Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
TITLE
2-FLUORO-2-ALKYL ALKANOAMIDES WITH ANTICONVULSANT ACTIVITY
FIELD OF THE INVENTION
This invention relates to 2-fluoro-2-alkyl alkanoic acid amides, to the
preparation of
these compounds, and to the use thereof as anti-convulsive therapeutic agents.
The
anticonvulsant fluorinated amides of the invention have an improved
therapeutic ratio compared
to valproic acid and valpromide with respect to sedative effects, and have
reduced teratogenic
potential. In general, the fluorinated amides of this invention also exhibit a
more rapid onset of
action than the corresponding fluorinated acids. The invention provides
effective anti-epileptic
agents with a greater margin of safety than either valproic acid or
valpromide.
BACKGROUND OF THE INVENTION
Epilepsy affects roughly 1 % of the world's population. Among the drugs
employed for
control of epileptic seizures is valproic acid. Valproic acid (also referred
to as VPA, valproate,
or 2-propylpentanoic acid) is an effective anticonvulsant, but it has a short
duration of action.
More seriously, VPA suffers from serious side effects, among them sedation,
potentially fatal
hepatotoxicity, and teratogenicity. Hepatotoxicity is particularly a problem
in young children,
especially children on polytherapy. The VPA-induced hepatic fatality rate
among the latter
patient category is reported to be 1/500 (F. E. Dreifuss et al., Neurolo~v
(1987), 37, 379-385).
Valproic acid has been shown to induce neural tube defects in mice, and it is
estimated that the
risk of spina bifida among newborns of women taking VPA during pregnancy is 1-
2% (Centers
for Disease Control, Morbidit~and Mortality Weekly Report (1983), 32(33), 438-
439).
There has been a considerable effort to discover analogues of valproic acid
that are
equally effective, but that have a greater margin of safety. See, for example,
H. Nau et al., PCT
application WO 94/06743, wherein a variety of modifications to the alkyl
chains of valproic
acid are made, and the related U.S. Patent 5,786,380.
With regard to teratogenicity, it has been reported that introduction of a
triple bond into
the 4-position of valproic acid greatly increases teratogenicity, but that
this effect is largely
confined to the S-(-) enantiomer. Addition of a methyl group to the end of the
triple bond
abolished teratogenicity, while maintaining anticonvulsant activity (H. Nau,
R.-S. Hauck, K.
Ehlers, Pharmacologr~& Toxicology (1991), 69, 310-321.) These results
indicated that
separation of teratogenicity and anticonvulsive activity was possible.
Sedative side effects were
475049 f
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-2-
also separated from anticonvulsant activity in some analogues (M. Elmazar, R.-
S. Hauck, H.
Nau, J. Pharm. Sci. (1993), 82, 1255-1288.)
Alpha-branched carboxylic acids with an alpha-fluorine are little known. P.
Crowley et
al., in European patent application EP 468681, refers to 2-ethyl-2-
fluorobutanoic acid as a
fungicide intermediate, and a method for its preparation. Takeuchi refers to
several examples
of this class of compound in a publication relating to methods of preparing
tertiary alkyl
fluorides (Y. Takeuchi et al., J. Org. Chem. (1993), 58(13), 3483-3485).
The valproic acid analogue 2-fluoro-2-propyl-4-pentenoic acid has also been
reported.
The compound was used as a probe for studies of valproic acid hepatotoxicity
and metabolism.
(W. Tang et al., Chem. Res. Toxicol. (1995), 8(5), 671-G82; M. Jurima-Romet et
al.,
Toxicolo~v (1996), 112(1), 69-85; W. Tang and F. Abbott, Drue Metab. Dispos.
(1997), 25(2),
219-227.} In the above references, the presence of the 2-fluoro substituent
was reported to
reduce hepatotoxicity relative to 2-propyl-4-pentenoic acid. Anticonvulsant,
sedative or
teratogenic properties of the fluorinated compound were not disclosed.
Alpha-fluorinated valproic acid, 2-fluoro-2-propylpentanoic acid, has also
been reported
(Ph.D. thesis of Wei Tang, University of British Columbia, 1996). The
anticonvulsant activity
and pharmacokinetics of this compound were studied, and its pharmaceutical
potential was
speculated upon (F. Abbott, W. Tang, J. Palaty, J. Pharmacol. Ex~ Ther.
(1997), 282, 1163-
1172). The compound was reported to be less potent than VPA, and the
hepatotoxic, sedative,
or teratogenic properties were not disclosed.
Valproic acid analogues with terminal trifluoromethyl groups have been
reported: 5,5,5-
trifluoro-2-(3,3,3-trifluoropropyl) pentanoic acid (K. Yamaguchi and M.
Taninaka, Japanese
patent Application 4-21652 (1992), and 5,5,5-trifluoro-2-n-propyl pentanoic
acid (Hiroshima et
al., Japan J. Psychopharmacol. (1992) 12, 427). These compounds, too, are less
potent than
VPA.
Valpromide (VPD, the amide of valproic acid) is a widely-used prodrug of
valproic
acid. VPD is known to be rapidly metabolized in humans to valproic acid after
oral
administration (M. Bialer, Int. J. Pharm. (1985) 23 25-33). Interspecies
variations in the rate of
metabolism are considerable; in mice, for example, metabolic hydrolysis is
known to be
relatively slow. Certain beta-alkylated analogues such as 2-ethyl-3-methyl-
pentanoamide
(valnoctamide) are resistant to this route of metabolism. See A. Haj-Yehia, M.
Bialer et al., J.
Pharm. Sci., 79, 719-724 (1990), and references therein. Certain substituted
cyclopropane
carboxamides have also been found to be metabolically stable anticonvulsants:
M. Bialer et al.,
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-3-
Pharm. Res., 13, 284-289 (1996). The resistance to hydrolysis among these
compounds is
attributed to steric effects.
