,80 : mt : !OH ; 12,30 : se : 1H.
Préparation 2.17
Ester de 2,5-dioxopyrrolidin-1-yle de l'acide 3-(N-Boc)amino-3-biphényl-4-
ylpropionique.
3,41 g de l'acide décrit à la Préparation ci-dessus est placé dans SO ml de
dioxane et traité par 1,26 g d'hydroxysuccinimide en présence de 2,5 g de DCC.
On
laisse une nuit sous agitation à TA puis on essore et lave à l'acétone. Le
filtrat est
évaporé à sec puis repris dans l'isopropanol. Le solide est essoré, lavé à
l'éther et
séché pour donner 3,46 g du composé attendu, F = 161 °C.
RMN (DMSO) : 1,35 : s : 9H ; 2,80 : s : 4H ; 3,00-3,20 : mt : 2H ; 5,00 : qd
1H ; 7,30-7,75 : mt : IOH.
Préparation 2.18
Acide (1,2,3,4-tétrahydroisoquinolin-1-yl)acétique.
Ce composé est préparé d'après J. Org. Chem., 1987, 52, 616-622.
A) 3,4-dihydroisoquinoléine.
On dissout 9,4 ml de 1,2,3,4-tétrahydroisoquinoléine dans 200 ml de DCM et
on ajoute progressivement 14,7 g de NBS. On laisse sous agitation à TA en
refroidissant légèrement pour maintenir une température inférieure à
40°C pendant
minutes puis on ajoute 50 ml de NaOH !ON et laisse sous agitation 1 heure à
TA. La phase organique est décantée et lavée par 100 ml d'eau puis 2 fois par
100
ml d'HCI 4N. Les phases aqueuses sont lavées au DCM puis amenées à pH = 9 par
25 addition de NH40H concentrée. On extrait au DCM, sèche sur Na2S04 et
évapore
à sec. L'huile obtenue est distillée. On obtient 7,16 g du composé attendu, Eb
=
50-54°C sous 0,05 mbar.
B) Acide (1,2,3,4-tétrahydroisoquinolin-1-yl)acétique.
On triture dans un bain d'huile à 120°C un mélange contenant 6,62 g
de 3,4
30 dihydroisoquinoléine et 5,13 g d'acide malonique. Le mélange s'épaissit et
devient
entièrement solide, il est réduit en poudre à la spatule. La durée totale de
chauffage
est de 40 minutes environ. Le mélange est refroidi vers 60°C et traité
par 120 ml
MeOH et 20 ml d'eau, ce qui dissout le milieu. Un premier jet est obtenu à
froid
puis un second en évaporant le filtrat et en cristallisant le résidu dans l'
acétone.
RMN (DMSO + TFA) : 2,90-3,15 : mt : 4H ; 3,30-3,60 : mt : 2H ; 4,90 : t
1H ; 7,20-7,40 : m : 4H.
CA 02241788 1998-07-07
WO 97125315 39 PCT/FR97/00026
Préparation 2.19
Acide 3-amino-3-phénylpropionique, trifluoroacétate, isomére (R).
A) Ester de (2-isopropyl-5-méthyl)cyclohexyle de l' acide 3-(N-Boc)amino-3-
phényl
propionique, isomère (R).
Cette réaction est effectuée selon Tetrahedron Letters, 1988, 29, 6465-6466.
On
prépare une solution contenant 11,1 g d' acide 3-(N-Boc)amino-3-
phénylpropionique,
7,5 g de L(-)menthol et 2,1 g de DMAP dans 400 ml de DCM ; après agitation, on
introduit progressivement 11,2 g de DCC en solution dans 50 ml de DCM. Après
une nuit sous agitation à TA, on filtre, lave à l' acétone le DCU et évapore à
sec. Le
résidu est repris dans 150 ml d'heptane à 80-90°C ; l'insoluble est
filtré et la
solution est abandonné à TA pendant 4 heures. Le solide est essoré, lavé à
l'heptane
et séché à 40°C jusqu'à disparition de l'odeur de menthol. On obtient
4,25 g du
composé attendu.
a,D = -16,5° (c = 1 ; MeOH}
B) Acide 3-(N-Boc)amino-3-phénylpropionique, isomère (R).
On porte à reflux pendant 4 heures un mélange contenant 4,22 g du composé de
l'étape précédente, 15,7 ml de NaOH 1N dans 100 ml de méthanol. Le milieu
réactionnel est refroidi, traité par 15,7 ml d'HCl 1N puis évaporé à sec et
repris
dans l'heptane. Après abandon quelques heures à TA, le produit cristallise, il
est
essoré, lavé à l'heptane puis séché à 40°C jusqu'à disparition de
l'odeur de menthol.
On obtient 2,65 g du composé attendu,
aD = +42,1 ° (c = l, MeOH)
C) Acide 3-amino-3-phénylpropionique, trifluoroacétate, isomère (R).
On dissout 2,3 g du composé obtenu à l'étape précédente dans i5 ml de DCM
et on ajoute 15 ml de TFA. Après 35 minutes sous agitation à TA, on évapore à
sec.
Le produit est repris dans i'isopropanol, on évapore puis cristallise dans
Et20. On
essore, lave par Et20 et sèche pour obtenir 2,16 g du composé attendu.
Préparation 2.20
Acide 3-amino-4-phénylbutyrique, chlorhydrate, isomère (S).
A) Ester méthylique de l'acide 2-{N-Boc)amino-3-phénylpropionique, isomère
(S}.
On mélange 10, 8 g de chlorhydrate d' ester méthylique de l' acide (L) 2-amino-
3-phénylpropionique dans 150 ml de DCM et 7 ml de NEt3 et on ajoute
Progressivement 13 g de (Boc)20 dans 50 ml de DCM. Aprés 5 heures sous
agitation à TA, on lave le milieu réactionnel par KHS04IK2S04 puis une
solution
saturée de NaCI. On sèche pour éliminer (Boc}20 en excès, puis on dissout dans
CA 02241788 1998-07-07
WO 97/25315 40 PCT/FR97/00026
DCM et on ajoute 1,5 ml de N,N-diméthylpropanediamine. Après 4 heures sous
agitation, on lave par KHS04/K2S04 puis par une solution saturée de NaC'.1. On
sèche sur Na2S04 puis évapore à sec pour obtenir 11,9 g du composé attendu.
B} 2-(N-Boc)amino-3-phénylpropanol isomère (S).
Cette étape et les 2 suivantes sont effectuées selon Tetrahedron, 1994, Sa
(31),
9457. On dissout 10 g du composé de l'étape précédente dans 120 ml de THF' et
on
refroidit au bain de glace ; on ajoute 3;17 g de chlorure de lithium puis 2,8
g de
borohydrure de sodium et, progressivement 170 ml d'EtOH. Après une nuit sous
agitation à TA, on ajoute lentement 70 ml de KHS04 1 M et on concentre presque
â
sec. On dilue dans le chloroforme et KHS04 1M puis on extrait par le
chloroforme.
On lave par une solution saturée de NaCI, sèche sur Na2S04 et évapore à sec
pour
obtenir 8,65 g du composé attendu.
C) Ester 2-amino-3-phénylpropylique de l'acide méthanesulfonique isomère (S).
On dissout 7, 5 g du composé de l' étape précédente dans 40 ml de pyridine et
on
refroidit dans un bain de glace ; on ajoute en 15 ml 3,3 ml de chlorure de
mésyle
puis on laisse sous agitation 2 heures à TA. On ajoute en 5 minutes 15 ml
d'eau puis
on dilue le milieu à l'éther et lave par KHS04 1M (2 fois), de l'eau, puis une
solution saturée de NaCI ; on sèche sur Na2S04 puis on évapore à sec pour
obtenir
9,2 g du composé attendu.
a D = -24, 3 ° (c = 1, MeOH)
D) 3-(N-Boc)amino-4-phénylbutyronitrile, isomère (S).
On prépare une solution contenant 9,05 g du composé de l'étape précédente
avec 7,3 g d'éther couronne 18 crown b et 120 ml de DMSO et on ajoute sous
agitation au bain de glace 9 g de cyanure de potassium. Après 5 heures de
chauffage
à 50°C, on refroidit puis on ajoute 600 ml d'Et20. On lave à l'eau (3
fois) puis par
une solution saturée de NaCI ; on sèche sur Na2S04 puis on évapore à sec pour
obtenir b,58 g du composé attendu.
E) Acide 3-amino-4-phénylbutyrique, chlorhydrate, isomère (S).
Cette étape est effectuée selon Tetrahedron Letters, 1990, 31, 5153. On met en
suspension 4,1 g du composé de l'étape précédente dans 50 ml d' HCl 6N et on
chauffe à reflux pendant 5 heures. On concentre pour obtenir un premier jet du
composé attendu pur. Par évaporation à sec on obtient un deuxième jet du
composé
attendu 1,4 g souillé de NH4C1. Un échantillon est traité par (Boc)20 et on
mesure
son pouvoir rotatoire,
aD = -17° (c = 1, MeOH)
CA 02241788 1998-07-07
WO 97/25315 41 PCTIFR97/00026
littérature a D = -16 ° (c = 1, MeOH)
Préparation 2.21
Acide 3-amino-3-phénylpropionique, trifluoroacétate, isomère (S).
La préparation est identique à celle décrite en 2.19 mais en utilisant le D(+)
menthol.
Préparation 3.1
Acide 3-phényl-3-(2,4,6-trichlorobenzènesulfonamido)propionique.
On dissout 1,15 g d' acide 3-amino-3-phénylpropionique dans 25 ml de dioxane
et 7 ml de soude 1 N et on ajoute progressivement 1,95 g de chlorure de
2,4,6-trichlorobenzènesulfonyle dans 5 ml de dioxane en maintenant le pH à
1(),5-11
par addition de soude 1 N. Après 2 heures sous agitation à TA, on dilue par
100 ml
d'eau, extrait 2 fois par AcOEt et acidifie à pH = 2 par addition d'HCl 6N. Le
solide formé est essoré, lavé à l'eau et séché à 40°C. On obtient 2,09
g du produit
attendu, F = 218-219 ° C .
RMN (DMSO + TFA) : 2,60-2,95 : mt : 2H ; 4,75 : q : 1H ; 7,10-7,30 : m
5H;7,60:s:2H;8,85:d:1H.
Préparation 3.2
Acide 3-(napht-2-yl-sulfonamido)-3-phénylpropionique.
On dissout 4,13 g d' acide 3-amino-3-phénylpropionique dans 100 ml de dioxane
et 25 ml de soude 1 N et on ajoute par petites portions 5,6 g de chlorure de
naphtalène-2-sulfonyle en maintenant à pH = 10,5-11 par addition de soude N.
Après 2 heures sous agitation à TA, on dilue par 400 ml d'eau et ajoute HCl 2N
pour obtenir pH = 2. On extrait par AcOEt, Iave par un tampon KHS04/K2S04,
sèche sur Na2S04 et évapore à sec. Le résidu est trituré dans l'heptane,
décanté puis
séché sous vide. On obtient 7,33 g du produit attendu, F = 126-129°C.
RMN (DMSO + TFA) : 2,55-2,70 : mt : 2H ; 4,70 : t : IH ; 6,85-8,15 : m
12H.
Préparation 3.3
Acide 2-hydroxy-3-(napht-2-yl-sulfonamido)-3-phénylpropionique.
Ce composé est préparé selon Bull. Soc. Chim., France, 1940, 593-603.
En opérant selon les modes opératoires décrits dans les Préparations 3.1 et
3.2,
à partir des composés obtenus dans les Préparations 2 et du chlorure de
naphtalène
2-sulfonyle, on obtient les acides décrits dans le TABLEAU 4 ci-dessous.
CA 02241788 2004-10-28
42
TABLEAU 4
SOI--NH- i H-CHz-COOH
w
PrparationsR2 RMN F C
3.4 (DMSO) 145-150
\ ~ 2,80 :d:2H;4,90:q:1H;
\ ~ 7,15-8,20 : m : I4H
;
8,60 : d : 1H
3.5 (DMSO + TFA) 223-228
C! 2,00-2,20 : mt : 2H
;
4,45 : t : 1H ;
7, LO-8,25 : m : 11H
3.6 (DMSO) 253-255
OBn 2,50-2,75 : mt : 2H
4,60-4,80 : mt : 3H
6,65-8,15 : m : 16H
8,40 : se : 1H
3.7 OBn RMN (DMSO)
2,40-2,65 : mt : 2H
4,65 : s : 2H ; 4,75
: qd : 1H ;
6,40-8,05 : m : 16H
8,40 : se : 1H
Les acides décrits ci-dessus sont transformés par action du N-
hydroxysuccinimide en présence de DCC dans le DMF. Les composés obtenus sont
décrits dans le TABLEAU 5 ci-après.
