Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
PROPYLENE COPOLYMERS CONTAINING STYRENE UNITS
The present invention relates to random copolymers of propylene as main
recurring
units comprising recurring units deriving from styrene. The present invention
also relates to
functionalized copolymers and graft copolymers. The invention, moreover,
relates to
processes for the production of said copolymers.
The present invention lies in the technical field of the production of
thermoplastic
materials.
As it is well known, plastic materials based on isotactic polypropylene are
among the
most interesting ones from the technology viewpoint. In fact, they are not
only competitive
from a cost perspective, but are also suitable for various applications due to
suitable
chemical and physical modifications.
The chemical modification mostly used in the industry is the random
copolymerisation of propylene with small amounts of one or more comonomer(s),
generally
ethylene or butene-1. Said modification allows to obtain materials that have a
lower melting
temperature (above all used for producing films with thermoweldable layers),
lower
stiffness, higher impact resistance at low temperatures and a higher
transparency than the
isotactic propylene homopolymer.
The above mentioned variations in physical properties with respect to the
homopolymer are due to lower crystallinity and smaller size of crystallites
caused by the
comonomer units.
It is worth noting that ethylene and butene-1 recurring units have a sterical
hindrance
similar enough to propylene recurring units. Consequently, although they cause
a decrease in
packing energy, they are partially enclosed as defects in the crystalline
phase. As it is well
known, generally speaking, in semicrystalline polymeric materials one obtains
a more
efficient decrease in crystallinity and size of crystallites when one uses
comonomer units
with much higher hindrance than the basic monomer units, i.e. such that they
have inevitably
to be excluded from the crystalline phase.
In this connection, there is however the problem that hindered and cheap
comonomers, such as styrene, are not easily copolymerisable with propylene,
because
generally catalytic sites suitable for the isotactic polymerisation of
propylene are not capable
1
CONFIRMATION COPY
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
of polymerising styrene and vice-versa. In fact, generally speaking, catalytic
systems suitable
for the polymerisation of 1-alkenes to isotactic polymers, such as metallocene-
and
methylalumoxane-based catalysts, are not capable of polymerising styrene. On
the contrary,
styrene tends to act as a poison in such processes. It is worth noting that in
case of
heterogeneous catalysts, which typically contain different types of catalytic
sites, it is
possible to polymerise mixture of the said two monomers but mixtures of the
two
homopolymers are mostly obtained.
Another disadvantage of known random copolymers based on propylene produced by
heterogeneous catalysts is that macromolecules do not have a homogenous
content of the
cornonomer units, so that the fractions with a higher comonomer content are
more easily
extractable with solvents. This evidently limits their use for preparing
articles to be used in
contact with foods.
European patent application EP-A-872 492 discloses catalytic systems based on
stereorigid metallocenes that contain a metallic atom belonging to the IV
group of the
Periodic Table and whose substituted cyclopentadienyl groups are bridged
through a single
atom. Said metallocenes are capable of copolymerising olefins with vinyl
aromatic
compounds. As disclosed in the patent application, such catalyst systems,
however, produce
copolymers containing blocks of styrene units. This is, for instance, shown by
the Nuclear
Magnetic Resonance spectrum of Figure 29, therein.
It has now been produced a random copolymer of propylene that has a
homogeneous
distribution of recurring units deriving from styrene in the polymer chain.
Thanks to the homogeneous distribution of the styrene recurring units in the
polymer
chain, the copolymers of the instant invention essentially show no variation
of the glass
transition temperature compared with isotactic polypropylene. For example, in
the case of
propylene copolymers of the present invention, no increase of the glass
transition
temperature higher than 10° C compared with isotactic polypropylene is
observed, e.g. if TK
is measured by Differential Scansion Calorimetry at a rate of 10° K per
minute, its value
does not exceed 0° C.
