Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02263210 2004-09-30
26474-432
Process for the preparation of lithium borate complexes
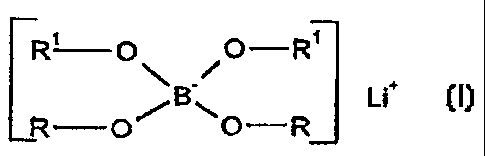
The invention relates to lithium complex salts of
the general formula (I)
R, O\ ./O--R'
ua
R O~ \O R ,
wherein R and R1 are identical or different,
are optionally joined together directly by a
single or double bond,
in each case individually are an aromatic ring
from the group phenyl, naphthyl, anthracenyl or
phenanthrenyl, which can be unsubstituted or monosubstituted
to tetrasubstituted by A or Hal, or
in each case together are an aromatic ring from
the group naphthyl, anthracenyl or phenanthrenyl, which can
be unsubstituted or monosubstituted to tetrasubstituted by A
or Hal, or
in each case individually are a heterocyclic
aromatic ring from the group pyridyl, pyrrole, 1,2-diazine,
1,3-diazine or 1,4-diazine, which can be unsubstituted or
monosubstituted to trisubstituted by A or Hal, or
in each case together are a heterocyclic aromatic
ring from the group pyrrole, 1,2-diazine or 1,3-diazine,
which can be unsubstituted or monosubstituted to
trisubstituted by A or Hal, and
Hal is F or C1, and
1
CA 02263210 2004-09-30
26474-432
A is alkyl having 1 to 6 C atoms, which can be monohalogenated
to tetrahalogenated,
to their use as electrolytes in secondary lithium batteries
and to a process for the preparation of these compounds.
Because of the low resting potential of lithium,
only aprotic compounds are suitable as solvents. Protic
compounds, such as alcohols, react with lithium-containing
anodes and produce hydrogen, which ultimately leads to
explosion of the cell.
Suitable organic solvents in secondary lithium
batteries are basically any of the solvents and solvent
mixtures known to those skilled in the art for this
application. Suitable solvents are, inter alia, both ethers
and esters, including cyclic organic carbonates, e.g.
propylene carbonate or ethylene carbonate. However, it is
possible to use not only liquids but also polymers as
solvents, the prerequisite in this case being that the
polymer used dissolves lithium salts and forms ionically
conducting mixtures therewith. One of the most commonly
used polymers is polyethylene oxide. The conductivity can
be increased by using mixtures of a polymer and one or more
solvents (Amalgier et al. in: Proceedings of the Symposium
on Primary and Secondary Lithium Batteries, Vol. 91-1,
131-141, (1991); K.M. Abraham and M. Salomon (editors), The
Electrochemical Society, Pennington N.J.). The use of
polymer electrolytes increases the operational safety of the
cell because the electrolyte is prevented from escaping and
thereby exposing the electrode surfaces if the cell
container is mechanically damaged.
The conducting salts used for lithium cells are
exclusively salts with large, negatively charged, inorganic
2
CA 02263210 2004-09-30
26474-432
or organic counterions. Conducting salts with small
counterions, e.g. lithium chloride, are unsuitable because
of the low solubility due to the high lattice binding
energy.
Lithium salts with fluorinated inorganic anions
are among the hitherto most frequently studied conducting
salts for secondary lithium cells. Solutions of lithium
tetrafluoroborate in various ethers give relatively low
cyclization yields on inert substrates. Poor yields have
also been achieved with LiBF4/polyethylene carbonate
solutions on carbon anodes (Maki Sato et al. in:
Proceedings of the Symposium on Primary and Secondary
Lithium Batteries, Vol. 91-3, 407-415, (1991); K.M. Abraham
and M. Salomon (editors), The Electrochemical Society,
Pennington N.J.). Lithium tetrafluoroborate is therefore
not ideally suitable for application in secondary lithium
cells. At potentials greater than the equilibrium potential
of the reaction of elemental lithium in Li+ + e-, lithium
hexafluoroantimonate is reduced inter alia to elemental
antimony and consequently cannot be used as a conducting
salt.