CA 02241788 2004-10-28
43
TABLEAU 5
~ SOz NH-i H-CH.~-COONSu
w w
Prparations R2 RMN (DMSO) FC
3.8 2,75 : s : 4H ; 3,20 : mt 165
: 2H ;
4,90: qd : 1H ; 7,20-8,10
: m : I4H ;
8,70 : d : 1H
3.9 -' 192
C1
3.10 187
OBn
3.11 OBn 2,80 : s : 4H ; 3,15 : mt 110
: 2H ;
4,65 : s : 2H ; 4,75 : qd
: 1H ;
6,45-7,95 : mt : 4H ;
7,25-8,20 : mt : 12H ; 8,60
: d : 1H
Préparation 3.I2
Acide 3-(2-hydroxyphényl)-3-(napht-2-yl-sulfonamido)propionique.
On dissout 1,1 g d' acide 3-amino-3-(2-hydroxyphényl)propionique,
chlorhydrate, préparé selon J. Agric. Food Chem., 1977, 25, 965, dans 25 ml de
dioxane et l'on ajoute 10 ml de NaOH N ; on ajoute progressivement 1,12 g de
chlorure de naphtalène-2-sulfonyle en maintenant à pH = 11,5-12 par addition
de
NaOH , 1 N. Après 2 heures sous agitation à TA, on dilue le milieu réactionnel
dans l'eau; lave par AcOEt (2 fois) puis on acidifie à pH = 1,5-2 par addition
d'HCl 6N. Le précipité blanc formé est essoré, lavé à l'eau et séché pour
obtenir
0,82 g du produit attendu, F = 190-200°C.
RMN (DMSO)
2,50-3,20 : mt : 2H ; 4,80 : mt : 1H ; 6,95-8,30 : m : 11H ; 8,75 : s : 1H.
Préparation 3.13
Acide 3-phényl-3-(quinol-2-ylsulfonamido)propionique.
CA 02241788 1998-07-07
WO 97/25315 q.4 PCT/FR97/00026
A) Chlorure de quinoléine-2-sulfonyle.
On met en suspension 3,2 g de 2-mercaptoquinoléine dans 40 ml d'eau
contenant 0,156 g de trichlorure de fer. On laisse refroidir dans un bain de
glace
puis on fait barboter pendant 1 heure du chlore à 4°C. On évapore puis
on reprend
dans un minimum d'eau, essore et sèche pour obtenir 2,30 g du composé attendu
sous forme sèche.
B) Acide 3-phényl-3-(quinol-2-ylsulfonamido)propionique.
On dissout 1,65 g du composé préparé à l'étape précédente dans 40 ml de
dioxane, on ajoute 20 ml de NaOH 1 N et, peu à peu, 2,27 g d' acide 3-alnino-3
phényl propionique, en maintenant à pH = 10. On laisse 18 heures sous
agitation à
TA puis on évapore à sec. Le résidu est repris par un mélange DCM-H20, on
décante puis on extrait à l'eau. On acidifie à pH = 1 par addition d'HCI puis
on
essore et sèche sur MgS04 pour obtenir 1,9 g du composé attendu.
RMN (DMSO + TFA) : 2,4-2,9 : m : 2H ; 4,9 : mt : 1H ; 6,9-7,1 : mt : 3H ;
7,2 : d : 2H ; 7,7-8,1 : m : SH ; 8,4 : d : 1H ; 8,8 : d : 1H.
Préparation 3.14
Acide 3-phényl-3-(quinol-8-ylsulfonamido)propionique.
Ce composé est préparé selon le mode opératoire décrit ci-dessus à partir du
chlorure de quinoléine-8-sulfonyle.
Préparation 3.15
Acide 3-(3-isopropyloxyphényl)-3-(napht-2-ylsulfonamido)propionique.
1,3 g du composé de la Préparation 2.7 sont mis en suspension dans 20 ml de
dioxane et traité par NaOH 2N puis NaOH 1N pour porter à pH = 12. On ajoute
par petites fractions 1,13 g du chlorure de napht-2-ylsulfonyle tout en
maintenant le
pH à pH = 10,5-11,5 par addition de NaOH 1N. On laisse sous agitation 2 heures
à
TA puis on dilue à l'eau, lave par AcOEt puis amène à pH = 2 par addition
d'HCl
2N. On extrait par AcOEt puis lave par un tampon KHS04/K2S04 et par une
solution saturée de NaCI. On sèche sur Na2S04 et concentre pour obtenir 900 mg
du composé attendu, F = 126°C.
RMN (DMSO) : 0,95 : d : 6H ; 2,40-2,55 : mt : 2H ; 4,10 : qt : 1H ;
4,60 : qd : 1H ; 6,20 - 8,05 : mt : 11H ; 8,30 : d : 1H ; 12,20 : se : 1H.
En suivant le mode opératoire décrit à la Préparation ci-dessus, et en
utilisant
comme produit de départ les composés des Préparations 2.8 à 2.15 on prépare
les
composés décrits dans le Tableau 6 ci-après.
CA 02241788 1998-07-07
WO 97/25315 45 PCTIFR97/00026
TABLEAU 6
SO,NH-ÇH-CHI-COOH
Prparation R2 RMN Fw C
3.16 "" (DMSO) 154
2,35-2,55 : mt : 2H ; 3,35
: s : 3H ;
OMe 4,60 : qd : 1H ; 6,20-6,85
: mt : 4H ;
7,40-8,0 : mt : 7H ; 8,30 :
d : 1H ;
12,20 : se : IH
3.17 - (DMSO) 2,35-2,60 : mt : 2H 139
;
OMe 3,35 : s : 3H ; 4,55 : qd :
IH ;
6,35 : d : 2H ; 6,90 : d :
2H ;
7,40-7,95 : mt : 7H ;
8,25 : d : 1H ; 12,20 : se
: IH
3.18 - (DMSO) 2>40-2,70 : mt : 2H 133
;
4,60 : mt : 1H ; 6,70-7,10
: m : 4H ;
CI 7,40-8,20 : m : 7H ; ; 8,45
: de : 1H ;
12,30 : se : 1H
3.19 - (DMSO) 2,40-2,70 : mt : 2H 164
;
4,70 : qd : 1H ;
CF3 7,00-8,00 : m : 11H ; 8,45
: d : 1H ;
I2, 20 : se : 1 H
3.20 (DMSO) 2,80 : de : 2H ; 5,60 189
i i : mt : IH
7,10-8,10:m:14H;8,65:d:1H;
12,30 : se : 1H
3.21 - F (DMSO) 2,45-2,65 : mt : 2H 167
; 4,70 : qd a
1H
6,60-7,05 : m : 4H ; 7,50-8,15
: m : 7H
8,50 : d : 1H : 12,30 : se
: 1H
CA 02241788 1998-07-07
WO 97125315 46 PCT/FR97/00026
3.22 (DMSO + TFA) 0, 80-1,60 : m I 98
: 11 H ;
1,85-2,30 : mt : 2H ; 3,40
: qd : IH ;
7,50-8,30 : m : 7H
3.23 - (DMSO) 1,85 : s : 3H ; 102
\ / 2,45-2,65 : mt : 2H ; 4,65
: qd : 1H ;
Me 6,60-6,90 : m : 4H ;
7,50-8,i5 : m : 7H ; 8,40 :
de : 1H ;
12,20 : se : 1H
Les acides décrits ci-dessus (Préparations 3.I5 à 3.23) sont transformés par
action du N-hydroxysuccinimide en présence de DCC dans le dioxane. Les
composés obtenus sont décrits dans le Tableau 7 ci-après.
I5
TABLEAU 7
SOZNH-ÇH-CH2-COONSu
Préparations R2 RMN F°C
3.24 - - 97
\ /
OiPr
3.25 -" - 142
OMe
3.26 - - 135
\ / OMe
3.27 '- - 163
CI
CA 02241788 1998-07-07
WO 97125315 47 PCT/FR97/00026
3.28 -- - 14 2
\ /
CF3
3.29 (DMSO + TFA)
192
i i ~ 2,70:s:4H;3,25:d:2H;
5,60 : t : 1H ; 7,05-7,95 : m : 14H
3.30 F (DMSO) 174
2,70 : s : 4H ; 2,95-3,10 : mt : 2H ;
\ / 4,75 : qd : 1H ; 6,50-6,90 : m : 4H ;
7,45-8,10 : m : 7H ; 8,60 : d : 1H
3.31 {DMSO + TFA) 0,60-1,60 : m : 11H 135
IS 2,40-2,80 : mt : 2H ; 2,70 : s : 4H ;
3,35 : mt : 1H ; 7,50-8,30 : m : 7H
3.32 - 142
\ /
Me
Préparation 3.33
Acide 3-(3,4-dichlorobenzènesulfonamido)-3-phényl propionique.
A) Chlorure de 3,4-dichlorophénylbenzènesulfonyle.
On met en suspension dans 60 ml d'eau, 5,4 g de (3,4-dichloro)thiophénol ; on
ajoute 0,234 g de FeCl3 et on laisse barboter du chlore pendant une heure à
une
température inférieure à 10°C. On évapore, essore, lave à l'eau puis
séche par
entraînement azéotropique pour obtenir 6,7 g du composé attendu.
B) Acide 3-(3,4-dichlorobenzènesulfonamido)-3-phényl propionique.
On procède ensuite selon le mode opératoire de la Préparation 3.1 pour obtenir
le composé attendu.
RMN (DMSO) : 2,5-2,9 : m : 2H ; 4,6-4,8 : q : 1H ; 7,1 : s : SH ; 7,5-7,7
mt : 3H ; 8,7 : d : 1H ; 12,4 : s : 1H.
Préparation 3.34
Acide 3-(5,6,7,8-tétrahydronapht-2-ylsulfonamido)-3-pllényl propionique.
A) 5,6,7,8-tétrahydronaphtalène-2-sulfonate de sodium.
CA 02241788 1998-07-07
WO 97/25315 4$ PCTIFR97/00026
On dissout 3,9 g de 1,2,3,4-tétrahydronaphtalène dans 10 ml de CC14 anhydre à
0°C ; on ajoute 4,3 g de dioxane dans 10 ml de CCl4 puis 6,1 ml
d'anhydride
sulfurique. On laisse sous agitation 18 heures à TA puis 3 heures à
80°C. On ajoute
un mélange eau/glace puis on extrait à l'éther. La phase aqueuse est ajustée à
pH =
6,5 par addition de NaOH SN. On essore, sèche sur Na2S04, puis par
entraînement
azéotropique. On obtient 10,6 g.
B) Chlorure de 5,6,7,8-tétrahydronaphtalène-2-sulfonyle.
On mélange 2 g du sulfonate obtenu à l' étape précédente et S g de
pentaclllorure
de phosphore et on chauffe à reflux pendant 6 heures. On évapore, jette sur un
mélange eaulglace puis on extrait au DCM, sèche sur Na2S04 et évapore pour
obtenir 1, 5 g du composé attendu.
C) Acide 3-(5,6,7,8-tétrahydronapht-2-ylsulfonamido)-3-phényl propionique.
On procéde ensuite selon le mode opératoire de la Préparation 3.1 pour obtenir
le produit attendu.
RMN (DMSO) : 1,6-1,8 : m : 4H ; 2,5-2,8 : m : 6H ; 4,5-4,7 : dd : 1H ;
7,7,2 : m : 7H ; 7,3 : d : 1H ; 8,1 : d : 1H ; 12,3 s : 1H.
Préparation 3.35
Acide 1-(napht-2-ylsulfonamido)-indane-2-carboxylique.
A) Ester méthylique de l'acide (1-oxo)indane-2-carboxylique.
La réaction est effectuée selon J. Med. Chem., 1970, 650. On mélange une
suspension de 6,75 g d'hydrure de sodium à 80 % dans l'huile et 45 g de
carbonate
de méthyle dans 120 ml de benzène. A 60°C, on ajoute en 1 heure et
demie 11,9 g
d'indan-1-one dissoute dans 100 ml de benzène puis on porte à reflux. Après I
heure, on distille de benzène. On ajoute du xylène et on porte à nouveau à
reflux.
Après 1 heure, on refroidit, ajoute 30 ml d'AcOH et verse sur 200 ml d'un
mélange
eau/glace contenant 30 ml d'HCl 1N. L'insoluble est éliminé par filtration
puis le
filtrat est extrait par Et20. On lave à l'eau, par une solution saturée de
NaHC03, à
l'eau, par une solution saturée de NaCl, puis on sèche sur Na2S04. On
chromatographie sur silice en éluant par AcOEt/hexane (1/3 ; v/v) pour obtenir
3,05
g du composé attendu.
RMN {DMSO) : 3,25-3,50 : mt : 2H ; 3,70 : s.d. : 3H ; 4,85-4,90 : mt : 1H ;
7,40-7,80 : m : 4H. Signaux multiples car le produit est en partie sous forme
énolique.
Remarque : en utilisant le THF comme solvant au lieu du benzène et du xylène,
on obtient un meilleur rendement : 67 % au lieu de 30 %.