SUMMARY OF THE INVENTION
This invention relates to 2-fluoro-2-alkyl alkanoamides, pharmaceutical
compositions
containing these compounds, and their use to treat or prevent convulsions.
This invention also
provides processes for the preparation of these compounds. The preferred 2-
fluorinated
carboxamides of the invention exhibit greatly reduced embryotoxicity and
teratogenicity, an
improved therapeutic ratio with respect to sedation, and a longer duration of
activity, when
compared to valproic acid or valpromide. The 2-fluorinated carboxamides of
this invention
unexpectedly provide a more rapid onset of activity than do the corresponding
2-fluorinated
carboxylic acids.
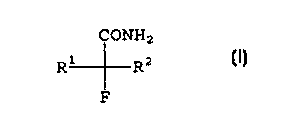
The invention provides compounds of formula I below:
R2 CONH2
(I)
R1 F
wherein R' and RZ are independently chosen from the group consisting of C3 to
CS alkyl, C3 to
CS cycloalkyl, (cyclopropyl)methyl, 1-(cyclopropyl)ethyl, 2-
cyclopropyl(ethyl), and
(cyclobutyl)methyl. The term "alkyl" as used herein is intended to include
both straight-chain
and branched alkyl groups.
The invention also provides a method for treating and/or preventing
convulsions due to
a variety of causes, by administering to an individual in need of such
treatment a therapeutically
or prophylactically effective amount of at least one of the compounds of this
invention.
In addition the compounds of this invention may be useful for treating and/or
preventing
affective disorders, especially the mania phase of bipolar depression; and
migraine.
An object of this invention is to provide compounds useful for preventing or
reducing
seizure activity in a mammal.
Another object of this invention is to provide anti-convulsant pharmaceutical
compositions comprising at least one compound of this invention.
Yet another object of this invention is to provide methods of preventing or
reducing
seizure activity by administering to an individual in need of such treatment a
pharmaceutical
composition of this invention.
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-4-
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1: Dose-effect curves of valproic acid and several 2-alkyl alkanoamides
and 2-
fluoro-2-alkyl alkanoamides.
3Me-VPD = 3-methyl valpromide, 3-methyl-2-propylpentanoic acid;
2F-VCD = 2-fluoro valnoctamide, 2-ethyl-2-fluoro-3-methylpentanoic acid;
VCD = valnoctaamide, 2-ethyl-3-methylpentanoic acid;
2F-VPD = 2-fluorovalpromide, 2-fluoro-2-propylpentanoamide;
VPD = valpromide, 2-propylpentanoamide;
VPA = valproic acid, 2-propylpentanoic acid.
I O Figure 2: Representative example of synthesis of compounds of the
invention:
2-fluoro-valnoctamide.
DETAILED DESCRIPTION OF THE INVENTION
The invention provides compounds having the following structure:
CONHZ
CI)
~F
15 where R~ and RZ are independently C3 to CS n-alkyl, C3 to CS branched
alkyl, C3 to CS
cycloalkyl, (cyclopropyl)methyl, 1-(cyclopropyl)ethyl, 2-cyclopropyl(ethyl),
or
(cyclobutyl)methyl. The term "alkyl" as used herein is intended to include
both straight-chain
and branched alkyl groups. In a preferred embodiment, Rl and RZ are
independently C3 to C4
alkyl, cyclopropyl, cyclobutyl, or (cyclopropyl)methyl. In another preferred
embodiment, R~ is
20 n-propyl, i-propyl, n-butyl, or 1-methylpropyl; and RZ is C3 to CS alkyl,
C3 to CS cycloalkyl,
(cyclopropyl)methyl, 1-(cyclopropyl)ethyl, 2-cyclopropyl(ethyl), or
(cyclobutyl)methyl. In a
more preferred embodiment, R~ and Rz are independently n-propyl, i-propyl, n-
butyl, or 1-
methylpropyl. Most preferably, the compound is selected from the group
consisting of 2-
fluoro-2-n-propylpentanoamide, 2-fluoro-2-ethyl-3-methylpentanoamide, and 2-
fluoro-2-(1-
25 methylpropyl)pentanoamide.
The compounds of this invention exhibit unexpected advantages over the
compounds of
the prior art with respect to their pharmacodynamics. While VPA and valpromide
have a very
rapid onset of activity, they suffer from the above-mentioned side effects of
sedation and
teratogenicity. While 2-fluoro-VPA has been found to be free of teratogenic
effects, it is much
30 less potent than VPA, and it does not reach maximal activity until about
one hour after
CA 02335598 2000-12-21
WO 99/67199 PCT/US99113941
-5-
administration. Also, the duration of activity of 2-fluoro-VPA is relatively
short, with in vfvo
activity lasting for only about 75 minutes (see Table 3).
In contrast to 2-fluoro-VPA, the compounds of this invention, as exemplified
by 2-
fluoro-2-propylpentanoarnide (example 5) exhibit a rapid onset, reaching
maximal activity
within 15 minutes (comparable to that of VPA and valpromide; see Table 3).
This is combined
with a long half life of 11.3 hours, versus 1.4 hours for VPA (see below), and
0.84 hours for
valpromide (in humans: M. Bialer et al., 1985, Int. J. Pharm., 23, 25-33).
Thus, 2-fluoro-2-
propylpentanoamide combines the rapid onset of VPA and valpromide with the
safety of 2-
fluorovalproic acid, and additionally has a remarkably and surprisingly long
half life. Such
properties have not been previously obtained with anti-convulsants of this
class.
There are two major reasons for employing a pro-drug form, which are to
improve
absorption/distribution properties, and/or to provide more prolonged
pharmacological effects.
Pro-drug forms must undergo metabolic conversion to their pharmacologically
active forms,
hence pro-drug forms will generally exhibit either a delayed onset of activity
relative to the
parent drug, or at best an onset of action comparable to that of the parent
drug.
