Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02252706 1998-10-27
WO 97/40854 PCT/US97tO7335
POLYPEPllDE CONlUGATES FOR TRANSPORnNG SUBSTANCES ACROSS CELL MEM-
8RANES
Field of the Invention
The invention relates to ~e use of polymeric compositions effective for delivering
compounds in living or~anis".s. The compositions exhibit solubility in both hydrophilic and
lipophilic envi-o,~"ents by undergoing a reversible pH-dependent transition from a low-pH,
lipophilic form to a high-pH, hydrophilic form.
10 References
Anderson, R.G.W. et al., Science 255:410411 (1992).
Atherton, Cameron, and Sheppard, Tetrahedron 44:843 (1988).
Aungst et al., Int. J. Pharmaceut. 33:225 (1986).
Blout, E.R. et al., J. Am. Chem. Soc. 78:497
Chen and Tai, Res. Comm. Molec. Path. & Pharm. 88:317 (1995).
Chopra, I., Parasitology_:25-44 (1988).
Clague, M.J. et al., J. Biol. Chem. 269:21-24 (1994).
Dawson et al., Science 226:776 (1994).
Deshpande, D. et al., Pharm. Res. 13(1):57 61 (1996).
Dixon, J.S., Scand. J. Gastroenterol. Suppl. 212:48-62 (1995).
Doty et al., J. Polymer Sci. 23:851 (1957).
Eberle and Nuninger, J. Org. Chem. 57:2689 (1992).
Fuchs, R. et al., J. Biol. Chem. 264:2212-2220 (1989).
Glupczynski, Y. et al., Am J. Gastroentrol. 85:1545-51 (1990).
Golden et al., J. Invest. Dermatol. 86 255 (1986).
Goodman and Barry, J. Pharm. Pharmacol. 37:80P (1985).
Goodman and Barry, J. Pharm. Pharmacol. 38:71P (1986). Gueritte-Vogelein et al., J.
Med. Chem. 34:992 (1991).
~n~b~sa et al., J. Polym. Sci. Polym. Lett. Ed. 22:559-564 (1984).
Hoes et al., Makromol. Chem. Suppl. 2: 175 (1985).
Tg~-i.chi, K. et al., Caries Res. 24(1):52-58 (1990).
K~me~ni et al, Chem Pharm Bull. 30:4545 (1982).
Kennedy, K.A. et al., Biochem. Pharm. _2:1-8 (1980).
Korting, H.C et al., Clin. Investig. 71(8):644-648 (1993).
Leo, Hansch, and Elkins, Chemical Reviews 71:525 (1971).
Ludwig, LeBorgue, and Hoflack, Trends in Cell Biol. 5:202 (1995).
CA 022=,2706 1998-10-27
W O 97/40854 PCTAJS97/07335
-2-
Mathew et al., J. Med. Chem. 35:I45-151(1992).
~Iathias, C.J. etal., J. Nucl. Med. 37(6):1003-1008(1996).
Nukui et al., MokTomol. Chem 192:2925(1991).
O'Connor, H.J., Eur. J. Gastroenterol. Hepatol. 6 (Suppl. 1):113-9(1994).
S Rapaport, E. et al., Proc. Natl. Acod. Sci USA, 93(2):709-713(1996).
Rehfeld and Elias, J. Invest. Dermatol. 79:1(1982).
Summerton, J.E. and Weller, D.D., U.S. Patent No. 5,185,444(1993).
Tannock, I.F. et al., Cancer ~es. 49:4373~384(1989).
Vaupel, P. et al., Cancer Res. 49:6449 ~ 65~1989).
van Houte, J. et ~., J. Dent. Res. 75(4):1008-1014(1996).
Yosipovitch, G. et al., Nephrol. Dial. Transplant. 8(10):1129-1132(1993).
Back~round of the Invention
Lipid layers, such as comprise cell membranes and the extr~c~ r matrix of the stratum
corneum, can constitute a formidable barrier to drug delivery. For optimal delivery, a drug
should freely dissolve in both the aqueous compartments of the body and the lipid layers which
enclose those compartments.
Although many low-molecular-weight compounds of low to moderate polarity can pass
directly through lipid layers, co~ ou~lds with greater polarity and/or higher molecular weight
generally enter eukaryotic cells only via endocytosis or related processes. In this process,
compounds are taken into the cell via progressive invagination of a region of the mel,lbl~.c,
eventually forming a closed vesicle, or endosome, within the cell. In most cases, the endosome
then merges with a Iysosome, resulting in exposure of the internalized compound to degradative
enzymes.
To f:~cilit~e more effective delivery of drugs and other compounds across lipid layers, it
would be desirable to provide a drug transporting composition which affords lipid solubility
under selected conditions and aqueous solubility under other conditions. It would also be
desirable to deliver colllpou--ds into the cell cytosol via a route which avoids exposure to
lysosomal enzymes.
Su~ r of the Invention
In one aspect, the invention provides a composition for transporting a compound from a
low-pH environment across a lipid layer to a higher-pH aqueous co~llpalL~.Ielll. The
composition includes a polypeptide having one or more pairs of carboxyl groups, where the
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 3 -
carboxyl groups of a pair are separated by zero, two or three amino acids, and a length of
between about 8 and about 100 amino acid residues, and preferably between lO and 50
residues. The polypeptide further contains an initiator moiety at one end region of the
polypeptide, to ~Cilit ~ç entry of said end region, and partitioning of the polypeptide, into the
5 lipid layer. Such a polypeptide is effective to undergo a reversible transition between a
lipophilic form, which is effective to partition from the low-pH envh~ol...,.,.ll into the lipid
layer, and a hydrophilic form, which is ~clive to partition p~er~lf~.lially from the lipid layer
into the higher-pH aqueous co",pa-~",cnt. The polypeptide is thereby able to traverse the lipid
layer from the low-pH en~i.o..~ t to the higher-pH compartment. The composition also
10 includes, covalently qt~q-rhf~d to the polypeptide, the compound to be transported.
In one preferred embo~limPnt 30-100 percent of the amino acid residues forming the
polypeptide, excluding the initiator moiety, are gh~tqmic acid. Pairs of glutarnic acid residues
in the polypeptide may be separated by other amino acid residues, which are preferably selected
from the group consisting of leucine, methionine, alanine, and 2-amino butyric acid.
15 Alternatively, the polypeptide may be polyglutamic acid having an initiator moiety at one end
region.
The initiator moiety may be an initiator amino acid sequenre having 3-12 amino acid
residues. The seq~rnre is effective to form an alpha helix at a pH higher than the pH at which
a same-length polygl-lt,q-nnic acid forms an alpha helix. Preferred amino acid residues in such
20 a sequenrf are glllt~q-nnic acid, leucine, methionine, alanine, 2-aminobutyric acid, norvaline, and
,l~-alanine, where the ratio of non-glutamic acid to glutamic acid residues is greater than 1.
Where the polypeptide component contains a sequPnre having less than about 50% glutamic
acid residues, the initiator sequence may be an end region of the polypeptide itself.
Alternatively, the may be a group linked covalently to the N or C terminnc which is
25 effective to eliminqtç a positive or negative charge at said tçrminnc~ and which has at least one
remote polar group effective to shield polar sites near the tf-l";."lc. A lipophilic substqnre at
the terminus, inrh~ a compound to be delivered, may serve as an initiator moiety. A single
polypeptide may also contain a combination of such initiator moieties.
The compound to be delivered may be attached to an end of the polypeptide component,
30 typically the end opposite the initiator seqnfnre The composition may have only one, or
multiple compounds per polypeptide CO~ On~ . The compound to be delivered may include
a comrol~n-l which is itself only ~alillgly soluble in free form in an injectable aqueous delivery
mf flinm, such as taxol, cyclosporin"qmphotf ricin B. In another embodiment, compound is a
sequence-specific nucleic acid binding polymer, e.g., an qnticPn.ce compound.
CA 022~2706 1998-10-27
WO 97/40854 PCTrUS97/07335
-4-
In another aspect, the invention provides a method of f:~rili~ing the transport of a
co.l.pùu.ld from a low-pH ellvilulllll~llL acrûss a lipid layer to a higher-pH aqueous
colllpdl~ n~. According to the method, the compound is covalently coupled to a polypeptide
as described above. The method is useful, for exarnple, in delivering a thclaptulic col--poulld
from an extracellul~r mP~ m~ having a given pH, to the cytosol of a ceJI, having a higher pH.
In one specific application, the method is used in Antitllrnor therapy, where the cell is a tumor
cell existing in an acidic extrac~ r m~flillm and the attached compound is an antineoplastic
agent. In other applications, the method is used in the ll.,aLIll~.lL of ~. pylor~ infection, where
the extracellular medium is stom~h fluid, the cell is an H. pylon bacterial cell, and the
compound is an Antih~rterial agent, or in the llcdL~ .l and prevention of tooth decay, where
the cell is an acid-producing cariogenic bacterial cell, and the attached compound is an agent
effective against cariogenic bacteria.
Further applications include trAncr~rmAI delivery of a compound through the stratum
comeum, wherein the lipid layer to be traversed Cu~ lists the extracellular matrix of the
stratum COIll-,~ll, and in llal~l~olling a compound across the brain-blood barrier. In the latter
case, the composition is transported within a transcytotic vesicle through an endothelial cell of
a capillary wall, and is taken up by a brain cell within an endocytotic vesicle.These and other and features of the invention will become more fully app~,nl when the
following detailed description of the invention is read in conju"clion with the accompanying
dld~
Brief Description of the Drawin~es
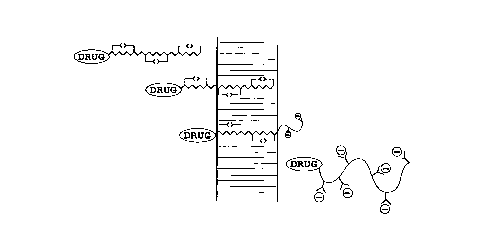
Figure lA-lB illustrate a pH-dependent transition between hydrophilic and lipophilic
collrolllldlions of side chain carboxyls of a polymer;
Figure 2A depicts the process of cytosolic entry, via endocytosis, of a polypeptide with
~tt~h~d coll.~uu..d, in accoldallce with one aspect of the invention;
Figure 2B shows the polypeptide-cclll~,oul-d complex in its high pH for~n, which exists in
the early-stage endosome;
Figure 2C shows the colll~lex in its low pH form, which exists in the late-stage ~ndQsnn~
30 clll~.illg the lipid l~ lbldne, and converting back to the high pH form upon contacting tne
higher pH cytosol;
Figure 3 depicts the lIAnC~C~ A~ JOII process of a polypeptide contAining an AttArhl~d
- co.... ~ ulid, where the complex passes from the extr~cell~ r lipid matrix to the a~lueous
co.ll~al~ .lL beneath the stratum corneum;
Figure 4 shows paired, spaced glutamic carboxyls of a polypeptide in an a-helix;
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 5 -
Figures SA-5C show representative C-terminal end structures of polypeptides, where the
structures in SB-SC have partially shielded or deleted terminal polar sites;
Figures 6A-6D shows representative N-terminal end structures of polypeptides, where the
structures in 6B-6D have shielded terminal polar sites;
S Figure 6E shows an "N-crown" structure which may be used to shield polar sites of N-
termin~l end structures in polypeptides, as shown in Fig. 6D;
Figure 7 shows a polypeptide functioning as a m()lecul~r engine, in accordance with an
embodim~nt of the invention;
Figures 8A-8C depicts polypeptides ~Cse~.~e~ for octanol solubility and cell entry, where
Fig. 8A shows a polyglutamic acid with unmodified termini, Fig. 8B illustrates the
incorporation of groups which delete or shield polar sites, and Fig. 8C shows further addition
of lipophilic amino acid residues at the termini;
Figure 9 illustrates representative sites of side-chain a~achm~-nt on a polypeptide, showing,
from left to right, amide and carbamate links to Iysine, an ester link to gh)t~mic acid, and
~iculfi~le, thioether, and amide links to cysteine;
Figure lO shows a method of pr~alion of polypeptides which may be used in
accordance with the invention;
Figures 1 lA-llB show representative polymer-compound linkages, such as may be used
in att:~hing a compound to a side chain or terminnc of a polypeptide;
Figure 12 illustrates çnh~n~em~nt of endocytosis, as shown in Figure 2A, by a lipid anchor
~t~rhed to a polycationic head group;
Figures 13A-C show procedures for linking a polypeptide of the invention to a Morpholino
~nti~ence oligomer; and
Figures 14A-14F show octanol/water partitioning properties of several exemplary
polypeptides, as a function of pH.
Detailed Description of the Invention
I. Definitions
The terms below have the following ~r ~ ;-.g~ umess otherwise noted.
The "low pH form" (also low pH collru~ n~ lipophilic form, or lipophilic confor-mation) of a polypeptide of the invention, or a segment of the polypeptide, is a .,.~h,l;."~ ly
non-ionic ~-helical cor~ru~ ion rendered lipophilic by hydrogen bonding between paired side-
chain or te~min~tl carboxyl groups.
The "high pH form" (also high pH conrul",alion, hydrophilic form, or hydrophilic35 cot~ru~ Qn) of a polypeptide of the i-.~e.~lioll, or a seg.~ of the polypeptide, is a
CA 022~2706 1998-10-27
W O 97/4~854 PCTAUS97/07335 -6-
con~ .alion in which the side-chain or terminal carboxyl groups are wholly or predu~ ly
in a non-hydrogen-bonded, ionic state.
A "reversible transition" between the lipophilic form and the hydrophilic form of a
polypeptide is a transition between a nonionic, a-helical con~u~ lion~ wherein side chain
carboxyls are engaged in intramolecular hydrogen bonding, favored by low pH, and a form
wherein side chain carboxyls are in ionic, non-hydrogen-bonded states, favored by high pH.
Such a transition may encoll,~ass an entire polypeptide, or it may occur at a localized region
of a polypeptide, particularly when the region is in the vicinity of an aqueous/lipid interface,
or has a composition that is especially lipophilic and/or especially favors the formation of an
10 cY-helix. Such a localized region of a polypeptide is often effective to initiate entry of the
polypeptide into a lipid phase, such as a membrane, even though regions of the polypeptide
more remote from the lipid phase may be in a hydrophilic conformation.
An "acid amino acid" or "acid side chain acid" is one which has a free carboxyl group
when incorporated into a polypeptide. FY~mrles are aspartic acid and ~hlt~mic acid.
A "non-acid amino acid" or "non acid side chain acid" is one which has no free carboxyl
group when incorporated into a polypeptide.
A "high glutamic" polypeptide is a polypeptide containing greater than about 50 % glut~mir
acid residues.
hn "initiator sequpn~e~ is a short (typically 3-12 arnino acid residues) sequence at a
t~ ;"~ of a polypeptide which readily forms an a-helix at pH's attainable in endosomes of
m~mm~ n cells, generally at a pH significantly higher than that at which the same length
sequence of glutamic acid residues would form an cY-helix. Such a sequence at the terminus
of a polypeptide initiates entry of the polypeptide into a lipid layer and promotes a-helix
form~ti-)n in immyli~t~.ly adjacent se~m~nt~ of the polypeptide.
A group which "deletes" or "shields" polar groups at or near the tPrmim~C of a polypeptide
is a covalently linked moiety which caps or replaces a carboxylate anion or plolonated amine
at the terminll~. Preferred groups further mask other polar sites near the ~P~ "~i."~ by non-
covalent associations, typically hydrogen bonding.
