Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-1-
OXAZOLE-ARYL-CARBOXYLIC ACIDS USEFUL IN THE
TREATMENT OF INSULIN RESISTANCE AND HYPERGLYCEMIA
BACKGROUND OF THE INVENTION
The prevalence of insulin resistance in glucose intolerant subjects has long
been
recognized. Reaven et al (American Journal of Medicine 1976, 60, 80) used a
continuous infusion of glucose and insulin (insulin/glucose clamp technique)
and oral
glucose tolerance tests to demonstrate that insulin resistance existed in a
diverse group
of nonobese, nonketotic subjects. These subjects ranged from borderline
glucose
tolerant to overt, fasting hyperglycemia. The diabetic groups in these studies
included
both insulin dependent (IDDM) and noninsulin dependent (1VIDDM) subjects.
Coincident with sustained insulin resistance is the more easily determined
hyperinsulinemia, which can be measured by accurate determination of
circulating
plasma insulin concentration in the plasma of subjects. Hyperinsulinemia can
be
present as a result of insulin resistance, such as is in obese and/or diabetic
(NIDDM)
subjects and/or glucose intolerant subjects, or in 1DDM subjects, as a
consequence of
over injection of insulin compared with normal physiological release of the
hormone by
the endocrine pancreas.
The association of hyperinsulinemia with obesity and with ischemic diseases of
the large blood vessels (e.g. atherosclerosis) has been well established by
numerous
experimental, clinical and epidemiological studies (summarized by Stout,
Metabolism
1985, 34, 7, and in more detail by Pyorala et al, DiabeteslMetabolism Reviews
1987,
3, 463). Statistically significant plasma insulin elevations at 1 and 2 hours
after oral
glucose load correlates with an increased risk of coronary heart disease.
Since most of these studies actually excluded diabetic subjects, data relating
the
risk of atherosclerotic diseases to the diabetic condition are not as
numerous, but point
in the same direction as for nondiabetic subjects (Pyorala et al). However,
the incidence
of atherosclerotic diseases in morbidity and mortality 'statistics in the
diabetic population
exceeds that of the nondiabetic population (Pyorala et al; Jarrett
DiabeteslMetabolism
Reviews 1989,5, 547; Harris et al, Mortality from diabetes, in Diabetes in
America
1985).
The independent risk factors obesity and hypertension for atherosclerotic
diseases are also associated with insulin resistance. Using a combination of
insulin/glucose clamps, tracer glucose infusion and indirect calorimetry, it
has been
demonstrated that the insulin resistance of essential hypertension is located
in peripheral
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-2-
tissues (principally muscle) and correlates directly with the severity of
hypertension
(DeFronzo and Ferrannini, Diabetes Care 1991, 14, 173). In hypertension of the
obese,
insulin resistance generates hyperinsulinemia, which is recruited as a
mechanism to
limit further weight gain via thermogenesis, but insulin also increases renal
sodium
reabsorption and stimulates the sympathetic nervous system in kidneys, heart,
and
vasculature, creating hypertension.
It is now appreciated that insulin resistance is usually the result of a
defect in the
insulin receptor signaling system, at a site post binding of insulin to the
receptor.
Accumulated scientific evidence demonstrating insulin resistance in the major
tissues
which respond to insulin (muscle, liver, adipose), strongly suggests that a
defect in
insulin signal transduction resides at an early step in this cascade,
specifically at the
insulin receptor kinase activity, which appears to be diminished (reviewed by
Haring,
Diabetalogia 1991, 34, 848).
Protein-tyrosine phosphatases (PTPases) play an important role in the
regulation
of phosphorylation of proteins. The interaction of insulin with its receptor
leads to
phosphorylation of certain tyrosine molecules within the receptor protein,
thus
activating the receptor kinase. PTPases dephosphorylate the activated insulin
receptor,
attenuating the tyrosine kinase activity. PTPases can also modulate post-
receptor
signaling by catalyzing the dephosphorylation of cellular substrates of the
insulin
receptor kinase. The enzymes that appear most likely to closely associate with
the
insulin receptor and therefore, most likely to regulate the insulin receptor
kinase
activity, include PTP1B, LAR, PTPa and SH-PTP2 (B. J. Goldstein, J. Cellular
Biochemistry 1992, 48, 33; B. J. Goldstein, Receptor 1993, 3, 1-15,; F. Ahmad
and
B. J. Goldstein Biochim. Biophys Acta 1995, 1248, 57-69).
McGuire et al. (Diabetes 1991, 40, 939), demonstrated that nondiabetic glucose
intolerant subjects possessed significantly elevated levels of PTPase activity
in muscle
tissue vs. normal subjects, and that insulin infusion failed to suppress
PTPase activity
as it did in insulin sensitive subjects.
Meyerovitch et al (J. Clinical Invest. 1989, 84, 976) observed significantly
increased PTPase activity in the livers of two rodent models of 1DDM, the
genetically
diabetic BB rat, and the STZ-induced diabetic rat. Sredy et al (Metabolism,
44, 1074,
1995) observed similar increased PTPase activity in the livers of obese,
diabetic ob/ob
mice, a genetic rodent model of NIDDM.
The compounds of this invention have been shown to inhibit PTPases derived
from rat liver microsomes and human-derived recombinant PTPase-1B (hPTP-1B) in
vitro. They are useful in the treatment of insulin resistance associated with
obesity,
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-3-
glucose intolerance, diabetes mellitus, hypertension and ischemic diseases of
the large
and small blood vessels.
DESCRIPTION OF THE INVENTION
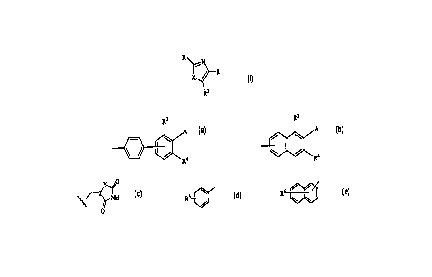
This invention provides a compound of formula I having the structure
R
X ~ R
R2
I
wherein
3 3
A A
R is ~ / ~ / or ~
~ Ra R4
Y~O
~'i
A is ORS, or ~~~ NH
O
R1 is alkyl of 1-6 carbon atoms, cycloalkyl of 3-8 carbon atoms, thienyl,
furyl, pyridyl,
i
R~ \ or R~ ~~ ;
l~ \ \
R2 is hydrogen, alkyl of 1-6 carbon atoms, or aryl of 6 to 10 carbon atoms;
R3 and R4 are independently halogen, hydrogen, alkyl of 1-12 carbon atoms ,
aryl of 6
to 10 carbon atoms; halogen, trifluoromethylof 1-6 carbon atoms, alkoxyaryl of
7-14 carbon atoms; nitro, amino, carboalkoxy, carbamide, carbamate, urea,
alkylsulfoamide, -NR~(CH2)mC02H, arylsulfoamide, cycloalkyl of 3-8 carbon
atoms, or heterocycle of 5 to 7 atom rings containing from 1 to 3 heteroatoms
selected from oxygen, nitrogen, or sulfur;
CA 02331118 2000-10-30
WO 99/58511 PCTNS99/10183
-4-
RS is hydrogen, alkyl of 1-6 carbon atoms, -CH(Rg)R9, -C(CHZ)~C02R1~,
-C(CH3)2C~2R1~~ 'CH(Rg)(CH2)nC02R1~, -CH(Rg)C6H4C02R1~, or -CH2_
tetrazole;
R6 is hydrogen, alkyl of 1-6 carbon atoms, halogen, alkyoxy of 1-6 carbon
atoms,
trifluoroalkyl of 1-6 carbon atoms or trifluoroalkoxy of 1-6 carbon atoms;
R~ is hydrogen or alkyl of 1 to 6 carbon atoms;
R8 is hydrogen, alkyl of 1-6 carbon atoms, aryl of 6-10 carbon atoms,
arylalkyl of 7-15
carbon atoms, cycloalkyl of 3-8 carbon atoms, phthalic acid,
02H
R1~~, , Ru-- I ~ , or R~~NH I ~ ;
N /
O
R9 is C02R12, CONHR12, tetrazole, POgRl2;
Rl~is hydrogen, alkyl of 1-6 carbon atoms, aryl of 6-12 carbon atoms, aralkyl
of 7-15
carbon atoms;
Rl ~ is alkylene of 1 to 3 carbon atoms;
R12 is hydrogen, alkyl of 1-6 carbon atoms, aryl of 6-12 carbon atoms, aralkyl
of 7-15
carbon atoms;
XisO,orS;
Y is O, N, or S;
Z is C, or N;
QisO,N,orS;
m = 1-3;
n = 1-6,
or a pharmaceutically acceptable salt thereof, which are useful in treating
metabolic
disorders related to insulin resistance or hyperglycemia.
