Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
INHIBITION OF TYPE 3 3a-HYDROXYSTEROID DEHYDROGENASE
FIELD OF THE INVENTION
The present invention relates to methods of treatment of sex steroid-dependent
diseases
based upon the use of inhibitors ~of enzymes involved in the biosynthesis of
sex steroids
from natural precursors. In particular, inhibitors that reduce the natural
production of
androgens such as testosterone and dihydrotestosterone, are disclosed.
BACKGROUND OF THE RELATED ART
Many androgen-sensitive disease, i.e. diseases whose onset or progress is-
aided by
androgenic activity, are known. They include but are not limited to prostate
cancer,
benign prostatic hyperplasia, acne, seborrhea, hirsutism, androgenic alopecia,
precocious
puberty, adrenal hyperplasia, a~ad polycystic ovarian syndrome. Estrogen
sensitive
diseases, i.e. diseases whose onset or progress is aided by estrogenic
activity are also
known. They include but arE~ not limited to breast cancer, endometrial cancer,
endometriosis, leiomyoma, and precocious puberty.
Androgenic and estrogenic activity may be suppressed by administering androgen
receptor antagonists ("antiandrog~ens") or estrogen receptor antagonists
("antiestrogens"),
respectively. See e.g. WO 94/26767 and WO 96/26201. Androgenic and estrogenic
activity may also be reduced by suppressing androgen or estrogen biosynthesis
or
SUBSTITUTE SHEET (RULE 26)
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-2-
sE~cretions by known methods. See e.g. WO 90/10462, WO 91/00731, WO 92/00733,
and
V~~O 86/01105. Type 517~i-hydroxysteroid dehydrogenase is described in WO
97/11162.
The molecular cloning and characterization of the human type 3 3a-
hydroxysteroid
d~~hydrogenase from human prostatic cDNA library have been described by Dufort
et al.,
Biochemical and Biophysical Research Communications 228, 474-479 (1996).
Inhibitors of human type 5 17(3-hydroxysteroid dehydrogenase enzyme are
disclosed in
United States Provisional Patent Application filed in March 11 1998, as serial
No
60/ 077,510.
Effective inhibitors of human type 3 3a-hydroxysteroid dehydrogenase enzyme or
ef~Eertive inhibitors of both human type 3 3a-hydroxysteroid dehydrogenase and
human
type 517~i-hydroxysteroid dehydro~;enase enzymes are provided by the present
invention,
as is the discovery that androgen formation can be suppress thereby. The prior
art is not
believe to have described or suggested that the inhibition of type 3 3a-
hydroxysteroid
dehydrogenase may play a beneficial role in reducing the amount of
testosterone and
dillydrotestosterone available in target tissues.
SUMMARY OF THE INVENTION
It is accordingly an object of the present invention to more selectively and
effectively
inhibit the conversion of 4-androstene-3,17-dione to testosterone and 5a-
androstane-3,17-
dione to dihydrotestosterone using; an inhibitor of human type 3 3a-
hydroxysteroid
de'.hydrogenase while preferably avoiding inhibition of type 2 or 4 17(3-
hydroxysteroid
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-3-
dehydrogenases, type 1 3a-hydroxysteroid dehydrogenase, or any other androgen
~~egradation enzyme.
la is another object of the present invention to more selectively and
effectively inhibit the
conversion of 4-androstene-3,17-dione to testosterone and 5a-androstane-3,17-
dione to
~~ihydrotestosterone using an inhibitor of both human type 3 3a-hydroxysteroid
dehydrogenase and type 5 17a-hydroxysteroid dehydrogenase while preferably
avoiding
inhibition of type 2 or 4 173-hydroxysteroid dehydrogenases, type 1 3a-
hydroxysteroid
~iehydrogenase, or any other androgen degradation enzyme.
lft is another object to provide treatment and prevention regimens for
prostate cancer,
benign prostatic hyperplasia and prostatitis.
la is another object to provide treatment and prevention regiments for
androgen-sensitive
skin diseases, particularly acne, seborrhea, hirsutism and androgenic
alopecia.
In one embodiment, the invention provides a method of inhibiting conversion of
4-
androstene-3,17-dione to testosterone or of 5a-androstane-3,17-dione to
dihydrotestosterone in a patient in need of such inhibition comprising
administering to
said patient a therapeutically effective amount of an inhibitor of human type
3 3a-
' llydroxysteroid dehydrogenase other than 17-lactone derivative compounds.
In another embodiment, the invention provides a method of inhibiting activity
of
human type 3, 3a-hydroxysteroid ~dehydrogenase comprising administering to a
patient in
need of such treatment a therapeutically effective amount of an inhibitor of
human type 3
3a-hydroxysteroid dehydrogenase having the following structure:
CA 02339368 2001-02-02
V1'~ 00/07576 PCT/CA99/00724
-4-
R17~
16a
R2 ~a
F
wherein the dotted line is an optional pi bond;
wherein R3 is a moiety selected from the group consisting of C~-Czo alkyloxy,
Cl-Clo
acyloxy, Ci-CZO alkoxycarbonyloxy, Ci-Czo alkyloxy alkyloxy, hydroxyl, (N-
alkyl or -H)
carbamate and a moiety transformed in vivo to hydroxyl;
wherein R2 and R4 are independently selected from the group consisting of
hydrogen,
cyano, fluoro, chloro, bromo, and nitro (wherein R2 and R4 are not
simultaneously
hydrogen).
wherein Rl~ is selected from the group consisting of hydrogen, a CrCi4 carbon
moiety substituted by a radical selected from the group consisting of
hydrogen, halogen,
carboxyl, amido, Ci-Cs alkoxy and Ci-C5 alkyl or Rl~° and Rl~ together
form a Cs-C~
lactone ring or is a ketonic oxygen;
wherein Rl~° is hydroxyl, acyloxy, alkoxy, alkenyloxy, (N-alkyl or H)
amido; or Rl~"
and R~~e together form a Cs-C~ lactone ring or is a ketonic oxygen;
wherein R16" and Rl~ are independently selected from the group consisting of
hydrogen, lower alkyl, and benz:yl, or R~6a and R~6'' together form a Cs-C6
cycloalkene.
CA 02339368 2001-02-02
W~ 00107576 PCT/CA99/00724
-5-
In another embodiment, the invention provides a method of inhibiting the
activity of
human type 3 3a-hydroxysteroid dehydrogenase comprising administering to a
patient in
need of such inhibition a therapeutically effective amount of an inhibitor of
human type 3
3a-hydroxysteroid dehydrogenase selected from the group consisting of:
~2
EM-1125
EM-01667-C
O
O
..~rll
N,
v
CA 02339368 2001-02-02
WO 00/07576
PCT/CA99/00724
-6-
EM-01645
EM-1834
EM-2359
EM-1926
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
EM-2132
EM-2318
EM-2330
N
In another embodiment, the invention provides a method for determining
effectiveness of a putative inhibitor of the conversion of 4-androstene-3,17-
dione to
testosterone and 5a-androstane-3,,17-dione to dihydrotestosterone, comprising
measuring
activity of type 3 3a-hydroxysteroid deydrogenase in the presence of said
putative
inhibitor and correlating effectiveness to a reduction in said activity relate
to activity of
said dehydrogenase in the absence of said putative inhibitor.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
_$_
l:n another embodiment, the invention provides a pharmaceutical composition
comprising
~~ pharmaceutically acceptable diluent or earner and a therapeutically
effective amount of
an inhibitor of human type 3 3a-hydroxysteroid dehydrogenase having the
molecular
structure:
RAW
16a
R2
~a
R
wherein R3 is a moiety selected from the group consisting of CrCzo alkyloxy,
Cl-Clo
acyloxy, Ci-Czo alkoxycarbonyloxy, Ci-Czo alkyloxy alkyloxy, hydroxyl, (N-
alkyl or -I~
carbamate and a moiety transformed in vivo to hydroxyl;
wherein RZ and R4 are independently selected from the group consisting of
hydrogen,
cyano, fluoro, chloro, bromo, and nitro (wherein R2 and R4 are not
simultaneously
Hydrogen).
wherein the dotted line is an optional pi bond;
wherein Rl~° is selected from the group consisting of hydrogen, a C2-
Ci4 carbon
moiety substituted by a radical selected from the group consisting of
hydrogen, halogen,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
_9_
carboxyl, amido, C1-Ca alkoxy and Ci-Cs alkyl or Rl~° and Rl~' together
form a Cs-C~
lactone ring or is a ketonic oxygen;
wherein Rl~ is selected from the group cosisting of hydroxyl, acyloxy, alkoxy,
alkenyloxy, (N-alkyl or H) amido; or Rl~°' and Rl~ together form a Cs-
C~ lactone ring or is a
ketonic oxygen;
In another embodiment, the invention provides a pharmaceutical composition
comprising a pharmaceutically acceptable diluent or carrier and a
therapeutically
acceptable amount of an inhibitor of human type 3 3a-hydroxysteroid
dehydrogenase
having the molecular structure:
OH ~ R~ 00
w
NC
HO
wherein R1°° is selected from the group consisting of hydrogen,
carboxyl, amido, Ci-
~~5 alkyl, halo, nitro, hydroxy, and 'Cl-Cs alkoxy.
