Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02243972 1998-07-21
WO 97/27195 - PCTlEP97/00229
- 1 - -
PROCESS FOR PREPARING PURINE DERTVATTVFS
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates to a process for
preparing a prodrug formulation of ganciclovir and its
pharmaceutically acceptable salts. More specifically, the
invention relates to a process for preparing the L-
monovaline ester derived from 2-(2-amino-1,6-dihydro-6-
oxo-purin-9-yl)-methoxy-1,3-propane-diol and its
pharmaceutically acceptable salts.
Background Information
British Patent 1523865 describes antiviral purine
derivatives with an acyclic chain in the 9-position. Among
those derivatives 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)-methoxy-ethanol with the INN name acyclovir has been
found to have good activity against herpes viruses such as
herpes simplex.
U.S. Patent 4,355,032 discloses the compound 9-[(2-
hydroxy-1-hydroxymethyl-ethoxy)-methyl]-guanine or 2-(2-
amino-I,6-dihydro-6-oxo-purin-9-yl)-methoxy-1,3-propane-
diol or 9-[(1,3-dihydroxy-2-propoxy)-methyl]-guanine
(DHPG) with the INN name ganciclovir. Ganciclovir is
highly efficacious against viruses of the herpes family,
for example, against herpes simplex and cytomegalovirus.
European Patent Application EP 0 375 329 discloses
prodrug compounds with the following formula
CA 02243972 1998-07-21
WO 97/27195 - PCqYEP97lO~D229
_ 2 _ ..
_ _
B~ _
O
~OR~
OR
wherein R and R1' are independently selected from a '
hydrogen atom and an amino acyl residue providing at least
one of R and R1 represents an amino acid acyl residue and
B represents a group of the formulae
H2(~ R2
O ~ HaN
in which R2 represents a C2_6 straight chain, C3-6
branched chain or C3_6 cyclic alkoxy group, or a hydroxy
or amino group or a hydrogen atom and the physiologically
acceptable salts thereof. These prodrug compounds are
described as having advantageous bioavailability when
administered the oral route, resulting in high levels of
the parent compound in the body.
Example 3 (b) European Patent Application EP 0 375
329 discloses the preparation of the bis(L-isoleucinate)
ester of ganciclovir as a white foam. Example 4 (b)
discloses the preparation of the bis(glycinate) ester of
ganciclovir as a white solid. Example 5 (b) discloses the
preparation of the bis (L-valinate) ester of ganciclovir
2o as a solid. Example 6 (b) discloses the preparation of the
bis(L-alaninate) ester of ganciclovir as a syrup
containing 90~ of the bis ester and 20~ of the monoester.
The bis-esters are prepared by reacting ganciclovir with
an optionally protected amino acid or functional
CA 02243972 2004-11-10
WU 9?/27195 ~ PCT/EP97/00229
- 3 -
equivalent thereof; the reaction may be carried out in a -
conventional manner, for example in a solvent such as
pyridine, dimethylformamide, etc., in the presence of a
coupling agent such as 1,3-dicyclohexylcarbodiimide,
optionally in the presence of a catalytic base such as 4-
dimethylaminopyridine. The described bis esters are non-
crystalline materials which are difficult to process for
the manufacture of oral pharmaceutical dosage forms.
British Fatent Application No. 8829571'is the
l0 priority patent application for European Patent
Application EP 0 375 329 and US Patent No. 5,043,339, and
discloses amino acid esters of the compounds of the
formula
R
N
'>
H2N N
O
OH
OH
(wherein R represents a hydroxy or amino group or -a
hydrogen atoms and the physiologically acceptable salts
thereof. Examples of preferred amino acids include
' aliphatic acids e.g. containing up to 6 carbon atoms such
as glycine, alanine, valine and isoleucine. The amino acid
esters include both mono and diesters. The preparation of
the diesters is identical to the preparation in European
Patent Application EP 0 375 329; however, this patent
application as well as
US Patent No. 5,043,339 do not disclose the
preparation of monoesters, or any data suggesting their
usefulness.
CA 02243972 1998-07-21
WO 97127195 - PCT/EP97IQ~229
_ ~ _
Leon Colla et. al., ~T. Med. Chem. (1983) 26, 602-604
disclose severa:L water-soluble ester derivatives of
acyclovir and their salts as prodrugs of acyclovir. The
authors indicate that acyclovir cannot be given as eye
drops or intramuscular injections because of its limited
solubility in water and have therefore synthesized ,
derivatives of acyclovir which are more water soluble than
the parent compound. The authors disclose the
hydrochloride salt of the glycyl ester, the hydrochloride
salt of the alanyl ester, the hydrochloride salt of the (3-
alanyl ester, the sodium salt of the succinyl ester, and
the azidoacetate ester. The alanyl esters were prepared
by conventional esterification methods, including reacting
acyclovir with the corresponding N-carboxy-protected amino
acid in pyridine, in the presence of 1,3-dicyclohexyl-
carbodiimide and a catalytic amount of p-toluenesulfonic
acid and subsequently catalytic hydrogenation to give the
alpha- and beta--alanyl esters as their hydrochloride
salts.
