Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02335799 2000-12-19
1
Novel compounds derived from a-D-xylose, preparation method and therapeutic
use.
Field of invention
The present invention concerns, as new industrial products, the derivatives of
a-D-
xylose defined by formula I below. It also concerns the process for the
preparation of these
compounds, as well as the therapeutic compositions containing them as active
ingredients.
Prior art
Derivatives of ~i-D-xylose, in particular derivatives of benzoyl- or a-hydroxy-
benzyl-
phenyl (i-D-xylosides, recommended in therapeutics for the treatment of venous
thromboses,
are already known, for example, according to EP-A-0051023.
Derivatives of benzyl-phenyl (i-D-xylosides, exhibiting a
hypocholesterolemiant
andlor hypolipidemiant activity, are also known according to EP-A-0133103.
Derivatives of the type p-D-phenylthioxylosides, used for their antithrombotic
activity,
are also known according to EP-A-0365397, EP-A-0290321, EP-A-0133103 and EP-A-
0051023.
The antithrombotic activity of a certain number of derivatives of ~3-D-xylose
has also
been reported and studied in the article of J. Med. Chem, 1993, 36, (no. 7)
pages 898-903.
Research carried out in the laboratory has shown that these derivatives of p-D-
xylose are
good substrates of galactosyl transferase 1. For this reason, these compounds,
active when
taken orally, initiate the synthesis of glycosaminoglycanes (GAGs). After
administration of
the compounds orally, the circulation rates of GAGs are appreciably increased
and
approximately 20% of the latter display an activity of the dermatan-sulphate
type capable of
inhibiting thrombin, via HC II (Heparin Cofactor II), this initiation of the
biosynthesis of GAGs
probably being responsible for the antithrombotic activity observed
experimentally for the
compounds mentioned previously. In correlation with the potential of these
compounds to
reduce the formation of venous thromboses, only the p configuration
derivatives of D-xylose
increase the, synthesis of GAGs. The other derivatives of the glycopyranoside
type have
proved to be inactive in this area, both from the biological standpoint on the
synthesis of
GAGs, as well as from the pharmacological standpoint on the reduction or
prevention of
venous thromboses.
CA 02335799 2000-12-19
2
The activity of certain derivatives of a-D-xylose, in particular estradiol p-D-
xyloside,
has been studied in the publication Journal of Biological Chemistry, 1991,
266, (No. 11)
pages 6674-6677 and the authors establish a relationship between this compound
and the
biosynthesis of heparane sulphate, as well as an initiator role of (3-D-
xyloside in the synthesis
of chondroitin sulphate. These studies confirm the benefit of derivatives of ~-
D-xylose for the
treatment or the prevention of venous thromboses.
Aim of the invention
According to the invention, it is proposed to provide a new technical solution
making
it possible to arrive at new products that are biologically beneficial with
respect to arterial
atheromatous platelets.
Subject-matter of the invention
According to the new technical solution of the invention, use is made of
products of
the type a-D-xylose or a-D-thioxylose.
It has in fact been found, in a surprising manner, that derivatives of D-
xylose or of 5
thio-D-xylose no longer displaying the ~i configuration, but the a
configuration on the anomeric
carbon, possess a particularly beneficial activity for the prevention or the
regression of arterial
atheromatous platelets.
The new products according to the invention, which are compounds of a-D-
xylose,
are characterised in that they are selected from the compounds of the general
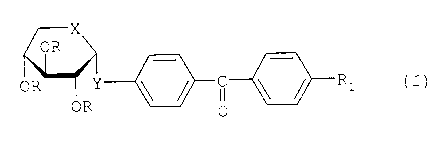
formula I:
X
OR
OR ORy ~ ~ I ~ ~ Ri
0
wherein
X and Y represent, independently of one another, an oxygen atom or a sulphur
atom,
R, represents a CN, CF3or SOzCH3 group, and
R represents a hydrogen atom or an aliphatic acyl group containing 2 to 5
carbon atoms.
According to another aspect of the invention, a composition is provided that
is
characterised in that it contains, in association with a physiologically
acceptable excipient, a
therapeutically effective quantity of at least one compound of the formula I.
It is also recommended to use the compound of the formula I as an
antiatheromatous drug.
