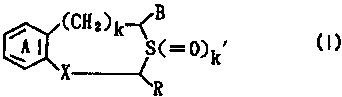Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
DESCRIPTION
COMPOSITION FOR TREATING CARTILAGE DISEASE
Technical Field
The present invention relates to a pharmaceutical
composition for prevention and/or treatment of a cartilage
disease, which comprise benzothiepine derivatives having
excellent chondrogenesis promoting effect, cartilage
destruction suppressing effect and cartilage cell
differentiation induction promoting effect.
Background Art
An articular disorder is a disease whose major
lesion is a degeneration of an articular cartilage.
Cartilage is the organization composed by collagen and
proteoglycan. Due to various causes, the synthesizing
ability of proteoglycan in this cartilage organization
declines , and proteoglycan starts to be released from the
organization. The release of type-I collagenase
(metalloprotease I) is simultaneously increased, and
collagen of the cartilage organization is resolved. The
destruction of the cartilage organization proceeds due to
a series of these responses. And it undergoes, depending
on the stage of the lesion, a hyperplasia of a synovial
membrane, a destruction of a subcartilaginous bone, a
hyperplasia or a neoplasia of a circumarticular cartilage,
which are followed by a deformation of the cartilage, which
may lead to dysfunction in a serious case. While the
articular disorder occurs most frequently in a knee joint ,
it occurs also in the joints of elbows, thighs, legs and
fingers . Among the articular diseases , the disease which
is observed in the largest number of patients is an
osteoarthritis, and it is considered to occur increasingly
in an elderlies-dominating society in near future, since
one of its causes is considered to be the aging of a human .
For treating this , an analgesic antiinflammatory agent or
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
2
a hyaluronic acid formulation is employed to remedy the pain
due to cartilage degeneration or subcartilaginous bone
destruction. However, all therapeutic methods are only
nosotropic, and exhibit no sufficient effects.
Suppression of cartilage destruction, promotion of
chondrogenesis and induction of cartilage cell
differentiation are considered to be effective in
prevention and treatment of a cartilage disease.
In the field of therapeutic and prophylactic agents
against cartilage diseases which are no more than
nosotropic currently, a novel cartilage disease preventing
and/or treating agent which is rather radical and excellent
in terms of the characteristics required in a useful
pharmaceutical (e. g., such as stability, absorption,
bioavailability) is demanded.
Disclosure of Invention
We made much effort to develop a pharmaceutical
capable of exerting a direct effect on a cartilage cell to
suppress a cartilage destruction and also capable of
promoting cartilaginous osteoanagenesis, and finally
discovered that Compound (I):
(CH2)k-~B ~ (I)
A ~ S(=~~k
X~R
wherein ring A is an optionally substituted benzene
ring; R is a hydrogen atom or an optionally substituted
hydrocarbon group; B is an optionally esterified or
amidated carboxyl group; X is -CH(OH)- or -CO-; k is 0 or
1; and k' is 0, 1 or 2, and its salt exhibits excellent
chondrogenesis promoting effect, cartilage destruction
suppressing effect and cartilage cell differentiation
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
3
induction promoting effect, and then we made a further
effort based on these findings, whereby establishing the
present invention.
Accordingly, the present invention relates to:
(1) a pharmaceutical composition for prevention
and/or treatment of a cartilage disease, which comprises
a compound (I) of the formula:
CCHz)k-~B
(I)
X--~g
wherein ring A is an optionally substituted benzene ring;
R is a hydrogen atom or an optionally substituted
hydrocarbon group; B is an optionally esterified or
amidated carboxyl group ; X is -CH ( OH ) - or -CO- ; k is 0 or
1; and k' is 0, 1 or 2, or its salt,
(2) a pharmaceutical composition according to (1),
wherein the ring A is a benzene ring which may be substituted
by 1 or 2 substituents selected from the group consisting
of a halogen, a C1_lo alkyl, a C1_lo alkoxy, an alkylenedioxy
group of the formula : -O- ( CHZ } ~-O- wherein n is an integer
from 1 to 3 and a Cl_lo alkylthio group; R is a hydrogen atom,
a C1_6 alkyl or a phenyl group ; B is -CON ( Rl ) ( Rz ) wherein R1
is a hydrogen atom or a C1_lo alkyl group and R~ is a phenyl
or a phenyl-C1_3 alkyl group which those groups may be
substituted by a halogen, a Cl_6 alkoxy, a mono- or di-C1_6
alkoxyphosphoryl, a mono- or di-C1_6 alkoxyphosphoryl-C1_
3 alkyl wherein two alkyl groups of these di-C1_6 alkoxy group
may bind together to form a Cl_6 alkylene group and C1_6
alkoxycarbonyl group,
(3) a pharmaceutical composition according to (1),
wherein the compound is an optically active compound ( II )
of the formula;
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
4
/Og4 _..,
,~~cONA ~ ~ ~2P\ 5 _. '
08
C ;~ s a
0 g3
wherein R' is a Cl_6 alkyl group; and R' and RS are
independently a C1_6 alkyl group or bind together to form
a C1_6 alkylene group,
(4) a pharmaceutical composition according to (3},
wherein R,, R~ and RS are independently a C1_4 alkyl group,
(5) a pharmaceutical composition according to (3),
wherein the optically active compound (II) is (2R,4S)-
(-)-N-[4-(diethoxyphosphorylmethyl}phenyl)-1,2,4,5-
tetrahydro-4-methyl-7,8-methylenedioxy-5-oxo-3-
benzothiepine-2-carboxamide,
(6) a pharmaceutical composition according to (1),
which is in the form of a sustained-release preparation
comprising a biodegradable polymer,
(7) a pharmaceutical composition according to (1),
which is for local administration,
(8) a pharmaceutical composition according to (1),
which is for oral administration,
(9) a pharmaceutical composition according to (1),
which is for injection,
( 10 ) a pharmaceutical composition according to ( 1 ) ,
which is for potent cartilage destruction suppressing agent,
chondorogenesis promoting agent or cartilage cell
differentiation induction promoting agent,
(11) a pharmaceutical composition according to(1},
which is for proteoglycan synthesis promoting agent, type
II collagen synthesis promoting agent, metalloprotease I
release suppressing agent or proteoglycan release
suppressing agent,
(12) a pharmaceutical composition according to (I),
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
which is for proteoglycan synthesis promoting agent or
metalloprotease I release suppressing agent,
(13) a pharmaceutical composition according to (1),
wherein the cartilage disease is a cartilage defect,
5 chronic rheumatoid arthritis and osteoarthritis,
(14) a method for prevention and/or treatment of
a cartilage disease, which comprises administrating
Compound ( I ) or its salts defined in ( 1 ) to mammal in need,
and
(15) use of Compound (I) or its salts defined in
( 1 ) for manufacturing a medicament for prevention and/or
treatment of a cartilage disease.
With respect to the Compound (I) or its salt, the
substituent of the substituted benzene represented by ring
A is exemplified by a halogen atom, a nitro group, an
optionally substituted alkyl group, an optionally
substituted hydroxyl group, an optionally substituted
mercapto group, an optionally substituted amino group, an
acyl group, a mono- or di-alkoxyphosphoryl group, a
phosphono group, an optionally substituted aryl group, an
optionally substituted aralkyl group and an optionally
substituted aromatic heterocyclic group. Of these
substituents , 1 to 4 , preferably 1 or 2 , whether identical
or not, may be present on the benzene ring.
The halogen atom includes fluorine, chlorine,
bromine and iodine.
The alkyl group of the optionally substituted alkyl
group includes an alkyl group having 1 to 10 carbon atoms
such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl,
sec-butyl, tert-butyl, pentyl, isopentyl, neo-pentyl,
hexyl, heptyl, octyl, nonyl and decyl, and a cycloalkyl
group having 3 to 7 carbon atoms such as cyclopropyi,
cyclobutyl, cyclohexyl and cycloheptyl. These alkyl
groups may be substituted by 1 to 3 substituents selected
from a halogen atom (e. g., fluorine, chlorine, bromine,
iodine), a hydroxyl group, an alkoxy group having 1 to 6
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
6
carbon atoms (e. g., methoxy, ethoxy, propoxy, butoxy,
hexyloxy) , a mono- or di-C1_6 alkoxyphosphoryl group ( a . g .
methoxyphosphoryl,ethoxyphosphoryl,dimethoxyphosphoryl,
diethoxyphosphoryl) and a phosphono group.
The substituted alkyl group includes
trifluoromethyl, trifluoroethyl, trichloromethyl,
hydroxymethyl, 2-hydroxyethyl, methoxyethyl, 1-
methoxyethyl, 2-methoxyethyl, 2,2-diethoxyethyl, 2-
diethoxyphosphorylethyl, phosphonomethyl and so on.
The substituted hydroxyl group includes alkoxy
group , an alkenyloxy group , an aralkyloxy group , an acyloxy
group , an aryloxy group and so on . Preferable alkoxy groups
is an alkoxy group having 1 to 10 carbon atoms ( e. g. , methoxy,
ethoxy,propoxy,butoxy, tert-butoxy,pentyloxy, hexyloxy,
heptyloxy, nonyloxy) and a cycloalkoxy group having 4 to
6 carbon atoms (e. g., cyclobutoxy, cyclopentoxy,
cyclohexyloxy}. Preferable alkenyloxy group is an
alkenyloxy group having 2 to 10 carbon atoms such as
allyloxy, crotyloxy, 2-pentenyloxy, 3-hexenyloxy, 2-
cyclopentenylmethoxy and 2-cyclohexenylmethoxy.
