Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02270963 1999-OS-03
WO 98/20009 PCT/LTS97/20036
(4R,SS,6S,7R)-HEXAHYDRO-1- [5-(3-AMINOINAZOLE)METHYL] -3-BUTYL-5,6-DIHYDROXY-
4,7-BIS
[PHAENYLMETHYL] -2H-1,3-DIAZEPIN-2-ONE, ITS PREPARATION AND ITS USE AS HIV
PROTEASE
INHIBITOR
FTET.D OF THE INVFNTTnN
This invention relates generally to a 1-(3-aminoindazol-
5-yl)-3-butyl-cyclic urea which is useful as inhibitors of
HIV protease, pharmaceutical compositions and diagnostic kits
comprising the same, and methods of using the same for
treating viral infection or as assay standards or reagents.
BACKGROL1ND OF THE INVENTION
Two distinct retroviruses, human immunodeficiency virus
(HIV) type-1 (HIV-1) or type-2 (HIV-2), have been
etiologically linked to the immunosuppressive disease,
acquired immunodeficiency syndrome (AIDS). HIV seropositive
individuals are initially asymptomatic but typically develop
AIDS related complex (ARC) followed by AIDS. Affected
individuals exhibit severe immunosuppression which
predisposes them to debilitating and ultimately fatal
opportunistic infections.
The disease AIDS is the end result of an HIV-1 or HIV-2
virus following its own complex life cycle. The virion life
cycle begins with the virion attaching itself to the host
human T-4 lymphocyte immune cell through the bonding of a
glycoprotein on the surface of the virion's protective coat
with the CD4 glycoprotein on the lymphocyte cell. Once
attached, the virion sheds its glycoprotein coat, penetrates
into the membrane of the host cell, and uncoats its RNA. The
virion enzyme, reverse transcriptase, directs the process of
transcribing the RNA into single-stranded DNA. The viral RNA
is degraded and a second DNA strand is created. The now
double-stranded DNA is integrated into the human cell's genes
and those genes are used for cell reproduction.
At this point, the human cell carries out its
reproductive process by using its own RNA polymerase to
-1-
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
transcribe the integrated DNA into viral RNA. The viral RNA
is translated into the precursor gag-pot fusion polyprotein.
The polyprotein is then cleaved by the HIV protease enzyme to
yield the mature viral proteins. Thus, HIV protease in
responsible for regulating a cascade of cleavage events that
lead to the virus particles maturing into a virus that is
capable of full infectivity.
The typical human immune system response, killing the
invading virion, is taxed because a large portion of the
virion's life cycle is spent in a latent state within the
immune cell. In addition, viral reverse transcriptase, the
enzyme used in making a new virion particle, is not very
specific, and causes transcription mistakes that result in
continually changed glycoproteins on the surface of the viral
protective coat. This lack of specificity decreases the
immune system's effectiveness because antibodies specifically
produced against one glycoprotein may be useless against
another, hence reducing the number of antibodies available to
fight the virus. The virus continues to reproduce while the
immune response system continues to weaken. Eventually, the
HIV largely holds free reign over the body's immune system,
allowing opportunistic infections to set in and without the
administration of antiviral agents, immunomodulators, or
both, death may result.
There are at least three critical points in the virus's
life cycle which have been identified as possible targets for
antiviral drugs: (1} the initial attachment of the virion to
the T-4 lymphocyte or macrophage site, (2) the transcription
of viral RNA to viral DNA (reverse transcriptase, RT), and
(3) the assemblage of the new virus particle during
reproduction (e.g., HIV aspartic acid protease or HIV
protease).
The genomes of retroviruses encode a protease that is
responsible for the proteolytic processing of one or more
polyprotein precursors such as the pol and gag gene products.
See Wellink, Arch. Virol.. S$ 1 (1988) . Retroviral proteases
most commonly process the gag precursor into the core
_ 2 _
CA 02270963 1999-OS-03
proteins, and also process the pot precursor into reverse
transcriptase and retroviral protease.
The correct processing of the precursor polyproteins by
the retroviral protease is necessary for the assembly of the
infectious visions. It has been shown that in vitro
mutagenesis that produces protease-defective virus leads to
the production of immature core forms which lack infectivity.
See Crawford et al., J. V~rol. 53 899 (198S); Katoh et al.,
Virology 1~5 280 (198S). Therefore, retroviral protease
inhibition provides an attractive target for antiviral
therapy. See Mitsuya, Nature 32S 77S (1987).
The ability to inhibit a viral protease provides a
method for bloc'.~i::g vi ral rep-i canon and t herefore a
treatment for viral diseases, such as AIDS, that mar have
1S fewer side e_-ects, be snore efficacious, ar_d be less prcne to
drug reSlS tanCe 'nlnen compar ea t0 Cl:rrent ty Batmen tS . .S a
reSUlt, three ~-i prOtedSe lnh~.:~itOrS, ROChe'S SaCs~.:inav--,
~.bb0 t t ' S '_'i'tCl:avlr , 'c._~.d ~e"C~'t' S _=1Qi~a~~'~r , ar a Curr
e:==~;i
being Tar~~CeteC a?'=C_ a ::' ~, er Oy pCter':tlal p~"~Otease ~~: h'~.x:~tO=S
are in clinical trials, e.g., -iertex's Va-478, Agouron's
,.
nel f i navir , Japan .=nerg~r' s i~'~1I-272 , and Ciba-Geiger' s C~;P
0175S.
As evidenced by the protease inhibitors presently
mar'.~ceted and in clinical trials, a wide variety of cempcur_ds
have been studied as potential rI'J protease inhibitors. Cne
core, cyclic areas, has received significant attention. =o.
example, in PCT Application Numbers rr1094i19329 and WO
93/07128, Lam et a1 generically describe c~rclic areas or the
fornula
O
R22 ~ R2s
\N N~
R ,R~
HO OH
- 3 -
AME1~ED SHEET
CA 02270963 1999-OS-03
and methods of preparing these ureas. Though the present
compounds fall within the description of Lam et al, they are
not specifically disclosed therein.