"Endocytosis" is a process by which extrarç~ r material is taken into a cell via an
invagination of the cell membrane, which closes to form a vesicle within the cell known as an
e~,-losome. Endocytosis may be lec~lor-mYli~tecl, where the extrace~ - compound binds
to a specific rece~tor on the cell surface, or eYtrarellnl~r co~ o~ ds may be imported
nonspecifically, by virtue of their presence near the cell membrane. The latter process is also
known as fluid-phase endocytosis or pinocytosis. A related process, potocytosis, takes
compounds into the cell via vesicles near the cell surface known as caveolae. In all of the
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 7 -
above processes, the vesicles enclosing the extracellular co~ uu..d become increasingly acidic
after formation. The compositions and methods of the invention are therefore applicable to all
of these methods of lla~ olL.
5 II. Polymer Composition
The polymer composition of the invention includes a polypeptide component capable of
undergoing a transition between a hydrophobic, c~-helical form, and a llydlolJhilic form; an
initiator moiety at one end region of the polypeptide, to f~Cilit~tP partitioning of the polypeptide
into a lipid layer through that end region, and the compound to be delivered, covalently
10 attached to the polypeptide.
As detailed below, the polypeptide component contains one or more pairs, and prtr~ ~ly
two or more pairs, of carboxyl groups, where the two carboxyls of a pair are separated by
zero, two or three amino acids. The composition and positioning of the amino acids of the
polypeptide are such that, in the presence of both an aqueous and a lipid-like phase, the
15 polypeptide undergoes, typically at a pH between about 4.0 and about 7.0, a reversible
transition between a high-pH form, comprising a multiply-ionized hydrophilic structure, and
a low-pH form, co.,.~lishlg a sul,s~dll~ially non-ionic cY-helical structure rendered lipophilic by
hydrogen bonding between the paired carboxyls.
The lipophilic, low-pH form partitions into a lipid en~dlolllllent, while the hydlophilic,
20 high-pH form partitions pr~rerel-Lially into aqueous solution. Figure lA illustrates the pH-
dependent transition between hydrophilic and lipophilic forms of a polypeptide, and Figure lB
illustrates this solubility transition for â specific sequenre of properly-spaced call o~yl-
co~ amino acid pairs.
25 A. Composition Transport Across a Lipid Layer
The polymer composition can be used to tldl~Oll an attached coll.pou-ld from theextr~c~ r compartment to the cytosolic compartment within cells. The process is illustrated
in Fig. 2A. The composition 10 is readily delivered, in an aqueous vehicle, to the extrarellnl~r
co"lpal~ll-cnt by virtue of the aqueous solubility hllp~ed by the high-pH form of the
30 polypeptide. Following endocytosis by a cell 12, the composition is enrlosecl within an
en~osomP 14. The pH within the en~losomp- decreases due to the action of ATP-driven proton
pumps within the endosomal membrane (see e.g. Clague, Fuchs). The composition COllvt~
in the increasingly acidic e~vi~mllent of the late stage endosome, shown at 16, to its low-pH
lipid-soluble form, which enters the endosomal lllell-blal1e.
CA 022~2706 1998-10-27
W O 97/40854 PCT~US97/07335
Upon contacting the cytosolic face of the endosomal membrane, the composition is actively
drawn into the cytosol by virtue of progressive ionization and solvation of the polypeptide chain
at the near-neutral pH of the aqueous cytosol, as shown at 18, resulting in delivery of the
attached co~ .uu.,d 20 into the desired subcellular co",~ ,..,nL. As ~icc~ ed further below,
a polypeptide constructed from L-amino acids will typically be degraded in the cytosol, as
shown, after the drug has been released. This unidirectional active transport process is
illustrated further in Figures 2B and 2C, where only a portion of the Pndosom~i membrane is
represented.
The above figures depict the polypeptide ~uming a completely hydrogen-bonded
conro""alion prior to entering the endosomal memhrane. Such is likely to occur when the
polypeptide is of a composition that readily forms an
~-helix at relatively high pH. However, for highly polar peptides, i.e. those containing a high
percentage of acid side chain residues, entry into the membrane may be initiated by a localized
lipophilic region of the polypeptide, even when other regions of the polypeptide are in a non-
I5 hydrogen-bonded, hydrophilic conrv""alion. Such entry is especially favored when this
lipophilic region is at a terminus of the polypeptide. Accordingly, a tç~ llc of the
polypeptide, especially a longer polypeptide containing a high concentra~ion of polar groups,
may comprise amino acid sequences which readily assume a lipophilic conro-",dLion at a
relatively high pH, or may be otherwise modified to render the l~""i"us more lipophilic. Such
modifications are ~i.ccll~sed further below.
An advantage of the composition of the invention, as can be seen from the above
description, is that endosome-to-cytosol transport may be achieved without disruption of the
endosomal membrane, thus avoiding leakage of Iysosomal enzymes into the cytosolic
compartment of the cell.
The polymer composition can also be used to transport an attached compound directly into
the cytosol of cells in cases where the extrac~ r medium has a pH less than the pH of the
cytosol of the cells, as described below. This allows, for example, selective delivery of
compounds into eukaryotic cells, bacteria or other target cells in acidic e lvirululle~
The polymer composition can further be used for tr~ncd~rm~l delivery of selectedcoll,~uu"ds, as described below, and as illustrated in Fig. 3. In this case, the composition 10
is cont:~te~, in its low-pH lipid-soluble form, with the surface of the epidermis, shown at 22,
leading to diffusion through the extracP~ - lipid matrix 24 of the stratum CO~ ulll, which
contains epithelial cells 26. Upon contact with the aqueous col"~l,l,enl underlying the stratum
col"eum, shown at 28, the composition is actively drawn into this col"p~l",ent by virtue of
progressive ionization and solvation of the polypeptide chain at the near-neutral pH of this
~C~/US~7~u7335 CA 02252706 1998-10-27 P~T
ANTIVIR~S l~'C- S;~3E~ R
Our Ref.: B 32~3 ec~ U,~C~EN
J a. JU~i 1998
companment. thereby effecting deli-e~ of ~he attached compound into the underlying tissues,
with subsequent distribution throughout the body.
The compositions are particularly useful for the deliverv of compounds which are only
sparingly soluble in free forrn in a~ueous delivery media. These compounds include~but 2~1
~20~ d to~ Ta~tol~, TaxolT~ analogs, cyclosporin analogs, and arnphotericin B. Also
contempla~ed is the delivery of sequence-specific nucleic acid binding polyrners.
B. PolvPeDtide ComT~onent
Two irnpontant strucrural fearures of the polypeptide component are that: a) it contains at
least one properly-spaced acid pair, and b) it corlt~in~ a number of amino acids sufilcient to
adopt a lipophilic a-heli~ at low pH. In selee~i~ a preferred polymer for a given application.
various factors are considered, such as the plup~lies of the compound to be delivered, the
C~ r ~ Ull.,lltS from and .into which the cc~ o-u~d is to be delivered, and the desirability of
including end m~ifir~tions to shield exposed polar sites and increase lipo,chilicity a~ the helix
15 termini.
B1. Acidic Amino Acids. To provide the free carboxyl groups which are able to inter-
hydrogen bond when the polypeptide is in an ~-helical conforrnation, carboxyl side chain arnino
acids, preferably aspartic acid and gl~lt~n~;c acid, are inrlude~ The preferred number and
frequency of these acids is tlicc~lcsecl below.
It will be dyylecidted that a free carboxyl group at either polypeptide terrninus rnay also
hydrogen bond with a properly situated carboxyl side chain arnino acid. For example, the
carboxyl ~rmin~ls rnay be prepared with-~-alanine, y-arnino butyric acid, or another omega-
amino carboxylic acid a its terrninal group, by initi~tin~ the synthesis of th~ polypeptide using
such an acid"as described in Exarnple 1. T~is results in an omega-carboxy alkyl amide at the
25 C-t~ninllc (as shown, for example, in Figs. 10 and SC), where the carboxyl group at the end
of the alkyl chain is available for hydrogen bonding. The N-terminus may be capped ~ith a
diacid, such as glutaric acid (as shown in ~ig. 6B) to provide an additional hydrogen-bonding
carboxyl group.
B2. Non-acidic arnino acids. The non-acid arnino acids of the polyrner, when present,
30 serve plinc.l~ally to provide proper spacing be~ween the two carboxyl groups of each hydrogen-
bonding pair, and to adjust the pH of the partitioning trarLsition to a selected value, as disc~csed
below. These amino acids should not contain moieties which are cationic when the pol-~ner
is in its low-pH ~-helical conformation (i.e., arginine, Iysine, and histidine). (An exception
is the use of low levels of arnino acids such as Iysine as sites for altachrner.t of the ~ransported
35 compound, as described in below.) Most of the non-acid amino acids of the poly~ner are
AMENDED SHEE~
IPEAIEP --
CA 022~2706 1998-10-27
W O 97/40854 PCT~US97/07335
- 10-
relatively non-polar and compatible with a-helix formation. Accoldillgly, pl~r~ non-acid
arnino acids for most applications are selected from the following~ lophall, phenylalanine,
leucine, isoleucine, methionine, valine, and alanine. Norvaline, ~-amino butyric acid, and side
chain esters of aspartic and ghlt~mir acid are also suitable as "non-acid" amino acids. Based
on cost, ease of use, and other practical considerations, leucine, methionine, alanine and ~-
amino butyric acid are particularly prer,,lled.
With the exception of nucleic acids, polymers with a high density of acid moieties are not
natural components of the interior of cells and so could prove toxic therein. However, such
toxicity is much reduced or prevented if, after carrying out its drug transport function, said
polymers are ~ cs~llhled into natural subunits çn~ mir. in the cytosol of cells. In this regard,
it is known that unstructured polypeptides composed of natural L-arnino acids can be rapidly
depolymerized in the cytosol of cells, primarily in complex multi-ring ~llu~;lulcs called
proteasomes.
For those applir~tion~ where it is desirable that the polymer be (iic~cs~ )led into
innocuous subunits within the cytosol of cells, the preferred amino acids of the polymer are
selected from natural L-amino acids. Conversely, for applications where it is desirable that the
polymer remain intact in the cytosol, such as those described below, the pref~ d amino acids
are selected from D-amino acids. In the latter applications, other non-natural amino acids are
also suitable, such as cY-amino butyric acid, norvaline, and norlèucine.
B3. Spacin~ of Acids. It is known that, in an ~-helix col-rullllalion, the amide moieties
of a polypeptide backbone are effectively 5hi~1ded from the solvent due to extensive
intramolecular hydrogen bonding. It was predicted that, in a polypeptide in an cY-helical
conrcllllalion, proper spacing of acid side chains would ~ub~ ially shield the polar sites of
the acid moieties from the solvent by virtue of formation of double hydrogen bonds between
paired acid moieties in their free acid state. Such .shi~ ing of the polar sites of the polyrner
from the envilu,l,l,~"l should result in improved lipid solubility. One such n,~,~,s~
structure, COIII~IiSi~lg paired glutamic acids separated by three amino acids, is shown in Fig.
4.
Studies with CPK molecular models were used to predict the pl~r~ ,d spacing of acid side
chains to provide the desired hydrogen-bonded ~llu~.lul~s. Subsequent octanollwater and
pentanol/water partitioning studies with suitable polypeptide seq-l~nrrs verified that good lipid
solubility can indeed be achieved at low pH if acid pairs, selected from aspartic and gl~ ..,.;r,
are so spaced. Table 1 lists these ~a -;..g.~ which provide the desired ~hie~ ng of polar sites
of acid pairs at low pH.
CA 022~2706 1998-10-27
W O 97/408~4 PCTAUS97/07335
- 11 -
Table 1
E-E
D-E
E-X-X-E
E-X-X-D
E-X-X-X-E
E-X-X-X-D
D-X-X-X-E~0 where: E = glu~micacid
D =aspanicacid
X = ~ninoacid
As can be seen from the table, spacings of zero, two, or three amino acids between
15 carboxyl side chain amino acids are effective. Of these, spacings of two or three arnino acids
between carboxyl side chain amino acids are prer~.led.
Polypeptides containing sequences wherein polar side chains alternate with nonpolar side
chains are known to adopt B-sheet and related conformations. Polypeptides with such alternat-
ing sequences greater than about 6 amino acids in length were found to be generally insoluble
20 in octanol at low pH, probably due to the formation of multi-chain complexes whose polar sites
are not adequately chielde-l from the solvent. Accordingly, the polymer of the invention should
be largely free of signifir~nt runs of alternating polar and nonpolar side chains.
B4. Length of Polvpeptide. In order to form an ~-helix with suitably spaced pairs of
carboxyl side chains, the polypeptides of the invention should be at least 8 amino acids in
25 length, and preferably at least 10 amino acids in length.
For delivery of particularly large and/or polar compounds, which do not diffuse across a
lipid membrane at a practical rate, such delivery is f~rilit~te(l when the polymer, in its ~-helical
collfulll.alion, is longer than the thickness of the membrane. In this case, at least a portion of
the polypeptide is able to enter the cytosol and convert to its high-pH form, a process
30 energetically favored by solvation and ionization, before the large and/or polar cu~ ,oul.d is
required to enter the lipid membrane~ as illustrated in Fig. 7.
In a polypeptide ~-helix, each arnino acid residue contributes approximately 1.5 A to the
axial length. Since lipid bilayers of cell ~..c.,lbldnes in eukaryotic cells are typically about 33
to 36 A thick, a preferred length for the polymer composilion is about 22 amino acids or more.
35 Several additional amino acids are preferably in~ludPd to assure that at least one acid side chain
enters the high-pH co,~ ll-e~lt to initiate conversion to the ionic high pH form. Thus,
polypeptides in the length range of about 24 to 100 amino acids, preferably 24 to 48 amino
acids, are preferred for endosome-to-cytosol transport of large and/or polar compounds.
CA 02252706 1998-10-27
0~1 1 ~I
For delivery of the small~r or less polar atlach-d compounds. shorter peptides may be
used. When the polypeptide is below, near, or even somewhat above its transition p~l, the
low-pH lipophilic form is present at an equilibrium concentration sufficient to effect difnlsion
of the polypeptide into and across the lipid membrane. Only for attached compounds which
5 significantly interfere with this dirrusion, as described above, is it neC~ss~ry for the pol~peptide
to completely span the membrane at any given time.
As ~icc~lcsed further below, entry into a cell membrane is most likely to be initi~red at a
terminus of the polypeptide, especially for more polar polypeptides. In longer pol,vpeptides,
such as those having lengths greater than about ~00 arrlino acids, the terrnini are at a low
10 co.lccnt.a~ion and are 5t~riC~ic~lly less likely to contact the cell membrane. In addition, longer
polypeptides, by virt8ud of size alone, rnay not be efficiently engulfed within an endosome,
which is typically abou~01lVin ~ m~ter. Preferred polypeptides, such as described above,
will thus have an average length of less than 200 arnino acids, and most preferably less than
about 100 amino acids.
B5. Polvelutamate and wlvaspartate homopolvmer comDonents.
High molecular weight, random-length polyglutamic acids, such as are con~ .iallyavailable from, e.g., Aldrich Che_ical Co., Milwau~;ee, WI, or readily polymerized from N-
carboxyanhydrides (Blout), precipitate from water, but do not partition into n-pentanol, at about
pH 4. Such polyrners also fail to cross cell membranes in direct-entry eA~e~i."~,lts, such as
20 described below and Exatnple 10A, at pH's attainable in endosomes of m~mm~lian cells.
Further, when a high molecular weight poly-L-glutamic acid was linkcd to various ~ntir~nn~r
drugs, such as adriamycin (Hoes, Nul;ui), in an effort to enhance endocytotic uptake of the
drug by increasing aqueous solubility, the polyglutamic acid component of the polypeptide~rug
conjugate neither entered the en~iosQm~l membrane nor L~n~ L~t the drug across the
25 ~ .,,b,~e. Rather, the carrier was dcgraded within the fused en-losom~-lysosome, and the
released drug thcn passively diffi~ced across the lysosom~l membrane into the cytosol.