Unexpectedly, the alpha-fluoro carboxamides described herein have been found
to
exhibit a significantly faster onset of action than the corresponding
carboxylic acids, even in the
absence of steric effects. This effect is observed upon parenteral
administration, and therefore
it is not due to more rapid absorption from the gastrointestinal tract. The
compounds are also
more potent than the corresponding acids, and have a longer duration of
activity.
The 2-fluoro carboxamides of this invention thus appear to possess intrinsic
anti-
convulsant activity. These compounds are not merely pro-drugs for the
fluorinated carboxylic
acids, but possess their own uniaue pharmacology. Accordingly, this invention
provides alpha-
fluorinated carboxamides, which are useful for treating or preventing
seizures, which are not
teratogenic, and which upon metabolism generate non-teratogenic metabolites
which are
themselves anticonvulsants.
The compounds of the invention can be prepared by methods known in the art for
the
preparation of other alpha-fluoro carboxylic acids, followed by conversion to
the amide. For
example, treatment of a 2-hydroxy-2-alkyl alkanoate ester with
diethylaminosulfur trifluoride
(DAST} provides the corresponding alpha-fluoro ester, which upon hydrolysis
provides the acid
(P. Crowley et al., 1992, European patent application EP 468681).
Alternatively, a 2-amino-2-
alkyl alkanoic acid can be subjected to diazotization in the presence of
fluoride ion, to effect a
deaminative fluorination (J. Barber, R. Keck, J. Retey, Tetrahedron Letters
(1982), 23, 1549-
CA 02335598 2000-12-21
WO 99167199 PCT/US99/13941
-6-
1552). The Reformatsky reaction can be carried out on a 2-bromo-2-
fluoroalkanoate ester (Y.
Takeuchi et al., J. Org. Chem. (1993), 58(13), 3483-3485) to introduce a 2-
alkyl group. In one
embodiment that is preferred for laboratory-scale synthesis, an ester enolate
or silyl enol ether
of a 2-alkyl alkanoic acid is fluorinated with a "positive fluorine" source,
such as an N-fluoro
pyridinium salt, N-fluoro amide, or N-fluoro imide (see, e.g., E. Differding,
G. Ruegg,
Tetrahedron Letters (1991), 32, 3815-3818). In the particular embodiment
exemplified below,
the lithium enolate of methyl 2-propylpentanoate is fluorinated with N-fluoro
benzenesulfonimide, but it will be understood that other methods of synthesis
are within the
scope of this invention.
The starting esters for the exemplified process are in many cases commercially
available; alternatively they may be obtained by methods known in the art, for
example the
malonic ester synthesis described in M. Elmazar, R.-S. Hauck, H. Nau, J.
Pharm. Sci. (1993),
82, 1255-1288. See also H. Nau et al., 1994, PCT International Application,
publication No.
WO 94/06743, and US patent application serial No. 08/344,810. The well-known
acetoacetate
variation of the malonic ester synthesis is also applicable. In the example
below, direct
alkylation of a straight-chain ester enolate is employed. Other methods of
synthesis will be
apparent to those skilled in the art, and this invention is not limited by the
particular synthetic
method exemplified herein.
In general terms, this invention provides a process for preparation of
compounds of
structure I, which comprises the steps of:
a) alkylating a compound of structure
COORS
R1 H (II)
COR4
wherein R~ is as defined above and R3 is lower alkyl (preferably C, to Ca
alkyl) or
benzyl, and R4 is lower alkyl (preferably C, to Ca alkyl), lower alkoxy
(preferably C~ to C4
alkoxy), or benzyloxy, with an alkylating reagent of structure
Rz X (III)
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
wherein X is a suitable leaving group, and wherein RZ is as defined above, in
the presence of a
suitable base, so as to obtain a compound of structure
COORS
R1 R2 C IV)
COR4
(b) in the cases where R4 is lower alkoxy or benzyloxy, hydrolysis,
decarboxylation, and
re-esterification if necessary of compound IV; or in cases where R4 is lower
alkyl, deacylation
S of compound IV, so as to obtain a compound of structure
COORS
R1 R2 (V)
H
(d) enolization of compound V with a suitable base, in an inert solvent;
(e) addition of a fluorinating reagent, so as to obtain a compound of
structure
COORS
R1 R2 (VI)
F
(f) hydrolysis of the ester moiety,
and
(g) conversion of the carboxylic acid to a carboxamide.
Suitable identities of group R'' will be apparent to those skilled in the art.
Preferred R4
groups are alkoxy groups which are readily saponified, such as methoxy or
ethoxy, or other
carboxylic acid protecting groups which are readily removed by other means,
such as tert-
butoxy, benzyloxy, and the like. It will be appreciated that the term
"hydrolysis" as used in
steps (b) and (f) is intended to encompass the deprotecting operations
appropriate to the nature
of R4, for example saponification in the case of lower alkoxy groups,
acidolysis in the case of
tert-butoxy groups or hydrogenolysis in the case of benzyloxy groups. Similar
considerations apply to the group R3, which is preferably lower alkyl such as
methyl or ethyl
but which may be another carboxy-protecting group, such as for example, tert-
butyl, benzyl,
and the like. It will be apparent that in step (b), where R3 is removed and
then re-introduced,
the practitioner will have the opportunity to change the identity of R3 if it
is desired to do so.
Where R4 is alkyl, the preferred groups are those which lend themselves to
deacylation
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
_g_
of the group COR4; in these cases R4 is most preferably methyl. Deacylation
may be
accomplished by treatment with, for example, sodium hydroxide or ammonia, or
other methods
known in the art.
Suitable leaving groups X may be selected from, but are not limited to, the
halogens
chlorine, bromine, or iodine, or sulfonate ester groups such as
methanesulfonyloxy or
toluenesulfonyloxy. Suitable bases for step (a) may be chosen from, but are
not limited to,
alkali metal alkoxides, calcium and magnesium alkoxides, alkali metal
hydrides, and the like.