L. M. Beauchamp et. al., Antiviral Chemistry &
Chemotherapy (1992), 3 (3), 157-164 disclose eighteen
amino acid esters of the antiherpetic drug acyclovir and
their effectiveness as prodrugs of acyclovir, evaluated in
rats by measuring the urinary recovery of acyclovir. Ten
prodrugs produced reater amounts of the
g parent drug in
the urine than acyclovir itself: the glycyl, D,L-alanyl,
L-alanyl, L-2-aatinobutyrate, D,L-valyl, L-valyl, DL-
isoleucyl, L-isaleucyl, L-methionyl, and L-prolyl ester.
According to the authors the L-valyl ester of acyclovir
was the best pradrug of the esters investigated. These
esters were prepared by methods similar to those employed
by Colla et. al.
European Patent Publication 308 065 discloses the
valine and isoleucine esters of acyclovir, preferably in
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/00229
_ 5 _
_ _
the L-form, as showing a large increase in absorption from
the gut after oral administration, when compared with
other esters and acyclovir. The amino acid esters are
prepared by conventional esterification methods, including
reacting acyclovir with an N-carboxy-protected amino acid
or an acid halide or acid anhydride of the amino acid, in
a solvent such as pyridine or dimethylformamide,
optionally in the presence of a catalytic base.
PCT Patent Application WO 94/29311 discloses a
process for the preparation of amino acid esters of a
nucleoside analogue, including acyclovir and ganciclovir.
This process comprises reacting a nucleoside analogue
having an esterifiable hydroxy group in its linear or
cyclic ether moiety, with a 2-oxa-4-aza-cycloalkane-1,3-
1S dione of the formula
O O
O
R2/N
R~
wherein R1 may represent hydrogen, Cl_4 alkyl or alkenyl
group or other amino acid side chains, and R2 may
_ represent hydrogen or a group COORS where R3 is a benzyl,
t-butyl, fluorenylmethyl or an optionally halo substituted
linear or branched C1-g alkyl group. Preferred RI groups
include hydrogen, methyl, iso-propyl and isobutyl,
yielding respectively the glycine, alanine, valine and
_ _ isoleucine esters of acyclovir or ganciclovir. Examples
1-3 of PCT Patent Application WO 94/29311 discloses only
the condensation of acyclovir with the valine-substituted
2-oxa-4-aza-cycloalkane-I,3-dione (Z-valine-N-carboxy-
anhydride) by conventional procedures. While the amino
acid esters of the PCT application include both the
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/Q0229
_ 6 _
_ _
acyclovir and ganciclovir esters, the application does not
disclose how to prepare the ganciclovir esters, much less
the mono-esters of ganciclovir.
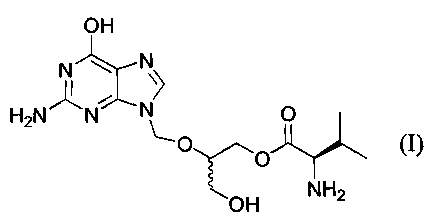
The L-monovaline ester derived from 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-yl}-methoxy-1,3-propane-diol and its
pharmaceutically acceptable salts are potent antiviral
agents and are described in European Patent Application EP
694 547 A. These compounds have been found to have
improved oral absorption and low toxicity. This patent
application also discloses certain processes for preparing
these esters, different from those described herein.
The present invention relates to an improved process
whereby ganciclovir is esterified with an L-valine
derivative to provide a di-valine ganciclovir
intermediate. Removal of one of the valine groups with a
lower alkyl amine, benzylamine or benzyl methylamine
provides the mono-valine ester compound of Formula I.
SUMMARY OF THE INVENTION
In a first aspect, this invention provides a process
for preparing the compound of the formula I:
OH
_ ,,, t N
HaN N~ ~ O
O
~O
O H H2N
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/Q0229
- 7 _ ..
and pharmaceutically acceptable salts thereof, which -
compound is named hereinafter 2-(2-amino-1,6-dihydro-6-
oxo-purin-9-yl)-methoxy-3-hydroxy-1-propyl-L-valinate or
mono-L-valine ganciclovir.
This process involves the di-esterification of
ganciclovir by an L-valine derivative, followed by removal
of one of the valine groups with a lower alkyl amine,
benzylamine or benzyl methylamine, and removal of any
protecting groups, to yield the prodrug of Formula I.
Optionally, the process can also include the formation of
salts of the prodrug of Formula I, the conversion of an
acid addition salt of the prodrug of Formula I into a non-
salt form, the optical resolution of a prodrug of Formula
I or the preparation of the prodrugs of Formula I in
crystalline form. Details of the process are described
below.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
Unless otherwise stated, the following terms used in
the specification and claims have the meanings given
below:
"Alkyl" means a straight or branched saturated
hydrocarbon radical having from one to the number of
carbon atoms designated. For example, C1_~ alkyl is alkyl
having at least one but no more than seven carbon atoms,
e.g. methyl, ethyl, i-propyl, n-propyl, n-butyl, n-pentyl,
n-heptyl and the like.
