Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02254557 2003-10-24
~nthesis of Long-Wavelength Absorbing Photosensitizers
The present patent application claims priority from Canadian patent
application
2,221,912 of Johnson et al., filed November 21, 1997, and entitled
"Photosensitizers with
Improved Biodistribution and Light-Absorbing Properties".
Field of the Invention
The present invention provides for novel therapeutic macrocycle compounds
useful in photodynamic therapy that are based on the chlorin ring system: The
macrocycle compounds have absorption spectra optimized for therapeutic use in
tissues, and are stabilized against oxidation by the attachment to the chlorin
ring of a
structure that comprises one or more exocyclic rings that also contribute at
least one
nitrogen atom. Protonation or covalent modification of this nitrogen atom, or
other
covalent modification of the one or more exocyclic rings, permits optimization
of
pharmacologically relevant properties including, for example, solubility.
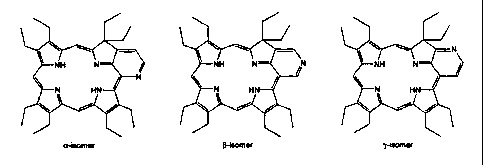
Representative of such chlorins are the "pyridochlorins" depicted as follows.
a-homer S-Isomer y-isomer
Background of the Invention
Photodynamic therapy ("PDT") generally involves administration of a drug
(photosensitizer) which, when irradiated with light, becomes electronically
activated
CA 02254557 2003-10-24
such that it may interact with molecular oxygen to generate reactive oxygen
species.
The reactive oxygen species are believed responsible for killing of targeted
cells,
which are generally those associated with an unwanted hyperproliferating
state.
Unless a photosensitizer compound is to be used solely for the treatment of
superficial skin diseases, it is important that the wavelengths) at which the
compound
absorb light be optimized. Preferably, the compound should absorb strongly in
the
red region of the spectrum (650-800 nm). Light scattering and the presence of
endogenous chromophores (such as hemoglobin) results in very poor penetration
of
tissues by light at wavelengths below about 600 nm. This means that the large
absorption band (the so called Soret band) displayed by porphyries in the
region of
400 nm is not available, practically speaking, for photosensitizer activation
in PDT.
Instead, the longer wavelength Q absorption bands must be used. However,
the longest wavelength Q band for Photofrin ~ (see U.S. Patent No. 5,059,619),
a first
generation porphyrin photosensitizer, is only at about 630 nm. Although this
wavelength is long enough to permit useful photodynamic therapy approaches for
some tumors, it is not ideal. Since light penetration of human tissues
typically
doubles between 630 and 750 nm, a photosensitizer absorbing at 750 nm would be
far
more effective at treating thick tumors than, for example, Photofrin ~ .
However,
increasing the absorption wavelength of photosensitizer compounds beyond about
800
nm (into the infrared), would not give rise to further improvements, since the
involved
energy transitions are insufficient to generate a sufficiently energetic
excited state,
and corresponding activated oxygen species.
Accordingly, there is a considerable medical need to develop classes of
photosensitizers having optimized light absorption properties. One such
improved
class of photosensitizers are the so called "green monohydrobenzoporphyrins"
which
are derived from natural porphyries by Diels-Alder type reactions at one of
the
functional groups attached to the porphyrin core. An example of such a
compound,
which is currently in phase III clinical trials, is BPDMA which shows
considerable
absorption at about 688 nm. In this regard, see for example, U.S. Patents
5,095,030;
5,171,749; 5,776,966 and the like. Finally, the porphyrin core structure is
characteristically non-polar, and such structures need to be modified by the
addition
2
CA 02254557 2003-10-24
of groups having sufficient polarity to improve the solubility and
amphiphilicity
properties of the compound, and to improve the rate of metabolism or clearance
in the
body.
The core structure known itself as porphyrin, as mentioned above, is presented
below in comparison with that of chlorin. In principle, the simplest way to
increase
the wavelength of absorption of a porphyrin would be by reduction of a double
bond
therein to give the corresponding chlorin (reduction of a porphyrin to a
chlorin results
in an increase in both the intensity and wavelength of the longest absorption
band -
providing a shift of about 25 nm and typically a substantial increase in
extinction
coefficient).
meso
' -
~ NH N
a meso meso
~ \
a
H N HN
\
PYRROLE ~ ~ r /
meso
PORPHYRIN
H
H
PORPHYRIN ~ H CHLORIN
Although reducing agents such as diimide are available that will
regioselectively reduce only the double bond targeted for simply conversion of
a
porphyrin to a chlorin, such reactions are typically reversible. Oxygen in the
air is
well known to oxidize chlorine back to porphyrins and this impacts not only
the
synthesis, but also the storage and clinical use of the resultant compound..
Accordingly, there are only a limited number of cases where such procedures
have
been used to synthesize chlorine (see for example R. Bonnet, Chem. Soc. Rev.,
1995,
3
CA 02254557 2003-10-24
p. 19, and R. Bonnet et al., Biochemical Journal, 261, p. 277, 1989, in
relation, for
example, to the synthesis of tetrakis (m-hydroxyphenyl) chlorin, "m-THPC'~.
Examples of known chlorins that contain unsaturated exocyclic rings fused to
the skeleton between a meso position and its adjacent (i-position include
purpurins
and benzochlorins. Fusion of such exocyclic rings has the advantage of
substantially
limiting oxidation of the prepared chlorin at the previously reduced pyrrole
ring.
A number of purpurins have been described. See, for example, R.B.
Woodward et al., J. Am. Chem. Soc., 82, p. 3800, 1960, and also J.H. Fuhrhop
et al.,
Angew. Chem. Int. Ec~ Engl., 14, p. 361, 1975 describing an octaethyl purpurin
which
absorbs at 695 nm. A number of purpurins have been shown to have photodynamic
activity, and the most effective member of this class of compounds may be a
tin
etiopurpurin formed by cyclization of meso-(/3-(2-ethoxycarbonyl)vinyl]
etioporphyrin, followed by metalladon with tin chloride. This compound is
currently
in clinical trials (A.R. Morgan et al., Photochem. Photobiol., 51, p. 589,
1990; A.R.
Morgan et ai., J. Med Chem., 32, p. 904, 1989).
The synthesis of a benzochlorin was first described by D.P. Arnold et al.
J.C.S Perkin I, p:1660, 1978, and sulfxnation of a benzo ring thereof was
effected in
concentrated sulfuric acid (see A.R. Morgan et al., Photochem. Photobiol. S5,
p.133,
1992 and B.C. Robinson et al., SPIE Optical Methods for Tumor Treatment and
Detection: Mechanisms and Techniques in Photodyrramic Therapy Y, 2675, p. 179,
1996). The resulting sulfonic acid group can be used as a platform for further
4
CA 02254557 2003-10-24
derivatization, such as to modify bioactivity. An additional pathway to effect
derivatization of a benzochlorin involves reaction of a metallated
benzochlorin at the
meso position adjacent to a gem-diethyl group to yield an iminium salt (D.
Skalkos et
al., Photochem. Photobiol. ,59, p. 175 1994). Photodynamic activity has been
demonstrated for various benzochlorins (see A.R. Morgan et al" Photochem.
Photobiol. 55, p.133, 1992, and A.R. Morgan et al., Tetrahedron Letters, 35,
p. 5347,
1994).
The preparation of a pyridinoporphyrin has been described, C. Alonso et al.,
Tetrahedron Letters, 38(15), pp. 2757-2758, 1997 wherein the pyrindinyl
nitrogen
atom, or the pyridinyl ring, may serve as a platform for further
derivatization.
However, such a compound is believed to lack the optimized absorption profile
characteristic of the pharmaceutically useful chlorine.
As aforementioned, the presence of an exocyclic ring fused to the core
structure of a chlorin at the site of the reduced pyrrole ring substantially
prevents re-
oxidation thereof. It would be further advantageous to derivatize the
exocyclic ring to
optimize biological properties such as solubility, physiological clearance, or
to
enhance amphiphilicity, that is the presence of both polar and non-polar
domains thus
enhancing interaction with both polar and non-polar environments. Although
derivatization has been described, for example, for benzochlorin by
sulfonation under
strongly acidic conditions, there is a clear need to develops more flexible
methods
whereby a large number of such pharmaceutically useful chlorine can be
prepared. As
described below, the present invention provides such methodology and resultant
compounds.
Summary of the Invention
The present invention provides for novel therapeutic macrocycle compounds
useful in photodynamic therapy that are based on the chlorin ring system. The
macrocycle compounds are stabilized against oxidation by the attachment to the
chlorin ring of a structure that comprises one or more exocyclic rings that
contribute
at least one nitrogen atom. Protonation or covalent modification of this
nitrogen
atom, or other covalent modification of the one or more exocyclic rings
(hereinafter
CA 02254557 2003-10-24
"derivatization'~ permits optimization of pharmacologically relevant
properties
including, for example, solubility. Optionally, the chlorin ring is
metallated.
In a preferred embodiment of the invention, compounds are represented by the
formula ( 1 )
wherein, for example,
R~', Rib are independently H or alkyl;
R2 is OH, halogen, alkoxy, OCO-alkyl, sulfonate, sulfate, or phosphate;
R3' is H, or a phenyl or other aryl or heteroaryl group optionally substituted
by one or
more groups, each independently selected, for example, from halogen, hydroxy,
alkyl,
alkoxy, cyano, and ester;
R3b is H, halogen, formyl, vitro, amino or cyano
R4 is H or alkyl;
M is a complexing metal, typically known in the art, or represents 2H; and
N, the nitrogen atom of the pyridine ring, is optionally in the form of an N-
oxide, or a
salt such as an alkyl or hydrogen halide.
In a further preferred embodiment of the invention, compounds are
represented by the formula (2)
6
CA 02254557 2003-10-24
(2)
wherein the substituents are generally as for compound ( 1 ) although certain
preferences or exceptions are described as follows:
R'' is not H, and is preferably methyl or ethyl, in which case R'' (formed
from an R'~
would be CH3CH= or CH2= ;
R'b is as for compound (1);
RZ is H or alkyl (and fixed at the position alpha to the N atom);
R3' is as for compound ( 1 );
R3b iS as for compound ( 1 );
The C-0 group may, optionally, be converted to a CHOH group, and the resultant
OH group may be further derivatized, such as via the RZ options described for
compound ( 1 );
It should be noted that since the exocyclic ring is not aromatic (comparing to
compound 1), the N-oxide or salt variations are inapplicable, although
compound (2)
can exist as a free base or a metal complex.
In an additional embodiment of the invention, compounds are represented by
the formula (3)
7
CA 02254557 2003-10-24
wherein, preferably,
R'' would be methyl or ethyl;
R~ b would be as for compound ( 1 );
R3' would be as for compound ( 1 );
R36 would be as for compound (1); and either
R~~, together with the carbon atom to which it is bonded, forms a C(Rl'xRi~
group
and Rz' and R2b together form a bond; or
Rl' is CH3CH or CH2;
RZ' 1S H; and
R2b 1S Rib.
With respect to the design of such compounds(3), the N-oxide or salts
mentioned for compound (1) are also within the practice of the invention, and
compound (3) can also exist in free base or as a dimetal complex.
As aforementioned, more than one exocyclic ring may be present in the
compounds of the invention, there being generally no structural limitation on
any such
combination of rings subject of course to practical considerations of
synthesis, and
that one or more suitable nitrogen atoms be appropriately placed therein.
Accordingly, additional representative compounds of the invention include:
8
CA 02254557 2003-10-24
R~ _~ R~ R~
R
~ N N-
.,
N
\ N H
R~ v ~ ~ R~
R~ R R~
3
R~ "..~~.'~. ~ ~ . N
R~-'
R~
R~ V R~ R~ N
R' ~ H N
~3
~ N HN
R~ ~ ~ i R~
R~ _ R~
9
i ,
CA 02254557 2003-10-24
R~ _~ R~ R~ -N
R1 \ ~
\ NH N
~3 /
\ N HN
R~ ~ ~ i R~
R~ _ R~
R~ _~ R~ R~
R~ \ H N -N
.s \ /
N HN
R~. ~ ~ i R~
R~ .. R~
R~ ~J R~ R~
R
\ NH N
N
~ N HN /
R~ ~ ~ i R~
R~ ,~ R~
R~ _,~ R~ R~
R~ . w w
~ NH N'
\ N HN / N
R~ ~ ~ i R~
R~ ,~ R~
14
i
CA 02254557 2003-10-24
1 _~ R
R~ 1=Nhl N
N HN ' LN
R~ v ~ R~
R, R R,
3
and the like, where it is additionally understood that more than one nitrogen
atom may
be present in the exocylic rings, which may be further substituted by other
functional
groups (such as hydroxyl or keto) and which permit further enhancements in
desireable properties such as solubility.
Additional preferred compounds include the "pyridochlorins" such as:
a-isomer p-isomer r-aomer
Additional preferred examples of the invention include:
11
CA 02254557 2003-10-24
(Zn-123)
Brief Description of the Drawings
Figure 1 depicts a synthesis of octaethylbenzochlorin (17).
Figure 2 depicts a vinylogous Vilsmeier reaction of metalled
octaethylporphyrin
Figure 3 depicts a possible synthesis of the a-imino precursor, species (92).
Figure 4 depicts the product expected from the cyclization of species (92).
Figure 5 depicts the synthesis of purpurins.
Figure 6 depicts the synthesis of australaochlorins.
Figure 7 depicts a postulated mechanism for the removal of the angular ethyl
group of
species (93) based an analogous reaction known for steroid ring A.
Figure 8 depicts a possible cyclization reaction for glycolaldehyde
condensation
product; species (96).
Figure 9 depicts the structure of amines (98) and (99), synthesis of which was
unsuccessful.
Figure 10 depicts a methylcarbamate condensation of (56) with subsequent
cyclization.
Figure 11 depicts a proposed route to meso-aminomethyloctaethylporphyrin (
104).
Figure 12 depicts a proposed use of (105) as a cyclization intermediate.
Figure 13 depicts the synthesis of Compound 106.
Figure 14 depicts a side-product from N-hydoxymethylation of (Ni-105,
nickelated
105).
Figure 15 depicts a proposed cyclization of N-formylamide compound (109).
Figure 16 depicts a synthesis of meso-isocyanooctaethylporphyrin (112).
Figure 17 depicts a Passerini reaction of ( 112).
12
CA 02254557 2003-10-24
Figure 18 depicts acid-catalyzed reaction of (112) with formaldehyde to give
(116).
Figure 19 depicts a rationale for the cyclization/lack of cyclization of the
two types of
hydmxymethylamide ( 106) and ( 116).
Figure 20 depicts a possible mechanism for the formation of the dimeric
structure (Ni-
119).
Figure 21 depicts a mechanism for the formation of the dimer (Ni-120).
