Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02332765 2000-11-14
WO 99/59952 PCTNS99/10709
TITI~F OF THE INVENTION
METHOD OF PREPARING ALKYL CARBOXYLIC ACIDS BY
CARBOXYLATION OF LOWER ALKANES METHANE
$Q.CKCRO IND OF THE INVENTION
Field of the Invention
The present invention relates to a method of preparing acetic and higher
carbon
number aliphatic acids directly by carboxylation of lower molecular weight
alkanes such as
methane using a solid, heterogeneous catalyst without the intermediate
formation of
synthesis gas, e.g., CH4 + COZ ~ CH3COOH.
Description of the Background
Organic acids are widely used as intermediates and as solvents in chemical
processing. One of the most widely used of these acids is acetic acid, which
is a high-value,
high-volume chemical currently produced at the rate of 6 x 106 tons/yr.
worldwide. Acetic
acid is widely used as a raw material in the production of vinyl acetate,
acetic anhydride, and
cellulose acetate, and as an industrial and pharmaceutical solvent.
Although prior art exists for the indirect conversion of methane and other
alkanes to
acids, no prior art is known to exist for the direct synthesis of acetic acid
from methane or
other lower molecular weight alkanes on solid catalysts. U.S. 5,659,077
describes a method
for production of acetic acid by subjecting a feed mixture consisting of (a)
methane gas and
(b) gaseous oxygen, air, or a mixture thereof to partial oxidation without
production of
synthesis gas in a reaction zone at elevated temperature and pressure to form
a reaction
mixture containing methanol, carbon monoxide, carbon dioxide, methane, and
water vapor.
At least a portion of the water vapor is removed from the reaction mixture,
and the remaining
partial oxidation reaction mixture is fed, together with additional methanol
from an external
source, through a carbonylation reaction zone at elevated temperature and
pressure to form a
reaction product containing acetic acid and/or methyl acetate and methanol.
The additional
-1-
CA 02332765 2000-11-14
WO 99/59952 PCT/US99/10709
methanol is added in an amount such that the additional methanol together with
the methanol
produced by partial oxidation is sufficient to convert substantially all of
the carbon monoxide
produced by partial oxidation. Excess methane and carbon dioxide are recycled
from the
carbonylation reaction zone back to the partial oxidation reaction zone, and
methanol in the
carbonylation reaction product is recycled back to the carbonylation reaction
zone and acetic
acid and/or methyl acetate is recovered as product. This process, in effect,
produces acetic
acid by the partial oxidation of methane to methanol, followed by
carboxylation to acetic
acid. Unlike the present invention, oxygen is required and methanol is
produced as a
separable intermediate product. Methanol is then caxbonylated to form acetic
acid, in manner
similar in principle to conventional commercial technology.
U.S. 5,510,525 describes a process for direct oxidative carbonylation of lower
alkanes to acids having one greater carbon atom. The process requires both CO
and oxygen
as reactants and uses a homogeneous metal salt catalyst system promoted by
halide ions
and/or a metal (with oxygen as the oxidant) in an aqueous medium. Although the
process
converts methane to acetic acid, again as in U.S. 5,659,077, oxygen is used
and
carbonylation is required as a separate step. Neither U.S. 5,659,077, nor
U.S.5,590,525
contemplate the use of COz and both absolutely require oxygen to react with
methane.
Additionally, U.S. 5,510,525 requires an aqueous homogeneous catalyst system,
unlike the
solid heterogeneous catalyst of the present invention.
U.S. 5,393,922 describes a process for direct catalytic oxidation of
hydrocarbons,
particularly C, - C6 alkanes and single ring aromatics to acids by hydrogen
peroxide (or
dihydrogen and dioxygen) under mild temperature conditions (70 - 200°C)
using liquid
phase metal or metal salt catalysts. This process is similar to that of U.S.
5,510,525, where
an aqueous metal salt catalyst system is used, and the presence of CO and
dioxygen is
required. Acetic acid is formed only from ethane, and methane reacts to form
formic acid.