It is important to note that possible random copolymers of propylene with
styrene or
substituted styrenes would present a substantial increase of the glass
transition temperature
2
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
(T~) in comparison with the propylene homopolymer, approximately according to
the Fox
relation:
1 /Tb = WP~oP/Tg P~~ + WS,y,JT~ scyr
where WP~oP and WS~,~ are respectively propylene and styrene fractions by
weight and Tg ~oP
and T~ S,y~ are respectively the glass transition temperatures of
polypropylene and polystyrene
homopolymers. Since the glass transition temperatures of the styrene polymers
are much
higher than that of polypropylene and the molecular mass of the styrenic units
is much Larger
than that of the propylene unit, a substantial increase of the glass
transition temperature
should be observed also in cases of a low content by mole of the styrenic
units and this
would make said materials unusable in applications which demand a low running
temperature.
As another advantage, some of the copolymers of the present invention can be
used
for preparing functionaIized polypropylene as well as graft copolymers.
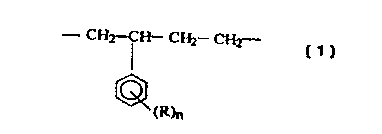
The present invention provides, therefore, new isotactic-polypropylene-based
copolymers having a homogenous distribution of recurring units of the formula
( 1 ):
- CH2-CH- CH2 CHz-
(R)n
where R is a hydrogen, halide radical or a hydrocarbyl radical optionally
containing an atom
selected from oxygen, nitrogen, sulphur, phosphorus and silicon and n is an
integer ranging
from 1 to 3.
The copolymers of the present invention contain the recurring units of formula
( 1 )
preferably in amounts ranging from 0.1 to 30% by weight.
Said copolymers have a '3C-NMR spectrum wherein the resonance signals
attributable to the links between different monomeric units fall around 30,
34, 35, 45 and 47
ppm and present intensities at least 2 times higher than the resonance signals
attributable to
styrene-styrene sequences around 41 ppm and 44-46 ppm (all chemical shifts are
relative to
tetramethylsilane). In particular, for the case R of formula ( 1 ) is
hydrogen, that is for styrene-
ethene comonomer units, the resonance signals attributable to the links
between different
monomeric units fall at 30.3, 33.9, 34.6, 44.8, 46.9 ppm.
3
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
The polymerisation degree of the copolymers of the present invention is
normally at
least 50.
When R is a substituent containing carbon atoms, it can be selected from C,-
C,o alkyl
radicals, linear or branched, C,-C,o cycloalkyl radicals and C6-Cz°
aryl radicals. The alkyl
radicals may be saturated or unsaturated radicals. The preferred radicals are
metyl, ethyl,
isopropyl, vinyl and allyl radicals.
Said substituent R may contain a functional group, such as -NRz, where R is an
alkyl
group as above defined.
Preferably the sequences of propylene recurring units are mainly isotactic.
Generally,
the content of meso diads (m) is higher than 80%.
The amount of the structural units of formula ( 1 ) in the copolymer may be
determined on the basis of the intensity of specific signals in the "C nuclear
magnetic
resonance spectra. For example, in the case of propylene copolymers with
styrene the
presence of said structural units is put in evidence by signals in the
aliphatic region at 33.9
and 25.2 ppm (chemical shift from tetramethylsilane, TMS) and the molar
fraction of the
styrenic units {XS), equal to the molar fraction of the connected ethylenic
units, can be
obtained by the following relation:
0. SA33.9+A25.2
XS -
{O.SA33.~+AZS.z) + (~.SA33.9+A25.2+A24.4) + {A44.8+0.SA34.6+A45.4+0.SA36.9)
where Ax is the intensity of the signal at x ppm.
Depending on the polymerisation conditions, random copolymers with various
compositions and polymerisation grades are obtained. In general the average
molecular
weight MW is between 3,000 and 1,000,000.
As said above, the copolymers of the invention have a homogeneous distribution
of
the comonomers. Such homogeneity is also proved by the impossibility by
solvent extraction
to obtain fractions of the copolymers with a XS value differing more than SO%
from the XS
value of the unfractionated sample.
4
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
The copolymers of the instant invention can be obtained according to the known
polymerisation methods.
