Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
1
Novel compounds and their use as positive AMPA receptor modulators
This invention relates to novel compounds useful as modulators of the AMPA
sensitive
glutamate receptors, pharmaceutical compositions comprising such compounds and
their use
in therapy
BACKGROUND OF TH1= INVENTION
~-Glutamate is the major excitatory neunoiransmitter in the mammalian central
nervous
system which activates several subtypes of ionotropic and metabotropic
receptors. The
ionotropic receptors can be divided into threE; subtypes, NMCrA, AMPA and
kainate receptors,
based on structural and pharmacological differences.
Impairment of glutamatergic neurotran:~rr~ission has been implicated in the
learning
and memory loss observed in numerous neurological disorders such as e.g.
Alzheimer's
decease, senile dementia: stroke (McEntee and Crook, F'sycopharmacology
111:391-401
(1993)). It is widely accepted that learning and memory is related to the
induction of long-term
potentiation (LTP) which is a stable increase in the synaptic strength
following repetitive high
frequency stimulations. Experimental studies have shown that increasing the
synaptic
response mediated by AMF'A receptors enhances the induction of LTP (Arai and
Lynch, Brain
Research, 598:173-184 (1992)). For the reason:; stated above, compounds that
stimulates
AMPA receptor response in the brain, may induce improvements in the
intellectual behavior
and performance.
Activation of AMI'A receptors with mglutamate or the selective agonist AMPA
leads to
a rapid receptor desensitization; i.e. tie receptor channel fails to open
despite the continued
presence of agonist. It i's therefore possible to obtain an increase of the
synaptic strength by
attenuating the AMPA, receptor desensitization normally elicited by the
endogenous
neurotransmitter ~-glutamate.
In 1990 Ito et al. reported (J. physial., 424:53;x-543) that the nootropic
drug
aniracetam (lV-anisoyl-2-p~yrrolidinor~e) increased AMPA induced currents in
oocytes injected
with rat brain mRNA. In another study, it has been shown that 1-(1,3-
benzodioxol-5-
ylcarbonyl)-1,2,3,6-tetrahydropyridine, a compound that enhances synaptic
transmission
mediated by AMPA receptors, is effective at improving mennary in experimental
animals at a
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
,7
very high dose 120 mg/kg (Stauble et al., Proc. Natl. Acad. Sci. USA, 91:11158-
11162
(1994)).
The benzothiadiazide cyclothiazide is a more potent and efficacious modulator
of
AMPA receptor current in-vitro than aniracetarn (Johansen et al.,
MoLPharmacol. 48:946-955
(1995)). The effect of cyclothiazide on tf~Ea kinetic properties of AMPA
receptor currents
appear to be by a different mechanism to that of aniracetam (Partin et al., J.
Neuroscience
16:6634-6647 (1996)). However, cyclothiazide has no therapeutic potential for
AMPA
receptor modulation <~s it can n~~t cross the blood-brain-barrier following
peripheral
administration. The low potency of know compounds also meets with higher
demands for a
high solubility due to the higher doses used for ;administration.
BACKGROUND ART
US 5,488,049 describe;; the use of benzothiadiazide derivatives to treat
memory and learning
disorders. The compounds are structurally closely related to the compounds of
the present
invention. However the compounds of the present invention shows greater
potentiation at
lower concentrations. (f=ig. 3 of US 5 488,049)
US 4,184,039 disclose:> benzothiadiazides for use in the promotion of hair
growth;
DE 1470316 describes a method for producing some benzothiadiazides for use as
additives
in galvanizing baths.
In Synthesis (10), 183, p. 851 a method for prE;paration of benzothiadiazine-
1,1-dioxides are
described. The compounds are described as useful as antihypertensive and
antimicrobial
reagents.
In J. Med. Chem. (15, no. 4), 1972. p. 400-403 bcanzothiadiazine-1,1-dioxides
are investigated
for their ~--substituents constants as structure activity study for anti
hypertensive activity.
WO 9812185 describes benzothiad~azines of different structure as the compounds
of the
present invention.
Object of the invention
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
3
It is an object of the present invention to provide positive AMPA modulators
which are useful
in the treatment of disorders or diseases in mammals, including a human, and
especially in
the treatment of diseases and disorders which are responsive to modulation of
the AMPA
receptors in the brain.
Another object of the present invention is tc:~ provide a method of treating
disorders or
diseases of a mammal, including a human, responsive to AMP,A receptor
modulators which
comprises administering to a mammal in need thereof a compound of the
invention.
A third object of the pnasent invention is to provide novel pharmaceutical
compositions for the
treatment of disorders. or diseasEa of mammals, including a human, responsive
to AMPA
modulators.
Other objectives of the present invention will be apparent to the skilled
person hereinafter.
Summary of thre invention
The invention then, inter alia, comprises the fotdo~wing, alone-; or in
combination:
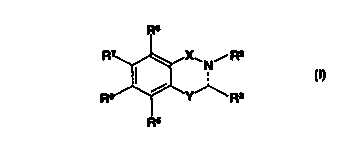
A compound represented by the general formula:
Ra
~.X.~ N, R2
R I~ .~.Y~ R
3
R5
wherein
the bond represented by the brokE;n line may be a single, a double bond or
absent;
and if the band is absent, then the nitrogen is substituted with a hydrogen
and R2;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
X represents S02 or C=O or CH~,;
Y represents -CH(IR4)-, -N(R4)- or -N(R°)-C~E~a-, O;
R2 represents hydrogen, alkyl, cycfoalkyl, ;aryl, benzyl;
CO-R9 wherein
R9 repre:>ents alkyl, cycloalkyl, benzyl, aryl; or
R2 together with R3 arid together with the atF.~ms to which they are attached,
forms a 4- to
7-rnembered rin~~ optionally substituted one or more times with substituents
selected
from halogen, alkyl, alkenyl, alkynyl, hydroxy, aikoxy, amino or thio and
optionally
containing one or more heteroatoms and optionally containing carbonyl groups;
R3 represents hydrogen, cycloalkyl, alkyl, cycloalkylalkyl, haloalkyl,
hydroxyalkyl,
cyanoalkyl, alko:Kyalkyl, alkoxy, haloalkoxy, acyl, alkyl-NR'3R'4 , alkyl-S-
R'3
wherein
R'3 and R'"' independE:ntly represents hydrogen, alkyl, cycloalkyl; or R'3 and
R'° together with the nitrogen to which they are attached forms a
3- to 8
memberE;d heterocyclic: ring structure;
A c:arbocyclic 7- to 12- membered ring ol7tionally substituted with halogen,
alkyl,
hydroxy or alkoxy; or
A heterocyclic 3- to 8 membered ring optionally substituted with halogen,
alkyl,
hydroxy or alkox:y; and optionally the hcaerocyclic rind is fused to an aryl;
Benzyl which is optionally substituted one or more times with substituents
selected
from the group consisting of halogen, cyc:loalkyl, alkyl, hydroxy, alkoxy,
amino or thio,
haloalkyl, hydroxyalkyl, alkoxyalkyl, alkylthio, alkylamino;
Aryl which is optionally substituted onE: or more times with substituents
selected from
the group consi~~ting of halogen, cycloalkyl, alkyl, hydroxy, alkoxy, amino or
thio,
haloalkyl, hydroxyalkyl, alkoxyalkyl, alkylthio, alkylamino; or
CA 02320354 2000-08-04
WO 99/42456 I'CT/DK99/00070
R3 together with R' or R4 and together with the atoms to which they are
attached, forms a
4- to 7- membered ring optionally substituted one or more times with
substituents
selected from halogen, alkyl, alkenyi, alkynyl, hydroxy, alkoxy, amino or thio
and
optionally containing one or more heteroatoms and optionally containing
carbonyl
5 groups.
R' represents hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, aryl,
-CO-R'°, or CCf2R'° wherein R'° represents hydrogen,
cycloalkyl, alkyl, aryl or benzyl;
or
R4 together with R'3 and together with the atoms to which they are attached,
forms a 4- to
7- membered ring optionally aubstitute~d c>ne or morEy times with substituents
selected
from halogen, alkyl, alkenyl, alkynyl, hycfroxy, alkoxy, amino or thio and
optionally
containing one or more heteroatom s and optionally containing carbonyl groups.
R5 represents hydrogen, halogen, alkyl, alkenyl, alkynyl, aryl,
-S02-NR"R'2 wherein
R" and R'' independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or
R" and R'z together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
halogen, alkyl, hydroxy, alkoxy, amino or thin, aryl, benzyl, SOz-alkyl, S02-
aryl,
S02-benzyl; and optionally the heterocyclic ring is fused to an aryl;
R6 represents hydrogen, halogen, alkyl, cyano, cyanoalkyl, nitro, alkoxy,
haloalkoxy,
haloalkyi, hydroxyalkyl, cycloalkyl, cyc:iohaloalkyl,
-NR'SR'6, NHSO2-R'~', NI-ISOd~-aryl wherein the aryl is optionally substituted
one or
more times with substituents selected from halogen, alkyl, cycloalkyl,
hydroxy, alkoxy,
arnino, thio, CF~3, OCF°, N02. aryl;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
6
Aryl optionally substituted one or more times with substituents selected from
the group
consisting of al~;yl, cycloalkyl, alkoxy, haloalkyl, haloalkoxy, hydroxyalkyl,
alkoxyalkyl
or amino;
HET optionally substituted oroe or more times with substituents selected from
the
group consisting of alkyl, cycloalkyl, alkoxy, halogen, haloalkyl, haloalkoxy;
-(alkyl)m-S-R'S; ..(alkyl)m-SO-R'S ; -(alkyl)r"-S02-R'S. _(alkyl)m-S020R'S, -
(alkyl)m-SOz-
NR'SR'6, -(alkyl)m-NHCOR'~', -(alkyl)",-C;C)PJR'SR'~', -(alkyl),~,-CR'=NOR", -
(alkyl)m-CO-
R'S, -(alkyl)m-CO 2-R'S wherein
misoorl;and
R' and F." independently represents hydrogen, alkyl, cycloalkyl, alkenyl,
alkynyl, ~~ryl, ben-r_yi; and
R'S and R'" independently reprcaents hydrogen, alkyl, cycloalkyl, benzyl,
aryl,
or
R'S and 'R''' together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered ~riracf structure optionally substituted with
halogen, alkyl, alkenyl, alkynyl, hydroxy, alko:xy, amino or thio, aryl,
benzyl,
S02-alkyl, SOz-aryl, S02-benzyl'; and optionally the heterocyclic ring is
fused to
an aryl;
R' represents hydrogen, halogen, alkyl, cyano, cyanoalkyl, vitro, nitroalkyl;
alkoxy,
haloalkoxy, haloalkyl, hydroxyalkyl, c:yc:loalkyl, cyclohaloalkyl,
-NR"R'8, NHSC)2-R", NHSO~-aryl wherEein the aryl is optionally substituted one
or
more times with substituents selected frcjrn halogen, alkyl, cycloalkyl,
hydroxy, alkoxy,
amino, thio, CF3, OCF~, NOz, aryl;
-(alkyl)m-S-R"; -(alkyl)m-SO-R" ; (alkyl)m..,;02_R"; _(alkyl)",-SO20R",
(alkyl)m-S02-
NR"R'8, -(alkyl)mNHCOR", -(alkyl),r,CONR"R'8, -(alkyl)m-CR'=NOR", -(alkyl)m-CO-
R'''
(alkyl)mC0?-R", wherein
misoorl;
and R' and R" independently reprEasents hydrogen, alkyl, cycloalkyl, alkenyl,
alkynyl, <~ryl, benzyl; and
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
7
R" and IR"~ independently represents hydrogen, alkyl, cycloalkyl, benzyl,
aryl,
or R" arid R'e together with the nitrogen to which they are attached forms a
heterocyclic :3- to 8 membered ring structure a~ptionally substituted with
halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thio, aryl,
benzyl,
S02-alkyl, SOz-aryl, SO2-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
HST optionally substituted one or more times with substituents selected from
halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino, thio, aryl, -S-
alkyl, -S-aryl,
SO-alkyl, SO-aryl, SO~-alkyl, S02-aryl, ;aO?NR"R'g;
Aryl optionally substituted one or more times with substituents selected from
the group
consisting of
alkyl, alk:enyl, alkynyl, hydroxy, alkoxy, hydroxyalkyl, halogen, haloalkyl,
amino,
NHCO-alkyl, nitro, OCF3, -SO~-NFi"R'~, wherein R" and R'e independently
represents hydrogen, alkyl, cycloalkyl, benzyl, aryl, or R" and R'8 together
with the nitrogen to which they .are attached forms a heterocyclic 3- to 8
membered ring structure optionally substituted with halogen, alkyl, alkenyl,
alkynyl, Ihydroxy, alkoxy, amino or thio, aryl, t>enzyl, SO2-alkyl, S02-aryl,
S02-
benzyl; anti aptionally the heterocyclic ring is fused to an aryl;
or
R' together with Rf' or together with RB forms a 5- to 7-membered ring having
the one of
the following structures -O-(CH2)n-O-; wherein ra is 1, 2 or 3; -SO~-NR-(CH2)n-
wherein n is 1
or 2; -SO-NR- (CH2)~- wherein n is 1 or 2; -SO2-(CH2)~- wherein n is 2 or 3; -
SO-(CH2)~-
wherein n is 2 or 3; -CO-C;H=CH-NH- ;-CO-CH=C;H-O-; -CO-(CH2)~-NH- wherein n
is 1 or 2; -
CO-NH-(C;H,~)~ wherein n cs 1 or 2; -CO- (CH2)~-f7-;-O-(CH2)rn-O-; wherein n
is 1, 2 or 3;
R8 represents hydrogen, alkyl, alkoxy, hydro;Kyalkyl, halogen, haloalkyl, CN,
cyanoalkyl,
nitro, nitraalkyl;
Aryl optionally substituted one or more times with substituents selected from
the group
consisting of halogen, CF;3, OCF3, N02, alkyl, cycloalkyl, alkoxy;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
8
HET optionally substituted one or more times with substituents selected from
the
group consisting of halogen, CF3, OCF-3, IV02, alkyl, cycloalkyl, alkoxy;
-(alkyl)rt,-S-R'9; .. (alkyl)m-SC7-R'9 ; -(alkyl)n,-S02-R''~; -(alkyl)m-
S020R'9, (alkyl)n,-SOZ-
NR'9R2°, -(alkyl)mNHCOR'9. -(alkyl)mCOf~IR'9R2°, -(alkyl)rn-
CR'=NOR", -(alkyl)m-CO-R'9;
(alkyl)m-C02-R''~, and m is CI or 1; and R' and R" independently represents
hydrogen,
alkyl, cycloalkyl" alkenyl, alkynyl, aryl, benzyl; and
R'9 and R2° indE:pendently represents hydrogen, alkyl, cycfoalkyl,
benzyl, aryl, or R'9
and R2° togethE~r with the nitrogen to which they are attached forms a
heterocyclic 3-
to 8 membered ring structure optionallyr substituted with halogen, alkyl,
alkenyl,
alkynyl, hydroxy, alkoxy, amir7o or thio, aryl, benzyl, SO?-alkyl, SO2-aryl,
S02-benzyl;
and optionally the heterocycli~; ring is fusE:d to an aryl;
provided that when the broken line in fon~nula I represents a double bond and
X
represents S02 and Y represent NH and the corrrpound is nnanosubstituted then
it is not
monosubstituted with FI3 representing OCH3, rnel:hyl, pentyl, t-butyl,
aminophenyl, 2-
phenylethylene, phene!lhyl, cyclopE~ntyl, cyclopentenyl, cyclohexyl,
cyclohexenyl, norbornene,
benzyl, thienyl, furyl, aryl, aryl substituted with 4-methyl, 4-methoxy, 4-
chloro, 4-nitro or 3-
nitro;
and when the compound is disubstitutc:d with R3 being methyl then R5 is not
CI, CH3;
or then R' is not F, CI, 13r, I, CH3 C:F,, nitro, SO;>N(CH3)2; or then R6 is
not CI, Br, CH3, CF3,
ethyl, methoxy;
or when R' is chloro then R3 is not ethyl, butyl, sec- butyl, t-butyl,
cyclobutyl, 2,2-
dimethylpropyl, phenyl;
and when the corrrpound is disubstitutE:d then it is not with R6=OMe,
R3=ethyl;
R6=methyl, R3=propyl; iR' =S02NH7, R~=Cl; R'=SOzNH2, R3=::phenyl; R'=Br,
R3=phenyl;
And provided that when the compouncl is trisubstituted then it is not R3=CH3 ,
R5=N02,
R'=CI; R3=CH3 , R6=NO 2, R'=CI; R3=CH3 , R5=:NI-i2, R'=CI;
and provided that when the broken IinE: in formula I represents a single bond
and X
represents S02 and Y represent NH
Then the compound is not a disubstituted compounds with R' or R6 being chloro
and
R3 being alkyl, cyclobutyl, cycfopropyl, cyclohexyl, cyclohexen, norbornenyl,
norbornanyl,
ethylthiomethyl, ethylo>;ymethyl, ethyloxyethyl, methyloxymE~thyl,
methylamino, 2-chloroethyl,
chloromethyl, dichloromethyl, trifluoromethyl, arnina;
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
9
and the compound is not a trisubstituted compound with R3 being CH3 and
R5=isopropyl , R'=F; R5=ethyl , R'=C:I; IR''=:propyl , R':=CI; R5=ethyl ,
R'=F; R5=methyl ,
R'=CI; R5=ethyl , R'=methyl; R5=CI , R'-=Methyl; RS=methyl , R'=CI; R4=methyl
,
R5=ethyl; or trisubstituted with R'=methyl, R5=methyi , R'---F;
A pharmaceutical composition comprising an therapeutically effective amount of
a
compound as above tol~ether with pharmaceutically acceptable carriers or
exipients;
The use of a compound represented by the general formula
R$
R~'
Ra
R5
wherein
the bond represented by the broken line may be a single, a double bond or
absent;
and if the bond is absent, then tt~e nitrogen is substituted with a hydrogen
and R2;
2o X represents S02 or C=O or C;H2;
Y represents -CH(R4j-, -N(R4)- or -N(R'1)-CI-i~~-, O;
R2 represents hydrogen, alkyl, cycloalkyl, aryl, benzyl;
CO-R9 wherein
R9 repre:>ents alkyl, cycloalkyl, benzyl, aryl; or
R2 together with R3 and together with the atoms to which they are attached,
forms a 4- to
7-membered ring optionally substituted c>ne or more times with substituents
selected
from halogen, alkyl, alkenyl, alkynyf, hydroxy, alkoxy, amino or thio and
optionally
containing one or more heteruatoms and optionally containing carbonyl groups;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
R3 represents hydrogen, cycloalkyl, alkyl, cyc;loalkylalkyl, haloalkyl,
hydroxyalkyl,
cyanoalkyl, alkoxyalkyl, alkoxy, haloalkoxy, acyl, alkyl-NR'3R'4 , alkyl-S-R'3
wherein
R'3 and R'~ independently represents hydrogen, alkyl, cycloalkyl; or R'3 and
R'° togei:her with the nitrogen to which they are attached forms a
3- to 8
membered heterocyclic ring structure;
A carbocyclic 7- to 12- membered ring optionally substituted with halogen,
alkyl,
hydroxy or alko>cy; or
A heterocyclic 3- to 8 membered ring optionally substituted with halogen,
alkyl,
hydraxy or alko>cy; and optionally the heterocyclic ring is fused to an aryl;
Benzyl which is optionally substituted one or more times with substituents
selected
from the group consisting of halogen, c:ycloalkyl, alkyl, hydroxy, alkoxy,
amino or thin,
haloalkyl, hydro:Kyalkyl, alkoxyalkyl, alkylthio, alkylamino;
Aryl which is optionally substituted one or more times with substituents
selected from
the group consistirng of halogen, cycloalkyl, alkyl, hydroxy, alkoxy, amino or
thin,
haloalkyl, hydro:Kyalkyl, alkoxyalkyl, alkylthio, alkylamino; or
R3 together with R2 or R4 and together with the atoms to which they are
attached, forms a
4- to 7- membered ring optionally substituted one or more times with
substituents
selected from halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thin
and
optionally containing one or more heteroatoms and optionally containing
carbonyl
groups.
R4 represents hydrogen, alkyl, cycloalkyl, cyc:loalkylalkyl, aryl,
-CO-R'°, or C02R'° wherein R'° repre:>ents hydrogen,
cycloalkyl, alkyl, aryl or benzyl;
or
R4 together with R3 and together with the .atoms to which they are attached,
forms a 4- to
7- membered ring optionally substituted one or more times with substituents
selected
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99100070
11
from halogen, alkyl, alkenyl, alkynyl. h~,rdroxy, alkoxy, amino or thio and
optionally
cantaining one ~or more heteroatoms and optionally containing carbonyl groups.
R5 represents hydrogen, halogen, alkyl, alkenyl, alkyny,l, aryl,
-S02-NR"R'2 wherein
R" and R''' independently represents hydrogen, alkyl, cycloalkyl, benzyl,
aryl"
or
R" and R" together with the nitrogen to whic::h they are attached forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
halogen, alkyl, hydroxy, alkoxy, amino or thin, aryl, benzyl, S02-alkyl, S02-
aryl,
S02-benzyl; and optionally the heterocyclic ring is fused to an aryl;
Rs represents hydrogen, halocfen, alkyl, oyano, cyanoalkyl, nitro, alkoxy,
haloalkoxy,
haloalkyl, hydro~xyalkyl, cycloalkyl, cycloh,aloalkyl,
-NR'SR'6, NHSO 2-R'S, NHSOa-aryl wherein the aryl i;~ optionally substituted
one or
more times with substituents selected from halogen, alkyl, cycloalkyl,
hydroxy, aikoxy,
amino, thio, CF=3, OCF3, NG2, aryl;
Aryl optionally :substituted one or more tunes with substituents selected from
the group
consisting of alkyl, cycloalkyl, alkoxy, haloalkyl, haloalkoxy, hydroxyalkyl,
alkoxyalkyl
or amino;
HET optionally substituted one or more times with substituents selected from
the
group consisting of alkyl, cycloalkyl, alkc~xy, halogen, haloalkyl,
haloalkoxy;
-{alkyl)m-S-R'S; -(alkyl)m-SG-R'S ; -(alkyl),-S02-R'S; ..(alkyl)m-SO20R'S, -
(alkyl)m-S02_
NR'SR'~, -(alkylh,-NHCOR'S, -(alkyi)m-(~ONR''R'6, -(alkyl)m-CR'=NOR", -
(alkyi)m-CO-
R'S; -(alkyl)m-CO 2-R~''' wherein
misoorl:and
R' and R" independently represents hydrogen, alkyl, cycloalkyl, alkenyl,
alkynyl, aryl, benzyl; and
CA 02320354 2000-08-04
WO 99/42456 PCT1DK99/00070
12
R'S and F~'6 independently represents hydrogE:n, alkyl, cycloalkyl, benzyl,
aryl,
or
R'S and F3'6 together with the nitrogen to which they are attached forms a
heterocyclic: 3- to 8 membered ring structure optionally substituted with
halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thio, aryl,
benzyl,
S02-alkyl, S02-aryl, SU2-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
R' represents hydrogE:n, halogen, alkyl, cyano, cyanoalkyl, nitro, nitroalkyl;
alkoxy,
haloalkoxy, haloalkyl, hydroxyalkyl, cycloalkyl, cyclohaloalkyl,
-NR"R'a, NHS02-R'', NHS02-aryl wherein the aryl is optionally substituted one
or
more times with substituents selected from halogen, alkyl, cycloalkyl,
hydroxy, alkoxy,
amino, thio, CF3, OCF;~, N02, aryl;
-(alkyl)m-S-R", -(alkyl)",-SO-R" ; (alkyl)m~-S02-R"; _(alkyl)~,-S020R",
(alkyl)m-SOz_
NR"R'e, -(alkyl)mNHCOR'7, -(alkyl)",CC>NR"R'e, -(alk:yl)m-CR'=NOR", -(alkyl)m-
CO-R";
(alkyl)mCOrR", wherein
misoorl;
20 and R' and R" independently represents hydrogen, alkyl, cycloalkyl,
alkenyl,
alkynyl, <~ryl, benzyl; and
R" and I~"' independently reprE;sents hydrogen, alkyl, cycloalkyl, benzyl,
aryl,
or R" and R'8 together with thE: nitrogen to which they are attached forms a
25 heterocyclic 3- to 8 membered ring structure optionally substituted with
halogen, alkyl, alkenyl. alkynyl, hydroxy, alkoxy, amino or thio, aryl,
benzyl,
SOralkyl, ;~02-aryl, S02-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
30 HFT optionally substituted one or more limes with substituents selected
from
halogen, alkyl, alkf:nyl, alkynyl, hydroxy, alkoxy, amiroo, thio, aryl, -S-
alkyl, -S-aryl,
SO-alkyl, SO-aryl, SOz-alkyl, SO?-aryl. ;a02NR"R'8;
CA 02320354 2000-08-04
WO 99J42456 PCT/DK99/00070
13
Aryl optionally substituted one or more times with substituents selected from
the group
consisting of
alkyl, alkenyl, alkynyl, hydroxy., aikoxy, hydroxyalkyl, halogen, haloalkyl,
amino,
NHCO-alkyl, nitro, OCF3, -SO~~-~NR"R'~, wherein R" and R'8 independently
represents hydrogen, alkyl, cycloalkyl, benzyl, aryl, or R" and R'e together
with the nitrogen to which they are attached forms a heterocyclic 3- to 8
member~ed ring structure optionally substituted with halogen, alkyl, alkenyl,
alkynyl, hydroxy, alk:oxy, amino ar thio, aryl, t>enzyl, S02-alkyl, S02-aryl,
S02-
benzyl; and optionally the heteroc:yclic ring is fused to an aryl;
or
R' together with R~' or together with R8 forms a 5- to 7-rnembered ring having
the one of
the following structures -O-(CH2~n-O-; wherein r7 is 1, 2 or 3: -SO~-NR-(CH2)n-
wherein n is 1
or 2; -SO-~NR- (CHZ)~- wherein n is 1 or 2; -SO2-(CH2)n- wherein n is 2 or 3; -
SO-(CH2)n-
wherein n is 2 or 3; -CC)-CH=CH-NH- ;-CO-CFI=C;H-O-; -CO-~(CH2)"-NH- wherein n
is 1 or 2; -
CO-NH-(CH~)~ wherein n is 1 or 2; -CO- (CH2)~-O-;-O-(CH2)r~-O-; wherein n is
1, 2 or 3;
Re represents hydrogen, alkyl, alkoxy, hy<iro:Kyalkyl, halogen, haloafkyl, CN,
cyanoalkyl,
nitro, nitroalkyl;
Aryl optionally substituted ane or more times with substituents selected from
the group
consisting of halogen, CF;3, OCF3, N02, alkyl, cycloalkyl, alkoxy;
HST optionally substituted one or more times with substituents selected from
the
group consisting of halogen, CF3, OCF~3, N02, alkyl, cycloalkyl, alkoxy;
-(afkyl)m-S-R'9; -~ (alkyl)m-SO-R'''' ; -(alkyl)m~-S02-R'''; ~~(alkyl)m-
S020R'9, (alkyl)rt,-S02-
NR'9R2°, -(alkyl)mNHCOR'9. -(alkyl)mCCJNR'9R2°, -(alkyl)n,-
CR'=NOR", -(alkyl)rt,-CO-R'9;
(ai'kyl)m-C02-R'''~, and m is U or 1; and f~' and R" independently represents
hydrogen,
alkyl, cycloalkyl. alkenyl, alkynyl, aryl, benzyl; and
R'9 and R2° independently represents hydrogen, alkyl, cycloalkyl,
benzyl, aryl, or R'9
and R2° together with the nitrogen to which they are attached forms a
heterocyclic 3-
to 8 membered ring structure optionally substituted with halogen, alkyl,
alkenyl,
alkynyl, hydroxy, alkoxy, amino or thio, aryl, benzyl, S02-alkyl, S02-aryl,
S02-benzyl;
and optionally the heterocyclic ring is fused to an aryl;
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
'I 4
for the manufacture of a medicament for the treatment of disorders or diseases
responsive to modulation of the AMPA receptor complex.
A method for the treatment of disorder, or diseases responsive to the
modulation of
the AMPA receptor complex wherein a therapeutically efficic.~nt amount of a
compound
represented by the general formula
R~'
Rz ~ ,~,X\N, R:>
Rsr ~ 'y'~'~. Ra
R5
wherein
the bond represented by the broken line may bE: a single, a double bond or
absent;
and if the bond is absent, then the nitrogen is substituted with a hydrogen
and R2;
X represents S02 or C=O or C;Hz;
Y represents -CH(R4)-, -N(R'')- or -N(R4)~-CH;z-, O;
R2 represents hydrogen, alkyl, cycloalkyl, aryl, benzyl;
CO-R9 wherein
R9 repre:;ents alkyl, cycloalkyl, bc:nzyl, aryl; or
R2 together with R3 and together with the atoms to which they are attached,
forms a 4- to
7-membered ring optionally substituted c>ne or more times with substituents
selected
from halogen, allkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thio and
optionally
containing one or more hetc:roatoms and optionally containing carbonyl groups;
R3 represents hydrngen, cycloalkyl, alkyl, cycloalkylalkyN, haloalkyl,
hydroxyalkyl,
cyanoalkyl, alkoxyalkyl, alkoxy, haloalkoxy, acyl, alkyl-NR'3R'4 , alkyl-S-R'3
wherein
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
R'3 and R'4 independently represents hydrogen, alkyl, cycloalkyl; or R'3 and
R'4 together with the nitrogen to which they are attached forms a 3- to 8
membered heterocyclic ring structure;
5 A carbocyclic 7- to 12- membered ring optionally sukastituted with halogen,
alkyl,
hydroxy or alko:Ky; or
A heterocyclic ;:~- to 8 membered ring optionally substituted with halogen,
alkyl,
hydroxy or alko:Ky; and optionally the heterocyclic ring is fused to an aryl;
Bc:nzyl which is optionally substituted orre or more times with substituents
selected
from the group consisting of halogen, c:ycloalkyl, alkyl, hydroxy, alkoxy,
amino or thio,
haloalkyl, hydroxyalkyl, alkoxyalkyl, alN;ylthio, alkylanoino;
Aryl which is optionally substituted one car more times with substituents
selected from
the group consisting of halogen, cycloalkyl, alkyl, hyciroxy, alkoxy, amino or
thio,
haloalkyl, hydroxyalkyl, alkoxyalkyl, alH;ylthio, alkylannino; or
R3 together with R'~ or R4 and together with the atoms to which they are
attached, forms a
4- to 7- membered ring optionally substituted one or more times with
substituents
selected from halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thio
and
optionally containing one or more heteroatoms and captionally containing
carbonyl
groups.
R4 represents hydrogen, alkyl, cycloalkyl, cyc:loalkylalkyl, aryl,
-t;0-R'°, or C02R'° wherein R'° represents hydrogen,
cycloalkyl, alkyl, aryl or benzyl;
or
R4 together with R'' and together with the ai:oms to which they are attached,
forms a 4- to
7- membered ring optionally substituted one or more times with substituents
selected
from halogen, alkyl, alkenyl, alkynyl, hyc~roxy, alkoxy, amino ar thio and
optionally
containing one or more heteroatoms ancj optionally containing carbonyl groups.