U.S. 5,393,922 thus contemplates no formation of a carbon-carbon bond, e.g.,
no formation
of acetic or higher acids from methane and is therefore unlike the present
invention in which
a given carbon number alkane, such as methane, reacts with COZ to form a
higher carbon
number aliphatic acid.
As an example of the current commercial technology used to produce aliphatic
acids
-2-
CA 02332765 2000-11-14
WO 99/59952 PCT/US99/10709
from alkanes, acetic acid is widely produced from methane in a series of
independent steps in
which a separate catalyst and reactor is typically used in each step:
Ni, etc.
(1)CH4+HZO ~ CO+3H
Cu, etc.
(1) CO + 2Hz ~ CH30H
Rh, etc
(3) CH30H + CO ~ CH3COOH
In step ( 1 ), methane is typically converted to synthesis gas, a mixture of
CO and
hydrogen using a nickel-based catalyst. This synthesis gas can also be
produced by
gasification of coal or other carbonaceous material using widely known
conventional
technology. Synthesis gas, in turn, is used to produce a number of chemicals,
including
methanol as shown in step (2), typically using a Cu-based catalyst. Finally,
methanol is
reacted with CO in a carbonylation step using a homogeneous Rh-based catalyst.
This three-
step process currently fulfills about 98% of the acetic acid market. Syngas
generation alone
typically accounts for at least 60% of the overall production cost of acetic
acid. Obviating
this step is clearly desirable to reduce the cost of synthesizing aliphatic
acids such as acetic
acid.
In step (3), this conventional process relies on the reaction of methanol and
CO to
form acetic acid using an expensive Rh catalyst dissolved in the liquid phase,
often using
iodine-based promoters. The economics of the process depends on successful
recovery and
recycle of the catalyst. As an example of the importance of developing a
solid,
heterogeneous catalyst, the cost of the separation unit can be more than 110
percent of the
cost of the reaction unit. Also, Rh-based catalysts are expensive, and I-based
promoters
(mostly CH3I) are toxic and corrosive, requiring expensive metallurgy, thus
resulting in
higher costs.
In theory, acetic acid might be made using a two-step reaction sequence where
syngas is first converted into methanol, and methanol is then carbonylated
into acetic acid in
the vapor phase using heterogeneous catalysts. Although vapor phase methanol
carbonylation has been a subject of intense lab-scale research for the past
several years, no
-3-
CA 02332765 2000-11-14
WO 99/59952 PCTNS99/10709
catalyst has been reported to be of industrial interest. The catalysts for
vapor phase reactions
have included RhCl3 supported by silica, alumina, and SiO,-A1,03. Ni is also
reported to be
an active catalyst for this reaction (Fujimoto et al., 1987). Further. nickel
supported on
activated carbon (AC) has been investigated for this reaction and tin has been
studied as a
promoter (Liu and Chiu, 1994a, 1994b). Although the catalyst was active.
significant
deactivation was observed via reduction of Ni to an inactive form and
reduction of Ni by the
AC support to form Ni carbide.
Thus, to date no industrially practical heterogeneous catalyst has been
developed for
the vapor phase carbonylation of methanol. Even if such a heterogeneous
catalyst were
developed, the overall process for the conversion of methane to acetic acid
would still be
indirect, requiring steps ( 1 ) and (2) above. Thus, there is a clear need for
a process based on a
solid heterogeneous catalyst for the direct reaction of lower alkanes (like
methane) with CO,
to form aliphatic acids such as acetic acid. This also provides an
environmentally benign
route to acetic acid based on inexpensive feedstocks.
Another conceptual route for the synthesis of acetic acid entails the reaction
of
methane, CO, and small amounts of oxygen. This type of oxidative conversion to
carboxylic
acids has been reported for a number of lower alkanes, such as methane and
ethane
homogeneous catalysts (Nishiguchi et al., 1992; Kurioka et al., 1995; Lin et
al., 1997; Sen
and Lin, 1996). For example, the reaction of CHQ (20 atm), CO (15 atm), in the
presence of
Pd(OAc)2; Cu(OAc)2 (0.05 mmol, ea), and KzS,Og (9 mmol) in trifluoroacetic
acid (TFA) (5
mL) at 80°C gives acetic acid in high yields. The results are
summarized in Table 1.