~fn another embodiment, the invention provides an inhibitor of human type 3
3a-hydroxysteroid dehydrogenase having the molecular structure:
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-10-
R1~9
16a
R2
R
R4
wherein R3 is a moiety selected from the group consisting of CrCzo alkyloxy,
Cl-Clo
acyloxy, CrC2o alkoxycarbonyloxy,, Ci-Czo alkyloxy alkyloxy, hydroxyl; (N-
alkyl or -I-~
c:arbamate and a moiety transformed in vivo to hydroxyl;
wherein R2 and R4 are independently selected from the group consisting of
hydrogen,
c:yano, fluoro, chloro, bromo, and nitro (wherein RZ and R4 are not
simultaneously
hydrogen).
wherein the dotted line is an optional pi bond;
wherein R» is selected frorn the group consisting of hydrogen, a CrCi4 carbon
moiety substituted by a radical selected from the group consisting of
hydrogen, halogen,
carboxyl, amido, Ci-Cs alkoxy and Ci-Cs alkyl or Rl'" and Rl's together form a
C5-C~
l.actone ring or is a ketonic oxygen;
wherein Rl'° is selected from the group consisting of hydroxyl,
acyloxy, alkyoxy,
a.lkenyloxy, (N-alkyl or H) amido; or Rl'" and Rl'~' together form a Cs-C~
lactone ring or is a
k:etonic oxygen;
wherein R16" and R~6p are independently selected from the group consisting of
hydrogen, lower alkyl, and benzyl, or Rib" and Rl~ together form a Cs-C6
cycloalkene.
CA 02339368 2001-02-02
- W O 00/07576
PCT/CA99/00724
-11-
In another embodiment, the invention provides an inhibitor of human type 3
3a-hydroxysteroid dehydrogenase :having the molecular structure:
OH ~ R100
CI
a
HO
wherein R~°° is selected from the group consisting of hydrogen,
carboxyl, amido, Ci-
Ca alkyl, halo, nitro, hydroxy, and C.'1-C3 alkoxy.
In another embodiment, the invention provides a method of treating, or
reducing the
risk of developing prostate cancer, comprising administering to a patient in
need of such
treatment or reduction a therapeutically effective amount of an inhibitor of
human type 3
3a-hydroxysteroid dehydrogenase other than 17-lactone derivative compounds.
In another embodiment, the invention provides a method of treating, or
reducing the
rusk of developing, benign prostatic hyperplasia comprising administering to a
patient in
need of such treatment or reduction, a therapeutically effective amount of an
inhibitor of
human type 3 3a-hydroxysteroid dehydrogenase other than 17-lactone derivative
a~mpounds.
In another embodiment, the invention provides a method of treating, or
reducing the
risk of developing, prostatitis comprising administering to a patient in need
of such
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-12-
treatment or reduction, a therapeutically effective amount of an inhibitor of
human type 3
3<x-hydroxysteroid dehydrogenase.
In another embodiment, the invention provides a method of treating or reducing
the risk
of developing acne, seborrhea, hirsutism or androgenic alopecia comprising
administering
to a said patient, in need of such treatment or reduction, a therapeutically
effective amount
of an inhibitor of human type 517~i-hydroxysteroid dehydrogenase activity of
human type
3 3a-hydroxysteroid dehydrogenase other than by administering a 17-lactone
derivative
compound.
The inhibitors of the invention are used for preventing and/or treating
certain diseases,
discussed herein, whose onset or progress is stimulated by androgenic
activity. One of the
mere surprising results of our laboratory work is the discovery that type 3 3a-
HSD which
is known for its catalytic activity of reactions affecting the 3 position of
steroids has now
been shown by Applicants to catalyze reactions affecting the 17 position. This
discovery
that type 3 3a-I-iSD participates in the formation of testosterone and DI-iT
from
androstenedione and androstanedione permits enhanced suppression of
biosynthesis of
these two important androgens b:y suppressing this new biosynthetic pathway
that
Applicants have discovered. It has been found that type 3 3a-hydroxysteroid
dehydrogenase displays activity similar to that of 17(3-hydroxysteroid
dehydrogenase
(catalyzing the conversion of 4-androstenedione-3,17-dione to testosterone and
5a-
androstane-3,17-dione to dihydrotestosterone), inhibitors which suppress the
17(3-
hydroxysteroid dehydrogenase activity of type 3 3a-hydroxysteroid
dehydrogenase, with
or without combination with inhibitors of type 5 173-hydroxysteroid
dehydrogenase
an~~/or inhibitors of 5a-reductase dinninish the production of androgens
catalyzed by these
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-13-
enzymes. Because androgens formed by reactions catalyzed by these enzymes are
precursors to estrogens, the invention also has applicability to diseases
whose onset or
progress is aided by estrogenic activity.
Vl~ith respect to all of the dosages recommended herein, the attending
clinician should
monitor individual patient response, and adjust dosage accordingly.
A patient in need of treatment or reducing the risk of onset of a given
disease is vne who
has either been diagnosed with such disease or one who is susceptible to
acquiring such
disease.
Except where otherwise stated, the preferred dosage of the active compounds of
the
invention is identical for both therapeutic and prophylactic purposes. The
dosage for each
active component discussed herein is the same regardless of which particular
disease is
being treated {or prevented).
As used in the methods of medical treatment of methods of reduction of risk of
onset of
diaease herein, an "inhibitor of type 3 3a-hydroxysteroid dehydrogenase" means
a
compound whose ICSO of inhibition for the enzyme in question (computed in the
same
m~~nner as described in connection with Table 1 herein) is no higher that 200
nM. It is
preferred that ICSO of such inhibitor be no higher than 50 nM, most preferably
lower than
nM. It is also preferred that undesirable inhibition of 3a-HSD type 1 and 173-
HSD type
2 be less than 90% at 3.10-~IVI, preferably less than 80%, and most preferably
less than 70°~6.
In some embodiments, it is preferred that androgenicity be less than 100% of
stimulation of
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-14-
~ihionogi cells at a concentration of 10-~IvI, preferably less than 50%, most
preferably less
than 20 % .
~Nhere two or more different active agents are discussed as part of a
combination therapy
herein {e.g. an enzyme inhibitor and an anHandrogen), a plurality of different
compounds
are administered rather than a single compound having multiple activities.
Except where otherwise noted or where apparent from context, dosages herein
refer to
v~eight of active compounds unaffexted by pharmaceutical excipients, diluents,
carriers or
other ingredients, although such additional ingredients are desirably
included, as shown in
tine examples herein. Any dosage form (capsule, tablet, injection or the like)
commonly
used in the pharmaceutical industry is appropriate for use herein, and the
terms
"<~xcipient", "diluent" or "earner" include such non-active ingredients as are
typically
included, together with active ingredients in such dosage forms in the
industry. For
e;Kample, typical capsules, pills, enteric coatings, solid or liquid diluents
or excipients,
flavorants, preservatives, or the Iike may be included.
As stated herein, the term of hydrocarbon moiety includes but are not limited
to straight or
branched alkyl, straight or branched alkenyl, straight or branched alkynyl,
phenyl,
pl:~enylalkyl, phenylalkenyl, phenylalkynyl.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-15-
BRIEF DESCRIPTION OF THE DRAWINGS
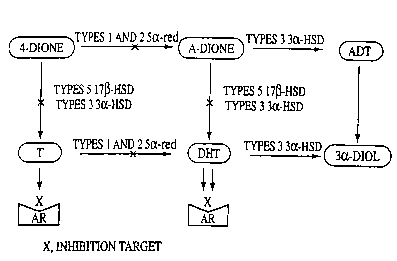
l~igure 1 shows a schematic biosynthesis pathway of active androgens in the
human
prostate.
1~igure 2 shows 17~i-HSD and 3a-I-iSD activity in intact 293 cells (ATCC CRL
1573) stably
transfected with type 3 3a-HSD, v1 culture. Cells stably transfected with type
3 3a-HSD
were seeded into 24-well plates at a density of 105 cells/well. 0.1 ~eM of
[14CJ-labeled 4-
dione and [14CJ-labeled DHT were added to freshly changed culture medium to
assess the
L7~3-HSD activity of type 3 3a-HSD~ enzyme [transform~+i~n of 4-dione to
testosterone (o)J
~~nd 3a-HSD activity of type 3 3a-HSD enzyme[transformation of DHT to 3a-diol
(O)J
a~ctiviiy of the transfected enzyme, respectively. Non transfected cells were
used as
control. After incubation for the indicated time periods, the media were
collected and
extracted, and assayed as described herein "Enzymatic assay for types 1, 2, 3,
and 5 17~-
fISD and types 1 and 3 3a-HSD".
Figures 3a to 3c show paraffin sections of normal human skin immunostained
with
antibody to type 3 3a-HSD. The prE~sence of type 3 3a-hydroxysteroid
dehydrogenase can
be seen in:
a) epithelium and fibxoblast
b) hair follicle
c) sudoriferous glands.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-16-
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Prostate cancer is a disease of the prostatic epithelium while benign
prostatic hyperpIasia
(E~PI-~ mainly involves the stroma.l compartment of the prostate. Prostatitis,
although
more common in the prostatic epithelium, can be found in both areas of the
prostate.
