Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02297301 2000-O1-18
"COMBINATION PROCESS FOR
ENHANCED LIGHT OLEFIN PRODUCTION "
FIELD
The present invention relates to an improved method for the
production of ethylene and propylene from a heavier olefin feedstock.
BACKGROUND
Ethylene, a light olefin hydrocarbon with two carbon atoms per
molecule, is an important building block petrochemical. The primary use for
ethylene is as a monomer for the production of polyethylene for both linear
low density polyethylene and high density polyethylene. Other uses include
the production of vinyl chloride, ethylene oxide, ethylbenzene and alcohols.
o Essentially all of the ethylene is produced by the steam cracking or
pyrolysis
of hydrocarbons. Hydrocarbons used as feedstock for ethylene plants include
natural gas, naphtha, and gas oils. The natural gas components are generally
paraffinic and include ethane, propane, and butane. Ethylene is co-produced
with propylene and butylenes. Depending upon the feedstock, the products of
is commercial ethylene plants can include higher olefinic and aromatic
hydrocarbons with more than 4 atoms per molecule.
An ethylene plant is a very complex combination of reaction and gas
recovery systems. The feedstock is charged to a cracking zone in the presence
of steam at effective thermal conditions to produce a pyrolysis reactor
2o effluent gas mixture. The pyrolysis reactor effluent gas mixture is
stabilized
and separated into purified components through a sequence of cryogenic and
1
CA 02297301 2000-O1-18
conventional fractionation steps. A typical ethylene separation section of an
ethylene plant containing both cryogenic and conventional fractionation steps
to recover an ethylene product with a purity exceeding 99.5% ethylene is
described in an article by V. Kaiser and M. Picciotti, entitled, 'Better
s Ethylene Separation Unit" HYDROCARBON PROCESSING MAGAZINE,
November 1988, pages 57-61.
Methods are known for increasing the conversion of portions of the
products of the ethylene production from a zeolitic cracking process to
produce more ethylene and propylene by a disproportionation or metathesis
0 of olefins. Such processes are disclosed in US-A-5,026,935 and US-A-
5,026,936 wherein a metathesis reaction step is employed in combination
with a catalytic cracking step to produce more ethylene and propylene by the
metathesis of CQ and heavier molecules. The catalytic cracking step employs a
zeolitic catalyst to convert a hydrocarbon stream having 4 or more carbon
~s atoms per molecule to produce olefins having fewer carbon atoms per
molecule. The hydrocarbon feedstream to the zeolitic catalyst typically
contains a mixture of 40 to 95 wt-% paraffins having 4 or more carbon atoms
per molecule and 5 to 60 wt-% olefins having 4 or more carbon atoms per
molecule. In US-A-5,043,522, the preferred catalyst for such a zeolitic
2o cracking process is selected from acid zeolites of the ZSM type and
borosilicates. Of the ZSM-type catalysts, ZSM-5 was preferred. Other zeolites
containing materials which could be used in this cracking process to produce
ethylene and propylene included zeolite A, zeolite X, zeolite Y, zeolite ZK-5,
zeolite ZK-4. synthetic mordenite, dealuminized mordenite, as well as
25 naturally occurring zeolites including chabazite, faujasite, mordenite, and
the
like.
EP-B-109,059 discloses a process for the conversion of a feedstream
containing olefins having 4 to 12 carbon atoms per molecule into propylene
2
CA 02297301 2000-O1-18
by contacting the feedstream with a ZSM-5 or a ZSM-11 zeolite having a
silica to alumina molar ratio less than or equal to 300 at a temperature from
400 to 600 °C. The ZSM-5 or ZSM-11 zeolite is exchanged with a hydrogen
or
an ammonium cation. This reference also discloses that, although the
conversion to propylene is enhanced by the recycle of any olefins with less
than 4 carbon atoms per molecule, paraffins which do not react tend to build
up in the recycle stream. An additional oligomerization step is also disclosed
wherein the olefins having carbon atoms less than 4 are oligomerized to
facilitate the removal of paraffins such as butane and particularly isobutane
o which is difficult to separate from C4 olefins by conventional
fractionation. In
a related EP-B-109060, a process is disclosed for the conversion of butenes to
propylene. The process comprises contacting butenes with a zeolitic
compound selected from the group consisting of silicalites, boralites,
chromosilicates and those zeolites ZSM-5 and ZSM-11 in which the mole ratio
~5 of silica to alumina is greater than or equal to 350. The conversion is
carried
out at a temperature from 500 to 600 °C and at a space velocity of from
5 to
200 kg/hr of butenes per kg of pure zeolitic compound.
Generally, the heavier olefins having six or more carbon atoms per
molecule which are produced in commercial ethylene plants are useful for the
2o production of aromatic hydrocarbons. Portions of the olefin product include
olefins with four carbon atoms per molecule. This portion includes both mono-
olefins and di-olefins and some paraffins, including butane and iso-butane.