These results may be explained on the premise that relatively polar polypeptides (e.g.,
con~ining over about 507O glu~mic acid residues) begin entty into cell membranes via one or
the other termini of the polypeptide, as diccncsed further below. Because high-glutamic
30 polypeptides with unmodified termini have a multiplicity of unshiclded polar site~s at both the
C-terminus and the ~I-terminus, they are apparently unable to efficiently initiate entry into the
nonpolar interior of a cell membrane.
In contrast to these results, relatively low molecular weight polypeptides, having 6~100%
glutamic acid residues. when modified to provide local lipophilicity at one or both ~errnini, as
35 described ~ below, crossed cell membrane~s in direct-cn~ry experimen~s, as described in
AMENDED SHEE7-
IPEA~EP
. CA 02252706 1998-10-27
0~5~" '' '
I j
E.~ample lOA. Such modified high-glutarnic polypeptides are also able to parlition into n-
pentanol from aqueous buffers at pH's in the range of 4.3 1.9.
Poiyaspartic acid is soluble in aqueous solution at acidic pH, failing to partition into n-
octanol or n-pentanol even when the pH of the aqueous phase is as low as 4. Polyaspartic acid
5 also fails to cross cell membranes in direct-entry experiments, such as described below. Such
lack of lipophilicity is expected on the basis of partitioning calcula~ions (Leo), and beeause
molecular modeling of a polyaspartic acid a-helis shows that the carboxyl groups are unable
to forrn the double-hydrogen-bonded pairs required to shield the polar acid si~es from the
environ,..en~.
lO C. Initia~or Moietv
Two general embodim~ntc of the initiator moiety are con~mrlated. The first is an~,liator polypeptide sequ~n~e, which rnay be an end-region ~x~ncio~ of a polypeptide
cv~ Cnt con-~inin~ less than about 50~ acidic residues, or, in the ease of a homopolyrner
of aeidic arnino acids, a more hydrophobic, alpha-helix forming region co"r~ less than
15 about ~150% acidie, e.g., glut~m~te residues. The seeond is a hydrophobic moiety that rnay
~e, for example, the compound to be a~lminictF~red~ or a eompound effective to elimin~ ?nd/or
r~ask end eharges and polar groups.
Cl. Initiator Seq-~nr~s Further addition of lipophilie arnino aeids near the C-terrninus
of the polyglut~rnic acid described above (i.e., leueines at residue positions C2, C3, and C~;
20 see Fig. 8C) afforded quite good transport aeross cell l,.c~b,~es. The polypeptide showed
good solubility in n-perltanol~ though not in n-oetanol, at acidic pH.
Sueh an arnino aeid sequenee at the polypeptide te~nin-lc is partieularly useful in that it
provides an initiator sequen~ effeetive to forrn an a-helix at a higher pH than would be
~e.~ ~l for a similar length scquence of, in the present example, ~ t~mic acid residues, to
25 ass~ne this eo,.f~"l,~tion. Cell entry and partitioning studies have rl~ ,..o"cl.dted that inelusion
of sueh an ~-helix-forrning initiator sequen~e ~nh~n~ c lipid solubility and eell eMry to a
5i~,.;f~ ly greater extent than lipophilic modifying groups which do not in thernselves initiate
a-helix forrnation. The initiator sequence, in forming an a-helix at relatively high pH, also
promotes subsequent a-helix propaga~ion in the adjaeent 5~,~""~ c of the polypeptide.
P~eft,.. ~d initiator sequences are three to twelve, and preferably five to ten, arnino aeid
residues in length, and preferably inelude glutamic acid, leucine, methionine. alanine, 2-
aminobutyrie acid, 3-arninobutyrie aeid, norvaline, and ,B-alanine, where the ra~io of non-acid
side chain residues to acid side chain residues is greater than l, and preferably greater than 2.
Exarnples of such initiator sequences are XEXX~3 and XEXXX,B (C-terminal) and glutaric acid-
35 XXEX and glutaric acid-XXXEX (N-terminal), where E is glutarnic acid, ,B is ~-alanine, and
AMENDED SHEEr
¦~'F ~ p
CA 022~2706 1998-10-27
W O 97/40854 PCTAUS97/07335
-14-
X is selected from the list above. One such C-terminal sequpnre~ leu-glu-leu-leu-,~, is shown
in Fig. 8C.
Such termin il sequences provide the clearest benefit in highly polar (e.g. high glutarnic)
polypeptides. They are also useful in defined-se~uenre polypeptides containing lower levels
S (i.e., less than about 50%) of acid side chain residues. Such polypeptides, however, generally
have a higher plo~ y toward ~-helix forrnation, and thus other initiator moieties, such as
a lipophilic drug at the terminus, or a shielding group as described above, may be effective to
initiate entry of such polypeptides into a IllClllb~dne.
C2. Shieldin~ or Removal of Polar Sites. A polypeptide in an a-helical collfo.l"d~iol-
10 typically contains multiple polar sites at both the C-terminus and the N-tennim-c which are not
~hiPlded by intramolecular hydrogen bonding. These nnchiplded polar and ionic sites conslilule
a s..l.s~ l bar to initiation of polypeptides into lipid layers, due to the presence of solvated
counterions and water of solvation, and the ~oper~iLy of the polar termini to assume a non-a-
helical confol~llaLion. Deleting the terminal charge and .chi~ltiing or removing one or more of
15 these polar sites can improve lipid solubility, particularly in the case of short or highly polar
polypeptides, such as high-glutamic polypeptides, described above.
A lipophilic ~lb~ e, such as a drug to be delivered, ~t~rhecl at a polymer terminus
elimin~tPs the charge at said tf~ i and provides local lipophilicity. Deleting the terminal
charge and chiP111ing or removal of polar sites can also be accomplished by hlcull oldling, at
20 the C-terrninus or the N-t~,....;...~c, a group which covalently bonds to, or includes, the terminal
carboxyl or amino group, and which further contains at least one remote polar group, such as
a hydroxyl or carbonyl group, which is effective to shield, by hydrogen bonding, one or more
additional polar groups at or near the polypeptide terminus. Specific examples follow.
C-Terminus chiPlrlin~. The C-terrnin~c of a polypeptide typically contains three carbonyl
25 groups and a negatively-charged carboxylate ion which are not chiPlrlPd by intramolecular
hydrogen bonding, as illustrated in Fig. SA. One method for reducing the number of these
Im~hiplded tP~Tnin~l polar sites is to i,lcc,~u,d~e an ~x-ester, preferably a 2-hydroxyethyl ester,
at the C-~-,--;----~, as illustrated in Fig. 5B. Polar sites may also be conv~.nie.llly P~
from the C-te~nim~s by initi~ting the synthesis of the polypeptide on a support resin with 13-
30 alanine, as shown in Fig. 5C, and as described in Example 1.
N-Tenninus chielAin~. The N-terrninllc of a polypeptide typically contains three amide
protons and a positively-charged protonated amine which are not ch~ pd by intramolecular
- hydrogen bonding, as illustrated in Fig. 6A. Polar sites may be conv~,.,;~,.,lly removed from
the N-~Prmimls by le..";~ i..g the polypeptide with a diacid, as illu~ ed in Fig. 6B.
35 Altematively, polar sites can be chiplded simply by acetylating the terminal amine,
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 15-
as ~ d in Fig. 6C. Hydlu~ bûnding occurs as shown when the polypeptide is in an
~-helix. More extensive shielding of the N-l~ s may be achieved by a nûvel structure,
referred to as an N-crown, designP~ to shield all of the normally-exposed polar sites at the N-
te~ ...;--.-c of cY-helices. Fig. 6D shows one such N-crown, whose structure is given in Fig. 6E,
5 in the H-bonded co~rn~ ;()n it is believed to adopt when linked to the N-tPrmim-~ of a
polypeptide existing in an a-helical cùllro~ on
A polyghlt~mic acid having such modifications is shown in Fig. 8B. The unmodified
polypeptide, shown in Fig. 8A, did not partition into n-octanol or n-pentanol, nor did it show
any ~ yûl~ across cell mel,~ es in direct entry studies.
Addition of an N-terminal carboxy-fluorescein, as shown in Fig. 8B, removes the positive
charge from the N-~ , partially shields N-terminal polar sites, and adds a large lipophilic
moiety (i.e., fluorescein in its low-pH form) to the N-terminus. In addition, the negatively
charged carboxylate, with two polar sites, was elimin~ted from the C-termin--~ by initiaring the
polypeptide synthesis with a ~-alanine, as shown in Fig. lO. These mo~lific~tions afforded
15 modest transport across cell membranes (Fig. 8B), and the polypeptide partitioned into n-
pentanol, but not into n-octanol.
In operation, once the initiator moiety at the terminus of a polypeptide has entered the
membrane, succeeAing segmPnt~ of the polypeptide are able to convert to a lipophilic,
hydrogen-bonded conro- ...~l ion. Such conversion is driven by the increasingly acidic
20 ellvilolllllent within the en~losomP~ as well as the increased local lipophilicity provided by the
adjacent cY-helical segmpnt~ of the polypeptide and the pluxilllily of the cell membrane.
After entry of the initiator moiety, a polypeptide having a large number of acid side chains
is able to partition into the membrane in a "stepwise" manner, in which an acid side chain
positioned immP~ t~ly adjacent to the membrane fûrms a hydrogen-bonded pair with a nearby
25 acid side chain, and the segmPnt of the polypeptide cont~ining this pair, having ~.m~P~ a
lipophilic cY-helical con~ûllllaLion, enters the ,,,~...hl.mP.. In this sense, the spacing between
carboxylic acid side chains is of particular i~ oll~lce. When a polypeptide enters a melllbla.~e
in the stepwise manner described abûve, a carboxylate side chain adjacent tû the mc~llblane
which is unable to pair with another carboxylate side chain is likely to block further entry. As
~ 30 noted above, sp~ring~ of zero, two or three amino acids are effective for such pairing.
Further, if drugs are to be ~tt~rhp~ at sites along the polypeptide chain, rather than only at the
termini, they must be located so as not to hlle,r~e with said spacing and pairing between side
chain carboxylates. Thus, random ~tt ~hmPnt of drugs, as well as random spacing of non-acid
side chain residues, inrhl-ling ~ ..ic esters, should be avoided.
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 16-
As described further below, additional motive force for unidirectional Ird~ ,oLI is provided
by ionization and hydration of the side-chain carboxyls once the polypeptide spans the
membrane and encounters the higher-pH cellular cytosol. Thus a polypeptide having a high
percentage of side chain carboxyls is expected to provide a high driving force to LlallS~o,l an
~tt.qrhed compound across the membrane.
Accordingly, one pref~l,ed class of polypeptides for use in the present invention includes
those having 30%-100%, and prereldbly 50%-100%, glutamic acid content, and having an
initiator sequenre as described herein at one ~ l;r--lc. (In ~is context, 100% glutamic acid
content refers to the central chain of the polypeptide, eYr~ inE the initiator moiety.)
~l~re~ably, the compound to be transported is attached at or near the other tPrmin-l~ of the
polypeptide.
D. Transition pH
The pH at which the polypeptide component, or a segment thereof, converts between its
hydrophilic and lipophilic forms, and is effective to partition from an aqueous to a lipid phase,
or vice versa, is referred to as the "transition pH". ~n general, for transporting a compound
from a low-pH environment across a lipid layer to a higher-pH aqueous compartment, this
conversion should occur at a pH greater than or approximately equal to the pH of the low-pH
environment, and less than the pH of the higher-pH aqueous cu",pa,Ll"ent.
It will be understood, however, from the previous discu~ion~ that local modifications of
a polypeptide can f~ilit~te entry and partitioning into a lipid layer at a pH greater than that at
which the polypeptide as a whole would convert to an ~-helical collru''''a~ion in the absence
of such modifications, or in the absence of a lipid layer. Transport through a thin lipid layer,
such as a membrane, to an aqueous co-"~alll"ent is also facilitated by ionization and solvation
as the composition converts back to a hydrophilic conformation upon contacting said aqueous
compartment. Therefore, the pH at which a composition err~ ely transports in vivo, or in
cell entry studies, may differ from the transition pH of the polypeptide molecule in two-phase
partitioning studies. The latter value thus serves as a useful guideline for initial selection of
polypeptides, but should be supplem~nt~d by direct entry studies, described below, particularly
for highly polar polypeptides.
Effective transition pH varies for different applications. For imt~rlre, the b~rteril-m H.
pylori, a major cause of ulcers, exists in a very acidic environment in the stomach. This
environment may be specific~lly targeted, as ~i~cl-~.ced below, by the use of a polymer
composition which is effective to partition into a lipid layer at a pH of about 4.5 or less.
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 17 -
In contr~ct for tr~n~dP,rm~l delivery, it is pl~r~ ed that: a) the polymer coll-po~ilion exists
in its lipophilic form at a pH safely achievable in the extr~cP1hll~r lipid matrix of the stratum
COll~eulll, and b) the polymer composition converts to its hydrophilic form on cont~ting the
aqueous environment underlying the stratum col ~e ~... A transition pH in the range of about
5 5 to 6.5 generally satisfies these two criteria.
In transport of relatively polar coll-poul1ds from the endosomP to the cytosol of eukaryotic
cells, a polymer composition as described herein can ~ln~tinn as a mnlec~ r engine, pulling
the relatively polar compound into and through the endosorn~1 membrane. The motive force
exerted by the engine is gene~dled as the non-ionic lipophilic a-helical polypeptide undergoes
10 ionization and solvation at the cytosolic face of the endosom~l membrane, as illustrated in
Figure 7. This motive force is, in part, a function of the difference between the transition pH
and the pH of the cytosol. Thus, the lower the transition pH, the more power such an engine
should exert, and hence the greater the load it can transport through the endosomal membrane.
For co~ ,oullds which are fairly small and/or of only moderate polarity, the polymer
15 composition used for endosome-to-cytosol transport may have a fairly high transition pH, such
as in the range of 6.4 to 6.8. For larger and/or more polar colllp~unds (i.e., greater loads),
the transition pH should generally be lower.
However, if the transition pH is too low, endosome/lysosome fusion can occur before the
polypeptide engine converts to its lipophilic form and enters the encompassing m~-..bl~c,
leading to enLynl~.lic degradation of the polypeptide (~cs~".;,.g it comprises L-amino acids)
before it can carry out its L~d~ Oll function. At the time of endosome/lysosome fusion, the
pH of the endosome is typically in the range of about 5 to 6. Thus, a minim~m transition pH
in tnis range is l)~ere,~ed for compositions assembled from L-amino acids and intellfled for
endocytotic entry.
From the above, it is seen that the optimal transition pH can vary substantially depending
on the compound to be delivered and the compartment from and into which it is to be
delivered. Several parameters, described below, can be varied to adjust the transition pH over
a broad pH range, thereby allowing o~ ion of a polymer composition for delivery of a
selected compound between selected cOil,pdlll"C.l~.
1.) Len~h. As shown in Table 2A, there is a modest increase in the transition pH,
as detPrminPd in an octanol/water partitioning system, as the length of the polypeptide is
h~creased.
2.) Acid Amino Acids. The results in Table 2B demonstrate that, as a rule, glutamic
acid residues provide a higher transition pH than do aspartic acid residues. Results in this table
CA 022~2706 1998-10-27
W O 97/40854 PCTAUS97107335
-18-
also d~ o.~ ale that the order and spacing (e.g., ELLDL versus ELL~D) also have a
5ignific~nt impact on the transition pH.