Applicants' recent results show that type 517(3-hydroxysteroid dehydrogenase
(type 517(3-
H;SD) and type 3 3a-hydroxysteroid dehydrogenase (type 3 3a-HSD) are present
in the
prostatic epithelium, mainly the lbasal cells, thus transforming
androstenedione into
tostosterone by the pathways shown in Figure 1. Such testosterone then
diffuses into the
lu:minal epithelial cells which are androgen-dependent, and where prostate
cancer grows.
Concerning the stromal compartment, type 3 3a-HSD and type 5 17~i-HSD are
mainly
for.~nd in the fibroblasts which are distributed among the muscle cells. It is
believed that
gr~~wth factors secreted by the fibroblasts stimulate the surrounding cells,
thus leading to
BF'H. The presence of estrogen rE~ceptors in these fibroblasts in the stroma
probably
provides the basis for the role of estrogens as well as androgens in BPH.
While Prostate
ca~icer, however, is essentially only an androgen-sensitive disease.
In the prior art, type 3 3a-HSD was known to convert DHT into androstane-3a,
17(3-diol,
an inactive metabolite. Applicants have recently discovered the surprising
role of 3a-HSD
in catalyzing the formation of testosterone and DHT from androstenedione and
androstanedione, respectively (See fiF;ure 1). The advantage of inhibiting
type 3 3a-HSD to
suppress formation of testosterone .and DHT is believed to substantially
outweigh any
reduction in androgen catabolism that might result when type 3 3a-HSD is
inhibited.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-17-
'T;he presence of numerous other enzymes which are essential for the formation
of
androgens in the prostatic tissues suggest the likelihood that combination
therapies
discussed herein (e.g., use of other inhibitors(s) of enzymatic activity,
antiandrogens
and/or castration) will Iead to superior results relative to use of one active
agent alone.
Tlus is especially believed true of those combinations that affect disease by
two or more
separate mechanisms (e.g., inhibiting two or more different synthetic
pathways, inhibiting
androgen formation in combination with blocking access to androgen receptors,
etc.).
Figure 1 applies to each of prostates cancer, benign prostatic hyperplasia,
and prostatitis,
although the cell types are different between prostate cancer and BPH. In
prostate cancer,
type 517(3-HSD and type 3 3a-HSD are mainly present in one cell type (basal
cells) while
5a;-reductases are present in the luminal cells, which are located just above
the basal cells,
thus permitting diffusion of testosterone from the basal to the luminal cells
and then
conversion into the more potent androgen DHT. For the fibroblasts located in
the stromal
compartment, the transformation of androstenedione to testosterone and then to
DHT
takes place in the same cells.
Type 1 3a-HSD is not present in a significant amount in the prostate but is
mainly a liver
enayme. Inhibitors used in the invention (e.g., inhibitors of type 3 3a-HSD,
inhibitors of
type 5 173-HSD, inhibitors of 5a-reductase, etc.) preferably have little or no
inhibitory
effect on type 1 3a-HSD which beneficially acts to inactivate androgens. Thus,
Applicants
prefer to avoid the inhibition of type 1 3a-HSD, when practicing the
invention, so as not to
delay the inactivation of androgens b:y the hepatic tissue.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-18-
V'Je have found that the human type 3 3a-hydroxysteroid dehydrogenase which
tz~ansforms 5a-androstane-3,17-dione and dihydrotestosterone to androsterone
and
a~ldrostane-3a, 17{i-diol, respectively, also transforms 4-androstene-3,17-
dione to
testosterone and 5a-androstane-3,1;x-dione to dihydrotestosterone. In
prostatic tissue, the
expression of human type 3 3a-hydroxysteroid dehydrogenase is much higher than
the
e~;pression of the human type ;i 17(3-hydroxysteroid dehydrogenase enzyme,
thus
implying that a significant propori:ion of androgens in the prostate are
formed by this
pathway (Figure 1).
In one of preferred embodiments, irihibition of androgen formation, as
illustrated in Figure
1, is performed by an efficient blockade of dihydrotestosterone (DH'I~
production with an
efi~ective inhibition of both, 5a-redu.ctase and type 5 17{i-hydroxysteroid
dehydrogenase
activities.
There are two types of 5a-reductase. Both types are expressed in the prostate,
type 2 5a-
reductase, however, is expressed at a higher level. A Merck product, Proscar
(finasteride,
MlC-906), inhibits mostly type 2 5a-reductase.
Compound GI 198745 (17(3-{N-:2,5-bis(trifluoromethyl) phenyl)carbarnoyl-4-aza-
5a-
androst-1-en-3-one) produced by Glaxo Wellcome, and EM-503 (17(i-(N,benzoyl,N-
phenyl)amino-4-methyl-4-aza-androstane-3-one), a compound of Endorecherche,
inhibit
efficiently both human types 1, and 2 5a-reductase, thus offering the more
efficient
po;~sibility of a blockade of DHT formation.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-19-
Inhibition of 5a-reductase activity, however, does increase testosterone (T)
levels.
Although weaker than DHT, T possesses also an androgenic effect that will keep
the
prostate growing to a variable extent.
:fn order to achieve a more efficient blockade of androgen formation, it is
useful to also
iinhibit the 173-hydroxysteroid dehydrogenase activity that converts 4-dione
to T or A-
~lione to DHT. This activity is catalyzed in the prostate by type 5 17(3-HSD,
and
:>urprisingly by type 3 3a-HSD. We have already developed a highly efficient
inhibitor for
type 517/3-HSD whose molecular structure and synthesis are set forth below:
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-20-
Synthesis of EM-1401, EM-1404
Scheme A
crfh~~
2,6-luGdine
DMAI'
CH~Cl=
rawneh
dppf
CO
DMSO
Pd (OAc}~ HZ, Pd/C
dppp EtOAe
EtjN. CO
ROH, DMF
n-nn
1. (COCIh, pyridine
2. HNR~R~, TF~ R:
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-21 -
3-trifluoromethanesulfonyloxy-1w't,5(10)-estratrien-17(R)-spiro-2'-(5',5'-
dimethyl-6'-
oxo)tetrahydropyran (b). Under argon atmosphere, a solution of compound a
(500. mg,
1.35 mmol), 2,6-lutidine (0.355 mL, 3.05 mmol) and 4-dimethylaminopyridine (33
mg, 0.27
nunol) in dry dichloromethane (25 mL) was cooled at 0 °C, treated with
t~~ifluoromethanesulfonic anhydride (0.308 mL, 1.83 mmol) and stirred for 45
min. The
reaction mixture was quenched with water and extracted with dichloromethane.
The
organic phase was washed with 2°~ hydrochloric acid, saturated sodium
bicarbonate and
~n~ater, dried over magnesium sulfate, filtered, and evaporated. The crude oil
was purified
by flash chromatography (hexaners-ethyl acetate 49-1 to hexanes-ethyl acetate
4-1) to
provide trifluoromethanesulfonate lb (EM-1399) {540 mg, 80%): IR (CHC13) 2957,
2872,1711,
1190, 1426, 1248, 1214, 1141, 926, 846, 621 cm-1; 1H NMR (300 MHz, CDC)3) 8
1.03 (s, 3H),
1.28 (s, 3H),1.29 (s, 3H),1.35-2.40 (gin, 17H), 2.88 (m, 2H), 6.98 (s,1H),
7.02 (d, J=8 Hz,1H),
7.33 (d, J=8.7 Hz, 1H); 13C NMR (7!i MHz, CDC13) b 14.32, 23.22, 25.48, 25.80,
26.89, 27.57,
27.68, 29.37, 31.49, 31.80, 34.69, 37.7;2, 38.46, 43.66, 47.10, 48.59, 93.43,
116.54,118.08,120.80,
121.07,127.05,139.31,140.43,147.46,,177.70.
3-carboxy-1,3,5(10)-estratrien-17(R)-spiro-2'-(5',5'-dimethyl-6'-
oxo)tetrahydropyran (EM-
15101). Method A: A mixture of compound b (560 mg,1.12 mmol), potassium
acetate (440
mg, 4.48 mmol), palladium acetate (12.6 mg, 0.056 mmol), and 1,1'-
bis(diphenylphosphino)ferrocene (1:25 rng, 0.255 mmol) in dimethyl sulfoxide
(20 mL) was
purged with carbon monoxide for 20 min and stirred over under a carbon
monoxide
balloon at 80 °C over a 3 h period. The reaction mixture was diluted
with 0.5 N
h3~drochloric acid and extracted with dichloromethane. The organic phase was
washed
with water, dried over magnesium :>ulfate, filtered, and evaporated. The
reaction mixture
w,as purified by flash chromatography (dichloromethane-methanol 19-1 to
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-22-
dichloromethane-methanol 4-1) to provide the carboxylic acid EM-1401 (300 mg,
68%): IR
(:KBr) 2937, 2872,1718,1676,1388,1.314,1230,1180,1160 cm-~;1H NMR (300 MHz,
CDC13 +
CLOD) 8 0.75 (s, 3H),1.01 (s, 6H), 1.I0-2.17 (m,17H), 2.65 (m, 2H), 7.09 (d,
J=8.1 Hz, 1H),
T.48 (s, 1H), 7.51 (d, J=8.5 Hz, 1H;1; 13C NMR (75 MHz, CDC13 + CDsOD) 8
13.71, 22.75,
24.98, 25.27, 26.65, 26.87, 28.76, 30.84, 3T .46, 34.21, 37.33, 38.22, 43.84,
46.74, 93.92, 124.84,
126.52,127.32,129.91,136.31,144.94,168.70,178.97.