Because the portion with four carbon atoms per molecule is generally less
valuable and requires significant processing to separate di-olefins from the
25 mono-olefins, processes are sought to improve the utilization of this
portion of
the ethylene plant product and enhancing the overall yield of ethylene and
propylene.
3
CA 02297301 2000-O1-18
Molecular sieves such as the microporous crystalline zeolite and non-
zeolitic catalysts, particularly silicoaluminophosphates (SAPO), are well
known
to promote the conversion of oxygenates to olefin-containing hydrocarbon
mixtures. SAPO catalysts are often employed in the conversion of oxygenates
s into light olefins, particularly light olefins having less than four carbon
atoms per molecule. In such processes, the ratio of ethylene to propylene
produced on a carbon basis varies from 0.1 to ten, and more typically, the
ratio of ethylene to propylene ranges from 0.8 to 2.5. Methods are sought to
alter the product distribution from the oxygenate conversion process for
o making light olefins to overcome equilibrium limitations of the SAPO
catalyst. These and other disadvantages of the prior art are overcome by the
present invention, and a new improved process for conversion of oxygenates
to hydrocarbons, specifically olefinic hydrocarbons, is provided.
SUMMARY
15 It is an objective of the present invention to provide a commercial
process for enhancing the production of ethylene and propylene from the
catalytic conversion of butene and heavier olefins.
It is an objective of the present invention to provide a process for
increasing the production of ethylene and propylene in a methanol-to-olefin
2o facility by the further conversion of butene and heavier olefins.
It is an objective to provide an economic route for enhancing the
production of ethylene and propylene from a catalytic cracking operation in a
petroleum refinery.
In the present invention, a novel use has been discovered for a group of
25 small pore zeolitic and non-zeolitic catalysts which can significantly
improve
the yield of ethylene and propylene in a process for the conversion of light
olefins having four carbon atoms per molecule and heavier. Specifically, in
4
CA 02297301 2000-O1-18
accordance with the invention, a C4 plus olefin stream from an ethylene
production complex is converted in a reaction zone over a selective catalyst
at
effective conditions to produce a product mixture containing ethylene and
propylene. Ethylene and propylene are separated from the product mixture
s and recovered. A portion of the remaining heavy hydrocarbons (C4 plus
olefins) and paraffins may be recycled to the reaction zone for further
conversion, or oligomerized to produce valuable downstream products. The
process of the present invention may be applied in commercial ethylene
plants, in petroleum refining catalytic cracking operations, and in processes
o for the conversion of oxygenates such as methanol-to-olefins. One aspect of
the present invention is a process step which removes a relatively low
yielding component of the butene stream, iso-butylene, which it was
surprisingly discovered was not converted to the same degree as linear mono-
olefins and thus reduced the effectiveness of the overall process. A
processing
~5 step for the selective removal of the iso-olefins provided the additional
benefit
of the simultaneous production of high value byproducts. The iso-olefin
removal step reduced the size of the butene conversion zone and improved the
overall value of the products from the process of the invention.
In one embodiment, the invention is a combination process for
2o producing ethylene and propylene. A feedstream is passed to an olefin
production zone and therein an olefinic product stream comprising ethylene,
propylene, and butylene and heavier olefins is produced. The olefinic product
stream is passed to an olefin separation zone where it is reported to provide
an ethylene product stream, a propylene product stream, a mixed butylene
25 and heavier stream, and a reject stream. The mixed butylene and heavier
stream comprising mono-olefins, such as normal and iso-olefins, paraffins,
and di-olefins is passed to a di-olefin removal zone which functions to remove
di-olefins and to provide a mono-olefin stream comprising normal olefins, iso-
olefins, and paraffins. At least a portion of the mono-olefin stream is
s
CA 02297301 2000-O1-18
converted in an olefin conversion zone containing an acid catalyst selective
for the conversion of iso-olefins in the presence of at least a stoichiometric
amount of an oxygen-containing compound to provide a conversion product
stream comprising oxygenates and, normal olefins. The conversion product
stream is passed to an oxygenate separation zone and therein separating
oxygenates from the normal olefins to produce a net butylene stream. At least
a portion of the net butylene stream is passed to a butylene cracking zone
and therein the net butylene stream is contacted with a small pore catalyst
selective for the cracking of the normal olefins at conditions effective to
o produce a light olefin stream comprising additional amounts of ethylene and
propylene which are recovered.
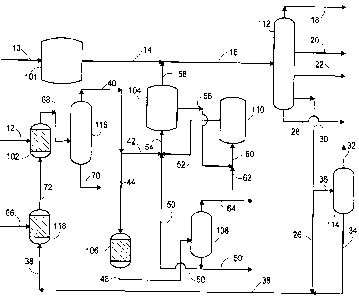
BRIEF DESCRIPTION OF THE DRAWING
~s Fig. 1 is a schematic block flow diagram of the process of the present
invention.