Pharmaceutically acceptable salts can be formed from organic and inorganic
acids, for example, acetic, propionic, lactic, citric, tartaric, succinic,
fumaric, malefic,
malonic, mandelic, malic, phthalic, hydrochloric, hydrobromic, phosphoric,
nitric,
sulfuric, methanesulfonic, napthalenesulfonic, benzenesulfonic,
toluenesulfonic,
camphorsulfonic, and similarly known acceptable acids when a compound of this
invention contains a basic moiety. Salts may also be formed from organic and
inorganic bases, preferably alkali metal salts, for example, sodium, lithium,
or
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-S-
potassium, when a compound of this invention contains a carboxylate or
phenolic
moiety, or similar moiety capable of forming base addition salts.
Alkyl includes both straight chain as well as branched moieties. Halogen means
bromine, chlorine, fluorine, and iodine. It is preferred that the aryl portion
of the aryl
or aralkyl substituent is a phenyl, naphthyl or 1,4-benzodioxan-5-yl group;
with phenyl
being most preferred. The aryl moiety may be optionally mono-, di-, or tri-
substituted
with a substituent selected from the group consisting of alkyl of 1-6 carbon
atoms,
alkoxy of 1-6 carbon atoms, trifluoromethyl, halogen, alkoxycarbonyl of 2-7
carbon
atoms, alkylamino of 1-6 carbon atoms, and dialkylamino in which each of the
alkyl
groups is of 1-6 carbon atoms, nitro, cyano, -C02H, alkylcarbonyloxy of 2-7
carbon
atoms, and alkylcarbonyl of 2-7 carbon atoms.
The compounds of this invention may contain an asymmetric carbon atom and
some of the compounds of this invention may contain one or more asymmetric
centers
and may thus give rise to optical isomers and diastereomers. While shown
without
respect to stereochemistry in Formula I, the present invention includes such
optical
isomers and diastereomers; as well as the racemic and resolved,
enantiomerically pure R
and S stereoisomers; as well as other mixtures of the R and S stereoisomers
and
pharmaceutically acceptable salts thereof.
Preferred compounds of this invention are those compounds of Formula I, X is
oxygen. More preferred compouds of this invention are those compounds of of
Formula I, wherein:
X is O;
Rl is phenyl substituted with R6;
R2 is alkyl of 1-6 carbon atoms; and
R3 and R4 are each, independently, hydrogen or halogen.
Specifically preferred compounds of the present invention are set forth below:
4-(4'-methoxy-biphenyl-4-yl)-S-methyl-2-(4-trifluoromethyl-phenyl}-oxazole
4-(4'-methoxy-biphenyl-3-yl)-5-methyl-2-(4-trifluoromethyl-phenyl)-oxazole
CA 02331118 2000-10-30
WO 99/58511 PCTNS99/10183
-6-
4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-of
3'-[5-methyl-2-(4-trifluoromethyi-phenyl)-oxazol-4-yl]-biphenyl-4-of
{4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-yloxy}-
acetic acid
{ 3'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-yloxy }-
acetic acid
2- { 4' -[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl ]-biphenyl-4-yloxy
} -3-
phenyl-propionic acid
2-{ 3'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-yloxy }-
3-
phenyl-propionic acid
3,5-dibromo-4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-
of
{ 3,5-dibromo-4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-
4-
yloxy}-acetic acid
2-{3,5-dibromo-4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-
4-
yloxy }-3-phenyl-propionic acid methyl ester
2-{ 3,S-dibromo-4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-
biphenyl-4-
yloxy}-3-phenyl-propionic acid
2-{4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-ylmethyl }-
( 1,2,4]oxadiazolidine-3,5-dione
2-{4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-3-ylmethyl
}-
[1,2,4]oxadiazolidine-3,5-dione
5-{4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-
yloxymethyl }-
1 H-tetrazole
or a pharmaceutically acceptable salt thereof.
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
The compounds of this invention can be prepared according to the following
schemes from commercially available starting materials or starting materials
which can
be prepared using to literature procedures. These Schemes show the preparation
of
representative compounds of this invention.
Scheme I
r
OH
2
RZ NH20H. HC_I ~ R R~COCI
B I ~ NaOAc B ~ pyridine R O R2
1 2 3
3
(HO)2B 3
CH3
R4 4 R~N f \ OCH3 BBr3
~''/\l~ia
Pd(PPh~4, Na2C03 2 '~ R CH2CI2
R 5
Rs ,~! N
R' _ OH R' ~NH
1 ) BrCH CN ~ ' '~./
/ \ / ' i 2- / \ / ' i
R2 ~--i ~R4 2) NaN3 RZ 'J Ra
6
1) (Br, or CI)(CH2)"C02R~2, NaH
H C02R12
2) NaOH, THF, MeOH ~) ~ , PPh3, Et02CN=NC02Et
Ra
2) NaOH, THF, MeOH
3
3
H
R ~ ~ / ~ / ~ 1..12) 'f ~2 R IN I \ I C02H
/ \ R
R
/ U R
R2 7
R2 $
In Scheme I commercially available ketones ( 1 ) were treated with
hydroxylamine in the presence of sodium acetate to produce oximes (2). Oximes
(2)
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
_g_
were converted to oxazoles by a known methodology [ref. Tet. Lett. 1980, 21,
2359-
2360], where oximes (2) were treated with acetyl chlorides in the presence of
pyridine
to produce oxazoles (3). Oxazoles {3) were coupled with aryl boronic acids of
general
structure (4; R', R4 are alkyl, aryl, trifluoromethyl, substituted aryl,
nitro, carbocyclic 5
to 7 carbon atoms rings or heterocyclic rings 5 to 7 atom rings with from 1 to
3
heteroatoms selected from oxygen, nitrogen, and sulfur) using the Suzuki
protocol [ref.