3-alkoxycarbonyl-1,3,5(10)-estratrien-17(R)-spiro-2'-(5',5'-dimethyl-6'-
oxo)tetrahydropyran (c). A mixtures of compound b, triethylamine (3.25 equiv),
palladium
acetate (0.07 equiv), 1,3-bis(diphenylphosphino)propane (0.06 equiv), and
alcohol (1.5
e~~uiv to large excess) in DMF (10% W/~ was purged with carbon monoxide for 20
min
and stirred under a carbon monoxide balloon at 90 °C over a 16 h
period. 'The reaction
mixture was cooled at room temperature, diluted with water and extracted with
dichloromethane. The organic please was washed with brine, dried over
magnesium
sulfate, filtered, and evaporated. The reaction mixture was purified by 3
flash
chromatographies (2 times with benzene-acetone 4-1 and hexanes-ethyl acetate 7-
3) to
provide compound c (e.g., EM-1398, R=benzyl, 70%): IR {CHC)3) 2938, 1716,
1293, 1262,
1J~77,1152,1130,1109, 732 cm-1;1H 1VMR (300 MHz, CDC''13) 81.02 (s, 3H),1.28
(s, 3H),1.29
(s, 3H),1.34-1.41 (m,17H), 2.91 (m, :?HJ, 5.35 (s, ZH), 7.33-7.45 (m, 6H),
7.79 (s,1H}, 7.83 (d,
J=~8.1 Hz, 1H); 13C NMR (75 MHz, CDCIs) b 14.39, 23.28, 25.55, 25.74, 27.14,
27.64, 27.75,
29.25, 31.56, 31.93, 34.75, 37.77, 38.56, 44.34, 47.16, 48.82, 66.42, 93.50,
125.34, 126.90,127.45,
12 8.05,128.10,128.52,130.23,136.22,136.81,145.49,166.55,177.75.
3-carboxy-T,3,5(10)-estratrien-17(R)-spiro-2'-(5',5'-dimethyl-6'-
oxo)tetrahydropyran (EM-
1901). Method B: A mixture of compound c (350 mg, 0.72 mmol) and 10% palladium
on
CA 02339368 2001-02-02
- WO UO/07576 PCT/CA99/00724
- 23 _
activated carbon (50 mg) in ethyl acetate (40 mL) was stirred under an
hydrogen balloon
over a 3 h period. The reaction mixture was filtered on celite and evaporated.
The crude
mixture was purified by flash chromatography (dichioromethane-THF 19-1 to
clichloromethane-THF 3-1) to provide the carboxylic acid EM-1401 (240 mg,
84°~6). A
sample was recrystallized in methanol-THF (the characterization was described
F~reviously).
3-carboxamido-1,3,5(10)-estratrien=17(R)-spiro-2'-(5',5'-dimethyl-6'-
oxo)tetrahydropyran
(d). Under argon atmosphere, a solution of EM-1401 and pyridine (15 equiv) in
dry
dichloromethane (1.6% W/~ was cooled at 0 °C, treated with oxalyl
chloride (6 equiv) and
stirred for 0.5 h. The reaction mixture was allowed to reach room temperature
and stirred
over a 4 h period. The reaction rnixture was evaporated, dissolved in dry THF
(1.6%
L'J/~, cooled at 0 °C, treated with :L0 equiv of amine and stirred for
15 min. The reaction
rr~ixture was quenched with water, extracted with dichloromethane, dried over
rr~agnesium sulfate, filtered, and evaporated. The crude mixture was purified
by flash
clromatography (hexanes-acetone '19-1 to hexanes-acetone 3-2) to provide
compound d
(e~.g., EM-1404, Rl=Rz=H, 65%): IR I;CHCl3) 3433, 3350, 2941, 2873, 1702,
1664, 1611, 1388,
1:510,1159 cm-1;1H NMR (300 MH2;, CDCl3 + CDsOD) 8 0.73 (s, 3H), 0.99 (s,
6H),1.10-2.16
(rn,17H), 2.64 (m, 2H), 7.08 (d, J=8.0 Hz,1H), 7.30 (s,1H), 7.32 (d, J~9
Hz,1H);13C NMR (75
NHz, CDCIs + CDsOD) 8 13.69, 22.72, 24.96, 25.27, 26.64, 26.84, 28.78, 29.09,
30.81,31.44,
3SE.19, 37.31, 38.27, 43.72, 46.74, 93.92, 124.20, 124.93, 127.70, 130.00,
136.45, 143.88, 170.63,
1 i 8.96.
Type 5 17ø-HSD and type 3 3a-HSD share 85.5% amino acid identity. The high
primary
structure homology between type 5 17ø-HSD and type 3 3a-HSD could explain a
minor
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-24-
1.7~i-HSD activity found in type ',3 3aHSD activity. However, since type 3 3a-
HSD is
Expressed at a much higher level i:han type 5 173-HSD, the unexpected 17~i-HSD
activity
contributed by type 3 3a-HSD plays a significant role in the prostate.
Inhibition of type 3
3a-HSD activity is thus necessary to have an efficient blockade of androgen
formation
(Figure 1).
hahibitors of type 3 3a-hydroxysteroid dehydrogenase may, in accordance with
the
W vention, be utilized alone or as part of a combination therapy with other
strategies (listed
below) which have beneficial effects on androgen-sensitive diseases through
different
mechanisms, thus providing synergistic combinations. These combination
therapies
include in addition to type 3 inhibitors of 3a-hydroxysteroid dehydrogenase
(and in some
embodiments in combination ~nrith an inhibitor of type 5 17(3-hydroxysteroid
d~~hydrogenase) one or more of the following strategies:
Strat_ egy 2: Suppression of ovarian or testicular hormonal secretion by
chemical or
surgical castration. This approach us useful for the treatment of diseases
which respond
adversely to estrogen or androgen, respectively. When surgical or chemical
castration is
utilized, chemical castration is preferred utilizing either an LHRI-I_agonist,
an L.HRH
antagonist and/or an inhibitor of type 3 17~i-hydroxysteroid dehydrogenase
(which as
discussed herein catalyzes some testicular androgen formation). Suitable LHRH
agonists
ar~~ reported in US Patent 4,659,695, but any LHRH agonust showing the ability
to induce
chemical castration can be used since they all act through the same mechanisms
as
originally described (Labrie et al., J. Androl. 1: 209-228, 1980). Dosages are
known in the
art. Some suitable LHRH antagonists are reported in U.S. Patent 4,666,885 but
any LHRH
antagonist is acceptable, if used according to the recommendation of the
manufacturer.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-25-
Sixategy 2: Utilizing androgen o~c estrogen receptor antagonists
("antiandrogens." or
"antiestrogens") to prevent activation of androgen or estrogen receptors by
androgens or
e~~trogens, respectively. Strategy 2 is useful against diseases that respond
adversely to
androgenic or estrogenic activity, respectively. Antiandrogens, and dosages
therefor, are
lalown in the art (e.g. Flutamide (N-[4-vitro-3-(trifluoromethyl)phenyl)]-2-
methyl
propanamide) at a dosage of 250 mg, 2 or 3 times a day, NiIutamide at a dosage
of I50
mg/day, Casodex at a dosage of 50 to 750 mg/day.
y1l'hen antiestrogens are used in accordance with the invention, either alone
or as part of
one of the combination therapies described herein, the attending clinician
should, at Ieast
initially, use the dosages recommended by the manufacturer. However, the
attending
clinician should monitor individual patient response and metabolism and adjust
patient
dosage accordingly. Indeed, that will be true of all of the strategies
discussed herein and
all of the active ingredients used in any of the combination therapies of the
invention. One
preferred antiestrogen is EM-800 reported in PCT/CA96/00097 (WO 96/26201) The
molecular structure of EM-800 is:
OCOC(CIi3)a
~N
O
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-26-
Another preferred antiestrogen of the invention is EM-01538
C1
n~-v . , vm-Lrn-,rmrrm.nc.v W - +4.,3 E3,3
~ ~CA 02339368 2001-02-02 3 ~!3U 8f321-. c c ~~UUA~Ce-." ..,
- 01-08-2000 '_ _ CA 009900724
A ~aelective estrogen receptor modulator ~SgRM; is a/compound
th~it either directly or through its active metabolite
fumctioris as an estzogen receptor anfiagonist ( "antiestrogera~~ >
in breast tissue; yet.provi8ea estrogen-like effect fln body
fat., on bone tissue and on strum cholesterol levels.