Preferable aralkyloxy group is an aralkyloxy group having
7 to 19 carbon atoms, with greater preference given to a
Ce-a aryl-Ci_, alkyloxy group ( a . g . , benzyloxy,
phenethyloxy). Preferable acyloxy group is an alkanoyloxy
group such as one having 2 to 10 carbon atoms (e. g.,
acetyloxy, propionyloxy, n-butyryloxy, hexanoyloxy).
Preferable aryloxy group is aryloxy group having 6 to 14
carbon atoms (e.g., phenoxy, biphenyloxy). Further, these
substituted hydroxy groups may be substituted by 1 to 3
substituents selected from the above-mentioned halogen
atom, a hydroxyl group, an alkoxy group having 1 to 6 carbon
atoms , a mono- or di-C1_6 alkoxyphosphoryl group, a phosphono
group, etc. Specifically, the substituted hydroxyl group
includes trifluoromethoxy, 2,2,2-trifluoroethoxy,
difluoromethoxy, 2-methoxyethoxy, 4-chlorobenzyioxy and
2-(3,4-dimethoxyphenyl)ethoxy, and so on.
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
7
The substituted mercapto group includes an
alkylthio group, an aralkylthio group and an acylthio group.
Preferable alkylthio group is an alkylthio group having 1
tolOcarbon atoms(e.g.,methylthio,ethylthio,propylthio,
butylthio, pentylthio, hexylthio, heptylthio, nonylthio)
and a cycloalkylthio group having 4 to 6 carbon atoms ( a . g . ,
cyclobutylthio, cyclopentylthio, cyclohexylthio).
Preferable aralkylthio group is an aralkylthio group having
7 to 19 carbon atoms , more preferably a C6_1~ aryl-C1_,
alkylthio group such as benzylthio and phenethylthio.
Preferable acylthio group is alkanoylthio group such as one
having 2 to 10 carbon atoms (e. g., acetylthio,
propionylthio, n-butyrylthio, hexanoyithio). Further,
these substituted mercapto groups may be substituted by 1
to 3 substituents selected from the above-mentioned halogen
atom, a hydroxyl group, an alkoxy group having 1 to 6 carbon
atoms , a mono- or di-Cl_6 alkoxyphosphoryl group , a phosphono
group, etc. Specifically, the substituted mercapto group
includes tri-fluoromethylthio, 2,2,2-trifluoroethylthio,
2-methoxyethylthio, 4-chlorobenzylthio, 3,4-
dichlorobenzylthio, 4-fluorobenzylthio, 2-(3,4-
dimethoxyphenyl)ethylthio, and so on.
As substituents of the substituted amino group,
there may be used 1 or 2 of identical or different
substituents selected from the above-mentioned alkyl group
having 1 to 10 carbon atoms , an alkenyl group having 2 to
10 carbon atoms (e.g., allyl, vinyl, 2-penten-1-yl, 3-
penten-1-yl, 2-hexen-1-yl, 3-hexen-1-yl, 2-cyclohexenyl,
2-cyclopentenyl, 2-methyl-2-propen-1-yl, 3-methyl-2-
buten-1-yl ) , an aryl group having 6 to 14 carbon atoms ( a . g .
phenyl , naphthyl ) and an aralkyl group having 7 to 19 carbon
atoms (e.g. benzyl, phenetyl). These substituents may be
substituted by the above-mentioned halogen atom, an alkoxy
group having 1 to 6 carbon atoms, a mono- or di-C1_6
alkoxyphosphoryl group, a phosphono group, etc.
Specifically, the substituted amino group includes
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
8
methylamino, dimethylamino, ethylamino, diethylamino,
dibutylamino,diallylamino,cyclohexylamino,phenylamino,
N-methyl-N-phenylamino, N-methyl-N-(4-
chlorobenzyl)amino and N,N-di(2-methoxyethyl)amino, and
so on.
The acyl group includes an organic carboxylic acid
acyl group and a sulfonic acid acyl group with a hydrocarbon
group having 1 to 6 carbon atoms ( a . g . , Cl_6 alkyl group such
as methyl, ethyl, n-propyl, hexyl) or phenyl. Useful
organic carboxylic acyl group is formyl, a C1_lo alkyl-
carbonyl group (e. g., acetyl, propionyl, butyryl, valeryl,
pivaloyl, hexanoyl, octanoyl, cyclobutanecarbonyl,
cyclohexanecarbonyl, cycloheptanecarbonyl), a Cz_lo
alkenyl-carbonyl group (e.g., crotonyl, 2-
cyclohexenecarbonyl), a C6_1, aryl-carbonyl group (e. g.,
benzoyl), a C,_19 aralkyl-carbonyl group (e. g.,
benzylcarbonyl, benzhydrylcarbonyl), a 5- or 6-membered
aromatic heterocyclic carbonyl group (e. g, nicotinoyl,
4-thiazolylcarbonyl) or a 5- or 6-membered aromatic
heterocyclic acetyl group (e.g., 3-pyridylacetyl, 4-
thiazolylacetyl ) . Useful sulfonic acyl group having 1 to
6 carbon atoms are methanesulfonyl and ethanesulfonyl, and
so on. These acyl groups may be substituted by 1 to 3
substituents selected from the above-mentioned halogen
atom, a hydroxyl group, an alkoxy group having 1 to 6 carbon
atoms, an amino group, etc. Specifically, the substituted
acyl group includes trifluoroacetyl, trichloroacetyl,
4-methoxybutyryl, 3-cyclohexyloxypropionyl, 4-
chlorobenzoyl and 3,4-dimethoxybenzoyl, and so on.
The mono- or di-alkoxyphosphoryl group includes a
mono-C1_6 alkoxyphosphoryl group such as methoxyphosphoryl ,
ethoxyphosphoryl, propoxyphosphoryl,
isopropoxyphosphoryl, butoxyphosphoryl,
pentyloxyphosphoryl and hexyloxyphosphoryl, and a di-C1_6
alkoxyphosphoryl group such as dimethoxyphosphoryl,
diethoxyphosphoryl, dipropoxyphosphoryl,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03I54
9
diisopropoxyphosphoryl, dibutoxyphosphoryl,
dipentyloxyphosphoryl and dihexyloxyphosphoryl, with
preference given to a di-CI_6 alkoxyphosphoryl group such
as dimethoxyphosphoryl, diethoxyphosphoryl,
dipropoxyphosphoryl, diisopropoxyphosphoryl,
ethylenedioxyphosphoryl, dibutoxyphosphoryl, etc.
The aryl group of the optionally substituted aryl
group includes an aryl group having 6 to 14 carbon atoms
such as phenyl, naphthyl and anthryl. These aryl groups
may be substituted by 1 to 3 substituents selected from the
above-mentioned alkyl group having 1 to 10 carbon atoms,
a halogen atom, a hydroxyl group, an alkoxy group having
1 to 6 carbon atoms, etc. Specifically, the substituted
aryl group includes 4-chlorophenyl, 3,4-dimethoxyphenyl,
4-cyclohexylphenyl and 5,6,7,8-tetrahydro-2-naphthyl.
The aralkyl group of the optionally substituted
aralkyl group includes aralkyl group having 7 to 19 carbon
atoms such as benzyl, naphthylethyl and trityl. These
aralkyl groups may be substituted by 1 to 3 substituents
selected from the above-mentioned alkyl group having 1 to
10 carbon atoms , a halogen atom, a hydroxyl group, an alkoxy
group having 1 to 6 carbon atoms , etc . on the aromatic ring .
Specifically, the substituted aralkyl group includes 4-
chlorobenzyl, 3,4-dimethoxybenzyl, 4-cyclohexylbenzyl
and 5,6,7,8-tetrahydro-2-naphthylethyl.
The aromatic heterocyclic group of the optionally
substituted aromatic heterocyclic group includes a 5- or
6-membered aromatic heterocyclic group having 1 to 4 atoms
of nitrogen, oxygen and/or sulfur, such as furyl, thienyl,
imidazolyl, thiazolyl, oxazolyl and thiadiazolyl. These
aromatic heterocyclic groups may be substituted by 1 to 3
substituents selected from the above-mentioned alkyl group
having 1 to 10 carbon atoms, a halogen atom, a hydroxyl group,
an alkoxy group having 1 to 6 carbon atoms, etc.
Provided that two alkyl groups are present as
mutually adjoining substituents on the benzene ring A, they
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
may bind together to form an alkylene group represented by
the formula : - ( CHZ ) m- wherein m is an integer from 3 to 5
(e. g., trimethylene, tetramethylene, pentamethylene).
Provided that two alkoxy groups are present as mutually
5 adjoining substituents on the benzene ring A, they may bind
together to form an alkylenedioxy group represented by the
formula: -O- ( CHZ ) "-O- wherein n is an integer from 1 to 3
(e.g.,methylenedioxy,ethylenedioxy,trimethylenedioxy).
In these cases, a 5- to ?-membered ring is formed in
10 cooperation with carbon atoms of the benzene ring.
With respect to the Compound (I), R is a hydrogen
atom or an optionally substituted hydrocarbon group.
The hydrocarbon group of the optionally substituted
hydrocarbon group represented by R is exemplified by the
above-mentioned alkyl group (preferably an alkyl group
having 1 to 10 carbon atoms such as methyl, ethyl, propyl,
isopropyl,butyl,isobutyl,sec-butyl,tert-butyl,pentyl,
iso-pentyl, neo-pentyl and hexyl), an alkenyl group
(preferably an alkenyl group having 2 to 10 carbon atoms) ,
an aryl group (preferably an aryl group having 6 to 14 carbon
atoms) and an aralkyl group (preferably an aralkyl group
having ? to 19 carbon atoms ) . Useful substituents on the
hydrocarbon group include the above-mentioned 5- or 6-
membered aromatic heterocyclic groupsuch as furyl,thienyl,
imidazolyl, thiazolyl, oxazolyl and thiadiazolyl , a
halogen atom, a di-C1_6 alkoxyphosphoryl group and a
phosphono group.