CA 02335799 2000-12-19
3
Detailed description of the invention
As presented in formula I above, the compounds according to the invention are
derivatives of a-D-xylose or a-D-thioxylose, substituted on the anomeric
carbon in the a
position by a substituted benzophenone group.
The hydroxyl functions of D-xylose or D-thioxylose can be free or substituted
by an
acyl group containing 2 to 5 carbon atoms, preferably the acetyl group.
Aliphatic acyl group containing 2 to 5 carbon atoms is understood here to mean
an
acyl group with a straight, branched or cyclised chain, such as in particular
CH3C0,
CH3CH2C0, (CH3)2CHC0, (CH3)3CC0, or cyclopropyl-carbonyl.
The compounds of the formula I can be prepared according to a method known per
se by using conventional reactive mechanisms. According to a preferred
operating method,
a benzophenone of the formula II is reacted (according to a coupling
reaction):
H Y ~ ~ I ~ ~ Ri (II)
0
wherein Y is the oxygen atom or the sulphur atom and R, represents CN, CF3 or
S02CH3,
with a compound of D-xylose (or of 5-thin-D-xylose) of the formula III:
X
OAc 0- Z ( I I I )
OAc
OAc
where-.","~ represents a bond of indeterminate configuration (a,a or a/ ).i
mixture), X is an
oxygen atom or a sulphur atom, Ac represents the acetyl group, Z represents an
acetyl group
or a trichloromethylimino group [-C(=NH)-CCI3], the reaction being conducted
in a solvent and
in the presence of a Lewis acid, in order to obtain a compound of the formula
I after
purification:
CA 02335799 2000-12-19
4
X
OR
OR y ~ ~ I ~ ~ Ri (I)
OR
wherein
X, Y and R~ retain the same significance as in the starting products and R
represents an
acetyl group.
The compounds of the formula I in which R is a hydrogen atom can be obtained
from
the preceding compounds, in which R is the acetyl group, by the action of a
base such as for
example sodium methylate or ammonia which permit the acetyl group to be
replaced by a
hydrogen atom.
The compounds of the formula I in which R is a C2-CS acyl group, such as
defined
above, can be obtained from compounds with formula I in which R is a hydrogen
atom, by
the action of chloride or acid anhydride corresponding to the desired C2-CS
acyl group, in the
presence of an aprotic base such as for example triethylamine or pyridine, and
in a solvent
such as for example dichloromethane.
Briefly, the process of preparation according to the invention comprises (a)
the II +
III coupling reaction and the purification of the compound I (R = Ac) thus
obtained, (b) if
necessary the hydrolysis (saponification) of said compound of the formula I (R
= Ac) so as
to obtain the deacetylated compound with formula I (R = H), and (c) if
necessary the
esterification of said compound of the formula I (R = H) so as to obtain any
other esterified
compound of the formula I (R = acyl, in particular an acyl group different
from Ac).
The compounds of the formula I are useful in therapeutics as active principles
of
drugs intended for the treatment or prevention of atherosclerosis.
According to the invention, it is recommended to use a product selected from
the
group consisiting of the compounds of the formula I above for the preparation
of an
antiatheromatous drug intended for use in therapeutics with respect to
atherosclerosis.
The preferred products according to the invention, in view of the beneficial
properties
with respect to atherosclerosis, are the compounds of the formula I where R,
is CN, and
among the latter the products in which X = Y = S or X = Y = O.
The following non-limiting examples of preparation permit the advantages of
the
invention to be better understood and appreciated.
CA 02335799 2000-12-19
Example 1
[4-(4-cyanobenzoyl)phenyl] 2,3,4-tri-O-acetyl-1,5-dithio-a-D-xylopyranoside
A suspension of 5 g (15 . 103 mol) of tetra-O-acetyl-5-thio-D-xylopyranose,
4.3 g (18
. 103 mol) of 4-(4-mercaptobenzoyl)benzonitrile and 5 g of 4 A molecular sieve
is prepared
5 in 100 ml of acetonitrile and 4 ml of boron trifluoride etherate (at 48%, d
= 1.13) is added at
0°C while stirring. The reaction mixture is allowed to return to room
temperature and stirring
is carried on for one hour. The mixture is then filtered, then concentrated
under reduced
pressure. The residue is dissolved in ethyl acetate and the solution is washed
with diluted
soda, then with diluted hydrochloric acid and finally with water until
neutrality. The organic
phase is dried and concentrated under reduced pressure. The residue is
purified by
chromatography on silica gel, eluting with a toluenelethyl acetate mixture
(6/1; vlv). 7 g of the
sought compound is thus obtained in a mixture with the ~3 isomer (the two
isomers are in the
ratio al [i = 60/40). This mixture is dissolved in 15 ml of ethyl acetate and
15 ml of ethyl ether
is added. The crystals obtained (essentially the [i isomer) are eliminated by
filtration and the
filtrate is concentrated under reduced pressure. 5.4 g of the sought compound
is thus
obtained containing 20% of the [3 isomer (yield = 63%). The sought compound is
obtained
with 98% purity in the a isomer after two recrystallisations in methanol.