Detailed Description of the Invention
The present invention provides for novel therapeutic macrocycle compounds
useful in photodynamic therapy that are based on the chlorin ring system. The
macrocycle compounds of the invention have relatively long wavelengths of
absorption, to permit penetration of tissues, such as about 670 nm, although
this may
vary with the particular compound based upon its exact cyclic structure and
ring
substituents.
The compounds are stabilized against oxidations, such as at the reduced ring
of the chlorin (which would cause the molecule to revert to a porphyrin
structure), by
the presence of one or more exocyclic rings in contact with the reduced
pyrrole ring.
In a typical embodiment, the exocyclic ring or rings are aromatic, although
they may
also be non-aromatic or have partial aromatic character. Most preferably the
exocyclic ring or rings comprise at least one nitrogen atom, or comprise at
least one
substituted carbon atom, or a carbon atom which is subject to substitution. As
a result
of this modification to the exocyclic ring(s), and/or further derivatizadon at
such sites,
physiologically relevant properties of the compound can be improved, such as
improved solubility, amphiphilicity, drug clearance, and the like.
Accordingly, there are provided compounds according to the structures
13
CA 02254557 2003-10-24
m
wherein the circle represents an exocyclic structure having one to about three
rings
and preferably at least one nitrogen atom which can be protonated or further
substituted. R' represents one or more substituents in the ring structure
which may
be selected (see below) to further enhance or modify the compound's
properties. As
depicted directly above, the exocyclic structure can be positioned on either
side of the
reduced pyrrole ring of the chlorin. Such positions could be considered as
symmetrical, except for the asymmetries introduced into the compounds by the
other
substituents (see also below).
Most preferably, the exocyclic structure contains from one to about three
rings, and from about 5 to about 25 carbon atoms, wherein preferably at least
some of
the carbon atoms are involved in double bonds. It is particularly preferred
that the
exocyclic structure contain one or more ring nitrogen atoms which, typically,
in
deriva'zed or protonated form, can be used to enhance the solubility in
aqueous
systems of the macrocycle. The exocyclic structure is subject to further
modification
by from one to many groups R' , which similarly to the other R gmups attached
to the
chlorin ring itself, may be used to modify the properties of the macrocyele.
Options
14
i ;.
CA 02254557 2003-10-24
for selection of groups R' are generally the same as for other R gmups present
in the
structure, although there would be exceptions as is readily understood in the
art.
Accordingly, representative exocyclic structures useful in the practice of the
invention include those depicted below:
3
R~ ' R~ R~
R1 ~ N H N-
.,-
R \ / N
N H
R~ ~ ~ i R~
R~ I R~
'J
R~ R~ R~
R~ ~~ ~ N
NH N /
N HN
R~ v ~ i R~
R~ _ R~
R~ ~ R~ R~
R '''
~ NH N
N HN
R~ ~ i ~ R~
R~ _ R~
i ,,
CA 02254557 2003-10-24
R~ _~ R~ R~
R '
\ NH N
3
\ N HN /
R~ ~ ~ i R~
R~ _ R~
R~ _~ R~ R~
'
R~ \ hl N-. -N
v
\ N HN /
R~ ~ ~ i R~
R~ _ R~
R~ _~ R~ R~
'
R~ \ NH N N
/ ~ s
N HN a
R~ v ~ ~ R~
R~ _ R~
R~ _~ R~ R~
'
R~ \ N N
\ HN / \N
N
R~ ~ ~ i R~
R~ ~ R~
16
i
CA 02254557 2003-10-24
R~ ~ R~ R~
w
~ NH N
N HN ' ~=N
R~ ~ ~ i R~
R~ I R~
In a preferred embodiment of the invention, compounds are represented by the
formula ( 1 )
wherein, for example,
Ria, R~b are independently H or alkyl;
R2 is OH, halogen, alkoxy, OCO-alkyl, sulfonate, sulfate, or phosphate;
R3a is H, or a phenyl or other aryl or heteroaryl group optionally substituted
by one or
more groups, each independently selected, for example, from halogen, hydroxy,
alkyl,
alkoxy, cyano, and ester;
R3b is H, halogen, formyl, vitro, amino or cyano;
R4 is H or alkyl;
M is a porphyrin-complexing metal, typically known in the art, or represents
2H; and
N, the nitrogen atom of the pyridine ring, is optionally in the form of an N-
oxide, or a
salt such as an alkyl or hydrogen halide.
It will be appreciated that the exact position of the nitrogen atom in the
exocyclic structure is subject to considerable variation, limited of course by
available
17
CA 02254557 2003-10-24
routes of synthesis, and thus numerous other compounds are preferred according
to
the practice of the invention. Representative compounds include
a.iaoma homer r.isomer
In connection with the design of such compounds, additional substituents may
be used as Long as they do not interfere with the intended therapeutic
activity, or
capacity of the compound to be formulated for use. As will be appreciated by
those
skilled in the art, additional derivatizing groups may be used, such as other
charged or
polar groups, to affect, for example,
compound solubility, the range of such substituents being limited of course,
by the
feasability of available synthesis routes.
The addition of further functional groups to the exocyclic structure provides
additional opportunities for derivatization and fine tuning of
pharmacologically
relevant properties. In a further preferred embodiment of the invention,
compounds
are represented by the formula (2).
R2
(2)
wherein the substituents are generally as for compound (1) although certain
preferences or exceptions are described as follows:
18
CA 02254557 2003-10-24
R1' is not H, and is preferably methyl or ethyl, in which case R1' (formed
from an Rla)
would be CH3CH= or CH2= ;
R~b is as for compound (1);
R2 is H or alkyl (and fixed at the position alpha to the N atom);
R3' is as for compound (1);
R36 is as for compound (1);
The C=O group may, optionally, be converted to a CHOH group, and the resultant
OH group may be further derivatized, such as via the R2 options described for
compound (1).
It should be noted that since the depicted exocyclic ring is not aromatic
(comparing to compound 1 ), the N-oxide or salt variations are inapplicable,
although
compound (2) can exist as a free base or a metal complex.
As aforementioned, the exact position of the nitrogen atom and any further
functional groups in the exocyclic structure can be varied considerably.
Accordingly,
representative related compounds of the invention include
(Zn-123)
19
CA 02254557 2003-10-24
In an additional embodiment of the invention, compounds are represented by
the formula (3)
wherein, preferably,
R'' would be methyl or ethyl;
Rlb would be as for compound (1);
R3' would be as for compound ( 1 );
R3b would be as for compound ( 1 ); and either
Rl°, together with the carbon atom to which it is bonded, forms a
C(R~8)(Rl~ group
and Ri' and R2b together form a bond; or
Rl° is CH3CH or CH2;
R2° is H; and
R2b is Rib.
With respect to the design of such compounds(3), the N-oxide or salts
mentioned for compound (1) are also within the practice of the invention, and
compound (3) can also exist in free base or as a dimetal complex.
It is again noted that generally the compounds of the present invention can
exist in a metallated form, or the metal atom of the chlorin rings) can be
replaced by
2H, as would be readily apparent to those skilled in the art.
zo
CA 02254557 2003-10-24
Pharmaceutical Administration
The compounds of the invention may be formulated in a variety of ways for
pharmaceutical use. Generally speaking, such formulations include any
excipients,
stabilizers, emulsifying agents, osmotic agents, solubilizing agents and the
like, that
are recognized as useful to deliver photosensitizes compounds to the body
whether
topically or internally in any way such as by intravenous, intraperitoneal, or
intramuscular injection, transmucosally, orally, transdermally by way of skin
patches,
salves and gels, and such. Liposomal formulations, as recognized in the art,
represent
a preferred form of formulation, including formulations with phosphatidyl
serine,
phosphatidyl glycerol, phosphatidyl choline, and the like. Additionally, it
will be
recognized that functional groups are typically added to the macrocycles of
the
present invention to facilitate their storage, preparation, solubility, and
physiological
utility.
Generally, reference may be made to Remington's Pharmaceutical Sciences,
Gennaro et al. (eds.), Mack publishing Company, Easton, PA ( 1990, 1998,
2000). Additional
information concerning generally acceptable formulations is provided in U.S.
Patents
5,095,030; 5,171,749; 5,776,966; 5,789,433; 4,512,762; 4,566,636; 5,399,583;
4,920,143
pertaining to photosensitizers for pharmaceutical use. The compounds of the
invention may generally be used for all of the therapeutic applications for
which
photosensitizes compounds have been recognized, as mentioned for example in
the
cited patents. As is recognized by medical practitioners, dosages vary
considerably
based on the mode of administration, formulation, condition of the patient,
condition
to be treated, and the like. For systemic administration, dosages on the order
of 10
microgram/kg to 100 mg/kg, preferably 100 microgram/kg to 10 mg//kg may be
preferred. With respect to topical administrations, suitable compositions may
range
from about 1 to 10% of the composition, or greater or Lesser, depending upon
the
application, as would be recognized in the art.
Examples
Synthesis Routes for Pyridochlorins
21
CA 02254557 2003-10-24
Although octaethylbenzochlorin (Arnold et al., 1978) proved resistant to many
kinds of derivatization reactions, its overall photochemical properties and
stability
suggested the desireability of synthesizing novel macrocylces that
incorporated some
of its features. By substituting an exocyclic pyridine ring for an exocyclic
benzene
ring, a compound would be provided that would permit nucleophilic substitution
reactions on the exocyclic pyridine ring, in contrast to mostly unsuccessful
electrophilic substitutions attempted on the benz~ochlorin benzene ring.
Representative of such "pyridochlorins" are the a-, ~-, and y- isomers of
"octaethylpyridochlorin" (17), as depicted below.
a-isomer ~-isomer y.NOmer
For demonstration purposes, an evaluation was made as to which of the three
isomers was likely to be the most easily synthesized. Here a comparison with
the
synthesis of octaethylbenzochlorin was made The formation of the latter is
achieved
by a vinylogous Vilsmeier reaction on metallated octaethylporphyrin (M-4) to
give the
acrolein-substituted metalloporphyrin (M-16 in Figure 1, Formation of
Octaethylbenzochlorin).
If a similar method were to be used for the synthesis of pyridochlorins, it
would seem logical first to make the nitrogen analogues of the meso-acrolein.
This
reaction pathway would rule out the synthesis of the 7-pyridochlorin as this
would
require nucleophilic attack by the (i-~i double bond on the nitrogen atom, a
highly
unlikely reaction. For this reason, efforts were concentrated on synthesizing
the a-
and ~i-pyridochlorins.
Example 1 The design and synthesis of a-pyridochlorin
22
CA 02254557 2003-10-24
In order to synthesize the a-pyridochlorin it was necessary first to make
(92),
the a-imino analogue of the acrolein derivative (16) used in the benzochlorin
synthesis (Figure 1). (M-16, "M" meaning metallated) is formed in two steps by
electrophilic attack on the porphyrin meso-carbon by the vinylogous Vilsmeier
reagent formed from 3-(dimethylamino) acrolein and phosphoryl chloride,
followed
by treatment with aqueous base to hydrolyze the iminium salt to the aldehyde
(Figure
2).
Clearly, another approach was necessary to create the a-nitrogen analog. One
well-known method of obtaining a porphyrin meso-substituted by a nitrogen
functionality is nitration. The vitro compound can be reduced to the
aminoporphyrin,
and such compounds are reported to react with aldehydes to give Schiff's bases
(A. W.
Johnson et al., J. Chem. Soc. (C), p. 794, 1966). Thus a reasonable route to
the
desired precursor from octaethylporphyrin would be to prepare the vitro
derivative
(90), reduce it, and then form the Schiffs base with glyoxaI (Figure 3).
The nitration was carried out in an ice-cold mixture of glacial acetic acid
and
concentrated nitric acid to give the mono-nitro compound (90) in 50 % yield.
The
reaction mixture had to be monitored carefully, in order to minimize the
formation of
poly-vitro compounds. The reduction of the vitro group to the amine was
effected
with stannous chloride dihydrate in concentrated hydrochloric acid and gave
the
desired product (91 ) in 85 % yield. Thus far, the synthesis proceeded as
described in
the literature. (A. W. Johnson et al., J. Chem. Soc. (C), p. 794, 1966; R.
Bonnett et al.,
J. Org. Chem., 30, p. 2791, 1965; A.W. Johnson et al., J. Chem. Soc. p. 4303,
1965).
The formation of Schiffs bases by condensing aminoetioporphyrin with
benzaldehyde and anisaldehyde has been described as occurring in good yield (>
80
%). However, these reactions were performed using the aldehyde as solvent,
while
glyoxal trimer dihydrate (the aldehyde necessary for this synthesis) is a
solid. Hence
a solvent system in which both the aldehyde and the porphyrin would have
reasonable
solubility was required. A number of small-scale experiments lead to the
finding that
a mixture of ethanol and THF was a suitable solvent, the ethanol dissolving
the
glyoxal trimer when heated, and the THF dissolving the porphyrin. Although the
condensations reported with benzaldehyde and anisaldehyde occurred at room
23
CA 02254557 2003-10-24
temperature, it was necessary to reflex the glyoxal/porphyrin mixture
overnight. This
might well be accounted for by the lower concentrations of reagents resulting
from
the use of a solvent. The optimized yield of the reaction was 69 %, or 80 %
based on
recovered starting material. Unsuccessful attempts were made to drive the
reaction to
completion by the addition of drying agents, but it is likely that the amount
of residual
water in the THF/ethanol solvent far outweighed the small quantity of water
produced
in the condensation. In any case, the product (92) could be separated from the
polar
starting material by column chromatography, and the latter could then be
reused.
Before attempting this condensation, it had been anticipated that dimer
formation,
resulting from the reaction of an aminoporphyrin with each of the two formyl
groups
of glyoxal, might occur. However, the use of a large excess of glyoxal
prevented the
dimeric product from being formed in any significant quantity.
(92) was stable enough to allow it to be chromatographed on silica, although
care was taken to elute it from the column as quickly as possible as some
decomposition to the amine was observed during chromatography. However, as it
appeared to be sensitive to acid, and the cyclization step was expected to
require the
use of such conditions, problems with this step of the synthesis were
envisaged.
Consequently, attempts were made to reduce the imine functionality to the
amine
using sodium cyanoborohydride, a reagent reported to reduce imines selectively
in the
presence of carbonyl groups. Unfortunately, any products formed in this
reaction
appeared to be very unstable, and decomposed during work-up to give the amine
(91).
In view of this result, the cyclization reaction was performed directly on
(92), in the
hope that conditions favoring cyclization over hydrolysis could be developed.