The copolymers can be produced by means of the homogeneous catalytic systems
used for the insertion polymerization of propylene which give an isotactic
homopolymer, e.
g. catalytic systems based on metallocene compounds. Suitable examples of said
metallocene
compounds are rac-ethylene-bis(1-indenyl)-ZrCl2, rac-isopropylidene-bis(1-
indenyl)-ZrCl2,
rac-dimethylsilyl-bis(1-indenyl)-ZrCI,, rac-dimethylsilyl-bis(2-methyl-I-
indenyl)-ZrCl2, rac-
dimethylsilyl-bis(2-methyl-4-isopropyl-1-indenyl)-ZrCl2, rac-dimethylsilyl-
bis(2-methyl-4-
phenyl-1-indenyl)-ZrClz, rac-dimethylsilyl-bis(2-methyl-bent[e]-1-indenyl)-
ZrClz, rac-
dimethylsilyl-bis(benz[e]-1-indenyl)-ZrCI, .
Suitable activating cocatalyst according to the process of the invention are
alumoxanes
or compounds capable of forming an alkyl metallocene cation.
Alumoxane useful as cocatalyst may be linear alumoxanes of formula (2):
R
R\Al-O--Al-O Al~
R~~ ~R
Y
wherein R' is selected from the group consisting of halogen, linear or
branched, saturated or
unsaturated C,-CZO alkyl, C,-CZO cycloalkyl, C6 CZO aryl, C7-CZO alkylaryl and
C7-Czo arylalkyl
radicals and y ranges from 0 to 40;
or cyclic alumoxanes of formula (3):
R
~ 1 O
-
Y
wherein R' has the meaning herein described and y is an integer ranging from 2
to 40.
The above alumoxanes may be obtained according to procedures known in the
state
of the art, by reacting water with an organo-aluminum compound of formula
A1R'3 or AIZR'6,
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
with the condition that at least one R' is not halogen. In this case, the
molar ratios of
AI/water in the reaction are comprised between 1:1 and 100:1. Particularly
suitable are the
organometallic aluminum compounds of formula (II) described in EP 0 575 875
and those of
formula (II) described in WO 96/02580. Moreover, suitable cocatalysts are
those described
in WO 99/21899 and in the European patent app. no. 99203110.4.
The molar ratio between aluminum and the metal of the metallocene is comprised
between about 10:1 and about 5000:1, and preferably between about 100:1 and
about 4000:1.
Examples of alumoxanes suitable as activating cocatalysts in the process of
the
invention are methylalumoxane (MAO), tetra-isobutyl-alumoxane (TIBAO), tetra-
2,4,4-
trimethylpentylalumoxane (TIOAO) and tetra-2-methyl-pentylalumoxane. Mixtures
of
different alumoxanes can also be used.
Not limiting examples of aluminum compounds of formula A1R8, or AIZRgb are:
tris(methyl)aluminum, tris(isobutyl)aluminum, tris(isooctyl)aluminum,
bis(isobutyl)aluminum
hydride, methyl-bis(isobutyl)aluminum, dimethyl(isobutyl)aluminum,
tris(isohexyl)aluminum,
ris(benzyl)aluminum, tris(tolyl)aluminum, tris(2,4,4-trimethylpentyl)aluminum,
bis(2,4,4-trimethylpentyl)aluminum hydride, isobutyl-bis(2-phenyl-
propyl)aluminum,
diisobutyl-(2-phenyl-propyl)aluminum, isobutyl-bis(2,4,4-trimethyl-
pentyl)aluminum,
diisobutyl-(2,4,4-trimethyl-pentyl)aluminum, Ms(2,3-dimethyl-hexyl)aluminum,
tris(2,3,3-
trimethyl-butyl)aluminum, tris(2,3-dimethyl-butyl)aluminum, tris(2,3-dimethyl-
pentyl)aluminum, tris(2-methyl-3-ethyl-pentyl)aluminum, tris(2-ethyl-3-methyl-
butyl)aluminum, tris(2-ethyl-3-methyl-pentyl)aluminum, tris(2-isopropyl-3-
rnethyl-
butyl)aluminum and tris(2,4-dimethyl-heptyl)aluminum.
Particularly preferred aluminum compounds are trimethylaluminum {TMA),
tris(2,4,4-trimethylpentyl) aluminum (TIOA), triisobutylaluminum (TIBA),
tris(2,3,3-
trimethyl-butyl)aluminum and tris(2,3-dimethyl-butyl)aluminum.
Mixtures of different organometallic aluminum compounds and/or alumoxanes can
also
be used.