Suitable bases for step (d) will be apparent to those skilled in the art,
since a number of
procedures for enolizing esters have been published. Preferred bases will be
those with a
sufficiently high pKa to substantially deprotonate the compound V, and which
are also non-
reactive with the functional groups of the compound V. Examples may be chosen
from, but are
not limited to, the lithium or sodium salts of hindered disubstituted amines,
such as lithium
diisopropylamide or lithium hexamethyldisilazide.
In an alternative process, compounds of formula (I) may be prepared by the
process
I S comprising the steps of:
a) alkylating a compound of structure
COORS
R1 H (VII)
H
wherein R~ is as defined above, and R3 is lower alkyl or benzyl, with an
alkylating
reagent of structure
RZ X (III)
wherein X is a suitable leaving group as described above, and wherein Rz is as
defined
above, in the presence of a suitable base, so as to obtain a compound of
structure
COORS
R1 Rz (V)
H
(b) enolization of compound V with a suitable base, in an inert solvent;
(c) addition of a fluorinating reagent, so as to obtain a compound of
structure
COORS
R1 Rz (VI)
F
CA 02335598 2000-12-21
WO 99/67199 PCTNS99/13941
-9-
(d) hydrolysis of the ester moiety, and
(e) conversion of the carboxylic acid to the carboxamide.
It will be apparent to those skilled in the art that modifications to the
exemplified
procedure can be made. For example, inert solvents other than THF may be
employed, such as
dioxane, di-alkyl ethers, dimethoxyethane and other polyethers, toluene,
heptane, and the like.
Additives such as hexamethylphosphoramide, tetramethylethylenediamine, or
tetramethylurea
can be employed in the fluorination reaction, and other bases such as alkali
metal hydrides,
alkoxides, or hexamethyldisilazide salts can be employed in the deprotonation
reactions. Such
modifications could be made for any reason, for example to improve the yield
or to reduce
process costs, without departing from the scope of this invention, and
experimentation to
determine the most desirable conditions for any given reaction would be a
routine matter to
those skilled in the art.
By way of example, a procedure for conversion of the carboxylic acid to the
carboxamide is also provided below. The carboxamides of this invention may be
prepared by
the method provided, or by any of a variety of methods, such as condensation
of 2-fluoro-2-
alkyl alkanoic acids with ammonia or ammonium salts in the presence of a
carbodiimide or
other condensation reagent, or by ammonolysis of the corresponding esters.
Such methods will
be apparent to those skilled in the art, and it will be understood that this
invention is not limited
by the exemplified means of synthesis.
By the methods provided herein, the following compounds of this invention may
be
prepared from the appropriate starting materials;
Example No. Compound
1 2-fluoro-2-ethylpentanoamide
2 2-fluoro-2-ethylhexanoamide
3 2-fluoro-2-ethyl-3-methylpentanoamide
4 2-fluoro-2-ethylheptanoamide
5 2-fluoro-2-n-propylpentanoamide
6 2-fluoro-2-n-propylhexanoamide
7 2-fluoro-2-n-propyl-3-methylpentanoamide
8 2-fluoro-2-n-propylheptanoamide
9 2-fluoro-2-i-propylpentanoamide
10 2-fluoro-2-i-propylhexanoamide
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
- 10-
11 2-fluoro-2-i-propyl-3-methylpentanoamide
12 2-fluoro-2-i-propylheptanoamide
i 3 2-fluoro-2-cyclopropylpentanoamide
14 2-fluoro-2-cyclopropylhexanoamide
S 1 S 2-fluoro-2-cyclopropyl-3-methylpentanoamide
16 2-fluoro-2-cyclopropylheptanoamide
17 2-fluoro-2-n-butylhexanoamide
18 2-fluoro-2-(1-methylpropyl)pentanoamide
19 2-fluoro-2-(1-methylpropyl)hexanoamide
20 2-fluoro-2-(1-methylpropyl)-3-methylpentanoamide
21 2-fluoro-2-(1-methylpropyl)heptanoamide
22 2-fluoro-2-cyclobutylpentanoamide
23 2-fluoro-2-cyclobutylhexanoamide
24 2-fluoro-2-cyclobutyl-3-methylpentanoamide
1S 2S 2-fluoro-2-cyclobutylheptanoamide
26 2-(cyclopropyl)methyl-2-fluoropentanoamide
27 2-(cyclopropyl)methyl-2-fluorohexanoamide
28 2-(cyclopropyl)methyl-2-fluoro-3-methylpentanoamide
29 2-(cyclopropyl)methyl-2-fluoroheptanoamide
30 2,2-bis(cyclopropyl)-2-fluoroacetamide
31 2-cyclobutyl-2-cyclopropyl-2-fluoroacetamide
32 2-fluoro-2-i-propyl-3-methylbutanoamide
33 2-fluoro-2-i-propyl-4-methylpentanoamide
34 2-fluoro-2-i-propyl-3,3-dimethylbutanoamide
3S 2-fluoro-2-i-propyl-3-methylhexanoamide
36 2-fluoro-2-i-propyl-4-methylhexanoamide
37 2-fluoro-2-i-propyl-S-methylhexanoamide
38 2-fluoro-2-i-propyl-3,3-dimethylpentanoamide
39 2-fluoro-2-i-propyl-4,4-dimethylpentanoamide
40 2-cyclopropyl-2-fluoro-3-methylbutanoamide
41 2,3-bis(cyclopropyl)-2-fluoropropionamide
42 2-cyclobutyl-2-fluoro-3-methylbutanoamide
43 2-cyclopentyl-2-fluoro-3-methylbutanoamide
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-11-
44 2-(cyclobutylmethyl)-2-fluoro-3-methylbutanoamide
45 2-(1-cyclopropylethyl)-2-fluoro-2-(i-propyl)butanoamide
46 2-(cyclopropylmethyl)-2-fluoro-3-methylbutanoamide
47 2-fluoro-2-(1-methylpropyl)-4-methylpentanoamide
48 2-fluoro-2-(1,1-dimethylethyl)-3-methylbutanoamide
49 2-fluoro-2-(1-methylpropyl)-3-methylhexanoamide
50 2-fluoro-2-(1-methylpropyl)-4-methylhexanoamide
51 2-fluoro-2-( I -methylpropyl)-5-methylhexanoamide
52 2-fluoro-3,3-dimethyl-2-(1-methylpropyl)pentanoamide
53 2-fluoro-4,4-dimethyl-2-(1-methylpropyl)pentanoamide
54 2-cyclopropyl-2-fluoro-3-methylpentanaamide
55 2-cyclobutyl-2-fluoro-3-methylpentanoamide
56 2-cyclopentyl-2-fluoro-3-methylpentanoamide
57 2-(cyclobutylmethyl)-2-fluoro-3-methylpentanoamide
58 2-(1-cyclopropylethyl)-2-fluoro-3-methylpentanoamide
59 2-(cyclopropylmethyl)-2-fluoro-3-methylpentanoamide
60 3-ethyl-2-fluoro-2-(1-methylpropyl)pentanoamide
61 3-ethyl-2-( 1-ethylpropyl)-2-fluoropentanoamide
62 3-ethyl-2-(1-ethylbutyl)-2-fluoropentanoamide
The above compounds are presented merely by way of example, and are not
intended to
limit the scope of the invention.