3.) Non-Acid Amino Acids. The results in Table 2C dr~ ale that selection of
non-acid amino acid moieties can also suhst~nti~lly affect the transition pH. For eY~mple,
S repl~ing leucines with phenyl~l~ninP~ reduces the transition pH. As shown in Table 2C, the
transition pH also is reduced further by each repl~rçmPnt in the series: leucine -> va}ine ->
alanine -> glycine (data for glycine not shown). Thus, one simple method of adjusting the
transition pH over a broad range is by progressively replacing leucine residues (which give
high transition pH values) with alanines. It is also seen that leucines give about the sarne
10 transition pH values as norleucines.
4.) Ratio of Acids to Non-Acids. It was found that the transition pH is generally
reduced as the proportion of acid amino acids, replacing lipophilic amino acids such as leucine,
is increased.
5.) End Structure. Masking or deleting polar end groups not engaged in hydrogen
15 bonding serves to increase the transition pH.
CA 02252706 1998-10-27
~50-~llJI
- 19-
Table 2
A. Len~h:
Repeatin~ Sequence Len_h TransitionpH'
-ELALE- ~5 5.83
-ELALE- 45 6.05
-ELALE- 65 6.14
B. Acid Amino Acids:
RepeatinP Seauence Len th TransitionpH'
-ELLLE- 50 6.92
-ELLLD- 50 6.51
-DLLLE- 60 6.45
-ELLEL- 49 6.85
-ELLDL- 49 6.29
C. Non-Acid Amino Acids:
Repeatin~ Sequence Len th Transition pH
2~
-ELEDLDLL- 54 5.50
-E~EDLDLL- 54 5.39
-ELLLE- 50 6.9'~
-ELVLE- 50 6.44
-ELALE- 50 6.14
-ELLLE- 30 6.83
-ELALE- 30 5.90
-EALAE- 30 5.10
-ELLLE- 30 6.93
_F.nl.n~ nT.F 30 6.90
A = alanine
D = aspartic acid
E = gluta nic acid
F = ph..,jl-' -
L = leucine
nL = no.lel~.;.,e
V = valine ~ As d~t~ d.. cJ in n-oc~nol/water
Table 2 shows a selection of lla~iliO~I pH's attained in an n-octanol/water partitioning
system by varying the structural features tliccl~ss~d above. By further adjusting these
50 pararneters, it is possible to prepare polypeptides with still higher or lower transition pH's.
AMEN~:)ED SHEET
IPEA./EP ~
CA 022~2706 1998-10-27
W O 97/40854 PCTAUS97/0733S
-20-
For example, a composition with a transition pH of 4 or lower may be prepared by inr.lllAing
a high proportion of aspartic acid and alanine subunits in a relatively short polypeptide.
As stated above, transition p~I's detPrmined from octanol/water or pentanol/water
partitioning studies provide a useful guideline for selecting a~?r~l ,iat~ polypeptides for deliveq
5 of a compound to a selected envirolu"e,lL. However, it will be ~precia~ed that ,~rtrliti~n~l
factors may influence the behavior of the polypeptide in a cellular environment. Studies have
suggested, for example, that polypeptides conlai~ g high levels of leucine may bind to serum
proteins. It has also been d~",onsl,dled herein that a polypeptide having local regions of
lipophilicity at the termini may undergo a transition effective for cell entry in vitro or in vivo
10 at a pH higher than that shown in bulk partitioning studies. Therefore, parthioning studies are
ideall~, followed by in vi~ro cell entry experiments, as described below, to further assess the
membrane transport properties of a composition. A c~rldid~te polypeptide may also be tested
for binding to serum proteins by performing electrophoresis on agarose gels with and without
added serum albumin.
E. Couplin~ of Compound to Polypeptide Component
One or more drugs or other compounds can be attached to the polypeptide through sites
at the termini and/or at a limited number of se1ected sites within the chain. As ~i~c~l~sed
above, random ~tt~rhmPnt or high loading of colllpoullds along the chain can impede
20 partitioning into the membrane, especially for high acid side chain polypeptides.
Methods for selective ~tt~rhmrnt are well known in the art, and several such methods are
described in Examples 2-7, below. Figure 9 shows representative linkage positions and types
which are both convenient and effective for a s11hst~nti~1 variety of co"l~oullds and polymer
compositions. Shown, from left to right, are amide and C~,~ulldl~ links to Iysine, an ester link
25 to g1ut~mic acid, and disulfide, thioether, and amide links to cysteine. In particular, linkages
selected from amide, ca~ l,aLe, ester, thioether, di~1fi-1e, and hydrazone are typically easy
to form and suitable for most applications. Ester and digu1fi(1e linkages are especially pl~l.ed
if the linkage is to be readily cleaved in the cytosol after delivery of the compound.
Figure 11 illll~trates r~res~,L~ e starting materials and products of such linkages.
30 Examples 2-7 describe the linkage of representative co,,,~oullds, inr.ln-l ing cyclosporin, TaxolTM
(parlit~re1), and a Morpholino ~nti~çn~e oligomer (S~ on and Weller) to ,eL,lest.,La~ive
polymers via these linkage types. Cyclosporin is linked to a polypeptide via a disulflde linkage
(Example 3A), a thioether linkage (Example 4A), a c~la--laLe linkage ~xample 5), and an
amide or thioether linkage (Example 6). Also described are corresponding linkages of TaxolTM
.. ..
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 21 -
to a polypeptide using similar mPtho-ic (Examples 3B, 4B, 5, and 6B, and 7A) and an amide
linkage to a Morpholino ~nti~en.~e oligomer (Example 6C, shown in Fig. 13A-C).
In the above examples, TaxolTM is conveniently linked to the polypeptide via its C-7
hydroxyl group. For linking cyclosporin A, metabolite 17, which has a primary hydro~Lyl
S group (Eberle and Nuninger, 1992), is used. In both these cases, the linkage does not involve
known active sites of the molecule. Other drugs that may be used in applicAtion~ described
below, such as metronidazole or doxorubicin, also have primary hydroxyl groups for
convenient ~tt~ArhmPnt In general, numerous functional groups (hydroxyl, amino, halogen,
etc.) may be used for ~t:~hm~nt Groups which are not known to be part of an active site of
the compound are prefel,ed, particularly if the polypeptide or any portion thereof is to remain
~t~rhçd to the compound after delivery.
Example 8 describes representative mf~th()~l~ for purification of polymer-compound
products, as well as methods for structural analysis of said products.
The compound/polypeptide ratio is preferably 5:1 or less, and more prefeldbly 1:1 for
large and/or polar compounds. The compound is preferably attached at or near a terminus of
the polypeptide; most preferably, one tçrmin~ includes an initiator moiety and the other an
~tt~rhed co~ oulld.
For especially large and/or polar col"poullds, such as a nucleic acid binding polymer,
transport may be enhAnred by Att:~chin~ multiple, e.g. 2 to 5, polypeptide carriers of the
invention to a single compound molecule. The carriers are preferably a~t~rhed to the same
region of the molecule to be transported, e.g. at or near the nucleic acid binding polymer.
III. Assessment of Composition Properties
A. Partitioning Properties
The partitioning of a compound between water and n-octanol has colll,nollly been used in
ph~rmAreutir~l research to estimate the partitioning of that compound between an aqueous
co.l,L,~I.llent and a lipid bilayer of a cell membrane. Partitioning between n-octanol and a
series of buffers of varying pH was used to provide a ~lIIAIII ilhl ive measure of the pH-dependent
solubi~ity properties of polymer compositions of the invention having acid side chain acid
content of up to about 50% of the amino acids. For polymer co.nl)o~ilions having over 50%
acid side chain content, partitioning between n-pentanol and aqueous buffers was used. It
should be noted that transition pH values obtained in n-octanol/water are about 0.3 pH units
lower than those obtained in n-pentanol/water.
At its transition pH, a polymer partitions at equal concentrations in the aqueous and
35 organic phases. Transition pH values determin~d from these studies afford a simple
CA 022~2706 1998-10-27
W O 97/40854 PCT~US97/07335
-22-
4~ ive ~cse-csmpnt of the effects of different factors on polymer solubility properties.
Example 9 describes a convenient procedure for ca~yil g out such a parti~ioning study and
obtaining a transition pH value for a representative polymer composition. In this procedure,
a polypeptide tagged with fluorescein is p~liLioned between n-octanol or n-pentanol and
S aqueous buffers of various pH, and the abso~ ce of each phase is l"c~ul~d. Figures 14A-F
show plots of these absorbance values as a function of pH for six representative fluorescein-
tagged polypeptide compositions of the invention, where the six dir~enl polypeptides exhibit
octanol/water transition pH values ranging from S. 1 to 6.9.
Results from experiments with cultured cells, described in the next section, suggest that
for polymers with up to about 50% acid amino acid composition, these partitioning properties
are generally predictive of the polymer's ability to enter a lipid layer when the polymer is in
its low-pH conrol,l.aLion. Polymers with greater than 50% acid amino acid composition, when
modified to produce end group lipophilicity as discussed above, tend to show more efficient
direct cell entry, at higher pH, than predicted from the transition pH shown in partitioning
studies.
B. Cytosolic Entry
Bl. Direct Entry. In screening polymer compositions for delivery of a selected compound,
direct tr~mm~rnhrane passage can be assessed by brief stepwise reduction of the pH of the
extr~c~llul~r m~inm, which e.m~ t~c the progressive pH reduction which occurs in an
endosome due to the action of proton pumps embedded in its membrane. The process is also
representative of direct entry in vivo when the pH of the extr~cPlh~l~r medium is lower than
the pH of the cytosol of the cells.
In a eukaryotic cell, when a fluorescent co~ oulld is restricted to the endosomal/lysosomal
coll,pall"lenl, fluorescence microscopy shows a perinuclear l~u~ e pattern in the cell. In
contrast, when a fluorescent coll,~oul.d enters the cytosol, one sees diffuse fluoresc~llce
throughout the cell. Accordingly, linking a fluorescent tag to the compound to be ~ Oll~d
into the cytosol allows one to readily assess subcellular loc~li7~ion of the polymer-compound
product after its addition to cultured cells. Ideally, the linked fluorescent tag should have
minim~l impact on the transport process. Specifically, it should be relatively small, and it
should be reasonably soluble in octanol and lipid layers at low pH and reasonably soluble in
aqueous solutions at neutral pH and above. Two lluol~c~ tags suitable for most such
applications are 5-carboxy-fluorescein and 7~imethylaminocowllalill~-acetic acid (obtained
from Molecular Probes Inc., Eugene, Oregon).
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 23 -
Example 10A describes a direct-cell-entry ~Ape,il-ltnl with cultured eukaryotic cells. In
this experiment, cells were treated for a few minutes with a representative polymer composition
of the invention tagged with 5-callJoxynuorescein. When the extrarP~ m~illm was neutral,
no cytosolic entry was observed. However, when the pH of the extra~P~ r m~lillm was
5 reduced stepwise to emulate the proglessive pH reduction which occurs in the endosome, the
polymer was seen to rapidly enter the cytosolic c.~lllpal Llllent, evidenced by diffuse fluorescence
throughout the cells.
B2. Entry via Endocytosis
After direct-cell-entry studies have dr~..n~cl ~ aled that one or more polymers are effective
10 for L~ brane delivery of a selected col.lpoulld, cell entry via endocytosis may be ~c.sPc~ed
by methods such as that described in Example 10.
Figure 2 illustrates the entry of a polymer-conlp~ulld composition of the invention into the
cytosol of a eukaryotic cell by endocytotic uptake. From the figure, it is seen that entry into
the cytosol of eukaryotic cells comprises multiple steps, principal of which are the initial
15 endocytotic uptake and the subsequent tr~ncmPmhrane passage from the acidihed endosome to
the neutral cytosol.
When a fluorescent-tagged polypeptide is endocytosed into cells, observation of a
perinuclear punctate pattern in~ir~tes that the tagged material is localized in the en-lnsom~ or
endQsom~l/lysosomal co~ ~Lmelll. Thus, a diffuse fluorescence throughout the cell could
20 indicate that the polypeptide achieved the desired en~iosQmP, to cytosol l~d-lsl,olL. However, in
the case of a polypeptide assembled from L-amino acids, such a pattern could also indicate that
the polypeptide was degraded by Iysosomal enzymes and only the fluorescent tag diffuse-d into
the cytosol. Therefore, it is prererable that cytosolic entry of such polypeptides via an
endocytotic route be ~ccP~csed by a fi~nrtion~l assay for the drug component of the polypeptide-
25 drug product, as desrrihed inExample 10. Alternatively, initial endocytotic entry studies,directed toward d~ ol~clld~ii.g and O~J~illli~illg endocytotic delivery of selected polypeptides,
can be carried out with polymers assembled from D-amino acids, which are not degraded by
Iysosomal enLy---es. Studies in support of the h.~..lion have shown that D- and L-polypeptide.
having the same se(luenre exhibit the same partitioning and ,..t..,bral.e transport properties.
In this Example, the compound linked to the polypeptide is a sequPnce-specific nucleic
acid-binding polymer, specific~lly~ a nonionic ~nticpn~e oligomer (Su-l-,..tl~on and Weller)
targeted against a specific mpc.cpnger RNA for firefly luciferase, coded by a plasmid co-ll~h.ed
in the treated cells. If the ~ntiC~Pme oligomer gains access to the cytosolic co--.l)all.l-ent of the
r~cled cells, a cignifir~n~ re~hlrtion in luciferase activity upon d~ h~corlP induction,
35 relative to untreated cells, should be observed.
CA 022~2706 1998-10-27
WO97/40854 PCr/US97/07335
- 24 -
lhe polymer-co~ ,ùund composition was contacted with cells for a period of time
a~lu~ t; for endocytotic uptalce, about 5 hours. Also tested were the polypeptide alone, the
~ntiS~n~e oligo alone, and the medium alone (control). As described in the example, the
~nlicP..ce oligo alone failed to inhibit its targeted m~-sse~ger RNA, p,~u..,ably because it was
5 unable to enter the cytosol of the cells. In contrast, the same ~ntisen~e oligo linked to the
polymer of the invention inhibited luciferase activity by about 31% relative to the control,
suggesting successful delivery of the co~ nJulld into the cytosolic Colll~alLI~
Enhancement of Endocytosis by an Attached Affinity Moiety. The polymer compositions
of the invention carry a relatively high density of negative charges in the ec.~enti~lly neutral
10 extracP~ r mP~ m Further, due in s~bst~nti~l part to the sialic acid residues on the
glycocalyx, the outer surface of a eukaryotic cell also typically carries a sl~kst~nti~l density of
negative charges. Possibly because of electrostatic repulsion between the like-charged polymer
and cell surface, the rate of cell entry of some polymer-compound products via fluid-phase
endocytosis appears to be relatively slow.
Further experimental results, described below, suggest that the rate of endocytosis can
often be Pnh~nred by using a moiety with an affinity for cell surfaces, such as a lipid anchor
(i.e., a lipophilic molecuie such as a fatty acid, long-chain alkyl amine, long-chain alcohol,
etc.) linked to or complexed with the polymer-col"l,uund co",~osiLion. Such lipid anchors
likely serve to increase the concentration of the polymer-col"pound at the cell surface, such that
20 upon invagination of the cell l"e"~ e to form the endosome, a larger amount of polymer-
col"~uul,d is enveloped therein than would otherwise be the case.
Where the lipid anchor is bound to the polymer via ele-,llusl~ic attraction, such attractiûn
will be elimin~tçd at low pH, i.e. within the late stage endosome. In cases where the lipid
anchor is covalently linked to the polymer-compound, it is generally desirable that the linkage
25 be cleavable in the cytosol of cells so that the polymer-compound is released free in the cytosol
rather than if~ 5h~ ,e Iinked to the lipid anchor embedded in the ~n~osom~l lllell,blane.