Lithium hexafluoroarsenate gives very high
cyclization yields in most solvents and solvent mixtures on
inert substrates. Cyclization yields of over 96o have been
found for a solution of LiAsF6 in 2-methyltetrahydrofuran
(Goldman et al., J. Electrochem. Soc., Vol. 127, 1461-1467
(1980)). The toxicity and poor environmental compatibility
of lithium hexafluoroarsenate and its secondary products are
an obstacle to large-scale application (Archuleta, M.M.,
J. Power Sources 54, 138 (1995)).
LiPF6-containing solutions based on organic
carbonates have also been tested in cells with lithium
3
CA 02263210 2004-09-30
26474-432
anodes. An essential disadvantage of these systems is the
low thermal stability of LiPF6. Partial dissociation into
LiF and PFS takes place in solution, which can lead to
cationic polymerization of the solvent initiated by the
Lewis acid PFS (Koch et al. in: Proceedings of the Symposium
[lacuna) Lithium Batteries, Vol. 81-4, 165-171, (1981), The
Electrochemical Society, Pennington N.J.; H.V. Venkatasetty
(editor) ) .
To avoid the dissociation of inorganic fluorinated
counterions, organic lithium salts with perfluorinated
organic radicals have also been tested, examples being
lithium trifluoromethanesulfonate, lithium
bis(trifluoromethanesulfonyl)imide and lithium
tris(trifluoromethanesulfonyl)methide. Because of their
high thermal stability, these salts are used principally in
sonically conducting polymers. The last two salts mentioned
have an appreciably higher conductivity than the first and
are substantially stable to oxidation. They have been used
successfully in cells with carbon anodes and nickel oxide
cathodes (Dahn et al., J. of Electrochem. Soc., Vol. 138,
2207-2211 (1991)). A serious disadvantage, however, is the
high price due to the manufacturing process. Because of the
high fluorine content of these compounds, there is also a
risk of exothermic reactions with lithium.
The use of lithium organoborates has also been
studied by Horowitz et al. (in: Proceedings of the
Symposium on Lithium Batteries, Vol. 81-4, 131-143, (1981),
The Electrochemical Society, Pennington N.J.;
H.V. Venkatasetty (editor)). Because of the low anodic
stability, the safety problems associated with the formation
of triorganoboranes and their high price, tetraorganoborates
are not used in lithium cells.
4
CA 02263210 2004-09-30
26474-432
A synthesis of lithium borate complexes, starting
from boric acid in aqueous solution is described in
WO 94/27335 and J. Barthel et al. in J. Electrochem. Soc.
142/8, 2527 (1995). A decisive criterion for electrolytes
which can be employed in Li ion batteries is, in addition to
the high purity, in particular the absence of water. It is
not possible to obtain the products from this synthesis in
an absolutely anhydrous manner.
The use of lithium chloroborates has also been
studied (Johnson, J.W.; Brody, J.F.; J. Electrochem. Soc.,
Vol. 129, 2213-2219 (1982)). However, the processes for the
preparation and purification of these compounds are very
expensive and solutions of these salts tend to undergo phase
separation. Their high chlorine content makes them
unstable.
One object of the invention was therefore to
provide environmentally compatible, stable lithium complex
salts with improved properties, which are economic to
prepare and, in appropriate solvents, are suitable as
electrolytes for the manufacture of secondary lithium
batteries: Another object of the invention is to provide a
process for the preparation of these lithium complex salts.
It has been found by experiment that the object of
the invention can be achieved by means of lithium complex
salts of the general formula (I)
u'
wherein R and R1 are identical or different,
5
CA 02263210 2004-09-30
26474-432
are optionally joined together directly by a
single or double bond,
in each case individually are an aromatic ring
from the group phenyl, naphthyl, anthracenyl or
phenanthrenyl, which can be unsubstituted or monosubstituted
to tetrasubstituted by A or Hal, or
in each case together are an aromatic ring from
the group naphthyl, anthracenyl or phenanthrenyl, which can
be unsubstituted or monosubstituted to tetrasubstituted by A
or Hal, or
in each case individually are a heterocyclic
aromatic ring from the group pyridyl, pyrrole, 1,2-diazine, '
1,3-diazine or 1,4-diazine, which can be unsubstituted or
monosubstituted to trisubstituted by A or Hal, or
in each case together are a heterocyclic aromatic
ring from the group pyrrole, 1,2-diazine or 1,3-diazine,
which can be unsubstituted or monosubstituted to
trisubstituted by A or Hal, and
Hal is F or C1, and
A is alkyl having 1 to 6 C atoms, which can be monohalogenated
to tetrahalogenated,
and especially by means of lithium complex salts in which
R and R1 are identical or different, are optionally joined
together by a single or double bond and are each phenyl or
pyridyl. Very particularly suitable lithium complex salts
of the general formula (I) are
lithium bis[2,2'-biphenyldiolato(2-)-O,O']borate(1-) and
lithium bis[2,3-naphthalenediolato(2-)-0,0']borate(1-).