R5 represents hydrogen, halogen, alkyl, aNkenyl, alkynyl~ aryl,
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
-I 6
-SOrNR"R'2 wherein
R" and R'' independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or
R" and R''' together with the nitrogen to whit..~.h they are attached forms a
heterocyclic 3- to 8 membered rind structure optionally substituted with
halogen, alkyl, hydroxy, alkoxy, amino or thio, aryl, benzyl, SOz-alkyl, SOz-
aryl,
S02-ber~zyl; and optionally the helerocyclic ring is fused to an aryl;
R6 represents hydrogen, halogen, alkyl, cyano, cyanoalkyl, nitro, alkoxy,
haloalkoxy,
haloalkyl, hydroxyalkyl, cyciaalkyl, cyclohaloalkyl,
-NR'SR'~, NHSO2-R'S, NHSO,-aryl wherein the aryl i:~ optionally substituted
one or
more times with substituents selected from halogen, alkyl, cycloalkyl,
hydroxy, alkoxy,
arnino, thio, CF~3, OCF3, NO 2, aryl;
Aryl optionally substituted one or more times with substituents selected from
the group
cansisting of alH;yl, cycloalkyl, alkoxy, haloalkyl, haloalkoxy, hydroxyalkyl,
alkoxyalkyl
or amino;
HET optionally substituted one or more times with substituents selected from
the
group consistin~~ of alkyl, cycloalkyl, alkoxy, halogen,, haloalkyl,
haloafkoxy;
-(alkyl)m-S-R'S; ..(alkyl)m-SO-R'S ; -(alkyl)",-S02-R'S; -(alkyl)m-SOzOR'S, -
(alkyl)m-S02_
NR'SR'6, -(alkyl)m-NHCOR"', -(alkyl),n-C:C)IVR'''R's, -(alkyl)m-CR'=NOR", -
(alkyl)n,-CO
R'S; -(alkyl)m-CO 2-R'S wherein
m is o or 1; and
R' and Ft" independently repre:>ents hydrogen, alkyl, cycloalkyl, alkenyl,
alkynyl, aryl, bent yl; and
R'S and R"' independently represE:nts hydrogen, alkyl, cycloalkyl, benzyl,
aryl,
ar
R'S and R't' together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
halogen" alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thin, aryl,
benzyl,
CA 02320354 2000-08-04
WO 99/4245fi PCT/DK99/00070
1%
S02-alkyl, S02-aryl, SO~-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
R' represents hydrogen, halogen, alkyl, c;yano, cyanoalkyl, nitro, nitroalkyl;
alkoxy,
haloalkoxy, haloalkyl, hydroxyalkyl, cycloalkyl, cyclohaloalkyl,
-NR"R'8, NHSO2-R", NHSO~-aryl wherein the aryl is optionally substituted one
or
more times with substituents selected from halogen, alkyl, cycloalkyl,
hydroxy, alkoxy,
amino, thio, Cf=3, OCF3, NO?, aryl;
-(alkyl)m-S-R"; -(alkyl)m-SG-R" ; (alkyl),-S02-R"; -(alkyl)m-S020R", (alkyl)m-
S02-
NR''R'~, -(alkyl)mNHCOR", -(alkyl)mCONR"R'e, -(alkyl)n,-CR'=NOR", -(alkyl)rt,-
CO-R";
(a.lkyl)mC02-R", wherein
misoorl;
and R' and R" independently rE~presents hydrogen, alkyl, cycloalkyl, alkenyl,
alkynyl, aryl, benzyl; and
R" and R'e independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or R" and R'H together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
halogen, alkyl, alkenyi, alkynyl, hydroxy, alkoxy, amino or thio, aryl,
benzyl,
S02-alkyl, S02-aryl, S02-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
HFT optionally substituted one or more times with substituents selected from
halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino, thio, aryl, -S-
alkyl, -S-aryl,
SO-alkyl, SO-aryl, S02-alkyl, S02-aryl, ',.~OZNR"R'8;
Aryl optionally substituted one or mare times with substituents selected from
the group
consisting of
alkyl, al~;enyl, alkynyl, hydroxy, alkoxy, hydroxyalkyl, halogen, haloalkyl,
amino,
NHCO-alkyl, nitro, C>CF3, -SO,~--NR"R'8, wherein R" and R'8 independently
represents hydrogen, alkyl, cycloalkyl, benzyl, aryl, or R" and R'8 together
with the nitrogen to which they are attached forms a heterocyclic 3- to 8
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
18
memberc:d ring structure optionally substituted with halogen, alkyl, alkenyl,
alkynyl, hydroxy, alkoxy, amino or thio, aryl, benzyl, S02-alkyl, S02-aryl,
SOZ-
benzyl; a,nd optionally the heteroc:yclic ring is fused to an aryl;
or
R' together with R6 or together with Rg forms a 5- to 7-membered ring having
the one of
the following structures -O-(CH2)n-O-; wherein n is 1, 2 or 3; -S02-NR-(CH2)n-
wherein n is 1
or 2; -SO-NR- (CH2)~- wherein n is 7 ~r 2; -SO2-(CH2)~- wherein n is 2 or 3; -
SO-(CH2)~-
wherein n is 2 or 3; -CO-CH=CH-NH- ;-CO-CH=CH-O-; -CO-(CH2)~-NH- wherein n is
1 or 2; -
CO-NH-(C;H2)~ wherein n is 1 or '~; -CO- (CH2)~:-(J-;-O-(CH2)n-O-; wherein n
is 1, 2 or 3;
Re represents hydrogen, alkyl, alkoxy, hydrc~xyalkyl, halc:~gen, haloalkyl,
CN, cyanoalkyl,
nitro, nitroalkyl;
Aryl optionally substituted one or more times with substituents selected from
the group
consisting of halogen, CF3, OCF3, N02, alkyl, cycloalkyl, alkoxy;
HE:T optionally ~~ubstituted one or morE~ tirnes with substituents selected
from the
group consisting of halogen, CF3, OCF3, NO2, alkyl, c;ycloalkyl, alkoxy;
-(alkyl)m-S-R'9; - (alkyl)m-SO-R'9 ; -(alkyl),n-S02-R'9; -(alkyl)m-S020R'9,
(alkyl)m-S02-
NR'9R2°, -(alkyl)imNHCOR'q, -(alkyl)rnCONR'9R2°, -(alkyl)m-
CR'=NOR", -(alkyl)m-CO-R'9;
(alkyl)m-C02-R'9, and m is 0 or 1; and Fi' and R" independently represents
hydrogen,
alkyl, cycloalkyl, alkenyl, alkynyl, aryl, benzyl; and
R''~ and R2° independently represents hydrogen, alkyl, cycloalkyl,
benzyl, aryl, or R'9
and R2° togethE~r with the nitrogen to which they are attached forms a
heterocyclic 3-
to 8 membered firing structure optionally substituted with halogen, alkyl,
alkenyl,
alkynyl, hydroxy, alkoxy, amino or thio, aryl, benzyl, S02-alkyl, S02-aryl,
S02-benzyl;
and optionally the heterocyclic ring is fused to an aryl,
is administered.
Detailed disclosure of the invention
The invention provides novel compounds of formula f as shown above.
Preferred embodiments of the invention are compounds of formula I as above
wherein
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
19
R2 represents
hydrogen, alkyl, c;ycloalkyl, phenyl, benzyl;
or
R2 together with F13 and together with the atoms to which they are attached
forms a 5- to
5-membered ring optionally substituted one or more times with substituents
selected
from halogen, ~~Ikyl, hydroxy, alkoxy, arnino or thio; and aptionally
containing one or
more heteroatoms and optionally containing carbonyl groups;
R3 represents
hydrogen, cycloalkyl, cycloalkylalkyl, alkyl, haloalkyl, alkoxy, a carbocyclic
7- to 10-
membered ring; a heterocyclic 5- to E3 rnErmbered ring; benzyl; aryl;
or R3 together with Rz or R4 forrns a 5- to 6- membered ring;
R4 represents
hydrogen, alkyl,
or R4 together with R3 and together with the atoms to which they are
attached, forms a 5- to 6- membered ring; optionally substituted one or
more times with substituents selected from halogen, alkyl, alkenyl,
alkynyl, hydroxy, alkoxy, amino or thin and optionally containing one or
more heteroatoms and optionally containing carbonyl groups.
RS represents
hydrogen, halogen, alkyl, alkenyl, alkynyl, phenyl
-S02-NR"R'2 wherein
R" and R'2 independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or R" and R" together with the nitrogen to which they are attached forms a
heterocyclic 5- to 6- memberecl ring structure;
R~ represents
hydrogen, Br, F, I, cycloalkyl, alkyl, alkoxy, alkoxyalkyl,
Phenyl optionally substituted one or more times with substituents selected
from the
group consisting of
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
2Ci
alkyl, alhoxy;
HET;
-S-R,s; -SO-Ris _ -S02-R,s; -S020R's, -;aO2-NR'sR,s _NHCOR's, -CONR'sR,s _
CR'=NOR", -CO-R'~'; -CO2-R's, wherein
f3' 4~nd R" independently represents kaydrogen, alkyl, cycloalkyl,
phenyl, benzyl; and
f3''' and R's independently represents hydrogen, alkyl, cycloalkyl,
benzyl, aryl, or R's and F3's together with the nitrogen to which they
are attached forms a hetE:rocyclic 3- to 8 membered ring structure
optionally substituted with halogen, alkyl, alkenyl, alkynyl, hydroxy,
alkoxy, amino or thio., phenyl, benzyl, S02-alkyl, S02-aryl, S02-benzyl;
ancf optionally the hetero,;yclic ring is fused to an aryl;
R' represents
hydrogen, Br, F, I, alkyl, cyano, cyanoalkyl, nitro, nitroalkyl, alkoxy,
haloalkoxy,
haloalkyl, hydroxyalkyl, cycloalkyl, cyclohaloalkyl, -(alkyl)m-NR"R'e, NHS02-
R", -S-
R'", -SO-R" ; -302-R"; -SO 2OR", -SO 2-NR"R'°, NfvCOR", CONR"R'8,
CR'=NOR", -
CcJ-R"; -C02-R ",
wherein R' and R" independently represents hydrogen, alkyl, cycloalkyl,
phenyl, I~enzyl; and
R" and R"' independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or R" and R'E' together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
alkyl,
SOZ-alkyl, S02-aryl, S02-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
HET optionally substituted one or morE: tunes with substituents selected from
halogen,
alkyl, phenyl, S(J2NR"R'8;
Phenyl optionally substituted one or rru~re times with substituents selected
from the
group consisting of
alkyl, hydroxy, alkoxy , halogen, Inaloalkyl, amino, NHCO-alkyl, nitro, OCF3, -
S02-NR"R's wherein R" and F;"' independently represents hydrogen, alkyl,
cycloalkyl, benzyl, aryl, or R~' and R'e together with the nitrogen to which
they
are attached forms a heterocyclic: ;3- to 8 menobered ring structure
optionally
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
21
substitui:ed with halogen, alkyl, SO 2-alkyl, SU2-aryl, S02-benzyl; and
optionally
the heterocyclic rind is fused to an aryl;
or
R' together with R6 or together with Re forms a 5- to '7-membered ring having
the
one of thc: following structures -O-(CH2)n-O-; wherein n is 1, 2 or 3; -S02-NR-
(CH2)n- wherein
n is 1 or 2; -SO-NR- {CH2)~- wherein n is 1 or ~; -S02-(CH~),~- wherein n is 2
or 3; -SO-(CH2)~-
wherein n is 2 or 3; -CO-C;H=CH-NH-- ;-CO-CH=:C;H-O-; -CO-(CHz)~-NH- wherein n
is 1 or 2; -
CO-NH-{CH2)~ wherein n is ~ or 2; -C;O- (CH?)2-O-;-O-(CH2)n-O-; wherein n is
1, 2 or 3;
Rs represents hydrogen, alkyl, alkoxy. hydroxyalkyl, halogen, haloalkyl, CN,
cyanoalkyl,
nitro, nitroalkyl;
Phenyl optionally substituted one or more times with substituents selected
from the
group consisting of
alkyl, cycloalkyl, alkoxy;
H F_T"
-S-R'9; -SO-R'9 ; -S02-R'9; -S020R'9, -~~Oz-NR'9R2°, NHCOR'9, -
CONR'9R2°,
CR'=NOR", -CC)-R'9; -CO?-R'9, wherein
R' and Fi" independently represents hydrogen, alkyl, cycloalkyl, phenyl,
benzyl;
and
R'9 and R2° independently represents hydrogen, alkyl, cycloalkyl,
benzyl, aryl,
or R'~ and R'" together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered rind structure rcptionally substituted with
halogen, alkyl, alkenyl, alkynyl, hydroxy, aikoxy, amino or thio, phenyl,
benzyl,
SO?-alkyl, S02-aryl, S02-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
provided that when X represents S02 and Y represents N and the broken line
3o represents a single bond then neither of R' or R6 are chlaro when R2, R4,
R5, Ra and
the remaining of R6 and R' are all hydrogen; and provided that R3 can
represent CH3
only when RS is hydrogen or R' is not sulfamoyl;
and provided that when X represents S02 and Y represents N and the broken line
represents
a double bond then neither of R' or R6 are chloro when R2, R4, R''', R8 and
the remaining of R6
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
2,c'.
and R' are all hydrogen; and provided that R', R4, R5, R6, R' and R8 are not
all hydrogen; and
provided that the compound is not disubstituted with R3 is being CH3 when R'
is fluoro,
bromo, iodo, CF3, CH3, NO,, SO2N(CH3)2, or R'' is broma, C; F3, CH3, ethyl,
methoxy; or RS is
chloro, CH3; or R8 is chloro; and provided that: the compound is not
3-ethyl-f3-methoxy-1,2,4-benzothi<~diazine-1,1-dioxide;
3-propyl-6-methyl-1,2,4-benzothiadiazine-1,1-dioxide;
3-ethyl-fi-methoxy-1,2,4-benzothiadiazine-1, ~i -dioxide;
3-phenyl -I-bromo- t ,2,4-benzott~iadiazine-1,1 ~-dioxide;
3-phenyl-7-sulfamoyl-1,2,4-benzothiadiazine-1,1-dioxide;
5-bromc-7-chloro-3~-methyl-1,2,4-benzothiadiazine-1,1-dioxide;
5-iodo-7-chloro-3-methyl-1,2,4-benzothiadiazir~e-1,1-dioxide;
5-vitro-T-chloro-3-methyl-1,2,4-~benzothiadiazine-1,1-dioxide;
6-vitro-7-chloro-3-methyl-1,2,4-~berrzothiadiazir~e-1,1-dioxide; or
6-amino-7-chloro-3-methyl-1,2,4-~benzothiadiazine-1,1-dioxide;
A more preferred embodiment of the invention is a compound of formula I as
above , wherein
Rz represents hydrogen, alkyl, cycloalkyl;
or R2 together with R3 forms a 5- tc> 6-membered ring; optionally substituted
one or more
times with substituents selected from halogen, alkyl, alkenyl, alkynyl,
hydroxy, alkoxy, amino
or thio and optionally containing one or more heteroatoms and optionally
containing carbonyl
groups;
And a preferred embodiment is wherein
R3 represents
hydrogen, cyclo;alkyl, alkyl, haloalkyl, alkoxy, a carbocyciic 7- to 10-
membered ring; a
heterocyclic 5- t~o Ei membered ring; benzyl; aryl;
or
R3 together with R2 or R' forms a 5- to 6- rnembered rind; optionally
substituted one or
more times with substituents selected from halogen, alkyl, alkenyl, alkynyl,
hydroxy,
aikoxy, amino or thio and o~7tionally containing one or more heteroatoms and
optionally containing carbonyl groups.
And another preferred Embodiment is wherein
R° represents
hydrogen, alkyl, or R4 together with R3 ;end together with the atoms to which
they are
attached, forms ,~ E>- to 6- membered ring optionally substituted one or more
times with
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
substituents selected from halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy,
amino or
thio and optionally containing one or rnorc~ heteroatoms and optionally
containing
carbonyl group:>.
And another preferred embodiment is wherein
R5 represents
hydrogen, halogen, alkyl, alkenyl, alkyynyl, phenyl,
-S02-NR" R'2 wherein
R" and R'' independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or R" and R'' together with the nitrogen to vvhich they are attached forms a
heterocyclic 3- to 8 membered ring structure;
And another preferred .embodiment is wherein
R6 represents
hydrogen, halogen, cycloalkyl, alkyl, alk<axy, alkoxyalkyl,
Aryl optionally substituted one or more times with substituents selected from
the group
consisting of alH;yl~ alkoxy;
H ET;
-S-R,s; -SO-R,s ; -S02-R,s; -S020R'S, .-S02_NR'SR'6, -NHCOR'S, -CONR'SR,s -
CR'=NOR", -CC>-R'S; -CO2-R'S, wherein
R' and R" independently represents hydrogen, alkyl, cycloalkyl, phenyl,
benzyl;
and
R'S and R'~' independently represents hydrogen, alkyl, cycloalkyl, benzyl,
aryl,
or R'S and ~i'~ together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered rir7g structure optionally substituted with
halogen, alkyl, alkenyl, alkynyl, hydroxy, alko.xy, amino or thio, phenyl,
benzyl,
S02-alkyl, SOz-aryl, SO2-benzy~l; and optionally the heterocyclic ring is
fused to
an aryl;
And another preferred embodiment is wherein
R' represents
hydragen, halogen, alkyl, cyano, cyanoalkyl, nitro, alkoxy, haloalkoxy,
haloalkyl,
3o hydroxyalkyl, cycloalkyl, cyc;lohaloalkyl" -(alkyl)m-NR"R'8, NHS02-R",
_5._R"; -SO-R" ; -SOz-R"; -SO20R", -.SC>2-NR"R,B, NHCOR'', CONR"R'8,
CR'=NOR", -CC~-R", -CO~-R'', wherein
R' and R" independently represents hydrogen, alkyl, cycloalkyl, phenyl,
benzyl; ~~nct
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99100070
2~
R" and R'e independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or R" and R'e together with the nitrogen to which they are attached forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
alkyl,
S02-alkyl, S02-aryl, SO~-benzyl; and optionally the heterocyclic ring is fused
to
an aryl;
HET optionally substituted one or more times with substituents selected from
h<~logen, alkyl, phenyl, SO,~NR"R'~;
1o Phenyl optionally substituted one or more times with substituents selected
from the
group consisting of
alkyl, hydroxy, aikoxy , halogen, haloalkyl, amino, NHCO-alkyl, nitro, OCF3,
S02-NR"R'e wherein R" and R"' independently represents hydrogen, alkyl,
cycloalkyl, benzyl, aryl, or R" ~~nd R"' together with the nitrogen to which
they
are attached forms a heterocyclica 3- to 8 mernbered ring structure optionally
substituted with halogen, alkyl, SC>2-alkyl, SG2-aryl, S02-benzyl; and
optionally
the heterocyclic ring is fused to a.n aryl;
or
R' together with Rf' or together with R8 forms a 5- to 7-rnembered ring having
the one of
the following structures
-O-(CH2)n-O-; wherein n is 1, 2 or 3; -S~G2-NR-(CH2)n- wherein n is 1 or 2; -
SO-NR-
(CH2)n- wherein n is 1 or 2; -S02-(CH2)n-'wherein n is 2 or 3; -SO-(CH2)n-
wherein n is
2 or 3; -CO-CH=:CH-NH ;-CO-CH=CH-tJ~-; -CO-(CH2)"-NH- wherein n is 1 or 2; -CO-
NH-(CH2)~ wherein n is 1 or 2; -CO- (CH,~)>-O-; O-(CH2)~-O-; wherein n is 1, 2
or 3;
And another preferred Embodiment is wherein
R8 represents
hydrogen, alkyl, alkoxy, hydroxyalkyl, halogen, haloaikyl, CN, cyanoalkyl,
nitro,
nitroalkyl;
Phenyl optionally substituted one or more times with substituents selected
from the
group consisting of alkyl, cycloalkyl, alkoxy;
H ET;
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
-S-R"3; -SO-R'9 ; -S02-R''3; -S020R''~, -SO2-NR'9R2°, NHCOR'9, -
CONR'9R2°,
CR'=NOR", -CO-R'9; -C02-R'°, wherein
R' and Ft" independently repre:;ents hydrogen, alkyl, cycloalkyl, phenyl,
5 benzyl; and
R'9 and R 2° independently represents hydrogEyn, alkyl, cycloalkyl,
benzyl, aryl,
or R'9 and R~° together with the nitrogen to which they are attached
forms a
heterocyclic 3- to 8 membered ring structure optionally substituted with
halogen, alkyl, alkenyl, alkynyl, hydroxy, alkoxy, amino or thio, phenyl,
benzyl,
1o S02-alkyl, S02-aryl, SC>2-benzyl; and optionally the heterocyclic ring is
fused to
an aryl;
An especially preferred embodiments is a compounds of formula I as above
wherein
X represents S02; Y represents N and the broken fine represents a single band;
R2
represents H;
15 R3 represents cycloalkyl, a carbocyclic ~- to 10- membered ring; a
heterocyclic 5- to 6
membered ring;
R4 represents H;
R5 represents H;
R6 represents hydrogen, alkyl or halogen;
20 R' represents
cyanoalkyl, nitroalkyl, haloalkyl,
-{alkyl)m-SO-R" ; {alkyl)m-SO 2-R"; (alkyl)"-S02-NR"R'e , -(alkyl)mCONR"R'B, -
(alkyl)m-
CR'=NOR", -(alkyl)",-CO-R"; {alkyl)mCC)2-R", wherein
misoorl;
25 R' and R" independently represents hydroger7, alkyl, cycloalkyl, phenyl,
benzyl; and
R" and FI'e independently represents hydrogen, alkyl, cycloalkyl, benzyl,
aryl,
or R" and R'8 together with the nitrogen to which they are attached forms a
heterocyc;lic 3- to fi mernbered ring structure optionally substituted with
alkyl,
3o S02-alkyl, 502-aryl, S02-benzyi; and optionally the heterocyclic ring is
fused to
an aryl; or
H ET;
or
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
2.6
R' together with Fis or together with R8 forms a 5- to 7-membered ring having
the one of
the following structures
-O-(CH2)n-O-; wherein n is 1, 2 or 3; -SO2-NR-(CH2)n- wherein n is 1 or 2; -SO-
NR-
(CH2)n- wherein n is 1 or 2; -S02-(C;H~)n~- wherein n is 2 or 3; -SO-(CH2)n-
wherein n is
2 or 3; -CO-CH=CH-NH~-;-(:,O-CH=CH-O-~; -CO-(CH~~)"-NH- wherein n is 1 or 2; -
CO-
NH-(CH2)~ wherein n is '1 er 2; -CO- {C;E~~)2-O-; O-(CH2)~-O-; wherein n is 1,
2 or 3;
R8 represents alkyl, halogen, cyanoalkyl, nitroalkyl, haioalkyl,
-~(alkyl)m-SO-R" ; (alkyl)",-SO2-R"; (alkyl)",-S02-NR''R'e , -
(alkyl)mCONR"R'8, -(alkyl)m-
CR'-NOR", -(alkyl),-CO-R"; {alkyl)mC-O2-R", wherein
t0 misoorl;
R' and R" independently reprE;sents hydrogen, alkyl, cycloalkyl, phenyl,
benzyl; and
R" and R'~ independently represents hydrogen, alkyl, cycloalkyl, benzyl, aryl,
or R" and R'8 together with the nitrogen to which they are attached forms a
heterocyclic 5- to 8 membered ring structure optionally substituted with
alkyl,
S02-alkyl, S02-aryl, SOZ-benzyl; and optionaliy the heterocyclic ring is fused
to
an aryl; or
H FT;;
Special embodiments of the invention are the following all referring to
formula I as above.
An embodiment of the invention is wherein
R3 represents
hydrogen, cyclopropyl, cyclopentyl, cyc:lohexyl, methyl, ethyl, propyl,
isopropyl, CF3,
ethoxy, norbornene, norbornane, adamantane, benzyl; phenyl;
or
R3 together with R2 or R4 and together with the atoms to which they are
attached forms a
5-membered ring;
Another embodiment of the invention is wherein
R4 represents
hydrogen, methyl, ethyl; or R4 together with R3 and together with the atoms to
which
they are attached, forms a 5-membered ring;
And another embodiment of the invention is wherein
RS represents
hydrogen, chloro, bromo, methyl, phenyl, -S02NH~;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
2%
And another embodiment of the invention is wherein
R6 represents
hydrogen, 2-methoxyphenyl, 2-pyridyl, ;f-pyridyl, methyl, methoxy, chloro or
bromo;
And another embodiment of the invention is wherein
R' represents
hydrogen, chloro, broma, methyl, 1-hydroxyethyl, acetyl, -~(CH3)C=N-OH, CONH2,
CO 2-ethyl, cyano, phenyl, 2-N-acetylarninophenyl, 2-nitrophenyl, 2-
methoxyphenyl, 4-
trifluaromethyl-~~-methoxyphenyl, 2,4-dimethoxyphenyl, 2-N,N-
dirnethylsulfamoylphenyl, 2- chlorophenyl, 2-fluorophenyl, 3-hydroxyphenyl, 2-
pyridyl,
3-pyridyl, 2-pyrimidyl, 2-furyl, 3-furyl, 2-~tY~ienyl, 2-(N-methyl)-
imidazolyl, 5-triazolyl, 4-
phenyl-triazol-5-~yl, 5-methyl-1,2,4-oxadiazol-3-yl, CH3CONH-, CH3S02NH-, N02,
SO 2OH, phenyl-SOz-, sulfamoyl, N,N-dimethylsulfamoyl, N,N-diethylsulfamoyl, N-
phenyl-N-methyl-sulfamoyl, N-cyclohea;yl-sulfamoyl, -502-heterocyclic ring,
wherein
thc: heterocyclic rings are selected from the group of piperidine,
pyrrolidine, 1,2,5,6-
tetrahydropyridine, tetrahydroquinoline, N-methylpiperazine, N-sulfonylmethyl-
piperazine, morpholine;
And another embodiment is wherein
R8 represents
hydrogen, methyl, hydroxymethyl, 2-methoxyphenyl, 3-methoxyphenyl, 2-pyridyl,
methoxy;
Especially preferred embodiments are a compounds represented by formula I as
above
wherein X is S0~2 and Y is N and the broken line represents a single bond
and R2 represents hydrogen or CH3;
and R3 represents cyclohexyl, cyclopentyl, norbornene, norbornane, adamantane,
phenyl, ethoxy;
and R4 represents hydrogen or CH3;
and R5 represents hydrogen, CH3, phenyl, sulfamoyl, chloro, bromo,
and R6 represents hydrogen, CH3, 2-methoxyphenyl, methoxy, chloro, bromo, 2-
pyridyl, 3
pyridyl;
and R' represents
hydrogen, chloro, bromo, methyl, 1-hydroxyethyl, acetyl, -(CH3)C=N-OH, CONH2,
C02-
ethyl, cyano, ph~anyl, 2-N~-acetylaminophenyl, 2-nitrophenyl, 2-methoxyphenyl,
4-
trifluoromethyl-2-methoxyphenyl, 2,4-dirnethoxyphenyl, 2-N,N-
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
28
dirnethylsulfamoylphenyl, 2-chlorophenyl, 2-fluorophenyl, 3-hydroxyphenyl, 2-
pyridyl,
3-pyridyl, 2-pyrimidyl, 2-furyl, 3-furyl, 2~-thienyl, 2-(N-methyl)-imidazolyl,
5-triazolyl, 4-
phenyl-triazol-5~-yl, 5-methyl-1,2,4-oxadiazol-3-yl, CH3CONH-, CH3S02NH-, N02,
SO 20H, phenyl-SOz-, sulfamoyl, N,N-dimethylsulfarnoyl, N,N-diethylsulfamoyl,
N-
phenyl-N-methyl-sulfamoyl, N-cyclohexyl-sulfamoyl, -S02-heterocyclic ring,
wherein
the heterocyclic rings are selected from the group of piperidine, pyrrolidine,
1,2,5,6-
tekrahydropyridine. tetrahydroquinoline, N-methylpiperazine, N-sulfonylmethyl-
piperazine, marpholine;
R8 represents methyl, hydroxymethyl, 2-methoxyphenyl, 3-methoxyphenyl, 2-
pyridyl,
methoxy,
Another especially preferred embodiment of the invention is a compounds of
formula I as
above wherein X is S02 and Y is N and the broleen line represents a double
bond
and R3 represents CH3 or CF3 or R3 together vvith R4 and together with the
atoms to which
and R4, R6 and RB are all hydroger3;
and RS is hydrogen or halogen;
and R' is N-methylsulfamoyl, N,N-dimethylsulfarnoyl, N-cyclohexylsulfamoyl,
tetrahydropyridyl-sulfonyl; S020H, sulfamoyl;
Another especially preferred embodiment of the invention is a compounds of
formula I as
above wherein X is C=c~ and Y is N, O or CH; and
R2 represents hydrogen; and
R3 represents hydrogen, C~H3, CF3, cyclohexyl, norbornene, phenyl, ethyl; and
R' represents hydrogen, N,N-dimethylsulfamoyl, N-cyclohexylsulfamoyl,
tetrahydropyridyl-
sulfonyl, rnorpholino-sulfonyl sulfarnoyl, bromo; and
RS represents hydrogen or bromo; and
R4, R6 and RB all represent hydrogen;
Another especially preferred embodiment of the invention is a compounds of
formula I as
above wherein X represents CH2 and Y is N; and
R3 represents cyclohexyl or narbornene; and
R5 represents hydrogen or bromo; and
R' represents bromo or sulfamoyf; arid
R2, R4, R~' and R8 all represent hydrogen;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
29
Another especially preferred embodiment of the invention is a compounds of
formula I as
above wherein X represents SO, and Y represents NH; and the broken line is
absent and R2,
R4, R5 and R~ all represent hydrogen;
R3 represents cyclohexyl, methyl or hydrogen; and
R' represents N,N-dimethylsulfamc>yl. tetrahydropyridyl-sulfonyl, bromo;
and R6 represents bromo or hydrogen;
Another especially pref~srred embodiment of the invention is a compounds of
formula I as
above wherein X is SO;~ and N is -NHCH2-; arid R3 represents 3-methylbut-2-yl,
phenyl or
cyclohexyl; and R'reprEaents 1-piperidino-sulfonyl.