However, the reaction times are 20 to 40 h. Further, homogeneous catalysts
such as
Cu(OAc)Z, Pd(OAc)z, and K,SZOB are required and are used in a reaction medium
(such as
trifluoroacetic acid, TFA). The use of a homogeneous reaction medium where the
product
(acetic acid) is soluble in the reaction mixture requires the use of cost-
intensive and energy-
intensive separations, which increase the cost and complexity of the overall
process design.
-4-
CA 02332765 2000-11-14
WO 99/59952 PCT/US99/10709
TABLE 1. Acetic Acid Synthesis From CH4 and CO'
Run K,SZOBJmmol Time (h) AcOH Yield (%)b
1 _- 20 240
2 _- 40 410
-_ 20~ __
4 9a 20 120
CH, (20 atm), CO ( 15 atm), O= ( 15 atm), Pd(OAc)Z = Cu(OAc)~ = 0.05 mmol, TFA
(5 mL),
80°C.
b Yield based on Pd metal content.
' No catalyst used.
° No oxygen used.
Although the results demonstrate the possibility of this route, reaction times
of 20 to
40 h make these catalysts commercially impractical. In addition, this differs
from the present
invention which uses solid, heterogeneous catalysts.
1 S Direct conversion of methane to acetic acid using oxygen as an oxidant,
with
no added CO; has also been studied by other researchers (Lin et al., 1997; Lin
and Sen, 1994;
Sen and Lin, 1996). Oxidative carbonyiation of methane to selectively produce
acetic acid
was carried out at 95°C in a glass-tines stainless steel (SS) bomb. CH4
at 800 psi (54 atm),
CO at 150 psi (10 atm), and oxygen at 50 psi (3.b atm) were added to the bomb
which
contained RhCl3, HCI, and HI, and 5 mL of DSO as the solvent. No methanol was
formed in
the reaction products after 420 h, only acetic acid was recovered along with
trace quantities
of formic acid, as determined by nuclear magnetic resource (NMR) spectroscopy.
The results
clearly suggest that CO and OZ can be used to carboxylate CH4 but, again,
require
homogeneous catalysts. The use of heterogeneous catalysts to activate methane
has not been
demonstrated experimentally.
In related work, the reaction of an adsorbed methyl group with CO, to form an
acetate was suggested as a possibility (Bowker, 1992) because the reverse
reaction (i.e., the
-5-
CA 02332765 2000-11-14
WO 99/59952 PCT1US99/10709
decomposition of an acetate into a methyl group) was observed over Rh(I10)
(Bowker and
Li, 1991 ). Formation of an acetate from dissociative adsorption of CH3I and
CO~ over
Ni(110) catalysts has also been demonstrated using high resolution electron
energy loss
spectroscopy (HREELS) (Wambach and Freund, 1994), although the formation of
acetate
was not clearly confirmed. The fact that heterogeneous catalysts can activate
methane and
form acetic acid has not been demonstrated prior to the present invention.
BRIEF DESCRIPTION OF THE DR AWINGS
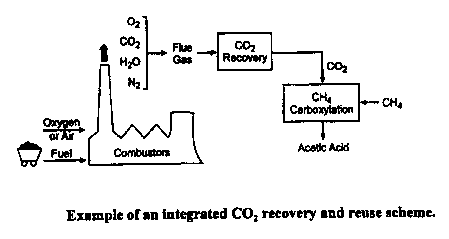
Figure 1 illustrates an integrated CO, recovery and reuse scheme.
Figure 2 illustrates conceptual economics of an integrated gasification
combined
cycle (IGCC) - acetic acid (AA) co-production scheme.
Figure 3 illustrates FTIR spectra over a 5% Pd/C catalyst after exposure to
acetic acid
under variable temperatures.
