Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
2-SUBSTITUTED BENZOYL-CYCLOALKYL-l.-CARBOXYLIC ACID DERIVATIVES
The present invention relates to 2-(substituted benzoyl)-
cycloalkyl-1-carboxylic acid derivatives, to a process for
their preparation, to pharmaceutical compositions containing
them and to their use in therapy.
The compounds of the invention act as inhibitors of Kynurenine
3-hydroxylase (KYN-OH), an enzyme which forms part of the
metabolic pathway of kynurenine.
It is well known that through the kynurenine pathway,
tryptophan metabolism gives rise to the formation of 3-
hydroxykynurenine(3-OHKYN) and quinolinic acid (QUIN), on the
one side, and kynurenic acid (K~'NA), on the other side, as
shown in Figure 1. (The legend to Figure 1 is to be found on
the last page of the experimental part of the specification).
KYNA is endowed with neuroprotective properties (J. Neurosci.
1990,10,2965-2973), whereas QUIN is a potent neurotoxin which
has been implicated in the pathogenesis of a variety of
neurological disorders (Life Sci. 1984,35,19-32; Nature,
1986,321,168-171; Science, 1983,219,316-318).
Increased concentrations of QUIN have also been indicated as
responsible for neurological d'.isorders accompanying many
infections and inflammatory diseases including Acquired
Immunodeficiency Syndrome (AIDS) (Ann. Neurol. 1991,29,202-
209) .
One of the main strategies, aimed at altering the KYNA/QUIN
balance blocking 3-OHKYN and QUI:N production and increasing
KYNA production, entails inhibition of key enzymes of the
kynurenine (KYN) pathway, among which Kynurenine-3-hydroxylase
is of primary importance.
CA 02283184 1999-09-10
WO 98/40344 PGT/EP98/00883
- 2 -
Consequently, there is a need in therapy of compounds able of
inhibiting this enzyme.
The compounds of the present invention fulfill such a need.
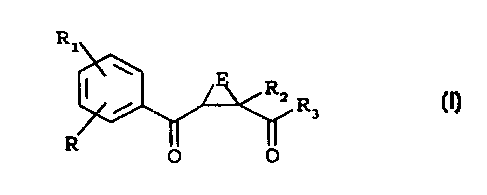
Accordingly, the present invention provides 2-(substituted
benzoyl)-cycloalkyl-1-carboxylic acid compounds of formula (I)
R1
W
RZ R3
R
O O (I)
wherein
E is a C1-C4 alkylene chain, in which a carbon atom is
optionally substituted by =CH2, one or two C1-Cq alkyl groups or
one or two halogen atoms;
each of R and R1, being the same or different, is hydrogen,
halogen, hydroxy, trifluoromethyl, cyano, nitro, phenyl,
benzyl, C1-C6 alkyl, C1-C6 alkoxy, C1-C6 alkylthio, SOR4 or SOZR4
in which R4 is C1-C6 alkyl or -N (RSR6) in which each of RS and R
is, independently, hydrogen, C1-C6 alkyl, formyl or C--C
alkanoyl;
RZ is hydrogen or -N(R7R8) in which each of R~ and R~ is,
independently, hydrogen, C1-C6 alkyl, benzyl, phenyl, hydroxy,
C1-C6 alkoxy, benzyloxy or one of R., and R8 is hydrogen and the
other is COR9 in which R9 is hydrogen, Cl-C6 alkyl, C1-C6 alkoxy,
phenyl or R9 is a group -N(R1oR11) in which each of Rlo and R1, is
independently hydrogen or C1-C6 alkyl;
R3 is hydroxy, C1-C6 alkoxy, phenoxy, benzyloxy or a group
-N(R1ZR13) wherein R12 and R13 are as R~ and R8 as defined above,
or one of RIZ and R13 is hydrogen and the other is a SOZRS group
in which R9 is as defined above; and pharmaceutically
acceptable salts thereof.
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 3 -
The alkyl and alkoxy groups may be branched or straight groups.
E as a C1-C4 alkylene chain is preferably a C1-Cz alkylene
chain, in particular a methylene~ (-CHz-) group. when such
alkylene chain is substituted, it is preferably substituted by
' S a =CHz group or one or two halogen atoms, in particular
fluorine or one or two methyl groups.
Representative examples of C1-C6 alkyl groups include C1-Cq
alkyl groups such as methyl, ethyl, n- and iso-propyl, n-,
iso-, sec- and tert-butyl.
Representative examples of C1-C6 alkoxy groups include C1-CQ
alkoxy groups such as methoxy and ethoxy.
Representative examples of C1-C6 a7_kylthio groups include C1-C4
alkylthio groups such as methylthio and ethylthio.
Representative examples of C1-C6 alkanoyl groups include C1-C4
alkanoyl groups such as acetyl and propionyl.
Representative examples of C1-C6 alkoxycarbonyl groups include
C1-C4 alkoxycarbonyl groups such as methoxycarbonyl and
ethoxycarbonyl.
A halogen atom is fluorine, brom_Lne, chlorine or iodine; i:
particular chlorine or fluorine.
Pharmaceutically acceptable salts of the compounds of the
invention include acid addition salts with inorganic, e.g.
nitric, hydrochloric, hydrobromic, sulphuric, perchloric and
phosphoric acids or organic, e.g. acetic, trifluoroacetic,
propionic, glycolic, lactic, oxalic, malonic, malic, malefic,
tartaric, citric, benzoic, cinnamic, mandelic and salicylic
acids, and salts with inorganic, e.g. alkali metal, especially
sodium or potassium bases or alkaline-earth metal, especially
calcium or magnesium bases, or with organic bases, e.g. acyclic
or cyclic amines, preferably methylamine, ethylamine,
diethlamine, triethylamine or piperidine.
CA 02283184 1999-09-10
WO 98/40344 p~/Epgg~Opgg3
- 4 -
The compounds of the invention have asymmetric carbon atoms and
therefore they can exist either as racemic mixtures or as
individual optical isomers (enantiomers). Moreover the
compounds of the invention can also be E- or Z- isomers or E-,
Z- mixtures thereof.
Accordingly the present invention also include within its scope
all the possible isomers and their mixtures and both the
metabolites and the pharmaceutically acceptable bio-precursors
(otherwise known as pro-drugs) of the compounds of the
invention.
Preferred compounds of the invention are the compounds of
formula (I) wherein:
E is a -CH2- or a -(CHZ)2- group, optionally substituted by
=CHz, one or two halogen atoms or one or two C1-CQ alkyl groups;
each of R and R1, being the same or different, is hydrogen,
halogen, hydroxy, trifluoromethyl, cyano, nitro, C1-C4 alkyl,
C1-C4 alkoxy, C1-C4 alkylthio, SORB, SOZR9 in which R.4 is C1-Cq
alkyl or -N(RSR6) in which each of RS and R6 is, independently,
hydrogen, C1-Cq alkyl or formyl ;
R2 is hydrogen or -N (R~Re) in which each of R.; and R~ is,
independently, hydrogen, C1-CQ alkyl, benzyl, phenyl, hydrox~~,
C1-C4 alkoxy or benzyloxy, or one of R, and R~ is hydrogen and
the other is COR9 in which R9 is hydrogen, C1-C,~ alkyl , C1-Cq
alkoxy, phenyl or R9 is a group -N(R1oR11) in which Rlo and R11
is, independently, hydrogen or C1-C~ alkyl;
R3 is hydroxy, C1-CQ alkoxy, benzyloxy, hydroxylamino or a group
-N (R1zR13) wherein one of R1z and R1, is hydrogen and the other is
hydrogen, C1-C4 alkyl , benzyl , phenyl or a SOZR9 group in which
R9 is phenyl;
and the pharmaceutically acceptable salts thereof.
More preferred compounds according to the invention are the
compounds of formula (I) wherein:
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 5 -
E is methylene, optionally substituted by =CH2, one or two
halogen atoms or one or two C1-CQ a:Lkyl groups;
- each of R and R1, being the same or different, is hydrogen or
halogen;
RZ is hydrogen;
R3 is hydroxy, C1-C9 alkoxy, hydrox:ylamino or a group -N (R12R13)
wherein one of R12 and R13 is hydrogen and the other is
hydrogen, C1-CQ alkyl, benzyl, phenyl cr a SOZR9 group in which
R9 is phenyl;
and the pharmaceutically acceptablE: salts thereof.
Most preferred compounds according to the invention are the
compounds of formula (I) wherein:
E is methylene, optionally substituted by =CH2, one or two
halogen atoms or one or two C1-CQ alkyl groups;
R and R1 are both halogen;
RZ is hydrogen;
R3 is hydroxy, C1-C4 alkoxy, hydro}:ylamino or a r~roup -N (R12R13)
wherein one of R12 and R13 is hydrogen and the other is
hydrogen, C1-Cq alkyl, benzyl, phenyl or a SO~R9 group in which
R9 is phenyl ;
and the pharmaceutically acceptable salts thereof.
Examples of preferred compounds of the invention are the
following:
2-(3-chlorobenzoyl)-cyclopropane-1--carboxylic acid;
2-(3-fluorobenzoyl)-cyclopropane-1~-carboxylic acid;
2-(3-bromobenzoyl)-cyclopropane-1-carboxylic acid;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-carboxylic acid;
2-(3,4-difluorobenzoyl)-cyclopropane-1-carboxylic acid;
2-(3,4-dichlorobenzoyl)-1-amino-cyclopropane-1-carboxylic acid;
2-{3,4-dichlorobenzoyl)-3-methylene-cyclopropane-1-carboxylic
acid;
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 6 -
2-(3,4-dichlorobenzoyl)-3,3-dimethyl-cyclopropane-1-carboxylic
acid;
2-(3,4-dichlorobenzoyl)-3,3-difluoro-cyclopropane-1-carboxylic
acid;
2-(3,4-dichlorobenzoyl)-3-methyl-cyclopropane-1-carboxylic
acid;
2-(3,4-dichlorobenzoyl)-cyclopentane-1-carboxylic acid;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-carboxamide;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-methyl-carboxamide;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-benzyl-carboxamide;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-phenyl-carboxamide;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-hydroxamic acid;
2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-phenylsulfonyl-
carboxamide;
2-(3,4-dichlorobenzoyl)-cyclobutane-1-carboxylic acid;
and, if the case, the C1-C6 , preferably C1-C4, alkyl esters
thereof; either as single E- or Z- isomer and/or as single
optical isomer or as a mixture thereof and, when appropriate,
the pharmaceutically acceptable salts thereof.
A further object of the present invention is also to
provide a 2-substituted benzoyl-cycloalkyl-1-carboxylic acid
compound of formula (I) as defined above, or a pharmaceutically
acceptable salt thereof, for use as an active therapeutic
substance, in particular as kynurenine-3-hydroxylase enzyme
inhibitor.
Object of the present invention is also the use of a
compound of formula (I), as defined above, or a
pharmaceutically acceptable salt thereof, in the manufacture of
a medicament for use as kynurenine-3-hydroxylase enzyme
inhibitor.