F = 144-146°C
[a]24o = + 328° (c = 0.42; CH2C12)
Example 2
[4-(4-cyanobenzoyl)phenyl] 1,5-dithio-a-D-xylopyranoside
A solution of 10.27 g (20 . 10-3 mol) of the compound obtained according to
example
1 is prepared in 100 ml of methanol and 35 ml of tetrahydrofuran. 1.15 ml (4 .
10-3 mol) of a
solution of sodium methylate is then added at 10-15°C.
After 20 min. while stirring at 10-15°C, the reaction mixture is
percolated on an IR 120 resin
(H+). The solution is decolourised by means of activated carbon, filtered and
concentrated
under reduced pressure. The residue is crystallised in methanol. After
recrystallisation in
methanol, 5 g of the sought product is obtained in the form of white crystals
(yield = 65%).
F = 142°C
[a]o 2°= + 508° (c = 0.42; CH30H)
Example 3
[4-(4-cyanobenzoyl)phenyl] 2,3,4-tri-O-acetyl-a-D-xylopyranoside
A mixture of 9 g (28.3 . 10-3 mol) of tetra-O-acetyl-D-xylose and 8 g (35.9 .
10-3 mol)
of 4-(4-hydroxybenzoyl)benzonitrile is prepared in 90 ml of dichloromethane. 8
ml (68.4 . 10-3
mol) of tin tetrachloride is then added drop by drop at room temperature, then
the reaction
CA 02335799 2000-12-19
6
medium is brought to 45°C, for 15 hours, while stirring. The mixture is
then poured onto ice
and diluted hydrochloric acid; extraction is then performed with ethyl acetate
in acid medium,
then the united organic phases are washed with a diluted solution of
hydrochloric acid, then
with water, with a diluted soda solution, then with water. The organic phase
is dried on
magnesium sulphate then concentrated under reduced pressure. The raw product
is purified
by chromatography on silica gel, eluting with the aid of a toluenelethyl
acetate mixture
(5/1 ; v/v). 3.3 g of the sought product is thus obtained in the form of a
white powder
(yield = 24%).
F = 75°C
[a]o 26 = + 146.3°(c = 0.36; CH30H)
Example 4
[4-(4-cyanobenzoyl)phenyl] a-D-xylopyranoside
4.3 g (8.94 . 103 mol) of the compound obtained according to example 3 is
added
to 50 ml of a saturated solution of ammonia in methanol, at 0°C, and
the mixture is held for
3 hours at 0°C while stirring, then for 2 hours at room temperature.
The reaction medium is
then concentrated under reduced pressure and the residue is purified by
chromatography on
silica gel, eluting with a dichloromethane/methanol mixture (10/1; v/v). The
pure fraction
obtained is concentrated to eliminate the solvents, then the product is put
into suspension in
distilled water; the mixture is congealed and freeze-dried. 2.15 g of the
sought product is
thus obtained in the form of a crushed white powder (yield = 67%).
F = 186-187°C
[a]26o = + 155° (c = 0.40; CH30H)
Example 5
[4-(4-cyanobenzoyl)phenyl] 2,3,4-tri-O-acetyl-1-thin-a-D-xylopyranoside
Operating in a similar manner to example 3, starting from 4-(4-mercapto-
benzoyl)benzonitrile, the sought product is obtained in the form of a white
solid (yield = 11 %).
F = 158-159°C
[a]26p = + 161 ° (c = 0.38; CH2C12)
Example 6
[4-(4-cyanobenzoyl)phenyl] 1-thin-a-D-xylopyranoside
Operating in a similar manner to example 4, starting from the compound
obtained
according to example 5, the sought product is obtained in the form of a cream
powder (yield
= 90%).
