Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
TITLE OF THE INVENTION
NOVEL BENZODIAZEPINE DERIVATIVES AS ANTIARRHYTHMIC
AGENTS
BACKGROUND OF THE INVENTION
Arrhythmias often occur as complications to cardiac
diseases such as myocardial infarction and heart failure. In serious
cases, arrhythmias may give rise to ventricular fibrillation which can
cause sudden death.
According to the classification of Vaughan-Williams,
there are four distinct classes of antiarrhythmic agents. Class I
antiarrhythmic agents are those agents which provide for sodium
channel blockade, including those compounds which exert a membrane
stabilizing effect. Exemplary of this class of compounds are quinidine,
procainamide, disopyramide, lidocane, tocainide, flecainide and
propafenone. Class II antiarrhythmic compounds are agents which
block sympathetic activity. Exemplary of this class of compounds are
propranolol and acebutolol. Class III antiarrhythmic agents are
compounds which prolong the effective refractory period without
altering the resting membrane potential or rate of depolarization.
Compounds such as amiodarone, bretylium and sotalol are considered
to be in this class. Class IV antiarrhythmic agents are effective in
calcium channel blockade. Exemplary of this class of compounds are
diltiazem and verapamil. Further definition of these classes can be
found in Pharma Projects, section C1B, May 1993.
Though various antiarrythmic agents are now available on
the market, agents which exhibit both satisfactory effects and high safety
profiles have not been marketed, For example, antiarrythmic agents of
Class I, which cause a selective inhibition of the maximum velocity of
the upstroke of the action potential (Vmax), are inadequate for prevent-
ing ventricular fibrillation. In addition, they have problems regarding
safety as they cause a depression of the myocardial contractility and
have a tendency to induce arrythmias due to an inhibition of the impulse
conduction. Beta-adrenoceptor blockers and calcium antagonists which
-1-
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/Z1597
belong to Class II and IV, respectively, have a defect in that their effects
are either limited to a certain type of arrhythmia or are contraindicated
because of their cardiac depressant properties in certain patients with
cardiovascular disease. Their safety, however, is higher than that of the
antiarrhythmic agents of Class I.
Antiarrythmic agents of Class III are drugs which cause
a selective prolongation of the duration of the action potential without
a significant depression of the Vmax. Drugs in this class are limited.
Drugs such as sotalol and amiodarone have been shown to possess Class
III properties. Sotalol also possesses Class II effects which can cause
cardiac depression and is therefore contraindicated in certain suscept-
ible patients. Amiodarone is also severely limited by side effects. Drugs
of this class are expected to be effective in preventing ventricular
fibrillations. Pure Class III agents, by definition, are not considered
to cause myocardial depression or an induction of arrhythmias due to
the inhibition of the action potential conduction as seen with Class I
antiarrhythmic agents.
U.S. Patent No. 5,658,901 discloses novel 2-oxo-1,4-
benzodiazepines which are potent Class III agents. Compounds in this
patent, while extremely potent, display extended half lives in animals,
resulting in a high potential for toxicity. The compounds of the present
invention show a decreased half life in animals.
U.S. Patent Nos. 5,595,900 and 5,631,251 disclose additional
antiarrhythmic benzodiazepines.
SUMMARY OF THE INVENTION
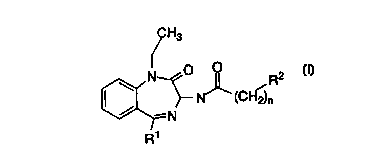
This invention relates to compounds of the general formula
H3
\ N O ~ R2
N~ cc~f j~
-N
R'
-2-
CA 02306960 2000-04-12
WO 99/Z0281 PCT/US98/21597
wherein
Rl is
1 ) phenyl,
2) substituted phenyl, with one or two substituents, selected
from the group consisting of
a) -hydrogen,
b) -C1, Br, F, or I,
c) -CFg,
d) -C1_3 alkyl,
e) -C1_3 alkoxy, or
3) cyclopropyl;
R2 is
1) phenyl,
2) substituted phenyl, with one or two substituents, selected
from the group consisting of
a) -hydrogen,
b) -C1, Br, F, or I,
c) -CFg,
d) -C1_g alkyl,
e) -C1_g alkoxy, and
n is 0, 1 or 2;
or a pharmaceutically acceptable addition salt and/or hydrate thereof, or
crystal form thereof, or where applicable, a geometric or optical isomer
or racemic mixture thereof.
The invention also relates to pharmaceutical compositions
containing said compounds for use in mammals and methods of
treating arrhythmia by the administration of one or a combination of the
novel compounds of the invention.