In a further e,~l e~ also described in Example 10, using ~e polypeptide-~nti~çnse
oligo composition described above, a tetracationic lipid anchor ~TI~1~CL~UII~, from ~lolueg~
Corp., Madison, WI, USA) was added to enhance the endocytosis step by complexing with the
30 polyanionic polymer composition. This is illustrated in Fig. 12, where the lipid anchor is
shown at 30. In this case, luciferase activity was inhibited by about 68% relative to the
control, compared to 31%, above, witbout the lipid anchor.
Figure 12 illustrates the likely role that a lipid anchor such as Tr~n~fect~m~ plays in
~nh~n~ing the initial endocytotic step, followed by its dissociation from the polymer when the
35 pûlymer COIlv~ to its low-pH non-ionic form in the late-stage endosome.
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 25 -
It should be ap~le-;ialed that nu,-le.ous compounds, such as lipophilic drugs (e.g., Taxol~M
and cyclosporin A), serve to enhance endocytosis due to their inherent lipophilicity. In such
cases a separate lipid anchor is generally not required.
Endocytosis may also be piol-,oled by ~ hmPnt of a ~c~tor signal, or ligand, to the
- 5 polypeeptide-co-"~-1ui-d composition. In r~c~lur-l~p~di~ed endocytosis, the ligand, ~tt~ch~d or
complexed to the composition targeted for uptake, is capable of binding to a ,~c~lor on the
cell surface. Such a ligand may be used to enhance general endocytotic uptake, or to target
specific cell types, as ~i~c~c~e~ further below.
IV. Applications
The polymers of the invention can be used to target delivery of attached compounds to
specific cells or locations within the body. The specificity of targeting may be controlled by
exploiting the pH differential between the cytosol and extracellular mPAillm or by the use of
targeted cell surface rece~,lo.s
A. Treatin~ H pylori h,reclion
If a target cell exists in an acidic environment, direct entry into the cell is f~ it~t~pd by
the pH dirre.enlial between the extrace~ e.-~i-ol----ent and the cytosol of the cell, without
requiring entry via endosomes. Such direct entry is generally much faster than endocytotic
entry. Examples of such targeting in low-pH e--~d.olJI--e-~ are given below.
The very acidic enviro~ JJl of the stomach may be speci~lcally exploited, as in l.e~ll"c.-
of H. pylori infection, by using a polymer composition effective to partition into a membrane
at a pH less than about 4.5 Other con~ ---ents in the body, inclutling the en~osom~l
compa.ll..e,-~ of eukaryotic cells, which have a pH in the range of 4.5-6.5, do not reach this
25 low pH and so fail to convert the polymer cor-~osilion to a lipophilic state required for passage
across their lipid layers.
Although a polypeptide with a higher transition pH, e.g. up to 6.5 or 7.0, would still enter
H. pylo~i cells, it could also enter other cells via endocytosis, with a ~~,.,ulli,,g decrease in
selectivity.
In order to penetrate a bacterial cell, a co.. ,po~ilion must be able to cross the outer
membrane (in gram negative bacteria), the cell wall and the inner plasma membrane The
bacterial cell wall generally eYrllldels entry of globular compounds of 2000 Da or more.
StudiP s in support of the invention indicate that the polypeptide co---l,o~ilions described herein,
though generally of higher mnleclll~r weight than 2000 Da, sllccP-~fi~lly pcnPl ,~le H. pylori
CA 022~2706 1998-10-27
W O 97/40854 PCT~US97/07335
-26-
cells. Rec~--ce the co-l-po~ ions, at low pH, are primarily linear rods rather than globular, it
is postnl~t~d that they pen~laL~ the cell wall via a reptation mPrh~nicm
Drugs that have been used to target H. pylori include amoxicillin, bismuth salts,
metronidazoline, olll~,p~azole, clarilhlon.ycin and tetracyclines. Therapy with a single drug has
5 not always been effective, due to pH effects and/or in~leql~te conce~ àlions of the drug at the
infection site. Triple-drug therapy is therefore fre~uently recol"...~ P-I using the combination
of a bismuth salt (e.g. bismuth subsalicylate), an anti-infective (e.g. metronidazole), and either
tetracycline or amoxicillin. (See e.g. Dixon, O'Connor, Glupczynski.) Because the present
method affords more selective l~rg~ing against the pathogen, higher doses of drug and/or more
10 cytotoxic drug may be delivered, thus providing greater efficiency of both single- and multiple-
drug re~i---~.ls, and a Irca~ e~ll period shorter than the 3~ weeks generally required for
eradication.
This application is one in which benefits could be gained by m~int~ining the polypeptide
intact after delivery of the compound. Specifir~lly7 the intact polypeptide-co.l,pou-ld
15 composition is unlikely to be absorbed into the stomach or i..~ l wall, thus preventing
delivery of the ~t~rhpd compound to non-targeted body sites. As noted above, a polypeptide
composed of D-amino acids is not degraded by proteolytic el~y.lles and thus would be
prerellcd for this purpose. A relatively stable compound-polypeptide linkage, such as an
amide, would also be prer~ ,d in this instance.
B. Tar~etin~ Tumor Cells
Most solid tumor masses contain cells which are hypoxic (oxygen deficient), due to
insufficient blood supply. This hypoxia makes such cells more lesi~la..l to both radiation and
drug therapy (see e.g. Kermedy). Delivery of ~ntir~nrPr drugs can also be h..~aire~ by the
25 limited v~cc~ -re.
The extracellular en~,hv.-n.c..l within such solid tumors has been shown to be more acidic
than normal tissue (e.g. Kennedy, Tannock, Vaupel) due to factors such as the production of
lactic acid under - -~.obic conditions. Measuled pH's are typically in the range of 6.0 - 7.0,
or, in general, about 0.3 to 1.0 units less than in the collc~.~ollding normal tissue (Vaupel).
The extr~pl~ r medium of tumor masses also appear to be of lower pH than the
intr~e~ r Colll~al L~ ,.lL of the tu nor cells themselves (Newell). For example, measul ~lll.,.ll~
of pH by 31p NMR, which Ill~.aSUl~ primarily the intr~ce~ r pH, showed that brain tumors
- and sarcomas had a higher intr~e~ r pH than the coll~ g nor nal tissues (Vaupel).
CA 022~2706 1998-10-27
W O 97/40854 PCT~US97/07335
-27-
ln accordance with the present method, hypoxic tumor cells, by virtue of their low pH
envi~o,u,-e.,l, may be targeted by an antineoplastic drug linked to a polypeptide which is
effective to partition into a membrane at the pH present in this extracellular e"~ilo"l,lent. Such
drugs include, for example, cis-platin, ~ntimet~llolites such as methotrexate and fluorouracil,
S topoisomerase inhibitors such as doxorubicin, alkylating agents such as cyclophnsph~mi-le and
chlo~ l,u(il, and tubulin-binding plant alkaloids such as vinhl~ctinP, vinrristin~, docetaxel,
and paclit~Ypl (TaxolT~.
A polypeptide effective for the targeting of hypoxic cells, as described above, could also
undergo pH-dependent transition to its lipophilic form in a late stage endosome, where the pH
10 range is typically about 4.5-6.5. However, direct cell entry is generally much faster than
endocytosis. In direct cell entry experiments using suitable pH dir~erenlials~ transport occurs
within a matter of minutes; see, e.g., Example 10. Appreciable endocytotic entry, on the other
hand, generally takes many hours. Therefore, endocytotic uptake by non-targeted cells would
be minimal relative to direct entry into the targeted cancer cells, driven by the existing pH
15 dirre~e..-ial between the extracp~ r medium and intracellular co",pa~ n~.
C. Treating/Preventing Tooth Decay
Tooth decay (dental caries) is promoted by the production of acid (acidogenesis) by
bacteria in breaking down carbohydlales. Studies of pH in sucrose-induced plaque (Igarashi)
20 showed a minimllm pH of 4.6 i 0.2 in 2-day-old plaque; the pH increased to 5.7 after 21
days. The dominant bacteria were Streptococci (>50~0 of total) and Actinomyces (> 10~ of
total). In another study, bacteria taken from caries-active sites produced a final pH of 4.2 or
less in sugar broth (van Houte).
,~ntih~rterial agents may be targeted to the site of decay or potential decay according to
25 the mpthot1~ described above, using polypeptides ~live to partition into a membrane at a pH
in the range of about 4.5 to 6.5, and preferably in the range of about 4.5 to 5.5. Suitable
~ntibactPvrials that have been used in the treatment of dental caries include chlorheYir~inP~
triclosan (FriPAm~n Bouwsma), xylitol, ~ntih~tPrial c.-Ly~~, and amine fluorides such as 9-
oct~decen-l-amine h-ydronuoride and 1-heAadecylamine hydlonuoride.
D. Targeting Cell Surface Receptors
Drug delivery via endocytosis may be ~.o,n()led by linking or complexing the polymer-
drug composition to a suitable ligand, or receptor signal. Use of such a ligand can provide
cell-~g~,.ing versatility because certain receptor signals, such as m~nn-)se-6-phosrh~P~ biotin,
35 folic acid, and other water soluble vitamins, afford delivery to many cell types (Ludwig) and
.
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/0733S
- 28 -
thus may be used to promote cell entry in many applications. Others may be used to focus
delivery into one or a few specific cell types, as ~iccussed further below.
It is generally desirable that the linkage between the polymer-drug composition and the
ligand be cleavable, so that after transport the polymer-drug can be released free in the cytosol.
Water-soluble vil~ullins such as riboflavin, ~hi~Tnine, nicotinic acid and folic acid can be
used to target cell surface lece~tol~. These co~ ,uullds are believed to be taken into the cell
by potocytosis, a variation of endocytosis that is speci~li7ed for the uptake of small molecules.
In potocytosis, receptors are present in small (approx. 50 nm ~i~mPter) pits or vesicles on the
cell surface, known as caveolae. These caveolae remain at or near the cell surface, going
through cycles of opening and closing. Upon closing, proton pumps within the membrane
produce a pH of about 6.0 within the caveola.
When the pH of the caveola becomes sufficiently low, the vitamin, with ~tt~hPd polymer,
is released from the receptor (Anderson). The polymer-compound composition then inserts into
the lipid ~-le,llb,alle, after undergoing a pH-dependent transition into a lipophilic con~olll.ation,
and thereafter transports into the cytosol, according to the mech~ni~m described herein.
Although this palllway does not typically involve as large a pH differential as the
endocvtotic paLhway~ it presents certain advantages. The caveolae do not merge with pre-
lysosomes, and thus potential exposure of the composition to degradative enzymes is avoided.
~urthermore, the water-soluble vitamin is generally released from the polymer when exposed
to low pH within the caveola, allowing free polymer-drug to enter the cytosol.
Cells that may be specifir~lly targeted by rec~lur signals include liver cells (hepatocytes),
whose surfaces contain receptors that specifically recognize g~ tose-t~-rmin~l glycoproteins.
Many m~lign~n~ cells o~ le~s certain receptors, and thus it may be possible to selectively
target such ce}ls, as has been reported using folate (Mathias) and epidermal growth factor
(Deshp~n~e). D-cycloserine has been reported to f~rilit~te transport through the cytoplasmic
l-lell.l.l~le of bacteria (Chopra, Rapaport).
E. Transport Across the Blood-Brain Barrier
The blood-brain barrier (BBB), which regulates the exchange of m~tP.ri~l~ between the
bloodstream and central nervous system, presents a formitl~ble barrier to drug Llau~OlL. The
endotltelial cells of cerebral capillaries contain "tight junctions", cir.;u.-.~elenLial bands around
a cell that are in close contact with adjacent cells. These jlm~tionc prevent transport between
cells, and thus, for effective transport, compounds must pass through the endothelial cells
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97107335
- 29 -
themselves. Studies directed to such l-a~ of peptides show that lipophilicity is probably
the most important factor in p.u~.,oling transport of a peptide across the BBB (Banga).
In general, compounds may be lla"~o-led across a cell by transcytosis. In the case of
polarized endothelial cells (i.e., cells having distinct apical and basolateral Ille...b.ànes) within
5 a capillary, the col,-~ound is first taken through the apical membrane in the inner capillary wall
into a transcytotic vesicle. Such a vesicle typically attains a pH of about 6Ø The vesicle
l~uls~l~ the col-.puund to the basolateral ll-e -~bral e of the endothelial cell, on the outer
capillary wall. The compound is then expelled from the transcytotic vesicle, thereby releasing
the co~,.poulld outside of the cell and the capillary.
In accordance with the present method, l,~sl,o-la~ion of a compound across the blood-
brain barrier may be effected by linking the compound to a polypeptide of the present invention
which is effective to partition into a membrane at a pH within a selected range, as described
below. The composition is preferably further linked to a rec~tor signal, e.g. hypoxanthine
or inosine, effective to bind to a rece~l(" on the surface of a cerebral endothelial cell. After
delivery to the cerebral bloodstream, the composition is l,~lspo,led across the capillary wall
via transcytosis, as described above.
The composition is then available to be taken up by a brain cell via endocytosis and
released into the cell cytosol, according to the mech~ni.cm~ described herein. In the latter
process, the endosome is expected to have a pH of about 5Ø
From the above description, it can be seen that the pH at which the polymer is effective
to traverse a cell membrane should be between 5.0 and 6.0, or, more generally, below the pH
of the transcytotic vesicle in an endothelial cell and above that of the endocytotic vesicle in the
ultimate target cell. If the pH were above 6.0, in this case, the polymer would assume its
lipophilic collrol...a~ion within the transcytotic vesicle and penetrate its ~"e",b~ e, thus entering
25 the endothelial cell instead of the targeted brain cell.
F. Transdermal Delivery
_The principal permeability barrier of the skin, the stratum corn~m consists of cornified
epithelial cells surrounded by an extrac~ lipid matrix. The eYtrac~ r lipid matrix,
30 which CO.~ ,S the principal route for p~sage of col-,youllds through the skin, consists of
lipids (ceramides, cholesterol, free fatty acids, and cholesteryl sulfate) ordered in multiple
sheets of lipid bilayers, with water and other polar compounds dispersed between the polar
faces of the stacked bilayers. This structure of multiple ~ g polar and nonpolar layers
presents a formidable barrier to penetration of both hydrophilic and lipophilic compounds.
35 Each of the multiple lipid bilayers acts as a sllbst:~nti~l barrier to passage of hydrophilic
CA 022~2706 1998-10-27
W O 97/40854 PCTrUS97/07335
-30-
co~llpoul-ds, while each of the layers of water and other polar co~ ou-lds between the polar
faces of the stacked lipid bilayers acts as a substantial barrier to passage of lipophilic
compounds.
It is known that the stacked bilayers of the extrace~ r lipid matrix can be disordered by
S a variety of penetration enh~n~P~s~ such as Azone (I-dodecyl-azacycloheptan-2-one),
unsaturated long-chain alcohols, and unsaturated long-chain fatty acids, such as oleic and
linoleic acid (Aungst et al., 1986; Rehfeld and Elias, 19~2; Golden et al., 1986; Goo~lm~n and
Barry, 1985,1986).
While treatment of the skin with p~clldLion enh~n~ers affords improved delivery of a
10 variety of relatively :Imphiphilic drugs, simple and effective tr~n~derrn~l delivery of many
lipophilic and hydrophilic colllpoullds is still not readily achieved by mPthodc known in the art.
In accordance with the present invention, a suitable polypeptide is contacted with the
surface of the epiderrnis in its low-pH lipid-soluble form. Because the pH of the skin surface
is typically in the range of 5.0 -5.5 (see, for example, Yosipovitch, Korting), polypeptides with
transition pH's in this range or higher will exist pre~ ly in their low-pH form on the
skin surface. The peptide, in its lipophilic forrn, is able to diffuse through the lipid layer of
the extracellular matrix of the stratum corneum.