Older preferred SERMs o~ th~_ invrt~tion include Tamoxi~en ((Z)-2 (4-('~,2
Biphenyl 1-
b~tt~enyl)1 N,N-di.methyle~e~ (,ay~able from Zeneca, UK~, Tore~,f~,e
(available
from Onion Farmos Pharrnaceutida, Finland, or Schering plough), Drolo_~cifene
a_-~d CP
336,,56 (cis-11~-~~'-PS'n'olidino-ethoxyphenylj..25-phenyl-6-hydroxy-1,2,3,4,-
o tetrv~hydronagthalene 'p-~ )-tartrate salt) (Pfizer Inc., USA). Raloxifene
Eli Lilly and Ca.,
US~~), LY 335563 and LY 35338T (Eli Lilly and Co., LTSA), Iodoxifene (S~e g~.
US~~), Levormeloxi~ene (3,4-traps-2,2-dzmethyl-3-phenyl-~-j4-(2-(2-(pyrralidin-
1-
yl)efihnxy)PhenYll-7 met-hoxyclhromaziy (Novo Noxdisk. ASS, Den~;rk) which zs
disclosed
in Sb~alir~i et al. WC'3 97j25034, Wp g7/'2503~a, WO 9?/Z5037,W0 97/25038 ;
and Karsga~rd
et 2:i. yITCJ 37/25036), GW5638 (described by Willson at aL, Er_docrinology,
138(9), 3901-
3917!, 199 and indole derivatives (disclosed by Miller et al. EP 0$02188A1)
and 'fSE 424
dem'loped by Wyeth Ayers (USA} and disdased in JIa20036347 (American home
pzoducts
caryoz2.tion) and nonsteroidal estacogex, derivatives described in Ydp
g7/g~g37.
'There are two types of 5a-reductase.Both types are expressed at the prostatE,
type 2 5a-
red~zctase, however, is expressed at a :higher letreZ. A Merck product,
Pxoscar (finasteride,
MK-906), inhibits mostly type 2 5as-reductase.
Cozr.pound GI ~ x98?45 (I7~i-~'N-2,;i-bi$(t~ifluoromethyl) phenyl,}carbamoyl-
~axa.-5a-
andzost 1-en-3-one) produced by Cilaxo Wellcome, and EM-503 (J.?~3-
(Iv;benzoyl,N_
pheryllamiya-e~.methyl-~-aza-andxostane-3-one), a compound of Endorecherche,
ir~ibit
efficiently both human types 7., and 2 5oc-reductase, thus afEering an,
efficient possibility of a
b:or.~;ade of DHT formation,
AMENDED SHEET
CA 02339368 2001-02-02
WO 00/07576
PCT/CA99/00724
_2g_
Strategy 3: Suppression of conversion of the androgen testosterone to the more
potent
androgen dihydrotestosterone (DH'T) by inhibiting the activity of testosterone
5a-reductase
(E~.g. by administering Proscar, available from Merck Sharp and Dohme Canada,
at the
rExommended dosage). Any other potent 5a-reductase inhibitor can be used. The
dosage
u;>ed can be 2 to 20 mg daily orally. The dosage should be the one recommended
by the
manufacturer. Strategy 3 is useful against diseases that respond adversely to
androgenic
activity.
Strategy 4: Utilizing an aromatase inhibitor to reduce estrogen production.
Strategy 4 is
useful against diseases that respond. adversely to estrogenic activity or
estrogen receptor-
mediated exacerbation of the tyrpe of androgen-sensitive diseases that are
also
estrogen-sensitive diseases (e.g. benign prostatic hyperplasia). Aromatase
inhibitors (and
antiestrogens) may also be used to reduce unwanted estrogenic effects that
result from
increased estrogenic levels that may occur during some treatments of androgen-
dependent
di;>eases. When aromatase inhibitors are used in accordance with the
invention, either
alone or as part of one of the combination therapies described herein, the
attending
cli;rucian should initially use the dosage recommended by the manufacturer.
When
administered orally, the dosage which is usually effective to provide the
desired serum
levels is between 1.0 mg and 20 mg of active ingredient per day per 50 kg of
body weight.
Fo:r example, Arimidex (Zeneca) is i:aken at the oral dose of 1 mg daily.
However, the
attending clinician should monitor individual patient response and metabolism
and adjust
spfxific patient dosage accordingly. Some aromatase inhibitors include, for
example,
molecular structures set forth in US patent 5,227,375. Aromatase inhibition
may also be
acl-ueved, for example, by administering Arimidex (2,2'-[5-(1H-1,2,4-triazol-1-
yImethylr
1,3--phenylene bis (2-methylpropiono:nitrile)) available from Zeneca, UK, at a
dosage of 1
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-29-
mg/day. Any other aromatase inl~ubitor can be used according to the
recommendations of
the manufacturer.
:fn general, for both androgen-sensitive diseases and estrogen-sensitive
diseases,
simultaneous treatment with inhibitors of sex steroid biosynthesis inhibitors
(inhibitors of
enzymes which catalyze one or more steps of estrogen or androgen biosynthesis
or
biosynthesis of estrogen or androgen precursors), and with estrogen receptor
antagonists
and/ or androgen receptor antagonists, are believed to have additive rather
than
redundant effect because they are acting in a beneficial manner by a different
mechanism.
likewise, the activity of two different enzyme inhibitors (enzymes which
catalyze one or
more different steps of sex steroid: biosynthesis) are believed to provide
additive effect,
f~specially where the inhibitors affect more than one synthetic pathway. Such
an approach
is believed to achieve a more complete effect.
7.'he type 3 3a-hydroxysteroid dehydrogenase inhibitors and inhibitor of type
5
17(3-hydroxysteroid dehydrogenase of the invention may be used in anX
combination with
a~ of the strategies 1-4 above whose effect (increasing or decreasing
androgenic or
estrogenic activity) is consistent with a desirable effect on the disease in
question. With
that in mind, set forth below are a list of representative diseases which may
be treated, or
the risk of which may be reduced, in accordance with the present invention.
Beneath each
disease, are indicated several preferred therapies or combination therapies
for treatment,
or risk reduction, of that particuaar disease. However, these combinations may
be
svppIemented using one or more of the four strategies listed above, limited
only by
mhether a particular disease responds favorably or adversely to estrogenic
activity and/or
to androgenic activity.
CA 02339368 2001-02-02
- WO 00/07576
-3a-
PCT/CA99/00724
Vii)
Prostate
cancer
(responds
adversely
to
androgenic
activity)
1. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase
2. Inhibitor of type 3 3a-hydroxysteroid dehydrogenaseinhibitor of
+ type 5
17~-hydroxysteroid dehydrogenase
3. Inhibitor of type 3 3a-hydroxysteroid dehydrogenaseinhibitor of
+ type 5
17(3-hydroxysteroid dehydrogenase + LHRH-agonist
(or antagonist).
4. Inhibitor of type 3 3a-hydroxysteroid dehydrogenaseinhibitor of
+ type 5
17~-hydroxysteroid dehydrogenase + inhibitor 17~i-hydroxysteroid
of type 3
dehydrogenase.
5. Inhibitor of type 3 3a-lhydroxysteroid dehydrogenaseinhibitor of
+ type 5
17(3-hydroxysteroid dehydrogenase + inhibitor17(3-hydroxysteroid
of type 3
dehydrogenase + LHRH agonist (or antagonist).
6. Inhibitor of type 3 3a-hydroxysteroid dehydrogenaseinhibitor of
+ type 5
17(3-hydroxysteroid dehydrogenase + LH12H-agonist(or antagonist)
+
antiandrogen
7. Inhibitor of type 3 3a-h,ydroxysteroid dehydrogenase + inhibitor of type 5
17~-hydroxysteroid dehydrogenase + inhibitor of type 3 17(3-hydroxysteroid
dehydrogenase + antiandrogen.
8. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + i~bitor of type 5
17(i-
hydroxysteroid dehydrol;enase + inhibitor of type 3 T7~i-hydroxysteroid
dehydrogenase + LHRH agonist (or antagonist) + antiandrogen.
9. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + i~bitor of type 5
17(3-
hydroxysteroid dehydrogenase + antiandrogen + 5a-reductase inhibitor +
LHRH agonist (or antagonist).
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-31 -
10. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17/3-
hydroxysteroid dehydro~;enase + LHRH agonist + 5a-reductase inhibitor.
11. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17~i-hydroxysteroid dehydrogenase + inhibitor of type 3 17(3-hydroxysteroid
dehydrogenase + 5a-reductase inhibitor.
12. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17~-hydroxysteroid dehydrogenase + inhibitor of type 3 17~i-hydroxysteroid
dehydrogenase + antiandrogen + 5a-reductase inhibitor.
13. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17~i-hydroxysteroid dehydrogenase + inhibitor of type 3 17(3-hydroxysteroid
dehydrogenase + LHRH ;agonist (or antagonist) + antiandrogen + 5a-reductase
inhibitor.
B) Benign prostatic hyperplasia (responds adversely to both androgenic
activity and
estrogenic activity)
1. Inhibitor of type 3 3a-hyd:roxysteroid dehydrogenase.
2. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + antiestrogen or
aromatase inhibitor.