"Lower alkyl" means an alkyl of one to six carbon
atoms.
CA 02243972 1998-07-21
WO 97/2X95 - PG'TIEP97/00229
g _ ..
_ _ _
"Aryl" means an organic radical derived from an
aromatic hydrocarbon by the removal of one hydrogen atom.
Preferred aryl radicals are aromatic carbocyclic radicals
having a single ring (e. g., phenyl) or two condensed rings
(e. g., naphthyl).
"Aralkyl" means an alkyl groups in which a hydrogen
atom is replaced by an above-defined aryl group.
"Acyl" means an organic radical derived from an
organic acid by the removal of the hydroxyl group; e.g.,
20 CH3C0- or acety:L is the acyl radical of CH3COOH. Other
examples for such acyl groups are propionyl, or benzoyl,
etc. The term "acyl" includes the term "alkanoyl" which is
the organic radical RCO- in which R is an alkyl group as
defined above.
3.~ "Lower alkoxy", "(lower alkyl)amino", "di(lower
alkyl)amino", "(Ioweralkanoyl)amino", and similar terms
mean alkoxy, alkylamino, dialkylamino, alkanoylamino, etc.
in which the or each alkyl radical is a "lower alkyl" as
described above.
20 "Halogen" or "halo" means fluorine, chlorine,
'bromine, or iodine.
- "Lower alkyl amine" means a straight or branched
organic radical R1N(R2)~ wherein R1 is lower alkyl and R2
is hydrogen or lower alkyl, and lower alkyl is as defined
25 above .
_- "Derivative" of a compound means a compound
obtainable from the original compound by a simple chemical
process.
"Activated derivative" of a compound means a reactive
30 form of the original compound which renders the compound
active in a desired chemical reaction, in which the
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/Q0229
_ g _ -,
original compound is only moderately reactive or non- _
reactive. Activation is achieved by formation of a
derivative or a chemical grouping within the molecule with
a higher free energy content than that of the original
compound, which renders the activated form more
susceptible to react with another reagent. In the context
of the present invention activation of the carboxy group
is of particular importance and corresponding activating
agents or groupings which activate the carboxy group are
described in more detail below. An example of an activated
derivative of L-valine is the compound of Formula II:
A
P2HN
H
O
wherein P2 is an amino-protecting group and A is a
carboxy-activating group, for example, halo, a lower
i5 acyloxy group, a carbodiimide group, such as 1-ethyl-3-(3-
dimethylaminopropyl)-carbodiimide (EDAC), an isobutyrate
group, and the like.
Of particular interest for the present invention is
- an amino acid anhydride which is an activated form of an
2o amino acid which renders the amino acid (especially L-
valine) susceptible to esterification. Amino acid
anhydrides are included in the compounds of Formula II,
above. Especially useful for the present invention are
- - the cyclic amino acid anhydrides of L-valine, described in
25 PCT Patent Application WO 94/29311, such as 2-oxa-4-aza-5
isopropyl-cycloalkane-1,3-dione of formula IIa:
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/0~229
- IO - ..
o
0
P2/N
a
in which P2 is an amino protecting group. Other examples
of the cyclic amino acid anhydrides are protected amino
acid N-carboxy anhydrides (NCAs) described in more detail
below.
"Protecting group" means a chemical group that (a)
preserves a reactive group from participating in an
undesirable chemical reaction; and (b) can be easily
removed after p~:otection of the reactive group is no
longer required. For example, the benzyl group is a
protecting group for a primary hydroxyl function.
"Amino-protecting group" means a protecting group
that preserves a reactive amino group that otherwise would
be modified by certain chemical reactions. The definition
includes the formyl group or lower alkanoyl groups with 2
to 4 carbon atoms, in particular the acetyl or propionyl
group, the trityl or substituted trityl groups, such as
the monomethoxyt:rityl group, dimethoxytrityl groups such
as the 4,4'-dimethoxytrityl or 4,4'-dimethoxytriphenyl-
methyl group, the trifluoroacetyl group, and the N-(9-
fluorenylmethox~~carbonyl) or "FMOC" group, the
allyloxycarbonyl. group or other protecting groups derived
from halocarbonates such as (C6-C22)aryl lower alkyl
carbonates (such. as the N-benzyloxycarbonyl group derived
~5 from benzylchlorocarbonate), or derived from biphenylalkyl
halo carbonates, or tertiary alkyl halo carbonates, such
as tertiary but~~lhalocarbonates, in particular tertiary
butylchloro-carx>onate, or di(lower)alkyldicarbonates, in
CA 02243972 1998-07-21
WO 97/27195 - PCTIEP97/OQ229
- 21 - -.
particular di(t-butyl)-Bicarbonate, the phthalyl group and
the triphenylmethyl halides such as triphenylmethyl
chloride, and trifluoroacetic anhydride.