DETAILED DESCRIPTION
2o The olefinic product stream produced in the first step of the present
invention may be derived from steam cracking, the catalytic cracking of
petroleum gas oils, or the conversion of oxygenates to olefins. In the steam
cracking of hydrocarbons such as ethane, liquefied petroleum gas, naphtha,
and gasoil, a steam cracking product is produced which comprises olefins
25 such as ethylene, propylene, butylene, and heavier hydrocarbons. The
composition of the heavier hydrocarbons from the stream cracking process
will vary according to the feedstock charged to the steam cracking reaction
zone. The lighter the feedstock, the more light olefins are produced. As the
steam cracking feedstock increases in carbon number, the more aromatics are
6
CA 02297301 2000-O1-18
formed among the heavier hydrocarbons. Generally, the C4 fraction,
produced by the steam cracking reaction may contain as much as 45 wt. % di-
olefins as butadiene, and 50 to 60 wt.% mono-olefins such as normal butenes
and iso-butenes. Approximately 15 to 25 wt.% of the C4 fraction comprises iso-
butylene. In fluid catalytic cracking of petroleum gas oils, the C4 fraction
comprises between 40 and 60% olefins, including both mono-olefins and di-
olefins. Paraffins in the product include isobutane, isopentane, normal
pentane, as well as propane and n-butane. The mono-olefins include butene-1,
butene-2, iso-butene, 2-methyl-2-butene, 2-methyl-1-butene, 3-methyl-1-
o butene, 1-pentene, 2-pentene, cyclopentene and propylene. The hydrocarbon
feedstream may also contain di-olefins such as 1,3-butadiene and 1,3-
pentadiene. Approximately 0.5 to 1 wt.% of the C4 components comprise
butadiene, and 15 to 20 wt.% of the C4 components from a fluid catalytic
cracking process comprise iso-butylene, 30 wt.% of the C4 fraction comprise
normal butenes - such as butene-1 and butene-2, and the balance are
paraffins. In the conversion of oxygenates such as methanol to olefins, the C4
fraction less than 1 wt.% butadiene, and less than 5 wt.% paraffins.
Preferably, the C4 fraction from a methanol-to-olefins reaction zone comprises
less than 20 wt.% iso-butene, and more preferably, the C4 fraction from a
2o methanol-to-olefins reaction zone comprises less than 15 wt.% iso-butene.
Central to the process of the present invention is a catalytic process for
the cracking of butenes to produce additional amounts of ethylene and
propylene. The butene cracking reaction zone contains a small pore size
catalyst. The preferred small pore catalysts are defined as having pores at
least a portion, preferably a major portion, of which have an average
effective
diameter characterized such that the adsorption capacity (as measured by the
standard McBain-Bakkr gravimetric adsorption method using given adsorbate
molecules) shows adsorption of oxygen (average kinetic diameter of 0.346 nm)
and negligible adsorption of isobutane (average kinetic diameter of 0.5 nm).
CA 02297301 2000-O1-18
More preferably, the average effective diameter is characterized by adsorption
of xenon (average kinetic diameter of 0.4 nm) and negligible adsorption of
isobutane, and most preferably, by adsorption of n-hexane (average kinetic
diameter of 0.43 nm) and negligible adsorption of isobutane. Negligible
adsorption of a given adsorbate is adsorption of less than 3 wt.% of the
catalyst
and adsorption of the adsorbate is over 3 wt.% of the catalyst. Certain of the
catalysts useful in the present invention have pores with an average effective
diameter of less than 5 Angstroms. The average effective diameter of the pores
of preferred catalysts is determined by measurements described in D. W.
o Breck, ZEOLITE MOLECULAR SIEVES by John Wiley & Sons, New York
(1974). The term "effective diameter" is used to denote that occasionally the
pores are irregularly shaped, e.g., elliptical, and thus the pore dimensions
are
characterized by the molecules that can be adsorbed rather than the actual
dimensions. Preferably, the small pore catalysts have a substantially uniform
pore structure, e.g., substantially uniformly sized and shaped pore. Suitable
catalyst may be chosen from among layered clays, zeolitic molecular sieves,
and non-zeolitic molecular sieves.
Zeolitic molecular sieves in the calcined form may be represented by the
general formula:
2o Mez"O:A1203:xSi02:yH20
where Me is a cation, x has a value from 2 to infinity, n is the cation
valence
and y has a value of from 2 to 10.
Non-zeolitic molecular sieves include molecular sieves which have the
proper effective pore size and are embraced by an empirical chemical
composition, on an anhydrous basis, expressed by the empirical formula:
(ELXAIYPZ)02
s
CA 02297301 2000-O1-18
where EL is an element selected from the group consisting of silicon,
magnesium, zinc, iron, cobalt, nickel, manganese, chromium and mixtures
thereof, x is the mole fraction of EL and is at least 0.005, y is the mole
fraction
of Al and is at least 0.01, z is the mole fraction of P and is at least 0.01
and x +
y + z = 1. When EL is a mixture of metals, x represents the total amount of
the
element mixture present. Preferred elements (EL) are silicon, magnesium and
cobalt with silicon being especially preferred.