3. Inhibitor of type 3 3a-hydraxysteroid dehydrogenase + antiandrogen.
4. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + antiandrogen + 5a-
reductase inhibitor + antiestrogen or aromatase inhibitor.
5. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + 5a-reductase
inhibitor.
6. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + antiandrogen + 5a-
reductase inhibitor.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-32-
7. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + 5a-reductase
inhibitor +
antiestrogen or aromata~~e inhibitor.
8. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17ø-hydroxysteroid dehydrogenase.
9. Inhibitor of type 3 3a-lhydroxysteroid dehydrogenase + i~bitor of type 5
17ø-hydroxysteroid dehydrogenase + antiestrogen or aromatase inhibitor.
10. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + i~bitor of type 5
17ø-
hydroxysteroid dehydrogenase + antiandrogen.
11. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17ø-hydroxysteroid dehydrogenase + antiandrogen + 5a-reductase inhibitor +
antiestrogen or aromatase inhibitor.
I2. Inhibitor of type 3 3a-tuydroxysteroid dehydrogenase + i~bitor of type 5
17ø-hydroxysteroid dehy~drogenase + 5a-reductase inhibitor.
13. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17ø-hydroxysteroid dehydrogenase + antiandrogen + 5a-reductase inhibitor.
14. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + ~bitor of type 5
17ø-hydroxysteroid dehydrogenase + 5a-reductase inhibitor + antiestrogen or
aromatase inhibitor.
C) Prostatitis (responds adversely to androgenic activity)
1. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + antiandrogen.
2. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + 5a-reductase
inhibitor.
3. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + antiandrogen + 5a-
reductase inhibitor.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-33-
4. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + i~bitor of type 5
17~i-hydroxysteroid dehydrogenase.
5. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17~-
hydroxysteroid dehydro;genase + antiandrogen.
6. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17(3-hydroxysteroid dehydrogenase + 5a-reductase inhibitor.
7. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
173-hydroxysteroid dehydrogenase + antiandrogen + 5a-reductase inhibitor.
L)) Acne, seborrhea, hirsutism, and androgenic aIopecia (responds adversely to
androgenic activity)
2. Inhibitor or type 3 3a-hydroxysteroid dehydrogenase.
2. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17~i-hydroxysteroid dehydrogenase.
3. Inhibitor of type 3 3a-hydroxysteorid dehydrogenase + antiandrogen.
4. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17~i-hydroxysteroid dehydrogenase + antiandrogen.
5. Inhibitor of type 3 3,x-hyddroxysteroid dehydrogenase + inhibitor of
5a-reductase.
6. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of type 5
17(3-hydroxysteroid dehydrogenase + inhibitor of 5a-reductase.
7. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + antiandrogen +
inhibitor of 5a-reductase.
8. Inhibitor of type 3 3a-hydroxysteroid dehydrogenase + inhibitor of
17~i-hydroxysteroid dehydrogenase + antiandrogen + inhibitor of 5a-reductase.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
'When type 3 3a-hydroxysteroid inhibitors are used in accordance with the
invention,
either alone or as part of one of tl-~e combination therapies described
herein, the attending
<:linician desirably will target patient serum concentration of the type 3
inhibitor between
0.5 ng/ml and 100 ng/ml, preferably between 1 ng/ml and 20 ng/ml, and most
preferably
hetween 1 ng/ml and 10 ng/ml. Serum concentration may be measured by LC/MS.
lNhen administered orally, the dosage which is usually effective to provide
the desired
~;erum levels is between 1.0 mg andl 1,000 mg of active ingredient per day per
50 kg of body
weight, preferably between 10 mg and 500 mg and most preferably between 10 mg
and 100
rng. However, dosage should vary with the bioavailability of the chosen
inhibitor and
with individual patient response. For example, when EM-01645, or EM-01667-C
are
chosen, oral dosage is preferably between 5 mg and 500 mg per day per 50 kg
body weight,
more preferably between 10 mg/d.ay and 300 mg/day, for example between 20
mg/day
and 100 mg/ day. The attending clinician should monitor individual patient
response and
serum levels, if judged appropriate, and adjust patient dosage accordingly.
When
administered by injection, a lesser dosage is usually appropriate, e.g. 10 mg
to 100 mg per
day per 50 kg of body weight.
V~Jhen type 5 17~i-hydroxysteroid inhibitors are used in accordance with the
invention, as
part of one of the combination therapies described herein, the attending
clinician desirably
will target patient serum concentrai:ion of the type 5 inhibitor between 0.5
ng/ m1 and 100
n;~/ml, preferably between 1 ng/mll and 20 ng/ml, and most preferably between
1 ng/ml
and 10 ng/ml. Serum concentration may be measured by LC/MS. When administered
orally, the dosage which is usually effective to provide the desired serum
levels is between
1. () mg and 1,000 mg of active ingredient per day per 50 kg of body weight,
preferably
bE~tween 10 mg and 500 mg and most preferably between 10 mg and 100 mg.
However,
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-35-
dosage should vary with the bioavailability of the chosen inhibitor and with
individual
patient response. For example, when EM-1404 are chosen, oral dosage is
preferably
hetween 5 mg and 500 mg per day per 50 kg body weight, more preferably between
10
mg/ day and 300 mg/ day, for example between 20 mg/ day and 100 mg/ day. The
attending clinician should monitor individual patient response and metabolism
(serum
levels, if judged appropriate) and .adjust patient dosage accordingly. When
administered
by injection, a lesser dosage is usually appropriate, e.g.10 mg to 100 mg per
day per 50 kg
of body weight.
Z'Vhen type 3 17~i-hydroxysteroid inhibitors are used in accordance with the
invention, as
frart of one of the combination therapies described herein, the attending
clinician desirably
will target patient serum concentration of the type 3 inhibitor between 0.5
ng/ml and 100
r~g/ml, preferably between 1 ng/rr~l and 20 ng/ml and most preferably between
1 ng/ml
and 10 ng/ml. When administered orally, the dosage is preferably between 1.0
mg and
1,000 mg of active ingredient per day per 50 kg of body weight, preferably
between 5 mg
and 500 mg and most preferably between 10 mg and 100 mg. However, the
attending
clinician should monitor individual patient response and metabolism and adjust
patient
dosage accordingly. Synthesis of such an inhibitor is described below.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
Synthesis of type 3 I7~i-HSD inhibitors
Scheme B
OTBDMS
TBOMSCI
Irtffdazols, DMF. rt
O O
a DHT f
RMpBr(Cp, THF a EtzO, 0'C
OTBDMS
R
As R' ~t~
9v R~ ~t~t~
a) MeOF4HCi (2%)
rt
e) da»s' rap.n, (2.7AAj
., rc
0
Rw
CS-213 R = CH=~ BTN-161-94
EM-13i'4-CS O~hC~~ BTN-194-82
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99100724
-37-
Protection of the 17(3-alcohol with TBDMS. To a solution of
dihydrotestosterone (DHT, e)
(.5g,17.2 mmol) in DMF was added imidazole (6 eq.) and TBDMSCI (5 eq.). The
reaction was
stirred overnight at room temperature. The mixture was poured onto ice and
filtered. The
resulting white precipitate was washed with water, dried over phosphorous
pentoxide
under reduced pressure for 24 h. .A 85 to 90 % yield was obtained.
17~i-[(tent butyldimethylsilyl)oxyJ-;5a-androstane-3-one (f). White solid; IR
(KBr)
v 1719 (C=O, ketone); 1H NMR (ClDCl3) S -0.001 and 0.005 (s, 6H, Si(CH3)2),
0.71 (s, 3H,
C:H3-18), 0.87 (s, 9H, SiC(CH3)3), 1.01 (s, 3H, CHg-19), 3.54 (t, J = 8.2 Hz,
1H, CH-17); 13C
1\fMR (CDC13) 8 -4.80 and -4.47, 11.41, 11.52, 18.11, 21.13, 23.56, 25.87,
28.98, 30.94, 31.36,
3:x.54, 35.78, 37.13, 38.21, 38.65, 43.36~, 44.74, 46.84, 50.55, 54.15, 81.79,
212.03.
A.lkylation of the carbonyl at position 3. To a solution of compound f (500
mg,1.23 mmol)
in dry THF (100 mL) at 0°C wa.s added dropwise 3 eq. of commercially
available
Grignard's reagent, in dry THF. ThE~ mixture was allowed to react for 3 h at
0°C, then left
o~~er night at room temperature. A ;solution of saturated NH4C1 was added and
the trade
product was extracted with EtOAc. The organic phase was washed with a
saturated NaCI
solution, dried over MgS04 and evaporated under reduced pressure. The 3(3-
alkylated
stereoisomer was easily separated from the 3a-alkylated stereoisomer by flash
chromatography on silica gel, using a mixture of hexanes and ethyl acetate as
eluent. When
th.e Grignard's reagent was generated in situ as in the case of ethylphenyl
magnesium
bromide, 5 eq. was prepared, by a well-known procedure, using the
corresponding
bromide, activated magnesium and iodide. The steroid was then dissolved in dry
diethyl
ether and added dropwise to the solution of reagent. The yields obtained were
around 60%
for the two stereoisomers.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-38-
3ø-benzyl-17ø[(tent-butyldimethyl~silyl)oxy]-3a-hydroxy-5a-androstane (g').