DETAILED DE~CRIPTIOhT OF THE INVEN~rTnN
This invention relates to compounds of the general formula
-3-
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
H3
\ N O ~ R2
N (C~~
=-N
R'
wherein
Rl is
1) phenyl,
2) substituted phenyl, with one or two substituents, selected
from the group consisting of
a) -hydrogen,
b) -Cl, Br, F, or I,
c) -CF3,
d) -C1-3 alkyl,
e) -C1_g alkoxy, or
3) cyclopropyl;
R2 is
1) phenyl,
2) substituted phenyl, with one or two substituents, selected
from the group consisting of
a) -hydrogen,
b) -C1, Br, F, or I,
c) -CF3,
d) -C1_3 alkyl,
e) -C1_3 alkoxy, and
n is 0, 1 or 2;
or a pharmaceutically acceptable addition salt and/or hydrate thereof, or
crystal form thereof, or where applicable, a geometric or optical isomer
or racemic mixture thereof.
-4-
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
The compounds of the present invention may have
asymmetric centers and occur as racemates, mixtures of enantiomers,
individual diastereomers, or as individual enantiomers with all
isomeric forms being included in the present invention.
A preferred embodiment of this invention is represented by
H3
O O
N ~ R2
N (C~"
-N
wherein
R2 is
1) substituted phenyl, with one or two substituents, selected
from the group consisting of
a) -hydrogen,
b) -Cl, Br, F, or I,
c) -CFg,
d) -C 1-g alkyl, and
n is 0, 1 or 2.
A second preferred embodiment of this invention is
represented by
-5-
CA 02306960 2000-04-12
WO 99!20281 PCT/US98/21597
H3
\ N O ~ R2
/ ~ N ~C~n
N
wherein
R2 is
1) substituted phenyl, with one or two substituents, selected
from the group consisting of
a) -hydrogen,
b) -Cl, Br, F, or I,
c) -CF3,
d) -C1-3 alkyl, and
n is 0, 1 or 2.
Unless otherwise stated or indicated, the following
definitions shall apply throughout the specification and claims.
When any variable occurs more than one time in any
constituent or in formula I, its definition on each occurrence is
independent of its definition at every other occurrence. Also,
combinations of substituents and/or variables are permissible only if
such combinations result in stable compounds.
The term "alkyl" is intended to include both branched-
and straight-chain saturated aliphatic hydrocarbon groups having the
specified number of carbon atoms, e.g., methyl (Me), ethyl (Et), propyl,
butyl, pentyl, hexyl, heptyl, octyl, nonanyl, decyl, undecyl, dodecyl, and
the isomers thereof such as isopropyl (i-Pr), isobutyl (i-Bu), sec-butyl
(s-Bu), tert-butyl (t-Bu), isopentane, isohexane, etc.
The term "halogen" or "halo" is intended to include
fluorine, chlorine, bromine and iodine.
-6-
CA 02306960 2000-04-12
WO 99120281 PCT/US98121597
As used herein, the term "composition" is intended to
encompass a product comprising the specified ingredients in the
specified amounts, as well as any product which results, directly or
indirectly, from combination of the specified ingredients in the specified
amounts.
The optical isomeric forms, that is mixtures of
enantiomers, e.g., racemates, or diastereomers as well as individual
enantiomers or diastereomers of the instant compound are included.
These individual enantiomers are commonly designated according to
the optical rotation they effect by the symbols (+) and (-), (L) and (D), (1)
and (d) or combinations thereof. These isomers may also be designated
according to their absolute spatial configuration by (S) and (R), which
stands for sinister and rectus, respectively.
The individual optical isomers may be prepared using
conventional resolution procedures, e.g., treatment with an appropriate
optically active acid, separating the diastereomers and then recovering
the desired isomer. In addition, the individual optical isomers may be
prepared by asymmetric synthesis.
Additionally, a given chemical formula or name shall
encompass pharmaceutically acceptable addition salts thereof and
solvates thereof, such as hydrates.
The compounds of the present invention, while effective
themselves, may be formulated and administered in the form of their
pharmaceutically acceptable addition salts for purposes of stability,
convenience of crystallization, increased solubility and other desirable
properties.
The compounds of the present invention may be
administered in the form of pharmaceutically acceptable salts. The
term "pharmaceutically acceptable salt" is intended to include all
acceptable salts. Examples of acid salts are hydrochloric, nitric,
sulfuric, phosphoric, formic, acetic, trifluoroacetic, propionic, malefic,
succinic, malonic, methane sulfonic and the like which can be used as a
dosage form for modifying the solubility or hydrolysis characteristics or
can be used in sustained release or prodrug formulations. Depending
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/Z1597
on the particular functionality of the compound of the present invention,
pharmaceutically acceptable salts of the compounds of this invention
include those formed from cations such as sodium, potassium,
aluminum, calcium, lithium, magnesium, zinc, and from bases such
as ammonia, ethylenediamine, N-methyl-glutamine, lysine, arginine,
ornithine, choline, N,N'-dibenzylethylenediamine, chloroprocaine,
diethanolamine, procaine, N-benzylphenethylamine, diethylamine,
piperazine, tris(hydroxymethyl)aminomethane, and tetramethyl-
ammonium hydroxide. These salts may be prepared by standard
procedures, e.g. by reacting a free acid with a suitable organic or
inorganic base, or alternatively by reacting a free base with a suitable
organic or inorganic acid.