F = 198°C
[a]26p = + 245° (c = 0.30. CH30H)
CA 02335799 2000-12-19
7
Example 7
[4-(4-cyanobenzoyl)phenyl] 2,3,4-tri-O-acetyl-5-thio-a-D-xylopyranoside)
A suspension of 2 g (9 . 10'3 mol) of 4-(4-hydroxy-benzoyl)benzonitrile is
prepared
in 40 ml of dichloromethane, 1 g of 4 A molecular sieve is added and the
mixture is cooled
to -30°C. 1.8 ml (8 . 10-3 mol) of triethylsilyl
trifluoromethanesulphonate is slowly added while
stirring, then a solution of 3.9 g (9 . 103 mol) of 2,3,4-tri-O-acetyl-5-thin-
a-D-xylopyranosyl
trichloroacetimidate in 20 ml of dichloromethane. The reaction mixture is
stirred for 4 hours
at -30°C then for 16 hours at 0°C. After neutralisation with the
aid of a collodine solution, the
reaction medium is filtered. The filtrate is diluted in 200 ml of ethyl
acetate and the organic
phase obtained is washed with a solution of diluted soda, then with water,
with the aid of a
diluted hydrochloric acidaolution, then with water and dried on magnesium
sulphate. After
concentration of the solution under reduced pressure, the residue is purified
by
chromatography on silica gel, eluting with a methylcyclohexane-ethyl acetate
mixture
(5/2; v/v). 0.86 g of the sought product is thus obtained in the form of a
beige powder
(yield = 19%).
F = + 85°C
(a]2'o = + 365° ( c = 0.3; CH2C12 )
Example 8
[4-(4-cyanobenzoyl)phenyl] 5-thin-a-D-xylopyranoside
Operating in a similar manner to example 4, starting from the compound
obtained
according to example 7, the sought product is obtained in the form of a clear
beige powder
(yield = 73 %).
F = 180°C
[a]24p = + 476° (c = 0.32; CH30H)
Example 9
[4-[4-(trifluoromethyl)benzoyl]phenyl] 2,3,4-tri-O-acetyl-a-D-xylopyranoside
A solution of 2.5 g (9.4 . 10'3 mol) of (4-hydroxy-phenyl)[4-
(trifluoromethyl)phenyl]
methanone and 3.5 g (11 . 10-3) of tetra-O-acetyl-D-xylose is prepared in 30
ml of acetonitrile
and 1 g of 4 A molecular sieve is added. The mixture is cooled to 0°C
and 7.2 ml (58 . 10-3
mol) of boron trifluoride etherate is added drop by drop while stirring. The
reaction medium
is then held at room temperature for 15 hours while stirring, then it is
filtered. The filtrate is
diluted with ethyl acetate and the organic phase is washed with a solution of
diluted soda, with
water, with a solution of diluted hydrochloric acid and again with water.
After drying on
magnesium sulphate, the organic phase is concentrated under reduced pressure
and the
residue is purified by chromatography on silica gel, eluting with the aid of a
CA 02335799 2000-12-19
g
methylcyclohexane-ethyl acetate mixture (512; vlv). 0.46 g of the sought
product is thus
obtained in the form of an amorphous beige solid (yield = 10%).
F = 65°C
(a]26p = + 141 ° ( c = 0.32; CH2C12 )
Example 10
[4-[4-(trifluoromethyl)benzoyl]phenyl] a-D-xylopyranoside
0.370 g (0.7 . 10-3 mol) of the compound obtained according to example 9 is
dissolved in 20 ml of methanol and 40 pl of a solution of sodium methylate at
25% in the
methanol is added . The solution is held for two hours while stirring, then
approx. 0.5 g of 120
IR resin is added in acidic form. After filtration, the filtrate is
concentrated under reduced
pressure. 240 mg of the sought product is thus obtained in the form of a beige
solid (yield
= 85%).
F = 188°C
(a)2'p = + 165° ( c = 0.27; CH30H )
Example 11
[4-(4-(trifluoromethyl)benzoyl]phenyl] 2,3,4-tri-O-acetyl-5-thio-a-D-
xylopyranoside
Operating in a similar manner to example 7, starting from (4-hydroxyphenyl)(4-
(trifluoromethyl)phenyl]methanone, the sought product is obtained in the form
of a beige
amorphous solid (yield = 19.5%).