In the catalyst system used in the process of the invention, both said
metallocene and
said alumoxane can be pre-reacted with an organometallic aluminum compound of
formula
A1R'3 or A12R'6, wherein R' has the meaning reported above.
6
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
Further activating cocatalysts suitable in the catalysts of the invention are
those
compounds capable of forming an alkylmetallocene cation. Examples are boron
compounds
tetrakis-pentafluorophenyl-borate is particularly preferred. Moreover,
compounds of formula
BAr3 can be conveniently used.
The catalysts of the present invention can also be used on an inert support,
by
depositing the metallocene, or the reaction product of the metallocene with
the cocatalyst, or
the cocatalyst and successively the metallocene, on the inert support, such as
silica, alumina,
magnesium halides, olefin polymers or prepolymers (i.e. polyethylenes,
polypropylenes or
styrene-divinylbenzene copolymers). The thus obtained supported catalyst
system, optionally
in the presence of alkylaluminum compounds, either untreated or pre-reacted
with water, can
be usefully employed in gas-phase polymerization processes. The solid compound
so obtained,
in combination with further addition of the alkyl aluminum compound as such or
prereacted
with water, is usefully employed in gas phase polymerization.
The molecular weight of the polymers can be varied by changing the
polymerization
temperature or the type or the concentration of the catalyst components, or by
using molecular
weight regulators, such as hydrogen, as well-known in the state of the art.
The polymerization process according to the present invention can be carried
out in
gaseous phase or in liquid phase, optionally in the presence of an inert
hydrocarbon solvent
either aromatic (such as toluene), or aliphatic (such as propane, hexane,
heptane, isobutane and
cyclohexane).
The polymerization temperature ranges from about 0° to about
250° C, preferably from
20° to 150° C, and more preferably from 40° to 90°
C.
The molecular weight distribution can be varied by using mixtures of different
metallocenes or by carrying out the polymerization in various steps differing
in the
polymerization temperature and/or in the concentration of the polymerization
monomers.
The polymerization yield depends on the purity of metallocenes in the
catalyst; the
metallocene according to the present invention may be used as such or may be
previously
subjected to purification treatments.
The metallocene and cocatalyst may be suitably contacted among them before the
polymerization. The contact time may be comprised between 1 and 60 minutes,
preferably
7
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
between 5 and 20 minutes. The pre-contact concentrations for the metallocene
are comprised
between 10''- and 10-g mol/1, whereas for the cocatalyst they are comprised
between 10 and 10'3
mol/1. The precontact is generally carned out in the presence of a hydrocarbon
solvent and,
optionally, of small amounts of monomer.
The copolymerization of propylene and styrene is carried out in the presence
of small
amounts of ethylene. In particular, propylene concentration may be between 0.1
M and 13
M, styrene concentration between 10'3 M and 9 M, ethylene concentration less
than one tenth
of the propylene concentration, catalyst concentration between 10-8 M and 10'z
M. The
polymerisation temperature is between -30° C and +1 SO° C,
preferably between 0° C and
100° C.
The copolymers of the present invention can be blended with other polymers,
preferably with isotactic propylene polymers.
Such polymer blends can be prepared by a mechanical blend of the polymers, at
least
at the soft temperature, preferably at the melting temperature, of the
polymers.
Alternatively, the blend can be carried out by way of a polymerisation that
can be
carried out in at least two sequential steps, wherein the polymers are
prepared in separate
subsequent steps, operating in each steps, except in the first step, in the
presence of the
polymer formed in the preceding step. The catalyst can be the same in all the
steps or
different. For instance, a Ziegler-Natta catalyst can be used in the first
step, while said
homogeneous catalyst systems can be used in the subsequent step(s).
The present invention also provides functionalized copolymers. As said above,
the
copolymers of the present invention are particularly useful for producing
functionalized
copolymers, which are technologically important to improve their adhesion to
and
compatibility with other materials. In fact, the comonomeric units of formula
( 1 ) can be
functionalized under various free radical, anionic and cationic processes, as
described in the
open and patent literature for random copolymers between ethene and styrene or
substituted
styrenes. For instance, benzylic protons can be oxidated, halogenated or
metallated, to form
desirable functional groups (COOH, CHzX , and CHzMt, respectively) bonded to
the phenyl
rings.