Also, in the case of an acid (-COOH) or alcohol group being
present, pharmaceutically acceptable esters can be employed, e.g.
methyl, ethyl, butyl, acetate, maleate, pivaloyloxymethyl, and the like,
and those esters known in the art for modifying solubility or hydrolysis
characteristics for use as sustained release or prodrug formulations.
The compounds of the present invention may have chiral
centers other than those centers whose stereochemistry is depicted in
formula I, and therefore may occur as racemates, racemic mixtures
and as individual enantiomers or diastereomers, with all such
isomeric forms being included in the present invention as well as
mixtures thereof. Furthermore, some. of the crystalline forms for
compounds of the present invention may exist as polymorphs and as
such are intended to be included in the present invention. In
addition, some of the compounds of the instant invention may form
solvates with water or common organic solvents. Such solvates are
encompassed within the scope of this invention.
The compounds of the invention are prepared by the
following reaction schemes. All substituents are as defined above unless
indicated otherwise.
_g_
CA 02306960 2000-04-12
WO 99/ZOZ81 PCT/US98/21597
Scheme 1
CHs
H O ~ O
N Et_I N
/ , N Cs2C03 ~ - N
DMF, RT
\ I \ I
0
O.S~Ns THF DS
CH3
O CHs
N ~ O
/ NH2 H2, Pd/C ( N3
''N ~ / -N
EtOAc
\I \I
O O
N
/ , N H NHCbz
I H2, Pd/C
D-Cbz-Phe-OH, EDC, HOBT
CH3
-9-
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
Scheme 1 ~~nt'd.
CH3
O O
N
E / .N H NH2
Separate Diastereomers
CH3 \ I CH3
O ~ O
N
I / H- D-Phe-H I / "" H- D-Phe-H
~N --N
\ I \ I
1 ) PhNCS
CH3 2) TFA CH3
O ~ O
N ~ N
NH2 ( "" NH2
/ -N / -N
\ / \ /
R(CH2)nCOCI or
R(CH2)~C02H, EDC, HOBT 1
CH3 ~CH O
O O N O
I N--~--(CH2)~ R I ' ""N~--(CH2)ri R
/ ,N H / ,N H
n=0,1, or 2 ''
\ I ~Y \ I
R = I j X X,Y = H, CI, CF3, Br
- 10 -
CA 02306960 2000-04-12
WO 99/20281 PCT/LJS98/21597
Scheme 2
NH2 D-MgBr ~ NH2
THF I ~ O
CN
1. BrCH2COBr
CH3 2. NH3
O H O
\ N Et-I ~ N
N Cs2CO3 I ~ ..- N
DMF, RT
O
KOtBu ~~ S~ N3
THF ~ ,
~CH3 i
O CHs
N ~ O
N3 H2, Pd/C \
2
N EtOH I / ~ N NH
RC02H
EDC
HOST
DMF
'CH3 'CH3
O ( O
\ N ~ Sep. \ N
....H R - ~ / H R
~N / N
- 11 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
The novel compounds of the present invention, have the
pharmacological properties required for antiarrhythmic agents of Class
III, namely the prolongation of the myocardial action potential ~_n vitro,
without a significant depression of the Vmax, and the prolongation of ,
QTc-interval in anesthetized dogs.
These compounds are effective in treating and preventing
all types of arrhythmias including ventricular and atrial (supra-
ventricular) arrhythmias. The compounds of the present invention are
especially useful to control reentrant arrhythmias and prevent sudden
death due to the ventricular fibrillation. These compounds are also
effective in treating and preventing impaired cardiac pump functions.
In the novel method of this invention of treating
arrhythmia, one of the compounds or pharmaceutically acceptable
salt thereof, is administered in an amount ranging from about 0.0001
to about 20 mg per kg of body weight per day, preferably from about 0.001
to about 5.0 mg per kg of body weight per day in a single dose or in 2 to 4
divided doses.
These compounds, or pharmaceutically acceptable salts
thereof, in the described dosages, are administered orally, intraperitone-
ally, subcutaneously, intramuscularly, transdermally, sublingually
or intravenously. They are preferably administered intravenously or
orally, for example in the form of tablets, troches, capsules, elixirs,
suspensions, emulsions, syrups, wafers, chewing gum, or the like
prepared by art recognized procedures. The amount of active compound
in such therapeutically useful compositions or preparations is such that
a suitable dosage will be obtained.
These compounds can be administered as the sole active
ingredient or in combination with other antiarrhythmic agents or
other cardiovascular agents, such as Class I, Class II or Class IV
antiarrhythmic agents, vasodilators, angiotensin converting enzyme
inhibitors, angiotensin II antagonists, diuretics or digitalis.