6
CA 02263210 2004-09-30
26474-432
The invention also provides a process for the
preparation of the compounds according to the invention and
a novel process for the preparation of monofluorinated to
tetrafluorinated aromatics hydroxylated on adjacent C atoms,
especially tetrafluorocatechol, which are required as
intermediates for the preparation of the lithium borate
complexes according to the invention.
According to one aspect of the present invention,
there is provided lithium bis(2,2'-biphenyldiolato(2-)-
0,0']borate(1-).
According to another aspect of the present
invention, there is provided lithium bis(2,3-
naphthalenediolato(2-)-0,0']borate(1-).
According to still another aspect of the present
invention, there is provided a process for preparation of
lithium complex salts of the general formula (I)
R~ O\ ./O-R,
g Lia
R O/ \O~R
wherein R and R1 are identical or different, and are
optionally joined together directly by a single or double
bond:
wherein, when R and R1 are not joined together,
each of R and R1 is an aromatic ring selected from phenyl,
naphthyl, anthracenyl and phenanthrenyl, which is
unsubstituted or monosubstituted to tetrasubstituted by A or
Hal; and
wherein, when R and R1 are joined together, R and
R1 together form an aromatic ring selected from naphthyl,
7
CA 02263210 2005-06-02
26474-432
anthracenyl and phenanthrenyl, which is unsubstituted or
monosubstituted to tetrasubstituted by A or Hal; or
wherein, when R and R1 are not joined together,
each of R and R1 is a heterocyclic aromatic ring selected
from pyridyl, p~~rrole, 1,2-diazine, 1,3-diazine and 1,4
diazine, which is unsubstituted or monosubstituted to
trisubstituted by A or Hal; and
wherein, when R and R1 are joined together, R and
R1 together form a heterocyclic aromatic ring selected from
pyrrole, 1,2-diazine and 1,3-diazine, which is unsubstituted
or monosubstituted to trisubstituted by A or Hal;
Hal is F or C1; and
A is alkyl having 1 to 6 C atoms, which is unsubstituted or
monohalogenated to tetrahalogenated; wherein
a) a lithium tetraalcoholatoborate is taken up
with an aprotic solvent,
b) equimolar amounts of a hydroxyl compound or a
1:1 mixture of two different hydroxyl compounds, dissolved
in an aprotic solvent, are added dropwise at a temperature
of 10 to 60°C, with stirring, optionally under an inert gas
atmosphere, and, optionally, the reaction mixture is
subsequently stirred at a temperature of 60 to 90°C,
c) optionally, alcohol formed during the reaction
is slowly distilled off by application of a slight vacuum at
a slightly elevated temperature,
d) the product formed is crystallized out,
optionally after concentration of the reaction mixture under
vacuum at a temperature of 0 to 10°C, and, optionally, is
separated off under an inert gas atmosphere, and
8
CA 02263210 2004-09-30
26474-432
e) the product which has been separated off is
dried by slow heating.
According to yet another aspect of the present
invention, there is provided a process as described herein,
wherein tetrafluorocatechol is used as the hydroxyl compound
for complexation; wherein the tetrafluorocatechol is
prepared by: a) reaction of pentafluorophenol with
potassium carbonate in aqueous solution to give potassium
pentafluorophenate~ b) etherification of the potassium
pentafluorophenate prepared in a) with ethylene oxide in
DMSO as solvent, under an inert gas atmosphere, to give a
cyclic diether, 5,6,7,8-tetrafluoro-(1,4)-benzodioxane, or
etherification of the potassium pentafluorophenate prepared
in a) with 2-bromoethanol and subsequent cyclization to the
cyclic diether in presence of potassium carbonate in DMF as
solvent; and c) cleavage of the ether in presence of
aluminium chloride with benzene as solvent, under an inert
gas atmosphere.
According to a further aspect of the present
invention, there is provided a process as described herein,
wherein mono-, di- or tricatechol, prepared in the same way
as tetrafluorocatechol as described herein, is used as the
hydroxyl compound for complexation.
According to yet a further aspect of the present
invention, there is provided a process as described herein,
wherein the lithium tetraalcoholatoborate used is lithium
tetramethanolatoborate, lithium tetraethanolatoborate or
lithium tetrapropanolatoborate.