8
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
Moreover, benzylic protons can be interconverted to stable anionic initiators
for graft
polymerizations. In particular, the metallated polymer (mainly lithiated) can
be suspended in
an inert organic diluent before addition of monomers, such as styrene,
substituted styrenes,
vinyl acetate, methylacrylate, methylmethacrylate, acrylonitrile. This
procedure can be
particularly relevant for the preparation of graft copolymers presenting
polystyrene branches
onto isotactic polypropylene backbones.
Another object of the present invention is the graft copolymers comprising
isotactic-
polypropylene-based copolymers as backbones. Examples of graft copolymers of
the present
invention are polystyrene or polyvinylacetate or polymethacrylate or
polymethylmethacrylate or polyacrylonitrile grafted onto isotactic
polypropylene.
Said graft copolymers may be obtained by the above-mentioned method.
These graft copolymers are mainly useful as compatibilizers in the preparation
of
normally incompatible polymer blends or alloys.
Examples of polymers to be blended with the propylene polymers in the presence
of
the graft copolymers are polystyrene, polyether, polyacrylate, such as
polymethylacrylate.
The following examples are given just to illustrate the instant invention and
not to
limit its scope.
Example 1
In a three necks 100 mL pyrex glass flask kept at -25°C are introduced,
in nitrogen
atmosphere, in the order: styrene (30 mL) and methylalumoxane (MAO) (300 mg);
after
removing the nitrogen, the liquid phase is saturated by causing a
propylene/ethylene mixture
(20/1 mol/mol) to bubble at atmospheric pressure, setting the flow at 0.3 L
per minute.
The reaction is started by injecting in the flask 3 mg of rac-ethylene-bis(1-
indenyl)
ZrCl2 catalyst dissolved in 2 mL of anhydrous toluene.
After a reaction time of 4 hours, the produced polymer is coagulated in 200 mL
of
ethanol acidified with HCI, filtered and dried in a vacuum oven.
The yield is of about 120 mg.
From the '3C-NMR analysis (Figs. 1 and 1 B) the product results to be composed
of
76% by moles of propylene units and 12% by moles of styrene units and 12% by
moles of
associated ethylene units (X, = 0.12), while significant amounts of blocks of
styrene units are
9
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
not observed. Fig. I B shows that the resonance signal at about 4I ppm, which
are
attributable to the styrene-styrene sequences, is nearly 6 times less intense
than the resonance
signal at 30.3 ppm and nearly 3 times less intense than the resonance signals
at 33.9, 34.6,
44.8, 46.9 ppm, all attributable to the links between different monomeric
units. This fact
confirms the statistical nature of the obtained product.
From the differential scanning calorimetric analysis, carried out ~.uth a
scanning rate
of 10 K/min., the polymer results to be characterised by a melting temperature
of 79° C (DH f
= 10 J/g) and a T~ ~ -9°C.
The weight average molecular mass measured by gel permeation chromatography is
of 3 x 103 u.m.a.
Comparative Example 1
A propylene-styrene polymerization is carried out with a catalyst system of
the same
kind as those described in the previously cited Arai et al. patent.
In a three necks 100 mL pyrex glass flask kept at 50° C are introduced,
in nitrogen
atmosphere, in the order: toluene (30 mL), styrene (5 mL), methylalumoxane
(MAO) (600
mg) and triisobutylaluminum (0.4 mL); after removing the nitrogen, the liquid
phase is
saturated by causing propylene to bubble at atmospheric pressure, setting the
flow at 0.3 L
per minute.
The reaction is started by injecting in the flask S mg of rac-isopropylidene-
bis(1-
indenyl) ZrCl2 catalyst dissolved in 2 mL of anhydrous toluene.
After a reaction time of 4 hours, the produced polymer is coagulated in 200 mL
of
ethanol acidified with HCI, filtered and dried in a vacuum oven.
The yield is of about 320 mg.
From the '3C-NMR analysis (Figs. 2 and 2B) the product results to be composed
of
13% by moles of styrene units.