These compounds can be administered as a method
- 12 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
of treating arrhythmia and impaired cardiac pump functions in
conjunction with defibrillators, including implantable defibrillators.
These compounds reduce the frequency of defibrillator firing.
Exemplary of vasodilators are compounds such as
papaverine and isosorbide dinitrat. Examples of angiotensin converting
enzyme inhibitors include enalapril, lisinopril and captopril. Examples
of diuretics include hydrochlorothiazide and acetazolamide. The
pharmaceutical agents listed herein are examples and do not represent
a complete listing of the many compounds in these classes which are
contemplated by this invention.
The activity of the compounds described herein as
antiarrhythmic agents is measured by their ability to block the IKs
and IKr currents as determined by the following test protocol.
Outward potassium currents are measured in single
guinea pig ventricular myocytes using a whole-cell voltage clamp
technique described in detail elsewhere (Sanguinetti and Jurkiewicz,
1990, Two components of cardiac delayed rectifier K+ current:
differential sensitivity to block by Class III antiarrhythmic agents.
J. Gen Physiol. 96: 195-215). Myocytes are isolated by enzymatic
(collagenase and protease) digestion of Langandorf perfused hearts.
Single cells are then voltage clamped using 1 mm square-bore pipettes
filled with 0.5 M Kgluconate, 25 mM KCI, 5 mM K(2)ATP. Cells are
bathed in a solution containing, in mN: 132 NaCl, 4KC1, 1.2 MgCl[2],
10 HEPES, 10, glucose: pH 7.2, temp. 35°C.
Each cell is maintained at a holding potential of -50 mV.
Test depolarizations are applied as voltage ramps from -85 to -50 mV,
and as steps to -10 mV (0.5 s) and +50 mV (1.0 s). IKI is measured as
peak outward current during the voltage ramp. IKr is measured as tail
currents upon repolarization from -10 mV to -50 mV. IKs is measured
as time-dependent current during the pulse to +50 mV. Currents are
measured during control, then after exposure to drug at two different
concentrations.
Employing this test the compounds described herein have
- 13 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
an IC50 of less than 1,000 nM as IKs blockers. The compounds of this
invention are at least 10 times more potent in the blockade of IKs than
the blockade of IKr.
The following examples illustrate the preparation of some. of
the compounds of the invention and are not to be construed as limiting
the invention disclosed herein.
~+)-2-t2,4-bis-Trifluoromethylnhenvl)-N-f2 3-di fro-1-et 1-2-oxo-5-
phenyl-1H-14-benzofel f ~, 4ldiazenin-3~r11 acetamide
CH3
O
N O i CF3
.,.. N ~ I
-.. N H
F3C
Step A: Preparation of 2,3-di ydro-'1-et vl-2-oxo-5-phenyl-1H-1, 4
benzofel f 1,41diazepine
A solution of 5-phenyl-1,4-benzo[e] [1,4]diazepine-2-one (23.6
g, 0.1 mole) in DMF ( 100 mL) was treated with potassium carbonate
(20 g, 0.14 mole) and ethyl iodide (19 g, 0.12 mole). The mixture was
stirred at room temperature for five hours. The reaction mixture was
then poured into water (1L) and extracted with ethyl acetate (3 X 300
mL). The combined ethyl acetate fractions were dried over anhydrous
magnesium sulfate, filtered and concentrated at reduced pressure to
give I7.2 g of the product.
IH NMR (CDC13) 8 ?.65-7.6(m, 2H), 7.50-7.35 (m, 3H), 7.35-7.20(m, 2H),
7.25-7.10(m, 2H), 4.79(d, J = 10 Hz, 1H), 4.30 (hex, J = 7.1 Hz, 1H), 3.75 (d,
J =10 Hz, 1H), 3.7 (hex, J = 7.1 Hz, IH), 1.05 (t, J = 7.I Hz, 3H)
- 14 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597 .
Step B: Preparation of 3-Azido-2,3-dihvdro-1-eth~rl-2-oxo-5- phen ~~1-
1H-1~4-benzofelf~,4ldiaze ine
To a solution of 2,3-dihydro-1-( ethyl)-2-oxo-5-phenyl-1H-
1,4-benzo[e] [1,4]diazepine (13 g, 0.049 mole) in THF (500 mL) at -70°C
.
was added a solution of potassium tert-butoxide in THF (57 mL of a 1 N
solution, 0.057 mole). The solution was then treated with a solution of
triisopropylbenzenesulfonyl azide (16.3 g, 0.052 mole) in THF (100 mL).
The reaction was stirred at -70°C for 10 minutes and then treated
with
acetic acid (1 mL in 5 mL of THF) and warmed to room temperature over
one hour. The reaction mixture was then poured into water (500 mL)
and extracted with ethyl acetate (3 X 250 mL) The combined ethyl acetate
fractions were washed with water (200 mL), and brine (200 mL). The
ethyl acetate solution was then dried over anhydrous magnesium
sulfate, filtered and concentrated at reduced pressure to afford 9.3 g of
the product.