Additional cyclic ureas are described in Lam et al, J.
S Med. Chem. 1996, 39, 351r3525. Though a variety of cyclic
ureas are dislosed in this publication, no indazolemethyl
containing compounds are described.
- 3A -
AME~r~ SHEET
CA 02270963 1999-OS-03
even with the current success of protease inhibitors, it
has been found that HIV patients can become resisv~:n- to a
single protease inhibitor. Thus, it is desirable to develop
additional protease inhibitors to further combat HIV
infection.
~Ly OF 'T'~ ~ T~1~TENTION
accordingly, one object of t:~e present invention is to
provide novel protease inhibitors.
It is another obi ec t of t:e present i nvention to = ro~r=de
pharmaceutical compositions with protease ir_hibiting act=~,-_ty
comprising a pharmaceutically acceptable carrier and a
theraoeuticallvn effective arnour.t of at 1 east one of t~:e
compour_ds of the present ir_ventvor_ or a pharnaceutical l yr
acceptable salt or prodrug form ~h ereof .
It is ar_other object of the present invention to prc~;ide
a novel method for treating HI'I ~___'cti on whi ch comprises
administering to a host in need of such treatment a
therapeutically effective amount of at least one of the
compounds o' the present invention or a pharmaceutically
acceptable salt or prodrug form thereof.
It is another object of the present invention to provide
a novel method for treating H=V infection which comprises
administering to a host in need thereof a therapeutically
effective combination. of (a) one of the compounds of the
present invention and (b) one or more compounds selected form
the group consisting of HIV reverse transcriptase inhibitors
and HIV protease inhibitors.
It is another object of the present invention to provide
a method of inhibiting HI',l present in a body fluid sample
which comprises treating the body fluid sample with an
effective amount of a compound of the present invention.
It is another object of the present invention to provide
a kit or container containing at least one of the compounds
- 4 -
AMEi~I~ED SHEET
CA 02270963 1999-OS-03
WO 98/20009 PCT/U597/20036
of the present invention in an amount effective for use as a
standard or reagent in a test or assay for determining the
ability of a potential pharmaceutical to inhibit HIV
protease, HIV growth, or both.
These and other objects, which will become apparent
during the following detailed description, have been achieved
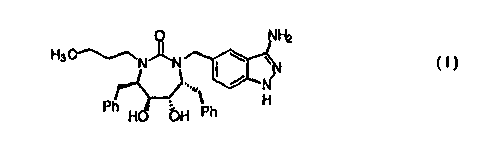
by the inventors' discovery that compounds of formula I:
NH2
H~~N~ ~ v
N
Ph H bH ,' Ph H
I
or pharmaceutically acceptable salts or prodrug forms
thereof, are effective protease inhibitors.
DETAILED DESCRIPTION OF PREFERRED EMBODIMFNTS
Thus, in a first embodiment, the present invention
provides a novel compound of formula I:
NH2
H3~N~ ~ v
.... ~ / NTI
Ph HO bH Ph H
I
or a pharmaceutically acceptable salt or prodrug form
thereof .
In a second embodiment, the present invention provides a
novel pharmaceutical composition comprising a
pharmaceutically acceptable carrier and a therapeutically
effective amount of a compound of formula I or a
pharmaceutically acceptable salt or prodrug form thereof.
In a third embodiment, the present invention provides a
novel method for treating HIV infection which comprises
administering to a host in need of such treatment a
- 5 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
therapeutically effective amount of a compound of formula I
or a pharmaceutically acceptable salt or prodrug form
thereof .
In a fourth embodiment, the present invention provides a
novel method of treating HIV infection which comprises
administering, in combination, to a host in need thereof a
therapeutically effective amount of:
(a) a compound of formula I; and,
(b) at least one compound selected from the group
consisting of HIV reverse transcriptase inhibitors and HIV
protease inhibitors.
In another preferred embodiment, the reverse
transcriptase inhibitor is a nucleoside reverse transcriptase
inhibitor.
In another more preferred embodiment, the nucleoside
reverse transcriptase inhibitor is selected from AZT, 3TC,
ddI, ddC, and d4T and the protease inhibitor is selected from
saquinavir, ritonavir, indinavir, VX-478, nelfinavir, KNI-
272, CGP-61755, and U-103017.
In an even more preferred embodiment, the nucleoside
reverse transcriptase inhibitor is selected from AZT and 3TC
and the protease inhibitor is selected from saquinavir,
ritonavir, and indinavir.
In a still further preferred ebodiment, the nucleoside
reverse transcriptase inhibitor is AZT.
In another still further preferred embodiment, the
protease inhibitor is indinavir.
In a fifth embodiment, the present invention provides a
pharmaceutical kit useful for the treatment of HIV infection,
which comprises a therapeutically effective amount of:
- 6 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
(a) a compound of formula I; and,
(b) at least one compound selected from the group
consisting of HIV reverse transcriptase inhibitors and HIV
protease inhibitors, in one or more sterile containers.
In a sixth embodiment, the present invention provides a
novel method of inhibiting HIV present in a body fluid sample
which comprises treating the body fluid sample with an
effective amount of a compound of formula I.
In a seventh embodiment, the present invention to
provides a novel a kit or container comprising a compound of
formula I or II in an amount effective far use as a standard
or reagent in a test or assay for determining the ability of
a potential pharmaceutical to inhibit HIV protease, HIV
growth, or both.
DEFINT_TIONS
As used herein, the following terms and expressions have
the indicated meanings. It will be appreciated that the
compounds of the present invention contain an asymmetrically
substituted carbon atom, and may be isolated in optically
active or racemic forms. It is well known in the art how to
prepare optically active forms, such as by resolution of
racemic forms or by synthesis, from optically active starting
materials. A11 chiral, diastereomeric, racemic forms and a11
geometric isomeric forms of a structure are intended, unless
the specific stereochemistry or isomer form is specifically
indicated.