CA 02241788 1998-07-07
WO 97/25315 49 PCT/FR97/00026
B) Estèr méthylique de l'acide 1-hydroxyiminoindane-2-carboxylique.
La réaction est effectuée selon J. Heterocycl. Chem., 1974, 11, 982. 3,Ci4 g
du
composé de l'étape précédente dans 9 ml de MeOH sont ajoutés en 10 minutes à
un
mélange de 2, 21 g d' acétate de sodium et 3,1 g d' hydroxylamine chlorhydrate
dans
3 ml d' eau. Ensuite on chauffe à reflux pendant 1 heure 30 minutes puis on
refroidit
et extrait par AcOEt. On lave à l'eau, par une solution de NaHC03 3/4 saturée,
à
l' eau, par une solution saturée de NaCI. Après évaporation d' AcOEt on
obtient 3 ,28
g du composé attendu sous forme solide.
C) Ester méthylique de l'acide 1-aminoindane-2-carboxylique, chlorhydrate.
On dissout 3,27 g du composé de l'étape précédente dans 80 ml d'EtOH, on
ajoute 1,2 g de Pd/C à 10 % puis 20 ml d'éthanol chlorhydrique 1M et on laisse
sous agitation sous pression d'hydrogène (5 bars), à TA, pendant une nuit. Le
catalyseur est filtré, le filtrat est concentré sous vide puis repris par
AcOEt et de
l'eau. On décante l'eau puis on amène à pH = 8,5 par addition de NaOH 2N. On
extrait par AcOEt, lave à l'eau, par une solution saturée de NaCI, sèche sur
Na2S04
et évapore. Le résidu est repris dans 20 ml de MeOH et 6 ml de HCI 2,5N. On
concentre sous vide puis le résidu est trituré dans Et20. On obtient 2,71 g du
composé attendu sous forme de poudre.
D) Ester méthylique de l'acide 1-(napht-2-ylsulfonamido)-indane-2-
carboxylique.
On place 0,91 g du composé précédent dans 10 ml de chloroforme puis on
ajoute par petites portions 0,95 g de chlorure de naphtalène-2-sulfonyle et
0,68 ml
de DIPEA par petites portions pour maintenir à pH = 7-8. Après 3 heures, le
milieu
réactionnel est extrait par AcOEt puis on lave par NaOH O,OSN, HCI 0,25N, de
l'eau et une solution saturée de NaCI. On obtient 0,99 g du composé attendu
sous
forme d' un solide blanc rosé.
RMN (DMSO + TFA) : 2,80-3,30 : mt : 2H ; 3,40 : s : 3H ; 3,50 : q : 1H ;
5,10 : d : 1H ; 6,40-8,50 : mt : 11H.
E) Acide 1-(napht-2-ylsulfonamido)-indane-2-carboxylique.
On place 0,98 g du composé de l'étape précédente dans 10 ml de MeOH, on
ajoute 1 ml de KOH 8,36 N et on chauffe 3 heures à 50°C puis on rajoute
C),25 ml
de KOH 8,36 N. Après 2 heures, on dilue le milieu par addition d'eau et
d'AcOEt
et on amène à pH = 2,5 par addition d'HCl 1N. On décante la phase organique,
lave par H20, une solution saturée de NaCI puis on sèche sur Na2S04 et
évapore.
On obtient 0,80 g du composé attendu sous forme de mousse.
Préparation 3.36
Acide [2-(naphtalène-2-sulfonyl)-1,2,3,4-tétrahydroisoquinolin-1-yl]acétique.
CA 02241788 1998-07-07
WO 97/25315 $0 PCT/FR97/00026
1,9I g d'acide (1,2,3,4-tétrahydroisoquinolin-1-yl)acétique (Préparation 2.18)
est mis en suspension dans 2$ ml de dioxane, on ajoute 10 ml de NaOH, 1N,
puis,
par petites fractions, 2,3 g de chlorure de naphtalène-2-sulfonyle et on
maintient à
pH = 10,5-12 par addition de NaOH 1N. On maintient sous agitation à pH
constant
pendant 2 heures à TA. Le milieu réactionnel est dilué par 100 ml d'eau puis
lavé 2
fois par AcOEt et amené à pH = 1,2 par addition d'HCI 6N en présence d'AcOEt.
On extrait par AcOEt, lave par une solution saturée de NaCI, sèche sur Na2S04
et
évapore à sec. Le produit attendu cristallise dans l'heptane, il est essoré,
lavé à
l' heptane puis sèché pour donner 3,14 g, F = I 32 ° C .
RMN (DMSO + TFA} : 2,40-2,80 : m : 4H ; 3,40-3,90 : m : 2H ; 5,45 : t
1H ;
6,90-7,20 : m : 4H ; 7,50-8,40 : m : 7H.
Préparation 3.37
On prépare l' ester de 2, $-dioxopyrrolidin-1-yle de l' acide ci-dessus par
action
1$ de NSuOH en présence de DCC dans le dioxane.
RMN (DMSO + TFA) : 2,50-2,70 : m : 2H ; 2,75 : s : 4H ; 3,10-3,3~ : mt
2H ;
3,50-3,80 : mt : 2H ; $,50 : t : 1H ; 6,80-8,40 : m : 11H.
Préparation 3.38
Acide 3-(napht-2-ylsulfonamido)-3-phénylpropionique, isomère (R).
2 g du composé de l'étape 2.I9 sont placés dans 30 ml de dioxane en pxésence
de 7,2 ml de NaOH 2N ; on ajoute par petites portions 1,6 g de chlorure de
napht-2-
ylsulfonyle tout en maintenant à pH = 10,5-11,5 par addition de NaOH 1N. Après
2 heures sous agitation à TA on dilue par addition de 100 ml d'eau puis on
lave par
AcOEt (plusieurs fois). La phase aqueuse est diluée par AcOEt puis traitée par
HC1
2N jusqu'à pH = 2,2. On extrait par AcOEt, lave par une solution saturée de
NaCI,
sèche sur Na2S04 et évapore à sec. Le composé attendu cristallise dans
l'heptane, il
est essoré, lavé à l'heptane puis séché. On obtient 2,2 g, F = 123-
126°C.
aD = +67,9 (c = 1 ; MeOH)
Préparation 3.39
Ester de 2,$-dioxopyrrolidin-1-yle de l'acide 3-(napht-2-ylsulfonamido)-3-
phénylpropionique, isomère (R).
On prépare une solution de 2 g du composé précédent et 657 mg de N
hydroxysuccinimide dans 35 ml de dioxane et on ajoute progressivement 1,24 g
de
DCCI dans 10 ml de dioxane. Après 5 heures sous agitation à TA, le DCU est
CA 02241788 1998-07-07
WO 97/25315 51 PCT/FR97/0002b
essoré, lavé à l'acétone puis le filtrat est évaporé à sec. Le résidu est
repris dans
l'isopropanol. Le produit qui cristallise est essoré, lavé par Et20 puis
séché. On
obtient 2,18 g du composé attendu.
Préparations 3.40 et 3.41
On procède comme dans les 2 Préparations décrites ci-dessus à partir du
composé de la Préparation 2.21 pour obtenir l'acide 3-(napht-2-ylsulfonamido)-
3-
phénylpropionique, isomère (S) et son ester de 2,5-dioxopyrrolidin-I-yle.
Préparation 3.42
Acide 3-(4-chlorophényl)-3-(napht-2-ylsulfonamido)propionique, isomère (R).
A) Acide 3-{4-chlorophényl)-3-(phénylacétamido)propionique.
On dissout 9,6 g d'acide 3-amino-3-(4-chlorophényl)propionique dans '?00 ml
de dioxane et environ 50 ml de NaOH 1N pour atteindre pH = 10,5-11 >5. Le
milieu est refroidi à +5°C puis on ajoute progressivement 6,34 ml de
chlorure
d'acide phénylacétique en maintenant à pH = 10,5-11,5 et à une température
entre
+ 5 ° C et + 10° C. On maintient l' agitation pendant I heure et
demie puis on
concentre le milieu de moitié avant de diluer par 500 ml d' eau. On lave 2
fois par
AcOEt puis on acidifie à pH = 1-2 par addition d'HCl 6N. Le solide est essoré,
lavé à l' eau et séché pour donner 14 g du composé attendu .
B} Acide 3-Amino-3-(4-chlorophényl)propionique, isomère {R).
Cette étape est effectuée selon Syn. Letters, 1993, 339. On met en suspension
16,8 g du composé de l'étape précédente dans 300 ml d'eau et on ajoute NH4OH
IN jusqu'à obtention de pH = 7,5. On ajoute 0,6 ml de pénicillamidase~ Sigma
et
laisse sous agitation 72 heures à 40°C. Le composé attendu précipite.
Il est essoré,
lavé à l'eau, à l'acétone, puis séché à 40°C. On obtient 2,85 g.
~,D - _5° (c - 1, HCl 1N)
C) Acide 3-(4-chlorophényl)-3-(napht-2-ylsulfonamido)propionique, isomère (R).
I g du composé obtenu à l'étape précédente est dissous dans 15 ml de dioxane
et 5 ml de NaOH 1N pour atteindre pH = 10,5-I1,5. On ajoute progressivement
1,15 g de chlorure de (napht-2-yl)sulfonyle tout en maintenant le pH constant.
Après
2 heures sous agitation à TA, on dilue le milieu par addition d'un égal volume
d'eau
puis on lave 2 fois par AcOEt. Le milieu est acidifié à pH = 1,3 par addition
d'HC1
6N. On extrait par AcOEt, lave par une solution saturée de NaCI (plusieurs
fois).
On sèche sur Na2S04, évapore à sec puis le produit attendu cristallise dans
l'heptane. On obtient 1,31 g.
a D = + 92 ° (c = 1, MeOH)
CA 02241788 1998-07-07
WO 97/25315 52 PCT/FR97/00026
Préparation 3.43
On prépare l'ester de 2,5-dioxopyrrolidin-1-yle de l'acide ci-dessus selon la
technique habituelle.
Préparation 3.44
Acide 4-phényl-3-(napht-2-ylsulfonamido)butanoïque, isomère (S).
On dissout 1,08 g du composé de la Préparation 2.20 dans 30 ml de dioxane et
ml d'eau. On ajoute NaOH lON pour atteindre pH = 14 puis on ajoute par
petites fractions 1,13 g de chlorure de napht-2-ylsulfonyle en maintenant le
pH =
10, 5-11, 5 . On maintient l' agitation pendant 2 heures puis on dilue à l'
eau et lave par
10 AcOEt. On dilue à nouveau par AcOEt et acidifie à pH = 2 par addition d'
HCl 2N
et extrait par AcOEt ; on lave par une solution saturée de NaCl (plusieurs
fois} puis
on sèche et évapore à sec. Par addition d'heptane le produit attendu
cristallise. On
obtient 1,25 g.
a25 - -26,8 (c = 1 ; MeOH)
D
Préparation 3.45
On prépare l'ester de 2,5-dioxopyrrolidin-1-yle de l'acide ci-dessus en
utilisant
les techniques habituelles.
Préparation 3.46
Acide (2S, 4R) (4-(benzyloxy)-1-(napht-2-ylsulfonyl)pyrrolidin-2-yl}acétique.
A) Acide 4-{hydroxy)-1-(napht-2-ylsulfonyl)pyrrolidin-2-carboxylique.
On dissout 2,62 g de (2S, 4R) 4-hydroxyproline dans 15 ml d'eau contenant 5,9
g de Na2C03 et on ajoute 5,2i g de chlorure de napht-2-ylsulfonyle. Après 18
heures d' agitation énergique à TA, on essore puis on acidifie le filtrat à pH
=: 1. On
essore à nouveau, lave à l'eau et sèche pour obtenir 4,1 g du composé attendu.
B) Acide 4-{benzyloxy)-1-{napht-2-ylsulfonyl)pyrrolidin-2-yl acétique.
On dissout 1 g du composé de l'étape précédente dans 20 ml de DMF à
0°C
sous azote ; on ajoute 0,204 g d'hydrure de sodium à 80 % dans l'huile puis on
laisse sous agitation 1 heure à 0°C et 30 minutes à TA. On refroidit à
0°C puis on
ajoute 10 mg d'éther couronne 16 Crown 6 et 0,921 ml de bromure de benzyle. On
laisse sous agitation 1 heure à 0°C puis 18 heures à 50°C et on
ajoute 10 ml de
NaOH N. Après 18 heures sous agitation à TA, on dilue par de l'eau puis on
extrait
à l' éther et acidifie à pH = 2 par addition d' HCl 1 N . On extrait par
AcOEt, sèche
sur Na2S04 et évapore pour obtenir 0,8 g du composé attendu.
C} (2S, 4R) 4-Benzyloxy-2-hydroxyméthyl-1-(napht-2-ylsulfonyl)pyrrolidine.