The preparation of various ELAPOs are well known in the art and may
be found in US-A-5,191,141 (ELAPO); US-A-4,554,143 (FeAPO); US-A-
to 4,440,871 (SAPO); US-A-4,853,197 (MAPO, MnAPO, ZnAPO, CoAPO); US-A-
4,793,984 (CAPO), US-A-4,752,651 and US-A-4,310,440. Generally, the
ELAPO molecular sieves are synthesized by hydrothermal crystallization from
a reaction mixture containing reactive sources of EL, aluminum, phosphorus
and a templating agent. Reactive sources of EL are the metal salts such as the
chloride and nitrate salts. When EL is silicon, preferred sources include
fumed,
colloidal or precipitated silica. Preferred reactive sources of aluminum and
phosphorus are pseudo-boehmite alumina and phosphoric acid. Preferred
templating agents are amines and quaternary ammonium compounds. An
especially preferred templating agent is tetraethylammonium hydroxide
(TEAOH).
A preferred embodiment of the invention is one in which the element
(EL) content varies from 0.005 to 0.05 mole fraction. If EL is more than one
element, then the total concentration of all the elements is between 0.005 and
0.05 mole fraction. An especially preferred embodiment is one in which EL is
silicon (usually referred to as SAPO). The SAPOs which can be used in the
instant invention are any of those described in US-A-4,440,871; US-A-
5,126,308, and US-A-5,191,141. SAPO catalysts which are suitable for the
present invention include SAPO-11, SAPO-17, and SAPO-34. Of the specific
9
CA 02297301 2000-O1-18
crystallographic structures described in the '871 patent, the SAPO-34, i.e.,
structure type 34, is preferred. The SAPO-34 structure is characterized in
that
it adsorbs xenon but does not adsorb isobutane, indicating that it has a pore
opening of 4.2 E~. Another SAPO, SAPO-17, as exemplified in Examples 25 and
26 of the '871 patent, is also preferred. The SAPO-17 structure is
characterized
in that it adsorbs oxygen, hexane, and water but does not adsorb isobutane,
indicating that it has a pore opening of greater than 4.3 A and less than 5.0
E~.
The preferred oxygenate conversion catalyst may be, and preferably is,
incorporated into solid particles in which the catalyst is present in an
amount
to effective to promote the desired hydrocarbon conversion. In one aspect, the
solid particles comprise a catalytically effective amount of the catalyst and
at
least one matrix material, preferably selected from the group consisting of
binder materials, filler materials, and mixtures thereof to provide a desired
property or properties, e.g., desired catalyst dilution, mechanical strength,
and
the like to the solid particles. Such matrix materials are often, to some
extent,
porous in nature and may or may not be effective to promote the desired
hydrocarbon conversion. The matrix materials may promote conversion of the
feedstream and often provide reduced selectivity to the desired product or
products relative to the catalyst. Filler and binder materials include, for
2o example, synthetic and naturally occurring substances such as metal oxides,
clays, silicas, aluminas, silica-aluminas, silica-magnesias, silica-zirconias,
silica-thorias, silica-berylias, silica-titanias, silica-alumina-thorias,
silica-
alumina-zirconias, aluminophosphates, mixtures of these and the like.
If matrix materials, e.g., binder and/or filler materials, are included in
the catalyst composition, the non-zeolitic and/or zeolitic molecular sieves
preferably comprise 1 % to 99%, more preferably 5% to 90%, and still more
preferably 10% to 80% by weight of the total composition. The preparation of
solid particles comprising catalyst and matrix materials is conventional.
CA 02297301 2000-O1-18
During the oxygenate conversion reaction, a carbonaceous material, i.e.,
coke, is deposited on the catalyst. The carbonaceous deposit material has the
effect of reducing the number of active sites on the catalyst which thereby
affects the extent of the conversion. During the conversion process a portion
of
the coked catalyst is withdrawn from the reaction zone and regenerated to
remove at least a portion of the carbonaceous material and returned to the
oxygenate conversion reaction zone. Depending upon the particular catalyst
and conversion, it can be desirable to substantially remove the carbonaceous
material e.g., to less than 1 wt %, or to only partially regenerate the
catalyst,
to e.g., to from 2 to 30 wt % carbon. Preferably, the regenerated catalyst
will
contain 0 to 20 wt % and, more preferably, from 0 to 10 wt % carbon.
Additionally, during regeneration there can be oxidation of sulfur and in some
instances nitrogen compounds along with the removal of metal materials from
the catalyst. Moreover, regeneration conditions can be varied depending upon
~5 catalyst used and the type of contaminant material present upon the
catalyst
prior to its regeneration.