It will be apparent to those skilled in the art that most of the compounds of
this
invention may exist in enantiomeric and diastereomeric forms. Pure enantiomers
may be
resolved from the racemate by methods well-known in the art, for example by
fractional
recrystallization of diastereomeric amine salts of the corresponding 2-fluoro-
2-alkyl alkanoic
acids, by chromatography of diastereomeric derivatives, or by chiral column
chromatography.
Alternatively, enantiomeric forms may be prepared by chiral synthesis, for
example by
alkylation or fluorination of chiral hydrazones (R.-S. Hauck, H. Nau, 1989,
Toxicology Letters,
49, 41-48) or by alkylation or fluorination of chiral oxazolidinone
derivatives (H. Nau et al.,
1994, PCT International Application, publication No. WO 94/06743, US patent
application
serial No. 08/344,810). Chiral starting materials may also be employed in the
synthesis of the
compounds of this invention. Individual diastereomers, R and S enantiomers,
racemates, and
non-racemic mixtures of enantiomers or diastereomers of structure I are all
contemplated to be
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-12-
within the scope of this invention.
Another object of this invention is to provide a method of treating
individuals with
epilepsy, or others in need of anticonvulsant therapy, with compounds of
formula I. Mammals,
and in particular humans, who would benefit from this method of treatment
include those
exhibiting, or at risk for exhibiting, any type of seizure. For example, the
methods of this
invention are useful for treating individuals with idiopathic generalized
seizures such as
absence, myoclonic and tonic-clonic seizures and partial seizures. Individuals
suffering from
epilepsy, in particular, are expected to benefit from administration of the
compounds of this
invention. The method of the invention comprises administering to an
individual a
therapeutically effective amount of at least one compound of formula I or a
salt or prodrug
thereof, which is sufficient to reduce or prevent seizure activity.
The dose of the compound used in the treatment of such disease will vary in
the usual
way with the seriousness of the disorder, the weight and metabolic health of
the sufferer, and
the relative efficacy of the compound employed. The preferred initial dose for
the general
patient population will be determined by routine dose-ranging studies, as are
conducted for
example during clinical trials. Therapeutically effective doses for individual
patients may be
determined by titrating the amount of drug given to the individual to arrive
at the desired
therapeutic or prophylactic effect while minimizing untoward side effects, as
is currently done
with valproic acid. Dosages may be similar to those used with VPA, however
they may be
adjusted appropriately, based on the potencies and kinetic parameters
disclosed herein or as
determined by routine methods.
For example, the compound 2-fluoro-2-n-propylpentanoamide would be expected to
be
useful at dosages which are about 25% of those used for VPA. A preferred
initial dose for this
compound, accordingly, may be estimated to be between about 1 and 60
mg/kg/day, more
preferably between S and 15 mg/kg/day. The initial dose may be varied so as to
obtain the
optimum therapeutic effect in the patient, and may be provided as a daily dose
or in a divided
dose regimen. In general, the compounds of this invention will be provided in
doses of between
1 and 150 mg/kg/day.
Administration of the compounds of this invention may be by any method used
for
administering therapeutics, such as for example oral, parenteral, intravenous,
intramuscular,
subcutaneous, or rectal administration.
This invention also provides pharmaceutical compositions useful for providing
anticonvulsant activity, which comprise at least one compound of the
invention. In addition to
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-13-
comprising at least one of the compounds described by formula I, the
pharmaceutical
composition may also comprise additives such as preservatives, excipients,
fillers, wetting
agents, binders, disintegrants, buffers, and/or carriers. Suitable additives
may be for example
magnesium and calcium carbonates, carboxymethylcellulose, starches, sugars,
gums,
S magnesium or calcium stearate, coloring or flavoring agents, and the like.
There exists a wide
variety of pharmaceutically acceptable additives for pharmaceutical dosage
forms, and selection
of appropriate additives is a routine matter for those skilled in art of
pharmaceutical
formulation.
The compositions may be in the form of tablets, capsules, powders, granules,
lozenges,
suppositories, reconstitutable powders, or liquid preparations such as oral or
sterile parenteral
solutions or suspensions.
In order to obtain consistency of administration it is preferred that a
composition of the
invention is in the form of a unit dose. Unit dose forms for oral
administration may be tablets,
capsules, and the like, and may contain conventional excipients such as
binding agents, for
1 S example syrup, acacia, gelatin, sorbitol, tragacanth, or
polyvinylpyrrolidone; and carriers or
fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol
or glycine.