According to still a further aspect of the present
invention, there is provided a process as described herein,
wherein the aprotic solvent is selected from acetonitrile,
9
CA 02263210 2004-09-30
26474-432
acetone, nitromethane, dimethylformamide, dimethylacetamide
and dimethyl sulfoxide.
According to another aspect of the present
invention, there is provided a process as described herein,
wherein equimolar amounts of the hydroxyl compound are added
dropwise at room temperature.
According to yet another aspect of the present
invention, there is provided a process as described herein,
wherein the alcohol formed is distilled off under slight
vacuum at a temperature of 50 to 60°C.
According to another aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein as a conducting
salt in an electrolyte for an electrochemical cell.
According to still another aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein as a conducting
salt in an electrolyte for a battery.
According to yet another aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein as a conducting
salt in an electrolyte in a secondary lithium battery.
According to a further aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein in combination with
one or more of another lithium salt and a borate complex in
an electrolyte of a secondary lithium battery.
According to yet a further aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein in combination with
CA 02263210 2004-09-30
26474-432
one or more other lithium salts selected from lithium
phenate and dilithium 2,2'-biphenyldiolate in an electrolyte
of a secondary lithium battery.
According to still a further aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein in a double-layer
capacitor or a supercapacitor.
According to another aspect of the present
invention, there is provided use of a lithium complex salt
of general formula (I) as defined herein in manufacture of a
switchable window or display.
Surprisingly, ligands which are not soluble in
water, e.g. pyridinediol, dihydroxybiphenyl or other,
preferably hydroxylated aromatic compounds, can also be
coordinated to the central boron atom by the process
according to the invention.
Because of the reaction conditions according to the
invention, the presence of water, which in aqueous media can
be bound to the central atom as a ligand to form B(OH)4-,
both during the complexation reaction and during the
isolation of the products prepared, which is carried out
inter alia by concentration of the solution, can have an
adverse effect due to the formation of by-products with B-0-B
bridges.
The process according to the invention influences
the position of the equilibrium of the complexation reaction
in such a way that the reaction solution can be evaporated
under mild conditions to give product yields of 1000.
One particular advantage of the process according
to the invention is the possibility of working under mild
11
CA 02263210 2004-09-30
26474-432
conditions without using aggressive reagents like BC13, BF3
or LiBH4, which result in the formation of HC1, HF or H2
during the complexation reaction. Furthermore, the process
according to the invention affords the elegant possibility
of incorporating a methanolic solution of LiB(OCH3)4 into a
polymer matrix, e.g. hydroxylated PEO, and preparing a
polymeric electrolyte in which the anion is fixed to the
polymer backbone.
The complex salts according to the invention are
prepared by placing a lithium tetraalcoholatoborate in an
aprotic solvent. This solution is heated slightly, if
necessary, to dissolve the borate.
Lithium tetraalcoholatoborates suitable for the
reaction are the derivatives of methanol, ethanol and
propanol, as well as those of other short-chain alcohols.
However, it is particularly preferable to use the methanol
or ethanol derivatives because the low boiling points of
these alcohols mean that they can be removed from the
reaction mixture at relatively low temperatures after
complexation.
For complexation a suitable hydroxyl compound or a
1:1 mixture of different suitable hydroxyl compounds is
dissolved in the same aprotic solvent as the lithium
tetraalcoholatoborate was previously dissolved in, and
slowly added dropwise to the lithium tetraalcoholatoborate
solution in an equimolar amount at a temperature of between
10 and 60°C, preferably at room temperature up to about
55°C, if necessary under an inert gas atmosphere. To
complete the reaction, the reaction solution is then
optionally stirred for some time at a temperature of between
60 and 90°C. The subsequent stirring can be dispensed with
in the case of very rapid complexation reactions.
12
CA 02263210 2004-09-30
26474-432
A solvent from the group comprising acetonitrile,
acetone, nitromethane, dimethylformamide, dimethylacetamide
and dimethyl sulfoxide can be used as the aprotic solvent.
It is preferable to use acetonitrile.
If the alcohol formed during the reaction
interferes with the subsequent isolation of the complex salt
prepared, it can be separated off by the application of a
slight vacuum and, if appropriate, by gentle heating to
about 50 to 60°C. Depending on the solubility of the
lithium complex salt prepared in the aprotic solvent used,
the reaction solution is concentrated or the solbent is
completely distilled off and, if the product does not
crystallize out spontaneously, the residue is cooled at a
temperature of 0 to 10°C for several hours. The crystalline
product is separated off in conventional manner and dried by
slow heating.