Upon contact with the aqueous col-l~allll.c"~ underlying the stratum corneum, the
composition is actively drawn into this compartment by virtue of progressive ionization and
20 solvation of the polypeptide chain at the near-neutral pH of this compartment. Both hydrophilic
and lipophilic compounds may be transported. A ple~ d polypeptide for this application
undergoes a reversible pH-dependent transition at pH between about 5 and 6.5.
The polymer composition of the instant invention is preferably used in concert with one
or more suitable penetration f ~h~nl.r 1~, as described above. Lipophilic acids, such as oleic or
linoleic acid, may be added to m~int~in the polymer in its low-pH lipophilic forrn during
passage across the lipid layer. These fatty acids also serve as pe~ ldlion enh~n~ers, as noted
above.
Example 11 illustrates the use of a ~ res~.lldLi~,re polymer col..~o~ilion of the invention
to enhance transdermal delivery of the a.lli-~,je~lion drug, cyclosporin A.
G. Disposition of Polvpeptide after Colll~uoulld DeliverY
As noted above, the polypeptides of the invention are likely decomposed by proteasomes
after entering the cytosol of the cell, thus freeing the transported drug or other c~JIIl~oul~d.
This release of the drug is shown, for example, in Fig. 2.
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 31 -
In some inct~n~es~ it may be desirable for the polypeptide to remain intact, in which case
it should be synthP-ci7ed from D-arnino acids. One example is in delivery to the stomach, as
described for the er~tlic~tion of ~1. pylori, above. Another is in the selective delivery of highly
toxic drugs, e.g. to tumor cells. Because the l~ s~ollhlg action of the polypeptide is
5 unidirectional, proceeding from lower to higher pH, the polypeptide ~ chPA to the drug
p~ n~ the drug from diffi~cinf~ back out of the target cell to which it was first delivered.
Undesirable side effects, resulting from access to non-targeted cells, are therefore ...i-~h~ ed
The intact polypeptide may also prevent or ~ e exportation of the delivered drug by
cellular processes. Many cells express a Plycv~,olein which serves as a drug-efflux pump,
10 ~ Ollillg drug co~ vullds out of the cell. The attached polypeptide will likely i~ ,rel~ with
this process and reduce or prevent eAI)o,l~lion of the attached drug.
As an alternative to m~int~ining the entire polypeptide attached to the drug, the polypeptide
may be prepared with one or more D-amino acids adjacent to the drug, and the remainder L-
amino acids. The L-amino acids will be removed by proteasomes, leaving a shortened
15 polypeptide "tail" of D-amino acids ~tt~rhed to the drug.
It should be noted, however, that when D- and L-amino acid segmentc are joined, a local
disruption of the a-helix is likely to arise at the junction of such segments. In this case,
hydrogen bonding and shielding of free ca~lJol~yls in the vicinity may be less effici~Pnt~ and the
a-helical form of the polypeptide less lipophilic, than if all one configuration were used.
H. Formulation and ~dmini~tration
Formulations containing the compositions of the invention may be in solid, semi-solid, or
liquid dosage forms, such as, for example, tablets, capsules, powders, sllct~in~pcl-release
formulations, solutions, suspensions, Pmlllcion~, suppositories, oi ~ , lotions, or aerosols,
25 plef,.ably in unit dosage forms suitable for simple ~tlmini~tration of precise dosages.
Such formnl~ions typically include a conventional ph~ ''e~ c~l carrier or excipient and
may additionally include other mpAicin~l agents, carriers, or adjuvants. Preferably, the
forml~ ion will contain about 0.5% to 75% by weight of a compound or compounds of the
invention, with the rrm~in~3er consisting of suitable ph~rm~elltic~l excipients. For oral admin-
30 istration,suchexcipientsinclude~h~rm~relltir~lgradesof m~nnitol, lactose,starch,m~gnPsjllmst~rate, sodium sacch~rinp7 talcum, ce~ ose, glucose, gelatin, sucrose, magnPcillm c~bollale,
and the like. If desired, the composition may also contain minor a,,,~ull~ of non-toxic
auxiliary compounds such as wetting agents, emulsifying agents, or buffers.
Liquid compositions can be plepared by dissolving or dispersing the polypeptide-compound
35 composition (about 0.5% to about 20%), and optional ph~ e.~l;c~l adjuvants, in a carrier,
CA 02252706 1998-10-27
.
such as, for example, aqueous saline, aqueous dextrose, glycerol, or ethanol, to form a solution
or suspens~on.
The composition may be administered to a subject by a variety of known routes, e.g.,
or~lly, transdermally, as describeci above, or parenterally, e.g., by i~travenous, subcu~neous,
5 intraperitoneal, or intr~m~sc~ injection.
For use in oral liquid preparation, the composition may be prepared as a solution,
suspension, emulsion, or syrup, being supplied either in liquid form or a dried forrn suitable
for hydration in water or normal saline.
Tr~nc~tn~l delivery typically involves the use of a tr~ncderm~l ~patch~ which allows for
10 slow delivery of compound to a selected skin region. Fs~mples of transderrnal patch delivery
systems are provided by U.S. Patent 4,655,766 (fluid--~u~ibi~g osmotically driven system), and
U.S. Patent ~,00~,610 (rate controlled tr~n~denn~l delivery system).
Fortr~ncdPrrn~ delivery, itmay bedesirableto includeperrnP~tion ~nh~nring compounds,
as described in above. Such for~nulations may be provided as occluded dressings to the region
15 of intere t, or may be provided in one or more of the transdermal patch configurations
described above.
~ :or pare.~t~al arlminictration~ an injectable composition for pare~te.al a~minic~-ation will
typically contain the composition of t'he ~e.lLion in a suitable IV solution, such as sterile
physiological salt solution. The composition may also be form~ ted as a s~spension in a lipid
20 or phospholipid~ in a liposomal ~uS~ChSiOu, or in an aqueous emulsion.
Methods for pl~;~J~U~g such dosage forms are known or will be apparent to those skilled
in the art; for example, see Remin~ton's Pl,a~ ac~L,Lical Sciences (1980).
The following examples ~ ctr7te b~ in no w~y ore ..~ ~d to 1;~ the present invention.
EXAMPLE 1
Re~resentative Methods for Prep~ ion of the Pol~mer Com~osition
A. Assembly
A synthesis resin is prepared so that ~-alanine will comprise the C-ternlinal residue of the
polypeptide, as illustrated in Figure 10. One gram of l'~o crosslinked polystyrene resin
cont~ining 0.7 mMol p-alkoxybenzyl alcohol, I (Cat. ~ A3039, Sigma Che;~s. Co., St. Louis,
MO) is dissolved in 8 ml of N-methylpyrrolidinone tNMP), and 0.6~ g of fluorenylmethoxy-
carbonyl (FMOC) ~-alanine ~ is added, followed by 316 ~1 of N,N'-diisopl opyl carbodiirnide
and 41 1~l N-methylimidazole. This slurry is inclJb2t~d with agitation at 37~~ for 100 minutes,
Al~llENDED SHEEl-i
ID~AlEP
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 33 -
then washed thoroughly with NMP, followed by CH2Cl2, drained, and dried. This affords a
resin 3 with a loading of about 250 ~mole l~-alanine-FMOC per gram of material. Suhlseq~1Pns
ition of protected/activated amino acids to extend the polypeptide can be carried out as per
Atherton et al. (1988). This method uses N-fluorenylm~thoxy- C~bOll~lpent~flllOrOphenyl
5 amino acid esters, as shown at 4. If desired, an end modifying sl,ucluL~ may be added, as
shown in Figs. 8B-C, where an activated ester 5 is reacted with the N-te~ c of the
polypeptide. The polypeptide is then cleaved and deprotected according to standard m~th~
When it is desired to attach the compound to be transported (typically a drug) at one or
more positions other than the terminus of the polypeptide, a suitably-protected Iysine or
10 cysteine is typically incorporated at the selected ~u.~ position(s). Following cleavage of
the completed polypeptide from the synthesis resin and si~lerh~in d~luleclion, the drug can be
~ttached to the resultant amine or sulfhydryl moiety, as described in Example 2, to afford
linkages illustrated in Figure 9.
Alternatively, certain drugs may be linked, suitably protected if necessary, to the gamma
15 carboxyl of glnt~mic acid, or the ~-carboxyl of aspartic acid, via an ester linkage. The
resultant amino acid is then incorporated into the polypeptide chain at one or more selected
positions. Such drugs must have structures which survive the conditions used to assemble the
polypeptide, to cleave the polypeptide from the synthesis resin, and to deprotect the side chains.
20 B. End Modifications
In cases where the compound to be transported is not linked through the N-terminal amine,
it is generally desirable to shield or delete at least some of the N-terminal polar sites. This is
readily achieved by cleaving the FMOC moiety from the N-tPrminl-c of the completed resin-
bound polypeptide and then treating with glutaric anhydride, acetic anhydride, or the
25 nitrophenyl ester of the succin~rni~e derivative shown in Fig. 6E, to give termin~l structures
such as illustrated in Figures 6B, 6C, and 6D, respectively.
C. Cleava~e from the Synthesis Resin. D~oro~clion and ~solation
The completed polypeptide can be cleaved from the resin by washing the resin with
30 CH2Cl2, draining, and adding, per gram of resin, a solution con,~ g 10 ml trifluoroacetic
acid ~FA), 10 ml CH2Cl2, and 400 mg dithioe~ ol. After 20 minutes the cleavagesolution is drained into a flask and the CH2CI2 removed by c;vdpGld~ion. Thereafter, 5 to 10
- ml TFA is added and the solution held at 43~C for 4 hrs to effect removal of p~l~live groups.
Workup of the d~role.;led polypeptide generally entails ether precipitation, thorough washing
35 of the pre~ a~e with ether, and drying.
CA 02252706 1998-10-27
W O 97/40854 PCT~US97/07335
-34-
It should be appreciated that there are a variety of other peptide assembly methods known
in the art which are also suitable for prepalillg the co~ osi~ion of the illvelllioll.
EXAMPLE 2
~tt~hin~ Coll,~Juu-~ds to Polypeptide on Column
For drugs or other compounds (e.g., a fluorescent tag for p~lilio~ g and cell entry
studies) which can survive the conditions used to cleave the polypeptide from the synthesis
resin and deprotect the sif~ef~h~in~, it is often desirable to cleave the FMOC from the N-
te~ of the completed resin-bound polypeptide and then link such compound to the terminal
amine. In such cases the co~ u~ld to be attached is typically activated, by methods known
in the art, to produce an active ester or active carbonate moiety effective to form an amide or
call)~l.d~e linkage, respectively, to the polypeptide, such as illustrated in Figure 9.
Examples 3-7~ following, describe representative methods for ~tt~hin~ compounds to
polypeptides after
removal from the column.
EXAMPLE 3
Disulfide Linka~e to Cysteine Residue of Polypeptide
In a convenient and well established method for linking a drug to a cysteine of the
polypeptide composition, the de~role-,led polypeptide is reacted with 2,2'-dipyridyl ~ ulfi~e,
and the drug containing a sulfhydryl moiety is added to form the desired ~ ulfirle link, as
illustrated in Figures 7 and 11. A particular advantage of such a disulfide linkage is that it is
relatively stable in the extracel]ul~r com~alLIllent and within endosomes, but after llal~oll
across the endosomal membrane it is readily cleaved in the cytosolic compartment. The
following specific examples illustrate applie~tion~ of this method.
3A. Prepald~ioll of a disulfide-linked polypeptide-cyclosporin conjugate. A polypeptide
with the se~u~nre (FMOC-ELLD-[LELLD]7LELL~ B-alanine) is assembled on the solid
phase support by the methods given in Example lA. The tçrmin~l FMOC group is cleaved by
.lel~l with 20% piperidine in NMP and a terminal S-tritylated FMOC-cysteine is introduced
by the method of Example IA. Following cleavage of the FMOC group and acetylation, the
polypeptide is cleaved from the column, d~role~L~d, precipitated, and washed as per F~r~mrle
lC. The polypeptide is dissolved in pH = 7.5 tris buffer containing 0.1 M diShiothreitol. The
solution is diluted with an equal volume of aceLorliLIile and treated with suffil~ient dilJylidyl
- ~li.cl-lfi~e to create a 0.4 molar solution. After stirring for 2 hours at room telll~elaLure, the
mixture is diluted with an equal volume of water and washed with sumcient water-saLulaLed
ethyl acetate to remove excess dipyridyl ~ ulfide. The solution is partially t;val,o.aLed to
CA 02252706 1998-10-27
W O 97/40854 PCT~US97/07335 -35-
remove residual ethyl acetate and the product isolated by passage of the solution through
Amberchrome ~TosoHaas) followed by elution with 0-80% acetonitrile with a 0.01% triethyl-
amine buffer followed by lyophili7~~inn.
In a separate flask, cyclosporin A metabolite 17, which has a primary hydroxyl group
5 (Eberle and Nuninger, 1992), is treated with succinic anhydride using the method of Chen and
Tai (1995), and the acid is conv~led to the activated ester with N-hydro~ .,ccinimide and 1-
(3-di-llelllyla"lillol)lo~yl)-3-ethylcarbodiimide m~thiodi(le (Aldrich). The product is dissolved
in DMF and treated with 2-aminoeth~nethiol hydrochloride and triethylamine. The reaction
mixture is diluted with dichlornm~-thAne and washed with 0.1M citric acid to remove excess
10 reagell~. The product is obtained by evaporation. The residue is dissolved in DMF and
treated with the pyridyl polypeptide (li.~ulfide from the previous p~la~h.
The product is isolated by evaporative removal of the DMF followed by reverse phase
purification on A.J,beLchrol.,e eluting with 0-80% ~celc-nill ile in a 0.01% triethylamine buffer
followed by lyophili7Ation. All~,..lalively, following ~vapolàlion of the DMF, the product is
15 dissolved in 0. lM Na HPO4 and washed well with water-saturaeed ethyl acetate. After partial
evaporation to remove the dissolved ethyl acetate, formic acid is added to precipitate the
product, which is washed well with water containing 0.1% formic acid, then dried thoroughly
under high vacuum.
3B. Preparation of a disulfide-linked polypeptide-TaxolTM conju~eate. A polypeptide with
20 the sequpnce (AcNH-CELLD-[LELLD],I,ELL,B; ~=~B-AIanine) with aterminal pyridyl di~ulfi~e
moiety on the cysteine is prepared as in the previous section.
In a separate vessel, TaxolTM is converted into 7-glutaryl TaxolTM by the method of
Gueritte-Vogelein et al. (199i). The carboxyl group of this species is activated as the N-
hydlo~y~uccinim~le ester with N-hydro~y~uccinim~e using 1-(3-dimethylami..op,o~yl)-3-
25 ethylcarbodiimide methiodide (Aldrich) and 4-dimethyla-..in~ylidine in dichloro-l .~lhAn~. The
product is dissolved in DMF and treated with 2-aminoeth~nPthinl hydrochlori-le and
triethylamine. The reaction mixture is diluted with dichlorom~th~ne and washed with O.lM
citric acid to remove excess reagents. The product is obtained by e~a~o~lion. The residue
is dissolved in DMF and treated with the pyridyl polypeptide ~i~ulfide from above. The
30 product is isolated as per the cyclosporin example.
EXAMPLE 4
Thioether LinkaFe to Cysteine Residue of Polypeptide
4A. Preparation of a thioether-linked polypeptide-cyclosporin co--ju~ale. Cyclosporin A
metabolite 17 (see Example 3A) is treated with chloroacetic anhydride in 1:1 dichloro~ r/
35 pyridihe to form an acid chloride (see Fig. 11). The excess reagent is qnen~h~d by the addition
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 36 -
of water, and the solvent is evaporated. The residue is dissolved in ethyl acetate and washed
with 0.1M citric acid, 0.1M sodium bicallJonà~, and brine, then evaporated. The residue is
redissolved in DMF.