The present invention also provides a method of treating a
mammal, including human, in need of a kynurenine-3-hydroxylase
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
inhibitor, such method comprising adminstering thereto a
therapeutically effective amount of a compound of formula (I),
as defined above, or a pharmaceutically acceptable salt
thereof .
- 5 The compounds of the invention and the salts thereof can be
obtained, for instance, by a process comprising:
a) reacting a compound of formula (II)
O
R C
~N
\ 2
R (II)
i
wherein
R and R1 are as defined above, with a compound of formula
(III)
Rz ~ Ra
(III)
CHz
wherein Rz is as defined above and R3 is C1-C6 alkoxy, to
obtain a compound of formula (:I) wherein R3 is Ci-C6 alkoxy
and E is an unsubstituted C1-alkylene (-CHz-) group; or
b) reacting a compound of formula (IV)
RizO C>
(IV)
O X
wherein E is as defined above; Rlz is C1-C6 alkyl and X is
halogen; with a compound of formula (V)
R
(V)
R1
CA 02283184 1999-09-10
WO 98140344 PCT/EP98/00883
_ g -
wherein R and R1 are as defined above, thus obtaining a
compound of formula (I) wherein R3 is C1-C6 alkoxy and RZ is
hydrogen; or
c) deacylating a compound of formula (VI)
O BOC p
I
N
R ~ ~ (VI)
COOCH3
R1
wherein R and R1 are as defined above and BOC means tert-
butoxycarbonyl, thus obtaining a compound of formula (I)
wherein Rz is -NHCOR9 in which R9 is tert-butoxy and R3 is
methoxy and E is an unsubstituted C1 alkylene chain; and, if
desired converting a compound of formula (I) into another
compound of formula (I), and/or, if desired, converting a
compound of formula (I) into a salt thereof, and/or, if
desired, converting a salt of a compound of formula (I) into
a free compound of formula (I), and/or, if desired,
separating a mixture of isomers of a compound of formula (I;
into the single isomers.
The above process-variants a), b) and c) are analogy processes
which can be carried out according to well know methods i:n the
art.
The reaction of a compound of formula (II) with a compound of
formula (III) can be carried out, for example, in a suitable
solvent such as, e. g. diethyl ether, in the presence of a
suitable metal complex, e.g palladium (II) diacetate, at a
temperature ranging from about -78°C to room temperature, for a
time ranging from about 1 hours to about 24 hours.
In a compound of formula (IV) the halogen atom X is preferably
chlorine or bromine.
The reaction of a compound of formula (IVj with a compound of
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- g _.
formula (V) can be carried out according to known methods; for
example, following the procedure z-eported in: Sommerville L.F.,
Organic Synthesis, Coll. Vol. 2, 81, (1943); Child R.G.,
Arzneim.-Forsch./Drug Res.,30, 695-702, (1980); QuaTlich G.J.,
J. Org. Chem., 55, 4971-4973 (1990); Thyes M., J. Med. Chem.,
26, 800-807 (1983); Hester J.B., J. Med. Chem., 34, 308-315
(1991); and De Saaqui-Sannes, Pharm. Acta Helv., 66, 7, 189-192
(1991) .
For example, this reaction can be performed in the presence of
a suitable Lewis acid catalyst, :in an inert solvent such as,
e.g., dichloromethane or 1,2-dichloroethane, or in an
appropriate aromatic hydrocarbon :such as, e.g., chlorobenzene,
nitrobenzene or in an excess of a compound of formula (V)
itself; optionally in the presence of a co-solvent, e.g.
nitromethane.
A suitable Lewis acid may be, e.g. anhydrous aluminium
trichloride, anhydrous zinc dichloride, typically anhydrous
aluminium trichloride.
Deacylation of a compound of formmla (VI) can be a.ccom~lished
by treatment with a suitable deacylating agent, e.g. hydrazine
hydrate, in a suitable solvent, e.g. anhydrous methanol. The
reaction may be carried out at a temperature ranging from about
0 to about 30°C, for a time between about 0.5 and about 24
hours.
Also the optional conversion of a compound of formula ( I ) into
another compound of formula (I) ca:n be carried out according to
known methods.
For example, hydrolysis of a compound of formula ( I ) wherein R3
is C1-C6 alkoxy to obtain a compound of formula ( I ) wherein R3
is hydroxy can be carried out according to well known methods
in the art . For instance, this reaction can be performed in an
aqueous or hydroalcoholic alkali solution, for example sodium
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 10 -
hydroxide, at a concentration ranging between 0.01 and 12N, at
a temperature ranging from -20°C to reflux temperature or by
acid hydrolysis, for instance, using an aqueous solution of
hydroholic acids, typically hydrochloric acid, in a suitable
solvent, e.g. acetic acid, at a temperature ranging from about
0°C to reflux temperature.
A compound of formula (I) wherein R3 is hydroxy, can be
converted in another compound of formula (I) wherein R3 is C1-C6
alkoxy, phenoxy or benzyloxy, by conventional alkylating
methods, e.g. by treatment with a suitable alkylating agent,
preferably a iodo derivative, in the presence of a base, e.g.
potassium bicarbonate, in a suitable solvent, e.g.
dimethylformamide, at a temperature ranging from about 0°C to
about 60°C.
A compound of formula (I) wherein R3 is hydroxy, can be
converted into another compound of formula (I), wherein R3 is
-N (R.,RB ) by conventional methods , a . g . methods employed usually
in the chemistry of peptides. In particular, a compound of
formula ( I ) wherein R3 is a N (R-,Re ) wherein R., and R~ are both
2G hydrogen can be converted into a compound of formula (I,'
wherein R~ and Re are, each independently, C1-C6 alkyl, benzyl or
phenyl, by alkylative procedures known in the literature.
The optional salification of a compound of formula (I) as well
as the conversion of a salt into the free compound and the
separation of a mixture of isomers into the single isomers may
be carried out by conventional methods.
As stated above, the compounds of the invention have asymmetric
carbon atoms and can have E/Z isomerism. Accordingly, they can
be synthesized either as a mixture of isomers and then the
desired isomer is separated by conventional techniques, or
synthesis can be carried out by known stereospecific processes
to obtain a single isomeric compound.
CA 02283184 1999-09-10
WO 98/40344 PG.i.~p9
- 11 --
A compound of formula (II), as defined above can be obtained,
for instance, by reacting a compound of formula (VII)
OH
R
,O
R (VII)
i
wherein
R and R1 are as defined above, with diazomethane, for example,
in a suitable solvent such as, e.g. diethyl ether, at a
temperature varying between -78°C and room temperature, for a
time ranging between 1 and 24 hour:.
Compounds of formula (III) and (VII) are known compounds or can
be prepared according to known procedures.
A compound of formula (IV) can he obtained by a multi-step
process comprising the reaction of a compound of formula (VIII)
E
i~ (VIII)
COOR12 COOR1~
wherein R1~ and E are as defined above, with an alkali agent to
obtain a compound of formula (IX)
(IX)
COORIZ COOH
wherein R12 and E are as defined above.
A compound of formula (IX) can then be reacted with a
halogenating agent to obtain a compound of formula (IV) as
defined above.
The reaction of a compound of formula (VIII) to obtain a
compound of formula (IX) can be accomplished by basic
hydrolysis, i.e using an alcoholic solution of an alkali metal
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 12 -
hydroxide, typically a potassium hydroxide solution in suitable
alkoholic medium, i.e. methanol, at a suitable temperature,
e.g. between 0 and 55°C, for a suitable time, e.g. 2-24 hours.
The reaction of a compound of formula (IX) to obtain-a compound
of formula (VI) can be accomplished in a suitable halogenating
agent, i.e. oxalyl chloride or bromide or thionyl chloride,
typically oxalyl chloride, in the presence or the absence of a
solvent, at a suitable temperature, e.g. 0-40°C, for a suitable
time, e.g. 1-6 hours.
The compounds of formula (V) and (VIII) are known compounds.
A compound of formula (VI) can be obtained by a multi-step
process comprising acid oxidation of a compound of formula (X)
O
R
w ~ (X)
R1
wherein R and Rl are as defined above, to obtain a compound of
formula (XI)
O
R
H
\ ~ (XI)
O
R1
wherein R and R1 are as defined above.
Acid oxidation of a compound of formula (X) can be accomplished
for instance by using a dimethylsulfoxide (DMSO) solution of
concentrated hydrobromic acid, e.g. aqueous 48o hydrobromic
acid.
The reaction may be carried out at a temperature ranging from
about 25 to about 100°C, for a time between about 2 and about
48 hours.
CA 02283184 1999-09-10
WO 98/40344 p~~p~8~
- 13 -
A compound of formula (XI) can be then converted into a
compound of formula (XII)
i
O N~ y
R
\ ~ (XII)
R1
wherein R and R1 are as defined above,by treatment with a
5 suitable hyppuric acid derivative in the presence of a suitable
acylating agent, e.g. acetic anhydride. The reaction may be
carried out at a temperature ranging from about 50 to about
200°C, for a time between about 0.5 and about 24 hours.
A compound of formula (XII) , if desired, can be converted into
10 its isomer of formula (XIIa)
0 O
O
R
N
(XIIa)
R1
The conversion of a compound of formula (XII) into a compound
of formula (XIIa) can be accomplished using a suitable
hydrohalic acid solution, e.g. 48% aqueous hydrobromic acid
saturated with anhydrous hydrogen bromide gas, at a suitable
temperature, e.g. between about -20 and about 25°C, for a
suitable time, e.g. 15 minutes to 24 hours.
Cyclopropanation of a compound of formula (XII) or (XIIa)
provides a compound of formula (XIII) and (XIIIa), respectively
CA 02283184 1999-09-10
WO 98/40344 PGT/EP98/00883
- 14 -
O
O N~ O O
R R _
I (XIII) ~ w ~ N (XIIIa)
O
/ R
R i
i
wherein R and R1 are as defined above.
The reaction can be accomplished by treatment of a compound of
formula (XII) or (XIIa), respectively, with an ethereal
solution of diazomethane. The reaction can be carried out at a
temperature ranging from about -78 and about 25°C, for a time
between about 2 and about 48 hours.
A compound of formula (XIII) or {XIIIa) is then converted into
a compound of formula (XIV) or (XIVa),
respectively
/
O O
R O HN O R O
(XIV) ~ N I ~ (XIVa)
O
Ri Ri
wherein R and R1 are as defined above.
The reaction can be accomplished by treatment with
dimethylaminopyridine (DMAP) in methanol. The reaction may be
carried out at a temperature ranging from about 0 to about
50°C, for a time between about 10 minutes and 48 hours.
Subsequent reaction of a compound of formula (XIV) or (XIVa)
with di-t-butyl-carbonate and DMAP in a suitable solvent, e.g.
anhydrous dichloromethane provides a compound of formula (VI),
which can be represented by the respective two isomeric
CA 02283184 1999-09-10
WO 98/40344
- 15 -
compounds of formula (XV) and (XVa.)
Boc O
O N O O
R O R -O
w ~ ~ (XV) y w (XVa)
O ~~ B
/ O / oc
R1 R~.
i
wherein R and R1 are as defined above.