CA 02241788 1998-07-07
WO 97125315 53 PCT/FR97/00026
Om dissout 2,8 g du composé de l'étape précédente dans 20 ml de THF: On
ajoute 1,12 ml de NEt3 puis 0,968 ml de chloroformiate d'éthyle en 30 minutes
à
TA, enfin on ajoute 0,762 g de borohydrure de sodium dans 5 ml d'eau. Après 20
heures sous agitation à TA, on évapore puis on reprend par 20 ml d'eau et on
acidifie à pH = 3 par addition de HCl 1N. On essore, lave à l'eau, sèche puis
on
précipité dans le mélange AcOEt/hexane pour obtenir 1,9 g du composé attendu.
D) (2S, 4R) 4-Benzyloxy-2-mésyloxyméthyl-1-(napht-2-ylsulfonyl)pyrrolidine.
On dissout 3 g du composé de l'étape précédente dans 100 ml de DCM à
0°C,
on ajoute 1,28 ml de NEt3 puis 0,72 ml de chlorure de mésyle. Après 30 minutes
sous agitation à 0°C, on rajoute 1,28 ml de NEt3 puis 0,72 ml de
chlorure de
mésyle puis on laisse sous agitation 30 minutes à TA. On lave à froid par
KHS04/K2S04 puis on filtre l'insoluble, sèche le filtrat et évapore. Le résidu
repris
par Et20 est utilisé tel quel à l'étape suivante.
E) (2S, 4R) 4-Benzyloxy-2-cyanométhyl-2-{napht-2-ylsulfonyl)pyrrolidine.
On dissout le produit de l'étape précédente dans 50 ml de DMSO, on ajoute 2 g
d'éther couronne 10 Crown 6 et on refroidit au bain de glace. On ajoute
ensuite 5 g
de cyanure de potassium et on chauffe pendant 4 heures à 50°C. Le
milieu est dilué
par 250 ml d'AcOEt puis on lave à l'eau, sèche et évapore. On reprend à
l'éther. Le
composé attendu cristallise et l'on obtient 1,7 g.
F) Ester éthylique de l'acide (2S, 4R) (4-(benzyloxy)-1-(napht-2-ylsulfonyl}
pyrrolidin-2-yl)acétique.
On dissout 1,6 g du composé de l'étape précédente dans 50 ml d'éthanol saturé
en HCl à 0°C. On laisse sous agitation 48 heures à +4°C puis on
évapore. Le
résidu est repris par EtOH et évaporé (2 fois) puis repris par Et20 et évaporé
(2
fois). On ajoute 20 ml d'eau bouillante puis on ajoute quelques ml de mélange
dioxane/EtOH pour homogénéiser. La solution est chauffé 15 minutes à 100"C. On
évapore, reprend par du dioxane puis on neutralise à pH = 6 par addition de
NaOH
1N. La solution est utilisé tel quel à l'étape suivante.
G) Acide (2S, 4R) (4-(benzyloxy)-1-(napht-2-ylsulfonyl)pyrrolidin-2-
yl)acétique.
A la solution de l'étape précédente on ajoute 2,4 ml de NaOH SN et on chauffe
à 50°C pendant 30 minutes. On évapore le solvant puis on acidifie à pH
= 3 par
addition d'HC1 concentré. On essore, lave à l'eau, sèche pour obtenir 1,44 g
du
composé attendu.
Préparation 4.1
A) N-(2-{4-{3,4-dihydroimidazol-2-yl)phényl)-1-((pyrrolidin-1-
yl)carbonyl)ét:hyl}-
3-(3,4-dichlorophényl)-3-(N-Boc)aminopropionamide.
CA 02241788 1998-07-07
WO 97125315 54 PCTIFR97/00026
On dissout 1,09 g de 1-[2-amino-3-(4-(3,4-dihydroimidazol-2-yl)phényl
propionyl]pyrrolidine, dichlorhydrate dans 15 ml de DMF, on ajoute 350 p1 de
NEt3 et on agite quelques minutes avant d'ajouter 835 mg d'acide 3-((N
Boc)amino}-3-(3,4-dichlorophényl) propionique (obtenu à la Préparation 2.61 et
515
S mg de DCC.
Après 3 heures sous agitation, on évapore à sec puis on reprend le résidu par
du
méthanol. On essore le DCU, concentre de moitié et dilue à l'acétone. On
filtre puis
évapore à sec. Le résidu est chromatographié en éluant avec un mélange
chloroforme/méthanol (de 100/2,5 à 100/20 ; v/v).
B) N-(2-(4-(3,4-dihydroimidazol-2-yl)phényl)-1-((pyrrolidin-1-
yl)carbonyl)éthyl)-
3-(3,4-dichlorophényl)-3-aminopropionamide.
Le produit brut obtenu à l'étape précédente est placë dans 20 ml d'une
solution
d'HCI 4N dans le dioxane et suffisamment de MeOH pour permettre la dissolution
complète. Après 1 heure à TA, on évapore à sec, triture le résidu 2 fois dans
l'éther
puis essore, lave à l'éther et sèche sous vide en présence de KOH. On obtient
225
mg du composé attendu.
Préparation 4.2
N-[2-(4-cyanophényl)-1-((pyrrolidin-1-yl)carbonyl)éthyl]-3-phényl-3-
aminopropionamide, trifluoroacétate, isomère (R,R).
A) N-[2-(4-cyanophényl)-1-((pyrrolidin-1-yl)carbonyl)éthyl]-3-phényl-3-(N-
Boc)aminopropionamide, isomère (R,R).
On prépare l'ester de 2,5-dioxopyrrolidin-1-yle de l'acide obtenu à la
Préparation 2.19, étape B. On ajoute 800 mg de ce composé à un mélange
contenant
790 mg du composé de la Préparation 1.13 dans 15 ml d' acétonitrile et 380 p1
de
DIPEA. On laisse sous agitation quelques heures puis on abandonne une nuit à
TA.
Le milieu est évaporé à sec, repris par KHS04/K2S04 puis extrait par AcOEt. La
phase organique est lavée par une solution saturée de NaCI puis séchée sur
Na2S04
et évaporée à sec. On obtient 1,1 g du composé attendu qui cristallise dans
l' heptane.
B) N-[2-(4-cyanophényl)-1-((pyrrolidin-1-yl)carbonyl)éthyl]-3-phényl-3
aminopropionamide, trifluoroacétate, isomère (R,R).
On dissout 1 g du composé de l'étape précédente dans 6 ml de DCM puis on
ajoute 6m1 de TFA et laisse sous agitation 35 minutes à TA. Le milieu
réactionnel
est évaporé à sec, on redissout dans I'isopropanol, puis on évapore, triture
dans
Et20, essore et sèche pour obtenir 0,88 g du composé attendu.
CA 02241788 2004-10-28
CI
I, HCl : R1= ~ ~ ~ , R2 = girl Cl ,
EXEMPLE 1
-N _C /NRs N
R~6 = R17 = R9 = g3 = ~ NRaRj ° ~~ ' '\~ N
H
Dans 10 ml de dioxane et 5 ml d'eau, on dissout 2I0 mg du composé de la
Préparation 4.1 et on ajoute de la soude , 1 N pour atteindre pH = 9,5. On
ajoute
en 3 fois 84 mg de chlorure de naphtalène-2-sulfonyle en maintenant à pH = 9-
9,5
par addition de soude O,SN. Après une nuit à TA, on dilue le milieu par de
l'eau et
acidifie à pH = 2,5 par addition d'HCI, 1 N. Le solide formé est
chromatographié
sur Sephadex~ LH20 en éluant par du méthanol. La~ fraction contenant le
produit
attendu est concentrée, traitée par une solution d' HCl 4N dans le dioxane
puis
évaporée à sec ; le résidu est repris dans l'éther, essoré et sèché. On
obtient 41 mg
du composé attendu.
MH+ : 692 : profil isotopique dichloré
RMN (DMSO + TFA)
1,40-1,70 : m : 4H ; 2,50-3,40 : m : SH ; 4,00 : sd : 4H ; 4,45-4,70 : m : 2H
6,60-8,00 : m : I4 H
EXEMPLE 2
20 I, HCl : RI = ~ ~ ~ , R~
Ris . Rl~ _ g9 - g3 = PI, NR4Rs = -N. ' , _C w _
~.~J N~ N
H
A)
,.
II : R1= ~ , Rz =
'. ~ ~ /
Ris=Rt7=Rs=gs=~ NRaRs= -N
On mélange 715 mg du composé de la Préparation 1. l, 15 ml de CH3CN, 250
p.1 de triéthylamine, 710 mg du composé de la Préparation 3.2 et 450 mg de DCC
et
CA 02241788 2005-08-22
56
on laisse 5 heures sous agitation à TA. On évapore à sec, reprend le résidu
dans
l' acétone, essore le DCU puis évapore à nouveau à sec. Le résidu est trituré
dans
l'éther puis décanté, plusieurs fois. On sèche sous vide en présence de P205
pour
obtenir 6I0 mg du composé attendu, F = 195-200°C.
RMN (DMSO + TFA)
1,40-1,70 : m : 4H ; 2,30-3,40 : m : 8H ; 4,40-4,60 : m : et 4,b0-4,70 : m
2H ; 6,75-8,15 : m : 16H
B)
On dissout 600 mg du produit obtenu à l' étape précédente dans 20 ml ~ de
solution d'éthanol saturée d'acide chlorhydrique à 0°C. Après une nuit
au
réfrigérateur, on évapore à sec puis sèche sous vide en présence de KOH. Le
produit
7 0 obtenu est dilué dans 25 ml d'éthanol anhydre et on additionne en
plusieurs fois 134
p.1 d'éthyiènediamine dans 2 ml d'EtOH anhydre. Aprés une nuit à TA, on
ëvapore
à sec puis chromatographie le résidu sur silice en éluant avec un mélange
chloroforme/méthanol (85/15 ; v/v). Les fractions contenant le composé attendu
sont réunies, évaporées puis reprises dans l'éther chlorhydrique dilué. Après
essorage et séchage sous vide en prësence de KOH, on obtient 180 mg du produit
attendu.
MH ~' : 624
RMN (DMSO + TFA)
1,40-1,80 : m : 4H ; 2,55-3,45 : m : 8H ; 4,00 : s : 4H ; 4,55 :.te : et 4,70
: te
2H ; 6,70-8,20 : m : 16H
20 ~ ''
I, 3TFA : Rl ° ~ ~ ~ '. Rz - \ /
ALE 3
Ris=Rm~ Rs~~=H~ NR4Rs ___ _N
30 r~ ~ N C~ ~ / C
- C~ - -C
\ /
CA 02241788 2004-10-28
56a
A)
On dissout à 0°C 800 mg du composé obtenu à l'EXEMPLE 2, étape A,
dans
20 rnl d' une solution saturée d' HCl dans EtOH anhydre. Après une nuit au
réfrigérateur, on évapore à sec, puis sèche le résidu sous vide en présence de
KOH.
Le résidu est dissous dans 30 ml d'EtOH anhydre et on ajoute 175 ~.l de NEt3
puis
CA 02241788 2004-10-28
57
350 mg de N-Boc-(4-aminométhyl)benzylamine. Après 48 heures à TA, on évapore
à sec puis le résidu est chromatographié sur Sephadexd LH20 en éluant par
MeOH.
On obtient 2 fractions contenant deux composés différents. Pour l'un (fraction
1,
570 mg), l'amidine est monosubstituée par un groupe Rg - -4((N-
Boc)aminométhyl)benzyle et R6 et R7 = H ; pour l'autre (fraction 2, 150 mg),
l' amidine est disubstituée par R6 = Rg = -4(N-{Boc)aminométhyl)benzyle et R7
=
H.
B)
On met en suspension dans 1 ml de DCM, 125 mg du composé de Ia fraction 2
obtenu à l'étape précédente, on ajoute 1 ml de TFA et on laisse sous agitation
40
minutes à TA. Après évaporation à sec du milieu, on dissout le résidu dans
l' isopropanol puis on évapore à sec à nouveau ; le résidu est trituré dans
Et20,
essoré, lavé par Et20 puis séché. On obtient 110 mg du composé attendu.
MH+ : 836
RMN (DMSO + TFA)
1,40-1,80 : m : 4H ; 2,40-3,30 : m : 8H ; 3,90-4,80 : m : lOH ;
6,80-8,45 : m : 24H
I; 3TFA : Rl = ~ ~ R= _ \
EXEMPLE 4
NH
R16 = R» - ~ - g3 = H~ ~4RS = -N~ , C~~6 - -C
/ C~H:
On met en suspension dans 8 ml de DCM, 535 mg du composé de la fraction 1
obtenue à l'EXEMPLE 3, étape A, on ajoute 8 ml de TFA puis on laisse 40
minutes
sous agitation à TA. Après évaporation à sec du milieu, le résidu est trituré
dans
l'éther puis séché sous vide en présence de potasse. On obtient 520 mg du
composé
attendu.