White solid
(;Z4°6); IR (KBr) v 3585 and 3460 (OH, alcohol); 1H NMR (CDCIg) 8 0.002
and 0.009 (s, 6H,
Si(CH3)2), 0.69 (s, 3H, CHg-18), 0.75 (s, 3H, CHg-19), 0.88 (s, 9H,
SiC(CHg)g), 2.71 (s, 2H,
C.'H2Ph), 3.54 (t, J= 8.2 Hz,1H, CH-:17), 7.20 to 7.34 (5H, Ph); 13C NMR
(CDCIg) b -4.82 and
-~E.50(SiC(CH3)g), 11.25, 11.40, 18.08, 20.62, 23.50, 25.85, 28.41, 30.91,
31.62, 33.27, 33.81,
3;5.60, 35.84, 37.19, 40.10, 40.84, 43.30, 50.43, 50.69, 54.43, 71.22, 81.82
{C-17), 126.37, 128.09
(a'.~,130.56 (2X),137.06.
3cx-hydroxy-3ø-(phenylethyl)-17øj(tert butyldimethylsilyl)oxy]-5a-androstane
(gb),
yl~hite solid (38°6); IR (film) v 3447 (OH, alcohol);1H NMR (CDCIg) 8
0.018 and 0.025 (s, 6H,
Si(C1-I3)2), 0.71 (s, 3H, CH3-18), 0.78 (s, 3H, CH3-19), 0.89 (s, 9H,
SiC(CH3)3), 273 (m, 2H,
Ph-CH2), 3.56 {t, J = 8.1 Hz,1H, CH-:L7), 7.18 to 7.31 (5H, Ph);13C NMR
(CDCxg) 8 -4.77 and
-4.46 (Si(CHg)g),11.28, 11.44,18.12 (SiC(CHg)3), 20.67, 23.54, 25.89
(SiC{CHg)3), 28.52, 29.60,
30.97, 31.66, 33.31, 33.92, 35.66, 36.04, 37.25, 40.03, 41.05, 43.35, 46.47,
50.76, 54.55, 71.50 (C-
3),, 8L86 (C-17),125.68,128.38 (4X),142.82.
Procedure for hydrolysis of TBDMS group and oxidation of the resulting
alcohol. The
sil;ylated ether was dissolved in a methanolic solution of HCl {2%, v/v) and
the resulting
mixture was stirred at room temperature for 3h. Water was then added and MeOH
evaporated under vacuum. The resulting white precipitate was submitted to
Jones'
oxidation without purification. To a stirred solution of crude alcohol in
acetone at 0°C,
Jones' reagent (2.7M chromic acid solution) was added dropwise. After 30 to 45
minutes,
thE~ reaction was completed. Isopropanol and water were added and acetone was
removed
in vacuo. The remaining aqueous layer was extracted with EtOAc. The combined
organic
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-39-
phases were washed with brine, dried over MgS04, filtered and evaporated under
reduced
pressure. The purification was done on silica gel, using HPLC grade solvents,
EtOAc and
hexanes as eluents.
3~~-benzyl-3a-Hydroxy- -5a-androstane-17 one (CS-213). White solid (88~ for
the two
steps); IR (KBr) v 3408 (OH, alcoho~l),1732 (C=O, ketone); 1H NMR (CDC13) 8
0.75 (s, 3H,
C~ig-19), 0.84 (s, 3H, CH3-18), 2.69 (s, 2H, CH2Ph), 7.18 to 7.32 (5H, Ph);13C
NMR (CDCl3)
8 11.18, 13.78, 20.20, 21.71, 28.16, 31).79, 31.52, 33.18, 33.70, 35.64,
35.79, 35.88, 39.97, 40.69,
47.75, 50.39, 51.41, 54.22, 71.12,126.40,128.09 (2X),130.51 (2X),136.93,
221.27
3a-hydroxy-3~-phenylethyl-5a-androstane-17-one (EM-1324-CS). White solid (82~
for the
tiIVO steps); IR (film) v 3486 (OH, alcohol), 1737 (C=O, ketone); 1H NMR
(CDC13) 8 0.79 (s,
3:H, CH3-19), 0.86 (s, 3H, CHg-18), :271 (m, 2H, Ph-CH2), 7.18 to 7.30 (5H,
Ph); 13C NMR
(('yDCIg) 811.21,13.82, 20.26, 21.76, .'8.26, 29.54, 30.87, 3T.58, 33.27,
33.80, 35.10, 35.84, 36.07,
3'x.89, 40.90, 46.43, 47.80, 51.49, 54.35, 71.42,125.69,128.31 (2X),128.39
(2X),142.70, 221.31.
All of the active ingredients used in any of the therapies discussed herein
may be
formulated in pharmaceutical compositions which include one or more of the
other active
ingredients. Alternatively, they may each be administered separately but
sufficiently
simultaneous in time so that a patient eventually has elevated blood levels or
otherwise
enjoys the benefits of each of the active ingredients (or strategies)
simultaneously. In some
preferred embodiments of the invention, for example one or more active
ingredients are to
bE~ formulated in a single pharmaceutical composition. In other embodiments of
the
invention, a kit is provided which includes at least two separate containers
wherein, the
contents of at least one container differs in whole or in part from the
contents of at least
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-40-
o:ne other container with respect to active ingredients contained therein. Two
or more
different containers are used in these combination therapies of the invention.
Combination
W erapies discussed herein also include use of one active ingredient of the
combination in
the manufacture of a medicament for the treatment (or prevention) of the
disease in
question where the treatment or prEwention further includes the other active
ingredients)
or strategy of the combination. Some embodiments of the methods of treating or
preventing disease discussed herein, utilize the specific type 5 17~i-
hydroxysteroid
dE~hydrogenase inhibitor and/or type 3 3a-hydroxysteroid dehydrogenase
inhibitors
discussed herein (i.e. the molecular structures discussed herein).
Ll-iRH agonists and LHRH antagonists may be used interchangeably to suppress
either
testicular or ovarian hormonal secreiions by known techniques, except where
preferences
are otherwise stated herein. It is desired that activation of glucocorticoid
receptors be
minimized when administering the active ingredients of the invention.
Inhibitors of type 3
17~i-hydroxysteroid dehydrogenase may be used to provide advantages similar to
those
provided by LHRH agorists or antagonists.
PREFERRED INHIBTTORS OF TYPE 3
3a-HYDROXY;iTEROID DEHYDROGENASE
Sei: forth in the tables below are lists of compounds which we have found to
be useful as
inlvbitors of type 3 3a-hydroxysteroid dehydrogenase. The tables also include
in many
instances further tests of a particular compound on other important parameters
such as
anc~rogenic and antiandrogenic activity and the effect of a compound on
androgen
receptors, proliferation of androgen-sensitive cells, and other effects more
fully explained
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
- '1-
below. In tables below that do not indude a "prime" (~ in their table number,
details of
moIecuiaz structure of preferred inhibitors (or comparison compounds) are set
forth The
corresponding tables with a ";prime" (') in heir table number shows
information about the
tonal efficacy of each tested compound. The numbers in the column headings
correspond to a desQiption at the end of all of the tables regarding what
information is
reported in each column acrd how it is determined. Entries Ieft blank are not
yet
determined.
SUBSTITUTE SHEET (RULE 26)
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-42-
Table 1
Laboratory Name STRUCTURE
EM-01645 °
EM-2330
»o
EM-1832 off
v
HO
EM-1831 O
HO
EM-1834 0
NC
HO
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-43-
Laboratory Name STRUCTURE
EM-1836
OH_
NC
0
HO
EM-1131
EM-01667-C
0
a
EM-01621
....
EM-1125-CS
....
EM-1126
_J
EM-1124
0
CA 02339368 2001-02-02
WO 00/075?6 PCT/CA99/00'724
- 44 -
n
~i m ~ N
WW
cn
wBM~3f wM~914~1 %
w
0
" iu ~, O u7
,e o
jy iu ~ O
d~
P!~~W wRl~4d1 X w
W o
' W ~- o
6 o th
iu M O
.un~y~sBoh1 wx w
we
"' W O O N
H ID O r
N ro~daa~ iu iu co
,..~uoBorpuro uo~9~yp
x
w
W a
' W O O N
m O
T'~W
He
X11 1~SL-NZ O
m ~
O
T'1
,R" - o n M
Y
O~ _ v
~
~ ~ ~ ~ o.
~il~!iod l6ouol4S
o M O c~1
$ y '
, D
,~
m
A
itl ~ O ~O
~
h A a~Lll w52 z
s
o
~~Ii~V ICihoC S ~ ~ N O~.~ _
1. ~I(1 w/~pp~ t_~ M v ~ M
~
v
s ~, gum ~ =.