In an effort to minimize the possibility of hydrolysis, initial experiments
employed nonacidic conditions, namely refluxing in toluene. Indications that
neutral,
high temperature conditions might be successful came during a synthesis of the
imine,
when the solvent inadvertently was allowed to boil away, and the reaction
mixture
was heated as a solid for several hours. On working up this product, no trace
of the
desired imine (92) was present; instead, in addition to the amine starting
material (91 ),
which was the major compound isolated, a few milligrams of a mixture of two
low
24
CA 02254557 2003-10-24
polarity green compounds (93) was obtained. By'H NMR and visible spectroscopy
these compounds appeared to be two isomers with a chlorin structure.
(93)
On refluxing the imine in toluene overnight this result was reproduced, the
isomeric mixture (93) being formed in 15 % yield. No starting material was
recovered, but a substantial quantity of the amine (91) was isolated; it seems
that the
cyclization and hydrolysis reactions are in competition with one another,
which would
suggest that obtaining the cyclized product in high yield might not be
possible.
Although the quantity of cyclic product (93) thus far obtained was small, and
it consisted of a mixture of two isomers, it was relatively simple to
determine the
structure using spectroscopic techniques: firstly, the visible spectrum showed
an
absorption peak at 722 nm (e = 10800), indicative of a reduced porphyrin
species
such as a chlorin (the longest-wavelength absorption of the precursor imine
was at
660 nm and very weak (E = 4400)). Secondly, in the 40.0 MHz'H NMR spectrum
there were some very distinctive peaks: two quartets at 7.45 ppm and 7.72 ppm
and
two doublets at 2.62 ppm and 2.81 ppm. These resonances are typical of the
ethylidene functionality CHCH3 attached via a double bond to the ~-position of
a
reduced pyrrole ring. There were also two singlets at 7.98 ppm and 8.1 S ppm,
in
addition to the expected meso-hydrogen signals, and these could be assigned to
the
imino CH of the two isomers. Finally there were two triplets at 0.10 ppm and
0.50
ppm, suggesting the presence of ethyl groups lying out of the plane of the
macrocycle
and hence experiencing a greater degree of shielding by the ring current than
those
lying in the plane. The two isomers (93) were present in a ratio of
approximately 4 to
CA 02254557 2003-10-24
1. This information lead to the two isomers (93) being assigned the structures
shown
above.
The formation of these products (93) from the cyclization reaction was
somewhat surprising, as it requires a formal oxidation: The expected product,
given
that the desired 1,2-diaikyl shift did not take place, and the carbocation
created by the
cyclization process was quenched instead by the loss of a proton from the
attached (3-
ethyl group, would be the imino-alcohol (94) (Figure 4). In (93), the carbonyl
group
of the ketone is in conjugation with the imine double bond, which may increase
its
stability with respect to the alcohol, but the mechanism by which this
transformation
occurs is unclear. However, a similar oxidation is seen during purpurin
synthesis (see
below) when the reaction mixture is refluxed open to the air; in that case
oxygen is
presumed to be the oxidant.
Despite the very close analogy to the octaethylbenzochlorin synthesis, the
replacement of a CH group with a nitrogen atom obviously changes the
cyclization
mechanism significantly, even though the N atom is several bonds removed from
the
site of reaction. There appears to be a closer similarity to purpurin
synthesis. These
compounds are produced by the acid-catalyzed cyclization of (metal-free) meso-
acrylic esters (Figure SA). There are two types of purpurins, named type A and
type
B. Type A purpurins possess an spa carbon atom on the reduced pyrrole ring,
adjacent to the exocyclic ring, while type B purpurins have an sp2 carbon at
this
position (Figure 58).
The formation of type B purpurins requires a formal oxidation after
cyclization. In some cases, the type A compound is produced when the
cyclization is
performed under nitrogen, while the type B arises when this step is performed
in a
vessel open to the air. However, this is not always true, and the outcome
varies
according to the particular system. Studies are on-going in this area to
elucidate the
mechanism controlling the formation of the two types of purpurin (for
background,
see K.M. Smith et al., J. Org. Chem., 48, p.500, 19$3 and A.R. Morgan et al.,
J. Org.
Chem., 51, p. 1347, 19$6) The cyclization of the imine displays
characteristics of a
26
CA 02254557 2003-10-24
type B purpurin synthesis, although the product contains a 6-membered
exocyclic
ring, reminiscent of a benzochlorin preparation.
Very recently a report of a system bearing similarity to our own appeared,
describing the synthesis of a new class of chlorine possessing 6-membered
exocyclic
rings, the "australochlorins" (D. Yashunsky et al., Aust. J. Chem., 50, p.
487, 1997).
These compounds are isomeric with the benzochlorins but have a [3-ethylidene
group
adjacent to the firsed ring, which is non-aromatic. They are obtained by the
thermolysis of the trimethylammonium salt as a mixture of two geometric
isomers in
a ratio of 10:7 (Figure 6). Spectroscopically, these compounds are typical
metallochlorins, absorbing at 643 nm, and hence are less interesting as
potential
photosensitizers than (93), which has a significantly red-shifted spectrum.
Although the cyclization experiments performed to this point had been
successful in producing quantities of the product (93) sufficient to allow its
structure
to be assigned, it obviously was desirable to attempt to improve upon the
yield of
15%. The reaction was repeated using Montmorillonite K10 clay as a solid acid
catalyst. This reagent has been used previously in acid-catalyzed reactions
such as
porphyrin syntheses, as it appears that the pore size of the clay is a good
fit for
allowing the entry of a porphyrin, while being small enough to hold such a
species in
a restricted conformation suitable for intramolecular cyclization reactions.
The
addition of a small amount of activated Montmorillonite clay to the toluene
solution
of the imine (92) doubled the yield of (93) to 30 % after ovenvght reflux.
However,
although the solvent and reaction time were varied, no further improvements
were
made to the efficiency of the cyclization, since the competing hydrolysis
reaction
limited the yield by destroying the imine starting material. Significant
quantities of
the aminoporphyrin (91 ) were recovered after each cyclization reaction, and
this could
be reused in the glyoxal condensation to regenerate (92).
In order to see if the cyctization would have a different outcome if performed
in the absence of air, in analogy with some purpurin syntheses, the reaction
was
performed under nitrogen. However, the same mixture of isomers was obtained as
previously. In an attempt to convert the isomeric mixture to a single
compound,
27
CA 02254557 2003-10-24
efforts were made to reduce the ethylidene double bond of (93) by catalytic
hydrogenation, but only decomposition products resulted. The mixture was then
subjected to diimide reduction, a reaction known to reduce symmetric double
bonds in
preference to double bonds between heteroatoms, but in this case,
surprisingly,
reduction occurred at the carbonyl group, giving the isomeric mixture of
alcohols (94)
(the same mixture that initially had been expected to result from the
cyclization
reaction, see Figure 4). The identity of this compound was confirmed by
reaction of
the original isomer mixhue with sodium borohydride, which gave the same
species as
its major product.
The alcohol product (94) gave the following selected resonances in the 400
MHz'H NMR spectrum (only signals for the major isomer given, for clarity): a
quartet at -0.23 ppm (methyl of the angular ethyl group), a doublet at 2.63
ppm
(methyl of the ethylidene group), a singlet at 4.50 ppm (CHOH) a quartet at
6.62 ppm
(methine of the ethylidene group), and a singlet at 7.88 ppm (CH=N). The
longest-
wavelength absorption peak was blue-shifted relative to (93) to 686 nm,
reflecting the
loss of the conjugated ketone.
As attempts to convert (93) to a single compound were unsuccessful, the two
isomers were separated by preparative thin layer chromatography, using a 0.5
mm
silica plate. There was sufficient differentiation between the two bands to be
able to
acquire the major isomer (93a) in good purity. The minor isomer (93b), owing
to
slight trailing of the faster moving major compound, as well as to its
presence in much
smaller quantity, was more difficult to isolate in a completely pure state,
but a
reasonable'H NMR spectrum of it was obtained. NOE experiments on (93a)
indicated that this compound has the Z-configuration, where the ethylidene
methyl
group is in the sterically less constrained position, pointing away from the
angular
ethyl group. This is analogous to the type B octaethylpurpurin situation,
where the Z-
geometry about the double bond was observed (although in that case no trace of
the
E-isomer was reported). The major isomer formed in the australochlorin
synthesis
also had the Z-geometry.
The intention had been to produce a compound with an aromatic exocyclic
ring. The major impediment to aromaticity in the cyclic product (93) is the
presence
zs
CA 02254557 2003-10-24
of the angular ethyl group. In steroid chemistry, an angular methyl group has
be
removed from androster-1,4-diene-3-one via a radical mechanism using zinc to
generate the aromatic A ring {Figure 7A, see also K. Tsuda et al., J. Org.
Chem. ,26,
p.2614, 1961; and K. Tsuda et al. J. Org Chem., 28, p. 795,1963).
As the exocyclic ring of (93) has a similar structure to the steroid A ring,
it
seemed possible this reaction might remove the angular ethyl group in an
analogous
way to yield the 3-hydroxypyridine structure (95) shown in Figure 7B. However,
on
refluxing the isomeric mixture (93) in pyridine in the presence of a large
excess of
zinc powder and a drop of water, only the zinc complex of the starting
material (Zn-
93) was obtained. At this point it appeared that the possibilities of this
compound had
been exhausted, and other strategies towards the desired goal were considered.
The replacement of glyoxal by glycolaldehyde in the condensation reaction
would yield an imine (96) which might be more likely to lead to the desired
product,
as under acidic conditions the hydroxyl group would be lost and the resulting
carbocation might easily lose a proton from the exocyclic ring to become
aromatic
(Figure 8). The analogous cyclization of the metal-free meso-(3-
hydroxypropenyl)octaethylporphyrin has been reported to give quantitative
yields of
octaethylbenzochlorin.
Unfortunately, despite several efforts, the condensation of glycolaldehyde
with
meso-aminooctaethylporphyrin (91 ) was not achieved. Possibly this is a
consequence
of the difficulty of depolymerizing the dimeric glycolaldehyde to its monomer,
although a reaction of this type with an aliphatic amine is reported to give
good yields
on stirring at room temperature in THF (see J.S. Davies et al., J. Chem. Soc.,
Perkin
Trans.,2, p. 201, 1991). In a different strategy to form the desired imine
alcohol.
attempts were made to condense glyoxalic acid to form the imine carboxylic
acid and
then reduce this to the alcohol (96). It appeared that the condensation
reaction was
successful, but on work-up, despite being careful to maintain very slightly
basic
conditions, the product reverted to the starting material (91 ), suggesting
auto-
hydrolysis of the imine double bond by the carboxylic acid functionality.
29
CA 02254557 2003-10-24
The next approach was to use the amine (91) as a nucleophile, to attack allryl
halides such as 1.2-dibromoethane and 1-bromo-2-(diethylacetal~thane, and give
the
amines (98) and (99) (Figure 9), which could then be used in cyclization
reactions.
However, in both of these cases no reaction occurred: it seems that the
nucleophilicity
of the nitrogen atom is low, presumably as a consequence of delocalization of
the lone
pair into the aromatic system. It was clear from these unsuccessful reactions
that a
different method was necessary to form the desired acyclic precursors with
nitrogen in
the a-position.
Efforts were then redirected to the ~i-pyridochlorins, with the expectation
that
they would in general be easier targets, and that the involved mechanisms
would
provide further insights into the efficient synthesis of a-pyridochlorins.
Example 22 S~~~s of ~-Ryridochlorin
In considering methods for synthesizing the acyclic precursor to the
~i-pyridochlorin., one might consider an approach very similar to that used
for the
a-pyridochlorin, and thus condensing the meso-formylporphyrin (5~ (Figure 10)
with
an appropriate amine, to obtain the [i-imine. Unfortunately, the necessary
amine
would be formamide, and amides do not possess the required nucleophilicity for
the
condensation. However, methylcarbamate, with greater nucleophilicity at the
nitrogen atom, would produce a condensation product (100) potentially useful
for the
cyclization (Figure 10). This condensation was attempted but despite the use
of a
various conditions, no reaction was observed, and the mesoformylporphyrin was
recovered unchanged.
Another potential way to make compounds with nitrogen in the desired
position would be to displace a leaving group on the meso-methyl group of the
porphyrin with a nitrogen nucleophile such as ammonia. The meso-hydroxymethyl
compound ( 1 O 1 ) can be made by sodium borohydride reduction of the formyl
group
of (56). Efforts were made to convert this first to the p-toluenesulfonate
ester, and
then displace the p-toluenesulfonyl group with ammonia gas. During the first
experiment, the intermediate ester was worked up, and an attempt was made to
isolate
CA 02254557 2003-10-24
it, but it appeared to be very reactive, and decomposed. Consequently, a
second
reaction was run, and this time the in situ formed p-toluenesulfonate was
reacted with
ammonia gas. However in this case, only the hydroxymethyl starting material
was
recovered. The next approach was a reductive amination of the meso- formyl
compound (56), using ammonium acetate and sodium cyanobomhydride, in another
attempt to form the meso-aminomethylporphyrin (102). Once again, no reaction
was
observed.
Having employed reactions well-known to work in typical organic systems,
but without success, literature methods that described reactions specific to
porphyries
were examined. There is one report of a condensation of an amine with meso-
formyloctaethylporphyrin (56), the amine being (3-alanine, and the yield was
only 15
(J-H. Fuhrhop et al., Liebigs Ann. Chem., p. 1537, 1976) As this synthesis had
been attempted with methylcarbamate without success, it was not pursued
further.
Another paper describes the condensation of meso-formyletioporphyrin with
aniline
and p-anisidine, with very good yields. However, this approach requires the
use of the
amine as solvent, and the compounds thus produced, being aromatic Schiffs
bases,
are structurally further from the desired product than the alanine conjugate
mentioned,
and it was felt that the latter was a more realistic model for the present
systems. A
more commonly used preparation of ~3-nitrogen-substituted porphyries involves
the
formation of the oxime of the meso-formylporphyrin using hydroxylamine
hydrochloride in refluxing pyridine (A. W. Johnson, J. Chem. Soc. (C), p. 794,
1966).
The oxime can be dehydrated to give the cyano group, which is reported to
yield the
meso-carboxylic acid by hydrolysis in sulfuric acid, although this latter
report has
been shown to be in error (P.S. Clezy et al., Aust. J. Chem.,27, p. 110, 1974)
and will
be discussed in more detail below.
The meso-cyanoporphyrin (103) provides a route to the meso-aminomethyl
compound ( 104) via reduction (Figure 1 I ). Hence ~neso-
cyanooctaethylporphyrin
was synthesized. and attempts were made to reduce it. However, this compound
was
extremely resistant to the many reagents used (lithium aluminium hydride,
aluminium
hydride, borane-dimethylsulfide complex, diborane, catalytic hydrogenation).