The butene and heavier material produced in the oxygenate conversion
zone, or methanol-to-olefins process, following separation from the first
reaction zone effluent can be converted in a secondary reaction zone to
produce
2o additional amounts of ethylene and propylene. Simply passing a portion of
the
reactor effluent which comprises methanol to the secondary reaction zone at a
higher temperature will not achieve the benefits of the instant invention
because the presence of methanol, a polar compound, will inhibit cracking
reaction by tying up acid sites on the catalyst. It was surprisingly found
that
25 the secondary reaction zone, wherein the butene and heavier material is
contacted with a catalyst at conditions effective to convert at least a
portion of
the butene and heavier materials to ethylene and propylene, favored the
production of propylene rather than the smaller molecule, ethylene. The
catalyst found to produce this conversion was the same small pore SAPO
a
CA 02297301 2000-O1-18
catalyst employed in the oxygenate conversion zone. The effective conditions
at
which the additional ethylene and propylene were produced when the butene
and heavier were contacted with the catalyst comprised a secondary reaction
temperature above 460 °C. Preferably, the secondary reaction
temperature
comprises a temperature between 460 to 700 °C, and more preferably, the
secondary reaction temperature is between 500 to 700 °C, and most
preferably,
the effective butene cracking reaction temperature is between 500 to 650
°C.
Preferably, the effective cracking pressure ranges from 140 to 700 kPa (20 to
100 psia) and the effective space velocity ranges from 0.05 to 10 hr 1.
to Conversion of the butene produced in the oxygenate conversion was evaluated
by measuring the conversion and the selectivity for the production of ethylene
in a fixed bed reactor. The fixed bed reactor contained a SAPO-34 catalyst. It
was found that conversion of 2-butene at temperatures ranging between 460
and 580 °C resulted in the production of primarily propylene and
heavier
olefins and initially produced ethylene at a 20% selectivity at short
residence
times. It was discovered that by increasing reactor temperature over 460
°C
and diluting the olefin feed with a nitrogen diluent, the product distribution
was shifted toward light olefins. Dilution of the feed with a diluent such as
steam appeared to have the same effect on product distribution as dilution
with nitrogen. Separate tests with a spray dried catalyst comprising 40%
SAPO-34, 40% kaolin clay and 20% Si-A1 binder gave the same results as the
100% SAPO-34 powder. It was further surprisingly discovered that there was a
significant difference in the conversion of butenes over the SAPO catalyst.
The
conversion of linear butenes occurred in the 60 to 70 percent conversion or
greater, while conversion of iso-butenes was less than 15 weight percent. By
the removal of the iso-butene by conversion to more valuable products, such as
tertiary butyl ethers, the overall profitability of the complex was improved
and
the size of the butene cracking zone was significantly reduced over schemes
processing the entire butene range of material.
12
CA 02297301 2000-O1-18
The conversion of butylene and heavier material can be accomplished by
separating this product fraction from the oxygenate conversion zone effluent
and by contacting the butylene and heavier stream or fraction (i.e. C4 plus
olefins) with the catalyst from the first reaction zone immediately following
s regeneration. Ideally, this conversion will take place in a secondary
reaction
zone between the regeneration and the first reaction zone and the catalyst and
the cracked lighter products (ethylene and propylene) will be transferred to
the
first reaction zone. To effect the cracking reaction favoring light olefins,
the
secondary reaction zone may be a riser cracking reaction zone with a short
to residence time to minimize hydrogen transfer reactions which would favor
production of paraffins such as ethane and propane. The cracking reactions can
also be carried out in a separate fluidized bed containing the oxygenate
conversion catalyst. With a separate fluidized bed, the catalyst circulation
can
be controlled to flow to and from the regenerator at effective catalyst
15 circulation rates which favor the production of ethylene and propylene in
the
secondary reaction zone and minimize production of the paraffins and
methane. When a separate fluidized bed reaction zone is employed as the
secondary reaction zone, a product gas stream comprising the cracked products
is passed to the oxygenate conversion zone or first reaction zone and catalyst
2o withdrawn from the secondary reaction zone is returned to the regenerator.
Because the cracking reaction is a slightly endothermic reaction, some heat of
the cracking reaction is provided by the regenerated catalyst. However, an
optional butene and heavier preheater may be required to vaporize the feed to
the secondary reaction zone when the catalyst circulation rates are very low
25 such as a catalyst to oil (mixed butylene and heavier stream) ratio between
1
and 20. In the second reaction zone, preferably the catalyst to oil ratio is
less
than 20, and more preferably in the second reaction zone, the catalyst to oil
ratio is less than 10. It is believed that the reaction in the secondary
reaction
zone over the SAPO catalyst proceeds initially by polymerizing some of the
13
CA 02297301 2000-O1-18
butylene and heavier hydrocarbons in addition to the cracking reactions which,
at an effective temperature, favors the production of propylene while also
producing ethylene.