With respect to the Compound ( I ) , B is an optionally
esterified or amidated carboxyl group.
The esterified carboxyl group represented by B is
exemplified by an alkoxycarbonyl group, preferably a C1_lo
alkoxy-carbonyl group (e. g., methoxycarbonyl,
ethoxycarbonyl, propoxycarbonyl, butoxycarbonyl), an
aryloxycarbonyl group, preferably C6_1, aryloxycarbonyl
group (e. g., phenoxycarbonyl), and an aralkyloxycarbonyl
group , preferably a C,_19 aralkyloxy-carbonyl group ( a . g . ,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
benzyloxycarbonyl).
The amidated carboxyl group represented by B is
exemplified by an optionally substituted carbamoyl group
represented by the formula: -CON( R1 ) ( RZ } wherein R1 and R2
independently are a hydrogen atom, an optionally
substituted hydrocarbon group or an optionally substituted
5- to 7-membered heterocyclic group.
The hydrocarbon group of the optionally substituted
hydrocarbon group represented by R1 or RZ is exemplified
by the above-mentioned alkyl group, preferably an alkyl
group having 1 to 10 carbon atoms (e. g., methyl, ethyl,
propyl, isopropyl,butyl,isobutyl,sec-butyl,tert-butyl,
pentyl, isopentyl, neopentyl, hexyl, heptyl, octyl, nonyl,
decyl}, an alkenyl group, preferably an alkenyl group
having 2 to 10 carbon atoms (e.g., allyl, vinyl, 2-
penten-1-yl, 3-penten-1-yl, 2-hexen-1-yl, 3-hexen-1-yl,
2-cyclohexenyl, 2-cyclopentenyl, 2-methyl-2-propen-1-yl,
3-methyl-2-buten-1-yl), an aryl group, preferably aryl
group having 6 to 14 carbon atoms ( a . g . , phenyl , naphthyl ,
anthryl) , and an aralkyl group, preferably aralkyl groups
having 7 to 19 carbon atoms ( a . g . , benzyl , naphthylethyl ,
trityl). These hydrocarbon groups may be substituted by
1 to 3 substituents selected from ( i ) a halogen atom ( a . g . ,
fluorine , chlorine , bromine , iodine ) , ( ii ) a hydroxyl group ,
(iii} an alkoxy group having 1 to 6 carbon atoms (e. g.,
methoxy, ethoxy, propoxy, butoxy, text-butaxy, pentyloxy,
hexyloxy), (iv) an amino group which may be substituted by
an alkyl group having 1 to 6 carbon atoms such as methyl,
ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl,
tert-butyl,pentyl,iso-pentyl,neo-pentyl and hexyl(e.g.,
amino, methylamino, ethylamino, dimethylamino,
diethylamino, dipropylamino), (v) an amino group which may
be substituted by an acyl group such as a C1_lo alkanoyl group
(e.g., acetylamino, propionylamino, benzoylamino), (vi) a
carbamoyl group which may be substituted by an alkyl group
having 1 to 6 carbon atoms (e. g., carbamoyl,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
i2
methylcarbamoyl, dimethylcarbamoyl, diethylcarbamoyl),
( vii ) a C1_6 alkoxy-carbonyl group ( a . g . , methoxycarbonyl ,
ethoxycarbonyl, propoxycarbonyl), (viii) a mono- or di-
alkoxyphosphoryl group (e. g. a mono- or di-C1_s
alkoxyphosphoryl group such as dimethoxyphosphoryl,
diethoxyphosphoryl, ethylenedioxyphosphoryl), (ix) a
mono- or di-alkoxyphosphorylalkyl group (e.g. a mono- or
di-Cl_6 alkoxyphosphoryl-C1_3 alkyl group such as
methoxyphosphorylmethyl, ethoxyphosphorylmethyl,
methoxyphosphorylethyl, ethoxyphosphorylethyl,
dimethoxyphosphorylmethyl, diethoxyphosphorylmethyl,
dimethoxyphosphoryethyl, diethoxyphosphorylethyl), (x) a
moiety:
0
-CH2 - P ~ (CHZ) P
0 0~
wherein p is an integer from 2 to 4 , ( xi ) a phosphono group,
(xii) the above-mentioned aromatic heterocyclic group,
etc.
The 5- to 7-membered heterocyclic group of the
optionally substituted 5- to 7-membered heterocyclic group
represented by R1 or R2 is exemplified by a 5- to 7-membered
heterocyclic group containing a sulfur, nitrogen or oxygen
atom, 5- or 6-membered heterocyclic groups containing 2 to
4 nitrogen atoms, and 5- or 6-membered heterocyclic groups
containing 1 or 2 nitrogen atom( s ) and a sulfur or oxygen
atom. These heterocyclic groups may be condensed with a
6-membered ring containing 2 or fewer nitrogen atoms, a
benzene ring or a 5-membered ring containing a sulfur atom.
As a substituent of the substituted 5- to 7-membered
heterocyclic group represented by Rl and Rz, there may be
used 1 to 4 of the same substituents as those for the
substituted hydrocarbon group represented by R1 and RZ
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
13
above.
Preferable examples of the 5- to 7-membered
heterocyclic group represented by R1 and RZ include 2-
pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, pyrazolyl,
imidazolyl,thiazolyl,oxazolyl,tetrazolyl,thiadiazolyl,
oxadiazolyl, triazinyl, triazolyl, thienyl, pyrrolyl,
pyrrolinyl, furyl, pyrrolidinyl, benzothienyl, indolyl,
imidazolidinyl, piperidyl, piperidino, piperazinyl,
morpholinyl, morpholino, pyrido[2,3-d]pyrimidyl,
benzopyranyl, 1,8-naphthyridyl, quinolyl, thieno[2,3-
b]pyridyl.
The moiety: -N(R1) (R2) may form a 5- to 7-membered
ring by binding together with Rl and R2. Such rings include
morpholine, piperidine, thiomorpholine, homopiperidine,
piperidine, pyrrolidine, thiazolidine and azepine.
The substituted alkyl group as preferable examples
of the optionally substituted hydrocarbon group
represented by R' and R2 include trifluoromethyl,
trifluoroethyl, difluoromethyl, trichloromethyl, 2-
hydroxyethyl, 2-methoxyethyl, 2-ethoxyethyl, 2,2-
dimethoxyethyl, 2,2-diethoxyethyl, 2-pyridylmethyl, 3-
pyridylmethyl, 4-pyridylmethyl, 2-(2-thienyl)ethyl, 3-
(3-furyl)propyl, 2-morpholinoethyl, 3-pyrrolylbutyl, 2-
piperidinoethyl, 2-(N,N-dimethylamino)ethyl, 2-(N-
methyl-N-ethylamino)ethyl, 2-(N,N-
diisopropylamino)ethyl, 5-(N,N-dimethylamino)pentyl,
N,N-dimethylcarbamoylethyl,N,N-dimethylcarbamoylpentyl,
ethoxycarbonylmethyl, isopropoxycarbonylethyl, tert-
butoxy-carbonylpropyl, 2-diethoxyphosphorylethyl, 3-
dipropoxyphosphorylpropyl, 4-dibutoxyphosphorylbutyl,
ethylenedioxyphosphoryimethyl, 2-phosphonoethyl and 3-
phosphonopropyl. The preferable substituted aralkyl
groups include 4-chlorobenzyl, 3-(2-fluorophenyl)propyl,
3-methoxybenzyl, 3,4-dimethoxyphenethyl, 4-ethylbenzyl,
4-(3-trifluoromethylphenyl)butyl, 4-acetylaminobenzyl,
4-dimethylaminophenethyl, 4-diethoxy-phosphorylbenzyl
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
14
and 2-(4-dipropoxyphosphorylmethylphenyl)ethyl. The
preferable substituted aryl groups include4-chlorophenyl,
4-cyclohexylphenyl, 5,6,7,8-tetrahydro-2-naphthyl, 3-
trifluoromethylphenyl, 4-hydroxyphenyl, 3,4,5-
trimethoxyphenyl, 6-methoxy-2-naphthyl, 4-(4-
chlorobenzyloxy)phenyl, 3,4-methylenedioxyphenyl, 4-
(2,2,2-trifluoroethoxy)phenyl, 4-propionylphenyl, 4-
cyclohexanecarbonylphenyl, 4-dimethyl-aminophenyl, 4-
benzoylaminophenyl, 4-diethoxycarbamoylphenyl, 4-tert-
i0 butoxycarbonylphenyl, 4-diethoxyphosphorylphenyl, 4-
diethoxyphosphorylmethylphenyl, 4-(2-
diethoxyphosphorylethyl)phenyl, 2-
diethoxyphosphorylmethylphenyl, 3-
diethoxyphosphorylmethylphenyl, 4-
dipropoxyphosphorylphenyl, 4-(2-phosphonoethyl}phenyl,
4-phosphonomethylphenyl and 4-phosphonophenyl. The
preferable substituted 5- to 7-membered heterocyclic
groups include 5-chloro-2-pyridyl, 3-methoxy-2-pyridyl,
5-methyl-2-benzothiazolyl, 5-methyl-4-phenyl-2-
thiazolyl, 3-phenyl-5-isoxazolyl, 4-(4-chlorophenyl}-5-
methyl-2-oxazolyl, 3-phenyl-1,2,4-thiadiazol-5-yl, 5-
methyl-1,3,4-thiadiazol-2-y1,5-acetylamino-2-pyrimidyl,
3-methyl-2-thienyl, 4,5-dimethyl-2-furanyl and 4-
methyl-2-morpholinyl.