In most
cases, the starting material was recovered unchanged, although in the case of
catalytic
31
CA 02254557 2003-10-24
hydrogenation over palladium on charcoal, the porphyrin ring was reduced at
one of
the meso-positions to give the green phlorin, which slowly reoxidized to the
porphyrin. Reduction of the nickel complex of the meso-cyano compound was also
attempted, as the reactivity of a metalloporphyrin can differ markedly from
that of the
corresponding free base. However, there was no improvement in reactivity; the
only
differences being a lack of reaction during catalytic hydrogenation, and the
partial
loss of the cyano-group during treatment with lithium aluminium hydride. These
disappointing results prompted the search for another strategy.
As aforementioned, it was reported that the hydrolysis of meso-
cyanoetioporphyrin in hot concentrated sulfuric acid gave the carboxylic acid
(A.W.
Johnson, J. Chem. Soc. (C), p. 794, 1966). Clezy et al. reinvestigated this
reaction
and discovered that the product was in fact the amide. The same reaction was
performed on the octaethylporphyrin analog with identical results, and despite
using
various forcing conditions, no further hydrolysis of the amide (105) was seen.
This
amide is a potentially useful intermediate in the synthesis of cyclization
precursors
(Figure 12), as it would lead via reduction to the desired meso-
aminomethylporphyrin
( 104). Unfortunately, reduction of the amide ( 105) using lithium aluminium
hydride
in dry THF under nitrogen resulted in formation of the meso-cyano compound
(103),
i.e. the dehydration product. This was formed in 35 % yield, with the
remainder of
the yield being decomposition products, suggesting the possibility that the
aminomethyl product was formed, but that it was too unstable to be isolated.
The
nickel complex of the amide was also synthesized in order to see if it would
be more
easily reduced by treatment with the same reagent. In this case, no reaction
occurred
at room temperature, but after refluxing, the major product was nickel
octaethylporphyrin. This follows the same trend as with the nickel cyano-
derivative:
the nickel complexes appear to readily lose their meso-substituents under
basic
conditions.
Thus, despite many efforts, no practical method for preparing the desired
aminomethyl-substituted porphyrin ( 104) had been found. However, the meso-
amide
(105) could be produced. Although no progress had boen made in reducing this
compound, it was hoped that it could be converted into a cyclic chlorin with
an
32
CA 02254557 2003-10-24
excocyclic amide functionality, and that this cyclic product would prove more
amenable to reduction (Figure 12).
In order to make the precursor to the cyclization step using the amide, it was
necessary to alkylate the nitrogen atom with a funetionalized methyl group
susceptible to nucleophilic attack under acidic conditions, ideally a formyl
or a
hydroxymethyl group. Amides can be hydroxymethylated at the nitrogen atom
using
paraformaldehyde and base. A small-scale test reaction was run using sodium
methoxide as base in refluxing THF: this lead to the formation of a more polar
compound with a ~H NMR spectrum consistent with the desired product (106).
Subsequent trials suggested that the quantity of base was crucial to the
success of the
reaction, as a large excess lead to the formation of a more polar compound
which was
initially attributed to the doubly alkylated species, produced by
deprotonating the
nitrogen atom of the N-hydroxymethyl compound and reacting with a second
equivalent of paraformaidehyde. As it evidently was important to control the
quantity
of base used, sodium methoxide was replaced by n-butyllithium, as this is
easier to
measure out in small quantities. Using 1.5 equivalents of this base in THF at
room
temperature, a 70% yield of the meso-(N-hydroxy
methyl)aminocarbonyloctaethylporphvrin (106) was obtained (Figure 13).
The nickel complex of the N-hydroxymethyl compound was also of interest,
as the cyclization outcome can vary depending on whether the free base or
metallated
species is used (for example, refluxing the metal-free meso-acrylaldehyde of
octaethylporphyrin in acetic acid leads to the purpurin, whereas the nickel
complex is
unaffected by these conditions, see D.P. Arnold et al., JCS Perkin l, p. 1660,
1978).
Therefore the metal-free form of (106) was subjected to the standard nickel
complexing conditions: addition of nickel acetate tetrahydrate in refluxing
DMF.
Unfortunately, upon work-up of this reaction, it was found that the
hydroxymethyl
group had been lost, and the product was the nickel amide (Ni-105). The
hydroxymethyl group was also lost under the conditions used for EI (electron
impact)
mass spectrometry (at 200°C), so FAB (fast atom bombardment) was used
for these
compounds. The metallation with nickel was run at 50°C, and a 70 %
yield of the
desired product (Ni-106) was obtained after 3 days (the remai.ader of the
recovered
33
CA 02254557 2003-10-24
material was the unchanged starting material). In view of the very slow
metallation at
the lower temperature necessary to preserve the integrity of the compound, it
seemed
more efficient to make the nickel derivative by hydmxymethylation of the
nickel
amide (Ni-105). This was found to work well, although the first trial using
1.5
equivalents of n-butyllithium resulted in a poor yield, or the desired
compound and a
large amount of more polar material.
Again, it was initially believed that this side-product was the result of bis-
hydroxymethyladon at the nitrogen atom, but in this case there was now enough
compound to study spectroseopically, and it was found to be the product of
hydroxymethylation at the N-hydmxymethyl oxygen atom (Ni-107, Figure 14). This
is an obvious result, given the relative pKa's of an alcohol (15 to 19)
compared with
an amide (25). Consequently, the reaction was repeated using 0.5 equivalents
of
n-butyllithium, to minimize formation of the side-product, and a 76 % yield of
the
target molecule was obtained. It is interesting to note the differences
between the
reaction using the metal-free amide and that with the nickel amide: the latter
appears
to require only catalytic amounts of base, whereas the former requires at
least
stoicheometric quantities. Presumably this is due to the central NH's in the
metal-free
compound, one of which is deprotonated by the base to give the monoanion-
apparently the conditions are not such that the dianion is formed, as fewer
than 2
equivalents of base are necessary.
Both the metal-free and the nickel-complexed N-hydroxymethylamides ( 106)
and (Ni-106) had been prepared, ready for cyclization studies. The first
experiment
entailed stirring the nickel complex in dichloromethane solution with a dmp of
the
Lewis acid, boron trifluoride etherate. This represents one method for making
inetallated benzochlorins from their acyclic precursors. On work-up after
three hours,
there appeared to be two major products (Ni-108, present in different
proportions),
both more polar than the starting material. An attempt was made to separate
these
compounds by chromatography, but only the less polar one was obtained pure, as
both
compounds had poor solubility and streaked throughout the column. A'H NMR
spectrum of the pure compound was run, but very broad signals resulted, and
this was
thought to arise from aggregation of the compound in solution. However, the
visible
34
CA 02254557 2003-10-24
spectrum did give cause for optimism, as it displayed a large absorption peak
at 646
nm, suggestive of a nickel chlorin.
The next cyclization attempt involved treatment of the nickel complex (Ni-
106) with concentrated sulfuric acid. A peak was seen in the visible spectrum
of the
neutralized reaction mixture at 680 nm, indicative of a metal-free chlorin. On
work-
up a green-brown product (108) slightly more polar than the starting material
was
isolated in 70 % yield. Although by TLC this appeared to be one compound, the
~H
NMR spectrum showed it to be a 1:1 mixture of two isomers. The visible
spectrum of
the isolated mixture showed a double long-wavelength absorption peak at
680/686
nm. The 1H NMR spectrum was reminiscent of that of the isomeric a-
pyridochlorin
mixture (93) made previously, and it was obvious that the two isomers arose
from the
same ethylidene functionality as in that case. The compound was assigned the
structure shown below.
The metal-free cyclization precursor (106) was treated with sulfuric acid.
with
identical results. The O-hydroxymethylated side-product (Ni-107) (Figure 14)
was
also subjected to the same conditions, resulting in isolation of the same
product.
Finally. the mixture of nickel lactams (Ni-108) from the boron trifluoride
reaction was
stirred with concentrated sulfuric acid. Again, the same l:l isomeric mixture
of metal-
free lactams (108) was obtained. It is interesting to note that although there
appears to
be a major and a minor isomer formed in the BF, reaction, on stirring with
sulfwic
acid, a l:l mixture results - obviously equilibration occurs under strong acid
conditions.
CA 02254557 2003-10-24
Once again, the products formed via loss of a proton froth the yl group
had been formed, rather than the desired compound that would result from a 1,2-
dialkyl shift. A number of different cyclization conditions were investigated
in the
hope of finding a method that would lead to the target compound. The nickel N-
hydmxymethylamide (Ni-106) was refluxed in chloroform with the acidic clay
Montomorillonite K10 (the successful catalyst in the earlier cyclization
reaction to
make the a-pyridochlorin-type structure). However, in this case, although a
trace of
cyclized material was detected, the major products were nickel
octaethylporphyrin
and the nickel amide, with only a small amount of starting material. It is
surprising to
see that the meso-substituent was lost so easily under acidic conditions, as
has already
been observed under strongly basic conditions. Another cyclization was run
using the
metal-free N-hydroxymethylamide ( 106) in trifluoroacetic acid, and adding
concentrated sulfuric acid dropwise until cyclization occurred. Here the
thought was
that one of the two isomers would be favoured, and the lower acid strength
would
prevent equilibration to the 1:1 mixture thus far seen. Unfortunately in this
case, no
cyclization occurred until a large quantity of sulfuric acid was added, and
the usual
isomeric mixture (108) was obtained.
The next line of approach was to synthesize the metal-free and nickel N-
formylamide analogues (109) and (Ni-109), as there was an interest in
determining if
these would cyclize in the same manner, or if they might give the
corresponding
cyclic imides (110) (Figure 15). The N-formylamides were produced by
tetrapropylammonium penuthenate ('TPAP) oxidation of the N-hydroxymethylamides
(see W.P. Grii~th, et al., J. Chem. Soc. Chem. Commun. p.1625, 198. The
oxidations proceeded smoothly to give the compounds in reasonable yields. The
nickel complex (Ni-109) was treated with concentrated sulfuric acid, but no
cyclization was observed, and the demeta.llated N-formylamide (109) and
demetallated amide (105) were recovered. Dichloromethane/boron trifluoride
etherate
conditions were tried, but yielded only nickel octaethylporphyrin and a small
amount
of starting material. The metal-free N-formylamide ( 109) was then treated
with
dichloromethanelboron trifluoride etherate, but on work-up only starting
material and
the porphyrin amide were obtained. Finally, cyclization of the metal-free
compound
36
CA 02254557 2003-10-24
( 109) in refluxing chloroform with Montmorillonite clay was attempted, and in
this
case the resulting compounds were starring material and octaet6vlporphnrin. In
conclusion. the acid treatment of the N-formylamides appears to lead
preferentially to
hydrolysis of the N-fonnyl moiety rather than to cyclization.
Returning to the lactam (108), efforts were made to reduce the amide
functionality which had resisted such a reaction before cyclization. However,
once
again, lithium aluminium hydride proved ineffective, both at room temperature
and in
refluxing TE. In each case, starting material was recovered.
Attempts were also made to remedy the fact that (108) was present as a
mixture of isomers, rather than as a single compound. Catalytic hydrogenation
over
palladium on charcoal was performed on the isomeric mixture. This reaction met
with
some success, although it proved difficult to obtain reproducible results.
During some
experiments, no reduction was seen, while in others, good results were
achieved. This
may be due to the presence of small amounts of impurities that poisoned the
catalyst.
One observation common to all experiments was the formation of a large
quantity of a
more polar pink compound which appeared from its visible spectrum to be non-
porphyrinic, possibly arising from reduction of the porphyrin ring.
Unfortunately, this
compound did not reoxidize to the chlorin on stirring in air, or even after
the addition
of an oxidizing agent such as 1,2-dichloro-4,5-dicyanoquinone (DDQ). Hence the
yields for this reaction, even when "successful", were prohibitive. However, a
single
porphyrinic reduction product ( 111 ) was produced, reaction presumably taking
place
only on the least hindered side of the molecule, i.e. that opposite the
angular ethyl
group (it should be noted that both the a- and the ~-pyridochlorins
synthesized so far
possess a chiral centre, and so exist as a mixture of optical isomers). The
reduced
compound exhibited a small blue-shift in the visible spectrum, the double peak
at
680/686 nm becoming a single peak at 670 nm, reflecting the loss of the
conjugated
double bond of the ethylidene group.
Since the nickel complex of the lactam (Ni-I08) gave very broad signals in the
'H NMR spectrum, there was interest in seeing whether other metal complexes
displayed similar behavior. Consequently the lactam free base (108) was
metallated
37
CA 02254557 2003-10-24
with zinc. The zinc complex (Zn-108) displayed the same broadness in the'H NMR
spectrum as previously seen with the nickel complex, and so it appears likely
to result
from strong association of the complexed metal from one molecule with the
amide
group of another molecule, leading to aggregation in solution.
During the course of these efforts to produce a 13-pyridochlorin, a report
came
to light detailing the synthesis of a meso-isocyanoporphyrin (112) (see P.S.
Clezy, et
al., Aust. J. Chem., 27, p. 1003, 1974). This was relevant to these studies as
it
provided an alternative approach to the formation of the a-pyridochlorin. The
following section describes the strategy developed using this compound, and
the
subsequent results.
Example 3 ~dditio~al a~pmach to s~mthe~is of a_pvridochlorin via meso-
isocyanooctaethylporphyrin
The ~neso-isocyanoporphyrin (112) is produced by the dehydration of the
corresponding meso-formamide (I 13), which in turn is formed by a Beckmann
rearrangement of the meso-acetoxime (114) in concentrated sulfuric acid
(Figure 16).
A Passerini reaction (I Ugi ed. Isonitriles, Academic Press, New York, 1971)
of the isocyanide with formaldehyde and acetic acid as attempted in an effort
to obtain
the alkylated amide ( 11 S) shown in Figure 17. The product of this reaction
would
have been set up for acid-catalyzed cyclization to give a chlorin. However, on
work-
up, only the formamide (113) was isolated, formed by acid hydrolysis ofthe
isocyanide.
The next strategy involved reacting the isocyanide with formaldehyde in the
presence of the Lewis acid, boron trifluoride etherate. It was envisaged that
the
nucleophilic isocyanide carbon atom would attack the carbocation of the
formaldehyde-BF3 adduct, to give the analog of the b-pyridochlorin cyclization
precursor, with the nitrogen in the a-position (116), Figure I8.
38
CA 02254557 2003-10-24
Formaldehyde gas was bubbled through a flask containing toluene, giving a
solution of formaldehyde, to which was added boron trifluoride etherate. This
solution was added to the solid isocyanide, and stirred at mom temperature.
After
work-up, two compounds were isolated: the major product was the formamide
(113),
formed by hydrolysis of the isocyanide by BF3. The other product was polar and
highly insoluble. Spectroscopy showod this latter compound to be the desired
species
(116). Test reactions were run to see if (116) was susceptible to cyclization
in acid,
but neither reaction in dichloromethane with BF3 catalysis, nor reaction in
concentrated sulfuric acid lead to any trace of a cyclized product, and the
starting
material was recovered unchanged. In order to see if the nickel complex would
be
more prone to cyclization, the C-hydroxymethylamide was metallated with
nickel,
and this product (Ni-116) was subjected to the same reaction conditions
described for
the metal-free compound. The only reaction seen to occur was demetallation.