Compounds which are particularly suitable for
complexation are aromatics hydroxylated in adjacent
positions, such as pyrocatechol, 1,2- or 2,3-
dihydroxynaphthalene, and also correspondingly hydroxylated
anthracene or phenanthrene. However, aromatics which are
joined together by a bond and have hydroxyl groups in the
direct vicinity of the bond, e.g. 2,2'-dihydroxybiphenyl,
are also suitable. Other compounds suitable for
complexation are corresponding heterocycles, e.g. pyridine-
2,3-diol and pyridine-3,4-diol, or correspondingly
hydroxylated bipyridyl. Other aromatics suitable for
complexation are diazines hydroxylated in adjacent
positions, e.g. 1,3-diazine-5,6-diol, 1,2-pyrazine-3,4-diol,
1,2-pyrazine-4,5-diol and 1,4-pyrazine-2,3-diol, or the
corresponding diols of pyrrole. Corresponding ligands
suitable for complexation can be monosubstituted or
polysubstituted, both on the heteroatom and on non-
13
CA 02263210 2004-09-30
26474-432
hydroxylated carbon atoms of the aromatic ring, by halogen
atoms or optionally halogenated alkyl radicals having 1-6 C
atoms. 1-Trifluoromethylpyrrole-2,3-diol is an example of
these ligands. However, not only the heterocyclic aromatics
but also the other suitable aromatics can be monosubstituted
or polysubstituted, i.e. up to tetrasubstituted, by halogen,
especially by fluorine or chlorine. Monoalkylated or
polyalkylated hydroxylated aromatics, especially
hydroxylated phenyl, naphthyl, anthracenyl or phenanthrenyl
substituted by methyl, ethyl, n- or i-propyl or n-, sec- or
tert-butyl, can also advantageously be used for
complexation.
The hydroxyl compounds suitable for complexation
also include those which are not commercially available, an
example being tetrafluorocatechol. It can only be prepared
in low yields by processes known from the literature.
Experiments have now shown that this dihydroxyl compound can
be prepared in high yields by reacting pentafluorophenol
with potassium carbonate to give potassium
pentafluorophenate, then reacting this with 2-bromoethanol
in DMSO to give the monoether and then reacting this with
potassium carbonate in DMF to give the cyclic diether, from
which the desired tetrafluorocatechol is then obtained by
cleavage of the ether in benzene [lacuna] presence of
aluminium chloride. However, the cyclic diether can also be
obtained in one reaction step by reaction with ethylene
oxide in DMSO at elevated temperature under an inert gas
atmosphere. Depending on how the reaction is conducted,
yields of 80 to 95% are obtained by this method. This
process can also be used to prepare mono-, di- or
tri-fluorinated dihydroxyaromatics.
In cyclization experiments, Li borate complexes of
this fluorinated dihydroxyl compound, according to the
14
w
CA 02263210 2004-09-30
26474-432
invention, have given particularly good results and have
proved particularly stable. In combination with other
salts, these complexes exhibit a synergistic stabilizing
effect towards oxidation. This effect seems to be dependent
on the number of fluorine atoms bound per ligand,
electrochemical measurements having shown a stabilization of
about 0.1 V/fluorine atom/ligand.
This means that these fluorinated borate complex
salts according to the invention, as well as the other Li
borate complexes according to the invention, are
particularly suitable for use in electrochemical cells, not
only in primary and secondary batteries but also in double
layer or super capacitors, and for the manufacture of
displays or electrically switchable windows.
The complex salts according to the invention can
be used on their own or in a mixture. However, they can
also be used in a mixture with other conducting salts known
to those skilled in the art for these applications. The Li
salts according to the invention can also be used in a
mixture with appropriate ammonium borate complexes or other
alkali metal or alkaline earth metal borate complexes.