The reaction can be carried out at a temperature ranging from
about 0 to about 50°C, for a time between about 1 and 24 hours.
The isomeric compounds of formula (XV) and (XVa), for
convenience, are herein represented as a compound of formula
(VI) .
The compounds of formula (X) and the above hyppuric acid
derivatives are known compounds.
When in the compounds of the invention and the intermediate
thereof groups are present which may interfere with the
reaction they may be protected be:Eore the reaction takes place
and then deprotected at the end of the reaction.
For instance, hydroxy, amino an.d/or carboxy groups may be
protected and then deprotected according to the common
techniques known from the peptide chemistry.
The compounds of the invention are active as kynurenine-3
hydroxylase enzyme inhibitors and therefore are useful in the
prevention and/or treatment of neuropathological processes,
related to a deranged production of quinolinic acid and/or 3-
hydroxykynurenine due to excessive activation of neuro-
transmission mediated by excitatory amino acid receptors and/or
oxidative stress. Examples of such neuropathological processes
CA 02283184 1999-09-10
WO 98/40344 pCT~~8~~g83
- 16 -
are neurodegenerative pathologies including, e.g. Huntington's
chorea, Alzheimer's disease, Parkinson's disease, olivoponto
cerebellar atrophy, non-Alzheimer's demential, including the
dementia like syndrome caused by Acquired Immunodeficiency
Syndrome (AIDS), multi-infarctual dementia, cerebral
amyotrophic lateral sclerosis, cerebral ischemia, cerebral
hypoxia, spinal and head trauma and epilepsy.
A human or animal in need of a kynurenine-3-hydroxylase
enzyme inhibitor can thus be treated by a method which
comprises the administration thereto of a therapeutically
effective amount of a compound of the invention or a salt
thereof. The condition of the human or animal can thereby be
improved.
The efficacy of the compounds of the invention in the
inhibition of the enzyme kynurenine-3-hydroxylase was evaluated
e.g., in rat liver mitochondrial extract following the method
reported below, according to the procedure described in
"Analytical Biochem. (1992), 205, 257-262", with minor
modifications.
The assay for kynurenine 3-hydroxylase is based on the
enzymatic synthesis c~ tritiated water during the hydroxylation
reaction. Radiolabeled water was quantified following selective
adsorption of the isotopic substrate and its metabolite with
activated charcoal.
Rat liver mitochor_drial extract was used as enzymatic
preparation for this assay.
The assay for kynurenine 3-hydroxylase activity was carried out
at 37°C for a time c~ 30 min. The reaction mixture of a total
volume of 301 was constituted of 44 ~.g of suspended extract,
100 mM Tris/Cl buffer pH 8.1, 10 mM EDTA, 100 mM KCl, 0.8 mM
NADPH, 0.025 mM L-Kynurenine, 0.3 uCi L-(3,5-3H)Kynurenine (10
CA 02283184 1999-09-10
WO 98J40344 PGT/EP9~00883
- 17 ._
Ci/mmol) and 3 ~l of different concentration of inhibitor
solutions. After the incubation, the reaction was terminated by
the addition of 300 ~1 of 7.5'% (W/v) activated charcoal,
vortexed and centrifuged for 7 min..
A 75 ~.1 aliquot of supernatant was transferred to optiplate and
200 ~1 of liquid scintillation added. The optiplates were
vortexed and the radioactivity counted in a scintillation
counter.
The obtained results, which have been reported in the following
Table 1, demonstrate the efficacy of a representative compound
of the invention (E)-2-(3,4-dichlorobenzoyl)-cyclopropane-1-
carboxylic acid (internal code PNU-165853).
Table
KYN-3-OH inhibition
Compound IC
so
PNU-165853 0.18 ~M
The dosage level, suitable for administration to a mammal,
e.g. to humans, depends on the age, weight, conditions of the
patient and on the administration route; for example, the
dosage adopted for oral administi.-ation, for instance for the
representative compound of the invention PNU 165853, may range
from about 10 to about 500 mg pro dose, from 1 to 5 times
daily.
The compounds of the invention can be administered in a
variety of dosage forms, e.g. orally, in the form of tablets,
capsules, sugar or film coated tablets, liquid solutions or
suspensions; rectally in the form of suppositories;
parenterally, e.g. intramuscolarly, or by intravenous and/or
intrathecal and/or intraspinal injection or infusion.
CA 02283184 1999-09-10
WO 98/40344 PG"T/EP98/00883
- 18 -
The invention includes also pharmaceutical compositions
comprising a compound of formula (I) or a pharmaceutically
acceptable salt thereof in association with a pharmaceutically
acceptable excipient (which can be a carrier or a diluent).
The pharmaceutical compositions containing the compounds of the
invention are usually prepared following conventional methods
and are administered in a pharmaceutically suitable form.
For example, the solid oral forms may contain, together with
the active compound, diluents, e.g. lactose, dextrose,
saccharose, sucrose, cellulose, corn starch or potato starch;
lubricants, e.g. silica, talc, stearic acid, magnesium or
calcium stearate, and/or polyethylene glycols; binding agents,
e.g. starches, arabic gum, gelatin, methylcellulose,
carboxymethylcellulose or polyvinyl pyrrolidone; disaggregating
agents, e.g. a starch, alginic acid, alginates or sodium starch
glycolate; effervescing mixtures; dyestuffs; sweeteners;
wetting agents such as lecithin, polysorbates, laurylsulphates;
and, in general, non-toxic and pharmacologically inactive
substances used in pharmaceutical formulations. Said
pharmaceutical preparations may be manufactured in known
manner, for example, by means of mixing, granulating,
tabletting, sugar-coating, or film-coating processes.
The liquid dispersions for oral administration may be e.g.
syrups, emulsions and suspensions.
The syrups may contain as carrier, for example, saccharose or
saccharose with glycerine and/or mannitol and/or sorbitol.
The suspensions and the emulsions may contain as carrier, for
example, a natural gum, agar, sodium alginate, pectin,
methylcellulose, carboxymethylcellulose, or polyvinyl alcohol.
The suspension or solutions for intramuscolar injections may
contain, together with the active compound, a pharmaceutically
acceptable carrier, e.g. sterile water, olive oil, ethyl
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98100883
- 19 -
oleate, glycols, e.g. propylene glycol, and, if desidered, a
suitable amount of lidocaine hydrochloride. The solutions for
intravenous injections or infusions may contain as carrier, for
example, sterile water or preferably they may be in the form of
sterile, acqueous, isotonic saline solutions or they may
contain as a carrier propylene glycol.
The suppositories may contain togesther with the active compound
a pharmaceutically acceptable <:arrier, e.g. cocoa butter,
polyethylene glycol, a polyoxyethylene sorbitan fatty acid
ester surfactant or lecithin.
The following examples illustrate but do not limit the
invention.
Example 1
Preparation of 3,4-d;rh~nrnhPn~ ~,~ chlor;d
A suspension of 3,4-dichlorobenzo:ic acid (10 g, 52.35 mmol) in
thionyl chloride (50 ml) was refluxed for 12 hours under
magnetic stirring and a nitrogen atmosphere. Thionyl chloride
was removed under vacuum and the solid was washed with
anhydrous benzene (3 x 50 ml? and dried under reduced pressure
(0.1 mm/Hg), giving 10.9 g of 3,4-dichlorobenzoyl chloride
(100%) .
Analogously, the following products can be prepared:
3-chlorobenzoyl chloride;
3-fluorobenzoyl chloride; and
3-bromobenzoyl chloride.
CA 02283184 1999-09-10
V1~0 98140344 PCT/EP98/00883
- 20 -
Example 2
-a-
A solution of 3,4-dichlorobenzoyl chloride (10.9 g, 52.35 mmol)
in anhydrous benzene (55 ml) was added over 1 h to a
magnetically stirred solution of diazomethane (370 ml
containing 287.9 mmol of diazomethane), maintained at a
temperature between -10°C and -15°C, under nitrogen atmosphere.
After 3 hours at -15°C the resulting pale yellow aolid was
filtered under reduced pressure and washed with anhydrous ethyl
ether (2 x 50 ml). 10.9 g of 3,4-dichlorophenyl-a-diazo-methyl
ketone were obtained (97.8%) (m. p. 94.5-95.5 °C); IR (CHC13;
umax = 2110 Cm 1 )
Analogously, the following products can be prepared:
3-chlorophenyl-a-diazo-methyl ketone;
3-fluorophenyl-a-diazo-methyl ketone; and
3-bromophenyl-a-diazo-methyl ketone.
Example 3
Preparation of t butvl (E) 2-(3,4-dichlorobenzoyl~-cvclopropyl
1 carboxyl ate and t-butyl ( Z) 2 - ( 3 4 -di~'~lorobenzoy' ; -.
~,y~l oprQp~l-1-carbox~rlate .
To a suspension containing dichloromethane (30 ml), t-
butylacrylate (5.96 ml, 40.71 mmol) and Pd(II)(OAc)z,
maintained under magnetic stirring and argon atmosphere, a
solution of 3,4-dichlorophenyl-a-diazo-methyl ketone (2.5 g;
11.63 mmol) in dichloromethane (200 ml) was added during 8 h.
After 3 h at room temperature, dichloromethane was removed
under reduced pressure and the mixture was submitted to flash
chromatography. Elution with light petroleum/diethyl ether
90:10 afforded t-butyl (E)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-Carboxylate (1 g; 27a). Following elution with light
CA 02283184 1999-09-10
WO 98/40344 p~.~~~~
- 21 -
petroleum/diethyl ether 80:20 t-butyl (Z)-2-(3,4-
dichlorobenzoyl)-cyclopropyl-1-car_boxylate (0.4, 11%) was
recovered.
1H-NMR (CDC13) [(E)-t-butyl-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-carboxylate]: 8 1.3-1.7 (m, 11H, COOt-Bu, 3-CHZ}; 2.3-2.5 (m,
1H, 2-CH, J=7 Hz); 2.9-3.1 (m, 11~, 4-CH, J=7 Hz); 7.5 (d, 1H,
5'-CH, J=7.5 HZ); 7.9 (dd, 1H, 6"-CH, Jo=8.5 Hz, Jm=2 HZ); 8.2
(d, 1H, 2' -CH, Jm=2 Hz) .
13C-NMR (CDC13) ((E)-t-:butyl-2-(3,4-dichlorobenzoyl)-
cyclopropyl-1-carboxylate]: 8 18.02; 24.68; 25.83; 28.06;
127.14; 130.06; 130.67; 133.27; 136.27; 137.92; 174.36; 195.04.
1H-NMR (CDC13) ((Z)-t-butyl-2-(3,4-dichlorobenzoyl)-cyclopropyl
1-carboxylate]: 8 1.3-1.7 (m, lOH, COOt-Bu, 3-(H)CH); 1.7-1.9
(m, 1H, 3-(H)CH, J=7 Hz); 2.3-2.~6 (m, 1H, 2-CH, J=7 Hz); 2.7
2 .8 (dd, 1H, 4-CH, J=8.4 Hz) ; 7 . 5 (d, 1H, 5' -CH, J=7. S Hz) ; 7. 8
(dd, 1H, 6' -CH, Jo=8.5 Hz, Jrt,=2 HZ) ; 8.1 (d, 1H, G' -CH, J~=2
Hz) .