' "Leaving group" means a labile group that is replaced
in a chemical reaction by another group. Examples of
- leaving groups are halogen, the optionally substituted
benzyloxy group, the isopropyloxy group, the mesyloxy
group, the tosyloxy group or the acyloxy group.
All the activating and protecting agents employed in
the preparation of the compound of Formula I must meet the
following qualifications: {1) their introduction should
proceed quantitatively and without racemization of the L-
valine component; (2) the protecting group present during
the desired reaction should be stable to the reaction
conditions to be employed; and (3) the group must be
readily removed under conditions in which the ester bond
is stable and under which racemization of the L-valine
component of the ester does not occur.
The process of the invention may also include the
optical. resolution of a prodrug of Formula I. Terminology
relating to the stereochemistry and optical resolution of
these compounds is described in European Patent
Application EP 694 547 A.
"Optional" or "optionally" means that a described
event or circumstance may or may not occur, and that the
description includes instances where said event or
circumstance occurs and instances in which it does not.
' - For example, "optionally substituted phenyl" means that
the phenyl may or may not be substituted and that the
description includes both unsubstituted phenyl and phenyl
wherein there is substitution; "optionally followed by
converting the free base to the acid addition salt" means
that said conversion may or may not be carried out in
CA 02243972 1998-07-21
WO 97!27195 - pC~'/Ep97/00229
- 12 _ ..
order for the process described to fall within the _
invention, and the invention includes those processes
wherein the free base is converted to the acid addition
salt and those processes in which it is not.
"Pharmaceutically acceptable" means that which is
useful in preparing a pharmaceutical composition that is -
generally safe and non-toxic and includes that which is
acceptable for veterinary use as well as human
pharmaceutical use.
1o "Pharmaceutically acceptable saltsn means salts which
possess the desired pharmacological activity and which are
neither biologically nor otherwise undesirable. Such
salts include acid addition salts formed with inorganic
acids such as hydrochloric acid, hydrobromic acid,
sulfuric acid, nitric acid, phosphoric acid, and the like;
or with organic acids such as acetic acid, propionic acid,
hexanoic acid, heptanoic acid, cyclopentane-propionic
acid, glycolic acid, pyruvic acid, lactic acid, malonic
acid, succinic acid, malic acid, malefic acid, fumaric
2o acid, tartaric acid, citric acid, benzoic acid,
o-(4-hydroxy-ben.zoyl)-benzoic acid,~cinnamic acid,
mandelic acid, methanesulfonic acid, ethanesulfonic acid,
1,2-ethane-disulfonic acid, 2-hydroxyethane-sulfonic acid,
benzenesulfonic acid, p-~chlorobenzenesulfonic acid,
2-naphthalenesulfonic acid, p-toluenesulfonic acid,
camphorsulfonic acid, 4-methyl-bicyclo[2.2.2]oct-2-ene-1-
carboxylic acid, gluco-heptonic acid, 4,4'-methylenebis(3-
hydroxy-2-naphthoic) acid, 3-phenylpropionic acid,
- - - trimethyl-acetic acid, tertiary butylacetic acid, lauryl
3o sulfuric acid, gluconic acid, glutamic acid, hydroxy-
naphthoic acids, salicylic acid, stearic acid, muconic
acid, and the like. Preferred pharmaceutically acceptable
salts are those formed with hydrochloric, sulfuric,
phosphoric acid, acetic or methanesulfonic acid, ethane-
CA 02243972 1998-07-21
WO 97/27195 ' PCT/EP97/OD229
- 13 - -
_ _
sulfonic acid, 1,2-ethanedisulfonic acid, 2-hydroxyethane-
sulfonic acid, benzene-sulfonic acid, p-chlorobenzene-
sulfonic acid, 2-naphthalenesulfonic acid, p-toluene-
sulfonic acid, and camphorsulfonic acid.
Synthetic Reaction Parameters
Unless specified to the contrary, the reactions
described herein take place at atmospheric pressure within
a temperature range from 5°C to 170°C (preferably from
10°C
to 50°C; most preferably at "room" or "ambient"
temperature, e.g., 20° - 30°C). However, there are clearly
some reactions where the temperature range used in the
chemical reaction will be above or below these
temperature ranges. Further, unless otherwise specified,
the reaction times and conditions are intended to be
approximate, e.g., taking place at about atmospheric
pressure within a temperature range of about 5°C to about
100°C (preferably from about 10°C to about 50°C; most
preferably about 20°C) over a period of about 1 to about
100 hours (preferably about 5 to 60 hours). Parameters
given in the Examples are intended to be specific, not
approximate.
Isolation and purification of the compounds and
_ intermediates described herein can be effected, if
desired, by any suitable separation or purification
procedure such as, for example, filtration, extraction,
crystallization, column chromatography, thin-layer
chromatography or thick-layer chromatography, or a
_ _ combination of these procedures. Specific illustrations
of suitable separation and isolation procedures can be had
by reference to the examples hereinbelow. However, other
~ equivalent separation or isolation procedures can, of
course, also be used.
CA 02243972 1998-07-21
WO 97127195 - PCTlEP97I0~229
- 14 - -.