The most preferred embodiment of the invention are compounds of formula I as
above
wherein the compounds are the following:
2-Cyclohexyl-4-oxo-1,2,3,4-tetrahydroquinazoline;
2-Phenyl-4-oxo-1,2,3,4-tetrahydroquinazoline;
2-Methyl-3,4-dihydro-1,3-benzoxazine-4-one;
2-Phenyl-3,4-dihydro-1,3-benzoxazine-4-one;
3-Bicyclo[2.2.1 ]hept-5'-E:n--2'-yl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Phenyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-i ,1-dioxide;
1,2,3,5,10,1 Oa-Hexahydrobenzo[e]pyrrolo[1,2-b]- t ,2,4-thiadiazine-5,5-
dioxide;
2-Ethyl-2-methyl-3,4-dihydro-1,3-benzoxazine-4~-crne;
3-Cyclohexyl-6-(2-methoxyphenyl)-1,2,3,4-tetr<~hydro-1,2,4-krenzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-6-(2-pyridyl)-1,2,3,4-tetrahydro-1,2,~1-benzothiadiazine-1,1-
dioxide;
3-Cyciohexyl-6-(3-pyridyl)-1,2,3,4-tEarahydro-1,2r4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-(1-hydroxyethyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-acetyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-(1-hydroxyiminoethyl)-1,2,3,4-tetrahydro-1,2,4~-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl~-7-carbamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-ethoxycarbonyl-1,2, 3,4-tetrahyciro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohe:Kyl-7-cyana-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Bicyclo[2.2.1 ]hept-5'-Eon-2'-yl-7-phenyl-1,2,3,~1-tetrahydro-1,2,4-
benzothiadiazine-1,1-
3o dioxide;
3-Cyclohexyl-~7-(2'-acetamidophenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-(2'-nitro~rhenyl)-1,2.,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-(2'-methoxyphenyl)-1,'?,3,4-tetr;~t~ydro-1,2,4-benzothiadiazine-
1,1-dioxide;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
3()
3-Cyclohexyl-7-(2'-methoxy-4'-trifluoromethylphenyl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-(2',4'-diimethoxyphenyl)-1,2,3, 4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide;
3-CycIohE:xyl-7-(2'-{N,~J-c!imethylsulfamoyl)phenyl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-(2'-chlorophenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;3-
Cyclohexyl-~-(2'-fluorophenyl)-1,2,3,4-tetrahynro-1,2,4-benaothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-(3'-hydroxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyf-7-(2'-pyrir~yl )-1,2,3,4-tetrahydro-1,2,4-benzoth iadiazine-1,1-
dioxide;
3-CycIohE~xyl-7-(3'-pyridyl )-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-CycIohE:xyl-7-(2'-pyrirnidinyl)-1,2,3,4-tetrahy~iro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-(2'-furyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-(3'-furyl)-1,2,3,4-tetrahydro-1,2,.4-~benzothiacdiazine-1,1-
dioxide;
3-CycIohE:xyl-7-(2'-thienyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-(1-methyl-1H-2-imidazolyl)-1,2,3,.4-tetrahydna-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-CycIohE;xyl-7-(1',2',3'-triazol-4'-yl)-1,2,3,4-teirrahydro-7 ,2,4-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-(5'-phenyl-1',2',3'-triazol-4'-yl)-1 ,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-(5'-methyl-1',2',4'-oxadiazol-3-yl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide;
3-CycIohE;xyl-7-acetamido-1,2,3,4-tetrahydro-1,'2.,4-benzothiadiazine- 1,1-
dioxide;
3-Cyclohexyl-7-methylsulfonylamino-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-vitro-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-phenylsulfonyl-1,2,3,4-tetrahycfro-1,2,4-benzothiadiazine-1,1-
dioxide;
2-Cyclohexyl-1,2,3,4-tetrahydro-6-qurnazoline sulfonamide;
3-Cyclohexyl-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-sulfamoyl-1,2,3,4-tetrahydro-1 ,2,4-benzothiadiazine-1,1-
dioxide;
3-Methyl-.7-dimethylsulfamoyl-1,2,3,4-tetrahyd ro-1,2,4-benzothiadiazine-1,1-
dioxide;
2-Cyclohexyl-1,2,3,4-tetrahydro-6-quinazoline N,N-climethylsulfonamide;
3-Cyclohexyl-7-dimethylaminosulfonyl-1,2,3,4-tetrahydro-1,2.,4-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-(N,N-diE~thylamino)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-pyrrolidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
31
3-Methyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclopropyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4~-benzothiadiazine-
1,1-dioxide;
3-Isopropyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-propyl-7-piperidinosulfonyl-1,2,~s,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Benzyl-7-piperidinos~ulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyciopentyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-piperidinasulfonyl-1,2,3,4-tetraloydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Bicycla[2-2.1 )hept-5'-en-2'-yl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-
1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-(1',2',3',6'-tetrahydropiperidin~o)sulfonyl-1,2,3,4-tetrahydro-
1,2,4-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-(N-methyl-N-phenylamino)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-(1'-(1',2',3',4'-tetrahydroquinolinyl))sulfonyl-1,2,3,4-
tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-7-(4'-me~thylpiperazino)sulfonyl-1,2 ,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-7-(4'-mel:hylsulfonylpiperazino)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyi-7-morpholinosulfonyl-1,2,3,4-tetr<~hydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Bicyclo[2.2.1 jhept-5'-err-2'-yl-7-bromo-1,2,1,4-tetrahydro-1,2,4--
benzothiadiazine-1,1-
dioxide;
2-Methyl-4-oxo-3,4-dihydro-6-quinazoline-N,N-dimethylsulfonamide;
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline sulfonamide;
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline N,N-dimethylsulfonamide;
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline-1',2',3',6'-
tetrahydropiperidinosulfonamide;
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline N-cyclohexylsulfonamide;
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline morpholinosulfonamide;
2-Cyclohexyl-4-oxo-3,~1-dihydro-6-quinazoline-N,N-dimethylsulfonamide;
3-Methyl-7-sulfamoyl-1,2~-dihydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Methyl-7-dimethylsulfarnoyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Methyl-7-(1',2',3',6'-tetrahydropiperidino)sulfonyl-1,2-dihydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Methyl--7-cyclohexyi~;ulfamoyt-1,2-dihydro-1,2 ,.4-benzothiadiazine-1,1-
dioxide;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
32
3-Trifluoromethyl-7-dimethylsulfamoyt-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide;
2-Trifluoromethyt-4-oxo-3,4-dihydro-6-quinazolinesulfonic acid;
3-Cyclohexyl-8-methyl-~1,2,3,4-tetrahydro-1,2,4-~benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-8-hydroxymethyl-1,2,3.4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-8-(2-methoxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-8-(3-methoxyphenyl)-1,2,3,4-tet~rahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-8-(2-pyrictyl)-1,2,3,4-tetrahydro--I ,2,4-benzothiadiazine~-1,1-
dioxide;
3-Cyclohexyl-8-methox:y-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
5,7-Dibromo-1,2-dihydro-1,2,4-benzothiadiazinc:-1,1-dioxide;
3-Cyclohexyl-2-methyl-7-morpholinosulfonyl-1,a?,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-4-methyl-7-morpholinosulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
7-Methyl sulfonylamino-~1,2,3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-
thiadiazine-5,5-
dioxide;
7-Sulfamoyl-1,2,3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-thiadiazine-5,5-
dioxide;
7-Methylsulfamoyl-1,2, 3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-
thiadiazine-5,5-
dioxide;
7-Cyclohexylsulfamoyl-~1,2,3,3a,4,5-hexahydrobenzo[ e] pyrrolo[2,1-c[-1,2,4-
thiadiazine-5,5-
dioxide;
7-Dimethylsulfamoyl-1,2,3,3a,4,5-hexahydrober~zo[e]pyrrolo[2,1-c]-1,2,4-
thiadiazine-5,5-
dioxide;
7-Methylsulfamoyl-1,2,:3,5-tetrahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-thiadiazine-
5,5-dioxide;
7-Dimethylsulfamoyl-1, 2,3,5-tetrahydrobenzo[e]pyrrolo[2,1-y-1,2,4-th
iadiazine-5,5-dioxide;
7-Cyclohexylsulfamoyl-1,2,3,5-tetrahydrobenzo[e]pyrrolo[2,'1-c]-1,2,4-
thiadiazine-5,5-dioxide;
7-(1',2',3',6'-Tetrahydropiperidino)sulfonyl-1,2,3,5-
tetrahydrobenzo[e]pyrrolo[2,1-c]-1,2,4
thiadiazine-5,5-dioxide;
3-Bicyclo[2.2.1 jhept-5'-en-2'-yl-5,7-dimethyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyt-7-(N,N~iiEahylsulphamoyl)-5-methyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide;
3-Bicyclo[2.2.1 jhept-5'-en-2'-yl-5,7~-diphenyl-1,2, 3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
33
3-Bicyclo[2.2.1 ]hept-5'-en-2'-yl-5,7-disulfamoyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Bicyclo[2.2.1 ]hept-5'-en~-2'-yl-5,7-dichloro-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
5-Bromo-3-cyclohexyl-~'-sulfamoyl-1,2,3,4-tetr,ahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
2-Bicyclo[2.2.1 ]hept-5'-en-2'-yl-6,8~-dibromo-1,2,3,4-tetrahydroquinazoline;
2-Bicyclo[2.2.1 ]kept-5'-~en-2'-yl-6,8-dibromo--4-oxo-1,2,3,4-
tetrahydroquinazoline;
3-Bicyclo[2.2.1 ]hept-5'-.en-2'-yl-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
5,7-Dibrorno-3-bicyclo[a?.2.1 ]heptan-2'-yl-1,2,3,4-tetrahydro-"I ,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-5,7-dibrorno-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Adamantyl-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Phenyl-;5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
75 3-Ethoxy-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide;
3-Methyl-5,7-dibromo-1,2-dihydro-7 ,2,4-benzothiadiazine-1,v-dioxide;
3-Cyclohexyl-6-methyl-7-(2'-pyridyl)-1,2,3,4-tetrahydro-1,2,4~-
benzothiadiazine-1,1-dioxide;
3-Cyclohexyl-6-methyl-;7-(4'-triazolyl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide ;
3-Cyclohexyl-6-methyl-;7-sulfamayl~-1,2,3,4-tetrahydro-1,2,4-h~enzothiadiazine-
1,1-dioxide;
3-Cyclopentyl-6-methyl-7-piperidinosulfonyl-1,;?,3,4-tetrahydro-1,2,4-
benzothia-diazine-1,1-
dioxide;
3-Cyclohexyl-6-methyl-;~-morpholinosulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-6-(2-meth~oxyphenyl)-7-methyl-1,2.3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-6-methoxy-T-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothia-
diazine-1,1-
dioxide;
3-Cyclohexyl-7,8-ethylenedioxy-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-cyclohexyl-6,7-ethylenedioxy-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;3-
Cyclohexyl-6-chioro-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Phenyl-Fi-chloro-7-sullramoyl-1,2,3,4-tetrahydr<>-1,2,4-benzothiadiazine-1,1-
dioxide;
3-Cyclohexyl-6-bromo-T-piperidinosulfonyl-1,2 ,3,4-tetrahydrc>-1,2,4-benzothia-
diazine-1,1-
dioxide;
2-cyclohexylmethylamino-5-N,N-dimethylsulfannaylbenzenesulfonamide;
CA 02320354 2000-08-04
WO 99,/42456 PCT/DK99/00070
34
2-Ethylamino-7-(1',2',3',6'-tetrahydropiperidino)sulfonylbenzene sulfonamide;
3-Isobutyl-8-(piperidinos;ulfonyl)-2,3,4, 5-tetrahydro-1,2,5-
benzothiadiazepine-1,1-dioxide;
3-Cyclohe:Kyl-7-cyclopentylsulfinyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-cyclopentylsulfinyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide;
3-Cyclohexyl-7-cyciopentylsulfinyl-1,2,3,4-tetrah~rdro-1,2,4-benzothiadiazine-
1,1-dioxide; or
3-Cyclohe:Kyl-7-cyclopentylsulfinyl-~1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
or a pharmaceutical acceptable salt thereof.
Pharmaceutically Acceptable Salts
The chemical c:ompounc~ of the invention may be provided in any form suitable
for
the intended administration. Suitable forms include pharmaceutically (i.e.
physiologically)
acceptable salts, and pre- or prodrug forms of the chemical c:;ompound of the
invention.
Examples of pharmaceutically accE;ptable addition salts include, without
limitation,
the non-toxic inorganic and organic acid addition salts such as the acetate
derived from
acetic acicl, the aconate derived from aconitic ,ac:id, the ascorbate derived
from ascorbic acid,
the benzenesulfonate derived from benzensulfonic acid, the benzoate derived
from benzoic
acid, the cinnamate derived from cinnamic acid, the citrate derived from
citric acid, the
embonate derived from embonic acid, the enantate derived from enanthic acid,
the formate
derived from formic acid, the fumarate derived from fumaric acid, the
glutamate derived from
glutamic acid, the glycolate derived from glycolic acid, the hydrochloride
derived from
hydrochloric acid, the hydrobromide derived from hydrobrornic acid, the
lactate derived from
lactic acid, the maleate derived from malefic acid, the malonate derived from
malonic acid, the
mandelate derived from mandelic acid, the methanesulfonate derived from
methane
sulphonic acid, the naphthalene-2-suiphonate derived from naphtalene-2-
sulphonic acid, the
nitrate derived from nitric acid, the perchlorate derived from perchloric
acid, the phosphate
derived from phosphoric: acid, the phthalate derived from phth~lic acid, the
salicylate derived
from salicylic acid, the sorbate derived from sorbic acid, the stearate
derived from stearic
acid, the :~uccinate derived from succinic acid, the sulphate derived from
sulphuric acid, the
tartrate derived from tartaric acid, the toluene-p-sulphonatEl derived from p-
toluene sulfonic
acid, and the like. Such salts may be formed by procedures well known and
described in the
art.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
W
Other acids such as oxalic acid, which may not be considered pharmaceutically
acceptable, may be u:>eful in the preparation of salts useful as intermediates
in obtaining a
chemical compound of the invention and its pharmaceutically acceptable acid
addition salt.
Metal salts of a chemical compound of the invention includes alkali metal
salts,
such as the sodium salt of a chemic<~I compouryd of the invc.~ntion containing
a carboxy group.
The chemical compound of the irrvention may be provided in dissoluble or
indissoluble forms together with a pharmaceutically acceptable solvents such
as water,
ethanol, and the like. Dissoluble forms may also include hydrated forms such
as the
monohydrate, the dihydrate, the hemihydrate, the trihydrate:, the
tetrahydrate, and the like. In
general, l:he dissoluble forms are considered ~ecfuivaient to indissoluble
forms for the purposes
of this invention.
Definitions of substituents:
Halogen is fluorine, chlorine, bromine, or iodine.
Alkyl means a straight chain or branched chain of from one to six carbon atoms
or cyclic alkyl
of from three to seven carbon atoms, including but not limited to, methyl,
ethyl, propyl,
isopropyl, butyl, isobutyl, t-butyl, pentyl, hexyl; methyl, ethyl, propyl and
isopropyl are
preferred groups.
Haloalkyl means alkyl as above substitutecl one or more times with halogen as
defined
above. Preferred embodiments arE: CF3, C2F5, (:;H2CI, CHCI;a, -CHFCH2F, -
CHCICH2CI;
Cycloalkyl means cyclic alkyl of from three to seven carbon atoms such as
cyclopropyl,
cyclobutyl, cyclopentyl, cyclahexyl, cycloheptyl.
Cycloalkylalkyl means cyclic alkyE as above arid alkyl as above wherein the
alkyl can be
regarded as a substituent on the cycloalkyl and vice versa . Preferred groups
are C3_s-
cycloalkyl and C,_4-alkyl such as -(CH2)-cyc:lopropyl, -cyclopropyl-(C,_4-
alkyl), -(CH2)~ -
cyclohexyl, -cyclohexyl-(C,.4-alkyl), (C,.4-alkyl)-cyclobutyl, -
cyclobutyl(C,_4-alkyl) -(C,_4-
alkyl)cyclopentyl, -cyclopentyl(C,_4-alkyl), -(C,_4-alkyl)cyclohE=xyl,
cyclohexyl(C,_4-alkyl);
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/000'70
36
Halocycloalkyl means cyclic alkyl as above which is substituted with one or
more halogen as
above, including but not limited to chlorocycfopropyl, fluorocyclopropyl,
iodocyclopropyl,
dichlorocyclopropyl, difluorocyclopropyl, chlorocyclobutyl, fluorocyclobutyl,
chlorocyclopentyl,
fluorocyclopentyl, iodocyclopenyl, chlorocyclohexyl, fluorocyclohexyl,
dichlorocyclohexyl,
difluorocyclohexyl, iodocyclohexyl. Preferred embodiments are mono- and di-
substituted
cycioalkyl of 3 to 1:> carbons, such as dichlorocyclopropyl,
difluorocyclopropyl,
chlorocyclohexyl, fluorocyclohexyl, iadocyclohexyl, chlorocyclopentyl,
fluorocyclopentyl.
Alkenyl means a straight chain ar branched chain of from two to six carbon
atoms containing
one double bond, including but not limited to ethenyl, 1-propenyl, 2-propenyl,
1-butenyl, 2-
butenyl, and 3-butenyl.
Alkynyl means a straight chain or branched chain of from two to six carbon
atoms containing
one triple bond, including but not limited to ethynyl, 1-propynyl, 2-propynyl,
1-butynyl, 2-butynyl, and 3-butynyl.
Alkoxy is O-alkyl, wherein alkyl is as defined above.
Alkoxyalkyl is -alkyl-O-alkyl, wherein alkyl is as defined above.
Hydroxyalkyl is alkyl as. defined above substituted with OH;
Amino is NH2 or NH-alH;yl or N-(alkyl), wherein alkyl is as defined above.
Alkylamino is alkyl as defined above whrch is substituted with amino as
defined above.
Preferred embodiments are -CHI-N(alkyl)z, -C;Fi-N(alkyl)2CH;~, -CH2CH2
N(alkyl)2, -CH2-NH2, -
CH-(NHS)-CH3, -CH2Chl2 NHS>~
Cyano is CN;
Cyanoalkyl is alkyl as clefined above substituted with CN;
Nitro is -N02;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
37
Nitroalkyl is alkyl as defined above subsituted with vitro as defined above;
Thio is SH or S-alkyl, wherein alkyl is as defined above;
Alkylthio is alkyl as above substituted with a thin group which is as defined
above.
Acyl is (C;=O)-R° or (C=S)-R° wherein R° is alkyl; phenyl
which may be substituted one or
more times with substituents selected from the group consisting of halogen,
CF3, N02, amino,
alkyl, alkoxy, phenyl and S02NR'R" wherein R' and R" each independently are
hydrogen or
alkyl or wherein R' and R" together ~s (CH~)n, wherein m is 2, 3, 4, 5 or 6;
or R° is benzyl; or
NR"' Rw wherein R"' an~~ R'v each independently are hydrogen or alkyl or
wherein R~° and R'v
together is (CH2)P wherein p is 2, 3, 4, 5 or 6.
Acylamino is acyl-NH- wherein aryl is as defined above.
Aryl is aromatic carbocycles such as phenyl or biphenyl and fused carbocycles
such as
naphtyl;
HET is an 5- to 6-membered cyclic heteroaryl and include: for example, oxazol-
2-yl, oxazol-
4-yl, oxazol-5-yl, isoxa:zol-3-yl, isoxazol-4-yl, iso:xazol-5-yl, thiazol-2-
yl, thiazol-4-yl, thiazol-5-
yl, isothiazal-3-yl, isolhiazol-4-yl, isothiazol-5--y1, 1,2,4-ox:adiazol-3-yl,
1,2,4-oxadiazol-5-yl,
1,2,4-thiadiazol-3-yl, 1,2,4-thiacfiazol-5-yl, 1,2,5~-oxadiazol~-3-yl, 1,2,5-
oxadiazol-4-yl, 1,2,5-
thiadiazol-3-yl, 1,2,5-thiadiazol-4-yl, 1-imidaz:olyl, 2-imidazolyl, 4-
imidazolyl, 1-pyrrolyl, 2-
pyrrolyl, 3-pyrrolyl, 2-furanyl, 3-furanyl, 2-thiE:nyl, 3-thienyl, 2-pyridyl,
3-pyridyl, 4-pyridyl, 2-
pyrimidinyl, 4-pyrimidinyl, 5-pyrirnidinyl, 3-pyrictazinyl, 4-pyridazinyl, 2-
pyrazinyl, 1-pyrazolyl, 3-
pyrazolyl, and 4-pyrazolyl, furanyl, tetrahydrofuranyl, pyrrolyl, pyrrolidyl,
imidazolyl,
oxadiazolyl, pyridyl, thieroyl, isooxazolyl, pyrimidyl, pyrazole, triazoiyl.
Especially preferred
heteroaryl of the invention are pyridyl, pyrimidyl, triazole, furyl, thienyl,
oxadiazolyl, imidazolyl;
A carbocyclic 7- to 12- membered ring structure includes mono- bi- and
tricyclic structures.
Preferred embodiments are 7- to 10 membered ring structures such as
CA 02320354 2000-08-04
WO 99I'42456 PCT/DK99/00070
$$
1~7 ~7:i~ ~J~ ~W L~ ~~
'~CW '~ ! ~~: ~
a heterocyclic 3- to 8 membered ring structure includes a partly or completely
saturated
heterocyclic ringstructure such as aziridine, pyrrcslidine, piperidine,
piperazine,
homopiperidine, homopiperazine, azacyclooctane, 1,.3-diazacyclooctane, 1,4-
diazacyclooctane, tetrahydrofuran, tetrahydrothiophene, mc:~rpholine,
tetrahydropyridine, and
compounds such as
N
N N
l
The preferred embodiments are 5- to 6-membered rings containing at least one
nitrogen such
as pyrrolidine, piperidin~~, piperazine, morpholinc;, tetrahydropyridine.
The described 4- to T-membered rings fused to the ring structure of formula I,
formed
between the substituents R' and R3 or R3 and R4 or RS and R6 or R6 and R' or
R' and R8 are
carbocyclic rings optionally containing a heteroatom and optionally containing
a carbonyl
group. PrE;ferred rings are 5- and F-membered c:arbocyclic rings;
The rings formed between the substituents R' and R6 or Rg are 5- or 6-membered
and
containing O, C=O, S=O, or S02-groups and optionally containg nitrogen.
Preferred rings are -O-(CH2)n-O-; wherein n is 1, 2 or 3; -SC)z-NR-(CH2)n-
wherein n is 1 or 2;
-SO-NR-((:,Hz)n- wherein n is 1 or a?; -S02-(CH2)n- wherein n is 2 or 3; -SO-
(CH2)n- wherein n
is 2 or 3; -CO-CH=CH-NH-;-CO-CH=CH-O-; -C;O-(CH2)~-Nlwl- wherein n is 1 or 2; -
CO-NH
(CH2)~ wherein n is 1 or 2; -CO- (CH2j2-O-;
The compounds of this invention rnay exist in unsolvated ,:~s well as in
solvated forms with
pharmaceutically acceptable solvents such as water, ethanol and the like. In
general, the
solvated forms are considered equivalent to tf-re unsolvated forms for the
purposes of this
invention.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Steric Isomers
The chemical compounds of the ;present invention may exist in (+) and (-)
forms
as well as in racemic forms. Thc: racemates of these isomers and the
individual isomers
themselves are within the scope of the present invention.
Racemic forms can be resolved intca the optical antipodes by known methods and
techniques. One way of separating the diastereomeric salts is by use of an
optically active
acid, and liberating thc: optically active amine compound k:~y treatment with
a base. Another
method for resolving racemates into the optical antipodes as based upon
chromatography on
an optical active matrix. Racemic cc>mpound:> of the present invention can
thus be resolved
into their optical antipodes, e.g., by fractional) crystallisation of d- or I-
(tartrates, mandelates,
or camphorsulphonate 1 salts for example.
The chemical compounds of the present invention rnay also be resolved by the
formation of diastereomeric amides by reaclion of the chemical compounds of
the present
invention with an optically active activated carboxylic acid such as that
derived from (+) or (-)
phenylalanine, (+) or (-) phenylglyc:ine, (+) or (-) camphanic acid or by the
formation of
diastereomeric carbamates by reaction of the chemical cc:~mpound of the
present invention
with an optically active chloroformate or the like.
Additional methods for the resolvinca the optical isomers are known in the
art.
Such methods include those described by Jaques J, Collet A, & tNilen S in
"Enantiomers,
Racemates. and Resolutions", John Wiley and Sons, New Fork (1981 ).
Moreover, Borne of thE: chemical compounds of the invention being oximes, may
thus exist in two forms., syn- and anti-form {Z- arid E-form), depending on
the arrangement of
the substituents around the -C=N- double band. A chE>mical compound of the
present
invention may thus be vthe syn- or the anti-forrn (a?- and E-form), or it may
be a mixture hereof.
A compound of the invention includes endo- and exo-forms and tautomers where
possible.
Pharmaceutical Comaositions
An aspect of the invention provides novel pharmaceutical compositions
comprising a therapeutically effective amount of the chemical compound of the
invention and
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
the use of compounds of the invention for the manufacture of a medicament for
the treatment
of specific: diseases or disorders;
White a chemical compound Gf the inventican for use in therapy may be
administered in the fornn of the raw chemical compound, it is preferred to
introduce the active
5 ingredient, optionally in the form of a physiologically acceptable salt, in
a pharmaceutical
composition together with one or more adju~dants, excipients, carriers,
buffers, diluents,
and/or other customary pharmaceutical auxiliarie s.
In a preferr~ad embodirnent, the invention provides pharmaceutical
compositions
comprising the chemical compound of the invention, or a pharmaceutically
acceptable salt or
10 derivative thereof, tagethc:r with one or mare taharmaceutically acceptable
carriers therefor,
and, optionally, other therapeutic and/or prophylactic ingredients. The
carriers) must be
"acceptable" in the sense of being compatible with the other ingredients of
the formulation
and not harmful to the recipient thereof.
Pharmaceutical compositions of the invention may be those suitable for oral,
15 rectal, bronchial, nasal, topical (including buc~aal and sub-lingual),
transdermal, vaginal or
parentera'I (including cutaneous, subcutaneous, intramusc:ular, and
intravenous injection)
administration, or those in a form suitable for administration by inhalation
or insufflation.
The chemical compound of the irtvE:ntion, together with a conventional
adjuvant,
carrier, or diluent, may thus be placed into the form of pharmaceutical
compositions and unit
20 dosages thereof. Such forms include solids, and in particular tablets,
filled capsules, powder
and pellet forms, and Liquids, in p;~rticular aducyous or non-aqueous
solutions, suspensions,
emutsions~, elixirs, and capsules filled with the same, all for oral use,
suppositories for rectal
administration, and sterile injectable solutions for parenteral use. Such
pharmaceutical
compositions and unit dosage forms thereof may comprise conventional
ingredients in
25 conventional proportions, with or without additional active ccampounds or
principles, and such
unit dosage forms may contain any suifab~le effective aamount of the active
ingredient
commensurate with the intended daily dosagE: range to be employed.
The chemical compound of the present invention can be administered in a wide
variety of oral and pare~nteral dosage forms. It will be obvious to those
skilled in the art that
30 the following dosage forms may comprise, as the activc:a component, either
a chemical
compound of the inveni:ior~ or a pharmaceutically acceptably-w salt of a
chemical compound of
the invention.
For preparing pharmaceutical c;ornpositions from a chemical compound of the
present invention, pharmaceutically acceptable carriers can be either solid or
liquid. Solid
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
41
form preparations include powders, tablets, pills, capsules, cachets,
suppositories, and
dispersible granules. A solid carrier can be one or mare substances which may
also act as
diluents, flavouring agents, solubilizers~ lubricants, suspending agents,
binders,
preservatives, tablet di~~integrating agents, or an encapsulating material.
In powders, the carrier is a finely divided solid which is in a mixture with
the finely
divided active component.
In tablets, tihe active component is mixed with the carrier having the
necessary
binding capacity in suitable proportions and compacted in the shape and size
desired.
The powdeirs and tablets preferably contain from five or ten to about seventy
percent of the active compound. Suitable carriers are m<~gnesium carbonate,
magnesium
stearate, talc, sugar, lactose, pectin, dexkrin, starch, gelatin, tragacanth,
methylcellulose,
sodium carboxymethylcellulose, a low melting wax, cocoa: butter, and the like.
The term
"preparation" is intended to include the formulation of the active compound
with
encapsulating material as carrier providing a capsule in which the active
component, with or
without carriers, is surrounded by a carrier, which is thus; in association
with it. Similarly,
cachets and lozenges are included. Tablets, powders, capsules, pills, cachets,
and lozenges
can be used as solid forms suitablE: for oral administration.
For preparing suppositores, a low rr~elting wax, such as a mixture of fatty
acid
glyceride or cocoa butter" is first melted and the <:~ctive component is
dispersed
homogenE~ously therein, as by stirring. The molten homogenous mixture is then
poured into
convenient sized moulds, allowed to cool, and thereby to solidify.
Compositions suitable for vaginal administration may be presented as
pessaries,
tampons, creams, gels, pastes, foams or sprays containing in addition to the
active ingredient
such carriers as are known in the art to be appropriate.
Liquid preparations include solutions., suspensions, and emulsions, for
example,
water or water-propyler7e glycol solutions;. For example, parenteral injection
liquid
preparations can be formulated as solutions in aqueous polyethylene glycol
solution.
The chemical compounc:i according to the f:~resent invention may thus be
formulated for parenteral administration {e.g. by injection, for example bolus
injection or
continuous infusion) and may be presented in unit dose form in ampoules, pre-
filled syringes,
small volume infusion car in mult~-dose c:oratainers with an added
preservative. The
compositions may take such forms as suspen.;ic>rrs, solutions, or emulsions in
oily or aqueous
vehicles, and may cont<~in formulation agents such as, suspending, stabilising
and/or
dispersing agents. Alternatively, the active ingredient may be in powder form,
obtained by
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
42
aseptic isolation of sterile solid or by lyophilization fronn solution, for
constitution with a
suitable vehicle, e.g. s~lerile, pyrogen-free wager, before use.
Aqueous solutions suitable for oral use can be prepared by dissolving the
active
component in water arid adding suitable colorants, flavours, stabilising and
thickening agents,
as desired.
Aqueous suspensions suitable for oral use cart be rnade by dispersing the
finely
divided active component in water with viscous material, such as natural or
synthetic gums,
resins, methylcellulose, sodium carboxymei:hylcellulose, or other well known
suspending
agents.
Also included are solid form preparations which are intended to be converted,
shortly before use, to liquid form preparations for oral administration. Such
liquid forms
include solutions, suspensions, and emulsion;. These preparations may contain,
in addition
to the active component, colorants, flavours;, stabilisers, buffers,
artificial and natural
sweeteners., dispersants, thickeners. solubilizing agents, arid the like.
for topical administration to the epidermis the chemical compound according to
the invention may be formulated as ointmenla, creams or lotions, or as a
transdermal patch.
Ointments and creams. may, for example, be formulated wish an aqueous or oily
base with the
addition of suitable thickening and/or gelling agents. Lotions may be
formulated with an
aqueous or oily base and will in general also contain one or more emulsifying
agents,
stabilising agents, dispersing agents, suspending agents, thickening agents,
or colouring
agents.
Compositions suitable for topicaG~ administration in the mouth include
lozenges
comprising the active agent in a flavoured base, usually sucrose and acacia or
tragacanth;
pastilles comprising the active ingredient in an inert base such as gelatin
and glycerine or
sucrose .and acacia; and mouthwashes comprising the active ingredient in a
suitable liquid
carrier.
Solutions or suspensions are applied directly to the nasal cavity by
conventional
means, for example with a dropper. pipette or spray. The compositions may be
provided in
single or multi-dose form. In the latter case of a dropper or pipette, this
may be achieved by
the patient administering an appropriate, predetermined volume of the solution
or suspension.
In the case of a spray, this may be achieved' for example by means of a
metering atomising
spray pump.
Administration to the respiratory tract may also be achieved by means of an
aerosol formulation in which the active ingredient is provided in a
pressurised pack with a
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
43
suitable propellant such as a chlorofluorocarbon (CFC) for example
dichlorodifluoromethane,
trichlorofluoromethane, or dichlorotetrafluoroEatt~ane, carbon dioxide, or
other suitable gas.
The aerosol may conveniently also contain a surfactant such as lecithin. The
dose of drug
may be controlled by provision of a metered valve.
Alternatively the active ingredients rnay be provided in the form of a dry
powder,
for example a powder mix of the compounc9 in a suitable powder base such as
lactose,
starch, starch derivatives such as hydroxypropylmethyl cellulose and
poiyvinylpyrrolidone
(PVP). Gonveniently the powder carrier will form a gel in the nasal cavity.
The powder
composition may be presented in unit dose forim for example in capsules or
cartridges of,
1o e.g., gelatin, or blister packs from which the powder may be administered
by means of an
inhaler.
In composil:ior7s intended for administration to the respiratory tract,
including
intranasal compositions, the compound will generally have a small particle
size for example of
the order of 5 microns or less. Such a particle size may be obtained by means
known in the
art, for example by microrrization.
When desired, compositions adapted to give sustained release of the active
ingredient may be employed.