As used herein, "HIV reverse transcriptase inhibitor" is
intended to refer to both nucleoside and non-nucleoside
inhibitors of HIV reverse transcriptase (RT). Examples of
nucleoside RT inhibitors include, but are not limited to,
AZT, ddC, ddI, d4T, and 3TC. Examples of non-nucleoside RT
inhibitors include, but are no limited to, viviradine
(Pharmacia and Upjohn 090152S), TIBO derivatives, BI-RG-587,
nevirapine, L-697, 661, LY 73497, and Ro 18, 893 (Roche) .
CA 02270963 1999-OS-03
WO 98I20009 PCT/LTS97/20036
As used herein, "HIV protease inhibitor" is intended to
refer to compounds which inhibit HIV protease. Examples
include, but are not limited, saquinavir (Roche, Ro31-8959),
ritonavir (Abbott, ABT-538), indinavir (Merck, MK-639), VX-
478 (Vertex/Glaxo Wellcome), nelfinavir (Agouron, AG-1343),
KNI-272 (Japan Energy), CGP-61755 (Ciba-Geigy), and U-103017
(Pharmacia and Upjohn). Additional examples include the
cyclic protease inhibitors disclosed in W093/07128, WO
94/19329, WO 94/22840, and PCT Application Number US96/03426.
As used herein, "pharmaceutically acceptable salts"
refer to derivatives of the disclosed compounds wherein the
parent compound is modified by making acid or base salts
thereof. Examples of pharmaceutically acceptable salts
include, but are not limited to, mineral or organic acid
salts of basic residues such as amines; alkali or organic
salts of acidic residues such as carboxylic acids; and the
like. The pharmaceutically acceptable salts include the
conventional non-toxic salts or the quaternary ammonium salts
of the parent compound formed, for example, from non-toxic
inorganic or organic acids. For example, such conventional
non-toxic salts include those derived from inorganic acids
such as hydrochloric, hydrobromic, sulfuric, sulfamic,
phosphoric, nitric and the like; and the salts prepared from
organic acids such as acetic, propionic, succinic, glycolic,
stearic, lactic, malic, tartaric, citric, ascorbic, pamoic,
malefic, hydroxymaleic, phenylacetic, glutamic, benzoic,
salicylic, sulfanilic, 2-acetoxybenzoic, fumaric,
toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic,
isethionic, and the like.
The pharmaceutically acceptable salts of the present
invention can be synthesized from the parent compound which
contains a basic or acidic moiety by conventional chemical
methods. Generally, such salts can be prepared by reacting
the free acid or base forms of these compounds with a
stoichiometric amount of the appropriate base or acid in
water or in an organic solvent, or in a mixture of the two;
generally, nonaqueous media like ether, ethyl acetate,
_ 8 _
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
ethanol, isopropanol, or acetonitrile are preferred. Lists
of suitable salts are found in Remington's Pharmaceutical
Sciences, 17th ed., Mack Publishing Company, Easton, PA,
1985, p. 1418, the disclosure of which is hereby incorporated
by reference.
The phrase "pharmaceutically acceptable" is employed
herein to refer to those compounds, materials, compositions,
and/or dosage forms which are, within the scope of sound
medical judgment, suitable for use in contact with the
tissues of human beings and animals without excessive
toxicity, irritation, allergic response, or other problem or
complication commensurate with a reasonable benefit/risk
ratio.
"Prodrugs" are intended to include any covalently bonded
carriers which release the active parent drug according to
formula I or other formulas or compounds of the present
invention in vivo when such prodrug is administered to a
mammalian subject. Prodrugs of a compound of the present
invention, for example formula (I), are prepared by modifying
functional groups present in the compound in such a way that
the modifications are cleaved, either in routine manipulation
or in vivo, to the parent compound. Prodrugs include
compounds of the present invention wherein the hydroxy or
amino group is bonded to any group that, when the prodrug is
administered to a mammalian subject, cleaves to form a free
hydroxyl or free amino, respectively. Examples of prodrugs
include, but are not limited to, acetate, formate, or
benzoate derivatives of alcohol and amine functional groups
in the compounds of formula I; phosphate esters,
dimethylglycine esters, aminoalkylbenzyl esters, aminoalkyl
esters and carboxyalkyl esters of alcohol functional groups
in the compounds of formula I; and the like. Additional
examples include compounds wherein the two hydroxy groups of
formula I join to form an epoxide; -OCH2SCH20-; -OC(=0)0-;
-OCH20-; -OC (=S) 0-; -OC (=0) C (=0) 0-; -OC (CH3) 20-;
-OC ( (CH2) 3NH2) (CH3) 0-; -OC (OCH3) (CH2CH2CH3) 0-; or -OS (=0) 0-.
_ g _
CA 02270963 1999-OS-03 .
"Stable compound" and "stable structure" are meant to
indicate a compound that is sufficiently robust to survive
isolation to a useful degree of purity from a reaction
mixture, and formulation into an efficacious therapeutic
S agent. Only stable compounds are contempleted by the present
invention.
"Substituted" is intended to indicate that one or more
hydrogens ~n the atom indicated in the expression using
"substituted" is replaced with a selection from the indicated
group(s), provided that the indicated atom's normal valer_cy
is not exceeded, and that the substitution results in a
stable compound. rnJhen a substituent is keto (i.e., =O)
group, then 2 hydrogens on the atom are replaced.