1 . _
Presently Preferred Embodiments _
While the :broadest definition of this invention is
set forth in the Summary of the Invention as a process for
preparing the compound of Formula I and its pharma-
ceutically accej~table salts, the (R, S) mixture and certain
salts are preferred.
The following acids are preferred to form pharma-
ceutically acceptable salts with the compound of Formula.
I: hydrochloric, sulfuric, phosphoric acid, acetic,
methanesulfonic, ethanesulfonic, 1,2-ethanedisulfonic,
2-hydroxyethanesulfonic, benzenesulfonic, p-chlorobenzene-
sulfonic, 2-naphthalenesulfonic, p-toluenesulfonic and
camphorsulfonic acid. Most preferred are strong inorganic
acids, such as hydrochloric, sulfuric or phosphoric acid.
I5 The most preferred compounds are 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-y1)-methoxy-3-hydroxy-1-propyl L-
valinate hydrochloride and acetate. These compounds can be
prepared as crystalline materials and therefore can be
easily manufactured into stable oral formulations.
2o In any of the processes described herein, a reference
to Formula I, II, III or IV refers to such Formulae
wherein P1, P2, and A are as defined in their broadest
- definitions set forth in the Summary of the Invention,
with the processes applying particularly to the presently
25 preferred embodiments.
Details of the Synthetic Processes
The process of the present invention is depicted in
the Reaction Sequence shown below:
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/0p229
- 15 -
_ _
OH OH -
N ~ II orIIa N ~ N
N
i i
P1HN ~ P~HN N ~ O
O
OH ~ O
. OH P2HN
NHP2
O
OH OH
H2N N N P~HN H N O
~O ~O
(n ~ O ~ ~ O
OH P2HN
OH H2N
wherein P1 is hydrogen or an amino protecting group, and
P2 is an amino protecting group.
2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-methoxy-1,3-
propanediol (ganciclovir) is esterified with an L-valine
derivative of Formula II or IIa to farm a di-valine ester
of ganciclovir (Formula III). Removal of one of the valine
groups with a lower alkyl amine, benzylamine or benzyl
methylamine, followed by removal of any protecting groups,
affords the compound of Formula I.
Compounds of Formula I can optionally be converted
into a pharmaceutically acceptable salt thereof. The
process can also include the conversion of an acid
addition salt of the prodrug of Formula I into a non-salt
form, the optical resolution of a compound of Formula I or
CA 02243972 1998-07-21
WO 9?/2?19S - PCT/EP97/Q0229
..
the preparation of the compound of Formula I in _
crystalline form.
The process for producing the compound of Formula I
may or may not involve protection of the amino group in
the 2-position of the guanine base (see the detailed
description below of Steps I through III for the case in
which the process is carried out without a protected amino
group). For the: case when the ganciclovir starting
material does have a protected 2-amino group, the
protecting group may be removed by conventional
procedures, well.-known in the art. For example, if the
amino-protecting' group is a lower alkanoyl group basic
conditions (pH between 8 to 11) are employed to remove the
protecting group. For example, 2-N-acetyl-ganciclovir is
treated with an alkaline reagent such as ammonium
hydroxide, sodium or potassium carbonate or sodium or
potassium hydroxide until the removal of the acetyl group
is complete. In general, this reaction will be conducted
in the presence of a suitable solvent such as a lower
2o alkanol. Preferably the starting material is dissolved in
methanol and a stoichiometric excess of ammonium hydroxide
is added. The reaction temperature is kept between 0 to
50°C, preferably at room temperature. After the reaction
is complete (which can be determined by TLC), another
solvent may be added to facilitate isolation of the de-
protected product, such as ethyl ether which leads to
precipitation of the de-acylated product which can be
filtered off and isolated using conventional separation
methods.
Starting Materials
All starting materials employed to make the compound
of Formula I are known, such as ganciclovir, and the
protecting and carboxylic-group- activating reagents.
CA 02243972 1998-07-21
WO 97/2?195 ' PCT/EP97/00229
_ 2~ -
Prior to carrying out Step I (esterification step),_
the amino group of the L-valine derivative must be
protected to avoid its interference with the
esterification by undesirable amide formation. The
various amino-protected L-valine derivatives useful in
this invention, such as N-benzyloxycarbonyl-L-valine, BOC-
L-valine~and FMOC-L-valine, N-formyl-L-valine and N-
benzyloxycarbonyl-N-carboxy-L-valine anhydride, are all
commercially available (SNPE Inc., Princeton, NJ, Aldrich
Chemical Co., Milwaukee, WI and Sigma Chemical Co., St.
Louis, MO), or are described in the literature, such as N-
allyloxycarbonyl-L-valine. Cyclic amino-protected L-
valine derivatives are also described in the literature,
as noted above. Of particular interest for the present
invention is the benzyloxycarbonyl valine-substituted 2-
oxa-4-aza-cycloalkane-1,3-dione (Z-valine-N-carboxy-
anhydride, or Z-Valine-NCA), which is also commercially
available (SNPE Inc., Princeton, NJ). Alternatively, the
protecting step may be carried out by conventional
methods.