Figure 4 illustrates an FTIR spectra evidencing formation of acetate with 5%
Pd/C
under variable temperatures.
erJh~h~tARy OF THE INVENTION
Accordingly, the present invention provides a process for the preparation of
lower
alkane -acids by carboxylation of C,-C,~ alkanes by utilizing CO, and these
alkanes to
directly produce acids of one higher carbon number using heterogeneous (solid)
catalysts.
r i DESCRIPTION OF TuF puFFFRRFp EMBODIMENTS
The present invention provides a process for synthesis of acids such as acetic
acid via
carboxylation of alkanes such as methane. As noted above, the conventional
route practiced
industrially for synthesis of acetic acid is an indirect route in which
methane or other carbon
sources) are first reformed into syngas, a mixture of hydrogen and carbon
monoxide (CO),
CO is then hydrogenated to methanol, and methanol is carbonylated with CO to
produce
acetic acid using homogeneous catalysts. Some of the benefits of the direct
route to acetic
acid provided by the present invention are as follows.
-6-
CA 02332765 2000-11-14
WO 99/59952 PCTlUS99/10709
First, the energy- and cost-intensive methane reforming step is not necessary.
This
step can contribute at least 60 percent to the overall production costs of
acetic acid.
Second, the rhodium-based homogeneous catalysts currently used in methanol
carbonylation (MC) are replaced by a heterogeneous catalyst, making separation
of
the product simpler and less expensive. Thus, no intermediate methanol is
required to be
formed, and no carbonylation is required.
Third, the MC step, using toxic, corrosive, and potentially hazardous iodine-
based
promoters like methyl iodide, is replaced.
Fourth, the direct route of this invention reduces emissions of the greenhouse
gases,
CO2, and, in the case of acetic acid, also utilizes the greenhouse gas
methane.
Fifth, the solid, heterogeneous catalysts of the present invention are much
more
amenable to high throughput industrial processes, and product separation is
simple and
relatively inexpensive.
Sixth, a principle environmental advantage of the present invention is the
reduced
risk and secondary pollution produced as compared to the current technology.
At least 55
percent of the worldwide acetic acid production uses the methanol
carbonylation (MC)
technology, which uses expensive Rh catalysts, employs toxic I-based
promoters, and
involves cost-intensive separations. The occupational and potential
environmental hazards of
the compounds provide a second clear environmental incentive to develop benign
manufacturing processes for acetic acid.
In accordance with the present invention, solid heterogeneous catalysts are
used for
the direct synthesis of alkyl carboxylic acids such as acetic acid from CO,
and alkanes, such
as methane.
One reaction of interest is shown:
CO,~s~ + CH4~6~ '~ CH3COOH~,~
nH° =-16.2 kJ/mol CO,
oG° = +55.7 kJ/mol CO~
The nG -°~9g for this reaction is +55.7 kJ/mol CO,, corresponding to an
equilibrium
conversion of COz and CH4 and equilibrium yield of acetic acid that are
extremely low.
_7_
CA 02332765 2000-11-14
WO 99/59952 PCTNS99/10709
Despite the is equilibrium limitation, the reaction can be carried out at non-
equilibrium
conditions to maximize the yield of acetic acid.
An alternative way to synthesize acetic acid from methane would be to use
carbon
monoxide and oxygen as oxidants, instead of or in addition to CO,. The
nG°29g from the
reaction, among methane, carbon monoxide, and oxygen to form acetic acid is in
fact
negative, at -212.2 kJ/mol, i.e., this reaction is thermodynamically
favorable. Of course, the
reactor design and catalyst choice can be used to maximize the yield of acetic
acid. The
calculations for nG°29sK for these two alternate routes to acetic are
summarized:
l, ~ynth~~is of acetic acid from CH~ and CO~:
1 O CH4cs> + COZC~~ ~ CH3COOH~,~
DG°~9sk = (G°r)CH3COOH - (G°,)CH4 - (G°f)CO~
_ +55.7 kJ/mol.
2. S~rnthesis of acetic acid from CH, ~O. and O,:
CH4(g) + CO~b~ +'/2 OZ~b~ ~ CH,COOH~6~
IS OG°298K = (G°e)C~COOH - (G°,)CH4 -
(G°,.)CO -'/2 (G° f)CO,
- -212.2 kJ/mol.