Syn. Comm. 1981, 11, 513-519] to produce biphenyls (5). The aryl boronic acids
are
either commercially available or can be prepared according to known
methodology [ref.
J. Org. Chem, 1984, 49, 5237-5243]. Biphenyls (5) converted to phenols (6) by
treatment with boron tribromide in dichloromethane [ref. J. Org. Chem. 1974,
39,
1427-1429]. Phenols (6)
were alkylated with bromo or chloro-alkylcarboxylates [(Br or Cl)(CHZ)nCO2R'2]
in the
presence of sodium hydride or potassium carbonate, using dimethylformamide or
acetonitrile as the solvent. Subsequent saponification with sodium hydroxide
in methyl
alcohol and tetrahydrofuran produced biphenyls (7). Coupling of biphenyls (6)
with
hydroxy-alkyl-carboxylates [HOCH(Rg)COZR'2] using the Mitsunobu protocol [ref.
Synthesis. 1981, 1-27], followed by saponification with sodium hydroxide in
methyl
alcohol and tetrahydrofuran produced biphenyls (8). Tetrazoles (9) were
prepared from
phenols (6) in a two step sequence. First the phenols (6) were alkylated with
bromoacetonitrile in the presence of sodium hydride, and secondly, the nitrite
was
converted to tetrazoles (9) with sodium azide.
Scheme II
R ~= Br2 R ~-..J~l CHs \ / \ / B(
S / AcOH~ S / gr
Pd(OAc)2, KzC03
R2 R2
10 11
3
R' OC H3
s ~ \ /
R2 R
12
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-9-
In Scheme II thiazoles ( 10) were brominated with bromine in the presence of
sodium acetate. The 4-bromo-thiazoles (11) were coupled with 4, 4'-methoxy
biphenyl
boronic acid using the Suzuki protocol [ref. Syn. Comm. 1981, I1, 513-519] to
give
biphenyls ( 12). Biphenyls { 12) were further converted to the desired
products in
substantially the same manner as described in Scheme I.
Scheme III
3 3
R
OH R1 ~ / OH
\ / \ / R4 Br2, KOAc, AcOH _ \ / \
/ / R
(R3, R4= H) 3 4_
R2 R2 (R , R _ H, Br, Br, Br)
13 14
(R3,R4-H, Br)
R1 \ (R3,R4=Br, Br)
\ / B(OH)~ ."
(dppf)PdCi2,
R3
R I- N ~ / OH R
~~~~~' R13 R13
X / ~ ~ \
R2
16
In Scheme III, the biphenyl compounds ( 13 ) can be monobrominated or
dibrominated using bromine, potassium acetate and acetic acid. One equivalent
of
bromine in a high dilution reaction mixture and low temperatures in the range
of 5-10 9C
afforded predominantly the monobrominated product (14; R3, R4 = H, Br). The
dibrominated product ( 14; R3, R4 = Br, Br) was obtained with two equivalents
of
bromine at room temperature. The Suzuki coupling protocol [ref. Syn. Comm.
1981,
11, 513-519] was used to generate the terphenyls 15 and 16. Coupling of the
monobromo compounds (14; R3, R4 = H, Br) with boronic acids R'3-Ar-B(OH)Z ;
(R'3
= halogen, trifluoromethyl, alkoxy, alkyl, nitro, amino, carboalkoxy) in the
present of
an inorganic base, for example KZC03, or Ba(OH)2, and palladium (0 or II)
catalyst,
for example Pd(PPh3)4, Pd(OAc)2, or (dppf)PdCl2, produced terphenyls (15; R3 =
H).
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
- 10-
Similarly, the dibromo compounds ( 14; R3, R4 = Br, Br) can undergo Suzuki
coupling
to afford either the di-coupled product ( 16) by using 2 equivalents of
boronic acid at
high temperatures ( 100 °C), or the mono-coupled-mono-bromo product (
15; R3, R° _
Br, Aryl-R'3). Both the bromo and dibromo compounds can afford in the same
synthetic manner products with various heterocyclic boronic acids, for example
thiophene, furan, oxazole, thiazole, pyridine.
Scheme IV
3
(HO)2B~
\ / CHO R~ 1 HO
4
R 4 ~/ \ / ~ ~ --..
/
R R2 Pd(PPh3)4, Na2C03 R2 R
3
17
3
R1
NOH NaBH3CN
NH20H. HCI
O c R2 ~-' R
Na A ~~ 4 HCI
1s
R3
R~
~N - ~ \ ~NHOH CICONCO
O ~ ~ ~ ~ / 4 THF
R
R2
19
R3
R ~J~ ~ ~ N
~NH
"-' R4 O
R2
20
In Scheme IV oxazoles (3) were coupled with aryl boronic acids of general
structure (4; R3, R4 are alkyl, aryl, trifluoromethyl, substituted aryl,
nitro, carbocyclic 5
to 7 carbon atoms rings or heterocyclic rings 5 to 7 atom rings with from 1 to
3
heteroatoms selected from oxygen, nitrogen, and sulfur) using the Suzuki
protocol [ref.
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-11-
Syn. Comm. 1981, 11, 513-519) to produce biphenyls (17). Biphenyls (17) were
converted to oximes (18) with hydroxylamine in the presence of sodium acetate.
Oximes ( 18) were reduced with sodium cyanoborohydride under acidic conditions
to
produce to hydroxylamines ( 19). The hydroxylamines ( 19) were treated with N-
(chlorocarbonyl)isocyanate to produce oxadiazolidinediones (20).
Thiazolidinediones
were prepared from benzaldehydes (17) using known methodology [ref. J. Med.
Chem., 1992, 35, 1853-1864).
The compounds of this invention are useful in treating metabolic disorders
related to insulin resistance or hyperglycemia, typically associated with
obesity or
glucose intolerance. The compounds of this invention are therefore,
particularly useful
in the treatment or inhibition of type II diabetes. The compounds of this
invention are
also useful in modulating glucose levels in disorders such as type I diabetes.
The ability of compounds of this invention to treat or inhibit disorders
related to
insulin resistance or hyperglycemia was established with representative
compounds of
this invention in the following two standard pharmacological test procedures
which
measure the inhibition of PTPase.
lnhihition of tri-phosnhorvlated insulin rece t~or, dodecaphosphope_ptide
~e_p~s~horylation bar rat hepatic protein-tyrosine phosphatasesSPTPases)
This standard pharmacological test procedure assess the inhibition of rat
hepatic
microsomal PTPase activity using, as substrate, the phosphotyrosyl
dodecapeptide
corresponding to the 1142-1153 insulin receptor kinase domain, phosphorylated
on the
1146, 1150 and 1151 tyrosine residues. The procedure used and results obtained
are
briefly outlined below.