1H NMR (CDC13) b 7.75-7.65(m, 2H), 7.65-7.56(m, 1H), 7.54-7.34 (m, 4H),
7.30-7.20(m, 2H), 4.52(s, 1H), 4.3(hex, J = 7 Hz, 1H), 3.8(hex, J = 7 Hz,
1H), 1.15 (t, J = ?.0 Hz, 3H).
Step C: Preparation of racemic 3-Amino-2;.3-dihydro-1- ethyl-2
oxo-5-8henvl-1H-1,4-benzofel f~ 4ldiazenine
To a suspension of 10% palladium on carbon (1 g) in ethyl
acetate (300 mL) was added a solution of 3-azido-2,3-dihydro-1-ethyl-2
oxo-5-phenyl-1H-1,4-benzo[e] [1,4)diazepine (9.1 g, 0.029 mole) in ethyl
acetate (25 mL) at room temperature. Hydrogen gas was then bubbled
into the reaction until the starting material was consumed (5 hr). The
catalyst was filtered ofF and the ethyl acetate was concentrated at
reduced pressure to give 7.1 g of the product.
1H NMR (CDC13) 8 7.65-7.50(m, 3H), 7.50-7.35(m, 4H), 7.35-7.20(m,
2H), 4.48(x, 1H}, 4.3(hex, J = 7.1 Hz, 1H}, 3.8(hex, J = 7.1 Hz, 1H), 1.15
(t, J = 7.1 Hz, 3H).
- 15 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
Step D: Preparation of (2R)-2-Amino-3-phenyl N-f2 3 dih- dv ro 1
ethyl-2-oxo-5 yl-1H-1,4-benzo(el ~1~41diaz,~3 yll
pronionamide
To a stirring solution of (~)-3-amino-2,3-dihydro-1-
( ethyl)-2-oxo-5-phenyl-1H-1,4-benzo[e][1,4]diazepine (7 g, 26.4 mmol) in.
dimethylformamide (150 mL) was added EDC (5.57 g, 29 mmol), HOBT
(2.35 g, 17.4 mmol) and N-Cbz-D-phenylalanine (8.68 g, 29 mmol). The
solution was stirred at ambient temperature for 2 h. The reaction was
diluted with saturated aqueous sodium hydrogen carbonate (1 L) and
extracted with ethyl acetate (3x 300 mL). The organic layers were
combined, dried with brine, anhydrous magnesium sulfate, filtered, and
evaporated under reduced pressure to give an oil (15.3g) The material
was dissolved in ethanol (100 mL) and added to a suspension of 10%
palladium on carbon. Hydrogen gas was then bubbled into the reaction
until the starting material was consumed (24 hr) The catalyst was
filtered off and the ethanol was concentrated at reduced. The residue
was chromatographed over silica (2.5 kg) with ethyl acetate). The faster
running diastereomer was recovered as a solid (5.0 g).
1H NMR, CDC13, 8 9.01 (d, J=8.4 Hz, 1H), 7.64-7.I9 (m, 14H), 5.55 (d,
J=8.4 Hz, 1H), 4.35 (hex, J=7.1 Hz, 1H), 3.80 (hex, J=7.1 Hz, 1H), 3.70 (dd,
J=3.9, 9.6 Hz, 1H), 3.35 (dd, J= 3.9, 13.9 Hz, 1H), 2.82 (dd, J=9.6, 13.9 Hz,
1H), 1.27 (t, J=7.1 Hz, 1H).
Step E: Preparation of (+)-3-Amino-2,~-~ r~hycLro 1 ethyl 2 oxo 5
p. ienvl-1H-1,4-benzo(el f~,,41 'aze ine
To a stirring solution of (2R)-2-amino-3-phenyl-N-[2,3-
dihydro-1 ethyl 2-oxo-5-phenyl-1H-1,4-benzo[e][1,4]diazepin-3-yl]
propionamide (5 g, 11.7 mmol) in methylene chloride (50 mL) was added
phenyl isothiocyanate (1.5 mL, 1.66 g, 12.3 mmol) and the resulting
solution was stirred for 16 h. The reaction mixture was cooled in an
ice/water bath. Trifluoroacetic acid (9 mL, 13.3 g, 117 mmol) was added
dropwise and the resulting solution was allowed to warm to ambient
temperature over 5 hours. The reaction mixture was concentrated cold
under reduced pressure to yield an oil which was chromatographed
- 16-
CA 02306960 2000-04-12
WO 99/20281 PCTNS98/21597
over silica (1 kg) with 90:10:1:1 methylene chloride:methanol:acetic
acid:water. The pure fractions were combined and concentrated at
reduced pressure and then partioned between ethyl acetate and
saturated sodium bicarbonate. The organic layers were combined,
dried with brine, anhydrous magnesium sulfate, filtered, and
evaporated under reduced pressure to afford 2.2 g of product as a foam.