The pharm<~ceutical preparations are preferably in unit dosage forms. In such
form, the preparation is subdivided into unit doses containing appropriate
quantities of the
active component. Thc~ unit dosage form can be a packaged preparation, the
package
containing discrete quantities of preparation, such as packaged tablets,
capsules, and
powders in vials or ampoules. Also, the unit do:~age form can be a capsule,
tablet, cachet, or
lozenge itself, or it can be the appropriate number of any of these in
packaged form.
Tablets or capsules for oral administration and liquids for intravenous
administration and continuous infusion are preferred compositions.
The dose administered must of course be carefully adjusted to the age, weight
and condition of the individual being treated, as well as the route of
administration, dosage
form and regimen, and the result desored.
The active ingredient rnay be adrninistered in one or several doses per day. A
satisfactory result can, in certain instances, bf; obtained at a dosage as tow
as 0.1 pg/kg i.v.
and 1 pg/kg p.o. The upper limit of the dosage r;~nge is presently considered
to be about 10
mg/kg i.v. and 100 mg/kg p.o. Preferred ranges are from about 0.1 pg/kg to
about 10
mg/kg/day i.v., and from about 1 yg/kg to about 100 mg/kg/day p.o.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
44
Method of Treating
The compounds of the present invention are ,AMPA receptor stimulators and
therefore useful
for the treatment of a range of disorders or diseases responsive to AMPA
receptor
modulators. As en embodiment of the invention the disease are responsive to
positive
modulation of the AMF'A receptor. The compounds may be used in the treatment,
prevention,
profylaxis or alleviation of a disease, disorder or condition crf the central
nervous system as
for example: neurodegenerative disorders, cognitive or memory dysfunction,
memory and
learning disorders, attention disorder, learning and memory disorders
resulting from ageing,
1o trauma, stroke, epilepsy; Alzheimer's disease, depression, schizophrenia,
memory loss,
AIDS-dementia, senile dementia, learning deficit, cognition deficit, sexual
dysfunctions,
psychotic; disorder, se>cual dysfuncaion, intellectual impairment disorders,
schizophrenia,
depression or autism, .attention deficit, or a disorder or disease resulting
from neurotoxic
agents, alcohol intoxication, substance abuse, cardiac bypass surgery or
cerebral ischemia;
Suitable dosage range are 0.1-X00 milligrams daily, and especially 10-70
milligrams daily,
administered once or twice a day, dependent as usual upon the exact mode of
administration, form in wtuch administered, th,e indication toward which the
administration is
directed, the subject involved and the body weight of the subject involved,
and further the
preference and experience of the physician or veterinarian in charge.
I.p. means intraperetoneally, which is a well known route of administration.
P.o. means peroral, which is a well known route of administration.
The invention then cornprises the following alone or in combination:
The use of a compound as above wherein the disease to be treated is responsive
to the
AMPA receptor modulation.
The use of a compound as above for the manufacture of a medicament for the
treatment of
disease which are responsive to the AMPA receptor modulation.
The use as above whE~rem the disease is memory and learning disorders,
psychotic disorder,
sexual dysfunction, intellectual impairment di:,c~rders, schizophrenia,
depression or autism;
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Alzheimer's disease, learning deficit, attention deficit, memory loss or
senile dementia; or a
disorder or disease resulting from trauma, stroke, epilepsy, Alzheimer's
disease, neurotoxic
agents, aging, neurodegenerative disorder, alcohol intoxication, substance
abuse, cardiac
bypass surgery or cerebral ischemia;
5
Bialoety
In vitro inhibition of ~sH-AMPA binding
1o L-glutamate (GLU) is the major excitatory neurotransmitter in the mammalian
central nervous
system. Frorn eiectrophysiological- and binding studies, there appear to be at
least three
subtypes of GLU receptors, tentatively named N-methyl-D-aspartate (NMDA)-,
quisqualate-
and kainate receptors. Gl_U receptor subtypes sE:nsitive to quisqualate and
kainate as a
group are often referred to as non-NMDA receptors. Receptor binding studies
using the
15 labelled agonists 3H-AMPA (a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic
acid) (for
quisqualate receptors) and ~H-kainate (for kainate receptors) have shown
different antagonist
selectivities and regional distribution. AMPA has been known for several years
to be a potent
and selective agonist at the traditionally named quisqualate receptors.
Activation of
quisqualate receptors by AMPA is associated with Na' influx and K' efflux
leading to
2o depolarizatian.
The non-IVMDA receptors have recently been re<;lassified to include the
quisqualate activated
metabotropic receptor hype, linked to the inositol triphosphate and
diacylglycerate me-
tabolism. AMPA does not interact with the metabotropic quisqualate receptor
but only the
25 ionotropic quisqualate receptor. SE:lective activation of the metabotropic
type has been
claimed for trans-ACPC). f-iecentfy, the potent and competitive non-NMDA
receptor
antagonists CNQX and NBQX have been described, and CNOX have been reported not
to
block the effect of quisc~ualate at the metabotropuc receptor subtype. 3H-AMPA
is a selective
radioligand for labelling the ionotropic quisqualate (AMPA) receptors.
TISSUE PREPARATION
Preparations are performed at 0-4"C unless otherwise indicated. Cerebral
cortex from male
Wistar rats (150-200 g) is homogenized for 5-1C) aec in 20 rrrl Tris-HCI (30
mM, pH 7.4) using
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
46
an Ultra-Turrax homogE~nizer. The suspension is centrifuged at 27,000 x g for
15 min and the
pellet is washed three times with buffer {centrifuged at 27,0()0 x g for 10
min). The washed
pellet is homogenized in 20 ml of buffer anct incubated an a water bath
(37°C) for 30 min to
remove endogenous glutamate and then centrifuged for 10 min at 27,000 x g. The
pellet is
then homogenized in buffer and centrifuged at: far 10 min at 27,000 x g. The
final pellet is
resuspended in 30 ml buffer and the preparation is frozen and stared at -
20°C.
ASSAY
The membrane preparation is thawed and centrifuged at 2°C for 10 min at
27,000 x g for 10
min. The pellet is washed twice with 20 ml 30 mM Tris-HCI containing 2.5 mM
CaCl2, pH 7.4
using an Ultra-Turrax homogenizes and centrifuged for 10 min at 27,000 x g.
The final pellet
is resuspended in 30 mM Tris-HCI containing a?..5 mM CaCl2 and 100 mM KSCN, pH
7.4 (100
ml per g of original tissue) and used for binding assays. Aliquots of 0.5(0.2)
ml are added to
25(20) NI of test solution and 25(20) pl of 3H-AMF'A (5 nM, final
concentration), mixed and
incubated for 30 min at 2"C. Non-specific binding is determined using L-
glutamate (0.6 mM,
final concentration). After incubation the 550 yl samples are added 5 ml of
ice-cold buffer and
poured directly onto Whatman GF/C glass fibre filters under suction and
immediately washed
with 5 ml ~of ice-cold buffer. The 240 pl samples are filtered over glass
fibre filter using a
Skatron cell harvrester. The filters are washed with 3 ml ice-cold buffer. The
amount of
2o radioactivity on the filters is determined by conventional liquid
scintillation counting. Specific
binding is total binding minus non-specific binding.
RESULT S
The test value will be given as the ICso (the concentration (NM) of the test
substance which
inhibits the specific binding of 3H-AMPA by 50°/a)
Test results
The compound numbers refer to the table below.
Compound -._ ICS( (pM)
_ - 22.0
14 45.0
15 ~ 39.0
~~ 44 ~ 3.5
47 _ ~ 5.3.
SUBSTITUTE SHEET (RULE 26~
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
~7
48 ~ 4.4
~
49 17.0
- i
~7 26.0
~
58 3.4~
~
Ei 1 8.5
Ei2 6.0
Ei3 ~ 6.0
~
113 13.(:)
114 ~ 33.(:1
~
115 7.0
F~otentiation ofi AMPA induced f3HIGABA release
from cultured cortical neurons
Neurons which express receptors for excitatory amino acids can be depolarized
by such
compounds and this depolarization will ultimately lead to a release of
transmitter substance
from the neurons. Cultured neurons obtained from 16-day-old mouse embryo
cortex are
mainly GABAergic andl express all types of excitatory aminc:~ acid receptors.
This means that
they can be stimulated by high potassium (55 mM) or by the excitatory amino
acids NMDA
(20 NM), AMPA (5 NM) and kainate (5 NM) to release their neurotransmitter
GABA.
3H-GABA may be used to label the GABA transmitter pool in the neurons and the
release of
3H-GABA from the neutrons may be used as a simple functional model for studies
of the
effects of excitatory amino acid receptor agonists, antagonists, and
modulators.
METHODS
Cell cultures
Cerebral cortices of 15-16 day-old NMRI mouse embryos are chapped in 0.4 x 0.4
mm cubes
and the tissue is dissociated by mild trypsinization (0.1 % (wt/vol) trypsin,
37°C, 10 min).
Subsequently the cell suspension (3 mill/ml) is inoculated into poly-L-lysine-
coated 30 mm
Petri dishes (3 ml/dish;l containing a slightly modified DMEM (24.5 mM KCI)
supplemented
with p-aminobenzoate (7 pM), insulin (100 mlJ/L) and 10% (vol/vol) horse
serum. Cells are
maintained in culture fnr 5-7 days with the addition of the antimitotic agent
cytosine
SUBSTITUTE SHEf=T' (RULE 26)
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
48
arbinoside (5 NM) from clay 2 in vitro to prevent glial proliferation. For
further details and
references, see Drejer et al. (Exp. Brao Res. 47, 259 (1982)).
Release experiments
Release experiments am performed using the model described by Drejer et al.
(Life Sci. 38,
2077 (198(i)). Cerebral cortex neurons cultured in Petri dishes (30 mm) are
added 100 mM y-vinyl-GAGA one hour before the' experiment: in order to inhibit
degradation of
GAGA in the neurons. 30 min before the experiment 5 uCi 3H-GABA is added to
each culture.
After this preloading period the cell monolayer ai the bottom of the dish is
covered with a
piece of nylon mesh to protect the cells against: mechanical damage and to
facilitate
dispersion of medium over the cell layer. The prE:loading medium is removed
and the Petri
dishes are placed in a s!uperfusion system consisting of a peristaltic pump
continuously
delivering thermostated 37°C superfusion mediium (HEPES buffered saline
(HBS): 10 mM
HEPES, 135 mM NaCI, 5 rnM KCI, 0.6 mM MgSO.,, 1.0 mM C;aCl2 and 6 mM D-
glucose; pH
7.4) from a reservoir to i:he top of the slightly tilted Petri dish. The
medium is continuously
collected from the lower part of the dish and dE:lirrc~red to a fr;~ction
collector. Initially, the cells
are superfused with HB;S for 30 min (tlow rate :? ml/min). Then cells are
stimulated for 30 sec
every 4 min by changinc) the superfusion medium from HBS to a corresponding
medium
containing 5 NM AMPA in the absence or presence of modulators.
Test subsl:ances are dissowed in 50% DMSO. 48"io ethanol. 'The final DMSO and
ethanol
concentration in the assay must not exceed 0.1 °>o
RESULTS
The induced release of 3H-GAGA (c;pm) is corrected for the mean basal release
(cpm) before
and after the stimulation and used ror calculation of the test value.
The potentiation of the ~4MPA response by a test substance is expressed
relative to the
potentiation of the AMPA response induced by c:yclothiazide (30 NM).
Results
The result of the test is shown in Figures 1 anc~ 2. The results show
significantly increased
Figur 1 shows potentiation of AMPA induced '"'H]GAGA release from cultured
cortical neurons
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
49
by compound 115. The patentiation is expressed relative to the potentiation
induced by 30
uM cyclothiazide.
Figur 2 shows potentiation of AMPA induced (''H]GABA release from cultured
cortical neurons
by compound 114. The patentiation is expressed relative to the potentiation
induced by 30
NM cyclothiazide.
Voltage Clamp.
METHODS
7 o Experiments were performed in voltage clamp using conventional whole cell
patch clamp
methods (Hamill et al., 1981 ), essentially as dE~scribed previausly
(Mathiesen et al., 1998).
The following salt solutions were used (mM): NaC;I (140), KC;I (4), CaCl2 (2),
MgCl2 (1 ),
Sucrose (30), Tetrodotaxin (0.0003), Bicuculline Methiodide (0.005) and HEPES
(10, pH 7.4).
Intracellular solution (mM): CsCI (120), CsF (20), MgCl2 (2), EGTA (10), HEPES
(10, pH =
7.2).
Cell cultures
Mouse neocortical neurons were cultured essentially as described by Drejer et
al. (1987).
Briefly, the forebrains from embryonic (E17} NMRI mice were removed under
sterile
conditions. The tissue was chopped in 0.4 rnrn ~aubes and the triturated with
trypsin (12.5
Ng/ml) and DNAse (2.5 ug/ml), 15 min, 37 °C. The cells were suspended
at a concentration of
1 x 106 cells/ml in a slil~htly modified DMEM which contained horse serum (10
% (vlv)),
penicillin (333 U/ml), paraaminobenzoic acid (1 mg/ ml), L-glutamine (0.5 mM),
insulin (0.08
U/ml) and KCI (23.8 mM). The cell suspension was subsequently inoculated into
poly-L-lysine
coated 35 mm Petri dishes (2 mlldish). Glass c<averslips (3.5 mm) were placed
in the dishes
before coating. After 24 hr in culture, the medium was replaced by freshly
made medium
containing 1 % N2 supplement instead of seruno.
The cells were kept in culture for 7-14 days at 37 °C (5% CO;>/95% O2)
before experiments
were carried out.
Electronics. programs and data acquisition: The amplifier used was the EPC-9
(HEKA-
electronica, Lambrect, Germany) run by a Power Macintosh G3 computer via an
ITC-16
interface. Experimentall conditions were set with the Pulse-software
accompanying the
amplifier. Data were lo~nr pass filtered and samptc~d directly to hard-disk at
a rate of 3 times
the cut-aff frequency.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Pipettes and electrodes: Pipettes were pulled from borosilicate glass
(Modulohm,
Copenhagen, Denmark) using a horizontal elecarode puller (Zeitz-(nstrumente,
Augsburg,
Germany). The pipette resistances were 1.7 - 2.4 MW in the salt solutions used
in these
experiments. The pipette electrode was a chloridized silver wire, and the
reference was a
5 silverchloride pellet electrode (In Vivo Metric, I-lealdsburg, USA) fixed to
the experimental
chamber. The electrodes were zeroed with the open pipette in the bath just
prior to sealing.
Experimental procedurE_ Coverslips were transferred to a l::p ml experimental
chamber
mounted on the stage of an inverted microscope I;IMT-2, Olympus) supplied with
Nomarski
optics. The neurons were continuously superfused with extracellular saline at
a rate of 2,5
10 ml/min. After giga-seal formation (1-5 GW, success-rate = 9t) %) the whole
cell configuration
was attained by suction.
The cells were held at a holding voltage of -60 mV and at the start of each
experiment the
current was continuously measured for at least ;30 sec to ensure a stable leak
current.
AMPA-containing solutions were delivered to thE: chamber through a custom-made
gravity-
15 driven flowpipe, the tip of which was placed approximately 50 pm from the
cell. Application
was triggered when the tubing connected to the flow pipe wcas compressed by a
valve
controlled by the Pulse-software. AMPA (30 NM) was applieri for 1 sec every 45
sec. After
obtainment of responses of a repeatable amplitude the compound to be tested
was included
in both the chamber and in the AMPA-containing solution. The compound was
present until
20 responses of a new repeatable was obtained.
The sample interval in <~II experiments was 310 Nsec.
All experiments were performed at room temperature (20 - 25 °C).
MATERIALS
25 Pregnant (9 days) NMRI mice were obtained from Bomholtgaard Breeding and
Research
Center, Ry, Denmark.
Horse serum, N2 supplement and culture media were purchased from Life
Technologies
(GIBCO), Roskilde, Denmark.
AMPA (a-amino-3-hydroxy-5-methyl-4-isoxazolE:propionic acid) was synthesized
at
30 NeuroSearch A/S. Tetnodotoxin was purchased from Alomone Labs, Jerusalem,
Israel and
Bicuculline Methiodide from RBI, MA, USA. Stacrose was from Fluka Chemie,
Buchs,
Switzerland. All other reagents were from SIGMA, USA.
Results
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
51
The results are shown in Fig. 3-I.
The compounds 56 (Fig. 3), 63 (Fig. 4), 111(F'ig. 6), 114(Fig. 7) and 115
(Fig. 5) all
potentiated the current induced by application of 30 NM AMPA. An example for
each
compour7d is shown below. It is seen that the potentiation in every case is
reversible, even
though the effect of 56 and 63 persit.s for several minutes after wash out of
the compounds.
The time between AMF'A stirnulations was 45 sec. Scalebars: 63: 200 pA/2 sec;
56: 500 pA/5
sec; 115: 50 pA/2 sec; 111: 400 pA/3 sec; 114: 40 pA/ 3 sec. In the
experiments shown the
concentration of the compounds was 3 NM (56. 63 and 114) or 10 NM (111 and
115)
The effect of the compounds were concentratlion-dependent, as exemplified for
114 below
(scale bars 200 pA/ 5 sec).(Fig. 8)
REFERENCES
Drejer J., Honore T. and Schousboe A. (1987; f~xcitatory amino acid-induced
release of 3H-
GABA from cultured mouse cerebral cortex interneurons. J. Neurosci. 7: 2910-
2916.
Hamill O.P., Marty A., f~eher E., Sakmann B. ~~r~d Sigworth F.J. (1981 )
Improved patch-clamp
techniques for high-resolution current recording from cells and cell-free
membrane patches.
Pflugers Arch. 39: 85-10C).
Mathiesen C., Varming T. and Jensen L.H. (1!398) In vivo arnd in vitro
evaluation of AMPA
2o receptor antagonists in rat hippc>campal neurones and cultured mouse
cortical neurones. Eur.
J. Pharmacol. 353: 15~~-1 fi7..
lontophoretic Application
PURPOSE
Evaluation of the in vivo effects of positive AMF'A modulators (PAMs) on the
AMPA evoked
spike activity in rat hipporampus.
PRINCIPLE
Hippocampai single neuron spike activity is strongly influenced by excitatory
input, and
iontophoretic application of AMF'A induces spike activity in vivo in a dose-
dependent manner
(Mathiesen ef al. 1998;1. The AMPA evoked spike activity is inhibited by
intravenous (i.v.)
administration of a wide range of AMPA receptor antagonists (Mathiesen et al.
1998), which
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
52
indicated that the excitation is primarily mediated via AMPA receptors. PAMs
potentate AMPA
receptor activation in vi~'ro, and if this mechani sm also operates in vivo,
the i.v. injection of an
PAM should enhance AMPA evoked spike activity. Thus, the aim of this study was
to test the
in vivo effect of a group of in vitro actme PAM;. This have been done by
studying their ability
to enhances AMPA evoked spike activity after i.v. administration.
PREPARATION
Experiments were performed on male Wistar rat (M & B, Denmark) weighing 280-
380 g,
housed in two per cage with free access to food and water. The rats were
anaesthetised with
mebumal (50 mg kg~' i.p.) and the femoral artE~ry was catheterised for the
purpose of
monitoring blood pressure and the vein for intravenous injection of drugs and
continuous
injection of 0.9% NaCI (O.p-1.U ml h-'~ and mebumal (5-10 mg h-'). Additional
anaesthetic was
given i.v. if the rat responded to a pinch of the back foot. The trachea was
cannulated and
the rats were placed in a stereotaxic frame arid ventilated by a rodent
ventilator (Ugo Basile,
Comerio-~Jarese, Italy). Core body temperature was maintained at 37.5°C
by a DC heating
pad. The left and dorsal part of the parietal bone was removed by craniotomy
and the dura
was withdrawn exposing the pia mater and underlying brain., covered with 0.9%
NaCI.
COMPOUNDS/REAGENTS
AMPA (Sigma, USA) was dissolved at 10 mM in 0.2 M NaCI. NMDA (Sigma, USA) was
dissolved at 100 mM in 100 mM NaCI. Both solutions were adjusted to pH 7.5-8.0
with NaOH.
COMPOUND 61 was diissolved in a'_O~J mM CH3;>O 3- Na' at <:r contration of 10
mM for
iontophoretic application (pH 5.7) and in isotor~i~; glucose (278 mM) for i.v.
administration.
Cyclothiazide, COMPOUND 63, COMPOUND 56, COMPOUND 115 and COMPOUND 114
were all dissolved at in 5°ro chrernophore solution at a concentration
of 5 mg ml~'.
PARAMETERS
Evoked neuronal spike activity was analyzed on-line by a ccamputer, saving
single spikes and
time of event. Neuronal spike activity (number of action potentials s-') was
monitored on a
pulse rate histogram together with indicators for AMPA, COMPOUND 61 and
vehicle
application.
PROCEDURE
ExtracellUlar recordings of single; hippocampal neuron spikes were made with
five-barrel glass
microelectrodes (5B120F-6, World Precision Instruments Inc., Sarasota,
Florida, USA) with a
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
53
tip diameter of 10-12 prn. The individual barrels were filled with 5 M NaCI
(recording), 400 rnM
NaCI (current balancing), 10 mM COMPOUNC> 61 in 200 mM CH~S03~ Na~ (pH 4.7),
200 mM
CH3SOs Na+ (pH 4.7, ~Jehicle), and the Vast k>;~rrc:l was filled with the
AMPA.
Experiments were performed on hippocampal neurons (A = 5.5-6.5 mm, L = 1.5-2.0
mm, H =
2.0-3.0 mm, according to Paxinos & Watson, 1 X386). Neuronal spike activity
was evoked by
iontophoretic: application of AMPA for periods of 10 to 15 s with 1.5 min
intervals. Single
neuron spike activity w;~s amplified 5000 times with a bandwidth of 0.3 and 3
kHz (CyberAmp
320 with a AI 402 x50 smartprobe, Axon Instrurnents, California, USA). On-fine
and off-line
analyses were performed by the Spike2 program with a 1401 plus interface
(Cambridge
Electronic Design Limited, England). The computer program also recorded mean
arterial
blood pressure and monitored and controlled iontophoretic rapplication.
AMPA was ejected into hippocampus in regular cycles of 1()0-105 s. When
neuronal
responses were stable (when the AMPA responses did not vary more than 10%,
measured
over a 10 s time period) for at least 3z hour, then a single dose of either
cyclothiazide,
COMPOUND 63, COMPOUND 56, COMPOUND 115, COMPOUND 61 or COMPOUND 114
(10 mg kg-') was injected into the lemoral vein. Recording of neuronal spike
activity was
continued for at least 45 min after intravenous: injection. The PAM reactivity
was tested by
microiontophoretic application of COMPOUND G1 (20 nA, Fig. ).
RESULTS
Fig. 8 shows that iontophoretic application of COMPOUND 61 enhanced AMPA
evoked spike
activity, whereas the vehicle did not influence the evoked spike activity.
Intravenous
administration cyclothiazide (10 mg kg-') did not enhance AMPA evoked spike
activity (Fig.
10).
Fig. 9. lontophoretic application of COMPOUND 61 enhanced AMPA evoked single
neuron
spike activity. The iontophoretic application of the vehicle did not influence
AMPA evoked
spike activity.
Fig. 10. Cyclothiazide 1;10 m kg-' i.v.) did not affect AMPA evoked spike
activity in
hippocampus. Shaded box above the AMPA1 trace indicatE, time of administration
(1500 s
3o after onset of registrating).
The in viwo effects of the PAMs on AMPA evoked spike activity was dependent on
the control
spike activity level. COMPOUND 63 enhanced small AMPA responses, evoked by low
CA 02320354 2000-08-04
WO 99!42456 PCT/DK99/00070
ti4
intensity AMPA stimulation, but had only marginal effect on large AMPA
responses, evoked
by high intensity AMPA. stimulation (Fig. 111. There was an initial inhibition
of the AMPA
responses and the onset of the enhancement occur 15 to 30 min after i.v.
administration (Fig.
11 ). Fig. shows an example of enhancement of AMPA responses by COMPOUND 56
(10 mg
kg-'). The onset occurred approximately 10 min after administration (Fig. 12).
Fig. 11. COMPOUND Ei3 (10 mg kg--1 i.v.) enhanced the low intensity AMPA
responses (12
nA), but had only marginal effect on high intensity AMPA responses (17 nA). 10
mg kg-1
COMPOUND 63 was given 1500 s after onset c>f recording. The time of injection
is marked by
a shaded box above the AMPA2 trace.
Fig. 12. COMPOUND Ei6 (10 mg kg~' i.v.) enhanced AMPA Evoked spike activity.
The
compound was injected 1250 s after onset of registration. The time of
injection is marked with
a shaded box above the AMPA trace.
Fig. 13 shows an example of enhancemenl of AMPA spike activity after i.v.
administration of
COMPOUND 115 (10 rng kg-'). 'The effect started 2 min aftE:r i.v.
administration and lasted for
more than 2 hours. COMPOUND 61 (10 mg kg' ) also enhanced AMPA responses in
hippocarnpus. The 3-field increase in AMPA evoked spike activity induced by
COMPOUND
61 started 20 minutes ~~fter administration and lasted for more than 2 hours
(Fig. 14).
COMPOUND 114 (10 mg kg-' ) induced a 10-fold increase of the AMPA responses,
when
control level of the AMIPA responses were low (from 21 to 209 spikes response-
', mean
response,Fig. 15), while only smaller enhancE;mc~nt observed with larger
control responses
(from 124 to 204 spikes response-',Fig. 16).
Fig. 13. COMPOUND '115 (10 mg kg~' i.v.) enhances AMPA evoked spike activity
in
hippocampus. The shaded bax indicate the time in i.v. injection, 1900 s after
onset of
registration. The effect of COMPOUND 115 IustE~d for more than 2 hours.
Fig. 14. COMPOUND Ei1 enhanced AMPA responses in hippocampus. 10 mg kg-' i.v.
was
given 1000 s after onset of registration (marked by a shaded box above the
trace). The effect
of COMPOUND 61 lasted for more than 2.5 hour.
Fig. 15. COMPOUND '114 (10 mg kg-' i.v.) enhanced AMPA evoked spike activity.
The
compound was given 11730 s after onset of registration, which is marked by a
shaded box
above the AMPA trace.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Fig. 16. COMPOUND 114 (10 mg kg' i.v.) approximately doubled the AMPA
responses. The
i.v. administration occurred at time 3900 s and is indicated by shaded box
above the AMPA
trace
The results show that C;yc~lothiazide did not show' any in viva effects after
i.v, administration.
5 However, COMPOUND 63, COMPOUND 56, COMPOUND 115, COMPOUND 61 and
COMPOUND 114 enhanced AMPA evoked s~>ike activity in an activity-dependent
manner.
References
MATHIESEN, C., VARMING, T. & JENSEN, L. H. (1998). In vivo and in vitro
evaluation of
AMPA receptor antagonists in rat hippocampal neurones and cultured mouse
cortical
10 neurones.. European Jr~urnal of Pharmacology 353, 159-r 67.
PAXINOS, G. & WATSON, C. (1986). The rat train in stereotaxic coordinates.
Second
Edition.
Passive avoidence
15 PURPOSE: To test the: pharmacological effect of compounds on associative
memory.
PRINCIPLE: A Mouse is placed in a light compartment with access to a dark
compartment. If
it enter the dark compartment it will receive a foot-shock {0.4 mA). After a
delay (24 hours)
the association to risk an unpleasant foot-shock by re-entering the dark
compartment is
2o tested.
ANIMALS: Female NMRI mice (Bomholdtgaard, DK) weighing 22-25 g were used. The
mice
were kept in Macrolon plastic cages with free access to food (Altromin, DK)
and tap water.
The mice were habituated to the laboratory for at least 3 days before testing
(light on
25 7:OOam/light off 7:00 pm).
EGZUIPMENT: The passive avoidance apparatus consisted of a modular test
chambers (ENV-
307, MED-Associates, US). The light and the dark compartment consisted of
plexiglas boxes
of equal size (15x17x13 cm; width x length x height) with metal grid floors. A
sliding guillotine
30 door was located at thc: aperture (4x4cm) connecting the two compartments.
A manual grid
scrambler (ENV-412, AHED-Associates, US) was used to provide the 0.4 mA foot-
shock.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99100070
~6
PARAMETERS: Entry I;atency (sec) to re-enter the dark compartment was
measured.
PROCEDURE: The mice are pre-treated (usually 30 min) before training with the
test
compound.
Training: One mouse is placed in the light compartment and the guillotine door
to the dark
compartment is openedl. When the mouse has Entered the dark compartment with
all 4 paws
it will receive one foot-shock (0.4 mAi and it will be taken to its home cage.
Test: After a delay
(24 hours) the mouse will re-enter the light compartment ancJ time latency to
enter the dark
compartment is measured with a time limit at ~~ minutes. Time latency is noted
as 2 minutes if
the mouse has not entered the dark compartment within maximal test time (2
minutes).
Vehicle: 10°/« Tween BCI.
Dosis vol.: 10 ml/kg
n=10;
RESULTS: Mean (~SEM) entry latency for each group is presented.
The result in Fig. 17 shows the memory enhancing effect of different
concentrations of the
compound 61;
2o Brief description of the drawings
Fig. 1 and Fig. 2 show; the potentiataon of AMPA induced [3H]GABA release from
cultured
cortical neurons by compounds of the invention.
Fig. 3-8 shows the voltage clamp experiments c>n compounds of the invention.
The
compounds 56 (Fig. 3), 63 (Fig. 4), 111 (Fig. 6), 114(Fig. 7) and 115 (Fig. 5)
all potentiated
the current induced by application of 30 NM AMPA.
Fig. 8 shows the concentration-dependent effect of a cornpc:~und of the
invention (114).
Fig. 9 - Fig. 16 are experiments of iontophoresis.
Fig. 9. lontophoretic application of COMPOUND 61 and the iantophoretic
application of the
3o vehicle;
Fig. 10. Cyclothiazide is applied in hippocampu:~.
Fig. 11. COMPOUND 6.3 is applied;
Fig. 12. COMPOUND 5.6 is applied.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
r~
Fig. 13 COMPOUND 115 is applied.;
Fig. 13. COMPOUND 115 in hippocampus.
Fig. 14. COMPOUND 6.1 in hippocampus;
Fig. 15. COMPOUND 114;
Fig. 16. COMPOUND 114;
Fig. 17. Passive avoidance test of compound 61.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
58
EXAMPLES
GENERAL TRANSFORMATION METHODS
Method A.
Suffamoylation in general.
i, CISCl"H 4 R'. OSp
w w
NR ii. HNR',, N~ ~l ~~ NRz
_..__.,.
~NR2 R, R~." NHa
The compound (32.5 rnmol) to be chlorosulfonated was dissolved in
chlorosulfonic acid (75
ml) and heated in an oil bath at 110 °C, until TLC indicated that the
reaction had gone to
completion'. The reaction mixture was poured onto ice and the precipitate
formed, was
isolated by filtration. The isolated solid was vvashed with a small amount of
water and dried
on the filter. The solid was dissolved in dry T'HF (200 ml) and added an
excess of the amine
(230 mmol) and the reaction mixture was left over night with stirring at rt.
The reaction mixture
was evaporated to dryness" then stirred with water to afford a solid which was
isolated by
filtration .and washed with EtOAc: on the fili:er. Further purification was
possible either by
column chromatography or recrystallization from EtOAc/hexane. Yields were
typically: 60-
90%.
* la small aliquot was taken out, added to ice in a test tube, neutralized
with Na2C03 and then
extracted with EtOAc. The aqueous phase was removed and the organic phase was
added piperidine
(xs) and left for some time. rLC; was taken from this small scale reaction
mixtureJ.