"Therapeutically effective amount" is intended to
1S include ar_ amount of a compour_d of the present invention or
an amount of the combination of compounds claimed effecti-ra
to inhibit ETV ir_~ection or treat t:~e symptoms of HIV
ir_fectior. in a host. The combination of compounds is
preferabl~r a synergistic combination. Synergy, as descri':ed
for e.{ample by Chou and Talalay, Adv. Enzyme Regul. 22:27-S.S
(1984), occurs wren the effect (in this case, inhibitior_ of
HIV replication) of the compounds when admir_istered in
combination is greater than the additive effect of the
compounds ~,vhen administered alone as a single agent. In
2S general, a synergistic effect is most clearly demonstrated at
suboptimal concentrations of the compounds. Synergy can be
in terms of lower cytotoxicity, increased antiviral effect,
or some other beneficial effect of the combination compared
with the individual components.
Other features of the invention will become apparent in
the course of the following descriptions of exemplary
embodiments which are given for illustration of the
invention.
Examples
Abbreviations used in the Examples are defined as
follows: "°C" for degrees Celsius, "d" for doublet, "dd" for
- 10 -
AMEVi~E'J SJ-BEET
CA 02270963 1999-OS-03
WO 98l20009 PCT/US97/20036
doublet of doublets, "eq" for equivalent or equivalents, "g"
for gram or grams, "mg" for milligram or milligrams, "mL" for
milliliter or milliliters, "H" for hydrogen or hydrogens,
"hr" for hour or hours, "m" for multiplet, "M" for molar,
"min" for minute or minutes, "MHz" for megahertz, "MS" for
mass spectroscopy, "nmr" or "NMR" for nuclear magnetic
resonance spectroscopy, "t" for triplet, and "TLC" for thin
layer chromatography.
'
Preparation of (4R,5S,6S,7R)-Hexahydro-1-[5-(3-
aminoindazole)methyl]-3-butyl-5,6-dihydroxy-4,7-
bis[phenylmethyl]-2H-1,3-diazapin-2-one (I).
O OMe
\ HN~NH ~ MeOTf, DCE ~ ~ HN~N _)
y ..~ ~ ~ v ..~ ~
O~O reflux '~ Q~O
A 1
Compound A can be prepared by known methods. For
example, preparation of compound A is shown in Scheme 1 of
Rossano et al (Tetr. Z,ett. 1995, 36(28), 4967, 4968), the
contents of which are hereby incorporated by reference. An
additional method of preparation of compound A is shown in
Example 6 of U.S. Patent No. 5,530,124, the contents of which
are hereby incorporated by reference.
PART A: To a suspension of compound 1 (10.0 g; 27.3
mmol) in 1,2-dichloroethane (100 mL) was added methyltriflate
(3.4 mL, 30 mmol). After refluxing overnight, the reaction
was washed with sat. NaHC03, sat. NaCI, dried (Na2S04) and
evaporated leaving 12.5 g of a yellow oil. Column
chromatography (flash Si02; 25~ EtOAc/hexane) gave 7.86 g of
compound 2 as a pale yellow oil which crystallized on
standing (75o yield). m.p.= 97-100 ~C. MH+ = 381.
- 11 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
~M a
N N _
/ \
O y0' \ /
2
PART B: To a solution of 1 (10.0 g, 26.3 mmol) in
anhydrous DMF (30 ml) was added sodium hydride (1.58 g, 65.8
mmol). The reaction mixture was stirred at room temperature
for 45 minutes followed by dropwise addition of a solution of
1-iodobutane (9.68 g, 52.6 mmol) in anhydrous DMF (10 ml).
After the addition, the stirring was continued at room
temperature overnight. The reaction mixture was cooled to 0 °C
and methanol (5 ml) was added to quench excess sodium
hydride. The mixture was partitioned between ethyl acetate
(200 ml) and water (150 ml). The organic phase was separated
and washed with water (4 x 100 ml), brine (100 ml) and dried
over sodium sulfate. Flash chromatographic purification (250
EtoAc/Hex.) gave n-butyl isourea 2 ( 10.5 g, 92$yd): MS(NH3-
CI/DDIP) (M+H+) 437.2 (100$) ; 1H NMR(300 MHz, CDC13, 25 ~C) S
7.23 (m, 10H) , 4 .19 (m, 3H) , 3. 64 (m, 1H) , 3. 44 (s, 3H) , 3 . 36
(m, 1H) , 3.02 (m, 2H) , 2.76 (m, 2H) ( 2.04 (m, 1H) , 1 .52 (s,
3H) , 1 .49 (s, 3H) , 1 .21 (m, 4H) , 0.82 (t, J=7.0 Hz, 3H) .
PART C: (4R,5S,6S,7R)-Hexahydro-1-[(3-cyano-4
fluorophenyl)methyl]-5,6-0-isopropylidene-4,7-bis-(4
phenylmethyl)-3-phenylmethyl-2H-1,3-diazapin-2-one (3).
F
CN
O
~N~N
/ \ .,,
Y
- 12 -
CA 02270963 1999-OS-03
WO 98/20009 PCTlUS97120036
To a solution of 2 ( 5.0 g, 11.5 mmol) in acetonitrile
(40 ml) was added 4-fluoro-3-cyanobenzyl bromide (3.68 g,
17.25 mmol). The reaction mixture was refluxed overnight.
After the solvent was removed under reduced pressure, the
residue was purified using flash chromatography ( 350
EtoAc/Hex.) to give cyclic urea 3 as a white solid (4.5 g,
71 o yd) : MS (NH3-CI/DDIP) (M+H+) 556.3 (100 0) ; 1H NMR(300 MHz,
CDC13, 25 °C) 8 7 . 41 (m, 1H) , 7.28 (m, 7H) , 7 .13 (d, J=9
.2Hz,
2H) , 7 . 05 (t, J=8 . 8 Hz, 1H) , 6. 95 (d, J=9 .2 Hz, 2H) , 4 .50 (d,
J=14.0 Hz, 1H), 4.07 (m, 2H), 3.70 (m, 3H), 3.44 (t, J=7.7
Hz, 1H), 2.90 (m, 4H), 2.12 (m, 1H), 1.50 (s, 6H), 1.26 (m,
4H) , 0 . 83 (t, J=7 . 0 Hz, 3H) .