Additives may include disintegrants, for example starch, polyvinylpyrrolidone,
sodium starch
glycolate or microcrystalline cellulose; preservatives, and pharmaceutically
acceptable wetting
agents such as sodium lauryl sulphate.
In addition to unit dose forms, mufti-dosage forms are also contemplated to be
within
the scope of the invention. Delayed-release compositions, for example those
prepared by
employing slow-release coatings, micro-encapsulation, and/or slowly-dissolving
polymer
carriers, will also be apparent to those skilled in the art, and are
contemplated to be within the
scope of the invention.
The solid oral compositions may be prepared by conventional methods of
blending,
filling, tabletting or the like. Repeated blending operations may be used to
distribute the active
agent throughout those compositions employing large quantities of fillers.
Such operations are
conventional in the art. The tablets may be coated according to methods well
known in normal
pharmaceutical practice, for example with an enteric coating.
Oral liquid preparations may be in the form of, for example, emulsions,
syrups, or
elixirs, or may be presented as a dry product for reconstitution with water or
other suitable
vehicle before use. Such liquid preparations may contain conventional
additives such as
suspending agents, for example sorbitol syrup, methyl cellulose, gelatin,
hydroxyethylcellulose,
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-14-
carboxymethylcellulose, aluminum stearate gel, and hydrogenated edible fats;
emulsifying
agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous
vehicles (which may
include edible oils), for example almond oil or fractionated coconut oil, oily
esters such as
esters of glycerine, propylene glycol, or ethyl alcohol; preservatives, for
example methyl or
propyl p-hydroxybenzoate or sorbic acid; and if desired conventional flavoring
or coloring
agents.
For parenteral administration, fluid unit dosage forms are prepared utilizing
the
compound and a sterile Garner, and, depending on the concentration used, can
be either
suspended or dissolved in the Garner. In preparing solutions the compound can
be dissolved in
water or saline for injection and filter sterilized before filling into a
suitable vial or ampoule and
sealing. Advantageously, additives such as a local anaesthetic, a preservative
and buffering
agents can be dissolved in the vehicle. Suitable buffering agents are, for
example, phosphate
and citrate salts. To enhance the stability, the composition can be frozen
after filling into the
vial and the water removed under vacuum. Parenteral suspensions are prepared
in substantially
1 S the same manner, except that the compound is suspended in the carrier
instead or being
dissolved, and sterilization cannot be accomplished by filtration. The
compound can be
sterilized by conventional means, for example by exposure to radiation or
ethylene oxide,
before being suspended in the sterile vehicle. Advantageously, a surfactant or
wetting agent is
included in the composition to facilitate uniform distribution of the
compound.
EXAMPLES
A. Preparation of the compounds.
Example 3: Synthesis of 2-fluoro-valnoctamide. Ethyl diethyl malonate (32.3 g,
0.172 mol) was dropped into a suspension of 7 g sodium hydride (60% in oil) in
200 ml dry
N,N-dimethylformamide (DMF). After complete deprotonation 28.8 ml (0.25 mol) 2-
iodobutane was added and the mixture was warmed up to 153° for 12 h.
The cooled mixture
was poured into ice/water and extracted with ether. The combined organic
layers were washed
several times with water and dried over sodium sulfate. The solvent was
evaporated and
distillation of the crude product yielded 16.5 g colorless liquid, boiling
point 68°C at 1 mbar.
The resulting dialkyl malonic ester (16.5 g) and potassium hydroxide (12 g)
were heated
to reflux in 30 ml water and 60 ml ethanol for 75 h. Ethanol was evaporated,
the residue was
diluted with water and extracted with ether. The water layer was then
acidified with
concentrated HCl and again extracted with ether. After drying and evaporation
of the solvent
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-15-
the crude product (13.9 g yellow oil} was heated to 175°C for
decarboxylation for 2 h.
Subsequent distillation yielded 7.2 g valnoctic acid as a colorless liquid,
boiling point 66-68°C
at 0.9 mbar.
Valnoctic acid (6.7 g) was warmed up to reflux with 100 ml methanol and 5 ml
concentrated sulfuric acid for 5 h. Methanol was distilled, the residue was
diluted with ether,
the phases were separated and the organic layer was washed with water and
dried over sodium
sulfate. The ester was distilled carefully and the crude product distilled
under reduced pressure
to provide methyl valnoctate as a colorless liquid (4.5 g), boiling point
46°C at 8 mbar.
At 4°C a solution of 0.0313 mol lithium diisopropyl amide (LDA) in 100
ml dry THF
was prepared from 4.4 ml diisopropylamine and 20 ml butyl lithium ( 1.5 M in
hexane). This
solution was cooled to -78°C and 4.Sg (0.0285 mol) valnoctic acid
methyl ester was dropped in.
The mixture was allowed to warm up to -20°C to make deprotonation
complete and then cooled
again to -78°C. lOg (l.l.equivalents) N-fluoro-benzenesulfonimide in 50
ml THF was added
and the stirred mixture was allowed to warm to room temperature over night.
After quenching
1 S with 100 ml water the layers were separated, the water layer was extracted
with either, the
organic layers were washed with aqueous sodium hydrogen carbonate and
saturated sodium
chloride, and dried over sodium sulfate. After careful evaporation of the
solvent the crude
product was distilled to provide 2-fluoro-valnoctic acid methyl ester (3.7 g)
as a colorless
liquid, boiling point 69-73°C at 8 mbar.
2-Fluoro-valnoctic acid methyl ester (3.7 g) was stirred with 1 g lithium
hydroxide (one
mole crystal water) in 80 ml methanol/water (3:1 ) at room temperature over
night. Methanol
was evaporated, the residue was diluted with water and extracted with ether.