Method I3.
o-Sulfamoylation of anilines.t
i. clsolNCo
ii. AICh D \ S ~'PJH HZSO4 (6 M, aq.) ~ ~ ">~
-_-_- ~ NHz
~NHFI R N' W, '1 /.~ NF-IR
I R
I-;
To a stirred solution of the aminobenzene derivative (250 mmol) in nitroethane
or
nitromethane (100 ml) at -50 "c:,, was addE:d a solution of CIS02NC0 (275
mmol) in
nitroethane or nitromei:hane (75 ml) so that the reaction temperature did not
exceed -30 °C.
The coating bath was removed and the thick reaction mixture allowed to heat up
to 0 °C.
Solid AIC;13 (300 mmol) was added in one portion. The clear brown reaction
mixture was
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/000'70
59
heated in an oil bath at 120 "C for 20 min, then cooled to rt. and poured into
a beaker with
stirred ice water (1 I). The precipitate formed, was isolated by filtration.'
The urea intermediate
(98 mmol) was suspended in a mixture of dioxane (250 ml) and 6 M H2S04 (or
only conc.
HCI) (500 ml) and healed to reffux over night. 'The reaction mixture was
cooled to rt., filtered
and dioxane was remrwed from thc: filtrale by evaporation. The aqueous
remanense was
neutralized to pH = 7-8 using 4 M iVaOH. The precipitate formed, was isolated
by filtration
and washed with water arod EtOAc.* Overall yields ranged from 10 % to 75 %.
t see also Girard Y., Atkinson J.G. and Rokach J., J. Chem. Soc., Perkm l,
(1979} 1043.
'(Meta substituted anilines gives rice '.:o a mixture of the two possible
isomers which at the stage of the
urea intermediate may be ::>eparated by crystallization from MeC:)E-I or at
the stage of end product by
crystallization from EtOAc/hexane or c:hromatography.l
Method G.
Trifluoroacetyl protection.
°wi' c~~ ° °ai°
S~nIHZ (CF3C0)p0 ~I~'~~~5'NFj PPA ~ ~ S'IVF
Ill .~l ,
~NH ~.% ~N_..f 1 / Ni
R ~ R ~F;
To a stirred solution of the 2-aminobenzenesulfonamide derivative (27 mmol) in
dry THF (75
ml) at 0 °C was added trifluoroacetic anhydride ouch that the reaction
mixture did not exceed
+10 °C. The reaction mixture was stirred ai rt. until all starting
material was consumed. The
reaction mixture was evaporated to dryness, atirred with water, filtered and
washed with
hexane. 'The isolated solid was added PPA (250 g) and heated in an oil bath at
140 °C for 2~h
h. The reaction mixture was cooled to 60~~70 "C; and poured onto a
mechanically stirred ice
water solution. The precipitated formed was isolated by filtration and air
dried. Overall yields
were typically 85-90%.
Method D.
Trifluoroacetyl deprotection.
c\c~o
'NR KoHiFr2o ~-\ :.>~n~r~,
I z
~yN:~~cF~ n ~ ~y~i~NH
i
F3
A stirred solution of the trifluoroac:etyl protected compouncl (3.6 mmol)
dissolved in 1 M KOH
(30 ml) was heated to 80 °C for 1 h. The reaction mixture was cooled to
rt. and pH adjusted to
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
~0
7 using cone. HCI (aq.). -The reaction mixture was cooled to 0 °C and
filtered. The isolated
solid was washed with water and air dried. Yields typically ratnged from 85-
95%.
Method E.
Formation of dihydrobenzothiadiazines-1,1-dioxide from 2-aminobenzene-
sulfonamides.
o
O~~O i. R'-COCI, DM AP, TEA
,S_
. S' Nl~p ii. t M NaOH. d, ~~ W N(t i)
NHR f?~.. H) R,
To a stirred solution of the 2-aminobenzenesulfonamide derivative (148 mmol),
triethylamine
(150 mmol), 4-(N,N-dimethylamino)pyridine (7.5 mmol) in 6t)U ml THF at 5
°C was added the
carboxylic acid chloride. The reaction mixturE, was left with stirring over
night and then
1o evaporateld to dryness. The crude material was stirred with water and
filtered. The isolated
solid was dissolved in 1 M NaOI-1 (250 ml) and heated to 80 °C for 3 h.
The reaction mixture
was coolE;d to rt. and pH adjusted to 7 using c~~nc. HCI. The precipitate
formed was isolated
and recrystallized from i-F~rOH. Overall yields ranged from 80-
90°,'°.
Method F'.
Reduction of dihydrobenzothiadiazines-1,1-dioxide to tetrahydrobenzo-
thiadiazines-1,1-
dioxide.
o~~o
~wi a
N(H) DIBALH S
NH
-W , ~ vi ~._N; .R,
R SH) R F~ FI
To a stirred solution of the dihydrabenzothiadiaz:ine-1,1-dioxide {19.4 mmol)
in dry THF (200
ml) at -70 °C was added a solution of DIBA~H 1.5 M in toluene (33 ml;
50 mmol). The
reaction mixture was left with stirring over night while the temperature
slowly increases from -
70 °C to -15 °C'. Water { 10 ml) was added tc~ the reaction
mixture followed by 1 M NaOH (5
ml). The reaction mixture was then warmed 1:o I~t. and extracted with EtOAc.
The combined
organic fractions were dried with MgS04 and evaporated to dryness. In some
cases the
product was further purified by column chromatography. Yields ranged from 45-
85 %.
'[The product aminal is further reduced to the ring opened alkylarnine if the
temperature is not controlled carefully.
Method G.
Formation of tetrahydrobenzothiadiazines-1,1-dioxides from 2-
3o aminobenzenesulfonamides.
CA 02320354 2000-08-04
WO 99/'42456 PCT/DK99/00070
61
0~~0 0~~~
S~NH~ R'-CHO, MgS04, ~ ~ ~J.'NH
__"
NHz '~ ~PJ~'~R,
Fi
A stirred solution of the 2-aminobenzenesulforr~mide derivative (7 mmol), an
aldehyde (10
mmol) and MgS04 (20 mmol) in dry THF or dry dioxane (~0 ml) was refluxed under
N2 until
TLC indicated comptet~a consumption of the 2:-aminobenzenesulfonamide
derivative (typically
12-36 h). The reaction mixture was filtered, at t:he precipitate washed
thoroughly with THF or
dioxane. The filtrate was evaporated to dryness, added water and extracted
with EtOAc. The
combined organic fractions were dried with MgS04 and evaporated to dryness.
Column
chromatography (EtOP,c/hexane) afforded the pure product. Yields typically
ranged from 25-
75%.
Method H.
Formation of aryl or hetaryl substituted compounds by use of Pd catalyzed
cross
coupling.
pd-cat.
X~ ~ ~-additives Fietaryl/Aryl~~,
I t Metalated Arene/Hetarene - -----.-»
R li
X=Br,l
Suzuki coupling: A stirred mixture of an arylh<~lide (2 mmol), an aryl or
hetarylboronic acid,
boronic acid ester or dialkylborane (6 mrnol), K2C03 (10 mmol), Pd(PPh3)4 (30
mg),
1,3-propanediol (10 moral), dimethoxyethane (50 ml) and E-120 (25 ml) under N2
was heated
to 70 °C for 3 h. The reaction mixture was cooled to rt. t=urther water
was added and the
reaction mixture was E>xtracted with EtOAc. -ft~e combined organic fractions
were dried with
MgS04 and evaporated to dryness. Column chromatography afforded the pure
product.
Yields ranged from 40-100 %.
Method I.
Formation of compounds containing triazolyl substitutian.
CA 02320354 2000-08-04
WO 99142456 P(.'.T/DK99/00070
fit
R R,
Pd-cat
additives ~ r~~, TMSN~ Nv
N-
.h I~' - H --. ~--.-,.. _-.-.. I I
I~ ~ R
X-Bi,l
R TMS
H TMSN,~
h;OhIIMeOH
0
R
Sonogasl~ira coupling: A mixture of an aryliodide or arylbrc7mide (2 mmol); an
acetylene (10
mmol); Pd(PPh3)2CI2 (1~0 rng; 0.2 mmol); Cul (40 mg; 0.1 mmol) and
triethylamine (10 ml)
under N2 was stirred at rt. (in the case of arylbromides prolonged heating at
60 °C was
necessary) over night. THF was added and the reaction mixture filtered through
celite and
the filtrate evaporated to dryness. Column chromatography gave the ethynylated
arene.
Yields ranged from 40-53"/° for arylbromides to 07% for
aryliodides.
Detrimelhylsilylation for R'--TMS: A solution of the ethynylated arene (1.7
mmol) in MeOH (8
ml) was added a solution of 1 M KOH in MeOFi (2 ml; 2 mmol) and stirred at rt.
for 2h. The
reaction mixture was diiluted with THF, adsorbed onto silica and
chromatographed to give the
desilylated ethynyl arene. Yields ranged from 61"/° to 73%.
Formation of trizoles: The ethynylarene (0.'7 rnrnol) and TMS-N3 (2 ml; 15
mmol) was heated
to 170 °C; in an ampule for 50 h. The reaction mixture was cooled to
rt. and evaporated to
nearly dryness (CAUTION ! evaporation to dryness may lead to explosion due to
the
presence of some HN3) and added MeOH. The reaction mixture was stirred for 1 h
(to
remove the TMS-group in the TMS-triazole), then adsorbed onto silica, and
purified by
chromatography. Yield; ranged from 20-72 °ra.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Ei3
SYNTHESIS OF INDIVIDUAL COMPOUNDS
Compound 1
2-Cyclohexyl-4-oxo-1,2,3,4-tetrahydroquinazaline
Anthranilamide was transformed by Method G (using cyclohexanecarboxaldehyde).
M.p. 172-
174 °C.
Compound 2
2-Phenyl-4-oxo-1,2,3,X1-tetrahydraquinazaline
Anthranilamide was transformed by Method G (using benzaidehyde). M.p. 221-222
°C.
Compound 3
2-Methyl-3,4-dihydro-1,3-benzoxazine-4-anE~
2-Hydroxybenzamide v~ras transformed by ME;i:hod G (using paraldehyde). M.p.
124-126 °C.
Compound 4
2-Phenyl-3,4-dihydro-1,3-benzoxazine-4-one
2-Hydroxybenzamide eras transformed by Mefhod G {using benzaldehyde). M.p. 157-
160 °C.
Compound 5
3-Bicycla[2.2.1]hept-5~'-en-2'-yl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Aminobenzenesulfonamide was transformed by Method G (using a racemic
endo/exo
mixture of 2-norbornencarboxaldehyde). M.p. 205-209 °C.
Compound 6
3-Phenyl-1,2,3,4-tetralhydro-1,2,4-benzothiadiazine-1,1-dioxide
2-Aminobenzenesulfonamide was transformecJ by Method G (using benzaldehyde).
M.p.
125.5-12!3.5 °C.
Compound 7
1,2,3,5,10,10a-Hexahydrobenzo[e]pyrrolo[1,2-b]-1,2,4-thiadiazine-5,5-dioxide
2-Aminobenzenesulfanamide was used a~~ starl:ing material for the following
transformation
sequence: Method E (using 4-c:hlorobutanayl chloride. The reaction mixture was
NOT
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
64
subjected to NaOH catalyzed ring closure, bui: dissolved in H2SU4 and heated
at 100 °C for
72 h and precipitated on ice). M.p. 149-154 °C.
Compound g
2-Ethyl-2-methyl-3,4-dihydro-1,3-benzoxazine-4-one
2-Hydroxybenzamide was transforrned by MetGnod G (using ?-butanone). M.p. 76-
78 °C.
Compound 9
3-Cyclohexyl-6-(2-methoxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
3-Bromo-aniline was transformed by Method h'~ (see corr7pound 121 ) to give 4-
bromo-2-
aminobenzenesulfonarrride.
A mixture of 4-broma-2-aminobenzenes,ulfonamide (140 mg, 0.56 mmol), 2-
methoxyphenylboronic acid (1 OG md, 0.70 rnmol), F'd(PPh3)2C12 (20 mg, 5 mol
%) in 1,2-
dimethoxyethane (30 mil) and Na2C;03 (2M, 3 ml, 6 mmol) was refluxed under NZ
for 4 h. The
solvents were removed under reduced pressure and the residue was treated with
saturated
NaHC03 (20 ml) and extracted with EtOAc (2' x 40 ml). The organic layer was
washed with
brine (20 ml), dried (Na2'30a) and tk7e solvent was removed under reduced
pressure. The
product was purified b~~ flash chromatography on SiO~ using EtOAc:n-hexane
(1:1, v/v) as
eluent, yielding 155 mg (100 ~;/°) of 2-amino-4-(2-methoxyphenyl)-
benzenesulfonamide as
colorless powder. Tl~e product was further transf<:~rmed by Method G (using
cyclohexanecarboxaldehyde). M.p. 219-221 °C.
Compound 10
3-Cyclohexyl-6-(2-pyri~dyl)-1,2,3,4-tetrahydra-'1,2,4-benzothiadiazine-1,1-
dioxide
3-Aminophenylboronic acid hemisulphate (5.58 g, 30 mmol), 2-bromopyridine (2.7
ml, 28
mmol), Pd(PPh3)2C12 (100 mg, 0.5 mol °/>), 2M K2C03 (50 ml) were
refluxed in 1,2-
dimethoxyethane (50 ml) for 24 h under N~. The mixture was diluted with CH2CI2
(100 ml) and
washed with sat. NaHCO i (50 ml). The organic layer was dried (Na2S04) and the
solvent was
removed under reduced pressure. Flash-chromatography with CH2CI2 as eluent
gave 3-(2-
pyridyl)aniline as a yellow oil, 1.0 g (~1 %). 3-(2-t'yridyl)aniline was
transformed by Method B
and Method G (using cyclohexanec;arboxaldel-Hyde). M.p. 21.:3-216 °C.
Compound 11
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
3-Cyclohexyl-6-(3-pyridyl}-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
3-Bromo-aniline was trar7sformed by Method E3 (see compound 121 ) to give 4-
bromo-2-
aminobenzenesulfonarnide.
A mixture of 4-brorno-2-aminobenzenesulfonamide (250 mg, 1.0 mmol), diethyl-3
5 pyridylborane (225 mg, i.5 mmol), Pd(PPh~)2(~I_. (35 mg, 5 rnol %) in 1,2-
dimethoxyethane (30
ml) and IVa2C03 (2M, 3 ml, 6 mmol) were refluxed under N2 for 4 h. The
solvents were
removed under reduced pressure and the residme was treated with saturated
NaHC03 (20 ml)
and extracted with EtOAc (2 x 40 m11. The organic layer was washed with brine
(20 ml), dried
(Na2S04) and the solvent was removed under reduced pre:rsure. The product was
purified by
10 flash chromatography on SiO~ using EtOAc:n-hexane (1:1, v/v) as eluent,
yielding 240 mg (96
%) of 2-amino-4-(3-pyridyl)-benzenesulfonarnsde as colorless powder. The
product was
further transformed by Method Ca (using cyclohexanecarboxaldehyde). M.p. 240-
243 °C.
Compound 12
15 3-Cyclohexyl-7-(1-hydroxyethyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Amino-.5-(1-hydroxyethyl)benzenesulfonamide: 5-Acetyl-a:e-
aminobenzenesulfonamide (see
compound 13) (0.95 g, 4 4 mmol) was suspended in 96% EtOH (50 ml) and NaBH4
(0.46 g,
12 mmol;l was added in one portion. The mixture was stirred at 25°C for
4 h and filtered
through Celite and the solvent was removf:d under reduced pressure. The
residue was
20 treated with sat. NaHCO;, (50 ml) and extracted with EtOAc (2 x 50 ml),
dried (Na2SOa) and
evaporated to dryness.. Flash-chromatography with EtOAc:n-hexane:Et3N
(200:100:4, v/v/v)
as eluent gave 0.35 c~ (37 %) of 2-amino-Ei-(1-hydroxyethyl)benzenesulfonamide
as light-
brown powder. M.p. 160-162 °C.
25 Compound 13
3-Cyclohexyl-7-acetyl-1,2,3,4-tetrahydro-1,2,4-~benzothiadiazine-1,1-dioxide
To a solution of ethylvinylether (5.8 ml, 60 mnu>Ij in dry Th-IF (50 ml) at -
78°C was added t-
BuLi (1.7 M in pentane, 25 ml, 40 mmol) and the yellow mixture was stirred for
1 h at -78°C.
3o The cooling bath was removed and the mixture was warmed slowly to
0°C and stirred for
another 30 min. The mixture was recooled to ~-78°C and a solution of
ZnCl2 (2M in THF, 20 ml,
40 mmol) was added slowly and the cooling bath was removed and varmed to
20°C. 5-lodo-
2-aminabenzenesulfonamide (see compound 37) (1.8 g, 6 mmol) and Pd(PPhs)4 (0.2
g, 3
mol%) was added and tt~e mixture was refluxc~d for 6 h. -i-he THF was
evaporated and the
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
66
residue w<~s boiled in 1 M hydrochloric acid (30 ml) and MeOH (30 ml) far 30
min. EDTA (14.6
g, 50 mmol) was added and made slightly basic (pH == 8-9) with 1 M NaOH
followed by
extraction with EtOAc (3 x 150 ml), drying (Na,S04) and evaporation of the
solvent gave a
brown solid. Trituratiort with r~-hexane gave 0.9E~ g (74 %) of 5-acetyl-2-
aminobenzenesulfonamide as aff-white powder. The product was further
transformed by
Method G (using cyclohexanecarboxaldehydej. fvl.p. 224-225 °C
(decomp).
Comaound 14
3-Cyclohexyl-7-(1-hyd~roxyiminoethyl)-1,2,3,4-tetrahydro-'1,2,4-
benzothiadiazine-1,1-
dioxide
To a suspension of 5-acetyl-2-<~minabenzenEasulfonamide (see compound 13)
(0.45 g, 2.1
mmol) in 96% EtOH (40 rnl) was added H2NOE~~HCI (0.28 g, 4 mmol) and 2M NaOH
(2 ml}.
The mixture was boiled fcar 2 h and the solvent was evaporated. The residue
was triturated
with water (25 ml) and the product was filtered off and dried, yielding 0.39 g
(81%) of 2-
amino-5-(1-hydroxyiminoethyl)benzenesulfona.mide as yellow powder. The product
was
further transformed by Method G (using cyclohexanecarboxaldehyde). M.p. 230-
233 °C.
Compound 15
3-Cyclohexyl-7-carbarnoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
2o A mixture of 5-cyano-2~-aminobenzenesulfonamicfe (see cornpound 37) (2 g;
10 mmol), conc.
H2S04 (4. ml) and abs. EtOH (4 ml) was heated to 80 °C for 5 h and over
night at 50 °C. The
reaction mixture was poured onto ice and extracted with EtOAc (1 x)". The
organic phase was
discarded and the aqueous phase neutralized with Na2C03 and extracted with
EtOAc (3x).
The organic phase was evaporated to drynt::;s and subjected to column
chromatography
(EtOAc/hE:xane=2/1 ) to give 200 mg (9 %) of the carboxamide. The carboxamide
was further
transformed by use of A/lethod G (using cyclohexanecarboxaldehyde}. M.p. 235-
237 °C.
'[The first extraction contains the ~itrile (starting material) and anothor
biproduct]
Compound 16
3-Cyclohexyl-7-ethoxycarbony!-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
A mixture of 5-cyano-2~-aminobenzenesulfonamicfe (see cornpound 37) (3 g; 15
mmol), cone
H2S04 (5 ml) and abs. EtOH (15 ml) was heated to 80 °C over night. The
reaction mixture
was poured onto ice. -i~hf~ precipitatkJ formed was isolated by filtration.
The precipitate was
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
b7
washed with EtOAc to ~~ive 2.01 r~ (55%) of the pure ethyl ester. The ester
was further
transformed by use of (Method G (using cyclohexanecarboxaldehyde). M.p. 234-
236 °C.
Compound 17
3-Cyclohexyl-7-cyano~-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
5-Cyano-2-aminobenzenesulfor7arnicte (see compound 3T) was transformed by
Method G
(using cyciohexanecar',ooxaldehyde) M.p. 231-2.37.
Compound 18
3-Bicyclo(2.2.1Jhept-fi'-en-2'-yl-7-phenyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide
5-Bromo-2-aminobenzenese~lfonamide: A stirred solution of 2-
aminobenzenesulfonamide
(1.72 g; 10 mmol) in AcOH (15 ml) was added ;:~ solution of Br2 (0.55 ml; 10.5
mmol) in AcOH
(5 ml). The reaction mixture was poured into H~,O (100 ml) .and filtered. The
isolated solid was
adsorbed onto silica and subjected to flash cloromatography to give 1.428 g
(57%) product
(and 650 mg (20%) of the 3,5-dibromo derivativE:).
3-Bicycloj2.2.1Jhept-5'-en-2'-.yl-7-phenyl-1,2,;a,.J-tetrahydro-1,2,4-
benzothiadiazine-1, J-dioxide
(78): 5-Bromo-2-amir~obenzenesulfonamide was transformed by Method H (using
2o phenylboronic acid) and Method Ca (using a rac;emic endo/E:xo mixture of
bicyclo[2.2.1 ]hept-5-
ene-2-carboxaldehyde). M.p. 190.5-195.0 "C.
Compound 19
3-Cyclohexyl-7-(2'-acetamidophenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-
dioxide
2-Nitrophenylboronic acid: A solution of phenyuboronic acrid (10 g; 82 mmol)
in acetic acid
anhydride (100 ml) at -15 °C was added Turning HN03 (5 ml; 120 mmol)
over 30 min such
that reaction temperature was kept belaw -10 "i:,. The reaction mixture was
allowed to warm
up to r1. and left with stirring over night. The reaction mixture was poured
onto ice and
concentrated to 50 ml. The remanense was tf~en re-evaporated 5 times from
additional H20
(100 ml) and finally filtered to givE 7.1 g crude product as a mixture of
isomers. Column
chromatography (CH2~C1~,/EtOH=1010.5) gave ~f.8 g (35°io) pure product
as an oil.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
fib
2-Acefamidaphenylbon~nic acid.. A mixture of .?-nitrophenyfboronic acid (2 g;
12 mmol) and
5% Pd/C (100 mg) in E.tGH (100 ml) was hydrogenated at 1 bar until TLC
indicated complete
conversion of starting rnaterial. The reaction mixture was filtered through
celite and the filtrate
evaporated to dryness. The remar~ense was washed with hexane and filtered to
give 900 mg
of (55%) 2-aminophenylbaronic acid.
A mixture of 2-aminophenylboronic acid (900 mg; 6.6 mmol), triethylamine (0.57
ml; 7 mmol)
and acetylchloride (0.5 rnl; 7 mmol) was stirrE:d at rt. fc7r 1 h. The
reaction mixture was
evaporated to dryness, stirred with H20 and filtered to give 750 mg (63%)
product.
3-Cyclohexyl-7-(2'-acefarnidophenyl)-1,2,3,4-fetr;~hydro-1,2,4-
benzothiadiazine-1,7-dioxide
(19): 5-lodo-2-aminobenzenesulfonamide (see compound 37) was transformed by
Method H
(using 2-Acetamidophenylboronic acid) and IVlethod G (using
cyclahexanecarboxaldehyde).
M.p. 245-249 °C.
Compound 20
3-Cyclohexyl-7-(2'-nitrophenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
5-lodo-2-aminobenzenesulfonamide (see compound 37) was transformed by Method H
(using 2-nitrophenylboronic acid (see compound 19)) and Method G (using
cyclohexanecarboxaldf:hyde). M.p. 204-207 °C.
Compound 21
3-Cyclohexyl-7-(2'-methoxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
5-lodo-2-aminobenzenesulfonamide (see compaund 37) was transformed by Method H
(using 2-methoxyphenylboronic acid) and CV9ethod G (using
cyclohexanecarboxaldehyde).
M.p. 219-222 °C.
Compound 22
3-Cyclohexyl-7-(2'-methoxy-4'-trifluoromethylphenyl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
4-Trifluon~mefhylanisole: Sodium (12.78 g; Ei55 mmol) was added to dry MeOH
(100 ml).
When the gas evolution ceased, the reactian mixture was evaporated to dryness
and dry
NMP (250 ml) followed by Cu (s) (35.3; 555 mmol) and 4-bromo-
trifluoromethylbenzene (25 g;
111 mmol) was added. The reaction mixture was heated tc:~ 130 °C for 4
h, cooled to rt. and
CA 02320354 2000-08-04
WO 99J42456 PCT/DK99100070
filtered. Water {500 ml) was added to the filtral:e and it was extracted with
Et20 {2x 200 ml).
The combined organic. fractions were washE~d with H20 (2x 100 ml), dried
(MgS04) and
evaporatE:d to dryness. Column chromatography gave 8.05 g {41 %) of product.
2-Methoxy-5-trifluorom~ethylphenylboronic acid: A solution c:~f 4-
trifluoromethylanisole (8 g; 45
mmol) in dry THF (80 rnl) at -30 °C under N2 was added a ;:solution of
2.5 M n-BuLi in hexane
(20 ml; 50 mmol). The reaction mixture was stirred for 1 h at -30 "C, then
cooled to -70 °C and
added B(Oi-Pr)3 (14.1 ml; 64 mmol). The reaction mixture was allowed to slowly
warm up to
rt. over night. The reaction mixture was addecJ 2 M HCI (40 mi) and THF was
removed by
evaporation.. The aqueous remanense was extracted with E~t20 (4x 20 ml) and
the combined
organic fractions were extracted with 1 M NaOI-i (5x 17 ml). The combined
aqueous fractions
were neutralized by 10 M HCI. The precipitai:e formed was isolated by
filtration and washed
with 1 M HCI to give 8.1 g (81 %;) product.
3-Cyclohcxyl-7-(2'-methoxy-4'-triouoromethylphenyl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-a'ioxide (22): 5-lodo-'~-aminobenzenesulfonamide (see
compound 37)
was transformed by Method H (using 2-methoxy-5-triflucaromethylphenylboronic
acid) and
Method G (using cyclohexanecarboxaldehyde). M.p. 255-2fs7 °C.
Compound 23
3-Cyctohexyl-7-(2',4'-dimethoxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothi
adiazi ne-1,1-
dioxide
5-lodo-2-aminobenzenesulfonamide (see compound 37) was transformed by Method H
(using 2,4-dimethoxypher~ylboranic acid) and Method G (using
cyclohexanecarboxaldehyde).
M.p. 208213 °C.
Compound 24
3-Cyclohexyl-7-(2'-(N,N-dimethylsulfamoyl)phenyl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
2-(N,N-dimethylsulfamoyl)phenylbor~nic acid.' A solution of
benzenesulfonylchloride (10 ml;
78 mmol) in THF (100 ml) was added a 40°/> solution of dimethylamine in
H20 (20 ml; 160
mmol) such that the reaction temperature w<~~; kept below 50 °C. The
reaction mixture was
stirred 1 h at rt. The reaction mixture was added H20 and THF removed by
evaporation. The
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
precipitate formed was isolated by filtration and air dried to give 14 g (97%)
of N,N-
dimethylbenzenesulfonamide.
A solution of N,N~tiimethylbenzenesulfonamide (!x.25 g; 50 mmol) in dry Et20
(150 ml) under
N2 was cooled to -70 "C and added a solution of 2.5 M n-BuLi in hexane (24 ml;
60 mmol)
5 such that the reaction temperature was kept below -60 °C.. The
cooling bath was removed
and the reaction mixture ~~Ilowed to slowly wa.rrn up to +20 "C. The reaction
mixture was re-
cooled to -70 °C and B(Oi-pr)3 (16.1 ml; 70 rnrrrol) was added. The
reaction mixture was left
cold and allowed to warm up over night, 1 M HCI (100 ml) was added and
stirring was
continued at rt. for 1 h. The reaction mixture was then extracted with Et20
(2x 50 ml) and the
~o combined organic fractions extracted with 1 M NaOH (4x 50 ml). The combined
aqueous
fractions were neutrali~:ed with 1 M HCI and extracted with Et20 (4x 100 ml).
The combined
organic fractions were cried (Na2S0,~) and evaporated to dryness, washed with
Et20/hexane
to give 3.4 g (30%) product.
15 3-Cyclohexyl-7-(2'-(N,N-dimethyisulfamoyl)~hc~rryl)-1,2,3,4-tetrahydro-
1,2,4-benzothiadiazine-
1,1-dioxide (24): 5-lodo-2-aminobenzenesulfonarnide (see compound 37) was
transformed by
Method H (using 2-(N,N-dimethylsulfamoyl)phenylboronic acid) and Method G
(using
cyclohexanecarboxaldehyde). M.p. 290-300 "(~.
2o Comaound 25
3-Cyclohexyl-7-(2'-chlorophenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
5-lodo-2-aminobenzen~esulfonamide (see compound 37) was transformed by Method
H
(using 2-chlorophenylboranic acid) and Method G (using
cyclohexanecarboxaldehyde). M.p.
233-236 "C.
Compound 26
3-Cyclohexyl-7-(2'-fl uorophenyl)-1,2,3,4-tetrahydro-1,2,4-benzoth iadiazine-
1,1-dioxide
5-lodo-2--aminobenzenesulfonamide (see compound 37) was transformed by Method
H
(using 2-fluorophenylboronic acid) and Method G (using
c:yclohexanecarboxaldehyde). M.p.
249-250 "C.
Compound 27
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
a1
3-Cyclohexyl-7-(3'-hydroxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
3-hydroxyphenylboronic acid: A stirred solution of 3-aminophenylboronic acid
hemisulfate (6.2
g; 33.3 rrimol) and 50°,~o H2S04 (3. i' ml; 33.~~ rnmol) in H20 (100
ml) at -2 °C was added a
solution of NaN02 (2.5 g; 36.3 mmol) in Hpt) (20 ml) over 1 h. The reaction
mixture was
slowly added to a stirred solution csf cone. h~>S04 (25 ml) in H20 (20 ml) at
reflux. After
complete addition, the reaction mixture was refluxed for 30 min., cooled,
added activated
charcoal, heated to reflux, cooled and filtered through celite. The filtrate
was saturated with
NaCI (s), filtered and extracted with Et20 (5x: '100 ml). The combined organic
fractions were
dried (Na2S04) and evaporated to dryness to give 4.3 g (94°io) product.
3-Cyclohexyl-7-(3'-hydroxyphen.yl)-1,2,3,4-tetiahydro-1,2,4-benzothiadiazine-
1,1-dioxide (27):
3-Hydroxyphenylboronic acid was transformed by Mei:hod H and Method G (using
cyclohexanecarboxaldc:hyde). M.p. 238-246 °C
Compound 28
3-Cyclohexyl-7-(2'-pyridyl)-1,2,3,4-tetrahydro-1,2,4-benzathiadiazine-1,1-
dioxide
4-(2'-Pyridyl)-2-aminobenzenesulfonamide: A stirred mixture of 5-iodo-2-
aminobenzenesulfonarnide (1 g; 3.3 mmoli), 2-tributylstannylpyridine (5.5 g;
15 mmol),
Pd(PPh3)4 (240 mg, 0.34 mmol) and Ag20 (780 mg; 3.3E~ mmol) in DMF (50 ml)
under N2
was heated at 100 °C for 6h and over night at rt. The reaction mixture
was evaporated to
dryness and resuspended and stirred in H20/E.tOAc and finally filtered. The
organic phase
was isolated and the aqueous phase extracted with EtC:>Ac (2x aq, vol.). The
combined
organic fractions were dried (Na2S04), ev<~porated to dryness and subjected to
column
chromatography to give 200 mg (24 %) product.