A detailed description of processes, including catalyst, processing
conditions, and product recovery, for the production of MTBE from iso-butylene
and methanol are provided in US-A-2,720,547 and US-A-4,219,678 and in an
article at page 35 of the June 25, 1979 edition of Chemical and Engineering
News. The preferred process is described in a paper presented at The American
Institute of Chemical Engineers, 85th National Meeting on June 4-8, 1978.
to Other etherification processes of interest are the production of tertiary
amyl
methyl ether (TAME) by reacting C5 iso-olefins with methanol, and the
production of ethyl tertiary butyl ether (ETBE) by reacting C4 iso-olefins
with
ethanol, the production of tertiary amyl ethyl ether (TREE) by reacting C5 iso-
olefins with ethanol, and the production of tertiary hexyl methyl ether (THME)
by reacting Cs iso-olefins with methanol. Etherification reactions are carried
out in the presence of an acid catalyst such as a sulfonated, macroporous
organic ion exchange resin in the liquid phase at temperatures between 30 and
100 °C.
Generally, the production of ethylene is accompanied by the production
of di-olefins such as butadiene. These di-olefins must be removed by any
means prior to the production of any ethers or prior to introducing the butene
and heavier stream to the secondary cracking reactor. Butadiene produced in
ethylene plants by the steam cracking process is present in amounts which
often justify the recovery of the butadiene by extractive distillation or
solvent
2s extraction. US-A-4,038,156 and US-A-4,128,457, disclose the use of a polar
solvent such as acetonitrile to recover butadiene by extractive distillation.
When C4 plus olefins are produced in fluid catalytic cracking and methanol to
olefins processes, the concentration of butadiene is significantly smaller
than
14
CA 02297301 2000-O1-18
produced by steam cracking. Butadiene found in such streams is generally
removed by selective hydrogenation in the presence of a solid catalyst
comprising nickel and a noble metal such as platinum or palladium or silver
as disclosed in US-A-4,409,410.
It is necessary to prevent the buildup of paraffins such as isobutane in
the feed to the secondary cracking reaction zone. However, butenes and
isobutane have close normal boiling points which makes it difficult - by
traditional fractionation methods - to reject isobutane from a stream
comprising butenes. One method of preventing the buildup of isobutane in
~o the recycle to the butene cracking reaction zone is to withdraw a portion
of
the butene and heavier fraction as a byproduct heavy olefin stream to be used
in motor fuel blending, fuel or for further conversion to petrochemicals.
Another method of preventing the buildup of isobutane in the butene
cracking reaction zone is to convert the butenes to heavier olefins by a
1s process known as oligomerization. Since the isobutane is not changed in
this
process, it is more easily fractionated from the oligomerization product. The
oligomerization product may be used to produce heavy alcohols or
plasticizers, or it may be returned to the butene cracking zone to increase
the
production of ethylene. Oligomerization reactions of olefinic hydrocarbons
2o having from three to six carbon atoms are disclosed in US-A-4,465,885 and
US-A-4,613,719. The oligomerization reactions are generally carried out in
the presence of a solid catalyst comprising copper and phosphate at effective
conditions well-known in the art.
2s DETAILED DESCRIPTION OF THE DRAWING
The process of the present invention is hereinafter described with
reference to the drawing which illustrates various aspects of the present
invention.
IS
CA 02297301 2000-O1-18
Referring to Fig. 1, a feedstream in line 10 is passed to an olefin
production zone 101 to catalytically or thermally convert the feedstream into
an olefinic product stream in line 14 comprising olefinic compounds
containing from 2 to 12 carbon atoms per molecule. The olefin conversion
reaction to olefins is typically conducted in the presence of steam. The
olefinic
product stream in line 14 is passed via lines 14 and 16 to a olefin separation
zone 112 for the separation of the olefinic product stream in the conventional
manner into a light ends stream in line 18, an ethylene stream in line 20, a
propylene stream in line 22. In olefin separation zone 112, the olefinic
1o product stream is further separated to provide a butylene and heavier
stream
(or C4 plus olefin stream) in line 30 which comprises butylenes, pentenes, and
hexenes. A reject stream comprising very heavy olefins such as C8 and
heavier olefins and aromatic hydrocarbons is withdrawn in line 28. This
reject stream may be used for fuel or passed to downstream processing for
further recovery. The butylene and heavier stream in line 30 may also
comprise dimes, such as butadienes. The butylene and heavier stream is
passed in lines 30, 26 and 38 to a selective hydrogenation reaction zone 118
wherein the butylene and heavier stream is contacted with a selective
hydrogenation catalyst in the presence of hydrogen supplied via line 66 to
2o convert the dimes to mono-olefins and produce a mixed mono-olefin stream
in line 72. The mixed mono-olefin product stream is passed to an iso-olefin
removal zone incorporating a fractionation step or a combination of a reaction
step and a fractionation step for the removal of iso-olefins from the mono-