MH+ : 717
RMN (DMSO + TFA) ; 1,30-1,70 : m : 4H ; 2,20-3,30 : m : 8H ; 3,90 : s
2H ; 4,40-4,70 : m : 4H ; 6,80-8,00 : m : 20H
EXEMPLE 5
'
I, HCl : R =
~-
CA 02241788 2004-10-28
58
'='~ i
Rls=Ri~= ~_ ~=H~ ~4Rs- _N~ ~ C\
H
On mélange 960 mg du composé de la Préparation 3.8 dans 20 ml de DMF avec
852 mg du composé de la Préparation 1.2 et 280 p,1 de NEt3, on ajuste à pH = 7-
7,5 par addition de NEt3. Après une nuit sous agitation, on évapore à sec puis
le
résidu est repris dans du méthanol et chromatographié sur Sephadex~ LH 20 en
éluant avec du méthanol. Les fractions contenant le produit sont réunies,
évaporées
puis reprises par 20 ml de butanol, 10 ml d'HCI, 1 N et 10 ml d'eau. Après
agitation et décantation, la phase organique est évaporëe. Le résidu est
chromatographié sur Sephadex~ LH 20 en éluant avec du méthanol et les
fractions
recueillies subissent le même traitement que précédemment. On obtient 180 mg
du
composé attendu.
MH ~' : 674
RMN (DMSO + TFA)
1,00-1,70 : m : 4H ; 2,30-3,20 : m : 8H ; 4,05 : sd : 4H ; 4,50 : rnt et 4,80
mt : 2H ; 7,05-8,10 : m : 18H
EXEMPLE 6
i
I, HCl : Rl = ~ ~ ~ , ~ _ ~ / Cl ,
N
Ris-Rv?-Rg-R3=H, NR4R5= -N~ ~ C\ _
s N
H
Ce composé est obtenu en opérant comme à l'EXEMPLE 5 à partir des
composés des Préparations 3.9 et 1.2.
MH+ : 658 : profil isotopique monochloré
RMN (DMSO + TFA)1,40-1,70 : m : 4H ; 2,40-3,30 : m : 8H ; 3,90 : sd
4H ;
4,40-4,65 : m : 2H ; 6,70-8,00 : m : 15H
EXEMPLE 7
~\ /,
I, HCI : RI = ~ ~ ~ , Rz = ~ / ~Bn ,
CA 02241788 2004-10-28
59
N
R16 - Rt7 = Rg = g3 = ~ ~4R5 = -~ ~ C~
N
H
On dissout 880 mg du composé de la Préparation 1.2 dans 15 ml de DMF et
300 N,1 de NEt3, on ajoute 1,12 g du composé de la Préparation 3.10 et laisse
sous
agitation 5 heures à TA. Après évaporation à sec du milieu, le résidu est
repris par
un rnélange d' HCl 1 N et de butanol puis on décante et évapore à sec la phase
supérieure. Le résidu est chromatographié sur Séphadex~ LH20 en éluant avec
MeOH. Le produit ainsi obtenu est repris par une solution d'HCl, 1 N dans le
butanol, puis on évapore à sec, triture le résidu dans l'éther, essore, lave à
l'éther et
sèche pour obtenir 330 mg du produit attendu.
MH+ : 730
RMN (DMSO + TFA) ; 1,45-2,25 : m : 4H ; 2,30-3,30 : m : 8H ; '3,85 : s
et 3,95 : s : 4H ; 4,50-4,70 : m : 4H ; 6,35-8,00 : m : 20H.
EXEMPLE 8
i i
I, HCl : R1= ~ , ~ _ '~,~ OFi
r
~--; N
R16 = Rt~ = R9 = R3 = H, NR4R5 = - i ~ Cys N
La réaction de débenzylation est effectuée selon J. Chem. Educ., 1987, 64,
1062.
On mélange 425 mg du composé de l'EXEMPLE 7 dans 20 ml de méthanol
avec 120 mg de formiate d'ammonium en présence de 500 mg de Pd/C à 10 %,
humide à 50 %. Après 24 heures de chauffage à reflux, on essore le palladium,
le
lave au méthanol et évapore le filtrat à sec ; le résidu est dissous dans du
méthanol
puis chromatographié sur Sephadex0 LH 20 en éluant avec du méthanol. Le
produit
obtenu est repris par un mélange d'une solution d'HCl 2N dans l'eau et de
butanol ;
après agitation, la fraction açueuse est extraite au butanol et les fractions
organiques
réunies sont évaporées à sec. Le résidu est repris à l' éther, essoré, lavé à
l' éther et
séché. On obtient 17I mg du produit attendu.
~ + ; 640
RMN (DMSO + TFA)
CA 02241788 2004-10-28
1,50-1,85 : m : 4H ; 2,40-3,30 : m : 8H ; 4,05 : s.d. : 4H ; 4,50-4,70 : m :
2H ; 6,40-8,20 : m : 15H
Les composés décrits dans le TABLEAU 8 sont préparés comme les
EXEMPLES 7 et 8 ci-dessus.
TABLEAU 8
10 ~ ~ , SOz~- i H-CH~z-CONH- ~CH-CON
w ~ Rz CHz
HCl
Cils
~+
P Z
Exem les R -C ~g~ ~MSO + TFA)
MH+ ; 730
20 N 1,45-1,?5 : m : 4H ;
2,40-3,40 : m : 8H ;
3,95:s:et4,00:s:4H;
H 4,50-4,70 : m : 4H ;
6,40-8,10 : m : 20H
MH+ : 640
10 ~N I,45-1,80 : m : 4H
2,25-3,30 : m . 8H
~ OH H 3,95 : s : 4H
4,45-4,70 : m : 2H
6,30-8,20 : m : 15H
CA 02241788 2004-10-28
61
MH+ : 683
11
1,45-1,80 : m :
4H
1,95-2,10 : m :
2H
2,75 : s.d. : 6H
2,50-3,60 : m :
12H
4,55 : m.t.: et
4,70 : mt:
2H 6,80-8,10 : m
: 16H
EXEMPLE 12
C1
,
I, HC1 : R1= ~ ~ Cl , R,, _ \
C1
NRs N
Ris _ R17 _ g9 =R3 - H, NR4Rs = _N ~ ~ C \ _
t--- NR~Rg N
H
On mélange 900 mg du composé de la Préparation 1.2 dans 10 ml de DMF et
550 p1 de NEt3 avec 1,04 g du composé de la Préparation 3.1 et l'on ajoute 1,3
g
de BOP et une quantité suffisante de NEt3 pour atteindre pH = 6-7. Après 5
heures
sous agitation en maintenent le milieu à pH = 6-7 par addition de NEt3, on
dilue le
milieu à l'éther puis on triture et laisse décanter (2 fois). On
chromatographie l'huile
sur Sephadex~ LH 20 en éluant avec du méthanol puis le produit obtenu est à
nouveau chromatographié sur silice en eiuant avec un um«i~~
chloroforme/méthanol (90/10 ; v/v). Le produit obtenu est lavé par AcOEt dans
de
l' eau, par AcOEt puis séché ; le solide est repris dans l' éther, essoré,
puis Iavé à
l'éther et séché. On obtient 313 mg du produit attendu.
MH-E' : 676 : profil isotopique trichloré
RMN (DMSO + TFA)
1,50-1,85 : m : 4H ; 2,70-3,20 : m : 8H ; 3,95 : s.e. : 4H ; 4,50-4,75 : m
2H ; 6,90-8,00 : m : 11H
On opérant comme à l' EXEMPLE 12, on prépare les composés décrit dans le
tableau ci-dessous.
CA 02241788 2004-10-28
62
TABLEAU 9
i : ~y SOZNH- ~ H- i H-CONH-~-CO
w w Rz R3 ~x
HCl
C~~~
Ws
Exemples R2 R3 ,C G~ MH+
\~N (DMSO + TFA)
MH "~ : 640
13 . H N 1,60-1,80 : m : 4H
~N 2,60-3,50 : m : 8H .
HO H 3,80 : s : et 4,00 : s : 4H
4,60-4,90 : m : 2H
6,90-8,70 : m : 15H
MH+ : 640
14 OH N 1,40-1,70 : m : 4H
2,50-3,20 : m : 8H
H 3,85 : s.d. : 4H
4,00 : d.d. : 1H
4,30-4,70 : m : 2H
6,55-8,10 : m : 16H
EXEMPLE I S
i i
I, HC1: R, = I , R2 =
N
i
Ris ' Rte - ~ ' ~ - H~ bars - -N~ ~ C.~ _
H C~
Ce composé est préparé â partir du composé préparé à I' EXEMPLE 2 étape A
et en opérant comme à l'EXEMPLE 2 étape B en utilisant la 2-méthylpropane-1,2-
diamine.
CA 02241788 2004-10-28
63
MH+ : 652
RMN (DMSO + TFA)
1,30 : s : 6H ; 1,30-I,70 : m : 4H ; 2,25-3,20 : m : 8H ; 3,65 : s : 2H ;
4,40-4,70 : m : 2H ; 6,70-8,05 : m : 16H
EXEMPLE 16
I, 2HC1 : R1= \ ~ ~ , R2 =
N
N
Rt6 = R17 = R9 = R3 = ~ bars = ._ N , C ~
NR~Rg N
On dissout 0,812 g du composé de la Préparation 3.13 et 0,810 g du composé
de la Préparation 1.2 dans 10 ml de DMF et on ajoute 1,22 g de BOP puis on
ajoute
à pH = 7 par addition de NEt3. Après I8 heures sous agitatiôn à TA, on évapore
à
sec. Le résidu est repris par AcOEt et une solution saturée de NaHC03 puis on
lave
par AcOEt, par le tampon KHS04/K2SO4, par une solution saturée de NaCI. On
sèche sur Na2S04 et évapore. Le résidu est chromatographié sur silice en
éluant par
le mélange CHC13/MeOH/ammoniaque concentré ( 10/0,5/0,1 ; v/v/v~. On obtient
0,390 g du composé attendu.
MH'~ : 625
RMN (DMSO) : 1,5-1,8 : mt : 4H ; 2,5-3,7 : m : 8H ; 4 : s : 4H ;
4,4-4,6 : mt : 1H; 4,7-4,9 : mt : 1H ; 6,8-7 : mt : 3H ; 7-7,1 : mt : 2H ; 7,4
: d
2H ; 7,7-8,1 : m : 5H ; 8,3-8,4 : m : 2H ; 8,7 : d : 1H ; 10,7 : s : 1H.
EXEMPLE 17
i
I, 2HCI : Rt = \ \ ~ , RZ =
N
-N C/~ N
NR,Rg N
H
Ce composé est préparé en suivant le mode opératoire de l' EXEMPLE
précédent à partir du composé de la Préparation 3.14 et de celui de la
Préparation
1.2.
MH+ : 625
RMN (DMSO) : 1,4-1,7 : mt : 4H ; 2,4-3,3 : m : 4H ; 4 : mt : 4H ; 4,5-4,7
mt : 2H ; 6,7-6,9 : mt : 2H ; 7,2-8,4 : m : 16H ; 9 : mt : 1H ; 10,7 : s : IH.
CA 02241788 2004-10-28
64
I, HC1 : R =
' ~~ ~ I ' R1= \
EXEMPLE 18 OiPr
/~ ~NRs N
Rls - R17 ° R9 = R~ - H, NRaRs _ -N~ , . C \ _ --~/
N
H
On place 740 mg du composé de la Préparation 1.2 dans 10 ml de DMF, on
ajoute 240 ~.l de DIPEA puis 740 mg du composé de la Préparation 3.24. Après
une
nuit sous agitation à TA, on ajoute de l'éther, décante, dilue à nouveau par
de
l'éther puis on triture et décante. La phase organique est chromatographie sur
Séphadex~ LH 20 en éluant par MeOH. Les fractions utiles sont reprises dans un
mélange contenant 5 ml de butanol, 5 ml d' HCl 1 N et 5 ml d' AcOEt. La phase
organique est lavée par 5 ml HCl IN et évaporée à sec. Le résidu est dissous
dans le
méthanol et précipité à l'éther. On obtient 470 mg du composé attendu.
MH + : 682
RMN (DMSO) : 1,00 : d : 6H ; 1,45-1,80 : m : 4H ; 2,30-3,20 : m : 8H ;
3,95 : sd : 4H ; 4,05-4,20 : m : 1H ; 4,50-4,70 : m : 2H ; 6,30-8,40 : m : 17H
(15H aromatiques + 2H amides) ; 10,70 : se : 1H.
En procédant selon Ie mode opératoire décrit ci-dessus on prépare les composés
selon l' invention du tableau ci-dessous à partir des composés des
Préparations 3.25 à
3.32.