The polypeptide pyridyl ~ -lfi-le prepared in Example 3A is dissolved in pH 9.0 borate
buffer. This is mixed with an equal volume of the above DMF solution (ratio of CS:polypep-
tide = 4:1), and TCEP (tris(carbu~-y~Ll-yl) phosphine hydrochloride) is added. After stirring
at room temperature, the product is isolated as in FY~mrle 3A.
4B. Preparâtion of a thioether-linked polypeptide-TaxolTM conjugate. A polypeptide with
the sequ~nre (FMOC-ELLD-[LELLD]7LELL~B; ~ -alanine) is pl~l,~ed on a solid support
as in example lA. The terminal FMOC group is removed and the column treated with acryloyl
chloride in dichloromPth~ne containing diisopropylethylamine, to form a terrnin~l acrylyl group
(Fig. ll). The po}ymer is removed from the column, d~role~;Led using ~,inuioaceLic acid
according to standard mPtho~, precipitated and washed. The product is dissolved in pH 9.0
borate buffer.
In a separate flask, TaxolTM is converted into a thiolated species by the methods in
Example 3A. The product is dissolved in DMF, mixed with the acrylamide species from the
paragraph above, and TCEP is added. After stirring at room temperature, the product is
isolated as in Exarnple 3A.
EXAMPLE 5
Calba,-,a~e Linkage to Amine Moiety of Polypeptide
The ether precipitate of a d~L,roLe-,Led polypeptide conLailli.Jg one or more amine moieties,
prepared as in example lC, is dissolved in aqueous 0.1 M Na2HPO4. Formic acid is added
sufficient to precipitate the polypeptide, and the precipitate is collected by centrifugation and
washed twice with 0.1% formic acid in water. The polypeptide precipitate is then dried under
high vacuum overnight.
The compound to be linked to the polypeptide, co~ at least one hydroxyl moiety,
is activated by reacting with 3 equivalents of bis-nikophenyl carbonate and 0.1 equivalent of
triethylamine in NMP. After activation, the excess bis-nitrophenyl calbonàLe is removed and
the active carbonate product (see Fig. 11) then reacted with 0.5 to 2 equivalent of the
polypeptide composition, prepared as described above, in NMP. Good coupling is generally
achieved by in~ub~ion for 12 to 72 hours at 43~C.
EXAMPLE 6
Amide Linkage to Amine Moiety
6A. P,el.ald~ion of an amide-linked polypeptide-cyclosporin conjugate.
CA 022~2706 1998-10-27
W O 97/40854 PCTrUS97/07335
- 37 -
1. Using a defined length peptide. A polypeptide with the seq~lPnce (FMOC-ELLD-
[LELLD]7LELL,~ -alanine) is assembled on the solid phase support by the methods given
in example lA. The terminal FMOC group is cleaved as per the method in example lA and
the resin treated with an excess of the bis-(4-nitrophenyl) ester of glutaric acid in NMP. ~ e
S ester is prepared from glutaryl chloride and nitrophenol using triethylamine in dichlo~ûl~.eLl.~
Following washing with sodium hydroxide solution to remove excess nitrophenol and ul~eacl~d
glutaric acid species, the evaporated product is recryst~lli7Pd ~rom toluene.) The activated
polypeptide is cleaved from the column, precipitated, and washed using the method in example
lC.
In a separate flask, cyclosporin A metabolite 17 (see Exarnple 3A) is treated with the
FMOC derivative of glycine and 1-(3-dimethyl~llhlopru~yl)-3-ethylcarbo~1iimi~1e mPthiodi~e
(Aldrich) in dichlorompt-h~np in the presence of 4-dimethyl~-lino~y,idine catalyst. The reaction
mixture is washed with acid and base to remove excess leage..Ls and the product chro~"~Lo-
graphed on silica gel using 0-2% meth~nol in chloroform. Removal of the FMOC group is
accomplished by ll~a~ llL of the FMOC-glycyl cyclosporin with 20% triethylamine in DMF
at 50 ~egrees C for one hour. The triethylamine is removed by evaporation under vacuum and
the free amino derivative mixcd with the activated polypeptide above. The solution is
evaporated in vacuo to a minimllm volume and allowed to sit at 42 degrees for 24 hours. The
product is isolated as in Example 3A.
2. Using a Random Len~h High Glutamic Acid Content Peptide. A high glutamic
acid content, random length peptide with C-terminal initiator region, is prepared as follows.
Chlorotrityl resin preloaded with N-Fmoc-~-alanine (Novabiochem, LaJolla CA) is reacted
with N-o~-Fmoc-L-glutamic acid ~-t-butyl ester and N-cY-Fmoc-L-leucine using HBTU
(Novabiochem) and diis-,plopylethylamine to produce FMOC-LeuGlu(OtBu)l el~T eu-
NHCH2CH2COO-Resin. The Fmoc group is then removed in the usual manner.
The resin bound initiator peptide is treated six times with the carbo~ya.~hyd,ide of L-
glut~mir acid ~-(4-methoxy benzyl) ester (Hanabusa et al., 1984) in DMF, using an anhydride
to initiator ratio of about 10 to 1, which is s~ffirient to add about 40-50 prole~Led glutamic acid
residues to the peptide. The N-l~ ...in~c is then reacted with excess bis-(p-nitrophenyl) ester
30 of glu~aric acid.
The peptide is cleaved from the resin and deprotected as described in Example lC. The
residue after evaporation is dissolved in dichlorompth~ne and washed with water to remove
- traces of L.inuoroacetic acid. The isolated polyacid is dissolved in DMF and treated with
TaxolTM-7-alanine (Mathew et al., 1992). The reaction mixture is diluted with pH 7.0
35 phosph~te buffer and the product purified by ion exchange chromatography on Q-Sepharose
CA 02252706 1998-10-27
W097/40854 PCT~S97tO7335
-38-
(ph~rm~ri~ Piscataway, NJ) using a 0-1 0 M NaCI gradient. The material is then isolated as
the sodium salt by adsorption to an Amberchrome SD (TosoHaas), elution with a 0-80%
q~etc)nitrile gradient, and lyophili7~tion
6B. Plel~aldlion of an amide-linked polypeptide-TaxolTM conju~ate A polypeptide with
5 the sequence (H2N-ELLD-[LELLD]7LELL~; ~B=~-alanine) is ple~a ed as in Examples 1 and
5 with a t~rmin~l amino group.
In a separate vessel, TaxolTM is col-v~lled into 7-glutaryl TaxollM by the method of
Gueritte-Vogelein et al. (1991). The calLO~yl group of this species is acLiva~ed a~s the N-
hydlo~y~uccinimde ester with N-llydloxy~uccinim~le using 1-(3-dimethylarninopiopyl)-3-ethyl-
10 carbodiimide methiodide (Aldrich) and 4-dimethylaminopyridine in dichlorometh~nP After
washing to remove excess reagents and byproducts, the ester is isolated by evaporation and
mixed with the polypeptide in DMF The solution is evaporated in vacuo to a minimllm vol-
ume and allowed to sit at 42 degrees for 24 hours The product is isolated as in Example 3
6C ~reparation of an amide linka~ee between a polypeptide and the S' te~ US of a 20-
15 mer Morpholino antisense oligo. Structures and subunit sequ~nr~-c of the Morpholino oligo and
polypeptide are as follows, with l~relence to Figure 13
Rl= -ELLDLELLDLELLDLELLDLELLDLELLDLELLDLELLDLELL~
where D = aspartic acid, E = glutamic acid, L = leucine"B = ,~-alanine
R2 = 5'-G-G UG G UUC-C-UUC UC~A G-UC-G G--acetyl
20 where
A- = Morpholino 6-benzoyl~nine
C = Morpholino 6-benzoylcytosine
G- = Morpholino 6-phenylacetylguanine
U = Morpholino uracil
Procedure 1 (Figure 13A) 17 mg (2 ~Mole) of base-plo~e~;led Morpholino antisense oligo
(MW 8361), shown in Figure 13A at 1, p~ ed as per Summerton & Weller (1993), is
suspended in 200 ~I NMP Bis(p-nitrophenyl) sllccin~te 2 (7 2 mg; 20 ~Mol) is added, and
the prepala~ion is inrllhated for 4 hours at 43~C. The ulllcacled surcin~te is removed by
30 p~eci~ i.lg the Morpholino-succinate product from 30 ml of ar~lon;l ile, centrifuging,
discarding the sup~ resuspending the pellet in 0 4 ml of NMP, adding to 30 ml of
ac~Lo~ ,ile, c~ .iru~ h g, discarding the supçrn~t~nt and drying ~e pelleted Morpholino-
succin~te product 3 under high vacuum
The Morpholino anticence oligo with ~uccin~te linlcer 3 (2 ~Mole), prepared above, is
3~ placed in a 0 7~ ml vial cont~ining a 3 mm m~gnPtic stir bar with 31 mg (6 ~Mole) of the
deprotected 44-amino acid polypeptide Rl-NH2, as defined above, shown at _, precipitated from
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 39 -
aqueous solution as described in Example 2B(iii), having a free amine moiety on the N-
~e ..,;..- c. DMF (150 ~L) is added and the mixture stirred in a warm water bath till dissolution
is complete. The reaction mixture is then incubated at 43 ~C for 72 hours, diluted with 200 ~L
NMP, and ~ f~l-ed to a 2 ml screw cap vial. 600 ~L of conc. NH40H is added and the
S solution ineub~ed 18 hours at 43~C to deprotect the purine and pyrimidine bases of the
Morpholino ~nticence oligo. The product 5 is purified as described in Example 8C.
Procedure 2 (Figure 13B): In this procedure, the activated sUccin~te linker is added to the
polypeptide, and the adduct is reacted with the Morpholino A~lict ~ce oligo. Accordi.l~ly, an
NMP suspension of 180 mg of synthesis resintpolypeptide, p~ed as in Example 1, is treated
10 with 20% piperidine in NMP, then washed repeatedly with NMP. Bis(p-nitrophenyl) succin~e
(150 mg) is dissolved in 0.9 ml NMP, added to a short fritted column containing the
resin/polypeptide p.~"lion and incubated 2 hours at 43~C. Excess succin~te linker is washed
out and the product cleaved from the synthesis resin, as described in Example lC, to give a
polypeptide-succin~te product (MW 5329).
Into a 0.75 ml vial with a m~nPtic stir bar are placed 32 mg (6 ~Mole) of the
polypeptide-succin ~e product prepared above and 17 mg (2 ~Mole) Morpholino antisense oligo
(1), co~ i..i.lg a 5' secondary amine moiety, and 150 ~L DMF is added. The mixture is
stirred in a warm water bath till dissolution is complete. The reaction mixture is then in~ batPd
at 43~C for 48 hours. Thereafter, the reaction mixture is diluted with 200 ~LL NMP and
t~ fe~led to a 2 ml screw cap vial. 600 ~L of con NH40H is added and the solution
int~ub~Pd 18 hours at 43~C to deprotect the purine and pyrimidine bases of the Morpholino
~nticPnce oligo. The product (O is purified as described in Exarnple 8C.
Procedure 3 (Figure 13C): Morpholino ~ntic~Pnce oligo is prepared wherein the first
subunit, col-l~illillg a 5'-SH, is linked to the synthesis resin via a dic~lfide bond, according to
mPtho~c known in the art. The 2vuthMorpholino oligo, with bases still prole~led, is cleaved
from ~he synthesis resin using N-methyl pyrroli~linone conlai~ g 1 % wt/vol dithiothreitol and
5% v/v triethylamine. The eluted oligo is precipitated with t-butylmethyl ether and the pellet
washed twice with t-butylmethyl ether and then dried under high vacuum to give base-protecled
Morphnlino oligo with 5'-SH and a 3'acetyl.
Polypeptide still on the synthesis resin, pl~aLed as illustrated in Figure 10, having the
se~uence E,~UF~M~.~ (N-te~...;.~..c to C-~e~ c)~ is capped on the N-t~-...i.mc using
chloroacetic anhydride in dichlorometh~ne, and the resin is washed thoroughly. The
- polypeptide is then cleaved and eluted from the resin with 49:49:2 TFA:CH2C12:H20, the
CH2Cl2 is removed under aspirator vacuum, 98:2 TFA:H2O is added, and the solution is
incuh3~ed from 4 hours at 43 ~C. The polypeptide is then precipitated with t-butylmethyl ether
CA 02252706 1998-10-27
W 097140854 PCT~US97/07335
-40-
and the pellet washed twice with t-bulyl~ lllyl ether and dried under high vacuum to give the
chloluace~ylated polypeptide shown in Figure 13C.
The Morpholino oligo and polypeptide are coupled by adding 5 ~mole 5'-SH Morpholino
oligo, 10 ~mole chloroacetyl polypeptide, and 400 ~mole N,N-dimethyl ethanolamine to 1 ml
S form~mi~lP" stirring until dissolution is complete, and then in~ubaLing at room l~,,,pelaluie for
2 hours to give polypeptide-Morpholino oligo joined by a thioether link, as shown in Procedure
3vuth, Figure 13C. The bases of the Morpholino oligo are d~iolecled by adding two volumes
of conrP~ dlion NH40H to the reaction mixture and jn. ukJ~ g 16 hours at 43~C. After
releasing excess NH3 the product is purified by anion exchange cl,ro,~àtdgraphy, as desrrihed
10 in Example 8C.
EXAMPLE 7A
Amide Linkage to Carboxyl Moiety:
This example describes the preparation of an amide-linked polypeptide-TaxolTM conjugate
with a random length, high glutamic acid content peptide.
A high glutamic acid content, random length peptide with C-terminal initiator region, is
first prepared as follows.
ChlordLIilyl resin preloaded with N-Fmoc-~-alanine (Novabiochem) is reacted with N-cr-
Fmoc-L-glutamic acid ~-t-butyl ester and N-a-Fmoc-L-leucineusing HBTU (Novabiochem) and
diisopropylethylamide to produce Fmoc-l.eu-Glu(OtBu)l ellT eu-NHCH2CH2COO-Resin. The
20 protected peptide is removed from the resin by ll~atlllell~ with 0.5% trifluoroacetic acid in
dichloromPth~ne. The residue is redissolved in dichloromPth~ne and treated with diphenyl-
diazometh~nP~K~mP.t~ni et al .) to produce the C-te~ al diphenylmet-h-yl ester, and the product
is precipitated with hexane. The residue is dissolved in dichlorompth~np and the product
precipi~l~d with ether/hexane. The precipitate is dissolved in DMF co..~ 20% (v/v)
25 pil,e.idi,.e. The solution is evaporated and the amino termin~ted initiator peptide is ready for
elongation.
The initiator peptide is dissolved in DMF and reacted witb the carboxyanhydride of L-
glllt~mic acid ~-(4-methoxy benzyl) ester (Hanabusa et a/.). The ratio of anhydride to initiator
was about 60 to 1, which is s~fflcipnt to add about 40-50 protected gl-~t~mic acid residues to
30 the peptide. The reaction was qu~onrhPd by the a ~ tinn of acetic anhydride, the solvent
~e.~ ved by evaporation, and the product purified by repetitive p~ iolls from dichloro-
mP,th~rlP using etherthexane.