Finally, attempts were made to oxidize the metal-free C-hydroxymethylamide
(11~
using TPAP to form the C-formylamide, in the hope that this might change the
reactivity of the chain in favor of cyclization; however this oxidation
reaction failed.
The lack of reaction of the C-hydroxymethylamide (116) compared with the
N-hydroxymethylamide ( 106) can be explained by the relative stabilities of
the
carbocations which must be formed in order for cyclization to occur (see
Figure 19).
In the case of the N-hydroxymethylamide, the carbocation is adjacent to a
nitrogen
atom, and hence can be resonance-stabilized by the lone pair on that atom This
stabilization allows the ration to exist long enough for the double bond to
attack it,
leading to cyclization. In the case of the C-hydroxymethylamide, the
carbocation
would be created adjacent to a carbonyl group, i.e., adjacent to a partially
positive
carbon. Formation of the carbocation would lead to the highly unstable
situation
where two positive (or partially positive) atoms exist side by side. The lack
of
cyclization suggests that such a species either does not form at all, or its
lifetime is too
short to allow nucleophilic attack on it, leading to cyclization.
It was also intended to make more nickel C-hydmxymethylamide (Ni-116) by
C-alkylating the nickel isocyanide (Ni-112) with paraformaldehydeJBF3. Instead
of
39
CA 02254557 2003-10-24
obtaining the expected more polar hydroxymethyl compound, one major non-polar
product was isolated, with an unusual visible spectrum (intense peaks at 406,
430,
496, and 680 nm and a low intensity peak at 814 nm). The ~H NMR spectrum
showed by the presence of a six hydrogen triplet at -0.05 ppm that this
compound
possesses a gem-diethyl group, which Lead to the suggestion that a cyclization
had
occurred i~ situ to give the unsaturated lactam (Ni-11~.
However, this structure was not consistent with the !H NMR spectrum, as
there were no signals corresponding to the proton a to the NH, even if the NH
signal
itself were too broad to be distinguished. Despite its low polarity, the
product was
very slow to dissolve, and had a melting point higher than most
octaethylporphyrin
derivatives. These facts, coupled with the atypical visible spectrum,
suggested that
the compound might be a dimer, and so it was submitted for FAB mass
spectrometry,
which confirmed this. A mechanism can be drawn for the formation of a dinner
(Ni-
119) consisting of two units of (Ni-118) joined at the carbon n to the
nitrogen atom
(Figure 20).
CA 02254557 2003-10-24
This mechanism requires the addition of two equivalents of H+ and two
electrons per molecule of dimer. Excess BF3 was used, aad accounts for the
acid
requirement, but reduction under these conditions might seem harder to
explain.
However, the forznation of nickel octaethylbenzochlorin (Ni-17, see also
Figure 1)
also requires a formal reduction to take place under similar conditions, and
it is
suggested that nickel porphyrin complexes can act as electron donors under
acidic
conditions (see D.P. Arnold et al., JCS Per~Ein, I, p. 1660, 1978). It should
be noted
that the best yields of this reaction were approximately 50%, and a large
quantity of
non-porphyrinic decomposition pmduct was always obtained, reinforcing the
possibility of electron donation from two porphyrin molecules to two others to
form
the dimer, followed by decomposition of the two porphyrin cation radicals.
Also, the
fact that the reaction only occurs when the nickel complex is used, and no
cyclizaton/dimerization is seen with the metal-free compound, suggests that
the nickel
is playing an important role. The proposed structure would be consistent with
the'H
NMR spectrum, the only signals not observed being the NH protons.
The final structure (Ni-120, Figure 20) was ascertained by X-ray
crystallography, and was significantly different to that proposed. This
structure
contains a pyrido[3,2-b]-pyridine unit linking the two chlorin molecules. It
possesses
two fewer hydrogen atoms than the proposed structure, and fits better with the
mass
spectrum (however, without the hindsight made possible by a crystal structure,
it
would have been difficult to be certain of the exact number of hydrogen atoms
in the
dimer simply by FAB mass spectrometry, as such an ionization method sometimes
41
CA 02254557 2003-10-24
gives rise to molecular ion peaks corresponding to the gain of one or more
hydrogens). A mechanism can be drawn for the formation of this diner which is
catalytic in acid, and has no need for the reduction step (Figure 21).
Finally, this
structure is completely consistent with the'H NMR spectrum, and there are no
"missing" signals.
This result was extremely interesting. The objective of this Example was to
create a chlorin with a fused pyridine ring, which so far was difficult to
erect. In this
reaction, finally this objective had been achieved, but "in duplicate," in the
form of a
diner. In all previous cyclizatioas, the alkyl 1,2-shift did not occur, and
instead the
ethylidene product resulting from the loss of a proton from the ethyl group
was
obtained. This did not appear to happen in this case. However, when the
dimerization reaction was run on a larger scale, and the product was very
carefully
isolated, another compound was seen running just behind the diner (Ni-120) on
TLG.
After purification by column chromatography, iH NMR and mass spectra of this
fraction were obtained, and it was revealed to be the ethylidene analogue of
the diner,
in which there is one ethylidene group, one angular ethyl group, and one ge»~-
diethyl
group (Ni-121). As there is now no symmetry in the molecule, the'H NMR
spectrum
is much more complex. Two noteworthy points about this minor product are (a)
that
it exists as one isomer, i.e., there is only one geometry about the ethylidene
bond
(although the presence of a chiral cenetre at the angular ethyl group gives
rise to
optical isomers) and (b) that its visible spectrum differs greatly from that
of the major
dimeric product, possessing a Soret band at 410 nm and very little absorption
at
longer wavelengths. The angular hydrogen atom at the position of fusion of the
two
pyridine rings prevents these rings from becoming aromatic, and this
presumably
prevents interaction between the two porphyrin moieties and accounts for the
more
porphyrin-like visible spectrum.
42
CA 02254557 2003-10-24
The major nickel diner (Ni-120) was demetallated in concentrated sulfuric
acid to give a very insoluble grey product (120). The'H NMR spectrum of this
product showed two new singlets at 4.97 ppm 5.82 ppm, exchangeable with 1>ZO,
but
no signals were seen upfield of 0 ppm, in the region where the central NH
protons are
usually observed. It is possiblc that the pyridine nitrogens are more basic
than the
central nitrogens, and that it is the protonated pyridine NH's that give rise
to the new
signals. High resolution FAB mass spectrometry confirmed that demetallation
had
occurred. Further confirmation was obtained by complexing the demetallated
compound with zinc; the zinc diner (Zn-120) thus obtained displays a'H NMR
spectrum very similar to that of the nickel complex. The nickel, zinc and
metal-free
compounds all possess similar visible spectra.
Exam~e 4 a-PyridQchlorin synthesis via cyclization of tie metallated
formylmethylimine
The differences in reactivity to acid of the nickel-complexed and metal-free
meso-isocyanoporphyrins prompted the review of all previous attempts at
synthesizing pyridochlorins, in order to be certain every logical cyclization
method
had been tried on both the metallated and metal-free substrates. The first
cyclization
that had been attempted, which gave rise to the iminoketone (93), rather than
the
desired a-pyridochlorin, had been performed only on the metal-free precursor
(92).
For the sake of completeness, further investigations using the metatlated
compound
(M-92) were conducted.
43
CA 02254557 2003-10-24
Metallation of the formylmethylimine (92, see Figure 3) was attempted under
mild conditions, by stirring with zinc acetate at room temperature. However,
this
resulted in cleavage of the imine, to give the zinc amine (Zn-91 ). In light
of this
result, it was necessary to go back one step, to synthesize the zinc
formylmethylimine
by condensation of the zinc aminoporphyrin with glyoxal. The condensation
occurred
in better yield than that with the metal-free amine: after refluxing overnight
approximately 80% of the desired product (Zn-92) was obtained. Test
experiments
were run using various acids to effect the cyclization of the zinc
formylmethylimine,
as initially it appeared to be more acid-stable than the metal-free analogue.
Unfortunately, acetic acid, trifluoroacedc acid and boron trifluoride etherate
all gave
rise to the metal-free amine (91 ), and no evidence of cyclization was
observed. Hence
the standard cyclization conditions of Montmorillonite K10 acidic clay and
refluxing
toluene were employed. After 24 hours reflux mostly starting material
remained,
according to TLC, but there was a small amount of more polar green product, as
well
as some zinc iminoketone (Zn-93) (this cospotted with an authentic sample
produced
by metallating the iminoketone (93) with zinc). Refluxing was continued for a
further
24 hours, then the mixture was worked up and purified by chromatography. The
polar
green material (Zn-123) was isolated in 10% yield, and 80% of the starting
material
was recovered. The visible spectrum of the new compound indicated it was a
chlorin,
with a prominent absorption peak at 678 nm. In contrast to the metal-free
cyclic
product (93), the 1H NMR spectrum of this product showed no ethylidene peaks,
but a
six hydrogen triplet at 0.33 ppm suggested that the cyclization had occurred
as
originally anticipated, to give an aromatic exocyclic pyridine ring and a
genre-diethyl
group at the adjacent ~-position. There were four peaks in the aromatic
region,
evidence of three rneso-hydrogens and one hydrogen on the exocyclic ring.
This,
coupled with the mass spectral data, showed that unlike in the benzochlorin
synthesis,
the hydroxyl group is retained on the ring. The exchangeable hydroxyl proton
was
not observed in the NMR spectrum, which had to be run in pyridine, as the
compound
was poorly soluble in other solvents. The new chlorin (Zn-123) was assigned
the
structure shown.
44
CA 02254557 2003-10-24
(Zn-123)
This compound was diffcult to obtain in a completely pure state, as it
streaked
badly during chromatography. It was demetallated in
dichloromethane/trifluoracetic
acid to give the metal-free species (123) in approximately 30°r6 yield.
It seems likely
that this poor yield is a reflection of the impurity of the starting material
rather than a
measure of the inefficiency of the demetallation. The metal-&ee compound (123)
was
easier to purify, as it is less polar and runs in a much narrower band on the
silica
column. It possesses a visible spectrum with a strong absorption at 672 nm;
the
diagnostic peaks in the ~ H NMR spectrum are a six hydrogen triplet at 0.40
ppm and
four aromatic peaks above 8 ppm, as well as a broad singlet at 3.06 ppm
corresponding to the OH proton.
The synthesis of one of the target compounds had finally been achieved, using
the very first method devised, but with one obviously crucial modification:
using the
metallated substrate for the cyclization rather than the free base. The (3-
hydroxypyrido) chlorin (123) appears to be fairly stable to acid (in that it
can be
demetallated by treatment with acid without destruction of the chromophore),
has
good spectroscopic properties, and possesses sites for possible further
derivation in its
hydroxyl group and basic N-atom. Unfortunately, so far efforts to improve the
yield
have failed - approximately 5 mg product is obtained from 100 mg zinc
formylmethylimine (Zn-92). However, the unused starting material is recovered
unchangcd, and can be recycled through the cyclization reaction many times.
CA 02254557 2003-10-24
Additional information concerning the synthesis of such compounds is as
follows. Octaethylbenzochlorin is synthesized via intramolecular cyclization
of the
metallated meso-acrylaldehyde substituted porphyrin under acidic conditions.
It
seemed logical to follow a similar route to prepare octaethylpyridochlorin.
Meso-
aminooctaethylporphyrin 1 was condensed with glyoxal in THF/ethanol. After 24
hours refluxing the formylmethylimine product 2 was obtained in 69% yield.
Intramolecular cyclization of 2 was achieved by overnight reflex in toluene in
the
presence of Montrnorillonite K10 acidic clay. The two geometric isomers 3a and
3b
were isolated in 31% total yield, in addition to a substantial quantity of the
hydrolysis
pmduct, 1. The cyclic product consisted of a 4:1 mixture of the two isomers,
which
were separable by preparative chromatography. The major component was examined
by NOE spectroscopy, which revealed it to be the Z-isomer 3a. The isomeric
mixture
was treated with acid, in an attempt to bring about a 1,2-diallcyl shift and
hence form
the desired pyridochlorin 4. However, this reaction was unsuccessful, leading
to
decomposition of the chromophore. Experiments intended to hydrogenate the
ethylidene double bond of the isomeric mixture to give a single isomer also
failed.
OEPNHz (~ .. pEPN=CHCHO (i ~ '~'
2
(~ CHCHO, THFIEtOH, n;
(ii) MontmoriHonite K10, toluene, D
Scheme 1. Synthesis of the isomeric chlorine 3a and 3b
The presence or absence of a centrally-eomplexed metal can have a marked
effect on
the outcome of intramolecular cyclization reactions of porphyries. Therefore
the
cyclization of the zinc complex of 2 was studied. Ze-2 was prepared by
condensation
of the zinc aminoporphyrin Zn-1 with glyoxal (metallation of 2 with zinc
acetate led
to hydrolysis of the imine, resulting in isolation of Zn-1). The cyclization
was
performed using the conditions described above (toluene, Montmonillonite K 10,
heat),
38 R=CH~,R'=H
3b R = H, R' = CH3
CA 02254557 2003-10-24
and did indeed proceed differently to that of the free base 2. In this case,
reaction was
much slower, little change being discernible until refluxing had continued for
3 or 4
days. At this point only a trace amount of the expected product (the zinc
complex of
3a/3b) was seen on TLC. A more polar green compound appeared to be the major
product of the reaction (in addition to unreacted starting material, Zn-Z,
present as
approximately 80% of the mixture). After work-up this compound was isolated in
20% yield and NMR analysis showed it to be the zinc complex of 4.
Demetallation
with trifluoroacetic acid gave the free base 4.
ZnOEPNHZ- C~ ZnOEPN=CHCHO
Zn-1 Zn-2
(i) CHCHO, THFIEtOH, A;
(ii) MontmoriHonite K10, toluene, 4
(iii) TFA
Scheme 2. Synthesis of pyridochlorin 4
1'yridochlorin 4 has a slightly red-shifted long-wavelength absorption (7~"~ =
672 nm), compared to octaethylbenzochlorin (7v""~ = 658 nm), and it possesses
additional characteristics that might improve its performance as a
photosensitiur over
the latter. It is known that amphiphilic macrocycles, i.e., those bearing both
hydrophobic and hydrophilic moieries display better tumour-localizing
properties than
photosensitizers without these properties. The presence of the polar
hydroxypyridyl
ring attached to the low polarity porphyrin skeleton increases the
amphiphilicity of 4.