TABLEAU 10
S02NH-CH-CHZ-CONH-ÇH-CO-N, '
~J
i
~ , HC1
N' NH
u
CA 02241788 1998-07-07
WO 97/25315 65 PCT/FR97/00026
Exemple R2 RMN MH +
19 - (DMSO + TFA) 1,50-1,80 : 654
m : 4H ;
\ / 2,25-3,25 : m : 8H ; 3,40
: s : 3H ;
4,00 : sd : 4H ; 4,45-4,75
: m : 2H ;
OMe
6,35-6,90 : mt : 4H ; 7,30-8,15
: m
11H
20 - (DMSO + TFA) 1, 50-1, 80 654
: m : 4H ;
\ / OMe 2,30-3,25 : m : 8H ; 3,40
: s : 3H ;
4,00 : sd : 4H ; 4,60 : qt
: 1H : 6,40
d : 2H ; 6,90 : d : 2H ;
7,25-8,05 : m
11H
21 - (DMSO + TFA) 1,50-1,80 : 658-660
m : 4H ; ~
\ / 2,35-3,40 : m : 8H ; 4,00
: sd : 4H ;
4,50-4,70 : m : 2H ; 6,75-7,00
: m
C1
4H ; 7,25-8,10 : m : 11H
22 - (DMSO + TFA) 1,40-1, 80 : 692
m : 4H ;
\ / 2,40-3,40 : m : 8H ; 3,95
: sd : 4H ;
4,50-4,65 : mt : 1H ; 4,75
: qd : 1H ;
CF3
6,90-8,00 : m : 15H
23 - (DMSO + TFA) 1,20- i , 60 674
: m : 4H ;
2,40-3,20 : m : 8H ; 3,95
: se : 4H ;
\ 4,25-4,50 : mt : 1H ; 5,45
: t : 1H ;
\ / 7,00-7,90 : m : 18H
24 - (DMSO + TFA) 1,50-1,75 : 64~'.
m : 4H ;
\ / 2,60-3,40 : m : SH ; 4,00
: se : 4H ;
4,50-4,75 : m : 2H ; 6,55-8,10
: m
F
15H
25 (DMSO + TFA) 0, 60-1, 70 630
: m : 15
H ; 1,80-2,20 : m : 2H .2,60-3,40
m : 7H ; 3,90 : s : 4H ;
4,30-4,60 : m
1H ;
7,25-8,25 : m : 11H
CA 02241788 2004-10-28
66
- ~(DMSO + TFA) 1,50-1,80 : 638 .
m : 4H ;
26
\ / 1,90 : s : 3H ; 2,20-3,30
: m : 8H ;
Me 4,05 : sd : 4H ; 4,55-4,75
: m : 2H ;
6,60-8,10 : m : 15H
EXEMPLE 27
I, HCl : Rl = \ \ I , ~ _ \ ~ \ / ,
R =R __ _ _~ ~ _N C%'~ _
17 ~ ~ ~ 4 '
N
H
A) N-(2-(4-cyanophényl)-1-(pyrrolidin-1-yl-carbonyl)éthyl)-3-(biphényl-4-yl)-3-
(N-
Boc)aminopropionamide.
On place 1,19 g du composé de la Préparation 1.1 dans 30 ml d' acétonitrile et
0,46 ml de NEt3 et on ajoute 1,46 g du composé de la Préparation 2.17. On
abandonne une nuit à TA puis on évapore à sec, reprend par KHS04/K2S04 et
extrait par AcOEt. On lave par une solution saturée de NaCI, sèche sur Na2S04
puis évapore à sec. Le composé attendu cristallise dans l'heptane, on essore,
lave à
l'heptane puis sèche pour obtenir 1,86 g.
B) Trifluoroacétate de N-(2-(4-cyanophényl)-1-{pyrrolidin-1-yl-carbonyl)éthyl)-
3-
(biphényl-4-yl)-3-aminopropionamide.
On laisse sous agitation à TA pendant 35 minutes un mélange contenant 1,82 g
du composé de l'étape précédente dans 15 ml de DCM et 15 ml de TFA. On
évapore à sec, dissout le résidu dans l'isopropanol et évapore à sec à
nouveau. Le
produit attendu cristallise dans Et20, il est essoré, lavé par Et20 puis séché
pour
donner I,37 g
C)
i ~ - _
u: R =
1 w w ( ' ~- \ / \ /
Ris - Rc~ ° R9 - R3 = H, NRaRs = _ N
CA 02241788 2004-10-28
67
On~place 1,34 g du composé de l'étape précédente dans 25 ml de DCM et 850 1z
1 de DIPEA, et on ajoute progressivement 522 g de chlorure de napht-2-
ylsulfonyle
dans 5 ml de DCM. Après 2 heures sous agitation à TA, on dilue par 60 ml de
DCM puis on lave par KHS04/K2S04, par une solution saturée de NaCl. On sèche
sur Na2S04, évapore â sec puis on chromatographie sur silice fine en éluant
par du
chloroforme contenant 1 % de MeOH. On obtient 0,802 g du composé attendu.
RMN (DMSO + TFA) : 1,45-1,85 : m : 4H ; 2,40-3,35 : m : 8H ; 4,50-4,90
mt : 2H ; 7,05-8,20 : m : 20H.
D)
On place 0,778 g du composé de l'étape précédente dans 50 ml d'EtOH anhydre
saturé en HCl à 0°C ; on laisse sous agitation au réfrigérateur jusqu'à
dissolution
puis on abandonne 48 heures au réfrigérateur. On évapore à sec et sèche
dessicateur
sous vide en présence de potasse pendant 2 heures. On dissout dans 80 ml
d'EtOH
anhydre, ajoute 206 itl de DIPEA et 93 p,1 d'éthylènediamine. Après 24 heures
à
TA, on évapore à sec puis on chromatographie sur Séphadex~ LH 20 en éluant par
le méthanol. Les fractions contenant le produit sont reprises par un mélange
de 6 inl
de butanol et 6 ml d'HCl IN. Après agitation et décantation, la phase-
organique est
lavée par 3 ml de HCl 1N et évagorée sous vide. On reprend par Et20, le solide
formé est essoré, lavé par Et20 et séché pour donner 0,495 g du composé
attendu.
MH + : 700
RMN : 1,40-1,80 : m : 4H ; 2,40-3,30 : m : 8H ; 3,80 : s : 2H ; 3,95 : s : 2H;
4,50-4,80 : mt : 2H ; 7,00-8,05 : m : 20H.
EXEMPLE 28
I, HCl : Rl ° I , Rz ~ '
w w \ /
_N C
- Rg - R3 - H' ~'c~s - ' ~ N
R16 - R17
~s
H
On dissout 0,790 g du composé obtenu à l'EXEMPLE 2, étape A dans 10 ml
d'EtOH saturé en HCl à 0°C et on laisse sous agitation 48 heures à
0°C. On
évapore à sec puis on reprend par EtOH et évapore (2 fois). On reprend par DCM
et
évapore (2 fois). On reprend dans 20 ml d'EtOH puis neutralise par addition de
NEt3. On ajoute 0,109 ml de diaminopropane et on laisse 24 heures à TA. On
CA 02241788 2004-10-28
68
évapore à sec, reprend par DCM, lave par KHS04/K2S04. On effectue ensuite une
chromatographie sur silice en éluant par un mélange
chloroforme/méthanol/ammoniaque concentré (10/1/1 ; v/v/v). On obtient 0,12 g
du
composé attendu.
MH'~ : 638
EXEMPLE 29
i i
I, HC1: Rt = \ \ I , g~ . ~ ~ ,
CH3
~NR6
R16 = RIS - g9 = g3 = g~ ~4~ ~ _.N ~ , C~ _
H
A)
~ i _
II : R1= \ \ ( ,
CH3
Ri6 RI7 ~ - ~ - ~ ~4~
C
On dissout 0,888 g du composé de la Préparation 3.2 et 0,957.g du composé de
la Préparation 1.4 dans 5 ml de DMF ; on ajoute 1,22 g de BOP puis on ajuste à
pH
= 7 par addition de NEt3. Après 18 heures sous agitation à TA, on évapore à
sec
puis on reprend par AcOEt. On lave par une solution de NaHC03, par
KHS04/K2S04, par une solution saturée de NaCI puis on sèche sur Na2S04 et
évapore. On reprend par de I' éther et quelques gouttes d' hexane puis on
essore et
sèche sur Na2S04 pour obtenir 1,1 g du composé attendu.
B)
On dissout 1 g du composé de l'étape précédente dans 20 ml d'EtOH saturé en
HCl à 0°C. Après 48 heures à +4°C, on évapore puis on reprend
par EtOH (3 fois)
puis on évapore et reprend par DCM (2 fois). Le résidu est repris par 20 ml de
EtOH, puis on ajuste à pH = 7 par addtion de NEt3 et on ajoute 0,122 ml
d'éthylènediamine. Après 18 heures sous agitation à TA, on évapore à sec puis
on
reprend par DCM et lave par 10 ml de KHS04/K2S04. On chromatographie sur
silice en éluant par un mélange chloroforme/méthanol/ammoniaque ( 10/ 1/0,1 ;
v/v/v) pour obtenir 0,640 g du composé attendu.
CA 02241788 1998-07-07
WO 97/25315 69 PCTIFR97/00026
MH + : 650
RMN (DMSO + TFA) : 0,8-1,2 : m : 4H ; 2,3-3,1 : m : 4H ; 4 : s : 4H ;
4,3-4,7 : m : 4H ; 5,4-5,7 : mt : 2H ; 6,8-7,9 : m : 15H ; 8,2 : d : 1H.
En opérant comme à l'EXEMPLE 12, on prépare les composés décrits dans le
Tableau 11 ci-dessous.
TABLEAU 11
Rl-S02NH- i H-CH2-CONH- i H-CO N
, HCl
N~.C~NH
u
Exemple R 1 R2 MH +
30 C1 - 642-644
CI \
\ /
31 ~ - 628
En opérant selon le mode opératoire de l'EXEMPLE 2, étapes A et B, on
prépare les composés selon l'invention du Tableau ci-après.
TABLEAU 12
S02NH-CH-CH'-CONH- i H-CONR4R5
i i
~ ~ I ~ I CHZ
w i
HCl
w
N~NH
CA 02241788 2004-10-28
Exemple -NR4R5 FC/RMN (DMSO + TFA) MH+
32 170C 640
- 2,40-3,45 : m : 12H ;4,00
: se : 4H ;
4, 60-4, 90 : m : 2H ;
6,80-8,15 : m : 16H
33 Me 0,6-0,9 : mt : 6H ; 2,2-3 626
: m : 7H ;
-N iPr 3,9 : s : 4H ; 4,3-4,9 :
m : 3H ;
6,7-7,8 : m : 15H ; 8,: d
: 1H
34 Me 0,6-1,6 : m : 8H ; 1,6-1,9 652
: mt : 2H ;
10 _N 2,4-3,1 : m : 4H ; 3,2-4
: m ; 6H ;
4,2-4,8 : m : 2H ; 6,8-7,9
: m : 15H ;
Me . 8:s: 1H
EXEMPLE 35
I, HCI : Rl = \ \ ~ , ~ -_ ~ ~ ,
N
~-Rn-SRS+Rts-'CH2' ~ ~4R5--N~ ' C\ s -
N
A)
i ~ -
r1: R1= 1 ~ ,
w w ' ~- \ /
~ = R17 = ~ ~ '~- Ris _ 'C~' ~ .NR4R5 = _N
On mélange 580 mg du composé obtenu à l'EXEMPLE 2, étape A avec 250 mg
de paraformaldéhyde et 60 mg d' acide paratoluène sulfonique monohydrate dans
25
ml de benzène. On porte à reflux dans un appareil Dean Stark pendant 1 heure.
Le
milieu réactionnel est ensuite lavé par une solution saturée de NaHC03, une
solution saturée de NaCl puis séché sur Na2S04 et évaporé à sec. Le résidu est
CA 02241788 2004-10-28
71
chromatographié sur silice fine en éluant par le chloroforme pour obtenir 165
mg'du
composé attendu.
RMN (DMSO + TFA) : 1,55-1,85 : m : 4H ; 2,40-3,40 : m : 8H ; 4,60-5,50
m : 4H ; 6,90-8,30 : m : 16H.
B)
On procëde comme à l'EXEMPLE 2, étape B pour obtenir le composé attendu
qui est purifié par chromatographie sur Séphadex~ LH 20 en éluant par MeOH.
MH+ : 636
RMN (DMSO + TFA) : 1,50-1,80 : m : 4H ; 2,40-3,30 : rn : 8H ; 3,95 : s
4H ;
4,90-5,45 : rn : 4H ; 6,90-8,10 : m : 16H.
EXEMPLE 36
I, HCl : Rt = \ ~ i ,
~~s
g3=Rts=RpH,Rs=Me,-NRaRs=_N~ ' C~ _
~rRa
A)
i -
II:R1= ' \ I , g
~=Rts=Rm_~~=Me~_~sRs=~hT
On mélange 1,05 g du composé de l'EXEMPLE 2, étape A avec 0,262 g de
carbonate de potassium dans 12 ml de DMF et on ajoute 423 p.1 d'iodure de
métl2yle. Le lendemain, on évapore à sec, puis on reprend par de l'eau et
AcOEt.