3-Cyclohexyl-7-(2'-pyri~alyl)-1,2,3,4-tetrahydro-1,2',4-benzothiadiazine-1,1-
dioxide (28): 4-(2'-
Pyridyl)-2-aminobenzenesulfonamidc~ was transformed by Method G (using
cyclohexanecarboxaldehyde). M.p. 222-224 "C.
3o Compound 29
3-Cyclohexyl-7-(3'-pyridyl)-1,2,3,4-tetrahydro-'1,2,4-benzothiadiazine-1,1-
dioxide
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
72
5-lodo-2-aminobenzencaulfonamide (see compound 37) was transformed by Method H
(using diethyl-3-pyridylk>orane) and Method G (using
cyclohexanecarboxaldehyde). M.p. 240-
242 °C.
Compound 30
3-Cyclohexyl-7-(2'-pyrimidinyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
4-((2,2-Dimethylpropanoyl)amino)phenylboror,~ic acid: To a solutian of 4-bromo-
N-
pivaloylaniline (1.56 g, 6 mmol) in dry THF (50 rril) at -78°C was
added t-BuLi (1.5 M in
pentane, 13.3 ml, 20 mmol) and the yellow mi:Kture was stirred for 1 h at -
78°C under N2. The
reaction was quenched with B(OCH3)3 (1.7 rnl, 15 mmol) and stirred for another
1 h at -78°C.
The reaction mixture w,~s then warmed to morn temperature and hydrolysed with
0.5 M
hydrochloric acid (50 ml) and extracted with EtC>Ac (3 x 80 ml), dried
(Na2S04) and
concentrated to ca. 40 ml n-Hexane (120 m!) was added slowly and the colorless
crystalline
product was filtered off and dried, yielding 1.26 g (95%).
N-(4-(2-pyrimidinyl)phenyl)-2,2-dimethylpropanamide: ~'v mixture of 4-((2,2-
dimethylpropanoyl)amino)phenylboranic acid (2.0 g, 9 mmol), 2-chloropyrimidine
(0.8 g, 7
mmol), Pd(PPh3)2C12 (100 mg, 2 mol %) 1,2-dimc~thoxyethane (40 ml) and Na2C03
(2M, 7 ml,
14 mmol) were refluxed under N2 for 5 h. The mixture was diluted with 10 %
Na2C03 (20 ml)
and extracted with EtOAc; (3 x 50 ml). The organic layer was dried (Na2SOa)
and the solvent
was removed in vacuo. The crude product was recrystallisE=d from MeOH/water
(1:1 ) yielding
0.52 g (85%) of N-(4-(c'-pyrimidinyi)phenyl)-2,:?-dimethylproi:~anamide as
colorless crystals.
4-(2-Pyrirnidinyl)aniline: N-(4-(2-pyrimidinyl)phenyl)-2,2-
dinnethylpropanamide (1.41 g, 5.48
mmol) was boiled in 6 IM hydrochloric acid (40 ml) for 2 h. The mixture was
cooled and made
strongly basic with NaOH (s) and extracted with CH2C12 (2 x 50 ml), dried
(Na2S04) and the
solvent vvas removed irn vacuo. Trituration with n-hexane gave 0.83 g (88%) of
4-(2-
pyrimidinyl)aniline as a light-yellow powder. ThE; product was further
transformed by Method B
and Method G (using cyclohexanecarboxaldehyde). M.p. 236-238°C.
Compound 31
3-Cyclohexyl-7-(2'-furyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
CA 02320354 2000-08-04
WO 99/42456 f'CT/DK99/00070
73
5-lodo-2-aminobenzenesulfonamide (see compound 37) was transformed by Method H
(using furyl-2-boronic acid) and Method G {usinct cyclohexanecarboxaldehyde).
M.p. 226-228
°C.
Compound 32
3-Cyclohexyl-7-(3'-furyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
5-lodo-2-aminobenzenesulfonamide (see cornpound 37) was transformed by Method
H
(using furyl-3-boronic acid) and Method G (using cyclohexanecarboxaldehyde).
M.p. 204-205
°C.
Compound 33
3-Cyclohexyl-7-(2'-thienyl)-1,2,3,4-tetrahydro-'1,2,4-benzothiadiazine-1,1-
dioxide
5-lodo-2-aminobenzenesulfonamide (see compound 37) was transformed by Method H
(using thienyl-2-boronic acid) and Method G (using cyclohexanecarboxaldehyde).
M.p. 234-
236 °C.
Compound 34
3-Cyclohexyt-7-(1-methyl-1 !-f-2-imidazolyt)-'1,2,3,4-tetrahydro-1,2,4-
benzothiad iazi ne-1,1-
dioxide
To a solution of 1-methyl~midazole (4.8 ml, 60 mmol) in dry THF (120 ml) at -
78°C was added
n-BuLi (2.5 M in hexane., 26 ml, 65 mmol) and the yellow mixture was stirred
for 45 min at -
78°C. A solution of ZnClz (2M in THF, 75 rrol, 150 mmol) was added
slowly and the cooling
bath was removed. The colorless solution was stirred for another 10 min at
0°C. 5-lodo-2-
aminobenzenesulfonamide (see compound 37) (2.1 g, 7 mmol) and Pd(PPh3)4 (0.5
g, 5
mol%) was added and the mixture was refluxed for 6 h. The THF was evaporated
and the
residue was treated with EDTA (53 g, 0.18 mol) and made slightly basic (pH = 8-
9) with 1 M
NaOH followed by extraction with EtOAc (3 x 150 ml), drying (Na2SO4) and
evaporation of the
solvent gave a dark oil. Flash-chromatography with 5% MeOH in CH2C1;~ as
eluent gave 1.33
g (75 "/°) of 2-amino-5-(1-methyl-1 H-2-irnidazolyl)-1-
benzenesulfonamide as colorless
crystals. The product was further transformEad by Method G (using
cyclohexanecarboxaldehyde). M.p. >>250 °(:, (decomp).
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
r4
Compound 35
3-Cyclohexyl-7-(1',2',3'-triazol-4'-yl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
5-lodo-2-aminobenzenesulfonamide (see compound 37) was transformed by Method I
(using
trimethylsilylacetyiene) and Method G (using cyclohexanecarboxaldehyde). M.p.
230-234 °C
(dec.).
Compound 36
3-Cyclohexyl-7-(5'-phenyl-1',2',3'-triazol-4'-yl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
5-lodo-2-aminobenzen~esulfonarnide (see compound 37} w<xs transformed by
Method I (using
phenylacetylene) and Method G (using cyclohexanecarboxaldehyde}. M.p. 231-232
°C (dec.).
Compound 37
3-Cyclohexyl-7-(5'-methyl-1',2',4'-oxadiazol-3-yl)-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
5-lodo-2-aminobenzenesulfonamide To a cold (0 °C) stirred solution of 2-
aminobenzenesulfonarnide (17.2 g; 100 mmol) in CHC13 1;200 ml) was added a
solution of
iodine manochloride (17.1 g; 105 mrnol) in CHc~l3 (50 ml) over 1 h. The
reaction mixture was
slowly warmed up to rt. and left with stirring aver night. ThE: reaction
mixture was filtered and
the isolated solid was washed on the filter with CHCI3 (3x 20 ml), NaHC03
(sat. aq., 1 x 20
ml), H20 (4x 50 ml). The isolated solid was air dried to give 27.3 g (92%) of
product.
5-Cyano-2-aminobenznnesulfonamide: A mixture of 5-iodo-2-aminobenzene-
sulfonamide
(17.9 g; 60 mmol), Zn(CN)2 (4.9 g; 41.9 mmol) and Pd(PPh3)4 (2.5 g; 2.2 mmol)
in DMF (150
ml) under N2 was heated to 80 °C for 2 h. ThE: reaction mixture was
poured into NaHC03
(sat. aq., 600 ml) and extracted with EtOAc (9x 200 ml). The combined organic
fractions were
washed with NaHC03 (sat. aq.} and NaCI (sat. aq.), dried (Na2S04), filtered
and evaporated
to dryness. The remanense was washed with water and hexane and filtered of to
give 10.9 g
(92%) of praduct.
5-(N-hydroxyamidino)-.2-aminobenzenesulfon.amide: A mixture of hydroxylamine
hydrochloride (764 mg; t 1 mmol} and NaOAAe (616 mg; '11.4 mmol) in MeOH (10
ml) was
stirred at rt. for 1 h and then added 5-cyano-2-aminobenzenesulfonamide (1 g;
5 mmol). The
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
reaction mixture was left with stirring for 48 h and poured into water and
extracted with EtOAc
(2x 50 mi). The combined organic fractions were dried (Na2S04), evaporated to
dryness and
purified by column chromatography to give 200 mg (17%) of product.
5 3-Cyclohexyl-7-(5'-methyl-7',2',4'-axadiazol-3-yl)-1,2,3,4-fetrahydro-1,2,4-
benzothiadiazine-
i, l-dioxide (37): A mixture of 5-(N-hydroxyamidino)-2-aminobenzenesulfonamide
(200 mg;
0.9 mmol), NaOMe (5C mg; 1 mmol), EtOAc (5 ml), crushed MS3A (2 g) in
anhydrous EtOH
(20 ml) was heated over night at 70 °C. 'rh~e reaction mixture was
evaporated to dryness,
stirred with water and extracted with EtOAc:. The combined organic fractions
were dried
10 (Na2S04) and evaporated to dryness to a brown oil, whici~ way: subjected to
transformation
by Method G (using cyclohexanecarboxaldehyde) to give 8 mg product after
chromatography.
M.p. 249-251 °C.
Compound 38
15 3-Cyclohexyl-7-acetamido-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
7-Amino-3-cyclohexyl-~1,2,3,4-tetrahydro-1,2,4-bc~nzothiadiazine-1,1-dioxide:
To a stirred
solution of 7-amino-~~-cyclohexyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide (0.5 g; 2
mmol) in dry THF (10 rnl) at -70 °C; was added a solution of 1.5 M
DIBALH in toluene (2.7 ml;
4 mmol) under N2. The reaction n-cixture was stirred for 2 h at -70 °C;
2 h at -40 °C and then
20 warmed to 0 °C. The reaction was quenched with H20 and stirred for
30 min. at 0 °C and left
over night without stirring at +5 °C. The mixture was evaporated to
dryness, resuspended in
MeOH and filtered. The isolated solid was washed thoroughly with MeOH and
filtered. The
combined filtrates was adsorped onto silica c~E;l., Column chromatography
(EtOAc) gave 200
mg of product.
7-Acetylamino-3-cyclohexyl-1,2,3,4-tetrahydro-1"2,4-benzothiadiazine-1,1-
dioxide (38): A
mixture ~of 7-amino-3-cyclohexyl--1,2,3,4-tetrahydro-1,2,4-benzathiadiazine-
1,1-dioxide (100
mg; 0.26 mmol), acetylchloride (20 pl; 0.29 rnol) and triethylamine (42 pl;
0.3 mmol) was
stirred fur 2 h at r.t. The reaction mixture was resuspended in H20 and
filtered. The isolated
solid was purified by column chrornatography (EtOAc) to give 22 mg 27a. M.p.
202-206 °C.
Compound 39
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
76
3-Cyclohexyl-7-methylsuffonylamino-1,2,3,4-tetrahydro-1 "2,4-benzothiadiazine-
1,1-
dioxide
3-Cyclohexyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide: 2-
Aminobenzenesulfonamide
was transformed by Me~rhad E (using cyclohexanecarbonyl chloride).
3-Cyclohexyl-7-vitro-1,2-dihydro-~y,2,4-benzothiadiazine-1,1-dioxide: To a
stirred solution of
KN03 (1.2 g; 11.5 mnnol) and cone. H2SO~i (f3 ml) at 5 °C was added a
solution of 3-
cyclohexyl-1,2-dihydro-1,2,4-benzoth~adiazine-1.11-dioxide in conc. H2S04 {8
ml). The
reaction mixture was allowed to warm up to r.t. ;end left with stirring over
night. The product
1 o was precipitated by slow addition of ice and isolated by filtration. The
crude product {4.3 g)
was used without further purification.
7-Amino-3-cyclohexyl-1,2-dihydro-~I,~n,4-benzothiadiazine-1, a-dioxide: A
stirred suspension of
3-cyclohexyl-7-vitro-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide (4.2 g; 14
mmol) and 10%
Pd/C (40G mg) in abs. EtOH (100 ml) was hydrogenated at 1 bar. After
consumption of the
calculated amount of H2, the reaction mixture was filtered through celite. The
celite was
washed twice with DfUIF {75 ml) and the comk>ined organic fractions were
evaporated to
dryness. The remanense was resuspended in E=tOAc/i-PrOhi and the precipitate
formed, was
isolated by filtration. The isolated solid was dissolved in 0.5 M NaOH (aq.)
and reprecipitated
2o with 4 M HCI (aq.). Filtr;~tion gave 1.6 g produca.
7-MethyJsulfonyJamino-3-cyclohexyl-l,2-dihyd~o-;1,2,4-benzothiadiazine-1,1-
dioxide: A stirred
solution of 7-amino-3-cyclohexyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide (1 g; 3.6
mmol) and triethylarnine (1.2 ml; 8 mrnol) in dry THF (25 ml) was added
methanesulfonylchloride {0.6 ml; 8. mmol) and stirred at rt. for 2 h. The
reaction mixture was
evaporated to dryness, resuspended in H2O/E=tOAc and filtered. The filtrate
was adsorbed
onto silica gel. Column chromatography (CH2C12:acetone = G:1 ) gave 210 mg of
product.
3-Cyclohexyl-7-methyls;ultonylamino-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
3o (39): 7-Methylsulfonylamino-3-cyciohexyl-1,2-dihydro-1,2,4-benzothiadiazine-
1,1-dioxide was
transformed by Method F. M.p. 255-258 °C.
Compound 40
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
?7
3-Cyclohexyl-7-nitro-f ,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
3-Trifluoromethyl-1,2,4-benzofhiadiazine-7, 1-dioxide: 2-
Aminobenzenesulfonamide was
trifluoroac:etyl protected by using Method G. A solution of this (1 g; 4 mmol)
in H2S04 (16 ml)
at 0 °C was added solid KN03 (4.4 mmol). The reaction mixture was
allowed to warm up to rt.
and stirred over night. The reaction mixture was poured into ice water (150
ml), filtered and
air dried to give 1.11 g (94%) of pure product as a yellow solid.
3-Cyclohexyl-7-nitro-1, 2, 3, 4-to tra h ydro-1,2, 4-berrzothiadiazine-1,1-
dioxide (40):
3-Trifluoromethyl-1,2,4-benzothiadiazine-1,1-dioxide was subjected to the
following
transformation scheme: Method U and Methoc:i F (using
cyclohexanecarboxaldehyde). M.p.
209-211 "C.
Comaound 41
3-Cyclohexyl-7-phenylsulfonyi-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
4-Phenylsulfonylaniline~ was used as starting material for the following
transformation
sequence: Method B and Method G fusing cyclohexanecark:roxaldehyde). M.p. 243-
245 °C.
Comaound 42
2-Cyclohexyl-1,2,3,4-tetrahydro-6-quinazoline sulfonamide
A solution of 2-aminok>enzylamine (3 g; 25 rnmol) in THF (50 ml) was added
trifluoroacetic
acid anhydride (3.8 nnl; 27 mmol) and stirrE~d at rt. for 2 h. The reaction
mixture was
evaporated to dryness, stirred with H20 and filtered. The <:rude product was
transformed by
Method A (using 25% NH,3 (aq.) as amine) and Method G (using
cyclohexanecarboxaldc:hyde). M.p. 178-180 "C.
Compound 43
3-Cyclohexyl-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazi ne-1,1-
dioxide
4-Sulfamoylanthranilarnide: A stirred solution caf CIS03H (20 ml) was added
anthranilamide
(7.5 g; 5 5 mmol) in small portions. The reacaior~ mixture was heated to 100
°C for 1 h and
3o then poured into ice water (300 mi). A precipitate formed, which was
isolated by filtration and
dried on the filter. The isolated solid was dissolved in 25°/~ NH3
(aq.) and stirred at rt. over
night. The aqueous phase was washed with LtOAc and concentrated to 20 ml. The
aqueous
phase was saturated with NaCI (s) and extracted with THF (3x 50 ml). The
combined organic
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
fractions were evaporated to dryness and subjected to column chromatography to
yield 13
mg product.
3-Cyclohcxyl-7-sulfamnyl-7,2,3,~f-tet~ahydro-l,~t,~4-benzofhia:rdiazine-1,1-
dioxide (43): 4-
Sulfamoylanthranilamicie was transformed by MEahod G (u;:>ing
cyclohexanecarboxaldehyde).
Fab+ 31 U. 'H-NMR (DiMSO-d6): 8.1 (1 H; br.), 8.0 (1 H; d), 7.58 (1 H; dd),
7.3 (1 H; br.), 7.05
{2H; br.), 6.79 (1 H; d), 4.5 (1 H; rn), 1.8-1.5 (6hi; rn), 1.2-1.0 {5H; m).
Compound 44
3-Cyclohexyl-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
2-Amino-5-sulfamoylbenzenesulfonamide (see compound 101 ) was transformed by
Method
G (using cyclohexanecarboxaldehyde). M.p. x!52-254 "C.
Compound 45
3-Methyl-7-dimethylsulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
3-Methyl-7-dimethylsul~farnoyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide
was reduced by
use of Method F. M.p. 210-212 "C.
Compound 46
2-Cyclohexyl-1,2,3,4-tetrahydro-6-quinazofine N,N-dimethylsulfonamide
A solution of 2-aminobenzylamine (3 g; 25 mmol) in THF (50 rnl) was added
trifluoroacetic
acid anhydride (3.8 rnl; 27 mmol) and stirred at rt. for 2 h. The reaction
mixture was
evaporated to dryness, stirred with H20 and filtered. The crude product was
transformed by
Method A (using dimethylamine as amine), Method D and Method G (using
cyclohexanecarboxaldehyde). M.p. > 300 °C. M;> (electrospray) M+ 323.
'H-NMR (DMSO-d6):
7.2 (1 H; dd); 7.1 (1 H; cl); 6.68 (1 H; br); 6.62 (11 H; d); 3.85 (1 H; s);
3.8 (2H; s); 2.2 (1 H; br); 1.8-
1.0 (11 H; m).
Compound 47
3-Cyclohexyl-7-dimethylaminosutfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-
dioxide
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
79
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using cyclohexanecarbonyl chloride), Method A (using
dimethylamine
as amine), Method F. Nl.p. 243-245 °C.
Compound 48
3-Cyclohexyl-7-(N,N-diiethylamina)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide
2-Aminobenzenesulfon.arnide was used as starting material for the following
transformation
sequence: Method E (using cyclohexanecarbonyl chloride), Method A (using
diethyiamine as
amine), Method F. M.p. 207-209 °C.
Compound 49
3-Cyclohexyl-7-pyrrolidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzoth iadiazine-
1,1-dioxide
2-Aminobenzenesulfon.amide was used as starting material for the following
transformation
sequence: Method E (u,~sing cyclohexanecarb~onyl chloride), Method A (using
pyrrolidine as
amine), Method F. M.p. 244-246 °C..
Compound 50
3-Methyl-7-piperidinosulfony!-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
2-Aminobenzenesulfon.amide was used as starting material for the following
transformation
sequence: Method C, Method A (using piperidine as amir7e), Method D, Method G
[using
paraldehyde and a cat. amount of 'TsOH]. Nl.p. 2!55-256 °C.
Compound 51
3-Cyclopropyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Aminobenzenesulfon.amide was used as starting material for the following
transformation
sequence: Method C, Method A (using pipericiine as amirne), Method D, Method G
[using
cyclopropancarboxaldehyde]. M.p. 228-231 °C.
Compound 52
3-Isopropyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method C, Method A (using piperidine as amirne), Method D, Method G
[using
isobutyraidehyde]. M.p. 2,'.37-239 "C.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Comaound 53
3-propyl-7-piperidinosulfonyi-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
5 sequence: Method C, Method A (u~~ing piperidine as amine), Method D, Method
G (using
butyraldehyde]. M.p. 14 7.4-151.:2 °C
Compound 54
3-Benzyl-7-piperidinosutfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
10 2-Aminobenzenesulfonanride was used as sl:arting material for the following
transformation
sequence: Method C, Method A (using piperidine as amine), Method D, Method G
(using
phenylacetaldehyde I. M.p. 242-244 °C.
Comaound 55
15 3-Cyclopentyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzathiadiazine-1,1-dioxide
Cyclopenfanecarboxaldehyde: A stirred solution of cyclopentanecarboxylic acid
(2.16 ml; 20
mmol) in dry THF (50 rnl) at rt. under N2 was added NaBH4 (2.28 g; fi0 mmol)
and left with
stirring for 20 min. The reaction mixture was cooled to 0 °(:; and
added BF30Et2 (10 ml; 80
mmol) over 1 h, while 'the reaction temperaturEa was kept below +3 °C.
The reaction mixture
20 was allowed to warm up to rt. and left with stirring aver night. The
reaction mixture was added
NaHC03 (sat., aq.), H20 and extracted with EtOAc. The combined organic
fractions were
washed with NaCI (sat., aq.), dried (Na2SO4) and evaporated to dryness to give
1.4 g of an
oil which was used without further purification.
The oil (1.4 g) was dissolved in CH2CI2 (75 rnl) and added PCC on AI203 (30 g;
30 mmol)'
25 and left with stirring for 1 h at rt. The reaction rnixture was filtered
and the filtrate evaporated
onto silica. Column chromatography afforded the pure aldehyde which was used
as a solution
in CH2C12.
'[see Cheng'~'.-S, Liu W.-L. and Chen S -H., Synthesis, (1980) 2.?3 J
30 3-Cyclopenty!-7-piperid'inosulfonyl-1,2,3,4-tetr,~hyrdro-1,2,4-
benzothiadiazlne-1,1-dioxide (55):
2-Aminobenzenesulfonamide was used as sl:arting material for the following
transformation
sequence: Method C, Method A (using piperidine as amine), Method D, Method G
[using
cyclopentanecarboxaldehydey. M.p. '~?58-260 "C..
CA 02320354 2000-08-04
WO 99!42456 PCT/DK99/00070
81
Compound 56
3-Cyclohexyl-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Aminobenzenesulfonamide was used as darting material for the following
transformation
sequence: Method E (using cyclohexanecarbonyl chloride), Method A (using
piperidine as
amine), Method F. M.p. 262-264 "C.
Compound 57
3-Bicyclo[2.2.1 )hept-5'-en-2'-yl-7-pi peridinosulfonyl-1,2,3,4-tetrahydro-
1,2,4-
benzothiadiazine-1,1-diaxide
2-Aminobenzenesulfonamide was used as sl:arting material for the following
transformation
sequence: Method C, Method A (using piperiidine as amine), Method D, Method G
[using a
racemic endolexo mixaure of 2-norbornencarboxaldehyde). Two separate
diastereomeric
mixtures were isolated with m.p. (A) ;?40-242 "C and m.p. (B) 234-238
°C.
Compound 58
3-Cyclohexyl-7-(1',2',3',6'-tetrahydropiperidina)sulfonyl-1,2,3,4-tetrahydro-
1,2,4-
benzothiadiazine-1,1-dioxide
2-Aminobenzenesulfonarrride was used as starting material for the following
transformation
2o sequence;: Method E (using cyclohexanecarbonyl chloride), Method A (using 7
,2,3,6-
tetrahydropyridine as amine), Method F. M.p. 23J-239 °C.
Compound 59
3-Cyclohexyl-7-(N-methyl-N-phenylamino)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using cyclohexanecarbonyl chloride)., Method A (using N-
methylaniline
as amine), Method F. M.p. 210-212 °C.
Compound 60
3-Cyclohexyl-7-(1'-(1',2',3',4'-tetrahydroquinolinyl))sulfonyl-1,2,3,4-
tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
$2
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using cyclohexanec:arbonyl chloride), Method A (using
1,2,3,4-
tetrahydroquinoline as amine), Method F. M.p. 218-220 °C.
Compound 61
3-Cyclohexyl-7-(4'-methyl piperazino)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method C, Method A (using N-methylpiperazinEa as amine), Method D,
Method G
(using cyclohexanecarboxaldehyde) M.p. 227-229 °C.
61 methane sulfonate salt: 61 (0.6 g; 1.4 mmol) was dissolved in 99% EtOH {30
ml) and
added a solution of 1 M C:,H3SO3H in 99°ia EtOH. The mixture was left
for precipitation for 2 h
and the salt isolated by filtration. The composition of thc~ sall was checked
by HPLC for
stability compared to the free base. The salt was found to be stable towards
hydrolysis under
these conditions and had a water solubility of 10 mg/ml.
Compound 62
3-Cyclohexyl-7-(4'-methylsulfonylpiperazi no)sulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
N-Mesylpiperazinium chloride: To a stirred solution of piperazine (4.3 g; 50
mmol) in CH2C12
(50 ml) at +5 °C was added a solution of CH3SO2C1 (4.25 ml; 55 mmol) in
CH2CI2 (15 ml).
The thick reaction mixture was stirred at rt. for 12 h, added CH2C12 (100 ml),
and extracted
with 1 M HCI (300 ml). A precipitate formed in the aqueous phase was filtered
of to give 2.66
g of N-mesylpiperazinium chloride (32 %).
3-Cyclohexyl-7-(4'-men"hylsulfonylpiperazino)suifnnyl-1,2, 3,~-tetrahydro-1,2,
4-
benzofhiadiazine-1,1-dioxide (62): 2--Aminobenzenesulfonamide was used as
starting material
for the following transformation sequence: AAethod E (using
cyclohexanecarbonyl chloride),
Method A (using N-Mesylpiperazinium chloride as amine (1.5 eq.), 3 eq. K2C03
was used for
neutralization), Method F. M.p. 272-274 °C.
Compound 63
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
83
3-Cyclohexyl-7-morpholinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using cyclohexanecarbonyl chloride), Method A (using
morpholine as
amine), Method F. M.p. 262-264 °C..
Compound 64
3-Bicyclo[2.2.1 ]hept-5'-en-2'-yl-7-bromo-1,2.,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide
5-Bromo~-2-aminobenzenesulfonarr~ide (see compound 18) was transformed by
Method G
(using a racemic endo/exo mixture of bicyclo[2.2.1 ]hept-5-E:ne-2-
carboxaldehyde). M.p. 200-
204 °C.
Compound 65
2-Methyl-4-axo-3,4-dihydro-6-quinazoline-N,N-dimethylsulfonamide
2-Methyl-4-oxo-3,4-dihydroquinazoline: A solution of anthranilamide (13.6 g;
100 mmol) in
acetic acid (100 ml) was refluxed for 60 h. The reaction mixture was
evaporated to dryness,
suspended in H20, filtered and washed thoroughly with Nal-IC03 until the
filtrate had a pH of
8-8.5.
2-Methyl-4-oxo-3,4-dihydro-6-quinazoline-N,N-~dirrrethylsulfonamide: 2-Methyl-
4-oxo-3,4-
dihydroquinazoline was transformed by Method A (using dirnethylamine as
amine). M.p. 264-
266 ° C.
Comaound 66
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoiine sulfonamide
2-Trilfluoroacetamidobenzamide (seE~ compound 68) was transformed by Method A
{Using
0.5 M NH,3 in THF as amine). M.p. 311-314 °C.
Compound 67
2-Trifluoromethyl-4-oxo-3,4-dihydra-6-quinazoiine N,N-dimethylsulfonamide
2-Trilfluoroacetamidobenzamide (see compound 68) was transformed by Method A
(Using
dimethylamine as aminE~). M.p. 257-258 °C.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
84
Compound 68
2-Trifluoromethyl-4-ox:o-3,4-dihydro-6-quinazoline-1',2',3'',6'-
tetrahydropiperidinosulfonamide
A stirred mixture of anthranilamide (13.6 g; 100 mmol) in ~~fHF (100 ml) at 0
°C was added
trifluoroacetic acid anhydride (15.2 rr7l; 110 mmol) and allowed to warm up to
rt. and left with
stirring over night. The reaction mixture was evaporated to dryness, suspended
in H20 and
filtered. The isolated solid was air dried to give 21.6 g (93°/«) 2-
trilfluoroacetamidobenzamide.
2-TriIfIuoroacetamidobE:nzamide was transformed by Method A (using 1,2,3,6-
tetrahydropyridine as amine). M.p. 2~'7-230 °C.
Compound 69
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline N-cyclohexylsulfonamide
2-Trilfluoroacetamidobenzamide (sere compouncl 68) was transformed by Method A
(Using
cyclohexylamine as amine). M.p. 261-263 °C.
Compound 70
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinazoline morpholinosulfonamide
2-Trilfluoroacetamidobenzamide (see compound 68) was transformed by Method A
(Using
morpholine as amine). M.p. 282-285 °C.
Compound 71
2-Cyclohexyl-4-oxo-3,4-dihydro-6-quinazoline-N,N-dimethylsulfonamide
Antranilamide was used as starting material for the following transformation
sequence:
Method A (Using 25% NF-13 (aq.) as amine. The reaction mixture contained both
the 5-mono
and 5,7-disulfonamide, which were separated by chromatography), Method G
(using
cyclohexanecarboxaldehyde. The arninal auto oxidizes to the aromatic
hydroxyquinazoline).
M.p. 306-310 °C.
Compound 72
3-Methyl-7-sulfamoyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide
3-Methyl-1,2-dihydro-1,2,4-benzothiadiazine-1,~!-dioxide (see compound 73) was
transformed
by Method A (using 0.5M NH3 in THi= as amine). M.p. 295-297 °C.
CA 02320354 2000-08-04
WO 99/42456 PC'.T/DK99/00070
Compound 73
3-Methyl-7-dimethylsulfamoyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide
2-Aminobenzenesulfonamide (17.2 g; 100 rnrnol) was refluxed in AcOH for 5
days. The
precipitate formed was isolated by filtration and washed with water to give
17.8 g (91 %) of 3-
5 methyl-1,2-dihydro-1,2,4-benzothiadcazine-1,1-dioxide. 3-Methyl-1,2-dihydro-
1,2,4-
benzothiadiazine-1,1-dioxide was transformed by Method A {using dimethylamine
as amine).
M.p. 260-261 °C.
Compound 74
10 3-Methyl-7-(1',2',3',6'-tetrahydropiperidino)sulfonyl-1,2-dihydro-1,2,4-
benzothiadiazine-
1,1-dioxide
3-Methyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide (see compound 73) was
transformed
by Method A (using 1,~~,3,6-tetrahydropiperidine .as amine). M.p. 265-268
°C.
15 Compound 75
3-Methyl-7-cyclohexyisulfamoyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide
3-Methyl-1,2-dihydro-1,2,4-benzothiadiazine-11,'1-dioxide (see compound 73)
was transformed
by Method A (using cyc:lohexylamine as amine). M.p. 239-242 °C.
2o Compound 76
3-Trifluoromethyl-7-dimethylsulfamoyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method C and Method A fusing dirnEahylamine as amine). M.p. 240-242
°C.
25 Compound 77
2-Trifluoromethyl-4-oxo-3,4-dihydro-6-quinaxolinesulfonic acid
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequencE~: Method C and Method A fusing Na~Of~i instead of an amine). M.p.
>330 °C.
3o Compound 78
3-Cyclohexyl-8-methyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
m-Toluidine was used as starting material for the following transformation
sequence: Method
B [2-amino-6-methylbenzenesultonamide was separated from 2-amino-4-
methylbenzenesulfonamide by recry:>tallization (EtOAc/hexane)]. 2-Amino-6-
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
86
methylbenzenesulfonamide was further purified by column chromatography and
transformed
by Method G (using cyclohexanecarboxaldehydca). M.p. 228-230 °C.