With respect to Compound ( I ) , ring A is preferably
a benzene ring which may be substituted by 1 or more, more
preferably 1 or 2 substituents selected from halogen atoms ,
an optionally substituted alkyl group, an optionally
substituted hydroxyl group, an optionally substituted
mercapto group and/or an optionally substituted amino
group.
More preferably, ring A is a benzene ring which may
be substituted by 1 or 2 substituents selected from the
above-mentioned halogen atom, an alkyl group having 1 to
10 carbon atoms ( furthermore preferably 1 to 5 carbon atoms ) ,
an alkoxy group having 1 to 10 carbon atoms (furthermore
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
preferably 1 to 5 carbon atoms), an alkylenedioxy group
represented by the formula: -O-(CHZ)n-O- wherein n is an
integer from 1 to 3, and/or an alkylthio group having 1 to
10 carbon atoms (furthermore preferably 1 to 5 carbon
5 atoms).
Most preferably, ring A is a benzene ring which may
be substituted by an alkylenedioxy group represented by the
formula : -O- ( CHZ ) n-O- wherein n is an integer from 1 to 3 .
R is preferably a hydrogen atom, an alkyl group
10 having 1 to 6 carbon atoms ( a . g . methyl , ethyl ) or a phenyl
group.
B is preferably an alkoxycarbonyl group or a group
represented by the formula : -CON ( Rl ) ( RZ ) wherein Rl and R2
independently are a hydrogen atom, an optionally
15 substituted hydrocarbon group or an optionally substituted
5- to 7-membered heterocyclic group.
With respect to R1 and RZ above , R1 is preferably a
hydrogen atom or an alkyl group having 1 to 10 carbon atoms
( a . g . methyl , ethyl , propyl ) , and RZ is preferably a phenyl
or phenyl-C1_3 alkyl group which may be substituted by a
halogen atom (e. g. fluorine, chlorine, bromine), a C1_6
alkoxy (e.g. methoxy, ethoxy), a mono- or di-
alkoxyphosphoryl (preferablly a mono- or di-C1_s
alkoxyphosphoryl such as dimethoxyphosphoryl,
diethoxyphosphoryl), a mono- or di-alkoxyphosphorylalkyl
(preferablly a mono- or di-Cl_6 alkoxyphosphoryl-C1_3 alkyl
such as dimethoxyphosphorylmethyl,
diethoxyphosphorylmethyl) wherein dialkyl groups of these
di-C1_6 alkoxy group may bind together to form a Cl_6 alkylene
group or a C1_6 alkoxycarbonyl (e. g. methoxycarbonyl,
ethoxycarbonyl) , or a 5- or 6-membered heterocyclic group
( a . g . pyridyl ) which may be substituted by a phenyl and that
contains 1 or 2 nitrogen atom( s ) or a nitrogen atom and a
sulfur atom.
More preferable example of R1 and Rz is a hydrogen
atom, and a phenyl group substituted by a mono- or di-C1_6
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
16
alkoxyphosphoryl-C1_, alkyl, respectively (e.g. 4-
diethoxyphosphorylmethylphenyl).
With respect to Compound (I), X is -CH(OH)- or -
CO-, preferablly -CO-.
With respect to Compound (I), k is 0 or 1, and k'
is 0, 1 or 2, preferablly k is 1, and k' is 0.
Compound (I) is preferably an optically active
benzothiepine derivertives represented by the formula
(II):
/Og4 _..
~CONH ~ ~ Cgzp '''
,. 11\Ogs_ (II)
C ,t s o
0 g3
wherein R' is a C1_6 alkyl group; and R' and R' are
independently a C1_6 alkyl group or bind together to form
a Cl_6 alkylene group .
The C1_6 alkyl group represented by R', R' or RS in
the Compound (II) is exemplified by alkyl groups such as
methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-
butyl,tert-butyl,pentyl,isopentyl,neo-pentyl and hexyl,
and preferably a Cl_~ alkyl group. R4 and RS may bind together
to form a C1_6 alkylene group. In this case, a moiety:
-p~0g 4 .',~~'~
n\
a
may represent a moiety:
0
P\ /(CHZ)p
0 0
wherein p is an integer from 2 to 4.
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
17
Preferable groups for R', R' and RS include alkyl
groups having 1 to 4 carbon atoms such as methyl and ethyl.
The compound (II) is an optically active compound
of the (2R,4S) configuration, and contains substantially
no compound of the (2S,4R) configuration. The compound
( II ) of which optical purity is nearly 100% is preferable.
Most preferably, the compound ( II ) is, for example,
(2R,4S)-(-)-N-[4-(diethoxyphosphorylmethyl)phenyl]-
1,2,4,5-tetrahydro-4-methyl-7,8-methylenedioxy-5-oxo-3-
benzothiepine-2-carboxamide {hereinafter also referred to
as compound A ) or its salt . The compound A is represented
as below.
CONH ~ ~ CHIP (0) (OC2H5) 2
0
S
p -CH3
The salt of Compound (I) is preferably a
pharmaceutically acceptable salt. Pharmaceutically
acceptable salts include salts with inorganic or organic
bases , salts with inorganic or organic acids , and basic or
acidic amino acids. Inorganic basic salts include alkali
metal salts (e.g., sodium salts, potassium salts) and
alkaline earth metal salts (e. g., calcium salts, magnesium
salts). Such organic basic salts include the salts with
trimethylamine, triethylamine, pyridine, picoline, N,N-
dibenzyl-ethylenediamine or diethanolamine. Such
inorganic acidic salts include the salts with hydrochloric
acid, hydrobromic acid, hydroiodic acid, phosphoric acid,
nitric acid and sulfuric acid. Such organic acidic salts
include the salts with formic acid, acetic acid,
trifiuoroacetic acid, oxalic acid, tartaric acid, fumaric
acid, malefic acid, methanesulfonic acid, benzenesulfonic
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
18
acid, p-toluenesulfonic acid and citric acid. Such the
salts with basic or acidic amino acids include the salts
with arginine, lysine, aspartic acid and glutamic acid.
Compound (I) or its salt included in the
pharmaceutical composition of the present invention can be
produced by the method described in Japanese laid-open
patent applications 232880/1991 (corresponding to EP-A-
0376197),364179/1992(corresponding to EP-A-0460488), and
231569/1996(corresponding to EP-A-0719782) or a
modification thereof.
It has been known that Compound (I) exhibits an
excellent alkaline phosphatase inducing activity, and that
it shows strong osteogenesis promoting effect (Japanese
laid-open patent application 231569/1996, and so on).
However, it has not been known that Compound (I)
normalizes the condition of the cartilage lesion by
excellent chondrogenesis promoting effect, cartilage
destruction suppressing effect and cartilage cell
differentiation induction promoting effect. The present
invention is made based on this fact. In this point the
present invention is totally different from the above
mentioned prior art. Therefore the pharmaceutical
composition comprising Compound (I) of the present
invention is useful for preventing or treating the
cartilage disease, that it is especially effective for
preventing or treating such disease before sickness reaches
bone itself . Therefore the composition can be used not only
for treating early stage of a cartilage defect, chronic
rheumatoid arthritis and osteoarthritis, but also for
preventing these desieses.
Since Compound (I) has potent cartilage destruction
suppressing effect, chondorogenesis promoting effect,
cartilage cell differentiation induction promoting effect,
proteoglycan synthesis promoting effect, type II collagen
synthesis promoting effect, metalloprotease I release
suppressing effect and proteoglycan release suppressing
CA 02334815 2000-12-05
WO 99!65474 PCT/JP99/03154
19
effect, and is more excellent in terms of clinically useful
characteristics such as stability, absorption,
bioavailability, it can be used for prevention and
treatment of a cartilage destruction in a joint in any of
various cartilage diseases such as cartilage defect,
chronic rheumatoid arthritis involving a cartilage,
osteoarthritis of a knee involving a cartilage as well as
disorders related thereto, in mammals (e. g., human, rat,
mouse, cat, dog, rabbit, cattle, pig, etc.).
The pharmaceutical composition comprising Compound
( I ) of the present invention can be administered orally or
non-orally, as formulated with a pharmaceutically
acceptable carrier, in the form of solid preparations such
as tablets, capsules, granules and powders, or liquid
preparations such as syrups and in~ectable preparations.
Pharmaceutically acceptable carriers are various
organic or inorganic carrier substances in common use as
pharmaceutical materials. They include excipients,
lubricants, binders and disintegrants for solid
preparations; and solvents, dissolution aids, suspending
agents, isotonizing agents, buffers and soothing agents for
liquid preparations. Other pharmaceutical additives such
as preservatives, antioxidants, stabilizing agents,
coloring agents and sweetening agents may be used as
necessary.
Preferable excipients include lactose, sucrose,
D-mannitol, starch, crystalline cellulose and light
silicic anhydride.
Preferable lubricants include magnesium stearate,
calcium stearate, talc and colloidal silica.
Preferable binders include binding cellulose,
pregelatinized starch, sucrose, D-mannitol, dextrin,
hydroxypropyl cellulose, hydroxypropylmethyl cellulose
and polyvinylpyrrolidone.
Preferable disintegrants include starch,
carboxymethyl cellulose, carboxymethyl cellulose calcium,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
croscarmellose sodium and carboxymethyl starch sodium,
low-substituted hydroxypropyl cellulose.
Preferable solvents include water for injection,
alcohol, propylene glycol, macrogol, sesame oil and corn
5 oil.
Preferable dissolution aids include polyethylene
glycol, propylene glycol, D-mannitol, benzyl benzoate,
ethanol,tris-aminomethane,cholesterol, triethanolamine,
sodium carbonate and sodium citrate.