PART D: (4R, 5S, 6S, 7R) -Hexahydro-1- [5- (3-
aminoindazole)methyl]-3-butyl-5,6-dihydroxy-4,7-
bis[phenylmethyl]-2H-1,3-diazapin-2-one (I)
To a solution of 3 ( 4.5 g, 8.11 mmol) in n-butanol (20
ml) was added hydrazine hydrate ( 0.81 g, 16.2 mmol). The
mixture was refluxed for 6 hr. The solvent and excess
hydrazine were removed under reduced pressure. The residue
was dissolved in anhydrous methanol (20 ml) followed by the
addition of 4 M HC1 in dioxane (2m1). The reaction mixture
was stirred at room temperature far 2 hr. Methanol was
removed and the residue was partitioned between ethyl acetate
(80 ml) and sodium bicarbonate (sat.) (50 ml) . The organic
phase was separated, washed with water (2x 50 ml) and dried
over sodium sulfate (anhydrous). Flash chromatographic
purification gave I (3.0 g, 72~ yd.) as a white solid: MP
129-131 °C; MS (NH3-CI/DDIP) (M+H+) 528.3 (100°s) ; HRMS calcd
for C31H37N503+1 528.2975, found 528.2958; 1H NMR(300 MHz,
CD30D, 25 °C) S 7 .19 (m, 12H) , 6 . 98 (d, J=1 .5 Hz, 2H) , 9 .
74
(d, J=13. 9 Hz, 1H) , 3. 85 (dd, J=10 .25, 4 .76 Hz, 1H) , 3 . 65 (m,
1H), 3.56 (m, 4H), 3.15 (m, 2H), 2.96 (m, 3H), 2.07 (m, 2H),
1.37 (m, 2H), 1.22 (m, 2H), 0.84 (t, J=7.0, 3H).
- 13 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
Utility
The compounds of formula I possess HIV protease
inhibitory activity and are therefore useful as antiviral
agents for the treatment of HIV infection and associated
diseases. The compounds of formula I possess HIV protease
inhibitory activity and are effective as inhibitors of HIV
growth. The ability of the compounds of the present
invention to inhibit viral growth or infectivity is
demonstrated in standard assay of viral growth or
infectivity, for example, using the assay described below.
The compounds of formula I of the present invention are
also useful for the inhibition of HIV in an ex vivo sample
containing HIV or expected to be exposed to HIV. Thus, the
compounds of the present invention may be used to inhibit HIV
present in a body fluid sample (for example, a serum or semen
sample) which contains or is suspected to contain or be
exposed to HIV.
The compounds provided by this invention are also useful
as standard or reference compounds for use in tests or assays
for determining the ability of an agent to inhibit viral
clone replication and/or HIV protease, for example in a
pharmaceutical research program. Thus, the compounds of the
present invention may be used as a control or reference
compound in such assays and as a quality control standard.
The compounds of the present invention may be provided in a
commercial kit or container for use as such standard or
reference compound.
Since the compounds of the present invention exhibit
specificity for HIV protease, the compounds of the present
invention may also be useful as diagnostic reagents in
diagnostic assays for the detection of HIV protease. Thus,
inhibition of the protease activity in an assay (such as the
assays described herein) by a compound of the present
invention would be indicative of the presence of HIV protease
and HIV virus.
As used herein "~.g" denotes microgram, "mg" denotes
milligram, "g" denotes gram, "~.L" denotes microliter, "mL"
- 14 -
CA 02270963 1999-OS-03
WO 98I20009 PCT/US97/20036
denotes milliliter, "L" denotes liter, "nM" denotes
nanomolar, "~1.M" denotes micromolar, "mM" denotes millimolar,
"M" denotes molar and "nm" denotes nanometer. "Sigma" stands
for the Sigma-Aldrich Corp. of St. Louis, M0.
DNA Plasmids and in vitro RNA t_ranscr,'_n~
Plasmid pDAB 72 containing both gag and pol sequences of
BH10 (bp 113-1816) cloned into PTZ 19R was prepared according
to Erickson-Viitanen et al. AIDS Research and Human
Retroviruses 1989, 5, 577. The plasmid was linearized with
Bam HI prior to the generation of in vitro RNA transcripts
using the Riboprobe Gemini system II kit (Promega) with T7
RNA polymerase. Synthesized RNA was purified by treatment
with RNase free DNAse (Promega), phenol-chloroform
extraction, and ethanol precipitation. RNA transcripts were
dissolved in water, and stored at -70°C. The concentration
of RNA Was determined from the A260
Probes:
Biotinylated capture probes were purified by HPLC after
synthesis on an Applied Biosystems (Foster City, CA) DNA
synthesizer by addition of biotin to the 5' terminal end of
the oligonucleotide, using the biotin-phosphoramidite reagent
of Cocuzza, Tet. Lett. 1989, 30, 6287. The gag biotinylated
capture probe (5-biotin-CTAGCTCCCTGCTTGCCCATACTA 3') was
complementary to nucleotides 889-912 of HXB2 and the pol
biotinylated capture probe (5'-biotin -CCCTATCATTTTTGGTTTCCAT
3' ) was complementary to nucleotides 2374-2395 of HXB2.
Alkaline phosphatase conjugated oligonucleotides used as
reporter probes were prepared by Syngene (San Diego, CA.).
The pol reporter probe (5' CTGTCTTACTTTGATAAAACCTC 3') was
complementary to nucleotides 2403-2425 of HXB2. The gag
reporter probe (5' CCCAGTATTTGTCTACAGCCTTCT 3') was
complementary to nucleotides 950-973 of HXB2. A11 nucleotide
positions are those of the GenBank Genetic Sequence Data Bank
- 15 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
as accessed through the Genetics Computer Group Sequence
Analysis Software Package (Devereau Nucleic Acids Research
1984, 12, 387). The reporter probes were prepared as 0.5 ~M
stocks in 2 x SSC (0.3 M NaCl, 0.03 M sodium citrate), 0.05 M
Tris pH 8.8, 1 mg/mL BSA. The biotinylated capture probes
were prepared as 100 ~M stocks in water.