13C-NMR (CDC13) ( (Z) -t-:butyl-2- (3, 4-dichlorobenzoyl) -
cyclopropyl-1-carboxylate]: b 12.1'7; 24.08; 26.09; 27.72; 81.11;
127.29; 130.26; 130.73; 133.15; 136.91; 137.52; 168.75; 192.23.
Analogously, the following products can be prepared:
(E)t-butyl-2-(3-chlorobenzoyl)-cyc:lopropyl-1-carboxylate;
(E)t-butyl-2-(3-fluorobenzoyl)-cyc:lopropyl-1-carboxylate;
(E)t-butyl-2-(3-bromobenzoyl)-cycl.opropyl-1-carboxylate;
(Z)t-butyl-2-(3-chlorobenzoyl)-cyc:lopropyl-1-carboxylate;
(Z)t-butyl-2-(3-fluorobenzoyl)-cyc:lopropyl-1-carboxylate; and
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 22 -
(Z)t-butyl-2-(3-bromobenzoyl)-cyclopropyl-1-carboxylate.
Example 4
prep~rarion of (E) -2- (3,4-dichlorobenz~rl) -c~rclopropyl1-
carboxylic acid.
To a solution of t-butyl (E)-2-(3,4-dichlorobenzoyl)-
cyclopropyl-1-carboxylate (0.297 g, 0.943 mmol) in ethyl
acetate (25 ml), maintained under magnetic stirring, 37%
hydrochloric acid (5 ml) was added. After 2 h at room
temperature, the mixture was diluted with water (5 ml). The
organic phase was separated, washed with brine (2 x 5 ml),
dried over anhydrous sodium sulfate and concentrated under
vacuum. The resulting mixture was then purified by means of
flash chromatography using dichloromethane/methanol 0=loo as
eluent. (E)-2-(3,4-dichlorobenzoyl)-cyclopropyl-1-carboxylic
acid (56.8 mg; 41.30) were obtained.
1H-NMR (CDC13) [(E)-2- (3,4-dichlorobenzoyl)-cyclopropyl -1-
carboxylic acid): 1.6-1.8 (t, 2H, 3-CH2, J=6 Hz) ; 2.3-2.5 (m,
b
1H, 2-CH, J=7 Hz); 3.1-3.2 (m, 1H, 4-CH, J=7 Hz); 7.5 (d, iH,
5'-CH, J=7 .5 Hz); 7.9 (dd,1H, 6'-CH, Jo=8.5 Hz, Jm=2 Hz); 8.2
(d, 1H, 2' -CH, J,~=2Hz) .
1~C-NMR (CDC13) ((E)-2-(3,4-dichlorobenzoyl)-cyclopropyl-1-
carboxylic acid): b 18.02; 24.68; 25.83; 127.14; 130.06; 130.67;
133.27; 136.37; 137.92; 174.36; 195.04.
Analogously, the following products can be prepared:
(E)-2-(3-chlorobenzoyl)-cyclopropyl-1-carboxylic acid;
(E)-2-(3-fluorobenzoyl)-cyclopropyl-1-carboxylic acid; and
(E)-2-(3-bromobenzoyl)-cyclopropyl-1-carboxylic acid.
CA 02283184 1999-09-10
WO ~PCT/EP98100883
- 23 -
Example 5
~reoaration of (Z) -2- (3,4-dichlnrnhPn~n; ~~_) -c~,~~ro~yl-1_
- carboxylic acid.
To a solution of (Z)t-butyl-2-(3,4-dichlorobenzoyl)
cyclopropyl-1-carboxylate (0.06 g, 0.188 mmol) in ethyl acetate
(5 ml), maintained under magnetic stirring, 37o hydrochloric
acid (1 m1) was added. After 2 h at room temperature, the
mixture was diluted with water (5 ml). The organic phase was
separated, washed with brine (2 x: 5 ml), dried over anhydrous
sodium sulfate and concentrated under vacuum. The resulting
mixture was then purified by means of flash chromatography
using dichloromethane/methanol 0-15o as eluent. (Z) 2- (3,4-
dichlorobenzoyl)-cyclopropyl-1-carboxylic acid (15 mg; 30.8%)
was obtained.
1H-NMR (CDC13) [(Z) -2- (3,4-dichlorobenzoyl) -cyclopropyl-1
carboxylic acid: b 1.3-1.7 (t, 2H, 3-(H)CH, J=6 Hz); 1.7-1.9
(m, 1H, 3-(H)CH); 2.3-2.5 (m, 1H, 2-CH); 2.6-2.8 (m, 1H, 4-CH);
7.4-7.5 (d, 1H, 5'-CHJ; 7.6-7.8 (m, 1H, 6'-CH); 8.2 (d, 1H, 2'
CH) .
13C-NMR (CDC13) ((Z) -2- (3,4-d_~chlorobenzoyl) -cyclopropyl-1-
carboxylic acid: 8 13.96; 24.08; 26.09; 127.79; 130.30; 130.69;
133.14; 136.63; 138.30; 171.15; 194.23.
Analogously, the following products can be prepared:
(Z)-2-(3-chlorobenzoyl)-cyclopropyl-1-carboxylic acid;
(Z)-2-(3-fluorobenzoyl)-cyclopropyl-1-carboxylic acid;
(Z)-2-(3-bromobenzoyl)-cyclopropyl-1-carboxylic acid; and
(Z)-2-(3,4-difluorobenzoyl)-cyclopropyl-1-carboxylic acid.
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 24 -
Example 6
( ~ R a,-phenylalXcm y1 ~ -c~arnoxamiae ana m r ~ 1~ . GJ I -a- »,~-
dichlorobenzoy~)-cyclonropyl-1-(1R-a-phenylalvcinovl)-
carboxamide.
(E)-2-(3,4-dichlorobenzoyl)-cyclopropyl-1-carboxylic acid
(0.175 g, 0.68 mmol) was dissolved in oxalylchloride (6 ml) and
left at room temperature for 2 h, under magnetic stirring and
argon atmosphere. Oxalylchloride was removed by rotary
evaporation and the corresponding 2-(3,4-dichlorobenzoyl)-
cyclopropyl-1-carboxylic acid chloride, dissolved in anhydrous
dioxane (1.5 ml), was added to a dioxane (3.5 ml) solution
containing R-(-)-phenylglycinol (0.093 g, 0.68 mmol) and
triethylamine (0.1 ml, 0.7 mmol), maintained under magnetic
stirring and argon atmosphere at 10°C. After 1 h at 10°C the
reaction mixture was treated with 37% hydrochloric acid (1 ml)
and ethyl acetate (10 ml). The organic phase was separated,
washed with water (3 x 10 ml), with brine (1 x 10 ml), dried
over anhydrous sodium sulfate and concentrated under vacuum.
The residue was submitted to medium pressure chromatography on
a silica gel Lobar column (60-43 ~.m) using light
petroleum/ethyl acetate 30:70 as eluent and two amides were
separated. According to the literature) , the first amide
eluted, ((a)DZO - -102.6, c = 0.5 CHC13) , can be assigned as (E) -
(1R,2R)-2-(3,4-dichlorobenzoyl)-cyclopropyl-1-)1R-a-
phenylglycinoyl)-carboxamide and the second one, ((a,]Dao - +56, c
- 1 CHC13) , as (E) - (1S, 2S) -2- (3, 4-dichlorobenzoyl) -cyclopropyi-
1-)1R-cx-phenylglycinoyl)-carboxamide (86%).
~ (a) G. Helmchen, G. Nill, D. Flockerzi, W. Schiihle, M.S.K. Youssef, Angew.
Chem. Inr. Ed. Engl., 18, 1979,
62-63. (b) G. Helmchen, G. Nill, D. Flockerzi, M.S.K. Youssef, Ange~s~. Chem.
Int. Ed Engl., 18, 1979, 63-6~.
(c) G. Hehnchen, G. Nill" Angew. C'henr. Int. Ed. Engl., 18, 1979, 65-66.
CA 02283184 1999-09-10
WO 98/40344 p~.~p~~
- 25 -
1H-NMR (CDC13) [(E)-(1R,2R)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-(1R-a-phenylglycinoyl)-carboxamide]: 8 1.4-1.5 (t, 2H, 3-
(H)CH); 1.5-1.6 (m, 1H, 3-(H)CH); 2.3-2.5 (m, 1H, 2-CH); 2.9-
3.2 (m, 1H, 4-CH and OH); ); 3.8-3.9 (d, 1H, a-CHZOH)); 5.0-5.1
(m, 1H, a-CH); 6.9-7.0 (bd, 1H, NH); 7.2-7.4 (m, 5H, a-C6H5);
7.2-7.4 (m, 1H, 5'-CH, J= 7.5 Hz); 7.9 (dd, 1H, 6'-CH, Jo=8.5
Hz, Jm=2 HZ) ; 8.2 (d, 1H, 2' -CH, Jm=2 Hz) .
13C-NMR (CDC13) ( (E) - (1R, 2R) -2- (3, 4-dichlorobenzoyl) -
cyclopropyl-1-(1R-a-phenylglycinoy:L)carboxamide]: 8 18.07;
25.54; 26.84; 29.64; 56.21; 66.29; 126.67; 127.31; 127.96;
130.25; 130.78; 133.46; 136.52; 138.07; 138.69; 170.72; 195.97.
1H-NMR (CDC13) ((E)-(1S,2S)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-(1R-a-phenylglycinoyl)-carboxamide]: 8 1.4-1.5 (m, 1H, 3-
(H) CH) ; 1 . 5-1 . 6 (m, 1H, 3- (H) CH) ; :2 . 35-2 .45 (m, 1H, 2-CH) ; 2 . 9-
3.1 (m, 1H, 4-CH and OH); 3.8-3.9 (d, 1H, a-CHzOH)); 4.9-5.0
(m, 1H, a-CH) ; 6. 9-7. 0 (bd, 1H, rdH) ; 7.1-7.3 (m, 5H, a-C6H~) ;
7.4 (d, 1H, 5'-CH, J= 7.5 Hz); 7.6-7.7 (dd, 1H, 6'-CH, J~=8.5
HZ, Jm=2 HZ) ; 7. 9 (d, 1H, 2' -CH, Jm=2 Hz) .
13C-NMR (CDC13) ( (E) - ( 1S, 2S) -2- (3, 4-dichlorobenzoyl ) -
cyclopropyl-1-(1R-a-phenylglycinoy:L)carboxamide]: 8 18.04;
25.44; 26.87; 29.64; 56.22; 66.29; 126.70; 127.31; 127.96;
128.88; 130.25; 130.74; 133.43; 136.49; 138.03; 138.76; 170.66;
195.92.
Example 7
carbox,rl_,'_c ac,'_d.