The peptide is deprole~;led as described in Example lC, and prepared for conjugation by
dissolution in 0.1 M Na2HP04 followed by precipitation witb formic acid, as described in
35 Example 5. The polyacid is dissolved in DMF and treated with TaxolTM-7-alanine (Mathew
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 41 -
et a/.) and 1-(3~h~leLl~yl~lhloprul~yl)-3-ethylcarbo(liimide mPthir)dide S~-mriPnt to ~vb~ d
5-30% of the acid side chains. The reaction mixture is diluted with pH 7.0 phr~srh~te buffer
and the product purified by ion exchange chro.llalography on Q-Sepharose (Ph~m~ ) using
a O-l.OM NaCl gradient. The material is then isolated as the sodium salt by adsorption to an
S Anbe~cLrol"e SD tTosoHaas), elution with a 0-80% açetonitrile gradient, and lyophili7~ion
EXAMPLE 7B
Amide Linkage to N-terminal Cysteine
Very efficiPnt coupling can be carried out between a drug or other compound C~"~ E
an active thioester and an N-terminal cysteine residue, according to the method of Dawson et
10 al. (1994). As in~lir~tPd in Figure 11, this coupling proceeds in two steps~ Attack of the
cysteine sulfhydryl on the initial thioester is followed by a rapid intramolecular attack by the
N-terminal amine on the product thioester to form an amide linkage.
EXAMPLE 8
Representative Methods for Purification and
Structural Analysis of Polymer-Compound Products
A. Silica Gel Chromatography
For many polypeptide-drug products, where the polypeptide component doll,h,~Les the
chromatographic properties of the composition, purification is readily achieved by silica gel
chro",alography. A mixture of isopropanol and 25% aqueous trimethylamine, in proportions
20 ranging from about 1:1 to 3:1, v/v, typically provides good chromatographic resolution.
B. Purification bv Partitioning
Polypeptide compositions which partition well into octanol at reduced pH (generally those
having less than 50% acid amino acid composition) can often be effectively separated from
undesired failure sequPnces generated during polypeptide assembly by partitionin~ between n-
25 octanol and an aqueous buffer having a pH about 0.2 pH units below the transition pH of thepolypeptide. The purified polypeptide can be recuvc~ed from the octanol phase by partitioning
into aqueous Na~HPO4 and then preci~ aLhlg with formic acid. Typically this purification
procedure is carried out before ~ rhmPnt of the drug. This "purification-by-function" method
has been found useful for pre~ard~ions of quite long polypeptides cont~ining a .cignific~nt
30 fraction of failure sequenres.
C. Ion F~çh~ngelReverse Phase Chromatographies
A particularly versatile method for puliryillg polypeptide-drug products employs ion
exchange chro".alography followed by reverse phase chlo-"aLography, where the latter stage
re",()v~s salt and provides additional purific$ion. Use of this purification method is described
CA 022~2706 1998-10-27
WO 97/40854 Pcr/uS97/07335
- 42 -
for the polypeptide-Morpholino ~nti.c~nce oligo products whose syntheses are described in
Example 6C and whose structures are shown in Figure 13.
Po}ypeptide-Morpholino oligo ple~a.a~ionislld-~r~llcd to a rotovap flask, and excess
al~l-ollia is removed under a~ alOr vacuum. Approx. 10 ml of Tris acetate buffer (0.1 M
Trizma base, acetic acid to pH 8) is added, and the solution is loaded onto a 2.5 cm by 15 cm
column of Macro Prep Q anion exchange resin (BioRad Corp.). After loading, the column is
washed (flow rate 5 ml/min) for 10 min. with Tris acetate buffer, followed by elution with Tris
acetate buffer, increased linearly from 0 to 1.0 M in NaCl, over 40 min. In this system, the
Morpholino ~nti~en~e oligo (monitored at 254 nm~ elutes in the first 15 min~t~s, and the
polypeptide-Morpholino product elutes at about 30 minutes.
The polypeptide-Morpholino peak is collected and then desalted on a 2.5 cm by 15 cm
column of 50 ~m polypropylene (Polysciences Corp.). This reverse phase column is washed
for 15 min. with 1% con NH40H and then eluted with a 0 to 80% acetonitrile gradient, 1%
in conc. NH40H, over 40 minutes at a flow rate of 5 ml/min. Fractions containing the
polypeptide-Morpholino oligo product are combined, rotovaped briefly to remove acetonitrile
and ammonia, and then freeze dried.
D. Mass Spectral Analysis
For mass spectral analysis, a portion of the polypeptide-Morpholino product is s.l~cllded
at a concentration of 20 ~M in 1% conc. NH40H. This material, co-cryst~l1i7ed with 3,4,5-
trihydroxyacetophenone/ diammonium citrate (1:2), is analyzed by laser-desorption time-of-
flight mass spectroscopy. In a representative analysis a mass of 11,881 was found, which is
in close agreement with the calculated mass of 11,861 expected for the polypeptide-Morpholino
product shown in Figure 13.
EXAMPLE 9
Asse~sment of Pa~ iO~ g Properties
In op~;,.,i,;t'ion of a polypeptide composition for a particular delivery application, it is
often desirable to first carry out partitioning studies on the polypeptide component alone. In
such studies it is generally useful to add a cl,-ullloL)hore or fluorophore tag to simplify
q~ntit~ti- n of the polypeptide's distribution between the octanol and aqueous phases. In this
regard, 5-carboxy fluorescein co~ ~ a tag which is easily q~ r.d and which does not
have an undue impact on the partitioning properties of polypeptides of reasonable length. This
tag has the added advantage of providing an easily vi~ i7ed signal suitable for cell entry
studies.
A representative partitioning study is described below. Stock buffer 1 {0.1 M citric acid
(pK~l = 3.14; pK~ = 4.77; pK~3 = 6.39), O.lM N-morpholin~eth~nesl~lfonic acid (MES; pK~
CA 02252706 1998-10-27
WO 97/40854 PCT/US97/07335
- 43 -
= 6.1) and 0.1 M phosphQric acid (pKa~ = 2.2; pK~ = 7.2)} and stock buffer 2 (0.1 M MES
sodium salt, 0.1 M tri~o~ium citrate, and 0.1 M disodium phr)Sph~te) are pre,ua~ed. Stock
buffer 1 is titrated with stock buffer 2 to form a sêries of buffers ranging from pH 4 to pH 8
in incre.l.mL~ of about 0.2 pH units.
A set of 0.75 glass ml vials, where each vial contains 0.28 ml of one buffêr of the set plus
0.3 ml of solvent, is p~ ed. The solvent is n-octanol for polypeptides conlai,li"g less than
about 50% acid amino acids and n-pentanol for polypeptides cOl.l~ g more than about 50%
acid amino acid. To each vial of this set is added 20 ~l of a 0.1 mM aqueous solution of
polypeptide having an attached carboxy fluorescein. Each of these vials is capped, shaken
thoroughly, and then centrifilged to separate the phases. Thereafter, 150 ,ul of the upper
octanol phase is added to 150 ~l of 85% isopropdnol/15% 1,8-diazabicyclo [5.4.0]undec-7-ene
(DBU), and the absorbance of this solution is measured at 500 nm. Next, 150 ~l of the lower
aqueous phase is added to 150 ~l of 85% isopropanol/15% DBU, and the absorbance is
measured.
lS Figures 14A-F show plots of these absorbance values as a function of pH for six
representative fluorescein-tagged polypeptide compositions of the invention, where the six
dir~u~enl polypeptides exhibit octanol/water transition pH values ranging from 5.1 to 6.9.
EXAMPLE 10
~ssçssment of Cytosolic Entry
A. Direct Entry
This Example describes a useful method for testing for transport of a fluorescein-tagged
polypeptide composition, with or without attached drug, from the extracellular m~illm directly
into the cytosol of adherent animal cells. It should be noted that appreciable cell entry via
endocytosis does not occur in the short time period (about 20 minutes) of this procedure.
Because mounting mPdil-m, which is generally used in fluorescence mic~oscopy, modifies both
the osmolarity and the pH of the cells' e~ u~en~, it is reco....; ~ded that lll~uu~ g medium
not be used in these cell entry studies. Instead, cells are viewed using a water-immersion
objective. Further, it is desirable that the culture medium in which the cells are viewed does
not contain phenol red, which is typically used as a pH indicator in culture mP~inm because
30 this co-ll~ound inle~LI;~ies with the fluol~sc~ l signal.
The procedure for a direct-cell-entry study is described below. MeAillm 1 co...l.,;c~-
~serum-free medium buffered with 20 mM MES (morpholine ethane sulfonic acid) adjusted to
- pH 6.5 with NaOH. Medium 2 comprises serum-free medium buffered with 50 mM MES
adjusted to pH 5Ø
CA 022~2706 1998-10-27
WO 97/40854 PCT/US97/07335
- 44 -
A 150 /11 aliquot of a 1 mM solution of fluorescein-tagged polypeptide composition is
diluted into 1.9 ml of Medium 1, and 0.5 ml portions are added to 2 cm x 2 cm wells in four
glass chambered slides in which Hela cells were plated 24 hours earlier. After 10 minutes 375
~11 of MeAillm 2 is added to wells 2, 3, and 4 (giving pH 6.0). After a further 10 minutes an
S additit nal 375 ~LI of M~i~lm 2 is added to wells 3 and 4 (giving pH 5.8). Ten minutes later,
a further 500 ~LI of Medium 2 is added to well 4 (giving pH 5.6). After each arl~ition the
slides are swirled gently to thoroughly mix the solutions. Ten minutes after the last a~l~ition
all wells are aspirated and each well washed with three 1 ml volumes of DME-F12 culture
m~lium plus 10% FBS, pH 7.4, and the cells are observed via fluorescence microscopy.
Cytosolic entry is generally evidenced by a diffuse fluorescence throughout the interior of
the cells. However, it should be noted that some compounds (e.g., antisense oligomers)
subsequently pass from the cytosol to the nucleus, where they plere,~ ially accum~ tP,. Thus,
nuclear ~cc~lm~ tion in direct entry experiments is also evidence for snrceccful cytosolic entry.
In addition to the above-described qualitative ~ccpccment of cytosolic entry by fluorescence
15 microscopy, it should be appreciated that qualitative and/or qu~ntit~tive functional assays
specific for the drug being transported can also be carried out following treatment of the cells
in reduced-pH mPAinm
The above procedure provides i~lfol...alion on how low the pH must be in order to effect
transmembrane ~ oll of a given polypeptide composition of the invention, in~ din~e~ when
20 desired, the a1t~ch~d co~-pou--d to be transported.
It should be noted that superior entry was generally obtained when the polymers were
neutralized during purification with triethylamine, rather than with an alkali metal base or
~mm~nli~ This effect may have been due to the relatively lipophilic counterion. Accordingly,
polypeptides to be tested should be purified in a consistent manner to ,.,i..;..,i~e any such effect
25 of the counterion on the test results.
B. Entrv Via Endocytosis
When a fluorescent-tagged polypeptide is endocytosed into cells, observation of a
periml~lP~ punctate pattern is indicative that the tagged material is localized in the
endosomal/lysosomal co---pa"---ent. However, a diffuse fluorescence throughout the cell could
30 indicate that the polypeptide achieved the desired endosome to cytosol t~SpOl~, or that the
polypeptide (if assembled from L-amino acids) was degraded by Iysosomal enzymes and only
the fluorescent tag diffused into the cytosol. Therefore, it is desirable that cytosolic entry via
an endocytotic route be co--rllllled by a fu~clional assay for the drug component of the
polypeptide-drug product. The two examples below utilize such a functional assay.
, . . -- ., .
CA 022~2706 1998-10-27
~50-~ll~l
1. Without Endocvtosis Fnh~nrer. Hela cells used in this functional assay were stably
transfected with a plasmid containing a mouse m~mm~ry tumor virus promoter (inducible with
dex~nn~h~cone) controlling a gene coding for the 5' untranslated region of rabbit ~-globin
rnRNA, followed by the coding sequence for firefly luciferase (Partridge, 1996). Cell-free and
5 in-cell scrape-load tr~n~l~tiorl studies have shown that the Morpholino ~nticence oligomer shown
in Figure 13 is highly effective in blocking the translation of luciferase from this gene construct.
Accordingly, if the ~ntiC~rlce oligomer of Figure 13 gains access to the cytosolic COlllpd~
of these l,d-~[e~;ted cells, it should effect a cignific~nt reduction in luciferase activi~y upon
de~m~th~cone induction, relative to untreated cells. Such a reduction in luciferase activity,
10 measured as relative light units in a I~ -in~ , would be indicative of succ~csful cytosolic
delivery of the Morpholino oligomer.
To test for cytosolic delivery, the polypeptide-Morpholino ~ ;cellce oligonllrl~oti~
product prepd-ed as in Example 6C was 5l~cpen~l~d in culture m~illnl at a concclllld~,on of 5
IlM. The above-described tldl~fcctcd Hela cells were treated with the s~lcpencit~n for ~ hours,
15 and then treated for 16 hours with dex~ h~cone to induce lucif~i.ase synthesis. In parallel,
cells in separate wells were treated with a) medium alone, b) Morpholino ~n~icence oligo, and c)
polypeptide.
Luciferase from the cells in these four wells was ql~ntir~trd~ and the relative light units are
given in Table 3.
TABLE 3
T,eat",c.,i Relative Lieht Units
medium alone 100
polypeptide 10~
polypeptide-Morpholino product 69
2~ Morpholino ~I;c~ ~c~ oligo 114
The inhibition of luciferase activity in the cells treated with the polypeptide-Morpholino
product sl~g~ctc that the polypeptide transported this ~ ce oligomer from the en~loso.,.r
into the cytosol of the Hela cells.
30 2. With endocvtosis enhancer. The e~--i",ent in the previous example was repeated
with the following changes: a) the concentlalion of the polypeptide-Morpholino product in
the mrrij~lnl was only 3aO nM, and b) Transfectam (Plo",ega Corp., Madison, WI) was
added at a conce.-~-ation of 20 ~g/ml. It was expected that the tetra-cationic Transfectam
would bind electrosr~ y to the polyanionic polypeptide (in its high-pH form), and the two
35 long-chain alkane moieties of the Transfectam would serve as a lipid anchor to substantially
increase the
AMENDED SHEEr
IPEAIEP
CA 022~2706 1998-10-27
W O 97/40854 PCTAUS97/0733S
-46-
effective co~ dlion of the complexed polypeptide-Morpholino product at the cell surface,
as illustrated in Figure 12, thereby substantially hlc,~a~illg the rate of entry into the cells.
Results from this experiment are given in Table 4.
TABLE 4
Tledllll~.~l Relative Li~eht Units
medium alone .................. 78
Tlan~r~l~ll .................. 97
Tldl~re~;l~ll + polypeptide-Morpholino product 35
The inclcased inhibition of luciferase activity in the cells treated with the co,llbil,dlion of
Tr~r~L~Il and polypeptide-Morpholino product again suggests that the polypeptide15 llan~l,olled this an~ic~nce oligo from the endosome into the cytosol of the Hela cells, and that
cytosolic entry can be increased by accelc.d~ g the initial endocytosis step.
EXAMPLE 1 1
Fnh~nr~ of Tr~n.cd~rm~l Delivery
This example illustrates the use of a representative polypeptide co,ll~o~iLion of the
20 invention to enhance transdermal delivery of a drug which normally exhibits very minimal
passage across the epidermis. Tritiated cyclosporin A metabolite 17 is linked to a suitable
polypeptide, as described in Examples 3 and 4, and the conjugate composition, with the
polypeptide moiety in its low-pH form, is suspended in a suitable pe.lc.rdlion enh~n-~er solution
which includes a lipophilic fatty acid (e.g., 10% linoleic acid/90% propylene glycol). Tritiated
25 cyclosporin A metabolite 17, without ~tt~hPd polypeptide, is suspended in the same penetration
enh~n~er solution, for comparative ~csescrnlont of the rate of transdermal passage of the
unmrldified drug.
Each cyclosporin-contaillillg solution is contacted with a defined area of skin on a nude
hairless mouse, and small aliquots of blood are withdrawn periodically for acc~ of the
30 amount of tritiated drug which has passed through the skin and into the circulatory system.
~ . . ~