F = 65°C
(a]o = + 320° (c = 0.34; CH2Clz)
Example 12
[4-(4-(trifluoromethyl)benzoyl]phenyl] 5-thio-a-D-xylopyranoside
Operating in a similar manner to example 10, starting from the compound
obtained
according to example 11, the sought product is obtained in the form of an
amorphous white
powder (yield = 80%).
F = 82°C
(a)22o = + 378° ( c = 0.25; CH30H)
PREPARATION I
Dimethylcarbamothioic acid, O-[4-(4-(trifluoromethyl)benzoyl]phenyl] ester
A solution of 10 g (37.6 .10-3 mol) of (4-hydroxy-phenyl)(4-
(trifluoromethyl)phenyl)
methanone is prepared in 100 ml of acetone and a solution of 2.94 g (45 . 10-3
mol) of
potassium hydroxide (at 80 %) in 80 ml of water is slowly added while stirring
and at room
temperature. A solution of 5.11 g (41.3 .10-3 mol) of dimethylthiocarbamoyl
chloride in 70 ml
of acetone is then added. The reaction mixture is held at room temperature for
8 hours while
CA 02335799 2000-12-19
9
stirring, then concentrated at reduced pressure. The raw product is put into
suspension in
60 ml of a 1 molll solution of potassium hydroxide and stirred for 20 min. at
10-15°C. The
solid is filtered, rinsed in suspension with water until a neutral pH , then
dried in the drier.
10.3 g of the product sought is thus obtained in the form of a beige powder
(yield = 78%).
F = 174°C
PREPARATION II
Dimethylcarbamothioic acid, S-(4-(4-(trifluoromethyl)benzoyl]phenyl] ester
10.3 g (29 .103 mol) of the compound obtained according to preparation I is
placed
in a flask, under an atmosphere of nitrogen, and the product is held at 250-
260°C for 1 hour.
After cooling, approx. 15 ml of ethyl acetate is added, the mixture is brought
to a slight reflux,
then left to cool down to 0°C. After approx. 10 hours at this
temperature, the crystallised solid
is filtered and the solid is rinsed with approx. 10 ml of cyclohexane. After
drying in the drier,
6.2 g of the product sought is obtained in the form of beige crystals (yield =
60%).
F = 140°C
PREPARATION III
(4-mercaptophenyl)-[4-(trifluoromethyl)phenyl]methanone
A suspension of 6.1 g (17.3 .10-3 mol) of the compound obtained according to
preparation II is prepared in 65 ml of methanol. After having deoxygenated the
medium by
bubbling-through with nitrogen, 10 ml (i.e. 34 .10-3 mol) of a solution of
sodium methylate in
methanol is added , then the reaction mixture is raised to 40°C while
stirring for three hours.
Partial concentration is then carried out (approx. 25 ml of methanol is
eliminated) under
reduced pressure and the solution is poured into a mixture of ice and diluted
hydrochloric
acid. The sought product precipitates. The solid product is separated by
filtration and washed
with water until neutral pH. After drying in the drier, 4.8 g of the product
sought is obtained
in the form of a clear green solid.
F = 150°C
Example 13
[4-[4-(trifluoromethyl)benzoyl]phenyl] 1-thio-2,3,4-tri-O-acetyl-a-D-
xylopyranoside
Operating in a similar manner to example 9, starting from the compound
obtained
according to preparation III, the sought product is obtained in the form of a
beige solid (yield
= 27%).
F = 60°C
[a]22p = + 145° ( c = 0.25; CH2C12)
CA 02335799 2000-12-19
Example 14
(4-[4-(trifluoromethyl)benzoyl]phenyl] 1-thio-a-D-xylopyranoside
Operating in a similar manner to example 10, starting from the compound
obtained
according to example 13, the sought product is obtained in the form of a cream
amorphous
5 solid (yield = 95%).
F = 172°C
(a]24p = + 211 ° (c = 0.35; CH30H)
Example 15
[4-[4-(trifluoromethyl)benzoyl]phenyl] 2,3,4-tri-O-acetyl-1,5-dithio-a-D-
xylopyranoside
10 Operating in a similar manner to example 9, starting from 1,2,3,4-tetra-O-
acetyl-5
thio-D-xylose and the compound obtained according to preparation III, the
sought product is
obtained in the form of a white powder (yield = 18%).