It has been found by experiment that the complex
salts according to the invention, especially
lithium bis[2,2'-biphenyldiolato]borate(1-), can be used in
conjunction with highly oxidizing cathode materials. These
compounds can also be used in a mixture with other lithium
compounds in order to guarantee overload protection. When
electrolyte mixtures are prepared, it is advantageous to
stabilize them by the addition of a lithium alcoholate when
lithium bis[perfluoro-1,2-benzenediolato(2)-0,0']borate(1-)
is used in a mixture with lithium bis[2,2'-
biphenyldiolato]borate(1-) or with other lithium borate
CA 02263210 2004-09-30
26474-432
complexes according to the invention. Stable mixtures of
lithium bis[perfluoro-1,2-benzenediolato(2)-0,0']borate(1-)
with other complex salts according to the invention are
obtained when complexes are added which have substituents
with electron donating properties, i.e. substituents with a
+I effect, on the aromatic ring. Examples of substituents
with a +I effect are alkyl groups such as methyl, ethyl,
propyl, i-propyl, n-butyl, sec-butyl and tert-butyl.
In particular, by electrochemical experiments
carried out with a 0.5 molar solution of lithium
bis[2,2'-biphenyldiolato]borate(1-) in a solvent mixture
consisting of 4:1 propylene carbonate/dimethylethylene glycol
(on stainless steel, 0.5 cm2, v = 20 mV/s), have shown that
oxidation of the anion only begins at a voltage over 4 V,
although only very weak currents flow. After the second
discharge cycle, however, oxidation can no longer be measured
above 4 V. From the first to the fourth cycle the resistance
of the protective coating increases with the deposition of
lithium and the yields of the cyclization are approximately
constant at 48 m/C to 65 m/C (74%) and are comparable to
other salts in PC/DME. If the inversion potential is then
raised from 4.5 to 5 V and 6 V, the resistance of the
protective coating increases further and the yields of the
lithium deposition fall. A particular feature to be
emphasized is the high anodic oxidation stability of the
protective coating formed. No oxidative current can be
observed even at an inversion potential of 8 V, although the
solvents are unstable above ca. 5 V (PC) and begin to
decompose.
Weak anodic currents were detected in the first
cycle in experiments with electrolyte solutions to which
lithium phenate had been added as well as lithium
bis[2,2'-biphenyldiolato]borate(1-), but no discolouration
16
CA 02263210 2004-09-30
26474-432
of the electrolyte occurred, so the oxidation which would
otherwise have occurred could be avoided by the addition of
lithium phenate. In fact, these experiments were performed
by starting from a resting potential of ca. 3000 mV,
increasing the potential to 4500 mV at a rate of 20 mV/s,
then reducing it to -500 mV and finally bringing it back to
the resting potential. The same positive result was
obtained in experiments in which the anodic inversion
potential was increased to 6000 mV. Electrolyte solutions
containing bis[1,2-benzenediolato)borate anions and the
solvent dimethylethylene glycol are also protected from
oxidation by Li phenate at potentials well above 4000 mV.
Dilithium 2,2'-biphenyldiolate exhibits the same
positive properties as Li phenate. Even small amounts added
to the electrolyte are sufficient to stabilize it.
Examples
Example 1
Lithium bis[2,2'-biphenyldiolato(2-)-O,O']borate(1-)
6.6 g (47 mmol) of lithium tetramethanolatoborate
are placed in 80 ml of acetonitrile at 55°C. A solution of
17.5 g (94 mmol) of 2,2'-dihydroxybiphenyl (purity >99~) in
100 ml of acetonitrile is added dropwise. All the lithium
tetramethanolatoborate has dissolved when 30 ml of the
biphenyl solution have been added. A large amount of
colourless product precipitates out rapidly when a further
20 ml have been added. The remaining 50 ml of solution are
added without the precipitate dissolving. The methanol
formed is removed from the mixture by gentle evacuation at
55°C over 2 hours. The residue is then cooled to room
temperature, filtered off under inert gas and washed with
acetonitrile. The colourless product which has been
17
CA 02263210 2004-09-30
26474-432
filtered off is dried by slow heating to a temperature of
180°C.
Yield: 3.2 g of lithium bis(2,2'-biphenyldiolato(2-)-
0,0']borate(1-) (17.70 of theory)
1H NMR (250 MHz, DMSO-d6) [b/ppm]
7.35(dd, 3Jg4/H3 7.5 Hz, 4~JHq/HZ= Hz,H4)
= 1.5
7 (td, 3Jg2/H1 7 Hz, 4Jg2/Hq= Hz,H2
. = . 1 )
25 5 .
5
6.99(td, 3Jg3/H1 7.4 Hz, 4JH3/H1= Hz,H3)
= 0.9
6 (dd, 3JH1/H3 7 Hz, 4tTH1/H3= Hz,H4
. = . 0 )
91 5 .