olefin product stream. In one embodiment, the iso-olefin removal zone
comprises passing the mono-olefin product stream in line 72 to an olefin
conversion zone 102 wherein an an oxygen-containing compound, or
oxygenate such as methanol, ethanol, or water in line 12 is combined with
the mono-olefin product stream in the presence of an acid catalyst to produce
a conversion product stream in line 68. In one aspect of the invention the
16
CA 02297301 2000-O1-18
olefin conversion zone 102 comprises an etherification zone and the
conversion product stream comprises an ether product stream. In the
etherification zone, which contains an acid catalyst, the mixed mono-olefin
stream is contacted with at least a stoichiometric amount of an alcohol such
as ethanol or methanol in the conventional manner to produce the ether
product stream. The conversion product stream comprises an oxygenate such
as an ether or an alcohol. Ethers produced in olefin conversion zone 102 can
include methyl or ethel tertiary butyl ether, or similar ethers derived from
iso-pentenes, or ethers derived from iso-hexenes to the extent these iso-
olefins are present in the mixed mono-olefin stream in line 72. Similarly, the
conversion product stream in line 68 may comprise alcohols such as tertiary
butyl alcohol and/or similar alcohols produced from iso-pentenes and iso-
hexenes. The alcohols or ethers are removed from the conversion product
stream in line 68 by passing the conversion product stream to an oxygenate
~5 separation zone 116 to provide an oxygenate product stream in line 70 and a
net butylene and heavier stream comprising a reduced amount of iso-olefins
in line 40 relative to the amount of iso-olefins in the mixed mono-olefin
stream in line 72. A portion of the net butylene and heavier stream in line 40
is passed in lines 42 and 54 to secondary cracking zone 104 wherein the
2o mono-olefins at effective conditions are contacted with a small pore
selective
catalyst for the conversion of the mono-olefins to produce additional amounts
of ethylene and propylene. A light olefin product stream comprising the
additional amounts of ethylene and propylene in line 58 is withdrawn from
cracking zone 104 and passed to olefin separation zone 112 via lines 58 and
25 16. During butylene cracking reaction within cracking zone 104, the
selective
catalyst rapidly deactivates by the formation of coke or carbon on the
catalyst. A portion of the deactivated cracking catalyst is withdrawn from
cracking zone 104 and passed via line 56 to a regenerator zone 110 wherein
deactivated cracking catalyst, containing from 3 to 20 wt.% coke, is
17
CA 02297301 2000-O1-18
regenerated by contacting the deactivated cracking catalyst with an oxygen-
containing stream in line 62 to produce a regenerated catalyst. Typically the
oxygen-containing stream comprises from 1 to 3 mol-percent oxygen. A
portion of the regenerated catalyst is withdrawn from the regeneration zone
110 in line 52 and is returned to cracking zone 104 via line 52 and line 54
where it is admixed with the mono-olefin product stream in line 54. The C4 +
olefin cracking reaction is carried out in the presence of a purge stream
comprising steam or inert diluent such as nitrogen, methane, or light paraffin
hydrocarbon, containing 2 to 4 hydrocarbons. In order to prevent the buildup
0 of light paraffins in cracking zone 104, at least a portion of the net
butylene
and heavier stream in line 40 is withdrawn as a drag stream in line 44. The
drag stream may be fractionated in a conventional manner (not shown) to
separate the paraffin portion from the olefin portion, wherein the olefin
portion is returned to the butylene cracking zone, or the drag stream 44 is
~s passed to oligomerization zone 106 wherein the low carbon number, normal
olefins, such as butene-1 and butene-2 combine to form heavier normal
olefins, having 8 carbon atoms per molecule to produce an oligomerization
effluent stream in line 48. The oligomerization effluent stream in line 48 is
passed to an oligomerization separation zone 108 to provide a light paraffin
2o stream, comprising butanes in line 64 and a heavy olefin stream in line 50.
The heavy olefin stream may be withdrawn in line 50' as a raw material for
plasticizer and alcohol production, or a portion of the heavy olefin stream in
line 50 may be returned to the butylene cracking zone via lines 50 and 54 to
produce additional amounts of ethylene and propylene.
25 In an alternate embodiment, a portion of the butylene and heavier
stream is processed in an alternate or additional di-olefin removal zone. In
this alternative the di-olefin removal zone comprises a butadiene extraction
zone which operates at effective conditions and employs a selective solvent to
extract the butadiene from the butylene and heavier stream to produce a
is
CA 02297301 2000-O1-18
raffinate stream essentially free of di-olefins. According to this alternate
operation, the butylene and heavier stream in line 30 is passed via lines 30
and 36 to butadiene extraction unit 114 wherein di-oelfins are extracted from
the butylene and heavier stream in the conventional manner and a butadiene
extract stream is withdrawn in line 32. The raffinate stream comprising
mono-olefins and paraffins in line 34, is passed to the selective
hydrogenation
reaction zone 118 via lines 34 and 38.
EXAMPLES
The following examples are only used to illustrate the present invention
and are not meant to be limiting.