Fig. 2B shows that the resonance signals at about 41 and 43 ppm, which are
attributable to the styrene-styrene sequences, have intensities comparable to
those of the
resonance signals attributable to the links between different monomeric units,
falling in the
region from 30 to 38 ppm, notwithstanding the fact that the content of styrene
units is lower
than in the product of Example 1 (8% vs. 12%). This fact indicates the
presence of blocks in
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
the polymer.
Example 2
The catalyst system employed, the operating method and the reaction conditions
are
identical with those of Example 1, except for the amounts of styrene (10 mL)
and toluene (20
mL) employed.
The yield is of about 200 mg.
From the '3C-NMR analysis the product results to be composed of 88% by moles
of
propylene units and 6% by moles of styrene units and 6% by moles of associated
ethylene
units (XS = 0.06).
From the differential scanning calorimetric analysis (DSC) the polymer results
to be
characterised by a melting temperature of 103° C (OHM = 24 J/g) and a
Ts ~ -13° C.
Example 3
The reaction is carried out at 0° C in a 250 mL autoclave containing
50 mL of
toluene, 3.2 ml of styrene, 0.9 g of MAO and 9 mg of the same catalyst
employed in
Examples 1 and 2, by feeding a gaseous mixture of ethylene/propylene ( 1 /4
mol/mol) at 2
atmospheres.
The reaction is stopped after 1 hour and about 600 mg of product is obtained
(conversion < 5%).
From the "C-NMR analysis the sample results to be characterised by a content
of
styrene units of 3% by moles (XS = 0.03), while the content of ethylene units
is of 7% and is
distributed among sequences of ethylene units contiguous to styrene units (3%)
and
sequences of ethylene units comprised between propylene units (4%).
The melting temperature of the polymer is 124° C (OHf = 53 J/g) and the
T~ ~ -17° C.
The homogeneous distribution of the comonomers is confirmed by the extraction
tests with hydrocarbon solvents: the polymer is fully soluble in boiling
hexane and fully
insoluble in boiling ethyl ether.
Example 4
In a three necks 100 mL pyrex glass flask kept at -25° C are
introduced, in nitrogen
atmosphere, in the order: toluene (28 mL), p-methyl-styrene (2 mL) and
methylalumoxane
(MAO) (460 mg); after removing the nitrogen, the liquid phase is saturated by
causing a
11
CA 02327781 2000-10-06
WO 00/47643 PCT/EP00/01219
propylene/ethylene mixture (124/1 mol/mol) to bubble at atmospheric pressure,
setting the
flow at 0.3 L per minute.
The reaction is started by injecting in the flask 6 mg of rac-ethylene-bis(1-
indenyl)
ZrCl2 catalyst dissolved in 2 mL of anhydrous toluene.
After a reaction time of 3 hours, the produced polymer is coagulated in 200 mL
of
ethanol acidified with HCI, filtered and dried in a vacuum oven.
The yield is of about 500 mg.
From the "C-NMR analysis the polymer results to consist essentially of
isotactic
polypropylene and to contain 0.7% by moles of p-methyl-styrene units and 0.9%
by moles of
ethylene units (of which 0.7% are associated with the p-methyl-styrene units,
XS = 0.07).
From the differential scanning calorimetric analysis, carried out with a
scanning rate
of 10 K/min, the polymer results to be characterised by a melting temperature
of 134° C (OHf
= 85 J/g).
It is interesting to note that the isotactic polypropylene, obtained under the
same
conditions with this catalyst system, shows a melting temperature of 151
° C (OHf = 95 J/g).
Example 5
The catalyst system employed, the operating method and the reaction conditions
are
identical with those of Example 4, except for the fact that divinyl benzene is
used instead of
p-methyl-styrene and the composition of the propylene/ethylene mixture is 75/1
mol/mol.
The yield is of about 500 mg.
From the '3C-NMR analysis the polymer results to consist essentially of
isotactic
polypropylene and to contain 0.7% by moles divinylbenzene units and 0.7% by
moles of
associated ethylene units (XS = 0.007).
From the differential scanning calorimetric analysis, carried out with a
scanning rate
of 10 KJmin, the polymer results to be characterised by a melting temperature
of 131 ° C (OH f
= 90 J/g).
12