StrP~tavid,'_n coated ply
Streptavidin coated plates were obtained from Du Pont
Biotechnology Systems (Boston, MA).
Ce1_1_s and virus stocks:
MT-2 and MT-4 cells were maintained in RPMI 1640
supplemented with 5o fetal calf serum (FCS) for MT-2 cells or
10~ FCS for MT-4 cells, 2 mM L-glutamine and 50 ~.g/mL
gentamycin, a11 from Gibco. HIV-1 RF was propagated in MT-4
cells in the same medium. Virus stocks were prepared
approximately 10 days after acute infection of MT-4 cells and
stored as aliquots at -70°C. Infectious titers of HIV-1(RF)
stocks were 1-3 x 107 PFU (plaque forming units)/mL as
measured by plaque assay on MT-2 cells (see below). Each
aliquot of virus stock used for infection was thawed only
once.
For evaluation of antiviral efficacy, cells to be
infected were subcultured one day prior to infection. On the
day of infection, cells were resuspended at 5 x 105 cells/mL
in RPMI 1640, 5~ FCS for bulk infections or at 2 x 106/mL in
Dulbecco's modified Eagles medium with 5% FCS for infection
in microtiter plates. Virus was added and culture continued
for 3 days at 37°C.
HIV RNA assay:
Cell lysates or purified RNA in 3 M or 5 M GED were
mixed with 5 M GED and capture probe to a final guanidinium
isothiocyanate concentration of 3 M and a final biotin
oligonucleotide concentration of 30 nM. Hybridization was
carried out in sealed U bottom 96 well tissue culture plates
- 16 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
(Nunc or Costar) for 16-20 hours at 37°C. RNA hybridization
reactions were diluted three-fold with deionized water to a
final guanidinium isothiocyanate concentration of 1 M and
aliquots (150 ~,L) were transferred to streptavidin coated
microtiter plates wells. Binding of capture probe and
capture probe-RNA hybrid to the immobilized streptavidin was
allowed to proceed for 2 hours at room temperature, after
which the plates were washed 6 times with DuPont ELISA plate
wash buffer (phosphate buffered saline(PBS), 0.05o Tween 20.)
A second hybridization of reporter probe to the immobilized
complex of capture probe and hybridized target RNA was
carried out in the washed streptavidin coated well by
addition of 120 ~tl of a hybridization cocktail containing 4 X
SSC, 0.66 Triton X 100, 6.66 deionized formamide, 1 mg/mL
BSA and 5 nM reporter probe. After hybridization for one
hour at 37°C, the plate was again washed 6 times.
Immobilized alkaline phosphatase activity was detected by
addition of 100 ~.L of 0.2 mM 4-methylumbelliferyl phosphate
(MUBP, JBL Scientific) in buffer S(2.5 M diethanolamine pH 8.9
(JBL Scientific), 10 mM MgCl2, 5 mM zinc acetate dehydrate
and 5 mM N-hydroxyethyl-ethylene-diamine-triacetic acid).
The plates were incubated at 37°C. Fluorescence at 450 nM
was measured using a microplate fluorometer (Dynateck)
exciting at 365 nM.
Micronlate based comt~oLnd evaluation in HIV-1 infected MT-2
Cap:
Compounds to be evaluated were dissolved in DMSO and
diluted in culture medium to twice the highest concentration
to be tested and a maximum DMSO concentration of 20. Further
three-fold serial dilutions of the compound in culture medium
were performed directly in U bottom microtiter plates (Nuns).
After compound dilution, MT-2 cells (50 ~,L) were added to a
final concentration of 5 x 105 per mL (1 x 105 per well).
Cells were incubated with compounds for 30 minutes at 37°C in
a C02 incubator. For evaluation of antiviral potency, an
appropriate dilution of HIV-1 (RF) virus stock (50 ~1L) was
- 17 -
CA 02270963 1999-OS-03
WO 98I20009 PCT/US97/20036
added to culture wells containing cells and dilutions of the
test compounds. The final volume in each well was 200 ~L.
Eight wells per plate were left uninfected with 50 ~.L of
medium added in place of virus, while eight wells were
infected in the absence of any antiviral compound. For
evaluation of compound toxicity, parallel plates were
cultured without virus infection.
After 3 days of culture at 37°C in a humidified chamber
inside a C02 incubator, a11 but 25 ~1L of medium/well was
removed from the HIV infected plates. Thirty seven ~.L of 5 M
GED containing biotinylated capture probe was added to the
settled cells and remaining medium in each well to a final
concentration of 3 M GED and 30 nM capture probe.
Hybridization of the capture probe to HIV RNA in the cell
lysate was carried out in the same microplate well used for
virus culture by sealing the plate with a plate sealer
(Costar), and incubating for 16-20 hrs in a 37°C incubator.
Distilled water was then added to each well to dilute the
hybridization reaction three-fold and 150 ~,L of this diluted
mixture was transferred to a streptavidin coated microtiter
plate. HIV RNA was quantitated as described above. A
standard curve, prepared by adding known amounts of pDAB 72
in vitro RNA transcript to wells containing lysed uninfected
cells, was run on each microtiter plate in order to determine
the amount of viral RNA made during the infection.