To a solution of (E){1R,2R)-2-(3,4-dichlorobenzoyl)-
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 26 -
cyclopropyl-1-(1R-a-phenylglycinoyl)-carboxamide (50 mg, 0.123
mmol) in dioxane (3.5 ml), maintained under magnetic stirring,
37o hydrochloric acid was added (1.5 ml) and the mixture was
left at 50°C for 7 h. After cooling, the reaction mixture was
diluted with ethyl acetate (40 ml). the organic phase was
washed with water (6 x 3 ml), brine (1 x 4 ml), dried over
anhydrous sodium sulfate and concentrated under vacuum. The
residue was submitted to flash chromatography using
dichloromethane/methanol 0-10 as eluent. (E)(1R,2R)-2-(3,4-
dichlorobenzoyl)-cyclopropyl-1-carboxylic acid, ([a]DZ° - -86, c
- 0.36, CHC13) was yielded (20 mg, 750).
1H-NMR (CDC13) [(E)(1R,2R)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-carboxylic acid]: b 1.5-1.7 (t, 2H, 3-CHI, J=6 Hz); 2.3-2.5
(m, 1H, 2-CH); 3.0-3.2 (m, 1H, 4-CH, J=7 Hz); 7.4-7.5 (d, 1H,
5'-CH, J=7.5 Hz); 7.7-7.9 (dd, 1H, 6'-CH, J;,=8.5 Hz); 7.9-8.1
(s, 1H, 2' -CH) .
13C-NMR (CDC13) [(E)(1R,2R)-2-(3,4-dichlorobenzoyl)-cycloprcpyl-
1-carboxylic acid]: b 18.62; 25.01; 26.22; 127.21; 130.16;
130.82; 133.52; 136.25; 138.28; 178.11; 194.59.
Example 8
Preparation of (E)(1S 2S)-2-(3~ 4-dichlorobenzoyl)-cyclopropyl-
1-carboxylic acid.
To a solution of (E)(1S,2S)-2-(3,4-dichlorobenzoyl)-
cyclopropyl-1-(1R-a-phenylglycinoyl)-carboxamide (50 mg, 0.123
mmol) in dioxane (3.5 ml), maintained under magnetic stirring,
37o hydrochloric acid was added (1.5 ml) and the mixture was
left at 50°C for 7 h. After cooling, the reaction mixture was
diluted with ethyl acetate (40 ml). the organic phase was
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
_ 27 __
washed with water (6 x 3 ml), brine (1 x 4 ml), dried over
anhydrous sodium sulfate and concentrated under vacuum. The
residue was submitted to flash chromatography using
dichloromethane/methanol 0=10 as eluent. (E)-(1R,2R)-2-(3,4-
dichlorobenzoyl)-cyclopropyl-1-carboxylic acid, (([a)DZ° - +77, c
- 0.65, CHC13) was yielded (13 mg, 49%).
1H-NMR (CDC13) [(E)(1S,2S)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-carboxylic acid]: S 1.6-1.8 (t, 2H, 3-CH2, J=7 Hz); 2.3-2.5
(m, 1H, 2-CH, J=7 Hz); 3.1-3.2 (m, 1H, 4-CH, J=7 Hz); 7.5 (d,
1H, 5'-CH, J=7.5 HZ); 7.8 (dd, 1H, 6'-CH, Jo=7.5 HZ, Jm=1.5 HZ);
8.1 (S, 1H, 2' -CH, Jm=1 . 5 Hz) .
isC-NMR (CDC13) [(E)(1S,2S)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
1-carboxylic acid): b 18.48; 24.68; 26.23; 127.25; 130.24;
130.86; 133.57; 136.38; 138.25; 177.61; 194.45.
Example 9
Prex>aration of (E) -1-methox~rcarbonyl -cyclopro~ne-2-carboxylic
acid.
A solution of potassium hydroxide (0.354 g, 6.32 mmol) in dry
methanol (4 ml) was dropped in one hour to a solution of
(E)1,2-cyclopropandicarboxylate dimethyl ester (1 g, 6.32 mmol)
in dry methanol (4 ml), maintained under magnetic stirring at
room temperature. The resulting solution was heated to 55°C
during 12 hours and then was poured into water ( l0 ml ) . After
acidification with 10% hydrochloris acid , the aqueous phase
was extracted with ethyl acetate: (10 x 10 ml). The organic
extracts were washed with brine (1 x 10 ml), dried over
anhydrous sodium sulfate and concentrated under vacuum to yield
an oil that was purified by flash.-chromatography. Elution with
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 28 -
dichloromethane/methanol (95:5) afforded the pure title
compound (0.556 g; 61%).
1H-NMR (CDC13) ; b: 1.5 (t, 2H, 2-CH2, J=8 Hz) ; 2.1-2.3 (m, 2H,
1-CH and 3-CH); 3.7 (s, 3H, COOCH3); 10.35 (bs, 1H, COOH).
13C-NMR (CDC13); 8: 14.94; 21.71; 22.24; 51.88; 171.74; 176.60.
Analogously the following compounds were prepared:
(E)-1-methoxycarbonyl-cyclobutane-2-carboxylic acid,
(E)-1-methoxycarbonyl-cyclopentane-2-carboxylic acid,
(E)-1-methoxycarbonyl-cyclohexane-2-carboxylic acid,
(E)-1-methoxycarbonyl-3-methylene-cyclopropane-2-carboxylic
acid,
(E)-1-methoxycarbonyl-3-methyl-cyclopropane-1-carboxylic acid,
(E)-1-methoxycarbonyl-3,3-dimethyl-cyclopropane-1-carboxylic
acid, and
(E)-1-methoxycarbonyl-3,3-difluoro-cyclopropane-1-carboxylic
acid.
Example 10
P~-P~arati~n of (E> -1-methoxvcarbonyl-c~rcloprobane-2-carboxvlic
acid chloride.
A solution of (E)-1-methoxycarbonyl-cyclopropane-2-carboxylic
acid (0.556 g, 3.857 mmol} in oxalylchloride (20 ml} was
stirred under an argon atmosphere at room temperature for 3
hours. then the solution was concentrated under vacuum for 2
hours and the oily residue containing the titled compound was
used in the next step without further purification.
Analogously, the following products were prepared:
(E)-1-methoxycarbonyl-cyclobutane-2-carboxylic acid chloride;
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 29 -
(E)-1-methoxycarbonyl-cyclopentane~-2-carboxylic acid chloride;
(E)-1-methoxycarbonyl-cyclohexane-2-carboxylic acid chloride;
(E)-1-methoxycarbonyl-3-methylene-cyclopropane-2-carboxylic
acid chloride;
(E)-1-methoxycarbonyl-3-methyl-cyclopropane-2-carboxylic acid
chloride;
(E)-1-methoxycarbonyl-3,3-dimethyl-cyclopropane-2-carboxylic
acid chloride and
(E)-1-methoxycarbonyl-3,3-difluoro-cyclopropane-2-carboxylic
acid chloride.
Example 11
Preparat,'_on of ( E ) me h;~. 2 - ( 3 4 --dichl_o-robenzo~ll~rcloprQp~.~
~ -carbox~rl_ate .
To a magnetically stirred solution of (E)-1-methoxycarbonyl-
cyclopropane-2-carboxylic acid chloride (0.627 g, 3.857 mmol)
in 1 , 2 -dichlorobenzene ( l0 ml ) , A1.C13 ( 1 . 542 g, 11 . 57 mmol ) was
added portionwise under an argon atmpsphere at 0°C. The
resulting solution was stirred 10 minutes at 0°C then warmed to
55°C for 2 hours. The reaction mixture was quenched by addition
of ice-water (10 ml) and l0a hydrochloric acid (10 ml). the
mixture so obtained was extracted with ethyl acetate (4 x 25
ml); the combined organic phases were washed with brine (1 x 10
ml), dried over an. sodium sulfate, concentrated under vacuum.
The oily residue was purified by flash-chromatography:
elutionwith light petroleum-ethyl acetate 100:0 to 85:15 thus
affording the titled compound as an oil (0.610 mg, 580).
1H-NMR (CDC13) : 8: 1.6 (dd, 2H, 2-CH2, J=6.9 Hz) ; 2.35-2.45 (m,
1-CH); 3.1-3.2 (m, 1H, 3-CH); 3.7 (s, 3H, COOCH3); 7.55 (d, 1H,
6' -CH, J°=8 HZ) ; 7.85 (dd, 1H, ~~' -CH, J°=8H2, Jm=2 HZ) ;
8.05
(dd, 1H, 2' -CH, Jm=2 HZ) .
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 30 -
isC-NMR (CDC13) : b . 17.78; 24.39; 25.49; 51.86, 127.00; 129.87,
130.44; 133.04; 136.25; 137.56; 171.88; 194.23.
Analogously the following products were prepared:
(E)-methyl 2-(3,4-dichlorobenzoyl)-cyclobutyl-1-carboxylate;
(E)-methyl 2-(3,4-dichlorobenzoyl)-cyclopentyl-1-carboxylate;
(E)-methyl 2-(3,4-dichlorobenzoyl)-cyclohexyl-1-carboxylate;
(E)-methyl-2-(3,4-dichlorobenzoyl)-3-methylene-cyclopropane-1-
carboxylate;
(E)-methyl-2-(3,4-dichlorobenzoyl)-3-methyl-cyclopropane-1-
carboxylate;
(E)-methyl-2-(3,4-dichlorobenzoyl)-3,3-dimethyl-cyclopropane-1-
carboxylate and
(E)-methyl-2-(3,4-dichlorobenzoyl)-3,3-difluoro-cyclopropane-1-
carboxylate.
Example 12
Pre~aratinn of (E) 2-(3,4-dichlorobenzoyl)-cvclonropane- -
carboxylic acid
To a magnetically stirred solution of (E)-methyl 2-(3,4-
dichlorobenzoyl)-cyclopropyl-1-carboxylate (70 mg, 0.244 mmol)
in dry dioxane (3.5 ml), a solution of sodium hydroxide (19.5
mg, 0.488 mmol) in water (1.5 ml) was added. After 2 hours at
room temperature, the mixture was diluted with water (10 ml
and extracted with dichloromethane (2 x 2 ml). The acqueous
layer was acidified with 10% hydrochloric acid, then extracted
with ethyl acetate (4 x 5 ml), the combined organic phases were
washed with brine (1 x 5 ml), dried over an. sodium sulfate and
concentrated under vacuum to obtain the titled compound (50 mg,
79%) as a white solid.
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 31 -
1H-NMR (Acetone-d6) : 8: 1.6 (m, 2H, 2-CH2) ; 2.35-2.45 (m, 1-CH) ;
3.25 (m, 1H, 3-CH) ; 7.75 (d, 1H, 6' -CH, Jo=8 Hz) ; 8 . 05 (dd, 1H,
5' -CH, J°=8 HZ, Jn,=2 HZ) ; 8.2 (d, 1H, 2' -CH, Jm=2 HZ) .
i3C-NMR (CDC13) : b . 18.16; 25.05; 26.38; 128.79; 130.91,
131.87; 133.55; 137.91; 173.01; 195.48.
Analogously the following products were prepared:
(E)-2-(3,4-dichlorobenzoyl)-cyclobutyl-1-carboxylic acid;
(E)-2-(3,4-dichlorobenzoyl)-cyclopentyl-1-carboxylic acid;
(E)-2-(3,4-dichlorobenzoyl)-cyclohexyl-1-carboxylic acid.