Octaethylbenzochlorin suffers from a lack of functional groups available for
further
derivatization; the preparation of analogues based on this compound requires
additional synthetic steps in order to introduce such a functionality. In
contrast,
chlorin 4 possesses "built-in" sites at the hydroxypyridine ring that allow
for more
direct analogue formation.
47
Zn-t M = Zn ~
4 M=2H
CA 02254557 2003-10-24
It will also be appreciated by those familiar with the art that the pyridinyl
hydroxyl group such as shown for compound 4 directly above can be replaced by
hydrogen under appropriate conditions, thereby providing an additions! pathway
to
compounds containing a simple (unsubstituted) pyridine ring as exocyclic
component.
3a:'H NMR (CDCI3) S: -1.78 (br s, 1H, NH), -0.74 (br s, 1H, NH), 0.50 (t,
J=7.3 Hz,
3H, CH3),1.65-1.90 (m, 18H, 6CH3), 2.81 (d, J=7.6 Hz, 3H, CH3=CH), 3.70-4.00
(m,
12H, 6CH2), 4.09-4.26 (m, 2H, CHZ), 7.45 (q, J=7.5 Hz, l H, CH~H3), 8.15 (s, l
H,
CH=N), 9.31, 9.51, 9.57 (3s, 3H, meso-H's); W-Vis in CHCH3, 7~ nm (s): 360
(4.33),
410 (4.82), 446 .(4.67), 584 (3.66), 672 (3.79), 722 (4.03); Analysis calc'd
for
C38H45NsO~H20: C, 75.34; H, 7.82; N, 11.56: found C, 74.89; H, 7.69; N,11.15.
3b: 'H NMR (CDCI3) 8: -1.14 (br s, 1 H, NH), -0.74 (br s, 1 H, NH), 0.10 (t,
J=7.3 Hz,
3H, CH3), 1.b8-1.84 (m,18H, 6CH3), 2.62 (d, J=7.5 Hz, 3H, CH3~H), 3.70-3.95
(m,
12H, 6CHz), 4.04-4.25 (m, 2H, CHz), 7.?2 (q, J=7.5 Hz, 1 H, CH~H3), 7.98 (s, 1
H,
CH=N), 9.16, 9.43, 9.53 (3s, 3H, »reso-H's).
4: 'H NMR (pyridine-ds) 8: 0.40 (t, J=7.3 Hz, 6H, 2CH3), l .b3-1.81 (m, I SH,
SCH3),
1.88 (t, J=7.3 Hz, 3H, CH3), 2.78-2.91 (m, 2H, CH2), 3.49-3.71 (m, IOH, SCHZ),
3.85
(q, J=7.5 Hz, 2H, CHZ), 4.43 (q, J=7.3 Hz, 2H, CH2), 8.40, 8.99, 9.58, 9.69
(4s, 4H,
3meso-H's and CH=N) 13.06 (br s, 1H, OH); W-Vis in CHCH3, 7~ nm (s): 412
(4.89), 490 (3.51), 522 (3.73), 556 (3.75), 616 (3.86), 672 (4.36); Analysis
calc'd for
C3xH4~N50~0.5H20: C, 76.22; H, 8.08; N, 11.70: found C, 75.96; H, 7.64; N,
11.67.
Example 5 Preparation and Characterization of Compounds
Presented below are details on the synthesis and characterization of
compounds used in the practice of the invention. In swnmary, these Examples
have
described efforts to synthesize a number of chlorine with fused pyridine
rings,
analogous to the benzochlorins. Varying yields were achieved, with one of the
preferred target molecules being prepared, while three other new types of
cyclic
chlorin were created.
48
CA 02254557 2003-10-24
The first new chlorin that was prepared, (93), possesses an exocyclic ring
containing a ~-carbonylimine functionality O=CCH=N, and an ethylidene group on
the reduced pyrrole ring. Its Longest-wavelength absorption in the visible
spectrum is
at 722 nm (E =10800). Both possible geometric isomers were produced, in a
ratio of
approximately 4:1, and these two isomers could be separated by chromatography,
although they wen not amenable to catalytic hydrogenation to give a single
isomer.
The carbonyl group could be reduced with sodium borohydride to give the
corresponding alcohol. Attempts to aromatize ring were unsuccessful. This
compound could be metallated with nickel or zinc under standard conditions.
The second type of chlorin synthesized, (108), contains an exocyclic lactam,
with an absorption at 686 nm (s = 34800). Again, an ethylidene group is
attached to
the reduced pyrrole ring, but in this case the ratio of geometric isomers is
1:1, and
these proved substantially inseparable by chromatography. The metal complexes
of
this compound appear to exist in solution as aggregates, indicating the high
affinity of
the centrally complexed metal for the lactam amide functionality of another
molecule.
Reduction of the amide group failed, but the ethylidene group underwent
catalytic
hydrogenation to give a single diastereomer. Metallation and demetallation
reactions
were successful.
The third class of compounds formed, ( 120), consists of a diner of two
chlorin
molecules possessing exocyclic pyridine rings, fused to each other through the
pyridine rings. These compounds display very unusual visible spectra, for
example,
the nickel complex possesses an absorption peak at 814 nm (s = 13600). The X-
ray
crystal structure shows that the molecule is essentially planar. Metallation
and
demetallation reaction were successful.
The fourth and final class of compounds synthesized; (123), contains an
exocyclic 3-hydroxypyridine ring, with the pyridine N-atom positioned a to the
porphyrin meso-position. The metal-free species has a visible spectrum with
its
longest-wavelength absorption at 672 nm (E = 22700).
49
CA 02254557 2003-10-24
Preparation and characterization of compounds
(92) 5-Formylmethyliminooctaethylporphyrin
N N
N 1 ChICHO
N
5-Aminooctaethylporphyrin (91 ) 194 ( 100 mg, 0.18 mmol) was dissolved in dry
THF (10 mL). Glyoxal trimer dihydrate (250 mg, 1.2 mmol) was dissolved in
ethanol
( 10 mL) with heating. The latter solution was added to the former, and the
mixture
was refluxed overnight. The solvent was removed in vacuo and the residue
purified by
chromatography (flash silica, dichloromethane eluent, polarity of eluent
increased to 5
ethyl acetate in dichloromethane after the product had been eluted to elute
starting
material), giving the product (74 mg, 69 %) and unreacted starting material
(24 mg).
1H NMR (200 MHz, CDCl3) 8 1.80 (t, J=7 Hz, 6H, 2 x CH3), 1.88-2.12 (m,
18H, 6 x CH3), 3.92-4.26 (m, 16H, 8 x CH2), 7.84 (d, J=9 Hz, 1H, CHCHO), 9.95
(s,
1H, 1 meso-H), 10.11 (s, 2H, 2 meso-H's), 10.45 (d, J=9 Hz, 1H, CHO); 13C NMR
(50 MHz, CDCI3) 8 16.21, 18.45, 18.51, 19.77, 22.15, 96.69, 98.19, 102.62,
128.26,
137.18, 140.30, 141.53, 142.36, 143.67, 143.89, 145.73, 169.16, 191.46; W-Vis
(CHCI3 (log s)) ~.t"~ 396 (4.96), 458 (4.67), 502 (3.98), 582 (3.91), 660
(3.64) nm;
MS (EI) »t/e 589 (M+, 100 %), 560 (M+-29, 32 %).
(93) Cyclic iminoketone (diastereomeric mixture)
N
N N
H
N
The formylmethylimine (92) (61 mg, 0.10 mmol) was dissolved in toluene (10
CA 02254557 2003-10-24
mL). Montmorillonite KI O acidic clay (50 mg) was activated by heating with a
heat-
gun in a test tube for 5 min, until all water had been driven off, and was
then added to
the solution. The mixtwe was refluxed overnight, then filtered and the solvent
was
removed in vacuo. The residue was purified by chromatography (silica,
dichloromethane eluent) to give the cyclized product (19 mg, 31 %) as 2
diastereomers and unreacted starting material (14 mg). The 2 isomers were
separated
by prep. TLC on a 0.5 mm thick silica plate, eluting with
dichloromethane:hexanes
1:1.
UV-Vis (CHC13 (log s)) ~,m~ 360 (4.33), 410 (4.82), 446 (4.67), 584 (3.66),
672 (3.79), 722 (4.03) nrn; MS (EI) mle calc'd for C38H45N50: 587.36243, found
587.36313; 587 (M+, 40 %), 558 (M+-29, 100 %); Singlet Oxygen Test: Negative.
(93a) Cyclic iminoketone (major isomer)
Present as approximately 80 % of the mixture.
m.p. 214-215°C; RF 0.36 (silica - CHzCl2:hexanes 1:1); 1H NMR (400 MHz,
CDCl3) b -1.18 (br s, 1H, 1 x NH), -0.74 (br s, 1H, 1 x NH), 0.50 (t, J=7.3
Hz, 3H,
CH3 of angular ethyl group), 1.65-1.90 (m, 18H, 6 x CH3), 2.81 (d, J=7.6 Hz,
3H,
CH3CH=), 3.70-4.00 (m, 12H, 6 x CH2), 4.09-4.26 (m, 2H, CH2 of angular ethyl
group), 7.45 (q, J=7.5 Hz, 1 H, CHCH3), 8.15 (s, 1 H, CH=N), 9.31 (s, 1 H, 1
meso-H),
9.5I (s, 1 H, 1 meso-H), 9.57 (s, 1 H, 1 meso-H); Analysis calc'd for
C3gH45N50~H20:
C, 75.34; H, 7.82; N, 11.56; found: C, 74.89; H, 7.69; N, 11.15.
(93b) Cyclic iminoketone (minor isomer)
Present as approximately 20 % of the mixture.
Rg 0.27 (silica - CH2CI2:hexanes 1:1); 1H NMR (400 MHz, CDCl3) 8 -1.14
(br s, 1 H, 1 x NH), -0.74 (br s, 1 H, 1 x NH), 0.10 (t, J=7.3 Hz, 3 H, CH3 of
angular
ethyl group), 1.68-1.84 (m, 18H, b x CH3), 2.62 (d, J=7.5 Hz, 3H, CH3=CH),
3.70-
3.95 (m, 12H, 6 x CHZ), 4.05-4.25 (m, 2H, CH2 of angular ethyl group), 7.72
(q,
J=7.5 Hz, 1 H, CH3=C~, 7.98 (s, 1 H, CH=N), 9.16 (s, 1 H, 1 meso-H), 9.43 (s,
1 H, 1
51
CA 02254557 2003-10-24
meso-H), 9.53 (s, l H,1 meso-H).
(Zn-93) Cyclic iminoketone-zinc (II) (diastereomeric mixture)
The cyclic product (93) (6 mg, 0.010 mmol) was dissolved in dichloromethane
(1 mL), and zinc acetate dihydrate (10 mg, 0.046 mmol) in methanol (0.5 mL)
was
added. The mixture was refluxed 2 hours, then the solvent was removed in vacuo
and
the residue purified by chromatography (silica, dichloromethane eluent) to
give the
metallated product (4 mg, 62 % yield).
R~ 0.64 (silica - CH2Cl2); 1H NMR (200 MHz, CDC13) (NB: only peaks for
major isomer listed) 8 0.50 (t, J=7.5 Hz, 3H, CH3 of angular ethyl group),
1.51-1.75
(m, 18H, 6 x CH3), 2.70 (d, J=7.5 Hz, 3H, CH3CH=), 3.49-3.75 (m, 12H, 6 x
CH2),
3.80-3.97 (m, 2H, CHZ of angular ethyl group), 7.35 (q, J=7.5 Hz, 1 H, CHCH3),
7.95
(s, 1 H, CH=N), 8.85 (s, 1 H, 1 meso-H), 9.05 (s, 1 H, 1 meso-H), 9.13 (s, 1
H, 1 meso-
H); UV-Vis (CH2C12) 7~t"~ 412, 454, 688 nm.
(94) Cyclic iminoalcohol (diastereomeric mixture)
N 9
H N
N
N
The cyclic product (93) (8 mg, 0.013 mmol) was dissolved in dichloromethane
(5 mL). Sodium borohydride (10 mg, 0.26 mmol) in ethanol (0.5 mL) was added,
and
the mixture was stirred at room temperature for 1 hour. The mixture was then
poured
into water and extracted with dichloromethane. After work-up, the residue was
purified by prep. TLC (0.2 mm thickness silica plate, dichloromethane eluent)
to give
the product, present as a mixture of diastereomers (2 mg, 25 % yield).
~H NMR (400 MHz, CDCl3) (NB: only peaks for major isomer listed) 8 -2.25
(br s, 1 H, 1 x NH), -1.69 (br s, 1 H, 1 x NH), -0.23 (t, J=7.4 Hz, 3H, CH3 of
angular
52
CA 02254557 2003-10-24
ethyl group),1.54-2.00 (m, 18H, 6 x CH3), 2.63 (d, J=7.5 Hz, 3H, CH3CH=), 3.70-
4.40 (m, 14H, 7 x CH2), 4.50 (s, 1H, CHOH), 6.62 (q, J=7.4 Hz, 1H, CHCH3),
7.88
(s, 1 H, CH=N), 9.42 (s, 1 H, 1 meso-H), 9.64 (s, 1 H, 1 meso-H), 9.71 (s, 1
H, 1 meso-
H); MS (EI) »t/e 589 (M+, 50 %), 587 (M+-2, 45 %), 560 (M+-29, 75 %), 558 (M~'-
31,
100 %); Singlet Oxygen Test: Positive.
(105) 5-Aminocarbonyloctaethylporphyrin
Prepared according to the method of Clezy et al. 20~
m.p. 296-299°C (lit.295-297); Rp 0.32 (silica - CH2Cl2 / 5 % AcOEt);
L1V-Vis
(CHZCl2) ~,~,~ 400, 502, 536, 572, 624 nm.
(106) 5-((N-Hydroxymethyl)aminocarbonyl)octaethylporphyrin
N N
N N CONiiCii ~Oli
The amide (105) (28 mg, 0.048 mmol) was dissolved in dry THF (10 mL)
under nitrogen. 1.6 M n-BuLi in hexanes (30 ~L, 0.048 mmol) was added, and the
mixture was stirred under nitrogen for 5 min. Paraformaldehyde (3 mg) was
added,
and the mixture was stirred at room temperature for 1 hour. Acetic acid (2
drops) was
added, then the solvent was removed in vacuo and the residue was purified by
chromatography (flash silica, eluent 5 % ethyl acetate in dichloromethane), to
give
unreacted starting material (6 mg) and the product (15 mg, 51 % yield).
m.p. >300°C; Rp 0.16 (silica - CH2C12 / 5 % AcOEt); ~ H NMR (200 MHz,
CDCl3) 8 -3.50 (v br s, 2H, 2 x NH), 1.33 (t, J=7.5 Hz, 6H, 2 x CH3), 1.78-
1.94 (m,
18H, 6 x CH3), 3.44-3.62 (m, 4H, 2 x CH2), 3.84-4.12 (m, 12H, 6 x CH2), 4.42
(d,
J =6 Hz, 2H, CH20H), 6.50 (t, J=6 Hz, 1 H, NH), 9.88 (s, 1 H, 1 meso-H), 10.10
(s, 2H,
2 meso-H's); 13C NMR (50 MHz, CDCl3) 8 17.55, 18.47, 19.57, 20.30, 65.18,
96.33,
53
CA 02254557 2003-10-24
97.02, 102.83, 110.80, 140.73, 141.48, 142.09, 142.25, 143.13, 144.58, 145.32,
172.10; LTV-Vis (CH2C12) 7~.n,~ 400, 502, 536, 570, 624 run; MS (FAB
(thioglycerol
matrix)) mle 608 (M+, 100 %).