1H NMR (CDCl3) S 7.65-7.50(m, 3H), 7.50-7.35(m, 4H), 7.35-7.20(m,
2H), 4.48(s, 1H), 4.3(hex, J = 7.1 Hz, 1H), 3.8(hex, J = ?.1 Hz, 1H),
1.15 (t, J = ?.1 Hz, 3H).
[a]D=+145.7° (c=0.35; MeOH)
Step F: Preparation of (+)-2-(2,4-bis trifluoromethylDheny~N-f2 3
dihvdro-1- ethyl -2-oxo-5-phenyl-1H-~,4-
benzofel f 1,4. ldiazenin-3-vllacetamide
To a stirring solution of (+)-3-amino-2,3-dihydro-1- ethyl-
2-oxo-5-phenyl-1H-1,4-benzo[e][1,4]diazepine (200 mg, 0.71 mmol) in
dimethylformamide (4 mL) was added EDC ( 150 mg, 0.783 mmol), HOBT
(95 mg, 0.71 mmol) and 2,4-bis trifluoromethylphenylacetic acid (215 mg,
0.78 mmol). This was stirred at ambient temperature for 0.5 h. The
reaction was diluted with saturated aqueous sodium hydrogen carbonate
(50 mL) and extracted with ethyl acetate (3x50 mL). The organic layers
were combined, dried with brine, anhydrous magnesium sulfate,
filtered, and evaporated under reduced pressure to give a colorless oil
which was chromatographed over silica with 25 to 50% ethyl acetate/
hexane. The resulting foam was crystallized from ethyl acetate/hexane
to give a solid (320 mg).
[a]D=+24.6° (c=0.45; MeOH), 1H NMR, CDCl3, S 7.95 (br s, 1H), 7.84-7.70
(m, 2H), 7.60-7.20 (m, 9H), 5.49 (d, J=8.2 Hz, 1H), 4.32 (hex, J=7.1 Hz, 1H),
3.90 (br s, 2H), 3.78 (hex, J=7.1 Hz, 1H), 1.12 (d, J=7.1 Hz, 3H). Anal.
Calcd. for C27H21F6N302 ~ 0.1 EtOAc ~ 0.35 H20:
C, 59.99; H, 4.13; N, 7.66.
Found: C, 60.61; H, 3.85; N, 7.65%.
- 17 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
EXAMPLE 2
The following compound was prepared in the same manner
as Example 1.
(+)-3,5-bis trifluoromethvl-N-f2 3-dihydro- -ethyl-2-oxo-
5-phenyl-1H-1,4-benzofel l1,41 diazepin-3-yll acetamide
CH3 CF3
O
N O .i
"~~ N \
-- N H CFs
[a]D=+23.4° (c=0.49; MeOH)
1H NMR, CDC13, 8 7.84-7.78 (m, 3H), 7.60-7.20 (m, 9H), 5.51 (d, J=7.9 Hz,
1H), 4.32 (hex, J=7.1 Hz, IH), 3.84 (br s, 2H), 3.79 (hex, J=7.1 Hz, 1H), 1.14
(d, J=7.1 Hz, 3H).
Anal. Calcd. for C27H21F6N302 ~ 0.05 EtOAc ~ 0.6 H20:
C, 59.54; H, 4.15; N, 7.71.
Found: C, 59.55; H, 3.95; N, 7.70%.
- 18 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98121597
EXAMPLE 3
(-)-2-(2,;,,4-ms's-trifluorometh,~~lnhenvl)-N-f 2a,3-dihydro-1-ethyl-2-
oxo-5-cvcloprop3~1-1H-1..4-benzofelfl,4ldiazepin-3-yllacetamide
rCH3
N O O
/ _
\ ~ ~ N H ~ ~ CFs
F3C
StepA 5-c.~lo~ropyl-1,4-benzofelf1,41diazepin-2-one
To a solution of anthranilonitrile (85 g, 0.720 mole) in
THF (1.0 L) at =10°C was slowly added a 1.6 M solution of
cyclopropyl
magnesium bromide in THF (1.55 L, 2.48 mole). The reaction was
allowed to stir overnite at room temperature then slowly quenched inta
a -10°C solution of 4N HCL (1.2 L). The mixture was stirred for 1 hour
at room temperature and the pH adjusted to 7.5 with lON sodium
hydroxide. The THF layer was removed, the aqueous layer washed
with ethyl acetate (800 mL), and the organic extracts concentrated
in uaccuo to an oil. The oil was dissolved in methylene chloride ( 1.2 L),
washed with water (500 mL), dried over sodium sulfate, and filtered.