Such a large negative free energy of reaction for 2 above corresponds to a
thermodynamically favorable reaction, with very high equilibrium conversions.
Several non-limitative examples herein below illustrate how the present
invention may be
20 utilized in the production of acetic acid.
~camn_ie 1: COz Removal from Power Plants. One way in which this process can,
W
principle, be used is to recover COz from conventional coal-fired power
plants, is shown in
Figure 1. The total CO, emissions in the United States in 1998 were
approximately 4,400
metric tones (Mt), with about 1,700 Mt CO, coming from the power plants. The
removal of
25 COZ on a large scale from industrial power plants is practiced
industrially; there are currently
two large coal-based power plants where CO~ is recovered in large quantities.
One is ABB
Lummus Crest's Shady Point, Oklahoma, operation where CO~ is recovered in
large
quantities. The cost of C02 production from such plants is estimated to be in
the range of
_g_
CA 02332765 2000-11-14
WO 99/59952 PCT/US99/10709
$20 to $30/ton CO~. This provides a useful application of the present
invention to removal of
CO~ from this type of combustion source.
Example 2' Conceptual Fconomiczs of COZ Removal. Some preliminary cost
e~tir:.ates shcwn in Figure 2 for CO~ removal scheme indicate that the process
is
economically viable, provided CO,/CH4 reaction can quantitatively form acetic
acid, in good
yields with extremely high selectivities: The conceptual economics of CO,
reuse from an
integrated gasification combined cycle (IGCC) power plant for acetic acid
production is
shown in Figure 2. The economics assumes a carbon tax of CO, emissions of
$50/ton of COZ
emitted. Although preliminary, these costs show the possibility of a
commercially practical
process.
~xa le 3: COZ~emoval from Natural Gas Streams. CO~ is also generated as a
byproduct in natural gas processing operations, with raw natural gas
containing up to 20 to
30 percent CO,. Such a gas can be used directly as a feedstock for the
reaction envisaged in
the present invention, reducing its cost, simplifying the process design, and
providing a
direct gas-to-liquids process of the type needed for remote gas field
operations.
As noted above, the present invention utilizes heterogeneous catalysts,
particularly
Group 8-11 transition metal catalysts, in a direct carboxylation of alkanes
such as methane to
form acids of one higher carbon number such as acetic acid. These
heterogeneous transition
metal catalysts may be prepared according to known preparatory procedures.
including
impregnation, incipient wetness and co-precipitation.
Generally, the transition metal catalyst contains one or more transition
metals from
the Periodic Table, however, of particular note are Group 8-11 transition
metals, such as Fe,
Ru, Os, Co, Rh, Ir, Ni, Pd, Pt, Cu, Ag and Au. These transition metal
catalysts may be
supported on an inert or acidic/basic support, such as carbon, silica, alumina
or even
diatomaceous earth. Generally, one or more transition metals are used in an
amount of 0.5 to
20% relative to the support. For example, 5% Pd/AC may be noted.
Generally, the reaction temperature may be from about 100°C to about
500°C. While
ambient pressures may be used, pressures of CO, and CHI of from about 0.5 to
about 200
atmospheres can also be used with higher equilibrium conversions at higher
pressures.
-9-
CA 02332765 2000-11-14
WO 99/59952 PCTNS99/10709
However, it is preferable to use a pressure of COZ and CH4 of from about 10 to
150
atmospheres.
Additionally, while any relative amounts of lower alkane and COZ may be used,
in
general, approximately equimolar amounts of each are used. By "equimolar" is
meant a
molar ratio range of from 0.1 to 10 of CH4/CO2. Generally, the amount of
catalyst used is
that typically used as a catalyst, and as is used for these known catalysts in
other reactions.
Generally, from about 10~ moles to about 0.5 moles per mole of each reactant
is used.
Preferably, the amount used is 0.1 mole or less of catalyst per mole of
reactant.