(Ni-105) (5-Aminocarbonyloctaethylporphyrinato)nickel(II)
The amide (105) (31 mg, 0.054 mmol) was dissolved in dimethylformamide (5
mL) and nickel acetate tetrahydrate (30 mg, 0.12 mmol) was added. The mixture
was
refluxed overnight, then poured into water, extracted with ethyl acetate, the
extracts
dried over sodium sulfate, and the solvent removed in vacuo. The residue was
purified
by flash chromatography (silica, eluent 5 % ethyl acetate in dichloromethane)
to give
the metallated product (28 mg, 82 % yield).
m.p. 272-274°C; Rp 0.44 (silica - CH2C12 / 5 % AcOEt); 1H NMR (200 MHz,
CDC13) 8 1.60 (t, J=7.5 Hz, 6H, 2 x CH3), 1.77 (t, J=7.5 Hz, 18H, 6 x CH3),
3.73-3.99
(m, 16H, 8 x CHZ), 5.75 (br d, J=2 Hz, 1 H, 1 x NH), 6.38 (br d, J=2 Hz, 1 H,
1 x NH),
9.52 {s, 1H, 1 meso-H), 9.60 (s, 2H, 2 meso-H's); 13C NMR (50 MHz, CDC13) S
18.02, 18.17, 19.58, 21.21, 96.36, 96.86, 139.24, 140.10, 140.81, 143.25,
143.47,
143.69, 145.61, 172.50; LTV=Vis (CH2C12) ~,m~ 400, 524, 558 nm; MS (FAB
(thioglycerol + CHCl3 matrix)) mle 634 (M+, 100 %).
(Ni-106) [5-((N-Hydroxymethyl)aminocarbonyl)octaethylporphyrinato]nickel(II)
The nickel amide (Ni-105) (30 mg, 0.047 mmol) was dissolved in dry THF
(20 mL) and 1.6 M n-BuLi in hexanes ( 15 ~L, 0.024 mmol) was added. This
solution
was stirred for 5 min under nitrogen, then paraformaldehyde (4 mg) was added.
The
mixture was stirred at room temperature for 1 hour, then acetic acid (2 drops)
was
added and the solvent evaporated. The residue was purified by chromatography
(silica, eluent 5 % ethyl acetate in dichloromethane) to give the pink product
(24 mg,
76 % yield).
Rp 0.25 (silica - CHZC12 / 5 % AcOEt); 1H NMR (200 MHz, CDCl3) 8 1.50 (t,
J=7.5 Hz, 6H, 2 x CH3), 1.68-1.85 (m, 18H, 6 x CH3), 3.61-3.92 (m, 16H, 8 x
CHZ),
54
CA 02254557 2003-10-24
5.08 (d, J=6.0 Hz, 2H, CHZOH), 6.58 (t, J=6.0 Hz, 1H, NH), 9.48 (s,1H, 1 meso-
H),
9.53 (s, 2H, 2 meso=H's); ~ 3C NMR (50 MHz, CDC13) 8 17.74,18.09, 18.17,19.54,
20.84, 65.67, 96.47, 96.88, 148.99, 135.29,140.10, 140.77, 143.22, 143.39,
143.46,
145.51, 171.77; I1V-Vis (CH2Cl2) ~,m~ 398, 522, 558 nm; MS (FAB (thioglycerol
+
CHC13 matrix)) »r/e 663 (M+, 30 %).
(109) 5-(N-Formylaminocarbonyl)octaethylporphyrin
N N
H CONICli O
N
The N-hydroxymethylamide (106) (5 mg, 0.008 mmol) was dissolved in
dichloromethane (2 mL) to which anhydrous magnesium sulfate (10 mg) had been
added. To this mixture was added N-methylmorpholine-N-oxide (1 mg), and the
solution was stirred for 5 min. Then tetrapropylammonium perruthenate (TPAP)
(0.5
mg) was added and the mixture was stirred for 30 min. The solvent was
evaporated,
and the residue purified by chromatography (silica, eluent dichloromethane) to
give
the pink product (4 mg, 80 % yield).
Rp 0.29 (silica - CH2C12); LJV-Vis (CH2Cl2) ~,,~~ 402, 504, 538, 572, 624
nm; MS (FAB (thioglycerol matrix)) mle 606 (M+, 100 %).
(Ni-109) [5-(N-Formylaminocarbonyl)octaethylporphyrinato]nickel(II)
The nickel N-hydroxymethylamide (Ni-106) (11 mg, 0.017 mmol) was
dissolved in dichloromethane (2 mL) to which anhydrous magnesium sulfate (20
rag)
was added. To this solution was added N-methylmorpholine-N-oxide (2 mg). The
mixtwe was stirred for S min, then tetrapropylammonium perruthenate (TPAP)
(0.5
mg) was added. The mixture was stirred for 30 min, then the solvent was
evaporated,
and the residue purified by chromatography (silica, dichloromethane eluent) to
give
CA 02254557 2003-10-24
the pink product (7 mg, 64 % yield).
RF 0.43 (silica - CH2Cl2); UV-Vis (CH2C12) ~"~ 398, 524, 560 nm; MS
(FAB (thioglycerol + CHC13)) mle 662 (M++1, 85 %).
(Ni-107) [5-((N-
Hydroxymethylmethylether)aminocarbonyl)octaethylporphyrinato]nickel-(II)
. ,
co~er~ ,oeH,oH
J ~ ~'
Formed as a side-product in the synthesis of (Ni-106), especially when
excesses of butyllithium were used.
1H NMR (200 MHz, CDC13) b 1.58 (t, J=7.5 Hz, 6H, 2 x CH3), 1.70-1.88 (m,
18H, 6 x CH3), 3.62-3.99 (m, 19H, 8 x CHZ + OCH20 + NH), 5.46 (d, J=8 Hz, 2H,
NHCH20), 9.58 (s, 1H, 1 meso-H), 9.64 (s, 2H, 2 meso-H's); MS (FAB
(thioglycerol
+ CHCl3 matrix)) mle 693 (M+, 16 %).
( 108) Unsaturated lactam (mixture of isomers)
N H N1
N _
N 0
The nickel N-hydroxymethylamide (Ni-106) (10 mg, 0.015 mmol) was
dissolved in concentrated sulfuric acid (1 mL), and stirred at room
temperature for 1.5
hours. The solution was then poured into water, extracted 4 times with
dichloromethane, washed successively with potassium carbonate solution and
water,
and the solvent evaporated. The residue was purified by chromatography
(silica,
eluent 20 % ethyl acetate in dichloromethane) to give the green-brown product
as a
56
CA 02254557 2003-10-24
1:1 mixture of isomers (7 mg, 79 % yield).
m.p. 293-294°C; Rg 0.44 (silica - CH2C12 / 20 % AcOEt); ~ H NMR (400
MHz, CDC13) 8 (selected peaks), -1.20 (br s, 2H, 2 x NH of one isomer), -0.62
(br s,
2H, 2 x NH of one isomer), 0.33 (t, 3H, Me of angular Et group of one isomer),
0.58
(t, 3H, Me of angular Et group of one isomer), 2.37 (d, 3H, CH3=CH of one
isomer),
2.60 (d, 3H, CH3~H of one isomer), 6.10 (q, 1H, CH=CH3 of one isomer), 6.66
(d,
1 H, NH of one isomer), 6.73 (d, 1 H, NH of one isomer), 7.17 (q, 1 H, CH~H3
of one
isomer), 8.86, 9.10, 9.39, 9.47, 9.60, 9.62 (6s, 6H, 6 meso-H's); UV-Vis
(CH2C12
(log s)) ~.~,~ 414 (5.15), 506 (3.95), 542 (3.57), 628 (3.57), 680 (4.53), 686
(4.54)
nm; MS (EI) m/e calc'd for C3gH4~N50: 589.37805, found 589.37825; 589 (M+, 100
%); Analysis calc'd for C3gH4~NsO: C, 77.38; H, 8.03; N, 11.87; found: C,
77.10; H,
7.96; N, 11.59; Singlet Oxygen Test: Positive.
(Ni-108) Nickel unsaturated lactam (mixture of isomers)
The nickel N-hydroxymethylamide (Ni-106) ( 14 mg, 0.021 mmol) was
dissolved in dichloromethane (5 mL) and boron trifluoride etherate (1 drop)
was
added. The colour changed from grey-green to bright green almost immediately.
The
solution was stirred for 3 hours, then the solvent was evaporated and the
residue
purified by chromatography (silica, eluent 10 % ethyl acetate in
dichloromethane) to
give 4 mg pure less polar isomer and 8 mg of a mixture of the two isomers.
Rg 0.18 and 0.08 (silica - CH2Cl2 / 5 % AcOEt); W-Vis (CH2C12) ~.m~ 414,
506, 562, 644 nm; MS (EI) mle 645 (M+, 100 %), 616 (M+-29, 61 %).
( 11 I ) Lactam reduction product
N N Mi
N
N O
CA 02254557 2003-10-24
The lactam (148) (5 mg, 0.008 mmol) was dissolved in THF (3 mL) and
triethylamine (1 drop). 10 % Pd on charcoal (2 mg) was added and the mixture
was
stirred overnight under a hydrogen balloon. The catalyst was filtered off and
air was
bubbled through the solution for 1 hour. The THF was evaporated and the
residue
purified by chromatography (silica, eluent 20 % ethyl acetate in
dichloromethane) to
give the desired product as a dark green solid (2.5 mg, 50 % yield).
m.p. >300°C; R~ 0.44 (silica - CH2Cl2 / 20 % AcOEt); 1H NMR (400 MHz,
CDCl3) 8 -0.66 (br s, 2H, 2 x NH), -0.22 (t, J=7.5 Hz, 3H, CH3 of angular
ethyl
group), 1.47-2.10 (m, 21 H, 7 x CH3), 2.60-2.75 (m, 1 H, CH of angular ethyl
group),
3.04-3.20 (m, 1H, CH of angular ethyl group), 3.50-4.00 (m, 13H), 4.05-4.25
(m, 3H),
4.63 (dd, J=12.5, 2.0 Hz, 1 H, angular H), 6.70 (d, J=5.22 Hz, 1 H, NH), 8.47
(s, 1 H, 1
meso-H), 9.38 (s, 1H, 1 meso-H), 9.57 (s, 1H, 1 meso-H); W-Vis (CHCI3 (log s))
~.~~ 406 (5.19), 502 (4.04), 534 (3.61 ), 670 (4.64) nm; MS (EI) m/e calc'd
for
C38H49NSO: 591.39374, found 591.39285; 591 (M+, 100 %).
(113) 5-Formamidooctaethylporphyrin
This compound was prepared in 87 % yield according to the procedure of
Clezy et al. 20~
m.p. 231-234°C; Rg 0.14 (silica - CH2Cl2); ~H NMR (200 MHz, CDCl3) 8 -
3.43 (br s, 2H, 2 x NH), 1.70 (t, J=8 Hz, 6H, 2 x CH3), 1.80-2.01 (m, 18H, 6 x
CH3),
3.95-4.22 (m, 16H, 8 x CH2), 8.61 (d, J=12 Hz, 1H, CHO), 9.36 (br d, J=12 Hz,
1H,
NH), 10.02 (s, 1H, 1 meso-H), 10.18 (s, 2H, 2 meso-H's); 13C NMR (50 MHz;
CDC13) b 17.50, 18.55, 19.82, 21.77, 96.84, 97.48, 102.81, 109.07, 140.57,
141.82,
142.55, 143.55, 143.75, 144.51, 145.74, 167.82; UV-Vis (CH2C12) 7~,r,~ 404,
504,
538, 572, 626 nm.
(112) 5-Isocyanooctaethylporphyrin
The formamide (113) (23 mg, 0.44 mmol) was dissolved in dry pyridine (5
58
CA 02254557 2003-10-24
mL). Phosphoryl chloride (200 p,L, 2.1 mmol) was added dropwise under
nitrogen,
and the mixture was stirred under nitrogen at 40'C for 2 hours. The solvent
was
removed in vacuo, and the residue purified by chromatography (flash silica,
dichloromethane eluent) to give the product (20 mg, 90 %).
m.p. 257-259'C; Rg 0.39 (silica -1:1 CH2C12:hexanes); 1H NMR (200 MHz,
CDCl3) 8 -3.41 (v br s, 2H, 2 x NH), 1.82-2.00 (m, 24H, 8 x CH3), 3.95-4.17
(m,
12H, 6 x CH2), 4.28 (q, J=8 Hz, 4H, 2 x CH2), 10.01 (s, 1H, 1 meso-H), 10.14
(s, 2H,
2 meso-H's); t3C NMR (50 MHz, CDCl3) S 17.22, 18.48, 19.70, 21.60, 97.96,
102.83,
114.30, 140.56, 141.01, 141.46, 141.92, 142.45, 143.76, 144.86, 145.38,
175.30; UV-
Vis (CH2C12) 7~m~ 408, 510, 546, 580, 634 nm.