9
11B NMR (128.38 MHz, 0.3 M DMSO-d6, Et20*BF3 ext. )
8.8 ppm (s)
Potentiometric titration:
The titration is performed with dilute HC1.
The theoretical and actual boron contents are
2.800 and 2.7910 respectively.
The purity of the substance is 99.90.
MS (NI-LISIMS; CH3CN):
37 9 . 0 ( 100 0, M-Li+)
Solubility:
The substance is soluble in EC/DME and also in PC
up to ca. 0.5 molal, although in pure PC solutions a solvate
precipitates out after a short time.
18
26474-432
Example 2
CA 02263210 2004-09-30
Lithium bis[1,2-benzenediolato(2-)-O,0']borate(-1)
6.19 g (43.6 mmol) of lithium tetramethanolatoborate
are placed in 100 ml of acetonitrile at 35°C. 9.61 g
(887.3 [sic] mmol) of pyrocatechol are added. A yellow
solution is formed immediately. It is heated at 80°C for one
hour and then concentrated under vacuum to a total volume of
40 ml. The solution turns brown. On cooling, colourless
rectangular platelets precipitate out of the solution below
50°C. Crystallization of the product is completed by keeping
the solution at a temperature of 5°C for a period of 12 hours.
The supernatant solution is then decanted off and the crystals
obtained are dried to constant weight under vacuum at 140°C to
give a grey powder.
Yield: 537 g of lithium bis[1,2-benzenediolato(2-)-
0,0']borate(-1) (23 mmol, 52.70 of theory)
Decomposition point: 270°C
1H NMR (250 MHz, DMSO-d6) [b/ppm]
6.48 ppm (s)
13C NMR ( 62 . 9 MHz, DMSO-d6)
151.6 (s, CI [sic],
ppm C2)
117.3 (s, C4, C5)
ppm
107.6 (s, C3, C6).
ppm
Example 3
Lithium phenate
2.26 g (326 mmol) of lithium are cut in a closed
hood equipped with manipulating gloves. 150 ml of
19
CA 02263210 2004-09-30
26474-432
tetrahydrofuran are added under an inert gas atmosphere
(Ar 6.0). A solution of 27.72 g (294.6 mmol) of phenol p.a.
in 75 ml of tetrahydrofuran is then added dropwise at 40°C
over 3 hours, with magnetic stirring. A colourless
precipitate is formed. The mixture is subsequently stirred
for 14 hours at room temperature and the precipitate
redissolves. Small amounts of unreacted lithium are
filtered off in the closed hood. The solvent is evaporated
off and the residual product is dried to constant weight
under an oil pump vacuum to give a product with a purity of
99.9.
Example 4
Dilithium 2,2'-biphenyldiolate
100 ml of methanol are added dropwise to 1.67 g
(0.241 mol) of lithium and the mixture is slowly heated. A
colourless solution forms at a temperature of 50°C. A
solution consisting of 100 ml of methanol and 22.4 g
(0.120 mol) of 2,2'-dihydroxybiphenyl is then added
dropwise, with stirring. The solvent is distilled from the
resulting reaction solution and the colourless product
obtained is heated slowly under vacuum to a maximum
temperature of 100°C and dried.
Yield: 100 of theory
Purity: 99.60 (determined by titration).
Example 5
Lithium bis[tetrafluoro-1,2-phenyldiolato(2-)-0,0']-borate
a) Potassium pentafluorophenate
A solution of 94.1 g of pentafluorophenol in water
is added slowly to 48.4 g of solid potassium carbonate, with
CA 02263210 2004-09-30
26474-432
stirring, giving rise to a substantial evolution of gas
(C02). Crystals separate out which and [sic) only dissolve
after a further 200 ml of water have been added and the
temperature raised to 95°C. This solution is cooled to
20°C. After 12 hours the crystals formed are separated off
and washed three times with 40 ml of water cooled to 0°C.
The crystalline product obtained is dried by raising the
temperature slowly to 150°C.
Yield: 93.70 of theory
Decomposition point: 240°C.
b) Lithium tetramethanolatoborate
9.92 g of lithium are placed in a PyrexTM flask under
an argon atmosphere in a closed hood equipped with
manipulating gloves. 250 ml of methanol are added slowly to
the lithium, with stirring and while cooling with an
ice/methanol mixture. A strongly exothermic reaction takes
place. A further 320 ml of methanol are then added gradually.