10 Preferable suspending agents include surfactants
such as stearyltriethanolamine, sodium lauryl sulfate,
lauryl-aminopropionic acid, lecithin, benzalkonium
chloride, benzethonium chloride and monostearic glycerol;
and hydrophilic polymers such as polyvinyl alcohol,
15 polyvinylpyrrolidone, carboxymethyl cellulose sodium,
methyl cellulose, hydroxymethyl cellulose, hydroxyethyl
cellulose and hydroxypropyl cellulose.
Preferable isotonizing agents include sodium
chloride, glycerol and D-mannitol.
20 Preferable buffers include buffer solutions of
phosphates, acetates, carbonates and citrates.
Preferable soothing agents include benzyl alcohol.
Preferable preservatives include p-oxybenzoic acid
esters, chlorobutanol, benzyl alcohol, phenethyl alcohol,
dehydroacetic acid and sorbic acid.
Preferable antioxidants include sulfites and
ascorbic acid.
The pharmaceutical composition of the present
invention can be produced by dissolving or dispersing
Compound ( I ) into an appropriate solvent and forming into
microcapsules, spheres, rods, needles, pellets, films or
the like, by an appropriate method.
In addition, the pharmaceutical composition of the
present invention can also be a sustained-release
preparation comprising a biodegradable polymer dispersed
Compound ( I ) by a method such as that described in Japanese
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
21
laid-open patent application 231569/1996.
The biodegradable polymer of the present invention
is a polymer that is poorly soluble or insoluble in water
and degradable in vivo in an appropriate period for
treatment. Examples of such polymers include fatty acid
polyesters such as polymers, copolymers and their mixture
of one or more kinds of c~ -hydroxycarboxylic acids ( a . g . ,
lactic acid, gly-colic acid, 2-hydroxybutyric acid, 2-
hydroxyvaleric acid, 2-hydroxy-3-methylbutyric acid, 2-
hydroxycaproic acid, 2-hydroxyisocaproic acid, 2-
hydroxycaprylic acid), hydroxydi-carboxylic acids (e. g.,
malic acid) and hydroxytricarboxyl-is acids (e. g., malic
acid), lactic acid caprolactones, valerolactones, etc.,
and derivatives thereof ( a . g . , block polymers of polylactic
acid, polyglycolic acid and polyethylene glycol), poly-
cx-cyanoacrylates, polyalkylene oxalates (e. g.,
polytrimethylene oxalate, polytetramethy-lene oxalate),
polyortho-esters, polyortho-carbonates, polycarbonates
(e. g., polyethylene carbonate, polyethylene-propylene
carbonate), polyamino acids (e.g., poly-r-benzyl-L-
glutamic acid, poly-L-alanine, poly-?'-methyl-L-glutamic
acid), hyarulonates, polystyrene, polymethacrylic acid,
acrylic acid-methacrylic acid copolymers,polyamino acids,
dakin stearate, ethyl cellulose, acetyl cellulose,
vitro-cellulose, malefic anhydride copolymers, collagen,
gelatin, fibrin and hydroxyapatite.
These biodegradable polymers may be in the form of
homopolymers or copolymers of two or more kinds , or these
mixtures.
Polymerization may be of the random, block or graft
type.
Preferable biodegradable polymers in-clude
aliphatic polyesters.
From the viewpoint of biodegradability and
biocompati-bility, polymers and copolymers synthesized
from one or more kinds of cx-hydroxycarboxylic acids are
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
22
preferred. Specifically, copolymers synthesized from one
or more kinds of lactic acid, glycolic acid, 2-
hydroxybutyric acid, 2-hydroxyvaleric acid etc., or
mixtures thereof are used.
The biodegradable copolymer for the present
invention can be produced by commonly known methods such
as that described in Japanese laid-open patent application
28521/1986 (EP172636), or a modification thereof.
Although the above-mentioned c~-hydroxycarboxylic
acids may be of the D-, L- or D,L-configuration, the
D,L-configura-tion is preferred.
Homopolymers of the above-mentioned a-
hydroxycarboxylic acids include homopolymers of lactic
acid, glycolic acid and 2-hydroxybutyric acid. The
preferable a-hydroxycarboxylic acid is lactic acid.
Copolymers of the above-mentioned cx-hydroxycarboxylic
acids include copolymers of glycolic acid and the other c~
-hydroxycarboxylic acids. Preferable cx-
hydroxycarboxylic acids are lactic acid and 2-
hydroxybutyric acid. Specifically, useful copolymers
in-clude lactic acid-glycolic acid copolymers and 2-
hydroxy-butyric acid-glycolic acid copolymers, with
preference given to lactic acid-glycolic acid copolymers ,
etc.
The average molecular weight of these biodegradable
polymers for the present invention is preferably chosen
from the range of about 2 , 000 to 800 , 000 , more preferably
about 5,000 to 200,000.
The weight-average molecular weight of a lactic acid
homopolymer (hereinafter also referred to as polylactic
acid) is preferably about 5 , 000 to 100 , 000 , more preferably
about 6 , 000 to 50 , 000 . A polylactic acid can , for example,
be synthesized by commonly known production methods such
as that described in Japanese laid-open patent application
28521/1986 (EP172636).
The content ratio of lactic acid and glycolic acid
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
23
in a lactic acid-glycolic acid copolymer is preferably
about 100/0 to 50/50 (w/w) , and more preferably about 90/10
to 50/50 (w/w) . The weight-average molecular weight of the
lactic acid-glycolic acid copolymer is preferably about
5,000 to 100,000, more preferably about 8,000 to 50,000.
The lactic acid-glycolic acid copolymer can be synthesized
by a commonly known production method such as that described
in Japanese laid-open patent application 28521/1986
(EP172636). The copolymer is preferably synthesized by
catalyst-free dehydration polymerization condensation.
With respect to the 2-hydroxybutyric acid-glycolic
ac-id copolymer, the content ratio is preferably such that
glycolic acid accounts for about 40 to 70 mol%, and 2-
hydroxybutyric acid accounts for the remaining portion.
The weight-average molecular weight of the 2-
hydroxybutyric acid-glycolic acid copolymer is preferably
about 5,000 to 100,000, more preferably about 8,000 to
50,000. The 2-hydroxybutyric acid-glycolic acid
copolymer can be synthe-sized by a commonly known
production method such as that described in Japanese
laid-open patent application 28521/1986 (EP172636). The
copolymer is preferably synthesized by catalyst-free
dehydration polymerization condensation.
The above-described 2-hydroxybutyric acid-
glycolic ac-id copolymer may be used in mixture with
polylactic acid. When the 2-hydroxybutyric acid-glycolic
acid copolymer is used in mixture with polylactic acid, the
mixing ratio of 2-hydroxybutyric acid/glycolic acid is
about 10/90 to 90/10 ( % by weight ) , preferably about 25/75
to 75/25 (% by weight).
In the present specification, weight-average
molecular weight is defined as that based on polystyrene
measured by gel permeation chromatography (GPC).
Measurements were taken using a GPC column KF804L X2
( produced by Showa Denko ) and an RI monitor L-3300 ( produced
by Hitachi Ltd.) with chloroform as a mobile phase.
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
24
The amount of biodegradable polymer is variable
according to the strength of the pharmacological activity
of Compound ( I ) , the speed and duration of drug release from
the biodegradable polymer and so on, as long as the desired
purpose is accomplished. For example, the biodegradable
polymer is used in amounts about 0 . 2 to 10 , 000 times ( ratio
by weight), preferably about 1 to 1,000 times, more
preferably about 1 to 100 times, for the amount of the
bioactive substance.
The pharmaceutical composition of the present
invention can be produced by ordinary methods of producing
a pharmaceutical composition, for example, it can be
produced by dispersing a non-peptide osteogenetic
promoting substance in a bio-degradable polymer, or by
filling a non-peptide osteogenetic promoting substance in
a previously shaped hollow biodegradable polymer.
Specifically, useful methods include the in-water drying
method, the phase separation method, the spray drying
method, and modifications thereof:
Example methods of producing microcapsules of the
present invention are described below.
(1) In-water drying method (o/w method)
In this method, an organic solvent solution
comprising a biodegradable polymer is first prepared. The
organic solvent used to produce the pharmaceutical
composition of the present invention preferably has a
boiling point of not higher than 120°C. Such organic
solvents include halogenated hydrocarbons (e. g.,
dichloromethane, chloroform, chloroethane,
dichloroethane, trichloroethane, carbon tetrachloride),
aliphatic esters (e. g., ethyl asetate, butyl asetate),
ethers (e. g., ethyl ether, isopropyl ether) and aromatic
hydrocarbons (e. g., benzene, toluene, xylene). These
solvents may be used in combination of two or more kinds
in appropriate ratios. The organic solvent is pref-erably
dichloromethane or acetonitrile. The organic solvent is
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
more preferably dichloromethane. The concentration of
biodegradable polymer in the organic solvent solution is
normally chosen over the range of about 0 . O1 to 80% (w/w) ,
preferably about 0. 1 to 70% (w/w) , and more preferably about
5 1 to 60% (w/w), although varying depending on molecular
weight of biodegradable polymer and organic solvent type,
etc.
Compound ( I ) is added and dissolved into the organic
solvent solution comprising the biodegradable polymer thus
10 obtained, if necessary after lyophilized or vacuum dried.
The amount of Compound (I) is about 0.001 to 90% (w/w),
preferably about 0.01 to 80% (w/w), and more preferably
about 0.1 to 50% (w/w), based on the concentration of
biodegradable polymer in the organic solvent solution,
15 although varying depending on drug type, mechanism of
action on cartilage destruction suppressing effect or
chondorogenesis promoting effect, effect duration, etc..