The water layer
was acidified with concentrated HC1 and extracted with ether. After drying and
evaporation of
the solvent 2.3 g crude acid were warmed in 10 ml thionyl chloride. Excess
thionyl chloride
was removed by distillation and the crude acid chloride was dropped with
stirnng into a cooled
solution of ammonia in water. The precipitate was filtered off and
recrystallized from
ethanol/water to provide 2-fluoro-valnoctamide ( 1.3 g} as colorless crystals,
melting point
110°C. ~H-NMR (CDCL3: 8=0.92 (9HM m, 3 x CH3), 1.2 (1H, m, CH), 1.86
(4H, m, 2 x CHZ),
5.75 and 6.34 (2H, broad, COHNz).
Example 5: 2-fluoro-2-n-propvlpentanoamide. A solution of 0.055 mol of lithium
diisopropylamide in 100 ml THF was cooled to -78°C under an inert
atmosphere, and 0.050
mol of methyl 2-propylpentanoate in THF was added dropwise with stirring. The
mixture was
allowed to warm to -20°C, then cooled again to -78°C. A solution
of N-fluoro
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-16-
benzenesulfonimide (16 g) in THF (SO ml) was added dropwise. The mixture was
allowed to
warm to room temperature overnight, and was quenched with saturated ammonium
chloride
solution. Aqueous 6N HCl (100 ml) was added, and the product extracted with
ethyl ether,
dried with anhydrous sodium sulfate, concentrated, and distilled in vacuo to
provide 7.3 g
S (83%) methyl 2-fluoro-2-propylpentanoate as an oil, by
73°C/8mbar.
The methyl ester (7.3 g, 0.041 mol) was saponified by dissolving it in 60 ml
of
methanol, adding 20 ml of water and 0.042 mol of lithium hydroxide, and
stirring at room
temperature for 24 hours. The methanol was removed in vacuo, and the residue
diluted with
water and extracted twice with ethyl ether. The aqueous solution was then
acidified with
hydrochloric acid and extracted again with ether. The ether extract was worked
up as above,
and after evaporation of solvent the crude 2-fluoro-2-propylpentanoic acid was
used as is for
the next step. The acid could be chromatographed on silica gel with 3:1 hexane-
ethyl acetate to
provide the title compound as an oil. The physical and spectroscopic
properties were as
reported (Ph.D. thesis of Wei Tang, University of British Columbia, 1996).
To 2-fluoro-2-n-propylpentanoic acid (7.0 g, .043 mol) was added thionyl
chloride (25
ml). The solution was warmed gently and maintained at 60°C until the
evolution of hydrogen
chloride ceased. Excess thionyl chloride was removed by distillation, and the
product was
distilled in vacuo to provide 7.3 g (94%) 2-fluoro-2-propylpropionyl chloride,
by 83°C/12
mbar, as an oil.
The acid chloride (7.3 g, 0.04 mol) was added dropwise with stirring to 25%
aqueous
ammonium hydroxide solution ( 100 ml), while maintaining the temperature below
0°C with an
ice/salt bath. The resulting precipitate was collected and recrystallized from
ethanol/water, to
provide the title compound (S.0 g, 77%) as a colorless crystalline solid, mp
131°C. 'H NMR
(CDCl3): b 0.92 (6H, t, 3=7 Hz), 1.2-2.4 (8H, m), 5.44 (1H, broad s) and 6.32
(1H, broad s).
Example 8: 2-fluoro-2-n-nroRvlheptanoamide. By the method described above,
methyl 2-propylheptanoate was converted into the title compound (37% overall),
a colorless
crystalline solid, mp 128°C. 'H NMR (CDC13}: b 0.9 (6H, 2t, 2CH3), 1.2-
2.02 (12H, m, 6CHz),
5.79 (1H, broad s, CONH) and 6.36 (1H, broad s, CONH).
Example 18: 2-fluoro-3-meth~lvalpromide. By either of the methods described
above,
the title compound could be obtained as a colorless solid. 'H NMR (CDC13): 8
0.94 (9H, m,
3CH3), 1.08-1.64 (4H, m), 1.64-2.0 (3H, m), 5.98 (1H, broad s, CONH) and 6.36
(1H, broad s,
CONH).
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-17-
B. Biological activities of the compounds.
The anti-convulsive activity of the compounds of the invention was determined
by the
PTZ convulsion test (E. Swinyard et al., 1969, "Laboratory Evaluation of
Antiepileptic Drugs,
Review of Laboratory Methods," Epilepsia 10, 107-119; E. Swinyard, J.
Woodhead, in
Antiepileptic Drugs, 2nd ed., D. Woodbury, J. Penry, C. Pippenger, eds., Raven
Press, New
York, 1982, 111-126). The compounds were suspended before administration in a
25%
aqueous solution of the castor oil-ethylene oxide derivative marketed under
the trade name
CREMOPHOR EL. Briefly, animals were dosed intraperitoneally with a suspension
of the
compound to be tested, and then challenged after 15 minutes with a
subcutaneous injection of
pentylenetetrazole (65 mg/kg). The number of animals that exhibited tonic
seizures lasting at
least five seconds was noted over the course of 30 minutes. Test groups of six
mice were used
at each dose level, and five dosages were used to calculate the EDSO values.
The calculated EDSo
values (the dose that protects 50% of animals form seizures) are presented in
Table 1 as
"Anticonvulsant Activity". The percentage of animals that were protected from
seizures is
reported in Table 2 as "Anticonvulsant Activity %".
The sedative activity of the compounds was determined by the "Rotorod" test
(Dunham,
Miya, 1957, J. Am. Pharm. Assoc., 46, 208-209). Groups of six animals were
dosed at five
dosage levels as above with a suspension of the compound to be tested, and
after 15 minutes
they were placed on the ROTOROD apparatus (Rotorod, Ugobasile, Italy). The
percentage of
animals that fell from the rod was recorded as "Sedative Activity %". The TDso
values (the
"Toxic Dose" at which 50% of animals fall from the Rotorod) was calculated as
above, and is
reported as "Sedative Activity TDSO" in Table 1 and as "Sedation TDSO" in
Table 2.