2-(3,4-dichlorobenzoyl)-3-methylene-cyclopropane-1-carboxylic
acid;
2-(3,4-dichlorobenzoyl)-3-methyl-cyclopropane-1-carboxylic
acid;
2-(3,4-dichlorobenzoyl)-3,3-dimethyl-cyclopropane-1-carboxylic
acid; and
2-(3,4-dichlorobenzoyl)-3,3-difluoro-cyclopropane-1-carboxylic
acid.
Example 13
Pre . ar tion of ~, 4-dichlorophen~rlgl
To a stirred solution of 3,4-dich:loroacetophenone (1 mmol) in
DMSO (3 ml), 48% aqueous hydrobromic acid (3 mmol) was added
slowly. The solution was stirred in an open flask at 55°C. when
the starting material was consumed (24 hours), the solution was
poured into ice. The crude product was extracted into Et=Ac,
the solution was washed with water,, dried over anhydrous sodium
sulfate and concentrated under vacuum. The titled arylglyoxal
was recovered in an essentially pux-e form.
CA 02283184 1999-09-10
WO 98140344 PCT/EP98/00883
- 32 -
Analogously, the following products can be prepared:
3-chlorophenylglyoxal;
3-fluorophenylglyoxal;
3-bromophenylglyoxal; and
3,4-difluorophenylglyoxal.
Example 14
oxazol-5-one.
A mixture of 3,4-dichlorophenylglyoxal (1 mmol), powdered dry
hyppuric acid (1 mmol), powdered freshly fused sodium acetate
(1 mmol) and high-grade acetic anhydride, is heated on an
electric hot plate with constant shaking in an apparatus fitted
with a calcium chloride tube. As soon as the material has
liquefied completely, the flask is transferred to a steam bath
and heated for two hours; during this time a part of the
product separates as crystals. At the end of the heating, ethyl
alcohol is added slowly into the flask, while maintaining the
temperature below 30°C. After allowing the reaction mixture to
stand overnight, the crystalline product is filtered with
suction,. The pure azlactone is collected as a white solid.
Analogously the following products were prepared:
(Z)-2-Phenyl-4-(3-chlorobenzoylmethylene)-oxazol-5-one;
(Z)-2-Phenyl-4-(3-fluorobenzoylmethylene)-oxazol-5-one;
(Z)-2-Phenyl-4-(3-bromobenzoylmethylene)-oxazol-5-one; and
(Z)-2-Phenyl-4-(3,4-difluorobenzoylmethylene)-oxazol-5-one.
CA 02283184 1999-09-10
WO ~PCT/EP98/00883
- 33 --
Example 15
azaspiro-~~, 4~~,~~-4-en-7-on
A solution of (Z)-2-Phenyl-4-(3,4-dichlorobenzoylmethylene)
oxazol-5-one (1 mmol) in anhydrous methylene was treated with
an ethereal solution of diazomethane (10 mmol, under magnetic
stirring. The mixture was allowed to stand at room temperature
overnight, treated with anhydrous calcium chloride to destroy
exces diazomethane, filtered and concentrated under vacuum. The
resultant oil was purified by flash-chromatography. Elution
with light petroleum-ethyl acetate afforded the pure titled
derivative.
Analogously the following derivatives were prepared:
(Z)-2-(3,4-difluorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4)-
hept-4-en-7-one;
(Z)-2-(3-chlorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4)-hept-4-
en-7-one;
(Z)-2-(3-fluorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4)-kept-4-
en-7-one; and
(Z)-2-(3-bromobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4)-hept-4-
en-7-one.
Example 16
Pre~a_rat,'_on of (Z>-Me hyl 2-(3,4-di hl robenzoyl)-~-
benzamidocyclo~ropane-1-carhnx~~late_
DMAP (1 mmol) was added to a suspension of (Z)-2-(3,4-
dichlorobenzoyl)-5-phenyl-6-oxo-4-a.zaspiro-[2,4)-kept-4-en-7-
one( 1 mmol) in absolute methanol and the resulting mixture was
magnetically stirred at room tE:mperature for 35 minutes.
Methanol was removed under vacuum, the crude product was
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 34 -
treated with a mixture 1:1 of dichloromethane and 5% aq. citric
acid. the organic phase was collected and the aqueous phase was
extracted with an additional amount of dichloromethane. The
organic extracts were combined, dried over an. sodium sulfate
and concentrated to give the titled compund.
Analogously the following derivatives were prepared:
(Z)-Methyl 2-(3,4-difluorobenzoyl)-1-benzamidocyclopropane-1-
carboxylate;
(Z)-Methyl 2-(3-chlorobenzoyl)-1-benzamidocyclopropane-1-
carboxylate;
(Z)-Methyl 2-(3-fluorobenzoyl)-1-benzamidocyclopropane-1-
carboxylate; and
(Z)-Methyl 2-(3-bromobenzoyl)-1-benzamidocyclopropane-1-
carboxylate.
Example 17
prPparat ion of ( Z ) -Methyl 2 - ( 3 ,~~ -dichlorobenzo~rl ) -1- ( N-benzoyl -
N-t- utoxycarbonylamino)-cyclopropane-1-carboxylate.
Di-t-butyl dicarbonate (2 mmol) and DMAP (1 mmol) were added to
a suspension of (Z)-Methyl 2-(3,4-dichlorobenzoyl)-i-
benzamidocyclopropane-1-carboxylate (1 mmol) in anhydrous
dichloromethane and the resulting mixture was kept under
magnetic stirring in a nitrogen atmosphere at room temperature
for two hours. After evaporation of the solvent, the crude
reaction product was dissolved in dichloromethane, washed with
5o citric acid, brine and dried over anhydrous sodium sulfate.
Evaporation of the solvent yielded the pure titled compound.
Analogously the following derivatives were prepared:
(Z)-Methyl 2-(3,4-dichlorobenzoyl)-1-(N-benzoyl-N-t-
CA 02283184 1999-09-10
WO 98/40344 p~~p9g~~
- 35 ~-
butoxycarbonylamino)-cyclopropane-1-carboxylate;
(Z)-Methyl 2-(3,4-difluorobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate;
(Z)-Methyl 2-(3-chlorobenzoyl)-1-(:N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate;
(Z)-Methyl 2-(3-fluorobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate; and
(Z)-Methyl 2-(3-bromobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate.
Example 18
Hydrazine hydrate (10 mmol) was added to a magnetically stirred
suspension of (Z)-Methyl 2-(3,4-dichlorobenzoyl)-1-(N-benzoyl-
N-t-butoxycarbonylamino)-cyclopropane-1-carboxylate in
anhydrous methanol at room temperature. Stirring was continued
for one hour after which the solvent was evaporated, while
maintaining the temperature of the water bath below 30°C. The
residue was then flash-chromatoc3raphed (eluent: chloroform-
methanol; 9:1) to afford the titled compound.
Analogously the following derivatives were prepared:
(Z)-Methyl 2-(3,4-difluorobenzoyl)-1-t-butoxycarbonylamino-
cyclopropane-1-carboxylate;
(Z)-Methyl 2-(3-chlorobenzoyl)-1-t-butoxycarbonylamino-
cyclopropane-1-carboxylate;
(Z)-Methyl 2-(3-fluorobenzoyl)-1-t-butoxycarbonylamino-
cyclopropane-1-carboxylate; and
(Z)-Methyl 2-(3-bromobenzoyl)-1-t-butoxycarbonylamino-
cyclopropane-1-carboxylate.
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 36 -
Example 19
PrP~aration of (Z)-Methyl 2-(3 4-dichlorobenzovl)-1-amino-
~~cl oprc~bane-1-carbox~rlate
12 N Hydrochloric acid was added to a solution of (Z)-Methyl 2-
(3,4-dichlorobenzoyl)-1-t-butoxycarbonylamino-cyclopropane-1-
carboxylate in ethyl acetate and the resulting mixture was
magnetically stirred at room temperature for 30 minutes. The
reaction mixture was then neutralised with saturated sodium
hydrogen carbonate, the organic phase separated and the aqueous
layer was extracted with ethyl acetate. The combined organic
phase were washed with brine and dried over anhydrous sodium
sulfate. Evaporation of the solvent gave the titled methyl
ester.
Aizalogously the following derivatives were prepared:
(Z)-Methyl 2-(3-chlorobenzoyl)-1-amino-cyclopropane-1-
carboxylate;
(Z)-Methyl 2-(3-bromobenzoyl)-1-amino-cyclopropane-1-
carboxylate;
(Z)-Methyl 2-(3-fluorobenzoyl)-1-amino-cyclopropane-1-
carboxylate; and
(Z)-Methyl 2-(3,4-difluorobenzoyl)-1-amino-cyclopropane-
1-carboxylate.
Example 20
Pr~aration of (Z) -2- (3,4-dichlorobenzoyl) -1-amino-
rvc-1 pane-~ -carboxylic acid
1 N Lithium hydroxide monohyrate was added to a solution of
(Z)-Methyl 2-(3,4-dichlorobenzoyl)-1-amino-cyclopropane-1-
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98100883
- 37 -
carboxylate in dioxane and the resulting mixture was kept under
magnetic stirring at room temperature overnight. The reaction
mixture was then evaporated to dryness, the residue diluted
with water and neutralised with 1 N hydrochloric acid. Ion
exchange chromatography on Dowex ~~Ox2 200 and elution with l0%
pyridine yielded the titled compound.
Analogously the following derivatived were prepared:
(Z)-2-(3,4-difluorobenzoyl)-1-amino-cyclopropane-1-carboxylic
acid;
(Z)-2-(3-chlorobenzoyl)-1-amino-cyclopropane-1-carboxylic acid;
(Z)-2-(3-fluorobenzoyl)-1-amino-cyclopropane-1-carboxylic acid;
and
(Z)-2-(3-bromobenzoyl)-1-amino-cyclopropane-1-carboxylic acid.
Analogously starting from the alkyl esters described in all the
preceding examples the respective free 1-carboxylic acids can
be obtained.
Example 21
A suspension of (Z)-2-Phenyl-4-(3,4-dichlorobenzoylmethylene)-
oxazol-5-one in 48o hydrobromic acid, kept at 0°C and
maintained under magnetic stirring, was saturated with
anhydrous hydrogen bromide gas for 30 minutes and left in a
refrigerator overnight. The product was poured into crushed ice
and the solid (E)-azlactone was filtered, washed with ice-water
and dried over phosphorous pentoxide.
Analogously the following derivatives were prepared:
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 38 -
(E)-2-Phenyl-4-(3-chlorobenzoylmethylene)-oxazol-5-one;
(E)-2-Phenyl-4-(3-fluorobenzoylmethylene)-oxazol-5-one;
(E)-2-Phenyl-4-(3-bromobenzoylmethylene)-oxazol-5-one; and
(E)-2-Phenyl-4-(3,4-difluorobenzoylmethylene)-oxazol-5-one.