EXAMPLE I
Generally, the conversion of oxygenates to light olefins, specifically
ethylene, can be improved by increasing the temperature at which the reaction
takes place. However, as temperature is increased, the catalyst life drops
significantly. To illustrate the degradation in catalyst activity, three
oxygenate
conversion pilot plant runs were conducted using a spray-dried metal
aluminophosphate catalyst comprising 40% SAPO-34, 40% kaolin clay and 20%
Si-Al binder. The catalyst was loaded into a 2.2 cm (7/8 inch) ID porcelain-
lined, stainless steel reactor and placed in a three-zone bronze block
furnace.
2o The reactor was heated to 435°C for Run A, 455°C for Run B,
and 475°C for Run
C under an Nz purge at 138 kPa (5 psig). These conditions were held for 1 hour
in each run to pretreat the catalyst. The N2 flow was stopped and a
methanol/water mixture (80/20 by weight) was introduced at 1 hr 1 MeOH
WHSV (weight hourly space velocity based on methanol) and continued until
the reactor effluent contained greater than 50% MeOH and DME. Table 1
summarizes the time on stream and product selectivities at the point where
19
CA 02297301 2000-O1-18
the overall conversion was 99% for each run. It can be seen by comparing run A
to run C that increasing reaction temperature by 40 °C increases the
ethylene/propylene product ratio from 1.5 to 2.0, but increasing reaction
temperature also decreases the catalyst life by 50%. At lower reactor
temperatures, larger amounts of butene and heavier compounds, primarily C4
C12 olefins and some small amounts of aromatic hydrocarbons are produced.
TABLE 1
SUMMARY OF OXYGENATE CONVERSION SELECTIVITIES FOR SAPO-
io _34
WITH INCREASING REACTOR TEMPERATURE
RUN A B C
Inlet Temp (C) 435 455 475
Pressure (kPa) 138 138 138
MeOH WHSV (hr') 1.0 1.0 1.0
Catalyst Life 4.3 3.3 2.3
(hr at > 99% Conv)
Selectivities
at 99% Conversion
(mole %)
C1
3.3 4.7 7.7
C2
0.6 0.6 0.8
C2
50.3 52.4 53.7
C3
0.4 0.4 0.5
C
33.5 30.8 27.0
C4S
9.0 7.9 6.9
CS
S
+ 2.0 3.1 3.2
C2-/C3 Ratio 1.50 1.70 1.99
EXAMPLE II
~5 By converting the butene and heavier compounds over the same catalyst
at effective conditions, it was proposed that improved ethylene yields would
result. The conversion of butenes over an aluminophosphate catalyst was
CA 02297301 2000-O1-18
evaluated with a catalyst containing 40 weight percent SAPO-34 and a binder
for a series of reactor temperatures to simulate riser reactor operation in a
fixed bed reactor. Approximately 20 g of catalyst were loaded into a 2.2 cm
(7/8
inch) ID porcelain-lined, stainless steel reactor and placed in a 3-zone
bronze
block furnace. The reactor was heated to temperatures between 460 and 580
°C
and purged with nitrogen for approximately 1 hour to precondition the
catalyst. The nitrogen flow was stopped and 2-butene was introduced at a rate
of 0.5 hr' WHSV on catalyst for a period of 5 hours.
According to the above procedure, the reactor was filled with the SAPO-
l0 34 containing catalyst and was heated to a temperature of 460 °C.
Following 1
hour of purging with nitrogen, vaporized 2-butene was introduced to the
reactor at a rate of 0.5 WHSV on catalyst. The conversion of the 2-butene
gradually decreased from an initial value of 66 percent to a value near the
end
of the run of 44 percent at 4.5 hours on stream. The selectivity to ethylene
began at 7% and decreased to 6% over the same period. The selectivity to
propylene began at 25% and increased to 30% over the same period.
EXAMPLE III
The procedure of Example II was repeated at a reactor temperature of
580°C. At 580 °C, the initial C4 olefin conversion was 75% and
decreased more
2o rapidly than in Example II, reaching a value of 30% after 4.5 hours on
stream. The selectivity to ethylene initially was 20% and decreased to 8%
after 4.5 hours on stream. The selectivity to methane initially was 10% and
increased to 17% after 4.5 hours on stream. The selectivity to propylene
initially was 35% and decreased to 32% after 4.5 hours on stream.
2s EXAMPLE IV
The procedure of Example II was repeated at a reactor temperature of
580 °C and after heating and purging the reactor with nitrogen for 1
hour, the
21
CA 02297301 2000-O1-18
2-butene vapor was introduced at 75% dilution with nitrogen. The conversion
of C4's was 70% initially and decreased to 5% after 4.5 hours on stream. The
selectivity to ethylene initially was 22% and decreased to 12 percent after
4.5
hours on stream. The selectivity to methane initially was 3 percent and
increased to 12 % after 4.5 hours on stream. The selectivity to propylene was
55 % initially and decreased to 45% after 4.5 hours on stream.
22