A preferred ganciclovir starting material for the
preparation of the compound of the invention is the
unprotected ganciclovir (2-(2-amino-1,6-dihydro-6-oxo-
purin-9-yl)methoxy-1,3-propanediol) which is described in
US Patent No. 4,355,032. Other ganciclovir starting
materials may have protection at the 2-amino group, such
as 2-(2-acyl-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-
2,3-propandiol.
- Preparation of Activated derivative of L-valine:
Prior to carrying out Step I (esterification step),
L-valine must also be activated. At Least 1 equivalent of
the protected amino acid and 1 equivalent of a suitable
coupling agent or dehydrating agent, for example 1,3-
CA 02243972 2004-11-10
WO 97127195 PCT/EP97/00229
- 18 -
dicyclohexylcarbodiimide or salts of such diimides with -
basic groups should be employed from the start. Other
carbodiimides such as N,N'-carbonyldiimidazole may also be
used. Further useful dehydrating agents are trifluoro-
acetic anhydride, mixed anhydrides, acid chlorides, 1-
benzo-triazolyloxy-tris(dimethylamino)phosphonium
hexafluorophosphate, benzotriazole-1-yl-oxy-trispyrroli-
dinophosphonium hexafluorophosphate, 1-hydroxybenzo-
triazole, 1-hydroxy-4-azabenzotriazole, 1-hydroxy-7-
1o azabenzotriazole, N-ethyl-N'-(3-(dimethylamino)-
propyl)carbodiimide hydrochloride, 3-hydroxy-3,4-dihydro-
4-oxo-1,2,3-benzotriazine, O-(benzotriazol-1-yl)-1,1,3,3-
tetramethyluronium hexafluorophosphate, 0-(7-azabenzo-
triazol-1-yl)-1,I,3,3-tetramethyluronium hexafluoro
phosphate, O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetra
methyluronium tetrafluoroborate, 0-(1H-benzotriazol-I-yl)
1,1,3,3-bis(tetramethylene)uronium hexafluoro- phosphate
or 4-(7-azabenzotriazol-1-yl)-1,1,3,3-bis(tetramethylene)-
uronium hexafluorophosphate. A description of these
coupling agents by L. A. Carpino can be found in J. Am.
Chem. Soc. 1993, 115, p. 4397-4398.
Also useful for this purpose are urethane-protected
amino acid N-carboxy anhydrides (UNCA's) which are an
activated form of an amino acid; these have been described
by William D. Fuller et al., J. Am. Chem. Soc. 1990, I12,
7414-7416.
Other protected aminb acid N-carboxy anhydrides are
described in PCT Patent Application WO 94/29311 discussed
above. In summary, any other reagent that produces an
3o anhydride or another activated derivative of the protected
amino acid under mild conditions can be used as the
coupling agent. . '
The amino-protected amino acid is dissolved in an
inert solvent'such as a.halogenated lower alkane,
CA 02243972 1998-07-21
WO 97127195 ' PCT/EP97I00229
- -
preferably dichloromethane under an inert atmosphere, for
example nitrogen, and the coupling agent is added
(preferably 1,3-dicyclohexylcarbodiimide). The reaction
mixture is stirred at temperatures between O and 50°C
preferably at about room temperature. The reaction mixture
is filtered and the reaction product (the anhydride of the
protected amino acid) isolated. The resulting product is
dissolved in a dry inert solvent such as dry
dimethylformamide and placed under nitrogen.
Preparation of Mono-L-valine Ganciclovir
Step I:
Ganciclovir with an optionally protected 2-amino
group is esterified with an L-valine derivative of Formula
II to give the di-valine ester of ganciclovir as an
intermediate (Formula III). Suitable amino-protecting
groups are lower alkanoyl groups with 2 to 4 carbon atoms,
in particular the acetyl or propionyl group. Other
suitable amino-protecting groups are the trityl or
substituted trityl groups, such as the monomethoxytrityl
group, and the 4,4'-dimetho
xytrityl group.
Suitable amino-protecting groups for the L-valine
derivative are the N-benzyloxycarbonyl group, the phthalyl
group, the tertiary butyloxycarbonyl group and the N-(9-
fluorenylmethoxycarbonyl) or "FMOC" group.
The di-valine ester of ganciclovir can be prepared by
conventional procedures, such as those described in
- European Patent 0 375 329.
Another example of a conventional procedure for
preparing the di-valine ester is as follows. A suspension
of ganciclovir is reacted with a solution containing
approximately equivalent amount of the L-valine
CA 02243972 1998-07-21
WO 97/27195 - 1'CT/EP97/OD229
- 20 _ ..
derivative, preferably Na-Boc-Valine-NCA, and an organic
base, such as triethylamine (TEA) at 10°-50°C, preferably
at ambient temperature for 10-90 hours, preferably about
24 hours. The reaction mixture is diluted, filtered,
washed and dried under vacuum.