Compound 79
3-Cyclohexyl-8-hydrox:ymethyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
5-Chloro-3-cyclohexyl-7,2-dihydro-t,~',4-benzothiadiazine-1,1-dioxide-8-
carboxylic acid: To a
solution of 5-chloro-3-cyclohexyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide (see
compound 80) (0.30 d, t .0 mmol) in dry THF' (15 ml) ,~t: -78 °C was
added s-BuLi in
cyclohexane (1.3 M, 1.E~ ml, 2.1 mmoi) under i'J2. 'The yellow mixture was
stirred for 15 min at -
78 °C and dry gaseous. C02 was bubbled throucth the solution for 30
min. The cooling bath
was removed and the rnixture was allowed to warm to 0 ''C. -The solvent was
removed under
reduced pressure and the residue was triturated with hydrochloric acid (0.2 M,
12 ml). The
crude product was rec:rystallised from 50 °~~ MeOH yielding 300 mg
(82%) of 5-chloro-3
cyclohexyl-1,2-dihydro-1,2,4-benzoth~adiazine-1,1-dioxide-8-carboxylic acid as
colorless
~ 5 crystals.
3-Cyclohexyl-1,2-dihydro-1,2,4-benzothiadiazine-l, l-dioxide-8-carboxylic
acid: 5-Chloro-3-
cyclohexyl-1,2-dihydro-1,a?,4-benzothiadiazine-1,1-dioxide-8~-carboxylic acid
(160 mg, 0.47
mmol) was dissolved in 90 % EtOH (50 ml) and hydrogenated using Pd/C (10 %, 10
mg) at 1
bar pressure for 24 h. NaOH (1 M, 12 ml) was added and the mixture was
filtered through a
pad of CeliteT"' and concentrated to 10 ml and concentrated hydrachloric acid
was added
slowly to precipitate the product yielding 11 l) mg (76%) of 3-cyclohexyl-1,2-
dihydro-1,2,4-
benzothiadiazine-1,1-dioxide-8-carboxylic acid as colorless powder.
3-Cyclohexyl-8-hydroxymeth.yl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide (79):
NaBH4 in triglyme (2M, 0.75 ml, 1.5 mmol) was dissolved in dry THF (20 ml) and
cooled to -50
°C under N2,. BF3-etherate (0.25 ml, 2.0 mmol) was added and the
mixture was stirred for 10
min at -50 °C. Solid 3-cyclohexyl-1,2-dihydro-1,,?,4-benzothiadiazine-1
,1-dioxide-8-carboxylic
acid (170 mg, 0.55 mmol) was added in one portion and the suspension was
stirred for 6 h at
-50 °C and overnight at 20 °C. The mixture was hydrolysed with
hydrochloric acid (1 M, 2 ml)
and the solvent was removed under reduced pressure. The residue was extracted
with EtOAc
(50 ml) and the organic; layer was washed with brine (10 ml), dried (Na2S04)
and the
evaporated to dryness. The product was purified by flash chromatography on
Si02 using
EtOAc:n-hexane (2:1, v/v) as eluerzt, yielding 1 C>C) mg (62 %;) of 3-
cyclohexyl-8-hydroxymethyl-
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
87
1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide as colorless needles. The
product was further
transformed by Method F. M.p. 220-223 °C.
Compound 80
3-Cyclohexyl-8-(2-methoxyphenyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
5-Chloro-3-cyclohexyl-1,2-dihydro-1,~,4-benzothiadiazine-1,-1-dioxide: 3-
Chloroaniline was
used as starting material for the following transformation sequence: Method B
followed by
Method E (using cycloh~sxylcarbanyl chloride).
5-Chloro-3-cyclohexyl-8-iodo-1,2-dihydro-1,~?,4-t>E~nzothiadiazine-~, i-
dioxide: To a solution of
5-chloro-3-cyclohexyl-1,2-dihydrc>-1,2,4-benzot:hiadiazine-1,1-dioxide (596
mg, 2 mmol) in dry
THF (20 ml) at -78 °C was added s-BuLi in c.yclohexane (-1.3 M, 3.8 ml,
5 mmol) under N2.
The yellow mixture was stirred for 15 min at -T8 °C and a solution of
12 (1.27 g, 5 mmol) in dry
THF (5 ml) was added. The cooling bath was removed and the mixture was allowed
to warm
to 0 °C. NaHS03 (5 °ro, 20 ml) was added and extracteca with
EtOAc (2 x 30 ml), dried
(Na2S04) and evaporated to dryness to give 0.76 g (90 %) of 5-chloro-3-
cyclohexyl-8-iodo-
1,2-dihydro-1,2,4-benzothiadiazine-1.1-dioxide.
5-Chloro-3-cyclohexyl-8-(2-methoxyphenyl)-l,a?-dihydro-1,2,4-benzothiadiazine-
1,1-dioxide: A
mixture of 5-chloro-3-cyclohexyl-8-iodo-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide (290
mg, 0.68 mmol), 2-methoxyphenylboronic acid {122 mg, 0.80 mmol), Pd(PPh3)2C12
(10 mg, 2
mol %) in 1,2-dimethoxyethane (50 rnl) and Na2C03 (2M, 2 rnl, 4 mmol) were
refluxed under
N2 for 2 h. The solvE~nts were removed under reduced pressure and the residue
was
extracted with EtOAc (:? x 40 ml) and the organic layer was washed with
saturated NaHC03
(20 ml), dried (Na2SOa; and the solvent was iremoved undc:~r reduced pressure.
The product
was purified by flash chromatography on SicJ; using EtOAc:n-hexane (1:2, v/v)
as eluent,
yielding 200 mg {73 °~~) of 5-chioro-3-cyclohexyl-8-(2-methoxyphenyl)-
1,2-dihydro-1,2,4-
benzothiadiazine-1,1-dioxide as colorless crystals.
3-Cyclohexyl-8-(2-methoxyphenyl)-1,2,3,4-tetrahydro-1,2,4-t~enzothiadiazine-
1,1-dioxide (80):
5-Chloro-3-cyclohexyl-8-(2-methoxyphenyl)-~1,;2-dihydro-1,2,4-benzothiadiazine-
1,1-dioxide
(190 mg, 0.47 mmol) was dissolved ~n 99 °~o EaOH (30 ml) and
hydrogenated using Pd/C (10
%, 10 mg) at 1 bar pressure. 'The mixture vva~~ filtered through a pad of
CeliteT"" and the
solvent was removed under reduced pressure yielding 174 mg (100 %) of 5-chloro-
3-
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
88
cyclohexyV-8-(2-methoxyphenyl)-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide
as colorless
crystals. The product w<~s transformed by Method F. M.p. 100-105 °C.
Compound 81
3-Cyclohexyl-8-(3-methoxyphenyf)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
Synthesis as for compound 80 (using 3-methoxyphenylbc:~ronic acid for the Pd-
cat. cross
coupling). M.p. 108-115 °C:;.
Compound 82
3-Cyclohexyl-8-(2-pyridyl)-1,2,3,4-tetrahydro-'1,2,4-benzothiadiazine-1,1-
dioxide
5-Chloro-3-cyclohexyl-b'-(dihydraxyboryl)-l,a?-a'ihydro-1,2,4-benzothiadiazine-
1,1-dioxide: To a
solution of 5-chlo~ro-3-cyclohexyl-1,2-dihydro-1,2,4-benzothiadiazine-1,1-
dioxide (see
compound 80) (0.60 d, 2.0 mmol) in dry Tf-IF (15 ml) at -78 °C was
added s-BuLi in
cyclohexane (1.3 M, 3.l3 ml, 5 mmol) under N,~. The yellow mixture was stirred
for 15 min at -
78 °C and B(OCH3)3 (0.57 ml, 5 mmol) was added. The ccaoling bath was
removed and the
mixture was allowed to warm to 0 "C and stirred for another 1 h. The mixture
was hydrolysed
with hydrochloric acid (0.5 M, 12 rnl) and extracted with EI:OAc (2 x 50 ml),
dried (Na2S04)
and evaporated to dryness to give 0.65 g {95 °,~o) of 5-chloro-3-
cyclohexyl-8-(dihydroxyboryl)-
1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide.
5-Chloro-;3-cyclohexyl-~~-(2-pyridyl)~-1,2-dihydra-P,2,4-benzofhiadiazine-1,1-
dioxide: A mixture
of 5-chloro-3-cyclohexyl-8-(dihydroxyboryl;i-1,2-dihydro-1,2,4-
benzothiadiazine-1,1-dioxide
(440 mg" 1.28 mmol), ~'--bromopyridine (0.14 ml, 1.50 mmol), Pd(PPh3)2Clz (10
mg, 2 mol %)
in 1,2-dirnethoxyethane (30 ml) and Na2C0;, (2M, 3 ml, 6 mrnol) were refluxed
under N2 for 24
h. The solvents were removed under reduced pressure and the residue was
treated with
saturated NH4C1 (10 ml) and extracted with EtOAc (2 x 40 nol). The organic
layer was washed
with water (20 ml), dried (NalS04) and the solvent was remcpved under reduced
pressure. The
product was purified by flash chromatography on Si02 using EtOAc:n-hexane
(2.1, v/v) as
eluent, yielding 280 mc~ (,58 °/>) of 5-chloro-3-cyclohexyl-8-(2-
pyridyl)-1,2-dihydro-1,2,4-
3o benzothiadiazine-1,1-dioxide as colorless crystals.
3-Cyclohexyl-8-(2-pyridyl)-1,2,3,4-fetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide (82): 5-
chioro-3-cycfohexyl-8-(a?-pyridyl)--1,2-dihydro-1,2,,4-benzothiadiazine-1,1-
dioxide (0.268 g,
0.713 mrnol) was dissolved in 99 % EtOH (50 rnl) and hycarogenated using Pd/C
(10 %, 10
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
89
mg) at 4 bar pressure for 24 h. The mixture was filtered through a pad of
CeliteT"" and the
solvent was removed under reduced pressure. The residue was dissolved in EtOAc
(50 ml)
and washed with phosphate buffer (pH=7, 10 ml), dried (NazSO4) and the solvent
was
removed under reduced pressure, yielding 20() mg (82 %) of 3-cyclohexyl-8-(2-
pyridyl)-1,2-
dihydro-1,2,4-benzothiadiazine-1,1-dioxide a:~ colorless crystals. The product
was further
transformed by Method F M.p. 200-203 °C.
Compound 83
3-Cyclohexyl-8-methoxy-1,2,3,4-tetrahyd ro-1,2,4-benzoth iadiazi ne-1,1-
dioxide
m-Anisidine was used as starting material for the following transformation
sequence: Method
B (2-amino-6-methoxybenzenesulfonamide was separated from 2-amino-4-
methoxybenzenesulfonamide by flash chromatography (EtC:)Ac/hexane)] and then
Method G
(using cyclohexanecarboxaldehyde). M.p. 221-223 °C.
Compound 84
5,7-Dibromo-1,2-dihydro-1,2,4-benzothiadiazine-1,1-dioxide
2-Amino-3,5-dibromobenzenesulfonamide (;see compound 125) was transformed by
Method
G {using ethylformiat and a catalytic amount of triethylamine). M.p. 289-292
°C.
Compound 85 and compound 86
3-Cyclohexyl-2-methyl-T-morpholinosulfonyi-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide (85) and 3-Cyclohexyl-4-methyl-7-morpholinosulfonyl-1,2,3,4-
tetrahydro-
1,2,4-benzothiadiazinc~-1,1-dioxide (86)
3-Cyclohexyl-7-morpholinosulfonyl-?~2-dihydro-?,,2,4-benzothiadiazine-?, ?-
dioxide: 2-
Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using cyclohexanecarborryl chloride) and Method A (using
morpholine
as amine) which gave 3-cyclohexyl-7-morpholinosulfonyl-1,,2-dihydro-1,2,4-
benzothiadiazine-
1,1-dioxide.
3-Cyclohsxyl-2-methyl-7-morpholinasulfonyl-1,2-dihydro-1,2,4-benzothiadiazine-
1,?-dioxide
and 3-cyclohexyl-4-methyl-7-morpholincrsulfonyl-?,2-dihydro-?,2,4-
benzofhiadiazine-1, ?-
dioxide: A mixture of 3-cyclohexyi-7-morpholinosulfonyl-1,2-dihydro-1,2,4-
benzothiadiazine-
1,1-dioxide (2 g; 5 mmol), DEAC> (2.35 ml; 15 mrnol), PPh3 (4 g; 15 mmol) in
dry THF (30 ml)
was cooled to 0 °C and added MeOH (1.25 ml; 30 mmol). The reaction
mixture was stirred
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
over night at rt., evaporated to dryness, stirred with EaOAc and filtered. The
isolated
precipitate was stirred with CH2C12 and filtered to leave the 2-methyl isomer
in the filtrate and
the 4-methyl isomer as the precipitatE:.
The 4-methyl was purified by recrystalizalion from DMSO/H2O. The 2-methyl
isomer was
5 purified by column chromatography (EtOAcl.
3-Cyclohexyl-2-methyl-7-rnorpholinosulfon yl-1,~, 3, 4-tetrahydro-1,2, 4-
benzothiadiazine-1,1-
dioxide (85): 3-Cyclohexyl-2-methyl-7-morpholir~osulfonyl-1,2-dihydro-1,2,4-
benzothiadiazine-
1,1-dioxide was reduced by use of method F. M.p. 243-245 "C.
3-Cyclohexyl-4-methyl-7-morpholinosulfonyl-1,2,3,4-tetrahyc:fro-1,2,4-
benzofhiadiazine-1,1-
dioxide (86): 3-CycIohE~xyl-4-methyl-7-morpholinosulfonyi-1,2-dihydro-1,2,4-
benzothiadiazine-
1,1-dioxide was reduced by use of method F. M.p. 207-210 °C.
Compound 87
7-Methylsulfonylamin~o-1,2,3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-
thiadiazine-
5,5-dioxide
1,2,3,5-tefrahydrobenz~[eJpyrralo/2,1-cJ-1,2,4-ihiadiazine-5,5-dioxide: 2-
Aminobenzene-
sulfonamide was transformed by Method E (uaing 4-chlorobutanoyl chlaride).
7-Nitro-1,2,3,5-tetrahydrobenzo~eJpyrrolo(2,1-cJ-1,2,4-thiadnzine-5,5-dioxide:
A stirred
solution of 1,2,3,5-tetr.ahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-thiadiazine-5,5-
dioxide (222 mg; 1
mmol) in H2S04 (2 ml) at 5 °C was added a solution of KNO3 (122 mg; 1.2
mmol) in H2S04
(2 ml). The reaction mixture was allowed to warm up to rt. and stirred for 2h.
The reaction
mixture was poured into ice water, filtered and air dried to give 190 mg {71
%) product.
7-Amino-1,2,3,5-tefrah.ydrobenzo[e~pyrrolo(2,1-~c)-1,2,4-thiadiazine-5,5-
dioxide: A stirred
solution of 7-nitro-1,2,3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c]-7,2,4-
thiadiazine-5,5-dioxide
(167 mg; 0.6 mmol) in dry THF (2 ml) at -50 °(~ was addeca LiAIH4 (115
mg; 3 mmol) in one
portion. The reaction mixture was allowed to warm up to rt, and stirred over
night. The
reaction mixture was quenched by addition of H20 and 10 M NaOH, stirred,
filtered through
celite and evaporated to dryness to give 150 mg product.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
X31
7-Methylsulfonylamino- i,2,3, 3a,4,:i-hexahydrobErnzo~e/pyrrolo(2, l -c(-1,2,4-
thiadiazine-5,5-
dioxide (87): A stirre~~ solution of 7-amino-1,2,3,3a,4,5-
hexahydrobenzo[e]pyrrolo[2,1-c~-
1,2,4-thiadiazine-5,5-dioxide (150 mg; 0.5 mrnol) and triethylamine (70 pl;
0.5 mmol) in THF
(1 ml) was added a solution of CH3S02C1 (4~0 lal; 0.5 mmol) in THF (1 ml) and
stirred at rt.
over night. The reaction mixture was evaporated to dryness, suspended in H20
and
extracted by EtOAc. The combined organic fractions were evaporated to dryness.
Column
chromatography gave ~10 mg product. M.p. 177-180 °C.
Compound 88
7-Sulfamoyl-1,2,3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c~-1,2,4-thiadiazine-5,5-
dioxide
2-Aminobenzenesulfonarnide was used a:~ stari:ing material for the following
transformation
sequence: Method E (using 4-chlorobutanoyl chloride), Method A (using 0.5 M
NH3 in THF as
amine), Method F (using LiAIH4 and rt.). M.p, 260-262 °C.
Compound 89
7-Methylsulfamoyl-1,:?,3,3a,4,5-hexahydrobenzo[e]pyrroio[2,1-c]-1,2,4-
thiadiazine-5,5-
dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using 4-chlorobutanoyl chloride), Method A (using
methylamine as
amine), Method F (using LiAIH4 and rt.). M p. 244-245 °C.
Compound 90
7-Cyclohexylsulfamoyl-1,2,3,3a,4,5-hexahydrobenzo[e] pyrrolo[2,1-c]-1,2,4-th
iadiazine-
5,5-dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using 4-chlorobutanoyll c:hloride), ME=thod A (using
cyclohexylamine as
amine), Method F (using LiAIH4 and rt.). M.p. 195-197 °C.
Compound 91
7-Dimethylsulfamoyl-1,2,3,3a,4,5-hexahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-
thiadiazine-5,5-
dioxide
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
92
2-Aminobenzenesuifonamide was used as startnng material for the following
transformation
sequence: Method E (using 4-chlorobutanoyl chloride), Method A (using
dimethylamine as
amine), Method F (using l_iAIH4 and rt.). M.p. ;?40-243 °C.
Compound 92
7-Methylsulfamoyl-1,2,3,5-tetrahydrobenzo[e]pyrrolo[2,1-c]-1,2,4-thiadiazine-
5,5-dioxide
2-Aminobenzenesulfonamide was used as si:arting materia:~l for the following
transformation
sequence: Method E (using 4~chlorobutanoyl c:hloride), Method A (using
methylamine as
amine). M.p. 244-247 °C.
Compound 93
7-Dimethylsulfamoyl-1,2,3,5-tetrahydrobenzo[e)pyrrolo[2,1-c]-1,2,4-thiadiazine-
5,5-
dioxide
2-Aminobenzenesulfonamide was transformed by Method E (using 4-chlorobutanoyl
chloride)
and Method A (using dimethylamine). M.p. 251 ~~2~53 °C.
Compound 94
7-Cyclohexylsulfamoyl-1,2,3,5-tetrahydrabenzo[e]pyrrolo[2,1-c]-1,2,4-
thiadiazine-5,5-
dioxide
2-Aminobenzenesulfonamide was used as starting material for the following
transformation
sequence: Method E (using 4-chlorobutanoyl <:hloride), Method A (using
cyciohexyiamine as
amine). M.p. 151-153 "C.
Compound 95
7-(1',2',3',6'-Tetrahydropiperidino)sulfonyl-1,2,3,5-
tetrahydrobenzo[e]pyrrolo[2,1-c]-
1,2,4-thiadiazine-5,5-dioxide
2-Aminobenzenesulfonarnide was used as starting material for the following
transformation
sequence: Method E (usinc) 4-chlorobui:anoyl chloride), Method A (using
1,2,3,6-
tetrahydropyridine as amine). M.p. 204-206 "c;.
Compound 96
3-Bicyclo[2.2.1]hept-!5'-en-2'-yl-5,7-dimethyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
93
2,4-Dimethylaniline was used as si:arting material for the following
transformation sequence:
Method B, Method G psing a racemic endoi'exo mixture of bicyclo[2.2.1 ]hept-5-
ene-2
carboxaldehyde. Column chromatography gave two diastereomeric fractions each
being a
mixture of two diastereomers~. Isomeric mixture b, m.p. 160-165 °C;
isomeric mixture B, m.p.
182-187 °C.
Compound 97
3-Cyclohexyl-7-(N,N-diethylsulphamoyl)-5-methyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
io 3-Cyclohexyl-7-(N,N-diethylsulpharno~yl)-5-formyl-1,2-dihydrc7-7,2,4-
benzothiadiazine-1,1-
dioxide: 2-Aminobenzenesulfonamide was used as starting material for the
following
transformation sequence: Method E (using cy<;Ic7hexanecarbonyl chloride) and
Method A
(using diethylamine as amine). The product from this transformation (0.60 g,
1.5 mmol)
dissolved in dry THF (1:5 rnl) at -78 °C was added s-BuLi in
c;yclohexane (1.3 M, 2.5 ml, 3.2
mmol) under N2. The yE:llow mixture was stirred for 25 min at ~-78 "C. The
reaction was
quenched with dry DMF (0.3 ml, 4 mrnol) and'rhe mixture was stirred for 20 min
at -78 °C. The
cooling bath was removed and the mixture was allowed to warm to 0 °C.
Hydrochloric acid
(0.5 M, 10 ml) was added and the mixture was extracted with EtOAc (40 ml). The
organic
layer was washed with brine (10 ml), dried (Na2SO4) and evaporated to dryness.
The residue
2o was dissolved in acetone (8 ml). Et2C> (30 ml) waa added and within few
minutes the product
crystallises, yielding O.~t1 g (64%) of 3-cyclahexy6'-7-(N,N-
diethylsulphamoyl)-5-formyl-1,2-
dihydro-1,2,4-benzothiadiazine-1,1-dioxide as colorless crystals.
3-Cyclohexyl-7-(N, N-di~efhylsulphamoyl)-5-me thyl-1,2,3, 4-tetrahydro-1,2, 4-
ben2othiadiazine-
1,1-dioxide (97): 3-Cyclohexyl-7~-(N,N-diethylsulphamoyl)-5-formyl-1,2-dihydro-
1,2,4-
benzothiadiazine-1,1-dioxide (0.20 g, 0.46 rnmol) was dissolved in 99 % EtOH
(60 ml). One
small drop of concentrated hydrochloric acid vvas added to ensure fully
hydrogenation. The
mixture was hydrogenated using PdiC (10 '%, 10 mg) at 4 bar pressure for 24 h.
The mixture
was filtered through a pad of CeliteT"" and evaporated to dryness. The residue
was dissolved
in EtOAc (50 ml) and washed with water (10 nnl}, dried (Na2SO4) and evaporated
to dryness
yielding 0.18 mg (95%) of 3-cyclohexyl-7-(N,hJ-~liethylsulphamoyl)-5-methyl-
1,2-dihydro-1,2,4-
benzothiadiazine-1,1-dioxide as colorless po~nrde~r. The product was further
transformed by
Method F'. M.p. 206-208 "C.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
94
Compound 98
3-Bicyclo[2.2.1 ]kept-5'-en-2'-yl-5,7-Biphenyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
3,5-Dibromo-2-aminobe;nzenesulfonamide (see compound 125) was transformed by
Method
H (using phenylboronic acid) and Method Ca (using a, racemic endo/exo mixture
of
bicyclo[2.2.1]hept-5-ene-2-carboxaidehyde). M.p. 222-225 °~~.
Compound 99
3-Bicyclo[2.2.1]hept-5'-en-2'-yi-5,7-disulfamoyi-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
2-Aminobenzenesulfonamide was transformed by Method A (using 25% NHg (aq.) as
amine)
and Method G (using using a racemic endo/exo mixture of bicyclo[2.2.1]hept-5-
ene-2-
carboxaldehyde). M.p. 172-180 "C.
Compound 100
3-Bicyclo[2.2.1 ]hept-5'-en-2'-yl-5,7-dichloro-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
2,4-Dichloroaniline was transformed by Method B and Method G (using a racemic
endo/exo
mixture of bicyclo[2.a?.1 Jhept-5-ene-2-carboxaldehyde. The product was
isolated as a
diastereomeric mixture). M.p. 149-151 °C.
Compound 101
5-Bromo-3-cyclohexyl-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
2-Amino-5-sulfamoylbenzenesulfonamide: A :>tirred suspension of 2-amino-4-
chloro-5-
sulfamoylbenzenesulfonamide (11.4 g; 40 mmol) and 10°/a Pd/C (750 mg)
in EtOH (300 ml)
was hydrogenated at 1 bar until hydrogen consumption ceased (24 h). The
reaction mixture
was evaporated to dryness, resuspended in THF and filtered through celite. The
filtrate was
evaporated to dryness and the isolated solid washed with boiling EtOAc (2x 150
ml) to give
9.58 g (95%) product.
2-Amino-3-bromo-5-sulfamoylbenzenesulfc>namide: A stirred solution of 2-amino-
5-
sulfamoylbenzenesulfonamide (3.77 g; 15 mmol) in AcOH (50 ml) was added a
solution of
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
Br2 (0.78 ml; 15 mmol) in AcOH (10 ml). The reaction mixture was heated to 70
°C for 6 days,
evaporated to dryness, resuspended in MeOf-I (85 ml) and added solid KOH (3.8
g; 68 mmol).
The reaction mixture was heated to 60 °C for 2.5 h (hydrolysis of 3-
methyl-, 3-bromomethyl-,
3-dibromomethyl and ~s-tribromomethyl-1,2-dihydro-i ,2,4-bEanzothiadiazine
isomers formed in
5 situ), filtered, neutralized and evaporated to dryness. Column
chromatography gave 3.1 g
(63%) product.
5-Broma-3-cyclohexyl-7-sulfamoyl-1,2,3,4-tef~ahydro-1,2,4-benzothiadiazine-1,1-
dioxide
(i01): 2-Amino-3-bromo-5-sulfamoylbenzene~sulfonamide was transformed by
Method G
10 (using cyclohexanecarboxaldehyde) M.p. 254-258 °C.
Comaound 102
2-Bicyclo[2.2.1 ]hept-5.'-en-2'-yl-6,8-dibromo-1,2,3,4-tetrahydroquinazoline
2-Amino-3,5-dibromob~enzylamine: A mixture of 2-aminobenzylamine (6.1 g; 50
mmol) in
15 CHC13 (100 ml) at 0 °(~ was added a salution Br2 (5.1 ml; 100 mmol)
in CHCI3 (45 ml) such
that the reaction temperature was kept below +2 °C. Cooling was then
removed and the
reaction mixture was stirred at rt. over night. The reaction mixture was
filtered and the
precipitate washed with EtOAc and purified by column chromatography.
20 2-Bicycloj2.2.1Jhept-5'-err-2'-yl-6,8-dibroma-1,2,;3,4-
tetrahydroquinazoline (102): 2-Amino-3,5-
dibromobenzylamine was transformed by Method G (using a racemic endo/exo
mixture of 2-
norbornencarboxaldehyde). M.p. 240 °C.
Compound 103
25 2-Bicyclo[2.2.1]hept-!i'-en-2'-yl-G,8-dibromo-4-oxo-1,2,3,4-
tetrahydroquinazoline
3,5-Dibramoanthranilamide: A stirred suspension of anthranilamide (13.6 g; 0.1
mol) in AcOH
(350 ml) was added a solution of Br2 (10.3 ml; 0.2 mol). l-he reaction mixture
was stirred at
45 °C for 120 °C, poured into H20 (1.5 I) and filtered.
Hecrystalization (including a warm
filtration) from 96% EtIJH (approx. 1 I) gave 23.6 g (80%) product.
2-Bicyclo(2.2.1Jhept-5'-en-2'-yl-6,8-dibroma-4-caxo-1,2,3,4-
tctrahydraquinazoline (103): 3,5-
Dibromoanthranilamide was transformed by Method G (using using a racemic
endo/exo
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
96
mixture of bicyclo[2.2.'I]hept-5-ene-2-carboxaldE:hyde). The: product was
separated into to
individual diastereomeric mixtures. M.p. (A) 213-215 °C, M.~r. (B) 209-
210 °C.
Compound 104
3-Bicyclo[2.2.1]hept-5'-en-2'-yl-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
2-Amino-3,5-dibromobenrenesulfonamide (see compound 125) was transformed by
Method
G (using a racemic endo/exo mixture of bicyclo[2.2.1 ]hept-5-ene-2-
carboxaldehyde). The
diastereomeric mixturE: was purified by column chromatography to give three of
the
~0 theoretically four possible diastereomers. M.p. (;A) 202-206 °C,
M.p. (B) 796-199 °C, M.p. (C)
180-184 °C.
Compound 105
5,7-Dibromo-3-bicyclo[2.2.1]heptan-2'-yl-1,2,3"4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide
Bicyclo[2.2.1 ]heptane-;?-carboxaldehyde: A stirred suspension of 2-
norbornylmethanol (0.5
ml; 5.8 mmol) and PC(:, on A1203" in CH2CIp (25 ml) was stirred at 2-3
°C for 1 h and then
allowed to slowly warim up to rt. The reaction mixture was filtered and the
solid material
washed with CH2C12 1;2x 25 ml). The combinE;d organic fractions were adsorbed
onto silica
and chromatographed to give 300 mg (42%) product as an oil.
'Jsee Cheng Y.-S, Liu W.-L. end Chen S.-H., Synthesis, (1980) 22".J
5,7-Dibromo-3-norbornanyt-1,2,3,4-tetrahydrc~-t,2,4-benzothiadiazine-1,1-
dioxide (105): 2-
Amino-3,5-dibromobenzenesulfonamide (see compound 125) was transformed by
Method G
(using bicyclo[2.2.1 ]heptane-2-carboxaldehyde). M.p. 182-183 °C.
Compound 106
3-Cyclohexyl-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
2-Amino-3,5-dibromobenzenesulfonamide (see compound 125) was transformed by
Method
G (using cyclohexanec;arboxaldehyde). M.p. 16E~-167 °C.
Compound 107
3-Adamantyl-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
97
1-Adamantanecarboxaldehyde: 1-Adamantylrnethanol was oxidized by the method
used for
2-norbornylmethanol (see compound 105)'.
'(see Cheng Y.-S, Liu W.-L. and (:hen S.-H., Synthesis, (1980) 223.
3-Adamantyl-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
(107): 2-
Amino-3,5-dibromobenzenesulfonamide (see compound 125) was transformed by
Method G
(using 1-adamantylcarboxaldehyde). M.p. 270-2.73 °C.
Compound 108
3-Phenyl-5,7-dibromo-~1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
2-Amino-3,5-dibromobE~nzenesulfonamide (see compound '125) was transformed by
Method
G {using benzaldehyde). M.p. 186-189 °C.
Compound 109
3-Ethoxy-5,7-dibromo-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-dioxide
A stirred mixture of 2-aimino-3,5-dibromobenzenesulfonamide (see compound 125)
(666 mg;
2 mmol), ethylorthoforrniate (15 mi; 90 mmol) and H2S04 {0.05 ml) was refluxed
over night.
The reaction mixture was evaporated to dryness and subjected to column
chromatography.
M.p. 96-98 °C.
Compound 110
3-Methyl-5,7-dibromo-1,2-dihydro-'1,2,4-benzothiadiazine-1,1-dioxide
A stirred solution of 2-<amino-3,5-dibromobenzenesulfonamide {see compound
125) (660 mg;
2 mmol) was refluxed in Ac20 (25 ml; 265 mmol) over night. The reaction
mixture was poured
onto ice and filtered. The isolated solid was washed with Et20. M.p. 287-289
°C.
Compound 111
3-Cyclohexyl-6-methyl-7-(2'-pyridyl)-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-
dioxide
To a solution of t-BuLi (1 7 M in pentane, 25 ml, 42 mmol) in dry THF at -
78°C was added 2-
bromopyridine (1.9 ml, 20 mmol) in such a rate that the temperature did not
exceed -70°C.