~gparation of Microsomal Fraction: Rats (Male Sprague-Dawley rats (Charles
River,
Kingston, NY) weighing 100-150 g, maintained on standard rodent chow (Purina))
are
sacrificed by asphyxiation with C02 and bilateral thoracotomy. The liver is
removed
and washed in cold 0.85°10 (w/v) saline and weighed. The tissue is
homogenized on ice
in 10 volumes of Buffer A and the microsomes are isolated essentially as
described by
Meyerovitch J, Rothenberg P, Shechter Y, Bonner-Weir S, Kahn CR. Vanadate
normalizes hyperglycemia in two mouse models of non-insulin-dependent diabetes
mellitus. J Clin Invest 1991; 87:1286-1294 and Alberts B, Bray D, Lewis J,
Raff M,
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-12-
Roberts K, Watson JD, editors. Molecular biology of the cell. New York:
Garland
Publishing, Inc., 1989 with minor modifications. The liver homogenate is
filtered
through silk to remove any remaining tissue debris and then is centrifuged at
10,000xg
for 20 minutes at 40C. The supernatant is decanted and centrifuged at
100,000xgfor 60
minutes at 40C. The pellet, microsomes and small vesicles, is resuspended and
lightly
homogenized in : 20 mM TRIS-HCl (pH 7.4), 50 mM 2-mercaptoethanol, 250 mM
sucrose, 2 mM EDTA, 10 mM EGTA, 2 mM AEBSF, 0.1 mM TLCK, 0.1 mM
TPCK, 0.5 mM benzamidine, 25 ug/ml leupeptin, 5 ug/ml pepstatin A, 5 ug/ml;HSB
antipain, 5 ug/ml chymostatin, 10 ug/ml aprotinin (Buffer A), to a final
concentration of
approximately 850 ug protein/ml. Protein concentration is determined by the
Pierce
Coomassie Plus Protein Assay using crystalline bovine serum albumin as a
standard
(Pierce Chemical Co., Rockford, IL).
Measurement of PTPase activit,~ The malachite green-ammonium molybdate method,
as described by Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA was used. An
improved assay for nanomolar amounts of inorganic phosphate. Anal. Biochem.
1979;100:95-97, and adapted for the platereader, is used for the nanomolar
detection
of liberated phosphate by rat hepatic microsomal PTPases. The test procedure
uses, as
substrate, a dodecaphosphopeptide custom synthesized by AnaSpec, Inc. (San
Jose,
CA). The peptide, TRDIYETDYYRK, corresponding to the 1142-1153 catalytic
domain of the insulin receptor, is tyrosine phosphorylated on the 1146, 1150
and 1151
tyrosine residues. The microsomal fraction (83.25 ul) is preincubated for 10
min at
37deg.C with or without test compound (6.25u1) and 305.5 ul of the 81.83 mM
HEPES reaction buffer, pH 7.4. Peptide substrate, 10.5 ul at a final
concentration of 50
uM, is equilibrated to 37deg.C in a LABLINE Multi-Blok heater equipped with a
titerplate adapter. The preincubated microsomal preparation (39.5 ul) with or
without
drug is added to initiate the dephosphorylation reaction, which proceeds at
37deg.C for
min. The reaction is terminated by the addition of 200 ul of the malachite
green-
ammonium molybdate-Tween 20 stopping reagent (MG/AMlTw). The stopping
30 reagent consists of 3 parts 0.45% malachite green hydrochloride, 1 part
4.2%
ammonium molybdate tetrahydrate in 4 N HCl and 0.5% Tween 20. Sample blanks
are prepared by the addition of 200 ul MG/AM/Tw to substrate and followed by
39.5 'ul
of the preincubated membrane with or without drug. The color is allowed to
develop at
room temperature for 30 min and the sample absorbances are determined at 650
nm
using a platereader (Molecular Devices). Samples and blanks are prepared in
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-13-
quadruplicates. Screening activity of 50 uM (final) drug is accessed for
inhibition of
microsomal PTPases.
Calculations: PTPase activities, based on a potassium phosphate standard
curve, are
expressed as nmoles of phosphate released/min/mg protein. Test compound PTPase
inhibition is calculated as percent of control. A four parameter non-linear
logistic
regression of PTPase activities using SAS release 6.08, PROC NLIN, is used for
determining IC50 values of test compounds. All compounds were administered at
a
concentration of 50 lrM. The following results were obtained using
representative
compounds of this invention.
'% hange from
Exam le Control
4 -28
6 -30
7 -74
8 -78
9 -20
10 -35
12 -68
-47
16 -20
17 -54
hen larsine (Reference)-57
Inhibition of Tri-Phosphorvlated Insulin Receptor Dodecaphosphopeptide
15 De~hosphorylation ~ hPTPIB
This standard pharmacological test procedure assess the inhibition of
recombinant rat protein tyrosine phosphatase, PTP1B, activity using, as
substrate, the
phosphotyrosyl dodecapeptide corresponding to the 1142-1153 insulin receptor
kinase
domain, phosphorylated on the 1146, 1150 and 1151 tyrosine residues. The
procedure
used and results obtained are briefly described below.
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
- 14-
Human recombinant PTP1B was prepared as described by Goldstein (see
Goldstein et al. Mol. Cell. Biochem. 109, 107, 1992). The enzyme preparation
used
was in microtubes containing 500-700 pg/ml protein in 33 mM Tris-HCI, 2 mM
EDTA,
10% glycerol and 10 mM 2-mercaptoethanol.
Measurement of PTPase activity. The malachite green-ammonium molybdate
method, as described (Lanzetta et al. Anal. Biochem. 100, 95, 1979) and
adapted
for a platereader, is used for the nanomolar detection of liberated phosphate
by
recombinant PTP1B. The test procedure uses, as substrate, a
dodecaphosphopeptide
custom synthesized by AnaSpec, Inc. (San Jose, CA). the peptide,
TRDIYETDYYRK, con:esponding to the 1142-1153 catalytic domain of the insulin
receptor, is tyrosine phosphorylated on the 1146, 1150, and 1151 tyrosine
residues.
The recombinant rPTPIB is diluted with buffer (pH 7.4, containing 33 mM Tris-
HCI,
2 mM EDTA and SO mM b-mercaptoethanol) to obtain an approximate activity of
1000-
2000 nmoles/min/mg protein. The diluted enzyme (83.25 mL) is preincubated for
10
min at 37°C with or without test compound (6.25 mL) and 305.5 mL of the
81.83 mM
HEPES reaction buffer, pH 7.4 peptide substrate, 10.5 ml at a final
concentration of 50
mM, and is equilibrated to 37°C. in a LABLINE Multi-Blok heater
equipped with a
titerplate adapter. The preincubated recombinant enzyme preparation (39.5 ml)
with or
without drug is added to initiate the dephosphorylation reaction, which
proceeds at
37°C for 30 min. The reaction is terminated by the addition of 200 mL
of the
malachite green-ammonium molybdate-Tween 20 stopping reagent (MG/AMlTw). The
stopping reagent consists of 3 parts 0.45% malachite green hydrochloride, 1
part 4.2%
ammonium molybdate tetrahydrate in 4 N HCl and 0.5% Tween 20. Sample blanks
are
prepared by the addition of 200 mL MG/AMlTw to substrate and followed by 39.5
ml
of the preincubated recombinant enzyme with or without drug. The color is
allowed to
develop at room temperature for 30 min. and the sample absorbances are
determined at
650 nm using a platereader (Molecular Devices). Sample and blanks are prepared
in
quadruplicates.
Calculations: PTPase activities, based on a potassium phosphate standard
curve, are
expressed as nmoles of phosphate released/min/mg protein. Inhibition of
recombinant
PTP1B by test compounds is calculated as percent of phosphatase control. A
four
parameter non-linear logistic regression of PTPase activities using SAS
release 6.08,
CA 02331118 2000-10-30
WO 99/5851 I PCT/US99/10183
-15-
PROC NLIN, is used for determining ICgp values of test compounds. The
following
results were obtained.