To the methylene chloride filtrate at 0°C was slowly added
bromoacetyl
bromide (168.0 g, 0.836 mole) followed by 3N sodium hydroxide (800mL).
The reaction was allowed to stir for 1 hour and the pH of the mixture
adjusted to 7.5 with concentrated hydrochloric acid. The methylene
chloride layer was removed and the aqueous layer washed with
methylene chloride (1.0 L). The methylene chloride extracts were
washed with 5% aqueous sodium bicarbonate, dried over sodium sulfate,
filtered and concentrated to an oil. The oil was dissolved in ethanol (1.5
L) added to a 50% solution of ethanol/ aqueous ammonium hydroxide (6.3
L) and allowed to stir for 48 hours. The reaction mixture was concen-
- 19 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
trated in vaccuo to 2.7 L and the pH adjusted to 12.0 with 50% sodium
hydroxide. After stirring at pH 12 for 1 hour, the reaction pH was
adjusted to 8.5 with concentrated hydrochloric acid and the solids were
filtered. The cake was washed with water (1.0 L), sucked dry and dried
in uacuo at 40°C to give 102.2 g of 5-cyclopropyl-1,4-
benzo[e][1,4]diazepine-2-one. MP- 192-193 °C
1H NMR (CDC13~ 300 MHz) b 9.45 (s, 1H) 7.84 (dd, J=8.0 and 1.6 Hz,IH),
7.45 (dt, J=8.0 and 1.6 Hz, 1H), 7.24 (dt, J=8.0 and 1.6 Hz,lH), ?.12 (dd,
J=8.0 and 1.6 Hz, 1H),4.04 (br s. 2H), 1.95 (m,lH), 0.9-1.2 (m, 4H)
Step B. Preparation of 2,3-dihvdro-1-ethyl-2-oxo-5-cyclopropy-1-
1H-1,4-benzofel f 1.41diazep~r~e
A solution of 5-cyclopropyl-1,4-benzodiazepine-2-one (50 g,
0.250 mole) in DMF (250 mL) was treated with cesium carbonate (122 g,
0.375 mole) , cooled to 0° C and iodoethane (47.2 g, 0.30 mole} added.
The
mixture was stirred at 25°C overnight. The reaction was cooled to room
temperature and filtered. The solids were washed with ethyl acetate
(2L) and the filtrates diluted with water (2L). The pH was adjusted to
7.6 with 10% aqueous potassium hydrogen sulfate and the organic layer
removed. The aqueous layer was washed with ethyl acetate (1L) and
combined ethyl acetate extracts washed with water (500mL). The ethyl
acetate extracts were dried over sodium sulfate, filtered and
concentrated in uczcuo to yield 57.5 g of an oil. The oil was taken up in
toluene (200mL} and concentrated. The oil was used in the next step
without further purification.
Step C. Preparation of 3-azido-5-cvcloprogvl-1-ethyl-1H-
benzofel f 1,41diaze~ine
To a stirring solution of 5-cyclopropyl-1-ethyl-lHbenzo[e) [1,4]
diazepine (6.4 g, 0.028 mol) in THF (125 ml) cooled to -78°C was added
potassium tert-butoxide (2.05 eq, 0.057 mol, 57.2 ml of a 1 M solution
in THF) dropwise over 15 min. A solution of 2,4,6-triisopropylphenyl-
-20-
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
sulfonylazide (9.5 g, 0.031 mol) in THF (100 ml) was added over 5 min.
The solution was stirred for 10 min, acetic acid (7.7 m1,0.123 mol) was
added and the reaction warmed to 30°C for 2 hour. The reaction was
concentrated the residue dissolved in dichloromethane (200 mL) and .