The organic solvent solution thus prepared is then
added to an aqueous phase to form an o/w emulsion using a
20 turbine type mechanical stirrer or the like. The volume
of the aqueous phase is normally chosen from the range of
about 1 to 10 , 000 times , preferably about 2 to 5 , 000 times ,
and more preferably about 5 to 2 , 000 times , for the volume
of the oil phase.
25 An emulsifier may be added to the aqueous phase . The
emulsifier may be any one as long as it is capable of forming
a stable o/w emulsion. Examples of such emulsifiers
include anionic surfactants, nonionic surfactants,
polyoxyethylene castor oil derivatives, polyvinyl
pyrrolidone, polyvinyl alcohol, carboxymethyl cellulose,
lecithin, gelatin and hyaluronic acid. These may be used
in combination as appropriate. The concentration of
emulsifier in the aqueous phase is preferably about 0.001
to 20% (w/w) , more preferably about 0.01 to 10% (w/w) , and
further more preferably about 0.05 to 5% (w/w).
Solvent evaporation from the oil phase can be
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
26
achieved by commonly used methods , including the method in
which the solvent is evaporated under normal or gradually
reduced pressure during stirring using a propeller stirrer
or magnetic stirrer, etc., and the method in which the
solvent is evaporated while the degree of vacuum is adjusted
using a rotary evaporator, etc.. The obtained
microcapsules are separated by centrifugal method or
filtration, after which they are washed with, for example,
water or heptane, several times to remove free Compound ( I ) ,
emulsifier, etc. adhering to the microcapsule surface.
The microcapsules are then again dispersed in distilled
water, etc. and lyophilized. To prevent particle
flocculation during washing, antiflocculants: water-
soluble sugars such as mannitol, lactol, glucose and
starches ( e. g . , corn starch ) , amino acids such as glycine
and alanine, and proteins such as gelatin, fibrin and
collagen may be added.
In the above-described o/w method, microcapsules
may be produced by the w/o/w method, in which Compound ( I )
is dispersed in an organic solvent solution comprising a
biodegradable polymer.
(2) In-water drying method (w/o/w method)
In this method, Compound ( I ) is first dissolved or
dispersed in water to obtain a concentration specified
above to yield an internal aqueous phase, if necessary with
dissolving or suspending by adding a drug-retaining
substance such as a protein ( a . g . , gelatin ) , seaweed ( a . g . ,
agar), polysaccharide (e. g., alginic acid), synthetic
high-molecular substance (e. g., polyvinyl alcohol), basic
amino acid (e.g., arginine, lysine) or the like. The
internal aqueous phase may be supplemented with an organic
acid such as acetic acid, oxalic acid or citric acid, an
inorganic acid such as carbonic acid or phosphoric acid,
an alkali metal hydroxide such as sodium hydroxide, a basic
amino acid such as. arginine or lysine or a salt thereof ( a . g . ,
salts with organic acids such as acetic acid, oxalic acid,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
27
citric acid or salts with inorganic acids such as carbonic
acid, phosphoric acid and hydrochloric acid) as a pH
regulator for keeping the stability and solubility of
Compound (I) or its salt thereof. As a stabilizer for
Compound ( I ) , there may be added a protein ( a . g . , albumin ,
gelatin), starch derivative (e. g. dextrin, pullulan),
organic acid {e. g., citric acid),
ethylenediaminetetraacetic acid alkali metal salt (e. g.,
sodium ethylenediamine-tetraacetate), sulfurous acid
hydrogen alkali metal salt ( a . g. , sodium hydrogen sulfite ) ,
synthetic high-molecular substance (e. g., polyethylene
glycol) or the like. Commonly preservatives may also be
added p-oxybenzoates (e. g., methyl paraben, propyl
paraben), benzyl alcohol, chlorobutanol and thimerosal.
The additional amount of compound ( I ) is about 0 . 001 to 90%
(w/w), preferably about 0.01 to 80% (w/w), and more
preferably about 0.1 to 50% (w/w), although varying
depending on drug type, mechanism of action on cartilage
destruction suppressing effect or chondorogenesis
promoting effect or effect duration, etc.
The obtained internal aqueous phase is added to a
solution (oil phase) containing the biodegradable polymer,
followed by emulsifying treatment, to yield a w/o emulsion.
This emulsification is achieved by a known dispersing
methods which include the intermittent shaking method, the
method using a mixer such as a propeller shaker or a turbine
shaker, the colloidal mill method, the homogenizer method
and the ultra-sonication method. The above-described
solution (oil phase) containing the biodegradable polymer
is a solution prepared by dissolving the biodegradable
polymer in an organic solvent. This solvent may be any
solvent as long as its boiling point is not higher than about
120°C and it is immiscible with water. Such solvents
include halogenated hydrocarbons (e. g., dichloromethane,
chloroform, chloroethane, dichloroethane,
trichloroethane, carbon tetrachloride), aliphatic esters
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
28
( a . g . , ethyl acetate , butyl acetate ) , ethers ( a . g . , ethyl
ether, isopropyl ether) and aromatic hydrocarbons (e. g.,
benzene , toluene , xylene ) . These solvents may be used in
combination of two or more kinds in appropriate ratios . To
prevent particle flocculation during washing,
antiflocculants: water-soluble sugars such as mannitol,
lactol, glucose and starches (e. g., corn starch), amino
acids such as glycine and alanine, and proteins such as
gelatin, fibrin and collagen may be added.
The produced w/o emulsion is then added to an aqueous
phase to yield a w/o/w emulsion, from which the oil phase
solvent is evaporated off, to yield microcapsules. The
specific procedure for this production is the same as that
described in (1) above.
(3) Phase separation method
In this method, a coacervating agent is gradually
added to the above-described w/o emulsion under the
stirring to precipitate and solidify the biodegradable
polymer. The coacervating agent can be used silicon oil,
vegetable oils and fats (e.g. , sesame oil, soybean oil, corn
oil, cotton seed oil, coconut oil, linseed oil), mineral
oils , hydrocarbons ( a . g . , n-hexane , n-heptane ) as long as
it is a polymeric, mineral oil or vegetable oil compound
which can be mixed with the solvent of the biodegradable
polymer and which does not dissolve the polymer for
encapsulation. These may be used in combination of two or
more kinds.
The obtained microcapsules are, after filtration
and separation of them, repeatedly washed with heptane, etc .
to remove the coacervating agent . The free drug and solvent
are then removed by using the same manner as in-water drying
method. To prevent particle flocculation during washing,
antiflocculants: water-soluble sugars such as mannitol,
lactol, glucose and starches (e. g., corn starch), amino
acids such as glycine and alanine, and proteins such as
gelatin, fibrin and collagen may be added.
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
29
(4) Spray drying method
For producing microcapsules by this method, the
above-described w/o emulsion is sprayed via a nozzle into
the drying chamber of a spray drier to volatilize the
organic solvent and water in the fine droplets in a very
short time, and microcapsules are obtained. The nozzle is
exemplified by the double-fluid nozzle, pressure nozzle and
rotary disc nozzle. To prevent microcapsule flocculation,
an aqueous solution of the above-described antiflocculant
may be sprayed via another nozzle, while the w/o emulsion
is sprayed. The microcapsules thus obtained may be warmed
under reduced pressure to facilitate the removal of the
water and solvent contained them.
When microcapsules are used as an injectable
suspension, for instance, their particle size is chosen
over the range from about 0.1 to 300 ,um of average particle
diameter, as long as the requirements concerning the degree
of dispersion and needle passage are met . Preferably, the
particle size is about 1 to 150 /.cm, more preferably about
2 to 100 ;um.
Methods of preparing microcapsules as a sterile
preparation include, but are not limited to, the method in
which the entire production process is sterile, the method
in which gamma rays are used as sterilant , and the method
in which an antiseptic is added.
In addition to the above-described microcapsules,
the sustained-release preparation of the present invention
can be produced by dissolving a biodegradable polymer
dispersed Compound (I) and forming the solution into
spheres , rods , needles , pellets , films or the like , by an
appropriate method.
In addition, the sustained-release preparation of
the present invention can also be produced by pulverizing
to appropriate particle size a biodegradable polymer
dispersed Compound ( I ) by a method such as that described
in Japanese laid-open patent applications 234656/1994,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
which employs a turbo counter jet mill pulverizes or an
ultrasonic jet pulverizes. Specifically, Compound (I) is
added to an organic solvent containing the biodegradable
polymer, and dissolved therein. The solid solution
5 obtained by vacuum drying is then coarsely pulverized $nd
sieved, followed by solvent removal, after which the coarse
particles are pulverized to controlled particle size using
an ultrasonicjet pulverizes to yieldthesustained-release
preparation of the present invention.
10 On the above mentioned preparation of the
pharmaceutical composition, the content ratio of Compound
( I ) based on the pharmaceutical composition is about 0 . O1
to 95% (w/w), and preferbly about 0.1 to 20% (w/w).
However, the pharmaceutical composition of the
15 present invention can be administered as an oral agent , a
non-oral agent for local administration (e. g., injectable
preparations of intramuscular, subcutaneous, organs or
joints, etc., solid preparations such as, indwellable
preparations, granules and powders, liquid preparations
20 such as suspensions, and ointments) is more preferable.
The practical injectable preparation can be
prepared as aqueous suspension by suspending Compound ( I )
in water, along with a dispersing agent (e.g. , surfactants
such as Tween 80 and HCO-60, polysaccharides such as
25 carboxymethyl cellulose, sodium alginate and hyarulonic
acid, and polysorbate), a preservative (e. g., methyl
paraben, propyl paraben), an isotonizing agent (e. g.,
sodium chloride,mannitol,sorbitol,glucose),buffer(e.g.
calcium carbonate), pH adjusting agent (e. g. sodium
30 phosphate, potassium phosphate), etc., and may be also
prepared as an oily suspension by dispersing Compound ( I )
in a vegetable oil such as sesame oil or corn oil with or
without a phospholipid such as lecithin, or a
moderatelength fatty acid triglyceride (e. g., MIGLYOL
812 ) .