Exam le IC50 (NM)
1 1.66
2 -47 (2.5 uM)
3 -56 (2.5 uM)
0.85
6 -47 (2.5 uM)
7 1.29
8 1.25
9 0.65
0.47
11 -40 (2.5 uM)
12 0.13
13 1.15
14 -65 (2.5 uM)
0.93
16 1.2
17 0.98
Phenylarsine oxide 39.7
(reference standard)
Sodium orthovanadate 244.8
(reference standard)
Ammonium molybdate 8.7
tetrahydrate
{reference standard)
5 The blood glucose lowering activity of representative compounds of this
invention were demonstrated in an in vivo standard procedure using diabetic
(ob/ob}
mice. The procedures used and results obtained are briefly described below.
The non-insulin dependent diabetic (NIDDM) syndrome can be typically
characterizes by obesity, hyperglycemia, abnormal insulin secretion,
hyperinsulinemia
10 and insulin resistance. The genetically obese-hyperglycemic oblob mouse
exhibits
many of these metabolic abnormalities and is thought to be a useful model to
search for
hypoglycemic agents to treat NIDDM [Coleman, D.: Diabetologia 14: 141-148,
1978].
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
- 16-
In each test procedure, mice [Male or female ob/ob (C57 BU6J) and their lean
litermates (obl+ or +/+, Jackson Laboratories) ages 2 to 5 months ( 10 to 65
g)] of a
similar age were randomized according to body weight into 4 groups of 10 mice.
The
mice were housed 5 per cage and are maintained on normal rodent chow with
water ad
libitum. Mice received test compound daily by gavage (suspended in 0.5 ml of
0.5%
methyl cellulose); dissolved in the drinking water; or admixed in the diet.
The dose of
compounds given ranges from 2.5 to 200 mg/kg body weight/day. The dose is
calculated based on the fed weekly body weight and is expressed as active
moiety. The
positive control, ciglitazone (5-(4-(1-methylcyclohexylmethoxy)benzyl)-2,4-
dione, see
Chang, A., Wyse, B., Gilchrist, B., Peterson, T, and Diani, A. Diabetes 32:
830-838,
1983.) was given at a dose of 100 mg/kg/day, which produces a significant
lowering
in plasma glucose. Control mice received vehicle only.
On the morning of Day 4, 7 or 14 two drops of blood (approximetly 50 ul) were
collected into sodium fluoride containing tubes either from the tail vein or
after
decapitation. For those studies in which the compound was administered daily
by
gavage the blood samples were collected two hours after compound
administration.
The plasma was isolated by centrifugation and the concentration of glucose is
measured
enzymatically on an Abbott V.P. Analyzer.
For each mouse, the percentage change in plasma glucose on Day 4, 7 or 14 is
calculated relative to the mean plasma glucose of the vehicle treated mice.
Analysis of
variance followed by Dunett's Comparison Test (one-tailed) are used to
estimate the
significant difference between the plasma glucose values from the control
group and the
individual compound treated groups ( CMS SAS Release 5.18).
The results shown in the table below shows that the compounds of this
invention are antihyperglycemic agents as they lower blood glucose levels in
diabetic
mice.
% Change Glucose
Exam le Dose (m ) from Vehicle
5 100 -40a
Ciglitazone100 -43
(reference
standard
a Statistically (p < 0.05) significant.
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-17-
Based on the results obtained in the standard pharmacological test procedures,
representative compounds of this invention have been shown to inhibit PTPase
activity
and lower blood glucose levels in diabetic mice, and are therefore useful in
treating
metabolic disorders related to insulin resistance or hyperglycemia, typically
associated
with obesity or glucose intolerance. More particularly, the compounds of this
invention
useful in the treatment or inhibition of type II diabetes, and in modulating
glucose levels
in disorders such as type I diabetes. As used herein, the term modulating
means
maintaining glucose levels within clinically normal ranges.
Effective administration of these compounds may be given at a daily dosage of
from about 1 mg/kg to about 250 mg/kg, and may given in a single dose or in
two or
more divided doses. Such doses may be administered in any manner useful in
directing
the active compounds herein to the recipient's bloodstream, including orally,
via
implants, parenterally (including intravenous, intraperitoneal and
subcutaneous
injections), rectally, vaginally, and transdermally. For the purposes of this
disclosure,
transdermal administrations are understood to include all administrations
across the
surface of the body and the inner linings of bodily passages including
epithelial and
mucosal tissues. Such administrations may be carned out using the present
compounds, or pharmaceutically acceptable salts thereof, in lotions, creams,
foams,
patches, suspensions, solutions, and suppositories (rectal and vaginal).
Oral formulations containing the active compounds of this invention may
comprise any conventionally used oral forms, including tablets, capsules,
buccal forms,
troches, lozenges and oral liquids, suspensions or solutions. Capsules may
contain
mixtures of the active compounds) with inert fillers and/or diluents such as
the
pharmaceutically acceptable starches (e.g. corn, potato or tapioca starch),
sugars,
artificial sweetening agents, powdered ceiluloses, such as crystalline and
microcrystalline celluloses, flours, gelatins, gums, etc. Useful tablet
formulations may
be made by conventional compression, wet granulation or dry granulation
methods and
utilize pharmaceutically acceptable diluents, binding agents, lubricants,
disintegrants,
suspending or stabilizing agents, including, but not limited to, magnesium
stearate,
stearic acid, talc, sodium lauryl sulfate, microcrystalline cellulose,
carboxymethyl-
cellulose calcium, polyvinylpyrrolidone, gelatin, alginic acid, acacia gum,
xanthan
gum, sodium citrate, complex silicates, calcium carbonate, glycine, dextrin,
sucrose,
sorbitol, dicalcium phosphate, calcium sulfate, lactose, kaolin, mannitol,
sodium
chloride, talc, dry starches and powdered sugar. Oral formulations herein may
utilize
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-18-
standard delay or time release formulations to alter the absorption of the
active
compound(s). Suppository formulations may be made from traditional materials,
including cocoa butter, with or without the addition of waxes to alter the
suppository's
melting point, and glycerin. Water soluble, suppository bases, such as
polyethylene
glycols of various molecular weights, may also be used.
It is understood that the dosage, regimen and mode of administration of these
compounds will vary according to the malady and the individual being treated
and will
be subject to the judgment of the medical practitioner involved. It is
preferred that the
administration of one or more of the compounds herein begin at a low dose and
be
increased until the desired effects are achieved.
The following procedures describe the preparation of representative examples
of
this invention.