In order to standardize the virus inoculum used in the
evaluation of compounds for antiviral activity, dilutions of
virus were selected which resulted in an ICgO value
(concentration of compound required to reduce the HIV RNA
level by 90~) for dideoxycytidine (ddC) of 0.2 ~.g/mL. ICgO
values of other antiviral compounds, both more and less
potent than ddC, were reproducible using several stocks of
HIV-1 (RF) when this procedure was followed. This
concentration of virus corresponded to ~3 x 105 PFU (measured
by plaque assay on MT-2 cells) per assay well and typically
produced approximately 75~ of the maximum viral RNA level
achievable at any virus inoculum. For the HIV RNA assay,
- 18 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
ICgO values were determined from the percent reduction of net
signal (signal from infected cell samples minus signal from
uninfected cell samples) in the RNA assay relative to the net
signal from infected, untreated cells on the same culture
plate (average of eight wells). Valid performance of
individual infection and RNA assay tests was judged according
to three criteria. It was required that the virus infection
should result in an RNA assay signal equal to or greater than
the signal generated from 2 ng of pDAB 72 in vitro RNA
transcript. The ICgp for ddC, determined in each assay run,
should be between 0.2 and 0.3 ~g/mL. Finally, the plateau
level of viral RNA produced by an effective protease
inhibitor should be less than 100 of the level achieved in an
uninhibited infection. A compound was considered active if
its ICgp was found to be less than lEtM.
For antiviral potency tests, a11 manipulations in
microtiter plates, following the initial addition of 2X
concentrated compound solution to a single row of wells, were
performed using a Perkin Elmer/Cetus ProPette.
Dotage and Formulation
The antiviral compounds of this invention can be
administered as treatment for viral infections by any means
that produces contact of the active agent with the agent's
site of action, i.e., the viral protease, in the body of a
mammal. They can be administered by any conventional means
available for use in conjunction with pharmaceuticals, either
as individual therapeutic agents or in a combination of
therapeutic agents. They can be administered alone, but
preferably are administered with a pharmaceutical carrier
selected on the basis of the chosen route of administration
and standard pharmaceutical practice.
The dosage administered will, of course, vary depending
upon known factors, such as the pharmacodynamic
characteristics of the particular agent and its mode and
route of administration; the age, health and weight of the
recipient; the nature and extent of the symptoms; the kind of
- 19 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
concurrent treatment; the frequency of treatment; and the
effect desired. A daily dosage of active ingredient can be
expected to be about 0.001 to about 1000 milligrams per
kilogram of body weight, with the preferred dose being about
0.1 to about 30 mg/kg.
Dosage forms of compositions suitable for administration
contain from about 1 mg to about 100 mg of active ingredient
per unit. In these pharmaceutical compositions the active
ingredient will ordinarily be present in an amount of about
0.5-95% by weight based on the total weight of the
composition. The active ingredient can be administered
orally in solid dosage forms, such as capsules, tablets and
powders, or in liquid dosage forms, such as elixirs, syrups
and suspensions. It can also be administered parenterally,
in sterile liquid dosage forms.
Gelatin capsules contain the active ingredient and
powdered carriers, such as lactose, starch, cellulose
derivatives, magnesium stearate, stearic acid, and the like.
Similar diluents can be used to make compressed tablets.
Both tablets and capsules can be manufactured as sustained
release products to provide for continuous release of
medication over a period of hours. Compressed tablets can be
sugar coated or film coated to mask any unpleasant taste and
protect the tablet from the atmosphere, or enteric coated for
selective disintegration in the gastrointestinal tract.
Liquid dosage forms for oral administration can contain
coloring and flavoring to increase patient acceptance.
In general, water, a suitable oil, saline, aqueous
dextrose (glucose), and related sugar solutions and glycols
such as propylene glycol or polyethylene glycols are suitable
carriers for parenteral solutions. Solutions for parenteral
administration preferably contain a water soluble salt of the
active ingredient, suitable stabilizing agents, and if
necessary, buffer substances. Antioxidizing agents such as
sodium bisulfate, sodium sulfite, or ascorbic acid, either
alone or combined, are suitable stabilizing agents. Also
used are citric acid and its salts, and sodium EDTA. In
- 20 -
CA 02270963 1999-OS-03
WO 98I20009 PCT/US97/20036
addition, parenteral solutions can contain preservatives,
such as benzalkonium chloride, methyl- or propyl-paraben and
chlorobutanol. Suitable pharmaceutical carriers are
described in Remington's Pharmaceutical Sciences, supra, a
standard reference text in this field.
Useful pharmaceutical dosage-forms for administration of
the compounds of this invention can be illustrated as
follows
A large number of unit capsules can be prepared by
filling standard two-piece hard gelatin capsules each with
100 mg of powdered active ingredient, I50 mg of lactose, 50
mg of cellulose, and 6 mg magnesium stearic.
Soft Gelatin Car~sules
A mixture of active ingredient in a digestible oil such
as soybean oil, cottonseed oil or olive oil can be prepared
and injected by means of a positive displacement pump into
gelatin to form soft gelatin capsules containing 100 mg of
the active ingredient. The capsules should then be washed
and dried.
Tablets
A large number of tablets can be prepared by
conventional procedures so that the dosage unit is 100 mg of
active ingredient, 0.2 mg of colloidal silicon dioxide, 5
milligrams of magnesium stearate, 275 mg of microcrystalline
cellulose, 11 mg of starch and 98.8 mg of lactose.
Appropriate coatings may be applied to increase palatability
or delay absorption.
Susr~ension
An aqueous suspension can be prepared for oral
administration so that each 5 ml contain 25 mg of finely
divided active ingredient, 200 mg of sodium carboxymethyl
- 21 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/LTS97/20036
cellulose, 5 mg of sodium benzoate, 1.0 g of sorbitol
solution, U.S.P., and 0.025 mg of vanillin.
_Iniectable
A parenteral composition suitable for administration by
injection can be prepared by stirring 1.5o by weight of
active ingredient in 10% by volume propylene glycol and
water. The solution is sterilized by commonly used
techniques.
Combination of components (a) and (b)
Each therapeutic agent component of this invention can
independently be in any dosage form, such as those described
above, and can also be administered in various ways, as
described above. In the following description component (b)
is to be understood to represent one or more agents as
described previously. Thus, if components (a) and (b) are to
be treated the same or independently, each agent of component
(b) may also be treated the same or independently.