The teratogenic potential of the compounds was determined by injecting
pregnant
animals on day 8 of gestation with a suspension of the compound to be tested,
and by
examining the fetuses on day 18 of gestation (H. Nau, 1985, Toxicol. Appl.
Pharmacol., 80,
243-250; H. Nau, W. Loscher, 1986, Fund. Appl. Toxicol., 6, 669). The percent
of fetuses
exhibiting exencephaiy is reported as "Teratogenic Activity %" in Table 1.
C. Pharmacokinetics
Anticonvulsant activity was measured as described above, with PTZ being
injected at
15, 30, 45, 60, and 75 minutes after dosing with either VPA, valpromide, 2-
fluoro-VPA, or 2-
fluoro-2-propylpentanoamide. The results are presented in Table 3.
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-18-
Pharmacodynamics were also studied in pregnant mice. Mice were injected with
3.0
mmol/kg of 2-fluoro-2-propylpentanoamide (Example 5) on day 8 of gestation.
Blood samples
were taken at 0.25, 0.5, l, 2, 4, 6, 8, 12, and 24 hours after injection. Drug
concentrations in
plasma were determined by GC-MS analysis after treatment with N-methyl-N-(t-
butyldimethylsilyl) trifluoroacetamide. Maximum drug concentration (57 t 15
pg/ml) was
reached after 1 hour. The half life was 11.3 hours (vs. 1.4 hours for VPA).
The maximum
concentration of the metabolite 2-fluoro-2-propylpentanoic acid was 6.5 t 3.6
pg/ml. Embryo
tissue concentrations were 60-70% of the maternal plasma concentrations (vs.
100% for VPA).
All in vivo assays with valproic acid (.VPA) were conducted similarly, except
that a
solution of the sodium salt of VPA was used for injections.
D. Results
Alpha-fluorination consistently reduced the sedative side-effect of the
carboxamides,
and in most cases increased the anticonvulsant potency as well. Therefore, the
2-fluorinated
carboxamides of the invention generally show improved anticonvulsant
properties, EDso-values,
and therapeutic ratios when compared to their non-fluorinated analogues. In
addition, they
exhibit greatly reduced teratogenic potential, a faster onset of action, and a
longer half life.
While the examples presented herein describe a number of embodiments of this
invention, it is apparent that the compounds, compositions, and methods of
this invention can
be altered to provide alternative embodiments which nonetheless utilize the
methods of this
invention. That such alternative embodiments may not have been expressly
presented is not to
be considered a disclaimer of those alternative embodiments. Therefore, it
will be appreciated
that the scope of this invention is not limited to the specific embodiments
which have been
presented above by way of example, and that such alternative embodiments will
be within the
literal scope of the claims or will be equivalent thereto.
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-19-
Table 1.
Anti-convulsive activity, sedation, therapeutic ratio, and teratogenic
potential.
i ANTICONVULSIVE SEDATIVE THERAPEUTIC TERATOGENIC
COMPOUND ACTIVITY ACTIVITY RATIO ACTIVITY,
~ (EXAMPLE EDSO, mmol/kg TDso, mmollkg(TDso/EDso) (dose, mmol/kg)
NO.)
2-Fluoro- 0.12 0.54 4.5 0 (3.0)
Valnoctamide
(Example
3)
2-Fluoro- 0.16 0.57 3.6 0 (3.0)
Valpromide
(Example
5)
0.12 0.45 3.8 0 (3.0)
2-Fluoro-
3-Methyl
Valpromide
(Example
18)
Valproic 0.61 1.83 3.0 37 (3.0)
acid
Valpromide 0.29 0.72 2.5 6.5 (3.0)
n.d. : not determined
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-20-
Table 2.
(see text for definition of % activity)
Substance Structure Dose Activity EDso Sedation Therapeutic
TDso
mmollkg (%) mmollkg ratio
mmollkg
Valpromide 0.25 16.7
CONH2 0.28 33.3 0.29 0.72 2.5
0.3 66.7
0.4 100
2-Fluoro- 0.06 16.7
Valpromide CONH2 0.12 16.7 0.16 0.57 3.6
(Example S) 0.20 50.0
F 0.26 83.3
0.52 100
V alnoctamide 0.1 20
CONHZ p,15 20 0.17 0.68 4.1
a 0.2 60
0.4 100
2-Fluoro- 0.1 16.6
Valnoctamide CONHZ 0,12 66.6 0.12 0.54 4.5
(Example 3) Ylu 0.14 100
IF
0.175 83.3
0.2 100
3-Methyl 0.05 0
Valpromide CONH2 O,Og 33.3 0:10 0.51 5.1
a ~ 0.1 66.6
0.2 83.3
0.3 100
2-Fluoro- CONH
3-methyl 2 0.12 0.45 3.8
V alpromide
(Example 18) F
2-Fluoro-
2-propyl- CONHZ 0.2 33.3 ~ 0,4 n.d. n.d.
heptanoamide 0.3 33.3
(Example 8) F 0.4 50
Valproic acid 0.5 16.7
COOH 0.63 50 0.61 1.83 3.0
0.75 83.3
1.0 100
n.d. : not determined
CA 02335598 2000-12-21
WO 99/67199 PCT/US99/13941
-21 -
Table 3.
Anticonvulsant activity at various times after administration
% OF
ANIMALS
PROTECTED
COMPOUND DOSE at
times
(minutes)
post-dosing
) (mmol/k
EXAMPLE NO )
. g
(
15 30 45 60 75 min
min min min min
Valproic acid 1.5 100 100
(VPA)
2-Fluoro-VPA 1.5 0 40 80 0
Valpromide 1.5 100
2-Fluoro-Valpromide
(Example 5) 1.5 100
2-Fluoro-Valpromide
(Example 5) 1.0 80 60 40