On lave à l'eau, par une solution saturée de NaCI puis on sèche sur Na2S04. On
obtient 0,6 g du composé attendu.
MH+ . 595
RMN (DMSO + TFA) : 1,50-1,80 : m : 4H ; 2,20-3,30 : m . 8H ; 2,55 : se
3H
4,35-4,65 : mt : 1H ; 5,45-5,60 : mt : 1H ; 7,10-8,45 : m : 16H.
B)
CA 02241788 2004-10-28
72
On 'procède comme à l' EXEMPLE 2, étape B, pour obtenir Ie composé attendu
qui est purifié par chromatographie sur silice DCM/MeOH (93/7 ; v/v) puis par
une
deuxième chromatographie sur Séphadex~ LH 20 en éluant par le méthanol, F =
154°C.
MH+ : 638
RMN (DMSO + TFA) : 1,20-1,80 : m : 4H ; 2,10-2,40 : mt : 2H ; 2,50 : se
3H ; 2,40-3,20 : m : 6H ; 4,00 : s : 4H ; 4,40-4,70 : mt : 1H ; 5,35-5,55 : mt
IH;
7,05-8,40 : m : 16H.
EXEMPLE 37
I, HC1: R =
i
Ri i
~ + Rl t - -CHz'~ ~ - Rts - Rte - H~ '~~Rs - ~ C ~ ~6 _ ~N
~ NRzRg '''~N
H
A)
i
II: R,= y w
g~ + Ri ~ - -CHZ-, Rg - Ri6 = Rm - H~ ~4~ _ -N
On prépare un mélange contenant.0,79 g du composé de la Préparation 3.35
0,79 g du composé de la Préparation 1.1 et 1,06 g de BOP dans 10 ml de DMF, on
ajoute du DIPEA pour obtenir pH = 7 et on laisse sous agitation 2 heures à TA.
On
extrait par AcOEt, lave par H20, une solution de NaOH, 0,25 N, une solution
d'HCl 0,25 N, de l'eau puis une solution saturée de NaCI. On sèche sur Na~S04
puis on évapore et Ie résidu est chromatographié sur silice en éluant par le
mélange
chloroforme/MeOH (9515 ; v/v).
B)
Le produit attendu est obtenu en procédant comme à l'EXEMPLE 2, étape B. II
est purifié par chromatographie sur Séphadex~ LH 20 en éluant par DCM/MeOH
(3/2 ; v/v)
MH+ : 636
CA 02241788 2004-10-28
73
RMN (DMSO + TFA) : 1,40-1,80 : m : 4H ; 2,60-3,60 : m : 9H ; 3,95 : s :
4H ; 4,60-5,20 : m : 2H ; 6,20-8,50 : m : 15H.
EXEMPLE 38
i ~
I, HCl : R~ _ \ \ I , g~ _ \ /
Rll
N
_N C~~6 /
Ra = Ris = Ris - ~ RS + Ri ~ _ -CI~-CHz-, -NR4R3 = \
N
H
On glace 533 mg du composé de la Préparation 3.37 dans 10 ml de DMF et 205
p.1 de DIPEA, on ajoute 573 mg du composé de la Préparation I.2 puis on
abandonne une nuit à TA. On dilue par 100 ml d'Et20 puis on chromatographie la
gomme formée sur Séphadex~ LH 20 en éluant par MeOH. Les bonnes fractions
sont réunies, essorées et le résidu est repris dans 6 ml de butanol et 6 ml d'
HCI. La
phase organique est décantée puis lavée avec 3 ml d'HCl 1N et évaporée à sec.
Le
produit cristallise dans Et20, il est essoré, séché pour donner 430 mg du
composé
attendu.
MH + : 650
RMN (DMSO + TFA) : 1,50-1,90 : m : 4H ; 2,50-3,80 : m : 12H ; 4,00 : sd
4H ;
4,45-4,80 : m : 1H ; 5,40 : qd : 1H ; 6,75-8,35 : m : 15H.
EXEMPLE 39
~~ ,i -
i, HCl : Rl = '~ , g~ -_ ,
\ /
NRs N
~'~ °RI~°~ Ris=CH3~ NR4R5= -N~, C~ -
NRrRg N
H
A)
/ i
II:R,° w w, ~ ' ~ \ / '
~ - Rs = Ri7 = ~ Ris = CH3, -NR4Rs = -N
On agite un milieu réactionnel contenant 7I0 mg du composé de la Préparation
3.2, 750 mg du composé de la Préparation I.8 et 0,98 g de BOP dans 12 ml de
CA 02241788 2004-10-28
74
DMF, ôn ajoute du DIPEA pour amener à pH = 6. Après 2 heures sous agitation à
TA, on extrait par AcOEt. Le produit brut obtenu est chromatographié sur
silice en
éluant par AcOEt/toluène (3/2 ; v/v). On obtient 0,52 g du composé attendu
sous
forme d'un solide blanc.
B)
Le composé attendu est obtenu en procédent comme à l'EXEMPLE 2, étape B.
Il est purifié sur Séphadex~ LH 20 en éluant par DCM/MeOH (3/2 ; vlv).
MH+ : 638
IuVIN (DMSO + TFA) : 1,40-,175 : m : 4H ; 2,30-3,20 : m : 11H ; 4,00 : s
4H ; 4, 60-5,40 : mt : 2H ; 6, 80-7,15 : m : 16H.
EXEMPLE 40
i
I, HC1 : R~ _
\ /
~NR6 N H
R3 = Rs = Ri6 = Ri7 = H, .NR4R5 = -N~~ C, _ /
A)
On traite 0,42 g du composé de I' EXEMPLE 2, étape A par 10 ~ ml d' éthanol
c~orhydrique saturé à -10°C. Après 72 heures à +4°C, on évapore
à sec puis on
sèche sous vide en présence de potasse. On obtient 0,51 g de produit non
purifié
sous forme de chlorhydrate.
B)
0,5 g de l'imidate d'éthyle obtenu à l'étape précédente sont refroidis à
0°C et
on verse, à 0°C, 10 m1 d'éthanol ammoniacal saturé. On laisse revenir à
TA. Le
lendemain on évapore à sec puis on effectue une chromatographie sur silice en
éluant par un mélange DCM-MeOH (90/10 ; v/v) puis on effectue une autre
chromatographie sur Séphadex~ LH 20 en éluant par MeOH. On obtient 0,15 g du
composé attendu, Fc = 180°C.
MH-~' : 598
RMN (DMSO) : 1,40-1,70 : m : 4H ; 2,25-3,10 : m : 8H ; 4,40-4,70 : m
2H; 6,80-8,30 : m : 18H.
CA 02241788 2004-10-28
EXEMPLE 41
i ~ -
I, HC1: R1= ( . R2 =
w
~°g9-Ri6=Riz=H, NR4R3= -N ,
,fis ' -C~NH _ ~~ N
C ~~ Z_~ ~~'NH_~CHz)3~~
4
N
A) 4-Aminobutyronitrile.
10 Ce composé est préparé selon J. Am. Soc., 1952, 74, 1836. Dans une bombe,
on place 6,7 ml de 4-bromobutyronitrile et 35 ml d'ammoniac liquide à
SO°C.
Après fermeture, la. bombe est laissée à TA pendant 48 heures. Le résidu est
repris
par une solution de NaOH à 50 % puis extrait à l' éther ; la phase organique
est
séchée sur Na2S04, évaporée à sec puis chromatographiée sur silice en éluant
,par
DCM/MeOH/NH40H (90/10/0,3 ; v/v/v}. On obtient 1,07 g du composé attendu.
B)
On traite 0,62 g du composé à l'EXEMPLE 2, étape A par 10 ml d'éthanol
chlorhydrique saturé à -10°C. Après 24 heures à +4°C, on évapore
à sec, puis on
sèche sous vide. On obtient 0,8 g de produit non purifié sous forme de
chlorhydrate.
C)
20 , , ,_
II', HCl : Rt = w w ~ ' Rz
~=~-R16=R~~=~.~4R5= -N
~NH r %NH
_ _C
C~ldH-Z-CN r \~"{~~3~
On dissout 0,8 g du composé gréparé à l'étape ci-dessus dans 10 mI d'EtOH et
on ajoute goutte à goutte 108 mg de 4-aminobutyronitrile dilué dans 10 ml
d'EtOH.
Le lendemain, on évapore à sec, reprend gar MeOH puis on ajoute quelques
gouttes
d' éther chlorhydrique. Après évaporation à sec, on chromatographie sur silice
en
30 éluant par DCM/MeOH (90/10 ; v/v) pour obtenir 0,38 g du composé attendu, F
=
I4~°C.
MH+ : 665
CA 02241788 2004-10-28
76
D)
A 0°C, on verse 10 ml d'éthanol chlorhydrique saturé à -10°C
sur 0,35 g du
composé obtenu à l'étape précédente. On laisse au réfrigérateur une nuit. Le
lendemain on évapore à sec et sèche sous vide en présence de KOH. Le produit
6btenu (0,35 g) est dissout dans 80 ml d'éthanol ; on ajoute goutte à goutte
43 ~1
d'éthylène diamine dans 10 ml d'éthanol. Après une nuit sous agitation, on
évapore
à sec puis on effectue une chromatographie sur silice en éluant par un mélange
DCM/MeOH (50!50 ; v/v). On obtient 0,075 g du composé attendu, F =
I95°C.
MH + : 708
RMN (DMSO + TFA) : 1,35-1,50 : m : 4H ; 1,80-2,00 : mt : 2H ; 2,40-3,20
: m : !OH ; 3,40 : mt : 2H ; 3,70 : s : 4H ; 4,40-4,70 : mt : 2H ; 6,70-8,05 :
m
16H.
EXEMPLE 42
. f .._ -
I, HCl : R, - w w ' ' Rz \ ! '
N
- RS - Ris -_ Ris ~ ~ -~4Rs = _N~t)2~ -C =
\'NR~Rg N
H
Ce composé est préparé en suivant le procédé décrit à l'EXEMPLE 12 à partir
des composés des Préparations 1.9 et 3.2.
MH+ : 626
EXEMPLE 43
i
I, HCl : R-, ° w. ~ ' ~ - \ / '
' ,~sNRs l
~ - ~ ~ Ris - RW H~ -bars - -' N' ' ~ -C ~
NRTRg N
H
isomère (R,R).
A)
II : R i = ~ , g~ . \ / ,
~ - ~ - Rts - Ry H~ _NR4Rs - - N
isomère (R,R).
CA 02241788 1998-07-07
WO 97/25315 77 PCTIFR97/00026
On mélange 1,44 g du composé de la Préparation 1.13 dans 40 ml d' acétonitrile
avec 700 p.l de DIPEA et 1,81 g du composé de la Préparation 3.39.
Le,précipité
formé est dissous dans du DCM puis lavé par KHS04/K2S04, une solution saturée
de NaHC03, une solution saturée de NaCI. On sèche et évapore à sec, Ie produit
obtenu ( 1, 57 g) est utilisé tel quel à l' étape suivante.
RMN (DMSO) : 1,40-1,60 ; m ; 4H ; 2,20-3,10 : m : 8H ; 4,40 : qd : 1H ;
4,60 : qd : 1H ; 6,80-8,35 : m : 18H.
B)
On met en suspension 1 g du produit de l'étape précédente dans 30 ml d'éthanol
anhydre saturé en HCl à 0°C et on laisse sous agitation 24 heures au
réfrigérateur.
On évapore à sec, sèche au dessicateur en présence de potasse. On place le
produit
dans 100 ml d'éthanol anhydre et on ajoute 273 p.1 de DIPEA et 136 ~l
d'ëthylènediamine. Après 24 heures sous agitation à TA, on évapore à sec,
reprend
dans un mélange butanol/chloroformeIHC1 1 N ( 1 / 1 / 1 ; v/v/v). On décante
puis on
lave la phase organique par HCl 1N et évapore à sec. Le résidu est
chromatographié
sur Séphadex~ LH 20 en éluant par MeOH. Le produit obtenu est traité par un
mélange butanol/chloroforme/HCl 1N (1/1/1 ; v/v/v) puis concentré. Après
cristallisation par Et20, les cristaux sont essorés, lavés par Et20 puis
séchés. On
obtient 198 mg du composé attendu.
a,D = +38,1 ° (c = l, DMF)
MH+ : 624
RMN (DMSO) : 1,50-1,75 : m : 4H ; 2,40-2,70 : mt : 2H ; 2,75-3,25 : m
6H ; 4,05 : s : 4H ; 4,55 : qd : 1H ; 4,75 : qd : 1H ; 6,95-8,20 : m : 16H ;
8,50 : t
: 2H ; 10,80 : se : 2H.