Example 22
Preparation of (E) -2- (3~4-dichlorobenzo~rl) -5-,~hen~rl-6-oxo-4-
azaspiro-f2, 4~-hept-4-en-7-one.
A solution of (E)-2-Phenyl-4-(3,4-dichlorobenzoylmethylene)-
oxazol-5-one (1 mmol) in anhydrous dichloromethane was treated
with an ethereal solution of diazomethane (10 mmol) under
magnetic stirring. The mixture was allowed to stand at room
temperature overnight, treated with anhydrous calcium chloride
to destroy excess diazomethane, filtered and concentrated under
vacuum. the resultant oil was purifird by flash-chromatography.
elution with light petroleum-ethyl acetate afforded the pure
titled derivative.
Analogously the following derivatives were prepared:
(E)-2-(3,4-difluorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4]-
hept-4-en-7-one;
(E)-2-(3-chlorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2, 4]-hept-4-
en-7-one;
(E)-2-(3-fluorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4]-hept-4-
en-7-one; and
(E)-2-(3-bromobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4]-hept-4-
en-7-one.
CA 02283184 1999-09-10
WO 98/40344 p~~~p9~g~
- 39 -
Example 23
DMAP (1 mmol) was added to a suspension of (E)-2-(3,4-
dichlorobenzoyl)-5-phenyl-6-oxo-4-azaspiro-[2,4J-hept-4-en-7-
one ( 1 mmol ) in absolute methanol and the resulting mixture was
magnetically stirred at room temperature for 30 minutes.
Methanol was removed under vacuum, the crude product was
treated with a mixture 1:1 of dichloromethane and 5% aq. citric
acid. The organic phase was collected and the aqueous phase was
extracted with an additional amount of dichloromethane. The
organic extracts were combined, dried over an. sodium sulfate
and concentrated to give the titled compound.
Analogously the following derivatives were prepared:
(E)-Methyl 2-(3,4-difluorobenzoyl)-:1-benzamidocyclopropane-1-
carboxylate;
(E)-Methyl 2-(3-chlorobenzoyl)-1-benzamidocyclopropane-1-
carboxylate;
(E)-Methyl 2-(3-fluorobenzoyl)-1-benzamidocyclopropane-1-
carboxylate; and
(E)-Methyl 2-(3-bromobenzoyl)-1-benzamidocyclopropane-1-
carboxylate.
Example 24
_r~pawaL~on oz c~~-metnvl ~-t.~,a-filchiorobenzoyG)-1-(N-benzoyl-
N-t-butoxycarbon~rl am, no) -c~~~e-1-carboxyl arP
Di-t-butyl dicarbonate (10 mmol) and DMAP (0.2 mmol) were added
to a suspension of (E)-Methyl 2-(3,4-dichlorobenzoyl)-1
benzamidocyclopropane-1-carboxylate (1 mmol) in anhydrous
tetrahydrofuran and the resulting mixture was kept under
magnetic stirring in a nitrogen atmosphere at room temperature
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 40 -
for 22 hours. After evaporation of the solvent, the crude
reaction product was dissolved in dichloromethane, washed with
5% citric acid, brine and dried over anhydrous sodium sulfate .
Evaporation of the solvent yielded a residue which was
submitted to flash chromatography: elution with light petroleum
ether-ethyl acetate afforded the pure titled compound.
Analogously the following derivatives were prepared:
(E)-Methyl 2-(3,4-dichlorobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate;
(E)-Methyl 2-(3,4-difluorobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate;
(E)-Methyl 2-(3-chlorobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate;
(Z)-Methyl 2-(3-fluorobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate; and
(E)-Methyl 2-(3-bromobenzoyl)-1-(N-benzoyl-N-t-
butoxycarbonylamino)-cyclopropane-1-carboxylate.
Example 25
prPp~rar;~n of (E)-Methyl 2-(3 4-dichlorobenzoyl)-1-t-
bmrox~yla~nino-c~rclopro~ane-1-carboxyl ate
Hydrazine hydrate (10 mmol) was added to a magnetically stirred
suspension of (E)-Methyl 2-(3,4-dichlorobenzoyl)-1-(N-benzoyl
N-t-butoxycarbonylamino)-cyclopropane-1-carboxylate in
anhydrous methanol at room temperature. Stirring was continued
for 15 minutes after which the solvent was evaporated on a
rotary evaporator while maintaining the temperature of the
water bath below 30°C. The residue was then flash-
chromatographed (eluent: chloroform-methanol; 9:1) to afford
the titled compound.
CA 02283184 1999-09-10
WO 98/40344 p~~p9g/OOgg3
- 41 -
Analogously the following derivatives were prepared:
(E)-Methyl 2-(3,4-difluorobenzoyl)-1-t-butoxycarbonylamino-
cyclopropane-1-carboxylate;
' 5 (E)-Methyl 2-(3-chlorobenzoyl)-1-t-:butoxycarbonylamino-
cyclopropane-1-carboxylate;
(E)-Methyl 2-(3-fluorobenzoyl)-1-t-:butoxycarbonylamino-
cyclopropane-1-carboxylate; and
(E)-Methyl 2-(3-bromobenzoyl)-1-t-butoxycarbonylamino-
cyclopropane-1-carboxylate.
Example 26
12 N Hydrochloric acid was added to a solution of (E)-Methyl 2-
(3,4-dichlorobenzoyl)-1-t-butoxycarbonylamino-cyclopropane-1-
carboxylate in ethyl acetate and the resulting mixture was
magnetically stirred at room tempE=_rature for 30 minutes. The
reaction mixture was then neutra=Lised with saturated sodium
hydrogen carbonate, the organic phase separated and the aqueous
layer was extracted with ethyl acetate. The combined organic
phase were washed with brine and dried over anhydrous sodium
sulfate. Evaporation of the solvent gave the titled methyl
ester.
Analogously the following derivatives were prepared:
(E)-Methyl 2-(3-chlorobenzoyl)-1-amino-cyclopropane-1-
carboxylate;
(E)-Methyl 2-(3-bromobenzoyl)-1-amino-cyclopropane-1-
carboxylate;
(E)-Methyl 2-(3-fluorobenzoyl)-1-amino-cyclopropane-1-
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 42 -
carboxylate; and
(E)-Methyl 2-(3,4-difluorobenzoyl)-1-amino-cyclopropane-
1-carboxylate.
Example 27
1 N Lithium hydroxide monohyrate was added to a solution of
(E)-Methyl 2-(3,4-dichlorobenzoyl)-1-amino-cyclopropane-1-
carboxylate in dioxane and the resulting mixture was kept under
magnetic stirring at room temperature overnight. The reaction
mixture was then evaporated to dryness, the residue diluted
with water and neutralised with 1 N hydrochloric acid. Ion
exchange chromatography on Dowex 50x2 200 and elution with l00
pyridine yielded the titled compound.
Analogously the following derivatived were prepared:
(E)-2-(3,4-difluorobenzoyl)-1-amino-cyclopropane-1-carboxylic
acid;
(E)-2-(3-chlorobenzoyl)-1-amino-cyclopropane-1-carboxylic acid;
(E)-2-(3-fluorobenzoyl)-1-amino-cyclopropane-1-carboxylic acid;
and
(E)-2-(3-bromobenzoyl)-1-amino-cyclopropane-1-carboxylic acid.
Example 28
Preparation of (El-Methyl. 2-(3~4-difluorobenzoyl)-
cyc 1 oorox~ane -1- c arbox~l at a
A solution of E-cyclopropane-1,2-dicarboxylate acid monomethyl
ester (1 g, 6.9 mmol) in oxalylchloride (20 ml) was stirred
under Argon atmosphere during 2 hours then concentrated under
vacuum. To a solution of the resulting residue in o
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 43 -
dichlorobenzene (4 ml), under magneting stirring and Argon
atmosphere at 0°C, aluminium chloride (2.76 g, 38.5 mmol) was
added portion wise during 30'. After fifteen minute the
reaction mixture was heated at 60°C for 90' and then poured
into ice-hydrochloric acid 3N. The aqueous layer was extracted
with ethyl acetate (4x50 ml). The organic phases were combined
washed with brine (1x50 ml), dried over sodium sulphate and
concentrated under vacuo. The o~_1 residue was purified on
silica gel by means of flash ~~hromatography. Using light
petroleum and light petroleum/ethyl. acetate 95:5 as eluent, 602
mg of methyl (E)-Methyl, 2-(3,4-d:ifluorobenzoyl)-cyclopropane-
1-carboxylate were collected (yield 36%).
1H-NMR (CDC13) 8: 1.6-1.7 (m, 2H, 3-CH2); 2.3-2.4 (m, 1H, 1-CH);
3 . 0-3 . 1 (m, 1H, 2-CH) ; 3 . 7 (s, 3H, COOCH3) ; 7 .2-7.4 (m, 1H, 5' -
CH); 7.8-7.9 (m, 2H, 6'-CH 2'-CH).
i3C-NMR (CDC13) 8: 17.9, 24.53; 25.68; 52.15; 117.35; 117.71;
125.31; 134.09; 147.84 (d); 151.13 (d); 152.83 (d): 156.24 (d);
172.32; 194.36.
Analogously the following compound; can be prepared:
(E)-Methyl, 2-(3,4-dichlorobenzoyl)-3-methylene-cyclopropane-1-
carboxylate.
m.p.= 81°C
1H-NMR (CDC13) 8: 3.1-3.2 (m, 1H, 1-CH); 3.7-3.8 (m, 4H, 2-CH
COOCH3); 5.5-5.7 (d, 2H, 3-exomet:hylene); 7.6 (d, 1H, 5'-CH,
J°=8. 5 Hz) ; 7.7 (dd, 1H, 6' -CH, J°=:8.4 Hz J",=2 HZ) ; 8. 1
(d, 1H,
2' -CH, Jm=2Hz) .
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 44 -
i3C-NMR (CDC13) b: 25.65; 29.96; 52.18; 105.25; 127.36; 130.21;
130.70; 133.30; 136.00; 137.96; 169.51; 190.82.
(E)-Methyl, 2-(3,4-dichlorobenzoyl)-3,3-dimethyl-cyclopropane-
1-carboxylate.
m.p.= 59-60°C
1H-NMR (CDC13)8: 1.2 (s, 3H, 3a-CH3) ; 1.3 (t, 3H, COOCHZCH3) ; 1.4
(S, 3H, 3~-CH3) ; 2.5 (d, 1H, 1-CH, Jtrans ='-~ ~ 7 HZ) ; 3 . 0 (d, 1H,
2-CH, Jtrans= 5.7 HZ) ; 4.0-4.2 (m, 2H, COOCHzCH3) ; 7.4 (d, 1H,
5' -CH, Jo=8.4 HZ) ; 7.7 (dd, 1H, 6' -CH, Jo=8.4 HZ Jm=2 HZ) ; 7. 9
(d, 1H, 2 ' -CH, Jm=2Hz) .
i3C-NMR (CDC13) 8: 14.16; 19.93; 20.20; 33.29; 33.59; 38.50;
60.78; 127.06; 130.01; 130.64; 133.27; 137.40; 137.58; 170.28;
193.47.