The present inventors carried out various experiments demonstrating the
formation of
the acetate group of acetic acid from a mixture of COZ and CH4. The following
examples are
provided solely for purposes of illustration and are not intended to be
limitative.
xa
Acetic acid was absorbed on a 5 % PdIAC catalyst to identify the infra-red
adsorption
bands corresponding to acetic acid on 5 % Pd/C as follows. The adsorption of
acetic acid
was carried out at 25 °C over a 5 percent PdIC catalyst, in a high
temperature environmental
chamber (HTEC). The catalyst was mixed with KBr powder (transparent to IR
radiation),
and loaded onto the sample cup in a HTEC. Helium was bubbled through an acetic
acid
impinger (maintained at 25 °C using a circulating coolant) and adsorbed
on the catalyst at 40
standard temperature and pressure (STP) mL/min for 60 min. The spectra were
collected
under flow-through conditions and under sealed conditions and ratioed to the
background
(Figure 3). Subsequent temperature programmed desorption (TPD) of chemisorbed
species
was carried out at 50°/min to 320°C, and spectra were collected
at each temperature, after
the spectra reached stable levels (after ca. 30 min).
Then, COz/CH4 were preadsorbed on the same catalyst and TPD-diffuse
reflectance
infrared Fourier transform spectroscopy (DRIFTS) was carried out and the
appearance/disappearance of spectral bands was matched with pure acetic acid
spectral data
on an identical catalyst (5% Pd/C)-KBr admixture.
-10-
CA 02332765 2000-11-14
WO 99/59952 PCTNS99/10709
In this case, the adsorption was carried out for 60 min and subsequent
desorption was
performed at 50°/min up to 420°C (Figure 4). The purpose of this
experiment was to
determine whether a 5% (5% Pd/C) 95% KBr admixture catalyzes the CO~/CH4
reaction to
acetic acid. The spectra at 25°C under flow-through (spectrum a) and
sealed (spectrum b)
conditions are identical. At 25°C characteristic bands at 3,729, 3,010,
2,362, and 1,301 cm''
are observed. The bands at 3,010 and 1,301 cm' can be assigned to gas-phase
methane
(Zhang et al., 1996) and a pronounced band at 2,362 cm' is due to CO,. A small
shoulder
band at 2,371 cm' visible in the adsorption spectra (spectra a, b) in Figure 4
is due to
naturally occurring ' 3CO2. (Burkett et al., 1990). The spectrum at 120
° C (spectrum c) is
similar to adsorption spectra, suggesting that no reaction has occurred among
the adsorbates.
However, at higher temperatures of 220 to 420°C (spectra d to fJ, small
but distinct peaks in
the 1,790 to 1,740 cm-' region corresponding to characteristics carbonyl
carboxylate
linkages are observed leading to the first evidence for synthesis of a
carbonyl species.
Further, small bands at 1,513 and 1,565 cm' can be assigned to an acetate
(CH3C00)
species (Viswanathan et al., 1990). These studies clearly evidence, for the
first time, the
synthesis of acetic acid from COZ and CH4 on a heterogeneous catalyst.
In another aspect of the present invention, higher carboxylic acids may be
prepared
from higher alkanes in accordance with the following scheme:
CnH2~ + COZ ~ CnH2~-~COOH
Specific examples of this reaction are the reactions of the use of ethane and
propane,
respectively, with COz:
CZH6 + CO, ~ CH3CH~COOH
C3H8 + COz ~ CH3CH~CH,COOH.
In order to effect this reaction, the same conditions and heterogeneous
catalysts are
used as described above. Generally, in the formulae above, n has a value of
from 1 to about
12, however, it is preferred that n have a value of from 1 to 8. This reaction
may be used
with CO~ and any of methane, ethane, propane, butane, pentane, hexane,
heptane, octane,
-11-
CA 02332765 2000-11-14
WO 99/59952 PCT/US99/10709
nonane, decane, undecane and/or dodecane. Of course, one may use any of the n-
, sec-, tert-
or iso- isomers of these alkanes.
Having described the present invention, it will now be apparent that many
changes
and modifications may be made to the above-described embodiments without
departing from
the scope of the present invenrion.
-12-