Exam In a 1
4-(4'-Methox~-_b_iphen~rl-4-yll-5-meth3rl-2-(4-trifluorometh3rl-phenvl)-
oxazole
Step a) 1-(4-bromo-phenyl-nronanone oxime
Sodium acetate (80.Og, 976 mmol) was added into a mixture of 1 (4-bromo-
phenyl)-propanone (52.0 g, 244 mmol), hydroxylamine hydrochloride (50.8 g,
732.3
mmol), ethyl alcohol (500 mL) and water ( 100 mL). The reaction mixture was
stirred
at 60 ° C for 1 hour, poured into water, and extracted with ethyl
ether. The organic
extracts were dried over MgS04: Evaporation and crystallization from ethyl
ether /
hexanes gave a white solid (49.6 g, 89% yield); MS mle 227(M+); Analysis for
C9H,o-BrNO: Calc'd: C, 47.39; H, 4.42; N, 6.14 Found: C, 47.42; H, 4.37; N,
5.99
Step b) 4-(4-Bromo-phen~)-5-methyl-2-(4-trifluoromethyl-nhenvl)-oxazole
Pyridine (3.55 mL, 43.86 mmol) was added into a mixture of 1-(4-bromo
phenyl)-propanone oxime ( lO.Og, 43.86 mmol) and toluene (20 mL). The reaction
mixture was stirred for 30 minutes, and then 4-trifluoromethyl-phenyl acetyl
chloride
(16.27 mL, 109.6 mmol) was added dropwise. The new mixture was stirred at 100
'~C
for 24 hours, and then w as poured into water and extracted with ethyl
acetate. The
organic extracts were dried over MgS04. Evaporation and purification by flash
chromatography on silica gel (hexanes/EtAOc 40:1) gave a white solid (7.3 g,
43%
yield): mp 82.84 °C; MS mle 381 (M+);
CA 02331118 2000-10-30
WO 99/58511 PCTNS99/10183
- 19-
Analysis for: C,~H"BrF3N0 Calc'd: C, 53.43; H, 2.90; N, 3.67 Found: C, 53.47;
H,
2.62; N, 3.43
Step c) 4-(4'-methoxv-biphenyl-4-vll-5-methyl-2-(4-~fluoromethyl-phenyl)-
oxazole
4-Methoxy-benzeneboronic acid ( 1.44 g, 7.19 mmol) in ethyl alcohol (5 mL)
was added into a mixture of 4-(4-bromo-phenyl)-5-methyl-2-(4-trifluoromethyl-
phenyl)-oxazole (2.5 g, 6.54mmo1), sodium carbonate (2N, 6.5 rnL), tetrakis-
(triphenylphosphine)palladium(0) (0.23 g, 0.196 mmol), and toluene (200 mL).
The
reaction mixture was refluxed for 12 hours, cooled to room temperature, and
treated
with hydrogen peroxide (30%, 5 mL) for 1 hour. Then, the mixture was poured
into
water and extracted with ethyl.acetate. The organic extracts were dried over
MgS04.
Evaporation and crystallization from hexanes/ethyl ether gave a white solid
(2.2 g, 82%
yield): mp 167-168 °C; MS mle 409 (M+);
Analysis for: C24H,$F3NO2 Calc'd: C, 70.41; H, 4.43; N, 3.42 Found: C, 70.14;
H,
4.32; N, 3.30
Example 2
4-l4'-Methoxy-biphenyl-3-y_1)-5-methy~4-trifluoromethyl-phen~)-oxazole
The title compound was prepared from 4-(4-bromo-phenyl)-5-methyl-2-(4-
trifluoromethyl-phenyl)-oxazole, and 4-methoxy-benzeneboronic acid in
substantially
the same manner, as described in Example 1 step c, and was obtained as a white
solid,
mp 93-94 °C; MS mle 409 (M+);
Analysis for: C24H,8F3NO2 Calc'd: C, 70.41; H, 4.43; N, 3.42 Found: C, 70.25;
H,
4.33; N, 3.34
Example 3
4'-[5-Methyl-2-(4-trifluoromethyl-phen~/11-oxazol-4-yll-biphen I
Boron tribromide ( 1.0 M, 3.91 mL, 3.9I mmol) was added dropwise into a
cold (-78 °C) mixture of 4-(4'-methoxy-biphenyl-4-yl)-5-methyl-2-(4-
trifluoromethyl-
phenyl)-oxazole ( 1.6 g, 3.91 mmol), and dichloromethane (20 mL). The reaction
mixture was allowed to come gradually to room temperature and stirred for 10
hours.
Then, the mixture was cooled to 0 °C and methyl alcohol (5 mL) was
added dropwise.
After stirring for 10 minutes the mixture was poured into water and extracted
with ethyl
ether. The organic extracts were dried over MgS04. Evaporation and
crystallization
CA 02331118 2000-10-30
WO 99/58511 PCT1US99/10183
-20-
from ethyl ether/hexanes gave an off white solid (1.4 g, 90% yield): mp 189-
191; MS
mle 396 (M+H)+;
Analysis for: C23H,6F3N0z x 0.3 HZO Calc'd: C, 68.92; H, 4.17; N, 3.50 Found:
C,
68.97; H, 4.23; N, 3.33
Example 4
3'-(5-Meth~4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-of
The title compound was prepared from 4-(4'-methoxy-biphenyl-3-yl)-5-methyl-
2-(4-trifluoromethyl-phenyl)-oxazole, in substantially the same manner, as
described in
Example 3, and was obtained as a white solid, mp 133-135 °C; MS m/e
395 (M+);
Analysis for: C23H,6F3NO2 X 0.3 H20 Calc'd: C, 68.92; H, 4.17; N, 3.50 Found:
C,
68.98; H, 3.83; N, 3.47
Examplg 5
{4'-j5-methyl-2-l4-trifluorometh r~l-phen~)-oxazol-4-yll-biphen~~Y}-acetic
acid
Sodium hydride (O.OSg, 1.26 mmol) was added into a mixture of 4'-[5-methyl-
2-(4-trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-of (0.5 g, 1.26 mmol),
and N,N-
dimethylformamide (5.0 mL). The reaction mixture was stirred at room
temperature for
1 hour. Methyl bromoacetate (0.18 mL, 1.89 mmol) was added dropwise into the
mixture. After stirrng for 30 minutes, the mixture was poured into water and
extracted
with ethyl acetate. The organic extracts were dried over MgS04. Evaporation
gave a
yellow oil (0.61 g). This residue was taken in methyl alcohol (20 mL) and
tetrahydrofuran (20 mL), and treated with NaOH (2.5 N, 5.0 mL) for 30 minutes.
The
new reaction mixture was then poured into water, acidified with HCl (2 N), and
extracted with ethyl ether. The organic extracts were dried over MgS04.
Evaporation
and crystallization form hexanes / ethyl ether gave an off white solid (0.42
g, 73 %
yield): mp 209-211; MS mle 454 (M+H)+;
Analysis for: CZSH,gF3N04 Calc'd: C, 66.23; H, 4.00; N, 3.09 Found: C, 65.97;
H,
3.93; N, 3.04
Example 6
~3'-f5-Methvl-2-(4-trifluoromethyl-phen rLl)-oxazol-4-yl]-biphenyl-4-ylox3r]~-
acetic acid
The title compound was prepared from 3'-[5-methyl-2-(4-trifluoromethyl-
phenyl)-oxazol-4-yl]-biphenyl-4-ol, in substantially the same manner, as
described in
CA 02331118 2000-10-30
WO 99/5$S1I PCT/US99/10183
-21-
Example 6, and was obtained as a light yellow solid, mp 178-1'~9 °C; MS
m/e 453
(M+);
Analysis for: CuH,8F3N04 x 0.3 H20 Calc'd: C, 65.44; H, 3.99; N, 3.05 Found:
C,
65.50; H, 3.93; N, 2.92
Example 7
2-{ 4'-j,5-Methyl-2-l4-trifluoromethyl-phenyl-oxazol-4-yll-biphen3rl-4-~v }-3-
phenvl-propionic acid
Diisopropyl azodicarboxylate (0.42 mL, 2.52 mmol) in benzene ( 10 mL) was
added dropwise into a cold (0 °C) mixture of 4'-[5-methyl-2-(4-
trifluoromethyl
phenyl)-oxazol-4-yl]-biphenyl-4-of (0.5 g, 1.26 mmol), 3-phenyllactic acid
methyl
ester (0.45 g, 2.52 mmol), triphenylphosphine (0.66 g, 2.52 mmol), and benzene
(20
mL). The reaction mixture was stirred at room temperature for 30 minutes,
poured into
water, and extracted with ethyl ether. The organic extracts were dried over
MgS04.