Step II:
The conversion of the di-valine ester of ganciclovir
to the mono-(L-~;ralinate)-ganciclovir is effected by
partial hydrolysis of one of the L-valine ester moieties
with a lower alkyl amine, benzylamine or benzyl
methylamine, preferably with n-propylamine in a nonpolar
aprotic solvent such as hexane. This results in cleavage
of one of the amino acyi residues.
For example the di-valine ester of ganciclovir is
3.5 treated with n-propylamine in a nonpoiar aprotic solvent,
preferably hexa~ze, and stirred at 10°-50oC, preferably at
ambient temperai:ure, for 1 hour to 10 days, preferably
from 1 to 7 days. The reaction mixture is evaporated
under vacuum and analyzed by HPLC.
Step III:
The amino ~?rotecting groups) of the product of Step
II is removed by a de-protection reaction, preferably in
an acidic medium or solvent, most preferably by
hydrogenolysis. De-protection under acidic conditions is
preferred, as this will ensure that the amino group
liberated in the de-protection reaction will be
protonated; that is, that the base of Formula I as it is
formed in the de-protection reaction will be captured by
an at least stoichiometric amount of acid present.
Isolating the compound of Formula I as an acid addition
salt will protect the desired stereoconfiguration of the
CA 02243972 1998-07-21
WO 97/27195 - PCTlEP97/00229
- 21 -
compound of Formula I. Therefore, those examples given _
below that show the de-protection step also show the
concomitant salt formation step.
= The de-protection reaction is carried by dissolving
the product of the previous step in an inert solvent,
preferably in an acidic solvent, using a hydrogenation
catalyst, such as platinum, or palladium hydroxide on
carbon or palladium on carbon, using elevated hydrogen
pressure between 1 and 2000 psi (0.07-240 atm), preferably
20 50 to 200 psi (3.5-14 atm), most preferably 5 to 20 psi
(0.35-1.4 atm). The completion of the reaction can be
monitored using conventional TLC analysis. The
hydrogenoiysis is continued until the conversion is
complete, if required with addition of further
hydrogenation catalyst. The catalyst is removed and
washed. The combined filtrates from filtration and the
washings are concentrated and lyophilized to isolate
ganciclovir L-valine ester. The purification of the
product and the isolation of a crystalline ester is
carried out by recrystallization or other purification
techniques, such as liquid chromatographic techniques.
If present, any protecting group at the 2-amino group
of the guanine group may be removed by conventional
- procedures, as described above.
If the tertiary butyloxycarbonyl group is being used
as amino-protecting group, its removal is effected with
acid, such as HC1 and isopropanol as a solvent or with
trifluoroacetic acid neat.
Alternatively if the.esterification step has been
carried out with a trityl or substituted trityl-protected
' ganciclovir derivative such protecting groups can be
removed by treatment with an aqueous alkanoic acid or
' trifluoroacetic or hydrochloric acid at temperatures
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/0_0229
22 -
between -20°C and 100°C, for example, aqueous acetic acid.
Preparation of Salts
One of ordinary skill in the art will also recognize =
that the compound of Formula I may be prepared as an acid
addition salt or as the corresponding free base. If
prepared as an acid addition salt, the compound can be
converted to the free base by treatment with a suitable
base such as amraonium hydroxide solution, sodium
hydroxide, potassium hydroxide or the like. However, it is
to important to point out that the free base of Formula I is
more difficult to characterize than its acid addition
salts. When converting the free base to an acid addition
salt, the compound is reacted with a suitable organic or
inorganic acid (described earlier). These reactions are
effected by treatment with an at least stoichiometric
amount of an appropriate acid (in case of the preparation
of an acid addition salt) or base (in case of liberation
of the free compound of Formula I). In the salt-forming
step of this invention, typically the free base is
2o dissolved in a polar solvent such as water or a lower
alkanol (preferably isopropanol) and mixtures thereof and
the acid is added in the required amount in water or in
lower alkanol. ~'he reaction temperature is usually kept at
about a° to 50°C, preferably at about room temperature.
The corresponding salt precipitates spontaneously or can
be brought out of the solution by the addition of a less
polar solvent, removal of the solvent by evaporation or in
a vacuum, or by cooling the solution.
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97J00229
- 23 - ..
Isolation of Stereoisomers and the Manufacture of
Crystalline 2-(2-Amino-1,6-dihydro-6-oxo-purin-9-
yl)methoxy-3-hydroxy-1-propyl-L-valinate
Y
From the Formula (I) it is apparent that the compound
of the invention has one asymmetric carbon atom (chiral
center) in the propanyl chain, in addition to the
asymmetric carbon atom in L-valine. Therefore, two
diastereomeric forms exist, the (R)- and (S)- form as
determined by the rules of Cahn et al. Suitable methods
for the separation of the diastereomers are described in
European Patent Application EP 694 547 A.
The compounds of Formula (I) may also be prepared in
crystalline form, which has many well-known advantages
over the non-crystalline form. Suitable methods for the
1.5 preparation of the compounds of the invention in
crystalline form are also described in European Patent
Application EP 694 547 A.