(116) 5-(C-Hydroxymethylformamido)octaethylporphyrin
N N
N N M~COCN~ON
Paraformaldehyde (100 mg) was placed in a 25 mL flask equipped with a
septum. Toluene (5 mL) was placed in another 25 mL flask equipped with a
septum
and a stir bar. The 2 flasks were connected by a canula and the flask
containing
toluene was also furnished with an empty balloon. The paraformaldehyde-
containing
flask was heated with a heat-gun and the resultant gaseous formaldehyde was
bubbled
through the toluene. The heating was continued for 5 min, after which the
canula was
removed, and the balloon filled with nitrogen. Boron trifluoride etherate (30
pL) was
added dropwise by syringe and the mixtwe was stirred for 5 min. The
isocyanoporphyrin (1 I2) (17 mg, 0.034 mmol) was added to the reaction mixture
and
stirring continued under nitrogen for 30 min. The mixture was powed into
water,
extracted with dichloromethane, the extracts dried over sodium sulfate and the
solvent
evaporated in vacuo. The residue was pwified by flash chromatography (silica,
eluent
59
CA 02254557 2003-10-24
% ethyl acetate in dichloromethane) to give the polar pink product (5 mg, 28
yield).
m.p. 272-280°C; RF 0.09 (silica - CH2Cl2 / 5 % AcOEt); 1H NMR (200 MHz,
CDCl3 + 1 drop TFA) 8 1.12 (t, J=7.5 Hz, 6H, 2 x CH3), 1.39 (t, J=7.5 Hz, 6H,
2 x
CH3), 1.60-1.80 (m, 12H, 4 x CH3), 3.39-3.68 (m, 4H, 2 x CH2), 3.79 (q, J=7.5
Hz,
4H, 2 x CHZ), 3.98 (q, J=7.5 Hz, 8H, 4 x CH2), 4.76 (s, 2H, CH20H), 14.10 (s,
l H, 1
meso-H), 10.26 (s, 2H, 2 meso-H's); W-Vis (CH2C12) ~,m~ 404, 504, 538, 572,
624
nm; MS (EI) m/e 607 (M+, 100 %), 576 (M+-CH20H, 72 %).
(Ni-I16) [5-(C-Hydroxymethylformamido)octaethylporphyrinato)nickel(II)
The C-hydroxymethylformamide (116) (5 mg, 0.008 mmol) was dissolved in
dimethylformamide (I mL) and nickel acetate tetrahydrate (30 mg, 0.12 mmol)
was
added. The mixture was refluxed for 2 hours, then poured into water, extracted
with
ethyl acetate, washed with water, dried over sodium sulfate and the solvent
removed
in vacuo. The residue was purified by flash chromatography (silica, eluent 5 %
ethyl
acetate in dichloromethane) to give the product (4 mg, 73 % yield).
RF 0.23 (silica - CHZCI2 / 10 % AcOEt); UV-Vis (CH2Cl2) ~,m~ 402, 526,
562 nm; MS (EI) mle 663 (M+, 100 %).
(Ni-113) [5-Formamidooctaethylporphyrinato)nickel(II)
The formamide (I I3) (32 mg, 0.055 mmol) was dissolved in DMF (5 mL),
and nickel acetate tetrahydrate (20 mg, 0.080 mmol) was added. The mixture was
iefluxed for 1 hour, then allowed to cool, poured into water, and extracted 3
times
with ethyl acetate. The organic phase was dried, the solvent was evaporated,
and the
residue was purified by chromatography (silica, eluent 5 % ethyl acetate in
dichloromethane), to give the metallated product (27 mg, 77 % yield).
m.p. 264-267°C; RF 0.27 (silica - CH2Cl2); 1 H NMR (200 MHz, CDCl3) b
1.61-1.86 (m, 24H, 8 x CH3), 3.82 (q, J=7.5 Hz, 16H, 8 x CH2), 7.68 (d, J=12
Hz, 1H,
CHO), 9.13 (br d, J=12 Hz, 1H, NH), 9.51 (s, 1H, 1 meso-H), 9.55 (s, 2H, 2
meso-
CA 02254557 2003-10-24
H's); 13C NMR (50 MHz, CDC13) S 17.42, 18.16, 18.21, 18.26, 19.51, 19.62,
22.04,
96.75, 97.25, 107.44, 138.32, 139.20, 140.31,141.05, 142.80,143.64,
143.76,145.72,
167.64; W-Vis (CHC13 (log s)) a.a,~ 400, 524, 562 nm.
(Ni-112) (5-Isocyanooctaethylporphyrinato)nickel(II)
The nickel formamide (Ni-113) (22 mg, 0.035 mmol) was dissolved in
pyridine (2 mL), and phosphoryl chloride (4 drops) was added to this solution.
The
mixture was stirred at room temperature for 30 min, then was added to water
and the
resulting solid was filtered off and washed with water, to give quantitative
product.
m.p. 282-284°C; RF 0.53 (silica - I :1 CH2Cl2:hexanes); 1 H NMR (200
MHz,
CDCI3) 8 1.58-1.83 (m, 24H, 8 x CH3), 3.62-3.83 (m, 12H, 6 x CH2), 3.99 (q,
J=7.5
Hz, 4H, 2 x CHz), 9.38 (s, 1H, 1 meso-H), 9.41 (s, 2H, 2 meso-H's); 13C NMR
(50
MHz, CDCl3) b 17.31, 18.27, 20.48; 22.08, 97.63, 97.79, 102.86, 136.10,
138.36,
140.78, 143.27, 143.59, 144.06, 146.12; W-Vis (CH2C12) ~,m~ 406, 536, 576 nm.
(Ni-120) Nickel dimer
The nickel isocyanide (Ni-112) (25 mg, 0.039 mmol) was dissolved in
dichloromethane (3 mL), and boron trifluoride etherate (25 pL) was added. The
mixture was stirred overnight, then pyridine ( 1 drop) was added and the
solvent was
evaporated. The residue was purified by chromatography (silica, eluent
dichloromethane:hexanes 1:1 ), then suspended in methanol and filtered, to
give the
green solid product (14 mg, 56 % yield).
m.p. >300'C; RF 0.64 (silica - 1:1 CH2C12:hexanes); 1 H NMR (400 MHz,
61
CA 02254557 2003-10-24
CDC13) 8 -0.05 (t, J=7.5 Hz, 12H, 4 x CH3 of gem-diEt), 1.40-1.60 (m, 36H,12 x
CH3), 2.64-2.76 (m, 4H, 2 x CH2), 3.10-3.30 (m, 16H, 8 x CH2), 3.42 (q, J=7.5
Hz,
4H, 2 x CH2), 3.64-3.76 (m; 4H, 2 x CH2), 4.I7 (q, J=7.3 Hz, 4H, 2 x CH2),
7.64 (br
s, 2H, 2 meso-H's), 7.96 (s, 2H, 2 meso-H's), 8.49 (br s, 2H, 2 meso-H's); LIV-
Vis
(CHCl3 (log s)) ~.m~ 408 (4.69), 500 (4.99), 680 (4.64), 816 (4.13) nm; MS
(FAB (3-
NBA + CHC13 matrix)) mle calc'd for C~4H8~N1p58Ni6~Ni: 1233.5757, found
1233.57831; 1233 (M++1, 42 %); Analysis calc'd for C~4Hg6Nlp~HCl: C, 70.02; H,
6.91; N, 11.03; found: C, 69.98; H, 6.85; N, 10.93; Singlet Oxygen Test:
Negative.
(120) Free base dimer
The nickel dimer (Ni-120) (5 mg, 0.004 mmol) was dissolved in concentrated
sulfuric acid (1 mL). The solution was stirred at room temperature for 2
hours, then
poured into water, and extracted 3 times with dichloromethane. The solvent was
dried
and evaporated, and the residue was suspended in methanol and filtered to give
the
product as a grey solid (3.5 mg, 77 % yield).
m.p. >300°C; RF 0.33 (silica - CH2ClZ:hexanes 1:1 ); 1H NMR (400 MHz,
CDC13) b 0.04 (t, J=7.5 Hz, 12H, 4 x CH3 of gem-diEt), 1.48-1.80 (m, 36H, 12 x
CH3), 2.88-3.02 (m, 4H, 2 x CH2), 3.23-3.46 (m, 16H, 8 x CH2), 3.67 (q, J=7.5
Hz,
4H, 2 x CH2), 4.00-4.23 (m, 4H, 2 x CHZ), 4.63 (q, J=7.5 Hz, 4H, 2 x CH2),
4.97 (br
s, 2H), 5.82 (br s, 2H), 7.69 (s, 2H, 2 meso-H's), 7.88 (s, 2H, 2 meso-H's),
8.67 (s, 2H,
2 meso-H's); LTV-Vis (CHZCl2) ~.m~ 400, 500, 572; 620, 678, 686, 742, 814 nm;
MS
(FAB (3-NBA + CHC13 matrix)) mle calc'd for C~4Hg1N1o: 1119.74262, found
11.19.74164; 1120 (Ni++1, 17 %); Singlet Oxygen Test: Positive.
(Zn-120) Zinc dimer
The free base dimer (120) (5 mg, 0.004 mmol) was dissolved in chloroform (5
mL) and zinc acetate dihydrate ( 10 mg, 0.046 mrnol) in methanol ( 1 mL) was
added.
The mixture was refluxed 1 hour, then the solvent was evaporated, the residue
was
suspended in methanol, and this suspension was filtered to give quantitative
dark
62
CA 02254557 2003-10-24
green solid product.
m.p. >300'C; RF 0.32 (silica - CHZC12); 1H NMR (200 MHz, CDCl3) b 0.02
(t, J=7.5 Hz, 12H, 4 x CH3 of gem-diEt), 1.46-1.88 (m, 36H, 12 x CH3), 2.84-
3.06
(m, 4H, 2 x CH2), 3.32-3.58 (m, 16H, 8 x CHz), 3.68 (q, J=7.5 Hz, 4H, 2 x
CH2),
4.01-4.24 (m, 4H, 2 x CH2), 4.67 (q, J=7.5 Hz, 4H, 2 x CH2), 7.88 (s, 2H, 2
meso-
H's), 8.23 (s, 2H, 2 meso-H's), 8.91 (s, 2H, 2 meso-H's); W-Vis (CH2Cl2) an,~
392,
466, 494, 518, 628, 680, 688, 728, 818 nm; MS (FAB (thioglycerol matrix)) role
1248
(M+, 100 %).
(Ni-121) Nickel ethylidene dimer
Formed in low yield as a side-product during the synthesis of (Ni-120).
RF 0.39 (silica - CH2CIZ:hexanes l :l); 1H NMR (400 MHz, CDCl3) b
(selected resonances), 0.27 (t, 3H, CH3 of one gem-diEt group), 0.91 (t, 3H,
CH3 of
one gem-diEt group), 2.82 (d, 3H, CH3=CH), 5.46 (s, 1H, angular CH), 8.33 (q,
1H,
CH=CH3), 8.84, 8.99, 9.03, 9.38, 9.42, 9.46 (6s, 6H, 6 meso-H's); IJV-Vis
(CH2Cl2)
~,m~ 410, 500, 626 nm; MS (FAB (matrix 3-NBA + CHC13)) role calc'd for
C~4Hg6N~p58N16~Ni: 1232.61802, found 1232.57458; 1233 (M+, 2 %).
(Zn-92) (5-Formylmethyliminooctaethylporphyrinato)zinc(II)
The zinc aminoporphyrin (Zn-91) (100 mg, 0.16 mmol) was dissolved in THF
(5 mL) and a solution of glyoxal trimer dihydrate (125 mg, 0.59 mmol) in
ethanol (S
mL) was added. The mixture was refluxed for 8 hours, then the solvent was
evaporated and the residue purified by chromatography (silica, eluent
63
CA 02254557 2003-10-24
dichloromethane) to give the product (73 mg, 70 % yield).
RF 0,74 (silica - CHzCl2); 1H NMR (200 MHz, CDCI3) 81.60-2.00 (m, 24H,
8 x CH3), 3.80-4.25 (m,16H, 8 x CH2), 7.57 (d, J=8.1 Hz, 1H, CH=CHO), 9.77 (s,
1H, I meso-H), 9.91 (s, 2H, 2 meso-H's), 10.30 (d, J=8.1 Hz, 1H, CHO); W-Vis
(CH2Cl2) a,m~ 408, 468, 544, 612 nm; MS (EI) m/e 6S 1 (M+, 100 %).
(Zn-123) [Octaethyl-(3-hydroxypyridoxhlorinatoJzinc(II)
The zinc formylmethylimine (Zn-92) (93 mg, 0.14 mmol) was dissolved in
toluene (10 mL) and activated Montmorillonite clay (SO mg) was added. The
mixture
was refluxed for 72 hours, then filtered and the solvent evaporated. The
residue was
purified by chromatography (silica, eluent initially dichloromethane,
increasing
polarity to S % methanol in dichloromethane) to give unreacted starting
material (S9
mg) and the slightly impure product (20 mg, 22 % yield).
RF O.SB (silica - 10 % AcOEt / CH2Cl2); 1H NMR (200 MHz, pyridine-ds) 8
0.33 (t, J=7.1 Hz, 6H, 2 x CH3 of gem-diEt), 1.S8-2.01 (m, 18H, 6 x GH3), 2.62-
2.82
(m, 2H, Z x CH of gem-diEt CH2), 3.43-3.92 (m, 12H, 6 x CH2), 4.45 (q, J=7.6
Hz,
2H, 2 x CH of gem-diEt CH2), 8.23, (s, 1 H, CH of pyridine ring), 9.1 S (s, 1
H, 1
meso-H), 9.60 (s, 1 H, 1 meso-H) 9.62 (s, 1 H, 1 meso-H); UV-Vis (CH2C12)
~.max
410, 422, 518, SS8, 574, 624, 678 nm; MS (EI) »r/e 6S1 (M+, 100 %); Singlet
Oxygen Test: Negative.
(123) Octaethyl-(3-hydroxypyrido)chlorin
The zinc chlorin (Zn-123) (20 mg, 0.030 mmol) was dissolved in
dichloromethane (S mL) and TFA (2 drops) was added. The solution was stirred
for 2
64
CA 02254557 2003-10-24
hours, then pyridine (5 drops) was added to neutralize and the solvent was
evaporated.
The residue was chromatographed (silica, eluent 5 % methanol in
dichloromethane) to
give the product (6 mg, 35 % yield).
Rp 0.70 (silica - 10 % AcOEt / CH2C12); ~ H NMR (440 MHz, pyridine-ds) 8
0.40 (t, J=7.3 Hz, 6H, 2 x CH3 of gem-diEt), 1.63-1.81 (m, 15H, 5 x CH3), 1.88
(t,
J=7.3 Hz, 3H, 1 x CH3), 2.78-2.91 (m, 2H, CH2), 3.49-3.71 (m, l OH, 5 x CH2),
3.85
(q, J=7.5 Hz, 2H, 2 x CH of gem-diEt CH2), 4.43 (q, J=7.3 Hz, 2H, 2 x CH of
gem-
diEt CH2), 8.40, 8.99, 9.58, 9.69 (4s, 3 meso-H's and CH=N) 13.06 (br s, 1 H,
OH);
LJV-Vis (CHC13 (log s)) ~,m~ 412 (4.89), 490 (3.51), 522 (3.73), 556 (3.75),
616
(3.86), 672 (4.36) nm; MS (EI) mle calc'd for C3gH4~N50: 589.37845, found
589.37791; 589 (M+, 100 %), 560 ((M-29)+, 65 %); Analysis calc'd for
C38H4~NSO~O.SH20: C, 76.22; H, 8.08; N, 11.70; found: C, 75.96; H, 7.64; N,
11.67; Singlet Oxygen Test: Positive.