The reaction mixture is heated to a temperature of 60°C and
148.6 g of trimethyl borate are slowly added dropwise to the
homogeneous solution formed. A white crystalline product is
formed. After standing for 24 hours at room temperature, the
product is filtered off and dried under reduced pressure.
Yield: 93.7 of theory
Decomposition temperature: 50°C.
c) 5,6,7,8-Tetrafluoro-(1,4)-benzodioxane
40.2 g of potassium pentafluorophenate are
dissolved in 150 ml of dried DMSO which has previously been
treated with highly purified argon. 100 ml of dried and
argon-treated DMSO are heated to a temperature of 175°C and
21
CA 02263210 2004-09-30
26474-432
75 ml of the potassium pentafluorophenate solution are added
dropwise over 30 minutes. 3.1 g of gaseous ethylene oxide
are simultaneously introduced. After 45 minutes at 175°C,
the solution turns brown and KF salt crystallizes out.
Further phenate solution (25 ml) is added slowly over
20 minutes and 3.1 g of gaseous ethylene oxide are
introduced. Over the next 45 minutes the remaining 50 ml of
phenate solution are added dropwise and a further 3.9 g of
gaseous ethylene oxide are introduced. More ethylene oxide
(1.9 g) is finally introduced over 60 minutes. The solution
is then stirred for a further 2 hours at a temperature of
175°C. A colourless precipitate forms. The crude product
is sublimed under reduced pressure (1 to 2 Torr) at a
temperature of between 60 and 70°C.
Yield: 950 of theory
Melting point: 78°C.
d) 3,4,5,6-Tetrafluorocatechol
The ether is cleaved by placing 41.6 g of ground
and dried aluminium chloride and 10.7 g of 5,6,?,8-
tetrafluoro-(1,4)-benzodioxane in a glass flask equipped
with a reflux condenser, under an argon atmosphere, and
dissolving them in 350 ml of toluene. The solution is
heated at a temperature of 80 to 110°C for two hours, with
stirring, and then heated at a reflux temperature of 110 to
118°C for six hours, during which time the reaction solution
turns black. After cooling, the reaction solution is poured
onto 400 g of ice. The aqueous phase is retained for
extraction. Toluene is distilled from the organic phase and
the residual crystals are extracted with hot water. Both
the aqueous phases, from the treatment with ice and the
extraction with water, are combined and extracted three
22
CA 02263210 2004-09-30
26474-432
times with 150 ml of diethyl ether. The ether is distilled
off to leave a green oily residue. At 50°C this gives a
colourless liquid which vaporizes at 60 to 80°C under
reduced pressure (14 - 16 Torr). The purified
tetrafluorocatechol is obtained by sublimation at this
temperature.
Yield: 63.5% of theory
M.p.. 68°C.
e) Lithium bis[tetrafluoro-1,2-benzenediolato(2-)-0,0']borate
5.24 g of lithium tetramethanolatoborate and
13.5 g of tetrafluorocatechol are dissolved in 11 g of
acetonitrile under an argon atmosphere. The solution is
heated to 50°C and 10 g of acetonitrile are distilled off to
leave a lilac-coloured viscous solution. Colourless
crystals have formed after three days at 5°C. The yield can
be increased by slowly heating the solution to 95°C under
reduced pressure. This leaves 15.6 g of a solid brown crude
product, which is recrystallized from a mixture consisting
of 20 ml of benzene and 5 ml of acetonitrile. Colourless
crystals are obtained which are filtered off and washed with
benzene.
Yield: 30.8% of theory
Decomposition point: 270°C.
Example 6
Lithium bis[2,3-naphthalenediolato(2-)-0,0']borate(1-)
90.0 g of 2,3-dihydroxynaphthalene, 17.4 g of boric
acid, 11.8 g of lithium hydroxide monohydrate and 100 ml of
water are placed in a glass flask. The mixture is heated to
55°C under an argon atmosphere. The addition of 300 ml of
23
CA 02263210 2004-09-30
26474-432
acetone gives a clear solution. Cooling to 5°C gives the
product in the form of colourless crystals. The crystals are
separated off and dried by slow heating from 10°C to 170°C
under reduced pressure. The product is purified by
recrystallization twice from 200 and 160 ml of acetone. The
product is then heated slowly to 170°C and kept at this
temperature.
Yield: 28.60 of theory
Decomposition point: 280°C.
24