The following preparations and examples are given to
enable those skilled in the art to more clearly understand
2o and to practice the present invention. They should not be
considered as limiting the scope of the invention, but
merely as being illustrative and representative thereof.
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
25 methoxy-1,3-propanediyl-bis-[N-(butyloxycarbonyl)-L-
valinate] and 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-1,3-propanediyl-bis-[N-(benzyloxycarbonyl)-L-
valinate]
'' 1A. Preparation of O,O,bis Na-Boc-Valine-ganciclovir
30 To a suspension of ganciclovir (4 mg) in
CA 02243972 1998-07-21
WO 97/27195 - PCT/EP97/00229
- 24 - -
dimethylformamide (25 ml) was added triethylamine (2.32_
ml) and Na-Boc--valine-NCA (11 mg) and stirred at room
temperature overnight. The reaction mixture was diluted
with water, filtered, washed with water and dried ,
overnight. The solid was dissolved in ethyl acetate and
washed with a sodium bicarbonate solution (5~), washed
with water, dried over sodium sulfate, filtered and
evaporated. The residue was dissolved in acetone and
precipitated by addition over petroleum ether and the
solid filtered and dried under vacuum overnight to give
10 mg of the product.
1B. Preparation of O,O,bis Na-Z-Valine-ganciclovir
To a suspension of ganciclovir (5 mg) in
dimethylformamide (30 ml) was added triethylamine (2.5 ml)
and the Na-Z-valine-NCA (7.5 mg), and half hour later
another portion. (7.5 mg). After 3 hours from the initial
addition, water was added and the reaction mixture was
extracted with ethyl acetate . The organic phase was
washed successively with sodium bisulfate solution (5~s),
2o water, sodium bicarbonate solution (5~s), water, and brine,
and then dried over sodium sulfate, filtered and
evaporated. The residue was dissolved in toluene and
- precipitated by hexane addition. The solid was filtered
washed with hexane, and dried under vacuum to give 15 mg
of product.
CA 02243972 1998-07-21
WO 97/27195 - PCTlEP97/0~229
- 25 - ..
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-hydroxy-1-propyl-N-(butyloxycarbonyl)-L-valinate
' and 2-(2-amino-2,6-dihydro-6-oxo-purin-9-yl}-methoxy-3-
hydroxy-1-propyl-N-(benzyloxycarbonyl)-L-valinate
2A. Preparation of O-(Na-Boc-Valine)-ganciclovir
O,O,bis Na-Boc-Vaiine ganciclovir {50 mg) in hexane
(10 ml) and n-propylamine (1 ml) was stirred at room
temperature for 7 days. The reaction mixture was
evaporated under vacuum and the product analyzed by HPLC
as a mixture of 11~ ganciclovir, 74~ O-(Na-BOC-valine)-
ganciciovir, and 155 bis-valine ganciclovir.
2B. Preparation of O-(Na-Z-Valine)-ganciclovir
O,O,bis Na-Z-Valine ganciclovir{2 mg) in n-
propylamine (20 ml) and hexanes (20 ml) was stirred at
room temperature; after 7 hours, 5 ml of hexanes were
added and the reaction mixture stirred for another 24
hours, evaporated. HPLC showed a mixture of 31~
ganciclovir, 57.5 O-(Na-Z-valine)-ganciclovir and 11.5
2o bis-valine ganciclovir.
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)methoxy-3-hydroxy-Z-propyl-L-valinate
A vigorously stirred suspension of Pd(OH)2/C (670 g)
in CH30H (23 1) was treated with H2 gas (7 psi [0.49 atm])
for 12 hours. To this suspension was added a solution O-
(Na-Z-Valine)-ganciclovir (6.7 kg, 23.7 moles) in CH30H
(34 1) containing concentrated HC1 (1.64 kg). The H2
atmosphere was maintained at 7 psi [0.49 atm], and
CA 02243972 2004-11-10
WOr97127195 ~ PCT/EP97/00229
- 26 -
replaced at 20 minute intervals. After 2.75 hours, the H2 -
atmosphere was replaced with nitrogen. The catalyst was
removed by filtration through Solka Floc The filtrate
was concentrated in vacuo to approximately 13 1, at which ,
time H20 (4 1) was added. The volume of the filtrate was
again reduced to approximately 13 L. The temperature of
the mixture was adjusted to approximately 38°C and
isopropyl alcohol (24 1) was slowly added. After
crystallization had occurred, the mixture was cooled to
21°C over a period of 2 hours. Additional. isopropyl
alcohol (24 1) was added and the mixture was stirred for
16 hours at 5°C. The solid was then collected by
filtration. The filtercake was washed with cold isopropyl ,
alcohol (19 1).and dried under a stream.of nitrogen for 3
days. The solid was placed in a vacuum oven (55oC,
nitrogen bleed, 25 ins vacuum). Isopropyl alcohol was
found to be 0.4~ after 24 hours. Weight of solid:'
4.35 kg. Purity: (HPLC) 98.6: MS: 355 (MH)+'
* Trade-mark