F = 70°C
[a]27o = + 215° (c = 0.45; CH2C12)
Example 16
[4-[4-(trifluoromethyl)benzoyl]phenyl] 1,5-dithio-a-D-xylopyranoside
Operating in a similar manner to example 10, starting from the compound
obtained
according to example 15, the sought product is obtained in the form of a white
powdery solid
(yield = 84%).
F = 104°C
[a]25o = + 399° (c = 0.50; DMSO)
PREPARATION IV
(4-hydroxyphenyl)[4-(methylsulphinyl)phenyl]methanone
A solution of 15 g (61.4 .10-3 mol) of (4-hydroxyphenyl)[4-(methylthio)
phenyl]methanone is prepared in 200 ml of methanol. This is cooled down to
approx. 5°C with
the aid of an ice bath and14.12 g (61.4 . 10-3 mol) of 3-chioro-
benzenecarboperoxoic acid
(mCPBA) is added by fractions titrating 75%. The reaction medium is kept
stirring for 15 min.
after completion of the addition and hydrolysis is performed on a diluted
solution of sodium
bicarbonate. Extraction is performed with the aid of ethyl acetate and the
phase obtained is
washed with water until neutrality, dried and concentrated under reduced
pressure. 13.2 g
of a white solid composed of the sought product is thus obtained in a mixture
with
(4-hydroxyphenyl)[4-(methylsulphonyl)phenyl]methanol.
PREPARATION V
(4-hydroxyphenyl)[4-(methylsulphonyl)phenyl]methanone
A suspension of 13.2 g of the compound obtained according to preparation IV is
CA 02335799 2000-12-19
11
prepared in 150 ml of methanol and 11.7 g (50.8 . 10-3 mol) of mCPBA is added
by portions
while stirring (titrating 75%). The reaction mixture is kept stirring for 30
minutes, then
hydrolysis is performed on a cold solution of sodium bicarbonate. Extraction
is performed with
the aid of ethyl acetate and the organic phase is washed with water until
neutrality, dried then
concentrated under reduced pressure. 13.6 g of the product sought is thus
obtained in the
form of a white solid (yield = 97%).
F = 144°C
PREPARATION VI
Dimethylcarbamotfiioic acid, O-[4-[4-(methylsulphonyl)benzoyl]phenyl] ester
Operating in a similar manner to preparation I, starting from the compound
obtained
according to preparation V, the sought product is obtained in the form of a
beige solid
(yield = 69%).
F = 150°C
PREPARATION VII
Dimethylcarbamothioic acid, S-[4-[4-(methylsulphonyl)benzoyl]phenyl] ester
Operating in a similar manner to preparation II, starting from the compound
obtained
according to preparation VI, the sought product is obtained in the form of
beige crystals
(yield = 91 %).
F = 172°C
PREPARATION VIII
(4-mercaptophenyl)[4-(methylsulphonyl)phenyl] methanone
Operating in a similar manner to preparation III, starting from the compound
obtained
according to preparation VII (adding dimethyl formamide to solubilise the
product), the sought
product is obtained in the form of a cream solid (yield = 95%).
F = 176°C
Example 17
[4-[4-(methylsulphonyl)benzoyl]phenyl] 2,3,4-tri-O-acetyl-1,5-dithio-a-D-
xylopyranoside
Operating in a similar manner to example 15, starting from the compound
obtained
according to preparation VIII, the sought product is obtained in the form of a
beige solid
(yield = 31 %).
F = 86°C
[a)2'o = + 293° (c = 0.44; DMSO)
Example 18
[4-(4-(methylsulphonyl)benzoyl]phenyl] 1,5-dithio-a-D-xylopyranoside
Operating in a similar manner to example 10, starting from the compound
obtained
CA 02335799 2000-12-19
12
according to example 17, the sought products is obtained in the form of a pale
yellow
solid (yield = 90.5 %).
F = 100°C
[a]Zbp = + 381° ( c = 0.58; DMSO)
The antiatheromatous activity of the compounds according to the invention has
been demonstrated on female mice, deficient in apolipoprotein E (homozygotes).
According to the report on this test, the product to be evaluated is
administered in the
food (standard diet) for 14 weeks. At the end of the experiment, the mice
undergo
euthanasia and sections are taken from the heart and the aortic arch. The
lesioned areas
are evaluated according to the method described in Arteriosclerosis, 1990, 10,
p. 316-323.