(E)-Methyl, 2-(3,4-dichlorobenzoyl)-3-methyl-cyclopropane-1-
carboxylate.
1H-NMR (CDC13) 8: 1.1-1.2 (d, 3H, 3-CH3); 2.0-2.2 (m, 1H, 3-
CH); 2.4-2.6 (m, 1H, 2-CH); 3.0-3.3 (m, 1H, 1-CH); 3.7 (s,3H,
COOCH3; 7.5 (d, 1H, 5'-CH, J°=8.4 Hz); 7.8-7.9 (dd, 1H, 6'-CH,
Ja=8.4 HZ Jm=2 HZ) ; 8.0-8.1 (d, 1H, 2' -CH, Jm=2Hz) .
(E)-Methyl, 2-(3,4-dichlorobenzoyl)-cyclopentane-1-carboxylate.
1H-NMR (CDC13) s: 1.6-2.2 (m, 6H, cyclopentane); 3.3-3.4 (m, 1H,
1-CH) ; 3.6 (s, 3H, COOCH3) ; 3.9-4.1 (m, 1H, 2-CH) ; 7.5 (d, 1H,
5' -CH, Ja=8 .7 HZ) ; 7.7 (dd, 1H, 6' -CH, Jo=8 . 3 HZ Jm=2 HZ) ; 8 . 0
(d, 1H, 2' CH, Jn,=2HZ) .
CA 02283184 1999-09-10
WO 98140344 PCT/EP98/00883
- 45 -
iaC-NMR (CDC13) 8: 25.73; 30.49; 31.36; 46.16; 49.47; 51.86;
127.56; 130.56; 130.65; 133.29; 136.05; 137.61; 175.37; 198.80.
(E)-Methyl, 2-(3,4-dichlorobenzoyl)-cyclobutane-1-carboxylate.
1H-NMR (CDC13) 8: 2.1-2.3 (m, 4H c~,rclobutane) ; 3.4-3.6 (m, 1H,
1-CH); 3.6 (s, 3H, COOCH3); 4.0-4.4 (m, 1H, 2-CH); 7.4 (d, 1H,
5' CH, J°=8 . 7 HZ ) ; 7 . 7 (dd, 1H, 6' --CH, J°=8 . 3 HZ Jm=2
Hz ) ; 8 . 0
IO (d, 1H, 2' -CH, Jrt,=2 HZ) .
Example 29
Prenara~ionof (E) -2- f3.4-difluorobenzo~ 1r_)-cyclop~nane-1-
carbox~rl_,'_c acid
To a solution of methyl (E)-2-(3,4-difluorobenzoyl)-
cyclopropane-1-carboxylate (522 mg, 2.17 mmol) in dioxane (7
ml), maintained under magnetic stirring, an aqueous solution (3
ml) of potassium hydroxide (182.5 mg, 3.26 mmol) was added. The
resulting solution was stirred at room temperature during 20'
then was diluted with water (30 ml). The aqueous layer was
extracted with ethyl acetate (3x10 ml), acidified with
hydrochloric acid 3N and extxracted with ethyl acetate (3x20
ml). These last organic phases are combined, washed with brine
(1x10 ml), dried over sodium sulphate and concentrated under
vacuo. 471 mg of (E)-2-(3,4-dif7_uorobenzoyl)-cyclopropane-1-
carboxylic acid as a white solid were collected (yield 96%).
m.p. - 81-82°C
1H-NMR (CDC13+CD30D)8: 1.6-1.7 (m, 2H, 3-CHZ); 2.4-2.5 (m, 1H,
1-CH); 3.1-3.3 (m, 1H, 2-CH); 7.2-7.4 (m, 1H, 5'-CH); 7.7(m,
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 46 -
2H, 6'-CH 2'-CH); 9.7 (bs, 1H, COOH).
iaC-NMR (CDC13+CD30D) 8: 18.27; 24.38; 26.15; 38.97; 117.45;
117.80; 125.39; 133.91; 147.88(d); 151.24(d); 152.86(d);
156.36(d); 177.96; 194.07.
Analogously the following compound can be prepared:
(E)-2-(3,4-dichlorobenzoyl)-3-methylene-cyclopropane-1-
carboxylic acid.
m.p.= 184°C dec.
1H-NMR (CDC13+CD30D} b: 3.1-3.2 (m, 1H, 1-CH} ; 3 .7-3 .8 (m, 4H, 2
CH); 5.5-5.8(m, 2H, 3-exomethylene, COOH); 7.6 (d, 1H, 5'-CH,
J°=8.5 Hz); 7.8 (dd, 1H, 6'-CH, Jo=8.4 HzJm=2Hz); 8.1 (d, 1H,
2' -CH, Jm=2Hz) .
i3C-NMR (CDC13+CD30D) 8: 25.94; 30.29; 105.57; 127.47; 130.42;
130.89; 133.55; 136.06; 138.33; 172.74; 191.29.
(E)-2-(3,4-dichlorobenzoyl)-3,3-dimetryl-cyclopropane-1-
carboxylic acid.
m.p. 148-150°C.
1H-NMR (CDC13+CD30D) b: 1.1 (s, 3H, 3a-CH3) 1.5 (s, 3H, 3~3-CH3)
; ;
2 . 6 (d, 1H, 1-CH, Jtrans=5 ~ 7 3 . 2 1H, 2-CH, Jtrans=5
Hz ) ; (d, ~ 7
Hz); 4.6 (bs, 1H, COOH); 8.0 (d, 1H, 5' -CH, J=8.4 Hz); 8.2
(dd, 1H, 6' -CH, Jo= 8.4 Jm=2HZ} ; (d, 1H, 2' -CH, Jm=2Hz) .
8 . 5
i3C-NMR (CDC13+CD30D) 8: 19.94; 20.21; 27.89, 28.30; 33.44;
38.72; 127.09; 130.01; 130.69; 133.28; 137.37; 137.64; 172.95;
193.78.
CA 02283184 1999-09-10
WO 98J40344 p~.~pgg~~~
- 47 -
(E) 2-(3,4-dichlorobenzoyl)-3-methyl-cyclopropane-1-carboxylic
acid.
m.p. - 129°C
1H-NMR (Acetone D6) 8: 1.0-1.1 (dd, 3H, 3-CH3) ; 2.0-2.2 (m, 1H,
3-CH); 2.2-2.3 (m, 1H, 2-CH); 3.1-3.2 (m, 1H, 1-CH); 7.5 (d,
1H, 5' -CH, J°=8.4 Hz) ; 7. 9-8. 0 (dcl, 1H, 6' -CH, Ja=8.4 HZ Jm=2
Hz); 8.0-8.1 (d, 1H, 2'-CH, Jm=2 Hz); 9.0 (bs, 1H, COOH).
13C-NMR (Acetone-D6) b: 11.03; 27.23; 28.81; 33.30; 128.71;
130.83; 131.82; 133.45; 137.66; 138.71; 173.17, 193.85.
(E) 2-(3,4-dichlorobenzoyl)-cyclobutane-1-carboxylic acid
m.p.= 129-131°C
1H-NMR (CDC13+CD30D) 8: 2.1-2.4 (m, 4H cyclobutanei; 3.5-3.7 (m,
1H, 1-CH); 4.1-4.3 (m, 1H, 2-CH); 4.9 (bs, 1H, COOH); 7.5 (d,
1H, 5' -CH, J°=8.4 Hz) ; 7.7 (dd, 1H, 6' -CH, J°=8 .4 Hz J~;=2
Hz) ;
8 . 0 (d, 1H, 2' -CH, J,~=2 HZ ) .
i3C-NMR (CDC13+CD30D) 8: 22.19; 23.23; 38.97; 44.30; 127.92;
130.84; 131.21; 133.71; 135.06; 138.23; 177.01; 197.63.
(E)-2-(3,4-dichlorobenzoyl)-cyclopentane-1-carboxylic acid
m.p.= 86°C (dec.)
1H-NMR (Acetone-D6) 8: 1.6-2.2 (m, 6H, cyclopentane); 3.3-3.4
(m, 1H, 1-CH) ; 4 .0-4 . 1 (m, 1H, 2-CH) ; 7. 7 (d, 1H, 5' -CH, J°=8. 7
Hz) ; 7. 9 (dd, 1H, 6' -CH, Jo=8.3 Hz Jm=2 Hz) ; 8. 1 (d, 1H, 2' -CH
Jm=2 Hz ) .
CA 02283184 1999-09-10
WO 98/40344 PCT/EP98/00883
- 48 -
iaC-NMR (Acetone-D6) 8: 25.73; 30.49; 31.36; 46.16; 49.47;
127.56; 130.56; 130.65; 133.29; 136.05; 137.61; 177.07; 198.80.
Example 30
Preparation of (E) -2- (3, 4-dichlorobenzoyl) -c~~~ropane-1-
carboxamide
3.5g (12.82 mmol) of (E)-Methyl-2-(3,4-dichlorobenzoyl)-
cyclopropane-1-carboxylate were dissolved in dioxane (50 ml)
and treated with 30% NH40H (140 ml) for 3 days at room
temperature.
The residue after evaporation was crystallized from i-propyl
ether to give 1.6g of (E)-2-(3,4-dichlorobenzoyl)-cyclapropane-
1-carboxamide (51%).
M.P. 188-191°C
Analogously the following compounds can be prepared:
(E)-2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-methylcarboxamide
m.p. 127-129°C;
(E)-2-(3,4-dichlorobenzoyl)-cyclopropane-1-hydroxamic acid
m.p. 61-62°C;
(E)-2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-benzylcarboxamide
m.p. 151-153°C;
(E)-2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-phenylcarboxamide
m.p. 156-157°C;
(E)-2-(3,4-dichlorobenzoyl)-cyclopropane-1-N-
phenylsulfonylcarboxamide, m.p. 172-173°C.
CA 02283184 1999-09-10
WO 98/40344 p~~p9g~ppgg3
- 49 -
Example 31
Capsule, each weighing 0.23 g and containing 50 mg of the
active substance can be prepared as follows:
Composition for 500 capsules:
(E)-2-(3,4-dichlorobenzoyl)-cyclopropyl-
-1-carboxylic acid 25 g
Lactose 80 g
Corn starch 5 g
Magnesium stearate 5 g
This formulation can be incapsul.ated in two hard gelatin
capsules of two pieces, each with each capsule weighing
0.23 g.
Example 32
Intr~m~scular injection of 50 ma/ml
A pharmaceutical injectable composition can be manifactured
dissolving 50 g (E)-2-(3,4-dichlorobenzoyl)-cyclopropyl-1-
carboxylic acid in sterile propyleneglycol (1000 ml) and sealed
in 1-5 ml ampoules.
Legend to Figure 1
IDO = Indolamineoxigenase
KYN = Kynurenine
KYN-OH = Kynurenine-3-hydroxylase
KYNA = Kynurenic acid
3-OHAA = 3-hydroxy anthranilic acid
KYNase = Kynureninase
QUIN = Quinolinic acid
3-HAO = 3-hydroxy anthranilic acid deoxygenase
KAT = Kynurenine amino transferase
3-OHKYN = 3-Hydroxy-kynurenine