The mixture was stirred for another 30 min at -78°C. A solution of
ZnCl2 (2M in THF, 30 ml, 60
mmol) was added slowly and the cooling bath was removed and warmed to
20°C. 5-lodo-2-
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
g8
aminobenzenesulfonamide (see compound 37) (1.0 g, 3.2 mmol) and Pd(PPh3)4 (0.3
g, 8
mol%) was added and the mixture was refluxed for 6 h. The THF was evaporated
and the
residue was treated wilh FDTA (5:3 g, 0.18 mol) and made slightly basic (pH =
8-9) with 1 M
NaOH followed by extraction with E=tC7Ac (3 x '100 ml), dryinc:l (Na;>SO4).
The organic layer was
concentrated to ca. 40 rnl and n-hexane was added slowly. The product was
filtered off,
yielding 0.72 g (87 %) of 2-amino-4-methyl-5-(2-pyridyl)-1-benzenesulfonamide
as light-yellow
crystals. The product was further transformed by Method G (using
cyclohexanecarboxaldehyde). M.p. 229-231 "c:,.
Compound 112
3-Cyclohexyl-6-methyl-7-(4'-triazolyl)-1,2,3,4-tetrahydro-1,2,4-benzoth
iadiazi ne-1,1-
dioxide
5-Iodo-4-methy!-2-aminobenzenesulfonamide: m-Toluidine was transformed by
method B and
the two isomers separated by column chromatography to give 4-methyl-2-
aminobenzenesulfonarnide.
A stirred suspension of 4-methyl-2-aminobenzenesulfonarraide (620 mg; 3.3
mmol) in CHCI3
(7 ml) at 0 °C was added a solution of iodine monochloride (1.6 g; 9.9
mmol) in CHCI3 (7 ml).
The reaction mixture was stirred at 0 °C until H-NMR indicated full
conversion of starting
material. The reaction mixture was filtered arid i:he isolated solid washed
with small volumes
of CHCI3, NaHC03 {sat. aq.), H2O and air dried to give 640 mg (62 %) of
product.
3-Cyclahexyl-6-methyl~~7-(4'-triazofyl)-1,2,3,4-a~etrahydro-1,2,4-
benzothiadiazine-1,1-dioxide
(112J: 5-lodo-4-methyl-2-aminobenz enesulfonarnide was transformed by Method I
(using
trimethylsilylacetylene) and Method G (using cyclohexanecarboxaldehyde). FAB+
348. 'H-
NMR (DMSO-d6): 8.0 (1 H; br); 7.65 (1 H; br); 7.25 (1 H; s;); 7.22 (1 H; s);
6.95 (1 H; br); 6.8 (1 H;
s); 4.45 (1H; dd); 2.35 (3F-1; s); 1.95-1.8 (2H; rn); 1.8-1.6 (31-I; m); 1.3-
1.0 (6H; m).
Compound 113
3-Cyclohexyl-6-methyl-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
N-Acetyl-3-methylaniline: m-Toluidine (10 mP; 93 mmol) was added to a stirred
solution of
acetic anhydride (30 ml). The reaction mixture was stirred for 1,5 h at rt.,
evaporated to
dryness, stirred with H,2O and filtered to give 13 g product (94%).
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
99
3-Cyclohexyl-6-methyl-7-sulfamoyl-7,2,3,4-tetiahydro-1,2,4-benzothiadiazine-1,
i-dioxide: N-
Acetyl-3-rnethylaniline was transformed by Method A (using 25% NH3 (aq.) as
amine.
Deacetylation was complete) and Method G (using cyclohexanecarboxaldehyde).
M.p. 231-
233 °C.
Compound 114
3-Cyclopentyl-6-methyl-7-piperidinosulfanyl-1,2,3,4-tetrahydro-1,2,4-benzothia-
diazine-
1,1-dioxide
Cyclopentanecarbonyl chloride: CyclopentanE:carboxylic acid (0.55 ml; 5 mmol)
was refluxed
in thionylchloride (1 ml) for 3 h. The reaction mixture was cooled to rt.,
evaporated to dryness
and used directly without any purification.
3-Cyclopentyl-6-methyl-7-piperidinosulfonyl-1,2, 3, 4-tetrahydro-1,2, 4-
benzothia-diazine-1,1-
dioxide (714): m-Toluidine was used as starting material for the following
transformation
sequence: Method B [2-amino-fi-rnethylbenzenE;sulfonamide was separated from 2-
amino-4-
methylbenzenesulfonamide by recrystalli~:ation (EtOAc/hexane)), Method E
(using
cyclopentanecarbonyl chloride), Method A (using piperidine as amine), Method
F. M.p. 229-
230 °C.
Compound 115
3-Cyciohexyl-6-methyl-7-morpholinosulfonyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
m-Toluidine was used as starting material for the following transformation
sequence: Method
B [2-amino-6-methylbenzenesulfanamidE: was separated from 2-amino-4
methylbenzenesulfonamide by recrystallization (EtOAc/hexane)), Method E (using
cyclohexanecarbonyl chloride), Method A (ush7c~ morpholirne as amine), Method
F. M.p. 268-
271 °C.
Compound 116
3o 3-Cyclohexyl-6-(2-methoxyphenyl)-7-methyl-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-
1,1-dioxide
3-Bromo-4-methylaniline was transformed by Method B (The isomers were
separated by
fractional crystallization from MeQH) to give :?-amino-4-bromo-5-
methylbenzenesulfonamide.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
100
A mixture of 2-amino-4-bromo-5-rrrethylbenzenesulfonamide {100 mg, 0.38 mmol),
2-
methoxyphenylboronic acid (76 mg, 0.5C) mmol), Pd(PPh,)2C12 (13 mg, 5 mol %)
in 1,2-
dimethoxyethane (20 ml) and NaZC03 {2M, 1 rnl, 2 mmol) were refluxed under N2
for 4 h. The
solvents were removed under reduced pressure and the residue was treated with
saturated
NaHCOs (10 ml) and extracted with EtOAc (~~ x 25 ml). The organic layer was
washed with
brine (20 ml), dried (Na2S04) and the solvent was removed under reduced
pressure. The
product was purified by flash chromatography on Si02 using EtOAc:n-hexane
(1:1, v/v) as
eluent, yielding 100 mg (90 %) of 2-amino-4-(2-rnethoxyphenyl)-5-
methylbenzenesulfonamide
as colorless powder. The product was further tran:~formed by Method G (using
cyclohexanecarboxalde:hyde). M.p. 172-175 °C.
Compound 117
3-Cyclohexyl-6-methoxy-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothia-
diazine-
1,1-dioxide
m-Anisidine was used as starting material for the following transformation
sequence: Method
B [2-amino-6-methoxybenzenesulfonamide was separated from 2-amino-4-
methoxybenzenesulfonamide by flash chromatography (EtOAc/hexane)), Method C,
Method
A (using piperidine as amine), Method D, Method G (using
cyclohexanecarboxaldehyde). M.p.
237-240 °C.
Compound 118 and compound 122
3-Cyclohexyl-7,8-ethylenedioxy-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
(122) and 3-cyclohexyl-6,7-ethylenedioxy-1,2,3,4-tetrahydro-1,2,4-
benzothiadiazine-1,1-
dioxide (118)
6-Amino-1,4-benzodio~;ane was used as startin<~ material and transformed into
a mixture of
ethylendioxy-2-aminob~enzenesulfonamide isomers by use of Method B. The two
isomers
were separated by column chromatography.
3-Cyclohexyl-6,7-ethylc~nedioxy-1,2,3,4-fefrah,yd~o-1,2,4-ber~zothiadiazine-
1,1-dioxide (118):
2-amino-5,6-ethylendioxybenzenesulfonamidc: was transformed by use of Method G
(using
cyclohexanecarboxylaldehyde). M.p. 196-200 °C.
CA 02320354 2000-08-04
WO 99142456 PCT/DK99/00070
1G1
3-Cyclohexyl-7,8-ethylenedioxy-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide (722):
2-amino-7,8-ethylendiox:ybenzenesulfonamide was transformed by use of Method G
(using
cyclohexanecarboxylaldehyde). M.p. '~68-270 "C..
Compound t 19
3-Cyclohexyl-6-chloro-7-sulfamoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Amino-4-chloro-5-sulf~arnoylbenzenesulfonarnid~e was transformed by Method G
(using
cyclohexanecarboxaldehyde). M.p. 2;'4-276 °~;.
Compound 120
3-Phenyl-6-chloro-7-sulfarnoyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-1,1-
dioxide
2-Amino-4-chloro-5-sulfamoylbenzc:nesulfonarnide was transformed by Method G
(using
benzaldehyde). M.p. 235-238 "C.
Compound 121
3-Cyclohexyl-6-bromo-7-piperidinosulfonyl-1,2,3,4-tetrahydro-1,2,4-benzothia-
diazine-
1,1-dioxide
m-Bromoaniline was used as starting material for the following transformation
sequence:
Method B [2-amino-6-bromobenzenesulfonamide was separated from 2-amino-4-
bromobenzenesulfonarnide by recrystallizatian (EtOAc/hexane)~, Method C,
Method A (using
piperidine as amine), Method D, Method G (u~;incl cyclohexanecarboxaldehyde).
M.p. 238-241
°C.
Compound 122
See compound 118.
Compound 123
2-cyclohexylmethylamino-5-N,N-dimethytsulfamoylbenzenesulfonamide
2-Aminobenzenesutfonamide was used as starting material for the following
transformation
sequence: Method E (using cyc:lohexanecarbonyl chloride;l, Method A (using
dimethylamine
as amine), Method F (the reaction mixture was Ic:ft over night with DIBALH at
rt. with stirring).
M.p. 123-125 °C.
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
1 C)2
Compound 124
2-Ethylamino-7-(1',2',3',6'-tetrahydropiperidino)sulfonylbenzene sulfonamide
3-Methyl-1,2-dihydro-1,a?,4-benzothiadiazine-1,1-dioxide {see compound 73) was
transformed
by Method A (using 1,2,3,6-tetrahydropyridine as amine) followed by Method F
(using LiAIH4
and rt.). M.p. 175-177 °(:,.
Compound 125
2-Amino-3,5-dibromobenzenesulfonamide
A stirred solution of 2-aminobenzenesulfonarnide (8.6 g; 50 mrnol) in AcOH
(100 ml) was
slowly added a solution of Br2 (5.13 ml; 100 mmol) in AcI.~H (20 ml). The
reaction mixture
was heated to 55 °C for 60 h, poured into ice vvater (800 ml),
filtered, adsorbed onto silica and
purified by column chromatography to give 11.1 g (67 %) product. M.p. 165-169
°C.
Compound 126
2-Acetamidobenzenesulfonamide
A stirred solution of 2-aminobenzenesulfonarnide (1.72 g; 10 mmol) and
triethylamine (1.53
ml; 11 mmol) in THF (25 rnl) at 0 °C was addE:d ,AcCI (0.85 ml; 12
mmol) and left with stirring
at rt. over night. The reaction mixture was filtered anri adsorbed onto
silica. Column
chromatography gave 1I .7 g (79 %) product. M.~>. 153.5-155.5 °C.
Compound 127
3-Isobutyl-8-(piperidinosulfonyl)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine-
1,1-dioxide
N-(3'-Methyl-1'-carboxybutyl)-2-nitrohenzenes~rlfonamide: A solution of 2-
nitrobenzenesulfonylchloride (11 g; 50 mmol) and NaOH (2.1 g; 53 mmol) in H20
(100 ml)
was added m-leucine (6.55 g; 50 mmol) and Is~ft over night with stirring at
rt. The reaction
mixture was added 4 M NaOH (12.5 ml) and filtered. The filtrate was acidified
with 1 M HCI
(50 ml) and extracted with EtOAc. The combined organic fractions were dried
(Na2S04) and
evaporated to dryness to give 6.7 g (42%) product.
3-Isobufyl-4-oxo-2,3,4,.5-tetrahydro-T,2,5-ben:~atrhiadiazepine-1,1-dioxide: A
stirred
suspension of N-(3'-methyl-1'-c;arboxybutyl)-2-nitrobenzencaulfonamide (6.7 g;
21.2 mmol)
and 10°ro Pd/C (200 mg) in abs. EaOH was hydrogenated at 1 bar. The
reaction mixture was
filtered through celite and evaporated to dryness. A solution of the crude
product in dry THF
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
103
(50 ml) at 0 °C was added N-hydroxysuccinimide (2.53 g; 22 mmol) and
DCC (4.54 g; 22
mmol). The reaction mixture was slowly warimed to rt. and left with stirring
over night. The
reaction mixture was filtered and the solid rnaierial was washed with THF. The
combined
organic fractions were evaporated to dryness. and the remanense added H20 and
extracted
with EtOAc. The combined organic fractions were dried (Na,2SO4) and evaporated
to dryness
to give 3 g (53%) product.
3-lsobutyl-8-piperidinos;ulfonyl-2,3,4,5-tetrahydra-1,2,5-benzathiadiazepine-
1,1-dioxide: A
solution of 3-isobutyl-4-oxo-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine-1,1-
dioxide (500 mg;
1.8 mmol) in dry THF (20 ml) under N2 at 0 °C ~rvas added a solution of
2M BH3~SMe2 (9.2
ml; 18 mmol) in THF. After complete .addition, the reaction mixture was warmed
to rt. and then
to reflux for 2h. The reaction mixture was cooled to rt. and carefully
quenched by addition of 6
M HCI (15 ml). The reaction mixture was made ;strongly alkaline by addition of
7.5 M NaOH
(aq.) and extracted by EtOAc. The combined organic fractions were dried
(Na2S04) and
evaporated to dryness to yield 320 mg (70'%) product. The product was
transformed by
Method A (using piperidine as amine). M.p. 2U~9-211 °C.
Compound 128
3-Cyclohexyl-8-(piperidinosulfonyl)-2,3,4,5-tetrahydro-1,2,5-
benzothiadiazepine-1,1-
dioxide
D~-Cyclohexylglycine: A solution of NaCN (15.91 c;; 0.32 mol) in H20 (60 ml)
was added NH4C1
{17 g; 0.32 mol) followE:d by a solution of cyc;lohexanecarboxaldehyde (37 ml;
0.31 mol) in
MeOH (60 ml). The reaction mixture was stirred vigorously for 2 h at rt. The
reaction mixture
was diluted with H20 (100 ml) and extracted with toluene (~:ex 70 ml). The
combined organic
phases were washed with H20 (2x 50 ml) and e~aracted with 6 M HCI (2x 90 ml).
The acidic
aqueous phase and th~a precipitate, which formed upon acidification, were
combined and
refluxed for 24 h. The reaction mixture was cooled to rt. and made slightly
alkaline (using
25% NH3 {aq.)). The prE:cipitate formed, was isolated by filtration, washed
with cold H20 and
air dried to yield 13 g (2fi°/a) of the free. amino acid.
Compound 128 was synthesized by the method used for compound 127 (using m-
cyclohexylglycine as amina acid).
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
104
Compound 129
3-Cyclohexyl-7-cyclopentylsulfinyl-1,2,3,4-tetrahydro-1,2,4-benzothiadiazine-
1,1-dioxide
2-Amino-5-cyclopentyh'hicbenzenesulfonamide: A mixture of 5-iodo-2-
aminobenzenesulfonarnide (1.192 g; 4 mmol), triethylamine (750 pl; 10 mmol),
Cul (76 mg;
0.4 mmol); cyclopentylrnercaptane (590 pl; 6 rnmol) and PdfPPh3)4 (462 mg; 0.4
mmol) in dry
dioxane (10 ml) under N2 was stirred in a screw cap ampule at 130 °C
over night. The
reaction mixture was coated to rt., diluted with H20, made alkaline (using 4 M
NaOH) and
filtered through celite. The filtrate was neutralized to pH 8.5 and evaporated
to dryness.
Column chromatography gave 327 mg (30 ~%) product.
3-Cyclohexyl-7-cyclopentylsulfin.yl-1,~?,3,4-tE~trah,ydro-7,2,4-
benzothiadiazine-1,1-dioxide
(129): 2-Amino-5-cyclopentylthiobenzenesulfonarnide was ring brominated (3-
position) and S-
oxidized under the conditions described by Ali and Bohnert (Synthesis, (1998)
1238), using 2
equivalents of Br2. The product was reduced at 1 bar using 5% Pd/C in 96% EtOH
and
transformed by Method G (using cyclohexar7ec;arboxaldehyde).
The following table summarises the compounds described:
CA 02320354 2000-08-04
WO 99/42456 PCTlDK99/00070
105
r1 Ov V~ ~ u.aN -
O
n
0 o
c; o ~ o ~ ~ o ~'
f (~
J
h 0 0
z z z o z z z o o z z c~
N
a
o. _ . . . . . .
a ~ ~ a ~ ~ C
cro ao o~ ~ro ~~ ac a4 m~asw ~ro p
a
x x ~ x x x x x x x
-~
t.
'
s ~ c..sn c.. N ~' ~ ~'n ~- w
cu x W ~ .-~~x ? ~ .".w
f ~ ~, 0. G ~ ~ X
' \
V ~c' ( n ~
7
~n
C C C
,
x x x ~ ~ a x ~ x x ~ ~ x x ~ x~
c c
_ - y _ ; c
3
o 0
C C C " C
C C ~ 1
3 C
G G GL iy O
x x x " x ~'
s_ ~ i x ~ x x x x x x ' W
C
~
.
. -t '
_ O. ?
- n
.._ - ~
n
~s .-
C ~ C ~ ~~ C','
~
~ \ 5 3 C
~ ~ x ~ x ~ x x x x x x
, _ l ~
~ C
C'. ,n
i
I
i
I ,
=' II I x x x x x x
I
x :~
~
i
x x x w I x x x x x x
:~
~I~I,
i
I
I
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
106
N N fJ fJ _ _ _ _
W N C',~ ~.u O~ J O~ Vv ~ W N C
Cn C/~ V1 (.n V7 fr'7 V7Cn C/7 (./~ V7 C/7
0 0 0 0 0 o a o 0 0 0 0
N N N ~J
N tJ r~.nN N N t.J N
z z z ~> :~ <: :zz z z z z n
N
cry cn cn cn cn c.~ ~ntn tn tn cn cn ,..,
_ a. C'
fr_~ cro_ tn_ no ti~a rrc'_dotro era oo crc cn
CD fL fD fD P fC ~,'DCD 6 (D fD ~ (D
~,
x x x ~- ~~ x: :xx x x x x x
<,
< ~< ~< ~- << y ~_ ~< .< < <
C_: n_ C_7 h C'J ~ C7 f7 r', f.
O C C O
C_ c:C
fD N CD J - ~~ - ~ ~ ~ ~' '-"
k x k >' ~ CDx ~ cro r fi
t
~< << ~ -< ~. ~ Jt X
... _ '< '< < v
_
__ _
x x x x x :~Ix x x x x
-,
i
x x x x x :~', x x x x
z
I
xl x x x :~ x x x x P
x .. x
I
I
_ / \ r N x
\ \ ~'_ ~ m > n io C7
/ ~ !, \
\ C>
-~' y
'~',
z:z
\ ~ ~ ; o ~~z,~ ,~o ~ 7,7
/ ~ " ~~ ' , s
o=~ , x
~
o
0
- -
l -
x x x x :C~x x x x x
x .~." i
~
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
107
W C.a W W ~ ~O OU tJ N N N
.F.. W tJ ~..7 OW n -F. O
C) 0 ~ ~ ~ ~ C~ ~ C) ~ 0
fJ CJ fJ IJ IIJ N CJ N fJ IJ N
z z z z z z 2 z z z z c
a'
ac ~ro uo ao ao cro Vo rro cro aro no O
v
c~ Wo iu cc in cu c'o co ~ ri ~ C
x x x x x x z x x x x
f: (~ (; f; C) f7 C: C C: n f7
~'G V ,< .,G 't < V
y
r: W C7 C: C7 W C: f: (~ C;
s
k x X ~ ~ k X x k ~ x
'G < 'G < <et '< ~~G '< <G '~G '<
x x x x x x x x x x x x
x x x x x x x x x x x x
x x w x x x x x x
t '~ o, /w,. -~ __ /- /=
'., i -
c7 z cn ,
~ Z
-' ~ z /-.:Z O .,
% '7' ' '7' ~ ~' ~, '/'-' c, z
~i
-- N
~~ x x x x! x x x x x x
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
1d)g
J O~ V .pW N ~
~a O vUo0,.1 a c.n O
Cn
O ~ o o ~ ~ o ~~o o .
0 0 0
N N tJ tJO pJI~.y,n h.>N Iv
N N N
1
z z. z z z z :y ! z z z z z N
z
cn tn v. m rnvi:n~ n y cn_
y y . ~ ~._.. :;. : . . ~ v; tn ,..,
. Q.
0~'= Or0 ~ a_ocro_rro_~ ~t'a_ p _ fro~ Qq ~~ -
h_a rro 0'OOO a o
o
_ _
!D CD (D fDCD(L~J!D ~ .'fDfGCD (D CD ~
~_--~ ~~ S~.
-_
.
x x x x x x:x :c ~ :xx x x x x
'< '< '-G~GK v y y%n n n !~ n
I
'G Y
!~ ~ r,c~n rS~i ~ o ~ !;
~
.U.z' ~- O O O cjC) O C C '~' . C
x J T
S
S ~T
C'J N w f (DcDo CD (D(D
G
k k Y~.~<>: >K~e CD
4 ~!
-.
x x x~ x x x x :~x x x x x
I
x x x'x xi w x :cx x x x x
~
', x
__
._ .
xx x x 3; ::x x x x x 7P
i x
i
I
-i __-
-
U
i Z Z z
x ~
t .'.~T.~ ~ O '''t'xfew Sz =zz'Z
-z z n i~ ~z
; z ~
~
,.o = ,,o~ ~ \ y o -~ ~ ~-J
cn\ cns ~ ~ NOc~=:c7 w rJO il _a
~ " ~, O '
% O \
O ,-r
'~ > . N '~ z
c '~', ~.f, 1~
o o
N
~ z x
_i
f
x x x x ~~x = x x x i x
x :x: ~: x ~
i ~
i
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
109
~"fT tin .p t~.. ~ ~ h O
O o
o
a o o a
a o 0 0 o a
N N IJ N IJ N N
:z z z z l z z z z a
._
I10 RA 00 00 f10 OC 00 "~,$
i rn ~
~ ro
x x x x x z x x x
~ .o .
o ~ .Q c
~ ~
T . , .~ z
~ b
n ~ ~ o .
b x
x x x~ ', x x x x z
xi
x
- -_ ~ r.' _
i -
a
x x x x~ x x z x
x
i
i
x x x x~ x x z
x
x
-. I
i
I
--.
_ _ .
m
,> %
z v ~ ~ m-z
' ~ ~
v ~o ~ ~O z .V _ _z ~ - --
v ,o z ~ z ~ ~ G~ J
_ z ~ O ~O
o '
~C~
Cn\ (17 lO (n. / , ; , '
o ' (O' ~ ~ '
~o , ~ ~
~
'
t~
~ ~~ ~ ~ .o 7
oi o ~o ~
~y.,
f~
7
i x x, x x x x x
xi x
x
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
~_ 7. 0
.i,w N ~~ C~ v,
C ~" ~ ~7 C
_- 7
n ~ ~ .~ ~ -
Il Q O -' ~ v; cry '
COI
O ~' ~' fJ rJ ~ O ~ o G
~ IJ CJ rJ fJ
I ._
z z z' r, '<'-z z z n
z N
I
c G _ ~ L~ (/ y:
~
f; ~ be -~ - - -: _
~ i rr_ rr~ rr' rr rr_: rrj ,,.C
I C . r ~
'G r~:- -
: -
c ~; ~; r:G
I ~ N
r: r,
C~ ;.a,- C I !
_ _
' Y r ; ~ ~ r: ..
" i ~: p
i
<<. ! f k ' v
;. . - v
=;
_-. _.--~. __. .
I
I,
_ = x ~- f ~- z
~ -'
I ~
i
I ~, _ _
i
I
.-
1
~
i
W
x w x~ H
J' C
I
-
n
~~ z ~ ---~ _
, , J\
z - C
~-z ~ - '
c
_ !-
in~ ~ % 'v ~ __z ~~z ~-
p _ O
p, ~; ,__ _ _z~ ~ ~p , ~~C ~,
~c,~ _1. _ , cn ;n.' ~o
' p '
G
'r 'j-, 4;.;
in~p rn; ,,~~p .~ ~C ~
, O o
' '~ y
i \p .0
.L ,
i r.
SUBS11TUT'E SHEET' (RULE 26)
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
111
J J ~J J ~.1 ~1 -..1
CT In ~~ W ~
CJ C~ ~O Cb CT O
c ~ ~' ~ ~ n n
0 o > o o ~'
o
f 0 0 0 0
z z :r z z z :y z z z z
o ~ ~ ~i c c c c n.
o a a n. c c a c c
o n. c n.
c ' 0 ~ a 0. c
- o ' c
'
(D (u W rn rv ~ ( ( c - ~
a_ _ o cr ~ o D y' o o r fD ~
.- ~ a' a
'
fD fD C~ CD ~ ~' " (D ~ _ _ ~
(b CD (G T' ~' (D
f ,ry
D
z ~ ' ~ ~ z ~~ z x z ~s x
~n x r7 ~ ~ ~n : ~ ~
'
r1 ~ ~ ~ ~
w" f rr r',
~ ~a ~D ..
x z ~c z ~ x r x x x x x
x x ~,: x x x :~ x x x x
x x ~ x x x x z x x x
n x x ~
m t ~ ~ ;' r.x
-Z z -<_ z z ___.~ ~ x
~ z
_ _ ~ ~ ~ xz _._z O cm
v s .
i
t
i~ n\ C O v~ \
' /O
~f,~'~~ ~.m v
(7
W
x x ~~ x x x := x :r x x x
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
112
!
z
a. cn ~. I w t.; o .c. oo J c
v~ c~ rn ! v: u. cn v~ co cn cn
O O O ' O C O O O O O
N ' N N tJ N N IJ tJ N N
z z z z :~ z z r z z
N
p ' c_n ~ ~ ~ cn v~ cn cm cn cn C a ,." C'
G ,~. ~ __ ~ O
00 00 CD d' 00 Cfa Op (Tf, ~ (14 ~ OQ ~ (: ~' ,~J
fC CD '-" ry (D iD~ C'~, N CA C4
x~~ ~ ~ 'x _ ~ ._ x . x x x z x
!
1~
n W ~ lV n o t7 n p;
~' ,,p Cue. t~ x '~ ~ C C O C C C W n
CD ~~..1 CC ~~l X X (S CSD N CSG ~ T,,
X X C. Y~ X Y 7<
'< ~ ~C :Z ;i;' ~ '< '< '< 'C 'G C':
rn ' (n C I C
c c c' ; c
c L-
y i .
~ c y c
:_ c. i _
x ~ x ,; ~x c. ~ x :_~ x x x x ~ x x
w t1 L_ ''' n A
t~ m
c c
O n ~ - _ ._
a
c
c
o ;~ c c
x G x !~ ~ $ x x x x x x ~ x
-,
.; '~ ~ .;
a c a
c '
.O. C_
i~ ~ n
C" tT C ~ C
x ~ x .~ x ~ ~ x x. x x ~- x ~, x
a i~,
tn In
A
a
_ .
.,~..
i
o ( o ~x x. x x x x
o
i
f i ,x ,~; _z yr ~ o ~ p O ~~ -,-
x x ~ n .r
i x
i
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
113
z
O~ tn t~ G~ t~> O vG oc ~ p
0 0 o c o o c o 0
ro la r~ r c r~ ro m r.~
'~
z z ~: ~: z: z z z z z ~?
._
j fJ IJ N c rJ y ~ ~, Cn (/~ '~
~ C ~
O
n_c n a a w p w no as cro rra
p p' ~ C
W '
c n rc r~ co ~ ~, a ~ ~
o
i
I , x x b x ~?
i zI
x
-_ -
I
~J1 1~. v.
7 I ~
n ~~ ~SS ~~' ~ U ~~ ~
'~ ~' ~'
T
C'
n m
r:
x C G ~
G
, c
n n
p -_~ ~ p
p G
C
~'
x x x z x z x~ x x
i i
__ ~ i .
j' O
v 1
n I
'
x x x x a x x x x: x x~
~
v G
V: I
I
_ _ ~~ ~ n
w x
_ '
_. ,
\ ~
x _z, ~,_..~_=z ~-~_ _-' _ z ~ x
w o xz s ~~\ ~ ~ =~: o cn O
C z
o
~ \ \ ' ~ o ~
r.~,~o,o ~, ~, ~ f ~ ~~o ro z
~<_ o ~ " ~.~o'
~ , '
\
'-,
o
I
I
x I x x I x x x
... i
x~ I
I
i
SUBSTITUTE= SliEFT (RULE 26)
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
~~ 14
O ~C oOC ~ ~ i,m .C W N O p ~ O~C J C
C/7 Cl~ Vi V7 Vl Vy/~
Q Q Q Q Q C~ Q
r.~ r,~ m r~ r.~ r..~ r~ x n~ ~ ~ 0 n
-_I .__ .
z~z ~ z z z z z z z
c a '~ -_ - ~- ._
~ o cn ;n rn ~ cn yr cn v~ ~ ~
c c ~. _ 'o' _ ~. = c _
a- rro rlw (JO rro v0 W cro ~ Uo rm r~ ~ _, ~ ~ =' < o
co rn o ~~ ~ ~ ~ ~ ~ ~ ~ ~ ~ v~ ~
x x x xix x x ,x x xx x x
I
' -- - --_ _
p kn
'' c
c
'.,
a x x z x~ x x :~x x x x x x
o ~_-~-- ___.~_ _
c
i
to r~ m ~ tx t~ ~c w to tz~ b y Z co TJ
r x ~' n
v~ -
Q, ~ w v,
r~
'
x~ x x z z; x a: x x x x x x
.'T'~, x m_
r~ r 'U
CU C7 ~ I~7 07 t~ 07 G; C7 2 ('~ Z ~ m-z/ IJ
,< ... cn s
'-~ c/~ ~ -. v ,O
Q
x x :~ x ~. w x. x ~ x x
j i
SUBSTITUTE SHEET {RULE 26)
CA 02320354 2000-08-04
WO 99/42456 PCT/DK99/00070
115
tJ tJ -- .~ ._, '_' _ -- .._,_
O vp oo ~1 0 O
v~ t~ i.ar.~
(n cn (~ cn n m ''' (r~ r.~
(n
0 (~
I J tJ rJ IJ
z z z z z z z z z z z
N
V7 (/7(n N ~ _._- .-
_ </: Cn V'1(n (/J
7 ~
~ ~ rr
ao vo rro ~ oa rro rro .~ ~ ~ LT
' ~ O
~ r~ ~ r ~ ~ ~ p
o r,
_
ii ~ ~=
C
x .~,x ~, .~ x x S. .~.~ x N
~.. N
~-
f7 ( y
r ~L7~ i r '~ k < 't ~; C;
<' ~
W i C r ~ ~' r t7
7 . - c r -
~ ;
J G ~ p _ ; r W
~ S ' ~l s C ~ C. O %
GO n C C,
~< ~ C ~
~
fir. N x .~ ~ D fD r, ~ ~ w
~< ~< < '
'~ << ,i ~< <t y .=.
_.
~
rn C
- -_ _ . C
.
C
x x x x x ;~ x x
w x ~ x
n
o . -~ _
.-
0 3
0
x x x x
F '~ x x x xi x r~
_
S
i
I ~f
.-- -~ 1
~._ .
n ~
a ~ ~ ~-~ n ~ n ~a n ~
x ~ ~ a
~ x G
I v'
o~
~\
(J
O _
_
.
p_
r x x ( ~ x z\ w
r 'z
N
. v z z'
= I
,a \ , _ \, ~ i
O U ~, ' ~~ _z ~ Y ,~z a
~~~ a \ .c) r
i
p ~~~, Q , ~
U
z x x x x ~: x ~x x x x
SUi3STITUTE SEiEET (RULE 26)
<IMG>