Table I shows the results obtained according to this test carried out with the
compound of example 2 at difference doses. The results are expressed as
percentage
reduction of the aortic sinus lesioned area, as compared with a group of
untreated control
mice.
Table I
dose administered 10 100 300
(m k
reduction (%) - 32 - 51 - 71 -
By way of comparison, the compound [4-(4-cyanobenzoyl)phenyl] 1,5-dithio-(3-
D-xylopyranoside, previously described in EP-A-0290321, was also tested. At
the dose of
300 mg/kg, this compound reduces the lesioned area by 30%. As shown in table
I, the
same biological result is achieved at only 10 mg/kg of the compound according
to
example 2. This demonstrates the superiority of the compound of the invention
in this
activity when the anomeric carbon of the xylose is in the a configuration.
During the 14 weeks of the test described above, aimed at
evaluating the antiatheromatous properties of the compound, the serum
cholesterol
level of the treated mice and the control mice was measured, and the result is
expressed in the form area-under-curve (AUC) of cholesterol during the whole
duration of the test. The results obtained with the compound of example 2 are
entered in
table Ilwhere AUC represents the area-under-curve (expressed in g.day.l-I) and
the variation (in percentage) is evaluated by reference to
CA 02335799 2000-12-19
13
the control group.
Table II
dose (mg/kg)0 10 100 300
(Ex 2) (control)
AUC 583 23 585 15 507 16 505 20
variation - 0 - 13 - 13
(%)
The analysis of the values observed reveals a cholesterol lowering effect
which
only appears over the period of 14 weeks at the 100 mg/kg dose whereas the
antiatheromatous effect is significant from the 10 mg/kg dose.
However, in view of the probability of a correlation between the antiathero
matous activity observed and a capacity of the compound to lower the serum
cholesterol
level, and in order to obtain a screening result as quickly as possible, the
compounds
according to the invention were evaluated according to their potential for
diminishing the
cholesterolaemia of mice subjected to a diet rich in fats. The test was
performed by
administration to female mice of strain C57BL/6J. The report is as follows: on
the first
day (JO), the mice undergo fasting from the 9.00 to 17.00 hours, a blood
sample being
taken at 14.00 hours. At 17.00 hours, a determined amount of food (fatty diet
containing
1.25% of cholesterol and 0.5% of cholic acid) is distributed. On the second
day (J 1 ), at
9.00 hours, the remains of the food are weighed and the mice undergo fasting
from 9.00
to 14.00 hours. At 14.00 hours, a blood sample is taken. For the groups of
treated mice,
the compound is administered by gavage, in suspension in a solution of arabic
gum, at 3
%, on the second day (J1) at 9.00 hours. The control groups only receive the
arabic gum.
The compounds were tested at the dose of 100 mg/kg. The serum total
cholesterol
is analysed and the results are expressed in percentage inhibition of the
increase in
cholesterolaemia as compared with the control group. The results obtained are
entered in
the column "Activity" of Table III. Moreover, it can be seen that the analysis
of the
cholesterol content of the various classes of serum lipoproteins reveals a
favourable effect
of the product on the HDL-cholesterol/total cholesterol ratio.
CA 02335799 2000-12-19
14
The products with formula I according to the invention can be administered,
preferebaly orally, in the form of tablets or capsules, each containing 20 to
500 mg of a
compound with formula I as the active drug, in association with eYCipients.
The posology
will be about 1 to 4 doses per day. These products will be prescribed for the
prevention or
treatment of atheromatous risk.
CA 02335799 2000-12-19
Table III
X
OR
OR Y ~ ~ I ~ ~ Ri
OR n
Ex R~ X Y R Activity
(%)
1 CN S S Ac - 40
2 CN S S H - 38
3 CN O O Ac - 46
4 CN O O H - 39
5 CN O S Ac - 41
6 CN O S H - 38
7 C N S O Ac - 26
8 CN S O H - 12
9 CF3 O O Ac -
10 CF3 O O H - 5
11 CF3 S O Ac - 23
12 CF3 S O H - 7
13 CF3 O S Ac - 24
14 CF3 O S H - 26
15 CF3 S S Ac - 17
16 CF3 S S H - 19
17 SO2CH3 S S Ac - 36
18 S02CH3 S S H -