En utilisant le mode opératoire décrit à l'EXEMPLE 43 et dans les Préparations
correspondantes, on prépare les différents isomères décrits dans le Tableau ci-
après.
TABLEAU 13
O
* * ii
~ ~ S02-NH-CH-CH2-CO-NH-ÇH-C- N
w w I ' ICHZ
~J
HN~N HC1
U
CA 02241788 2004-10-28
78
Exemple Isomre D5 - (c = l, DMF) MH+
FC
44 (S,S) -41,9 624
45 (R,S) +42,2 624
46 (S,R) F = 195-201C 624
g~N (DMSO)
de l'EXEMPLE
44 :
1,50-1,75
: m
: 4H
; 2,40-2,70
: mt
: 2H
;
2,75-3,25
: m
: 6H
; 4,05
: s
: 4H
; 4,55
: qd
: 1H
; 4,75
: qd
: 1H
; 6,95-8,20
m: 16H
; 8,50
: t
: 2H
; 10,80
: se
: 2H.
RMN (DMSO)
de l'EXEMPLE
45 :
1,65-1,90
: m
: 4H
; 2,40-3,45
: m
: 8H
;
4,05
: s
: 4H
; 4,55-4,85
: mt
: 2H
; 6,95-8,25
: m
: 16H
; 8,45
: t
: 2H
; 10,80
se :
2H.
RMN (DMSO
+ TFA)
de l'EXEMPLE
46 :
1,50-1,80
: m
: 4H
; 2,25-2,95
m : 4H
; 3,00-3,40
: m
: 4H
; 4,00
: s
: 4H
; 4,30-4,75
: mt
: 2H
; 6,80-8,15
: m
16H.
EXEMPLE
47
C1
I, HCl : R1= ~ ~ Cl , Rz = , ~ '
Cl
__ __ __ __ __ _
Ris Ri' R9 ~ ~ bars N ~ C ~ _
~rRa N-
H
isomère (R,R).
C!
II : R1= , ~ ~ Cl , R2 =
Cl
Ris - R" = Rg -R3 = H, NR4Rs = -N
isomère (R,R).
CA 02241788 2004-10-28
79
Ces 2 composés sont préparés en 2 étapes successives en suivant te mode
opératoire décrit à 1'BXEMPLE 27, étapes C et D à partir du composé obtenu à
la
Préparation 4.2 et du chlorure de (2,4,6-trichlorophényl)sulfonyle.
ï : a D _ -24 ° (c = 0, 5 : MeOH)
MH+ : 676 avec profil isotopique trichloré.
RMN (DMSO) : 1,50-1.65 : m : 4H ; 2,45-3,20 : m : 8H ; 3,95 : s : 4H ;
4,40-4,70 : mt : 2H ; 7,05 : s : 5H ; 7,45 : d : 2H ; 7,50 : s : 2H ; 7,90 : d
: 2H ;
8,45 : d : IH ; 8,80 : d : 1H ; 10,70 : se : 1H.
u : RMN (DMSO) : 1,50-1,60 : m : 4H ; 2,45-3,15 : m : 8H ; 4,40-4,55 : qd
1H ; 4,60-4.,75 : qd : 1H ; 7,05 : s : SH ; 7,40 : d : 2H ; 7,50 : s : 2H ;
7,70 : d
10 , 2H; 8,40 . d . 1H; 8,70 . d . 1H.
i
I, HCl : Ri = ~ ~ ~ , Ri = ~ ~ Cl ,
EXEMPLE 48
R16 = R17 = R3 = R3 ° ~ ~4R5 = - ~ , C ~ ~rRs N
H
isomère (R,R)
i
II : Rl = ~ ~ ~ , Ri ° \ ~ Ci
R16 = Rl~ = R9 = R3 = H, NR4R5 = --N
isomère (R,R)
Ce composé est préparé en 2 étapes en suivant le mode opératoire décrit à
l'bXEMPLB 43, à partir des composés des Préparations 3.43 et 1.13.
I : aD = +57° (c = 1 ; DMF)
MH+ : 658 et 660
RMN (DMSO + TFA) : 1,40-1,60 : m : 4H ; 2,25-3,10 : m : 8H ; 3,95 : s
4H ; 4,45-4,65 : mt : 2H ; 6,80-8,05 : m : 15H.
II : a D = +6 t ° (c = I ; DMF)
~N (DMSO) : I,40-1,60 : m : 4H ; 2,25-3,10 : m : 8H ; 4,35-4,60 : mt
2H ; 6,85-8,05 : m : 15H ; 8,25 : t : 2H.
CA 02241788 2004-10-28
EXEM~'LE 49
i i
I, HC1: R1= .~ .~ .~' ~ ='C~ \ / '
_ i.~s
Ris Ris Rs ~ ~ ~ N C
H
isomère (S), (R,S).
On mélange 800 mg du composé de la Préparation 1.2 dans 20 ml de DMF avec
250 p.1 de DIPEA et 933 mg du composé de la Préparation 3.45. Après une nuit
sous agitation à TA, on dilue à l' éther puis on décante. La gomme formée est
10 triturée dans l'éther puis on chromatographie sur Séphadex~ LH 20 en éluant
par
MeOH. Les bonnes fractions sont évaporées et le résidu est repris dans un
mélange
butanol/AcOEt/HCl 1N (1/1/1 ; v/v/v). La phase organique est décantée et
évaporée
à sec, le résidu est repris dans Et20, essoré et séché pour donner 770 mg du
composé attendu.
MH+ : 638
RMN (DMSO + TFA) : 1,55-1,85 : m : 4H ; 2,05-3,50 : m : 11H ; 3,75 : s
2H ; 4,00 : s : 2H ; 4,45-4,95 : mt : 1H ; 6,70-8,20 : m : 16H.
EXEMPLE 50
i i
I,HC1: Ri= ~ ~ ' ,R3=R16=R~9°H,
20 -i-fH-~ N
O'C~ \ / ' ~ = H~ NR4Rs ' _N~ . -~ \
NRiRg N
H
Ce composé est préparé à partir du composé de la Préparation 3.46 et du
composé de la Préparation 1.2 en suivant le mode opératoire de l' EXEMPLE 5 .
RMN (DMSO + TFA) : 1,5-1,8 : mt : 6H ; 2,7-3,6 : m : 6H ; 3,6 : mt : 3H ;
3,6-3,9 : mt 8H ; 4,6-4,8 : mt : 2H ; 6,5 : d : 2H ; 6,8-7 : m : 4H ; 7,4-8,1
: m
8H;
8,4 : d : 2H.
30 EXEMPLE 51
CA 02241788 2004-10-28
81
i
I, HCl : R~ _ ~ ~ ~ , ~ = Ris = Ri9 - H
N
N~6 -C.
-N_ i H_ _ _ N , ~ = H~ ~4Rs = _N~ , _~ \_ N
~Rz O
OCCF3
On dissout 0,4 g du composé obtenu à l'exemple précédent dans 8 ml de TFA et
0,2 ml de thioanisol. Après 24heures sous agitation à TA, on évapore puis le
résidu
est repris par Et20 et essoré. On lave plusieurs fois à l'éther puis on sèche
sur
Na2S04 pour obtenir 0,385 g du composé attendu.
MH'~' : 700
RMN (DMSO + TFA) : 1,7-2,1 : mt : 6H ; 2,5 : d : 2H ; 2,9-3,7 : m : 13 H ;
3,7-4:m:I3H;4,8:mt:1H;5,3:mt:1H;7,2-8,3:m:lOH;8,5:s:IH.
EXEMPLE 52
i .-
I, HCl : Ri = \ \ ~ , R3 = R~6 = R19 = H,
_N_CH_ - _ N ~ ~ = H ~4Rs = _N , _~ , NR6 _ N
OH
Le composé obtenu à l'exemple précédent est mis en solution dans 50 ml de
MeOH contenant 0,106 g de KOH. Après 18 heures sous agitation à TA, on
acidifie
à pH = 2 par addition d'une solution saturée d'HCl gaz dans le dioxane. On
évapore, reprend par 5 ml d'eau et triture. On essore, lave par de l'eau puis
sèche
pour obtenir 0,160 g du composé attendu.
MH + = 604
EXEMPLE 53
i
I, HCI : Rl = ~ ~ ~ , Rz -
NRs N
_ /
R16=R1=Rg-R3-H, NR4Rs= -N , Cy
~Rs
isomère (R,R)
CA 02241788 2004-10-28
82
Ce composé est préparé selon le mode opératoire décrit à l' EXEMPLE 43 mais
en utilisant le chlorure de napht-1-ylsulfonyle dans la Préparation 3.38.
a25 - -18° (c = 1 ; DMF)
D
MH + : 624
RMN (DMSO) : I,50-1,75 : m : 4H ; 2,25-3,20 : m : 8H ; 4,05 : s : 4H ;
4,50-4,75 : mt : 2H ; 6,85-8,70 : m : 18H (16H aromatiques + 2H).
EXEMPLE 5 4
i
I, 2HC1 : Rt = ~ ~ ~ , R2 = ~ ~ ,
Ris - ~ _ ~ - ~ Rm.= _CH3~ NR4R5 = I , C r~ -
~s NH
Ce produit est préparé en 2 étapes selon le mode opératoire décrit pour
l' EXEMPLE 2 en utilisant les composés issus des Préparations 3.2 et 1.14.
MH+ : 638
RMN (DMSO + TFA) : 0,95 : sd : 3H ; 1,30-1,60 : m : 4H ; 2,40-3,40 : m
8H ; 4,00 : sd : 4H ; 4,75 : mt : 1H ; 6,65-8,15 : m : 16H.
EXEMPLE 55
i i
I, 2HC1 : R~ _ ~ ~ ~ , Ra. _ \ ~ ,
NR i
Ris - Rv = ~ - Ru -H~ bars = -N J ~ C ~ 6 - -C~~-(C~)2_~z
isomère (R,R)
I,30 g du produit obtenu selon la mode opératoire de l'EXEMPLE 43, étape A
sont agités pendant I8 heures à 4°C dans 45 ml de méthanol contenant 22
g d'HC1
gaz. Le milieu réactionnel est concentré sous vide, reévaporé 2 fois par MeOH
et le
résidu séché sous vide en présence de KOH. A ce produit dissous dans 250 ml de
méthanol on ajoute en 90 minutes 0,18 ml d'éthylènediamine diluée dans 15 ml
de
méthanol puis on agite encore 18 heures. Le milieu réactionnel est concentré
sous
vide à 10 ml, on ajoute une solution Et20/HCl pour amener à pH = 3 et
concentre
à sec. Le résidu est purifié par chromatographie de partition sur Séphadex~ G
25 en
utilisant le système de solvants nBuOH/iPrOH/H20 (4/0,2/5 ; v/v/v). On isole
d'abord le produit avec l'amidine cyclisée (410 mg) telle que décrite à
l'EXEMPLE
43 puis Ie produit avec l'amidine ouverte attendu (410 mg).
CA 02241788 1998-07-07
WO 97/25315 8.~ PCT/FR97/00026
a.D = + 37,6° (c = O,S ; DMF)
RMN (DMSO + TFA) : 1,50-1,70 : m : 4H ; 2,30-3,15 : m : lOH ; 3,70 : t
2H ; 4,SS : t : 1H ; 4,70 : t : 1H ; 6,90-8,10 : m : 16H.
S
EXEMPLE S6 Glule dose 10 mg
Compos de l'EXEMPLE 43 (poids exprim en quivalent
sous forme non salifie) 10,0 mg
Lactose monohydrat 200 mesh QS
Mthylhydroxypropylcellulose 6 mPa.s 3,0 mg
Carboxymthylcellulose sodique rticule 4,S mg
Starate de magnsium 1, S mg
Eau purifie pour granulation humide
Pour une glule "blanc-opaque" taille n 3 remplie
1S0 mg
1S EXEMPLE 57 Comprim nu scable dos SO mg
Compos de l'EXEMPLE 43 (poids exprim en quivalent
sous forme non salifie) 50,0 mg
Lactose monohydrat 200 mesh QS
Cellulose microcristalline SO ~m 27,0 mg
Mthyihydroxypropylcellulose 6 mPa.s 3,6 mg
Carboxymthylcellulose sodique rticule S,4 mg
Starate de magnsium 1,8 mg
Eau purifie pour granulation humide
Pour un comprim nu scable termin 180 mg
2S E~MPLE S8 Glule dose Img
Compos de l'EXEMPLE 43 (poids exprim en quivalent
sous forme non salifie) 1,0 m~;
Lactose monohydrat 200 mesh
Mthylhydroxypropylcellulose 6 mPa. s 3 , 0 m~;
Carboxymthylcellulose sodique rticule 4,S m~;
Starate de magnsium 1, S m~;
Eau purifie pour granulation humide
Pour une glule "blanc-opaque" taille n 3 remplie 100 mg.
3S