Evaporation gave a yellow oil (0.6 g). This residue was taken in methyl
alcohol ( 15
mL) and tetrahydrofuran ( 15 mL) and treated with sodium hydroxide (2 N, 3.0
mL).
The reaction mixture was stirred for 30 minutes, poured into water, acidified
with HCl
(2 N), and extracted with ethyl ether. The organic extracts were dried over
MgS04.
Evaporation and crystallization from ethyl ether/hexanes gave a white solid
(0.38 g,
55% yield): mp 183-184; MS m/e 544 (M+H)+;
Analysis for: C3zHzaF3NOa Calc'd: C, 70.71; H, 4.45; N, 2.58 Found: C, 70.50;
H,
4.32; N, 2.53
Example 8
2-{3'-[5-Meth3rl-2-(4-trifluoromethyl-phenyl)-oxazol-4-~]-biphenyl=4~~3r~-3-
phen3r~_propionic acid
The title compound was prepared from 3'-[5-methyl-2-(4-trifluoromethyl-
phenyl)-oxazol-4-yl]-biphenyl-4-ol, in substantially the same manner, as
described in
Example 7, and was obtained as a white solid, mp 148-149 °C; MS mle
543 (M+);
Analysis for: C32HzaF3NOa Calc'd: C, 70.71; H, 4.45; N, 2.58 Found: C, 70.72;
H,
4.28; N, 2.50
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-22-
Example 9
3.5-Dibromo-4'-[5-methx_I-~4-trifluoromethyl-phenyl)-oxazol-4- l~phe~ 1
Bromine (0.73 mL, 14.18 mmol) in acetic acid (50 mL) was added dropwise
over a 30 minutes period into a cold (5 °C) mixture of 4'-(2-benzyl-
benzo[b]thiophen-
3-yl)-biphenyl-4-of (2.8 g, 7.09 mmol), potassium acetate (6.95 g, 70.9 mmol),
and
acetic acid (200 mL). After the addition, the mixture was poured into water.
The
precipitated solid was filtered, washed with water and dried to afford a white
solid (2.1
g; 61 °lo yield): mp 79-81 °C MS mle 551 (M+);
Analysis for: Cz3H,4Br2F3NO2 Calc'd: C, 49.94; H, 2.55; N, 2.53 Found: C,
49.78;
H, 2.46; N, 2.49
Example 10
~3.5-Dibromo-4'-f5-methyl-2-(4-trifluorometh rL1-phenyl)-oxazol-4-yll-biphenyl-
4-
yloxy}-acetic acid
The title compound was prepared from 3,5-dibromo-4'-[5-methyl-2-(4-
trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-ol, and methyl bromoacetate in
substantially the same manner, as described in Example 5, and was obtained as
an off-
white solid, mp 165-166 °C; MS mle 609 (M'");
Analysis for: CZSH,6Br2F3NO4 Calc'd: C, 49.13; H, 2.64; N, 2.29 Found: C,
49.24;
H, 2.58; N, 2.16
Example 11
2~ 3.5-Dibromo-4'-j5-methyl-2-(4-trifluoromethyl=phenyl)-oxazol-4-yll-biphenyl-
4-
ylox~r}-3,=phen~rl-pro~ionic acid methyl ester
The title compound was prepared from 3,5-dibromo-4'-[5-methyl-2-(4-
trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-ol, and 3-phenyllactic acid
methyl
ester in substantially the same manner, as described in Example 7, and was
obtained as
a white solid, mp 70-72 °C; MS mle 713 (M+);
Analysis for: C33HZaBraFsNO4 Calc'd: C, 55.41; H, 3.38; N, 1.96 Found: C,
55.01;
H, 3.21; N, 1.99
Example 12
2- j3.5-Dibromo-4'-[5-methyl-2-(4-trifluorometh ~~1-phenyl)-oxazol-4- 1~1-
biphenyl-4-
vloxy}-3-phenyl-propionic acid
The title compound was prepared from 2-(3,5-dibromo-4'-[5-methyl-2-(4-
trifluoromethyl-phenyl)-oxazol-4-yl]-biphenyl-4-yloxy}-3-phenyl-propionic acid
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-23-
methyl ester in substantially the same manner, as described in Example 7, and
was
obtained as a white solid, mp 241-243 °C; MS m/e 699 (M+);
Analysis for: C32H22Br2F3N~4 Calc'd: C, 54.80; H, 3.16; N, 2.00 Found: C,
54.54;
H, 3.03; N, 2.00
Example 13
~~~'-ji5-meth~rl-2-f4-trifluoromethyl-phenvll-oxazol-4.- l~l-biphenyl-4-
ylmethyl_}-
r 1.2.4]oxadiazolidine-3.5-dione
Step a) 4'-LS-Meth~4-trifluoromethyl-p~enyll-oxazol-4-yll-biphen,~rl-4-
carbaldeh
This compound was prepared from 4-(4-bromo-phenyl)-5-methyl-2-(4-
trifluoromethyl-phenyl)-oxazole, and 4-formylbenzeneboronic acid in
substantially the
same manner, as described in Example 1 step c, and was obtained as an off
white solid;
MS m1e 407 (M+);
Analysis for: Cz4H,6F3N0z Calc'd: C, 70.76; H, 3.96; N, 3.44 Found: C, 70.83;
H,
3.70; N, 3.42
Step b) 4' j5-Methy]-2 ~4-trifluorometh rl-1-phenyl)-oxazol-4-yll-biphen
carbalde yde oxime
This compound was prepared from 4'-(5-methyl-2-(4-trifluoromethyl-phenyl)-
oxazol-4-yl]-biphenyl-4-carbaldehyde, and hydroxylamine in substantially the
same
manner, as described in Example 1 step a, and was obtained as an off white
solid; MS
mle 422 (M+);
Analysis for: C24H,~F3N20z Calc'd: C, 68.24; H, 4.06; N, 6.63 Found: C, 68.10;
H,
3.82; N, 6.45
Step c) N-~(4'-f5-Methyl-2-(4-trifluoromethyl-phenyl-oxazol-4-~(-biphenyl-4-
xlmeth~rl_~,v dr roxylamine
Hydrochloric acid (4 N, in dioxane, 10 mL) was added dropwise into a mixture
of 4'-
[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-ylJ-biphenyl-4-carbaldehyde
oxime
( 1.5 g, 3.56 mmol), sodium cyanoborohydride ( 1.1 g, 17.81 mmol), methyl
alcohol
( 100 mL), and tetrahydrofuran ( 100 mL). The reaction mixture was stirred for
1 hour
poured into water, basified with sodium hydroxide (2 N), and extracted with
ethyl
acetate. The organic extracts were dried over MgS04. Evaporation and
purification by
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-24-
flash chromatography on silica gel (EtOAc/MeOH 20:1) gave an off white solid
(1.21
g, 80% yield); MS mle 424 (M+);
Analysis for: C24H~9F3NZO2 X Hz0 Calc'd: C, 67.06; H, 4.60; N, 6.52 Found: C,
67.10; H, 4.34; N, 6.69
Step d) 2-{4'-[5-Methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-3r1]-biphenyl-4-
ylmeth~I-f 1.2.4]oxadiazolidine-3.5-dione
N-(Chlorocarbonyl)isocyanate (0.2 mL, 2.6 mmol) was added dropwise into a
cold (-5 °C) mixture of N-{4'-[5-methyl-2-(4-trifluoromethyl-phenyl)-
oxazol-4-yl]
biphenyl-4-ylmethyl}-hydroxylamine (1.1, 2.6 mmol), and tetrahydrofuran (20.0
mL).