When the the pharmaceutical composition of the
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
31
present invention is administered locally to a joint of a
patient of osteoarthritis, the pharmaceutical composition
may be the preperation in which Compound ( I ) is dispersed
in the injectable hyalronic acid pharmaceutical
composition (e.g., KAKENSEIYAKU, trade name: *ALTZ). The
hyaluronic acids can be used as its pharmaceutically
acceptable salts. The salts include alkali metal salts
(e.g. , sodium salts , potassium salts ) and alkaline earth
metal salts (e.g., calcium salts, magnesium salts ),
prepherably sodium salts. The weight-average molecular
weight of the hyaluronic acid or a salt thereof about
200 , 000 to 5 , 000 , 000 , preferably about 500 , 000 to 3 , 000 , 000 ,
more preferably about 700,000 to 2,500,000.
The concentration of Hyaluronic acid or the sodium
salts in the dispersion medium dispersing Compound ( I ) is
less than 1% (W/v) , preferably about 0.02 to I% (W/v) , more
preferably about 0.1 to 1%(W/v) , because its viscosity is
proper to administrate by injection.
The dispersion medium can include pH regulators,
local anesthetics, antiiotics, dissolution aids,
isotonizing agents, adsorption preventing agents,
glycosaminoglycans, polysaccharides and the like, which
are conventionaly used in this field.
Preferable examples include mannitol, sorbitol,
sodium chloride, glycine, ammonium acetate or water-
soluble protein which is substantally inactive in the body.
Preferable glycosaminoglycan include hyaluronic
acid. condroitin, condroitin sulfate A, condroitin sulfate
C, dermatan sulfate, heparin, heparan sulfate, and the
like .
Preferable water-soluble protein include, which is
disolved in water or physiological salt solution, include
humanserum albmin,humanserum globulin, collagen, gelatin,
etc.
Preferable pH regulators include glycine, ammonium
acetate, citric acid, hydrochloric acid, sodium hydroxyde,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
32
etc.
Preferable local anesthetics include chlorobutanol,
lidocine hydrochloride, etc.
Preferable antibiotics include gentamicin, etc.
Preferable dissolution aids include glycerin,
polyethyleneglycol-400, etc.
Preferable isotonizing agents include mannitol,
sorbitol, sodium chloride, etc.
Preferable adsorption preventing agents include
polyoxyethylene sorbitan monooleate, etc.
The dose of the water-soluble protein may be about
0. 05 to 50 mg, preferably about 0. 5 to 20 mg, more preferably
about 0.75 to 10 mg per a injectable preperation.
The phosphate or its salts (e.g. , sodium phospate,
pottasium phosphate ) can enhance the activity of the
pharmaceutical composition of the present invention. The
concentration of sodium phosphate or potassium phosphate
in the in~ectable preparation is about 0.1 mM to 500 mM,
preferably about 1 mM to 100 mM.
The preferable preparation of the present invention
is as follows.
(A) a lactic acid-glycolic acid copolymer:
wherein the ratio of lactic acid/glycolic
acid is about 90/10 to 50/50 (w/w) and the weight-average
molecular weight is about 8000 to 50000,
(B) Compound (I}: (2R,4S)-(-)-N-[4-
(diethoxyphosphorylmethyl)phenyl]-1,2,4,5-tetrahydro-4-
methyl-7,8-methylenedioxy-5-oxo-3-benzothiepine-2-
carboxamide, and
(C) phosphoric acid or its salt: sodium
phosphate.
The content ratio of ( B ) based on ( A ) is about
5 to 30% ( w/w ) . The content ratio of ( C ) based on ( A ) and
(B) is about 0.1 to 20% (w/w).
The pharmaceutical composition of the present
invention is preferably a suspension as described above.
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
33
The pharmaceutical composition of the present
invention is preferably in the form of fine particles . This
is because said pharmaceutical composition is less likely
to cause excess pain to the patient when administered
through an injection needle for ordinary subcutaneous or
intramuscular injection.
The pharmaceutical composition of the present
invention is preferably an injectable preparation.
Methodsof preparing the pharmaceutical composition
of the present invention as a sterile preparation include,
but are not limited to, the method in which the entire
production process is sterile, the method in which gamma
rays are used as sterilant, and the method in which an
antiseptic is added.
Since the pharmaceutical composition of the present
invention has excellent chondrogenesis promoting effect,
cartilage destruction suppressing effect and cartilage
cell differentiation induction promoting effect, it can be
used in prevention and treatment of a cartilage disease ( for
example, chronic rheumatoid arthritis, osteoarthritis of
knee. Among these diseases, a lesion fixed and covered
frequently with a brace is especially applicable to a
sustained-release preperation formulation according to the
present invention, since it requires a promoted cure
continuously only by a single administration rather than
frequent administrations.
The pharmaceutical composition according to the
present invention may be used also in combination with other
pharmaceuticals for treating articular diseases. For
example, when Compound (I) is used as a cartilage
destruction preventing/cartilage cell differentiation
induction promoting agent, other pharmaceuticals for
treating articular diseases may be used in combination.
Examples of such pharmaceuticals employed in combination
are antiinflammatory steroidal agents (e. g. , prednisolone,
hydrocortisone, methylprednisolone, dexamethasone,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
34
betamethasone etc.), non-steroidal
antiphlogistic/analgesic agents (e. g., indomethacin,
diclofenac, loxoprofen, ibuprofen, aspirin, piroxicam,
sulindac, etc.) or hyaluronic acid formulations (e. g.,
sodium hyaluronate, etc.).
The pharmaceutical composition according to the
present invention is used as a safe and highly potent
formulation suitable to be used in prevention and treatment
of a cartilage disease as well as repair and regeneration
of a cartilage tissue. For example, it can exert a
chondrogenesis promoting effect locally and efficiently,
and improves the quality of life in a patient whose routine
activities are affected adversely by a pain due to the
wornout or the destruction of an articular cartilage.
The dose of the pharmaceutical composition of the
present invention may be an pharmacologically effective
amount of Compound (I), although depending on type of
Compound (I), the patient's condition, the way of
administration, releasing time of the active ingredient,
and subject hosts, etc. For example, when the
pharmaceutical composition of the present invention is
administered orally, administration dose is in the range
of about 5 to about 1000 mg, preferably about 30 to about
600 mg, based on the active ingredient content (e. g.,
compound (I)), per adult (weighing 50 kg), and it can be
administered once to three times a day dividedly. When the
pharmaceutical composition of the present invention is
administered non-orally, it may be administered at about
0.1 to 500 mg, preferably about 1 to 50 mg, based on the
active ingredient content ( a . g . , compound ( I ) ) , per adult
(weighing 50 kg) , and it can be administered once to three
times a day dividedly. When the sustained-release
preperation of the present invention is administered
non-orally, Compound ( I ) may be released about 0 .1 to about
100 mg a week.
In addition, Compound ( I ) can safely be used because
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
of its low toxicity. For example, 500 mg/kg/day of Compound
A given orally for 2 weeks in rats caused no abnormal
findings. Since Compound A is more excellent in terms of
absorption especially after oral administration when
5 compared with a corresponding racemate, it can
advantageously be used in an oral formulation.
Brief Description of the Drawings
Figure 1 indicates the results of the examination
10 for the effect on Interleukin-1-stimulated type-I
collagenase release in rabbit cartilage cell observed in
Experiment 1.
Figure 2 indicates the results of the examination
for the effect on Interleukin-1-stimulated proteoglycan
15 release in rabbit cartilage cell observed in Experiment 2.
Figure 3 indicates the results of the examination
for the effect on Interleukin-1-stimulated proteoglycan
synthesis observed in Experiment 3.
20 Best Mode for Carrying out the Invention
The present invention is hereinafter described in
more detail by means of the following working examples,
which are not to be construed as limitative.
Compound: (ZR,4S)-(-)-N-[4-
25 (diethoxyphosphorylmethyl)phenyl]-1,2,4,5-tetrahydro-4-
methyl-7,8-methylenedioxy-5-oxo-3-benzothiepine-2-
carboxamide (hereinafter also referred to as Compound A)
is prepared according to the method described in Example
1 of Japanese Patent Laid Open Publication 231569/1996.
Examples
Example 1
About 8 g of a lactic acid-valerolactone copolymer
(PLV 2500ML, produced by Taki Chemical, hereinafter also
referred to as PLV) or a glycolic acid-caprolactone co-
polymer (PGC 2500MG, produced by Taki Chemical, hereinafter
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
36
also referred to as PGC ) was placed in a centrifugal tube
and heated to about 50 °C in a water bath. About 80 mg of
compound A was mixed in each tube, followed by uniform
dispersion, to yield an ointment preparation, which was
stored at a cold place.
Example 2
500 mg of microcapsule prepared in the same manner
as Example 1 of Japanese Patent Laid Open Publication
263545/1997 was uniformly dispersed in two test tubes of
fibrinogen solution for Tisseel (produced by Nippon Zoki
Pharmaceutical). Thrombin solution of two test tubes for
Tisseel were gradually added. Subsequently, the mixture
was immediately aspirated into a plastic syringe. The
syringe was kept standing at 37 °C for 30 minutes to solidify
the content. After solidification, the content was
extruded from the syringe tip and cut using a razor into
pellets about 200 lcl in volume.