washed with satd. NaHC03 (200 ml). The aqueous layer was back
extracted with dichloromethane (100m1). The organic layers were
combined, dried with Na2S04 and evaporated to a foam. The foam was
chromatographed over silica eluting with 30% ethyl acetate:hexane. The
appropriate fractions were collected and evaporated under reduced
pressure to give 5.83 g of a powder. MP =158-160°C
Step D. Preparation of 3-amino-5-cyclo~roggl-1-ether
benzofel f 1.41diazenine
To a stirred solution of 3-azido-5-cyclopropyl-1-ethyl-1H-
benzo[e] [1,4]diazepine (5.8 g, 21.5 mmol) in ethanol (500 mL) under
argon was added 10 % palladium on charcoal (1.0 gm). The argon was
displaced with hydrogen and the mixture was vigorously stirred for
45min (1 atm hydrogen). The reaction was filtered and the filtrate
concentrated in vacuo to an oil (5.3 gm, 100 %) which solidified upon
standing. MP 93-95°C
Step E. Preparation of (+,-)-2-(2,4-bis-trifluorometl~vlphe~~1) -N-
f2.3-dihydro-1-ethyl-2-oxo-5-cyclouropyl-1H-1,4-
benzo f el f 1.41 diaze ip n-3-vll acetamide
To a stirring solution of 3-amino-5-cyclopropyl-1-ethyl-
1H-benzo[e][1,4] diazepine (0.10 g, 0.409 mmole) in DMF (2 mL) was
added 1-hydroxybenztriazole hydrate (88 mg, 0.573 mmol), 2,4-bis-
trifluoromethylphenylacetic acid (111 mg, 0.45 mmol), triethylamine
(80 L, 0.573 mmole), and (1-(3-dimethylaminopropyl-3-ethylcarbodiimide
(110 mg, 0.573 mmol). The solution was stirred at ambient temperature
for 2 hours. The reaction was diluted with 10% citric acid (20 mL} and
extracted with ethyl acetate (2 x 40 mL) . The combined organics were
washed with 10% sodium bicarbonate (20 mL) dried over Na2S04, and
evaporated to a foam. The foam was chromatographed using a
- 21 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
Chiralpak AD~ preparative HPLC column (Chiral Technology, 25x2 cm)
eluting with ethanol/hexane( 1/1, 6 mlJmin). The pure fractions were
collected and evaporated under reduced pressure to give 95 mg of the (+)
enantiomer and 95 mg of the (-) enantiomer as foams. The enantiomers
were crystallized from ether/hexane to give 80 mg (+) enantiomer and 80
mg (-) enantiomer as solids.
(+)enantiomer
1H NMR (CDC13, 300 MHz) 8 7.90 (s, 1H), 7.78 (d, J=8.6 Hz, 2 H),7.68 (d,
J=8.6 Hz,lH), 7.54(dt, J= 8.6 and 1.6 Hz, 1H), ?.32 (t, J= 8.6, 1 H), 7.29 (d,
J=8.6 Hz, 1H), 7.18 (br d, J=8.6 Hz, l H), 5.24 (d, J=8.6 Hz, l H), 4.15
(sextet, J=8.7 Hz, 1H), 3.89 (s, 2H), 3.75 (sextet, J=8.7 Hz, 1H), 1.95 (m, 1
H), 1.12 (t, J=8.5 Hz,3H), 0.8-1.1 (m, 4H).
Analysis Calcd. for C24H21F6N302:
C, 57.95; H, 4.26; N, 8.45;
Found: C, 58.13; H, 4.34; N, 8.12.
mp=124-125°C, [a]D= +19.2° (MeOH),
(-)enantiomer
1H NMR (CDCl3, 300 MHz) 8 7.90 (s, 1H), 7.78 (d, J=8.6 Hz, 2 H),7.68 (d,
J=8.6 Hz,lH), 7.54(dt, J= 8.6 and 1.6 Hz, 1H), 7.32 (t, J= 8.6, 1 H), 7.29 (d,
J=8.6 Hz, 1H), 7.18 (br d, J=8.6 Hz,l H), 5.24 (d, J=8.6 Hz,l H), 4.15
(sextet, J=8.7 Hz, 1H), 3.89 (s, 2H), 3.75 (sextet, J=8.7 Hz, 1H), 1.95 (m,
1 H), 1.12 (t, J=8.5 Hz,3H), 0.8-1.1 (m, 4H).
Analysis Calcd. for C24H21F6N302:
C, 57.95; H, 4.26; N, 8.45;
Found: C, 57.83; H, 4.25; N, 8.10.
mp=124-125°C, [a]D= -19.3° (MeOH),
Following procedures similar to those described above for
the preparation of Examples 1-3, the following compounds were
prepared:
- 22 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
~CH3
O
N-~
~ I ~N
-N
O
Ex. # R M.P. °C Stereochemist
4a ~ 124125 (+)
F3C / CF3
125-126 (-)
F3C / CF3
5a ~ CF3 160-161 (+)
/
CF3
5b ~ CF3 159-160 (-)
/
CF3
6a ~ 146-147 (+)
F3C
6b ~ 144-145 (-)
/
F3C
7a ~ CF3 118-120 (+)
?b ~ CF3 119-120 (-)
/
8a ~ 200-201 (+)
/
CFs
- 23 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
8b ~ 198-200 (-)
CF3
9a ~ 115-116 (+)
/
F3C CF3
9b ~ 114-115 (-)
/
F3C CF3
l0a ~ CF3 173-175 (+)
/
CF3
10b ~ CF3 174-175 (-)
/
CF3
lla ~ 115-116 (+)
CI / CI
llb ~ 115-116 (-)
CI / CI
12a ~ 130-132 (+)
/
-CI
CI
12b ~ 135-136 ~_
/
-CI
C)
13a ~ CI 157-159 (+)
CI
- 24 -
CA 02306960 2000-04-12
WO 99/20281 PCT/US98/21597
13b ~ CI 157-158 (-)
CI
14a ~ C F3 107-109 (+)
CH3
14b ~ CF3 107-108 (-)
CH3
- 25 -