The reaction mixture was stirred for 30 minutes, poured into water, acidified
with HCl
(2 N), and extracted with ethyl acetate. The organic extracts were dried over
MgS04.
Evaporation and purification by flash chromatography on acidic silica gel
(hexanes/EtOAc 2:1) gave a white solid (0.68 g, 53% yield): mp 196-198; MS mle
493
(M+);
Analysis for: CZ6H,8F3N3O4 Calc'd: C, 63.29; H, 3.68; N, 8.52 Found: C, 62.95;
H,
3.51; N, 8.40
Example 14
2-{4'-f5-MethXl-2~4-trifluorometh r~l-phem 11-oxazol-4- 1~1_-biphenyl-3-
ylmethyl}-
~.2.4joxadiazolidine-3.5-dione
This compound was prepared from 3-(4-bromo-phenyl)-5-methyl-2-(4-
trifluoromethyl-phenyl)-oxazole in substantially the same manner, as described
in
Example 1 steps a-d, and was obtained as a white solid, mp 216-218; MS mle 493
(M+);
Analysis for: C26H~8F3N3O4 Calc'd: C, 63.29; H, 3.68; N, 8.52 Found: C, 63.23;
H,
3.43; N, 8.48
Example 15
5-{4'-f5-Methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4- ly libiphen~~ymeth~}-
1 H-tetrazole
Sodium hydride (O.lg, 2.52 mmol) was added into a mixture of 4'-[5-methyl-
2-(4-trifluoromethyl-phenyl)-oxazol-4-yI]-biphenyl-4-of (1.0 g, 2.52 mmol),
and N,N-
dimethylformamide (5.0 mL). The reaction mixture was stirred at room
temperature for
1 hour. Methyl bromoacetonitrile (0.17 mL, 2.52 mmol) was added dropwise into
the
mixture. After stirring for 30 minutes, the mixture was poured into water and
extracted
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-25-
with ethyl acetate. The organic extracts were dried over MgS04. Evaporation
gave a
yellow oil (1.1 g). This residue was taken in N,N-dimethylformamide (20 mL),
and
treated with ammonium chloride (0.67 g, 12.6 mmol), and sodium azide (0.82 g;
12.6
mmol) at I20 °C for 10 hours. The mixture was then poured into water,
acidified with
HCl (2 N), and extracted with ethyl ether. The organic extracts were dried
over
MgS04. Evaporation and crystallization form hexanes / ethyl ether gave a white
solid
(0.49 g, 41 °lo yield): mp 226-227; MS mle 477 (M+);
Analysis for: C25H,8F3N502 Calc'd: C, 62.89; H, 3.80; N, 14.67 Found: C,
62.54; H,
3.63; N, 14.76
Examule 16
( 1-Bromo-6-[5-methyl-2-(4-trifluorometh ~~I-phenyl)-oxazol-4- ly 1-nanhthalen-
2-
ylox~~-acetic acid
Step a) 4~6-Methox~phthalen-2-~)-5-methyl-2-(4-trifluoromethyl-phen3rl)-
oxazole
This compound was prepared from 1-(6-methoxy-naphthalen-2-yl)propanone
oxime, and 4-trifluoromethyl-phenyl acetyl chloride in substantially the same
manner,
as described in Example 1 steps b, and was obtained as a white solid, mp 138-
139; MS
mle 383 (M'");
Analysis for: CZZH,9F3NO2 Calc'd: C, 68.93; H, 4.21; N, 3.65 Found: C, 68.83;
H,
4.25; N, 3.70
Step b) 6-f 5-Methyl-~-(4-trifluorometh,~~l-~~l-oxazol-4-~]-naphthalen-2-of
This compound was prepared from 4'-(6-methoxy-naphthalen-2-yl)-5-methyl
2-(4-trifluoromethyl-phenyl)-oxazole and boron tribromide in substantially the
same
manner, as described in Example 3, and was obtained as a white solid, mp 188-
191;
MS m/e 370 (M+H)+;
Analysis for: CZ,H,4F3NO2 Calc'd: C, 68.29; H, 3.82; N, 3.79 Found: C, 67.81;
H,
3.76; N, 3.66
Step c) 1-Bromo-6-f5-ether(4-trifluorometh~phenyl)-oxazol-4- l~phthalen-2-of
This compound was prepared from 4'-(6-hydroxy-naphthalen-2-yl)-5-methyl-2-
(4-trifluoromethyl-phenyl)-oxazole and bromine in substantially the same
manner, as
described in Example 9, and was obtained as an off white solid; MS mle 447
(M+);
CA 02331118 2000-10-30
WO 99/58511 PCT/US99/10183
-26-
Analysis for: CZ,H,3BrF~N02 Calc'd: C, 56.27; H, 2.92; N, 3.12 Found: C,
56.20; H,
2.66; N, 3.15
Step d) { 1-Bromo-6-f5-methyl-2-(4-trifluorometh~phen~)-oxazol-4-yll-
naphthalen-
2-vloxy~-acetic acid
This compound was prepared from 4'-(5-bromo-b-hydroxy-naphthalen-2-yl)-5-
methyl-2-(4-trifluoromethyl-phenyl)-oxazole and methyl bromoacetate in
substantially
the same manner, as described in Example 5, and was obtained as white solid,
mp 212-
214 °C; MS m/e 506 (M+H)+;
Analysis for: CZ3H,SBrF3N04 Calc'd: C, 54.57; H, 2.99; N, 2.77 Found: C,
54.17; H,
2.69; N, 2.76
Example 17
2-{ 1-Bromo-6-[5-methXl-2-(4-trifluorometh3rl-phenyll-oxazol-4-yl]-naphthalen-
2-
ylox~}-3-phenyl-propionic acid
This compound was prepared from 4'-(5-bromo-6-hydroxy-naphthalen-2-yl)-5-
methyl-2-(4-trifluoromethyl-phenyl)-oxazole and methyl bromoacetate in
substantially
the same manner, as described in Example 7, and was obtained as an off white
solid,
mp 195-197 °C; MS mle 596 (M+H)+;
Analysis for: C3oHz,BrF~N04 Calc'd: C, 60.42; H, 3.55; N, 2.35 Found: C,
60.31; H,
3.35; N, 2.42