Components (a) and (b) of the present invention may be
formulated together, in a single dosage unit (that is,
combined together in one capsule, tablet, powder, liquid,
etc.) as a combination product. When component (a) and (b)
are not formulated together in a single dosage unit,
component (a) may be administered at the same time as
component (b) or in any order; for example component (a) of
this invention may be administered first, followed by
administration of component (b), or they may be administered
in the reverse order. If component (b) contains more that
one agent, e.g., one RT inhibitor and one protease inhibitor,
these agents may be administered together or in any order.
When not administered at the same time, preferably the
administration of component (a) and (b) occurs less than
about one hour apart. Preferably, the route of
administration of component (a) and (b) is oral. The terms
oral agent, oral inhibitor, oral compound, or the like, as
used herein, denote compounds which may be orally
- 22 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/ITS97/20036
administered. Although it is preferable that component (a)
and component (b) both be administered by the same route
(that is, for example, both orally) or dosage form, if
desired, they may each be administered by different routes
(that is, for example, one component of the combination
product may be administered orally, and another component may
be administered intravenously) or dosage forms.
As is appreciated by a medical practitioner skilled in
the art, the dosage of the combination therapy of the
invention may vary depending upon various factors such as the
pharmacodynamic characteristics of the particular agent and
its mode and route of administration, the age, health and
weight of the recipient, the nature and extent of the
symptoms, the kind of concurrent treatment, the frequency of
treatment, and the effect desired, as described above.
The proper dosage of components (a) and (b) of the
present invention will be readily ascertainable by a medical
practitioner skilled in the art, based upon the present
disclosure. By way of general guidance, typically a daily
dosage may be about 100 milligrams to about 1.5 grams of each
component. If component (b) represents more than one
compound, then typically a daily dosage may be about 100
milligrams to about 1.5 grams of each agent of component (b).
By way of general guidance, when the compounds of component
(a) and component (b) are administered in combination, the
dosage amount of each component may be reduced by about 70-
80o relative to the usual dosage of the component when it is
administered alone as a single agent for the treatment of HIV
infection.
The combination products of this invention may be
formulated such that, although the active ingredients are
combined in a single dosage unit, the physical contact
between the active ingredients is minimized. In order to
minimize contact, for example, where the product is orally
administered, one active ingredient may be enteric coated.
By enteric coating one of the active ingredients, it is
possible not only to minimize the contact between the
- 23 -
CA 02270963 1999-OS-03
WO 98/20009 PCTIUS97/20036
combined active ingredients, but also, it is possible to
control the release of one of these components in the
gastrointestinal tract such that one of these components is
not released in the stomach but rather is released in the
intestines.
Another embodiment of this invention where oral
administration is desired provides for a combination product
wherein one of the active ingredients is coated with a
sustained-release material which effects a sustained-release
throughout the gastrointestinal tract and also serves to
minimize physical contact between the combined active
ingredients. Furthermore, the sustained-released component
can be additionally enteric coated such that the release of
this component occurs only in the intestine. Still another
approach would involve the formulation of a combination
product in which the one component is coated with a sustained
and/or enteric release polymer, and the other component is
also coated with a polymer such as a low viscosity grade of
hydroxypropyl methylcellulose or other appropriate materials
as known in the art, in order to further separate the active
components. The polymer coating serves to form an additional
barrier to interaction with the other component. In each
formulation wherein contact is prevented between components
(a) and (b) via a coating or some other material, contact may
also be prevented between the individual agents of component
(b) .
Dosage forms of the combination products of the present
invention wherein one active ingredient is enteric coated can
be in the form of tablets such that the enteric coated
component and the other active ingredient are blended
together and then compressed into a tablet or such that the
enteric coated component is compressed into one tablet layer
and the other active ingredient is compressed into an
additional layer. Optionally, in order to further separate
the two layers, one or more placebo layers may be present
such that the placebo layer is between the layers of active
ingredients. In addition, dosage forms of the present
- 24 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/iJS97/20036
invention can be in the form of capsules wherein one active
ingredient is compressed into a tablet or in the form of a
plurality of microtablets, particles, granules or non-perils,
which are then enteric coated. These enteric coated
microtablets, particles, granules or non-perils are then
placed into a capsule or compressed inta a capsule along with
a granulation of the other active ingredient.
These as well as other ways of minimizing contact
between the components of combination products of the present
invention, whether administered in a single dosage form or
administered in separate forms but at the same time or
concurrently by the same manner, will be readily apparent to
those skilled in the art, based on the present disclosure.
Pharmaceutical kits useful for the treatment of HIV
infection, which comprise a therapeutically effective amount
of a pharmaceutical composition comprising a compound of
component (a) and one or more compounds of component (b), in
one or more sterile containers, are also within the ambit of
the present invention. Sterilization of the container may be
carried out using conventional sterilization methodology well
known to those skilled in the art. Component (a) and
component (b) may be in the same sterile container or in
separate sterile containers. The sterile containers of
materials may comprise separate containers, or one or more
~25 multi-part containers, as desired. Component (a) and
component (b), may be separate, or physically combined into a
single dosage form or unit as described above. Such kits may
further include, if desired, one or more of various
conventional pharmaceutical kit components, such as for
example, one or more pharmaceutically acceptable carriers,
additional vials for mixing the components, etc., as will be
readily apparent to those skilled in the art. Instructions,
either as inserts or as labels, indicating quantities of the
components to be administered, guidelines for administration,
and/or guidelines for mixing the components, may also be
included in the kit.
- 25 -
CA 02270963 1999-OS-03
WO 98/20009 PCT/US97/20036
Obviously, numerous modifications and variations of the
present invention are possible in light of the above
teachings. It is therefore to be understood that within the
scope of the appended claims, the invention may be practiced
otherwise than as specifically described herein.
- 26 -