Example 3
4 mg of Compound A was filled in the bone defect
filler hollow hydroxyapatite (Boneceram P, produced by
Sumitomo Pharmaceuticals, 3 mm diameter, 14 mm length, 1
mm pore size). Both ends of the hollow were sealed with
clay.
Example 4
To microcapsule prepared in the same manner as
Example 1 of Japanese Patent Laid Open Publication
263545/1997 which contains compound A (content ratio 4%),
20~ pulverized gelatin (produced by Nitta Gelatin) was
added, to yield a microcapsule-containing tablet
preparation 5.5 mm in diameter and 125 mg in weight.
Example 5
Microcapsule containing compound A (content ratio
10~ ) was prepared in the same manner as Example 1 of Japanese
Patent Laid Open Publication 263545/1997, except that PLGA
having a lactic acid-glycolic acid content ratio of 85/15
(mold) and weight-average molecular weight of 14,900
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
3T
(produced by Wako Pure Chemical Industry). Mean particle
size was 31 lam.
Example 6
A dichloromethane solution containing 2.4 g of PLGA
( produced by Wako Pure Chemical Industry ) whose the lactic
acid/glycolic acid content ratio is 85/15 and the
weight-average molecular weight is 14, 900 and 0.1 g of the
compound A was prepared in the same manner as Example 1 of
Japanese Patent Laid Open Publication 263545/1997. And
0.2 g of estradiol was added into the solution. Further
PVA solution was put into the solution to obtain O/W
emulsion. Microcapsule containing the compound A and
estradiol was prepared. The mean particle size was 27 a
m.
Example 7
Preparation of capsule:
2 mg of hyaluronic acid (Q. P. Corporation, trade
name: HATMKW7001, MW: about 2,100, 000), 2 mg of sodium
chloride and 1 mg of Tween 80 ( NIKKO Chemical , trade name
*RHEODOL*TW-0120 INJ NF) were weighed and dissolved in 1
ml of distilled water for injection to form a dispersion
medium.
Separately, a microcapsule containing Compound A at
10 ~ was prepared by the method described in Example 6 in
Japanese Patent Application Laid-Open 263545/1997. 10 mg
of this microcapsule was weighed and dispersed in 0.2 ml
of the dispersion medium described above to form an aqueous
suspension. The suspension was used as a pharmaceutical
composittion for prevention and treatment of cartilage
disease which contained Compound A as an active ingredient .
Experiment 1
Effect on the release of type-I collagenase in rabbit
cartilage cell stimulated by Interleukin-1 (IL-1):
A cartilage cell was prepared and cultured according
to the method by Suzuki et al ( SHINSEIKAGAKUJIKKENKOZA 18 ,
Cell culture technology: 871-875, 1990). Thus, a Japanese
CA 02334815 2000-12-05
WO 99/65474 PCT/.TP99/03154
38
white rabbit (male, 400 to 500 g) just after weaning was
asphyxiated with C02 gas, and the whole body was sterilized
with 70 ~ ethanol and then a thorax was isolated aseptically
in a ventilated clean chamber. Each single costal
cartilage/bone margin was isolated to obtain a cartilage
piece free from circumferential soft tissues. This
cartilage piece was cut into pellets using a scalpel, washed
with Tyrode's solution free from Ca2' or Mg2', and then
suspended in a Dulbeco's modification of Eagle's medium
, (DMEM) containing 10 ~ calf fetal serum, and then inoculated
into a 12-well microplate at the cell density of 4 x 10'
cells/well. The cell was cultured for one week and the
culture medium was exchanged with a serum-free medium, and
then IL-1~ (30 ng/ml) and Compound A were added at
predetermined concentrations . After 48 hours , the culture
supernatant was recovered and the collagenase activity in
the ' culture supernatant was determined using a commercial
kit Type-I collagenase activity assay kit (COSMOBIO). The
results are shown in Figure 1 ( * : p<0 . 05, Dunnet test ) . In
Figure 1, the control is a sample containing no Compound
A.
As evident from the results shown in Figure 1,
Compound A exhibited type-I collagenase (metalloprotease
I ) release suppressing effect, and had an inhibitory effect
on a cartilage destruction due to arthritis.
Experiment 2
Effect on the release of proteoglycan in rabbit cartilage
cell stimulated by Interleukin-1
A cartilage cell was prepared and cultured according
to the method by Suzuki et al (SHINSEIKAGAKUJIKKENKOZA 18,
Cell culture technology: 871-875, 1990). Thus, a Japanese
white rabbit (male, 400 to 500 g) just after weaning was
asphyxiated with COZ gas , and the whole body was sterilized
with 70 ~ ethanol and then a thorax was isolated aseptically
in a ventilated clean chamber. Each single costal
cartilage/bone margin was isolated and circumferential
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
39
soft tissues were removed as far as possible and then the
margin between a bone and a cartilage was cut to obtain a
cartilage piece free from other tissues. This cartilage
piece was cut into pellets using a scalpel, and placed in
Tyrode's solution free from Ca2' or Mgz+, and treated
successively with EDTA (ethylenediaminetetraacetic acid)
( 0 . 1% , 20 minutes , twice ) , trypsin ( 0 .15% , 60 minutes ) and
collagenase ( 0 . 2% , 3 hours ) at 37°C , whereby dispersing the
cartilage cell. After washing the cell with the same
Tyrode's solution, the cell was suspended in Dulbeco's
modification of Eagle's medium (DMEM) containing 10 % calf
fetal serum, and then inoculated into a 96-well microplate
at the cell density of 1 x 10' cell/well. The cell was
cultured for 4 days and then cultured in a serum-
free/ ['SS ] sulfuric acid at 0 . 5 % ~Ci ( 1 x 106 dpm/well ) for
3 days, and then after washing twice it was cultured in
[ssS]sulfuric acid-free medium. After 6 hours, IL-1~ (3
ng/ml) and Compound A were added again at predetermined
concentration and then the culture was continued. 24 Hours
after this addition, the culture supernatant was recovered
and counted for the radioactivity. The cell was extracted
with 4M guanidine hydrochloride and then subjected to the
determination as described above. The results are shown
in Figure 2 . In Figure 2 , the control is a sample containing
no Compound A. "**" designates p<0.01 when compared with
IL-1 free sample ( Student t-test ) . "##" designates p<0 . O1
when compared with IL-1-supplemented sample (Dunnet test).
As evident from the results shown in Figure 2,
Compound A had a proteoglycan release suppressing effect,
thus a cartilage destruction suppressing effect.
Experiment 3
Effect on proteoglycan synthesis in rabbit cartilage cell
stimulated by Interleukin-1 (IL-1):
The cartilage cell prepared by the method similar
to that in Experiment 1 was suspended in Dulbeco's
modification of Eagle's medium (DMEM) and inoculated into
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
a 96-well microplate at the concentration of 1 x 10'
cells/well. After culturing for 4 days, the cell was
treated with IL-1 (0.3 ng/ml} in the absence of serum for
24 hours . Subsequently, the cell was washed and cultured
5 for 48 hours in the presence of ['SS] sulfuric acid. After
completion of the culture, the cell was washed with
physiological saline, extracted with 4 M guanidine
hydrochloride, and the resultant solution was counted for
the radioactivity, whereby assaying the synthesis of
10 proteoglycan. The results are shown in Figure 3. In
Figure 3, the control is a sample containing IL-1 (0.3
ng/ml) in the absence of test substance. Compound A was
added at 10 NM simultaneously with ['SS ] sulfuric acid ( ( 1 )
in Figure 3 ) . Insulin-like growth factor ( IGF ) -I was added
15 at 10 ng/ml simultaneously with ['SS ] sulfuric acid ( ( 2 ) in
Figure 3). (3) in Figure 3 indicates the addition of 10
~.M of Compound A and 10 ng/ml of IGF-I which was simultaneous
with the addition of ['SS ] sulfuric acid . In Figure 3 , the
legends mean as follows: *P<0.05, **P<0.01 based on Dunnet
20 test when compared with control; ##p<0.01 based on Student
t-test when compared with sample containing only IGF-I;
$$p<0.01 based on Student t-test when compared with sample
containing only Compound A.
As evident from the results shown in Figure 3,
25 Compound A had proteoglycan synthesis promoting effect.
Experiment 4
A mouse cell line ATDCS (purchased from RIKAGAKU
KENKYUSHO ) was inoculated into a 6-well microplate at the
density of 1 x 105 cells/well and cultured in DMEM/F-12 in
30 the presence of 5 ~ calf fetal serum and 10 ~,g/ml insulin.
On the 7th day of the culture, 10 N.M of Compound A was added
and the culture was continued. The culture medium was
replaced every 1 to 2 days. On the 14th day and the 21st
day, the cell was recovered and subjected to the extraction
35 of mRNA using STAT60 (COSMOBIO) followed by RT-PCR method
to quantify type II collagen mRNA expressed. As a control,
CA 02334815 2000-12-05
WO 99/65474 PCT/JP99/03154
41
actin mRNA expressed was determined.
The results are shown in Table 1. The control was
the culture in the presence of 10 ~g/ml insulin.
Table 1
type II collagenmRNA/ actin mRNA
I4cn 2lat
Control 0.0669 0.0325
IOI~M of Compound 0.1586 0.2876
A
As evident from the results shown in Table I,
Compound A had type II collagen mRNA synthesis promoting
effect.
Industrial Applicability
The pharmaceutical composition of the present
invention has potent cartilage destruction suppressing
effect, chondorogenesis promoting and cartilage cell
differentiation induction promoting effect, proteoglycan
synthesis promoting effect, type II collagen synthesis
promoting effect, metalloprotease I release suppressing
effect and proteoglycan release suppressing effect, and is
useful in prevention and treatment of cartilage disease.