0 N p ~
S ~ M !'1 ~ n
Z ~1 w~l9lY~N v u1 .r
v v
~~ r
M
r
a ~ w Ii ~ O c0 a ~ ~ a0 O~
C~ 'r
N i o~1 wfNVl4~I V V if~ +~
~
~e o c v,
a
o
r,
~: : ~'a:~
rn
I
g n
N
T''J
Nt-o anv
~
~
IB w0
iinV~l
~"
V
M C N
o '...-." O
W ~ ~ W W W
I t t7 tit
L
SUBSTITUTE SHEET (RULE 26)
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
- 45 -
", ,c N o 0
a ~
~.saa~ ~luaww x ,~
Wo
W O N ~ O
0
W O Y O
.o~a.~.a
n~rs~~n ~or~wra~t
x
W o
W O O O e-
~ O ~~ ~ N
.o~a.o.a
wwa.sw ~rslarrn x
jy o
' IL O If1 in n
M Y O I~.
ri o .-
~o~da'sa iu iu
wa~W ~til9I4W x
;" o
"' W N N
o O O O
W
n
W ~
Lil~ri~V i'St'aZ: O O O
o 0~0
O w
O
v-. O O
o __
0
U ~ '''
'~ 0 0
ISouo~
~ltud . 4S
o ~ O oD r
m iu r' ' r;
'
O A
u1 e'f O O
N
>:M'91i $ ~
A ~i ~14W1 ~?
n
_
~ N
A1!~ 1ZN'~i: b ~ iN4 ~' O
l.wRlltV~ ~ =
N m r c~
m
t~ ~A a z
0>tH'91i $ ~
Z ~i14i4wI ~ .~
I~11 GiH9L1 $ i
i ~l w~t9Nira ~3
_ t ~ Iff N a0
CSNtt H S' ~ A Ov p ~ o
~'
'r'~
t 1 ~t~IVI~Ni ~ ~ ~
,e ~
~ n oho
4c-o anw ~ " D .r
L7t119otlo~iot91~1) Z, +~
a
0~ H
WfH/ v_G N N N
_ ~ Y
O Y ~ r~
L
W W W W
SUBSTITUTE SHEET (RULE 26)
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-46-
Table 2
Laboratory Name STRUCTURE
EM-01678 o ff
-../
OpN
HO
EM-01666 OH
--./
HO
N02
0
HO
OH
-.,/
CI
0
HO
EM-01762
CI
HO
CI
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
Laboratory Name STRUCTURE
EM-01801
OH -
HO
Br
EM-OI807
EM-01812 OH
F
0
HO
EM-0I813 OH
(estradiol)
0
HO
~-1~ O
CI
HO
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
_ qg _
O
i
u
H u~s3 ~.
to C cnC N Q
0
~ f'7P7N ~ N
H Pi~R~~III~J IMO X
A
W o
'~ W N1 fWO t~N
W ~ N Q ~ N
w
!'tIloi/'~f1/'npla IIW
jy~ o
~ Q
W O O O -..
,
a a N N N
Y7 a
W W
~.s~p~, ~wru~w x
jy o
' W C O C O
a
e~ W
N~
W W
~ m
0 m
W
N
S
n
'~ - N
i6ouoi
~iM!t~l/
4S
.
iu ,..
' ~ i ' ~~
'
~ , ~ Y
fr 0 O ~ ~ N
~1 $ ~ v t
V
n ~1 ~ill~l~1 Y S
'~ O m
III lsl~t $ ~ a a i a a ~ cN~~
i ~l w11114~1 V S
W .~.IV V V
V
C ~l. ~1PM St ...
~ii~ 0~f'SLl ,s ~ ao a0
a ~0h
x..uct.vahni~ ~ =
v v v v
~L ~!. ~1111~Ir1
V =
O ~ Q'O~
O~
a! ~ ~ ~ n w ~ ~ ! f1
W
C ~Ri1N1~1 ~3 =
V a
v
~~i~~t! ~ 0
C
N
0 0
n ~ 0 0
a 0
O C O O O
O O O
V V ~ ~ ~
W W
ft ~ i1f 1 l
f. i t
i
SU9STITUTE SHEET (RULE 28)
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
- 49 -
R
,n ..ad.o.y
wtsups~ wpt9Nlui x
m
e!~
~wdxa.a
ri PI~R~M9 wRlht~! X
W a
W
~ a
M
W W
mdrwy
-, .y..n..sda uonn,t4w
x
we
h a
~ iu
~
I w6wpw uo
l4u1 X
iV
i~
c
iL
p.~H ao
u, iu
lilMi~V 1-Sl~liZ N
m
W
W
p O
p
A
~l~V lBouol4S
W
A
m A
W
~tN~Cll ." ~
A ~t wRNIPM v c
LiI~V Clf~! ~ ~ cN~fa
~ ~t wRl~IW1 ~? 5
.....
0lttlLl $ ~ n
wRI4lW! ~ ~ a
~O
V
alHltl ~
1 ~~l wliNllWI ~ =
t d'a
0>lIHC .o ~ W i o
o
C eRl91W1 ~3
N tn
F
4ro ~nv w
~tua~lt~=~t~ i~o 0
0
w w
sues sHe~r ~RU~ zs~
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-50-
LEGENDS TO TABLES
In column 1, the oral bioavailability of preferred type 3
3a-hydroxysteroid dehydrogenase inhibitors, expressed in ng/mL.h, was
determined as described below in " 1 n Vivo Assays of Bioavailability of
'.LO Human type 3 3a-hydroxysteroid dehydrogenase inhibitors". Higher
number are desirable. ND means that a determination was not done.
In column 2, the inhibition of human type 3 3a-hydroxysteroid
dehydrogenase activity expressed by the concentration which produce
:L5 50°r6 of inhibition of enzymatic activity (ICso in nM) is reported.
The
manner in which IC~o~ was determined is described infra in " II-
Enzymatic assay for types 1, 2, 3 and 5 I7~i-HSD and types 1 and 3 3a
HSD. Blank means that a determination was not done. In parentheses is
reported the percentage of inhibition of enzymatic activity by the
~!0 inhibitor at 3.10- and 3.1.0- M.
In column 3, the inhibition of human type 1 173-hydroxysteroid
dehydrogenase activity expressed by the concentration which produce
50°6 of inhibition of enzymatic activity (ICSO in nM) is reported. The
~~5 manner in which ICso ~Nas determined is described in " II- Enzymatic
assay for types 1, 2, 3 and 517/3-HSD and types 1 and 3 3a-HSD". Higher
numbers of ICso are desirable. Blank means that a determination was not
done. In parentheses is reported the percentage of inhibition of
enzymatic activity by the inhibitor at 3.10- and 3.10- M.
a~0
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-51-
In column 4, the inhibition of human type 2 17~i-hydroxysteroid
dehydrogenase activity expressed by the concentration which produce
50°~ of inhibition of enaymatic activity (ICso in nM) is reported. 'The
manner in which ICso was determined is described in " II- Enzymatic
assay for types 1, 2, 3 and 5173-HSD and types 1 and 3 3a-HSD". Higher
'.t0 numbers of ICso are desirable. Blank means that a determination was not
done. In parentheses is reported the percentage of inhibition of
enzymatic activity by they inhibitor at 3.I0-~ and 3.10- M.
In column 5, the inhibition of human type 3 17~i-hydroxysteroid
dehydrogenase activity .expressed by the concentration which produce
509'° of inhibition of en.rymatic activity (ICSO in nM) is reported.
The
manner in which ICSO was determined is described in " II- Enzymatic
assay for types 1, 2, 3 and 517(3-HSD and types 1 and 3 3a-HSD". Lower
:!0 numbers of ICso are desirable. Blank means that a determination was not
done. In parentheses is reported the percentage of inhibition of
enzymatic activity by the inhibitor at 3.10- and 3.10- M.
~!5 In column 6, the inhibition of human type 1 3a-hydroxysteroid
dehydrogenase activity expressed by the concentration which produce
50°~6 of inhibition of en2:ymatic activity (ICSO in nM) is reported.
The
manner in which ICso was determined is described in " II- Enzymatic
assay for types 1, 2, 3 and. 517~i-HSD and types 1 and 3 3a-HSD". Higher
?~0 numbers of ICso are desirable. Blank means that a determination was not
done. In parentheses is reported the percentage of inhibition of
enzymatic activity by the inhibitor at 3.10- and 3.10- M.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-52-
In column 7, the inhibition of human type 5 173-hydroxysteroid
dehydrogenase activity expressed by the concentration which produce
50% of inhibition (ICSO in nM) is reported (centered numbers). The
'.l0 manner in which ICSO svas determined is described in " II- Enzymatic
assay for types 1, 2, 3 and 5173-HSD and types 1 and 3 3a-HSD". Lower
numbers for ICSO are desirable. When ICSO was not determined, the
percentage of inhibition is reported in parentheses at 3.10-~IVI (left
number) and 3.10-~M (right number). In parentheses is reported the
7l5 percentage of inhibition of enzymatic activity by the inhibitor at 3.10-
and 3.10- M.
In column 8, the androgenic activity of preferred type 3
~!0 3a-hydroxysteroid dehydrogenase inhibitors expressed as the percentage
of stimulation of proliferation of Shionogi cells at concentrations of 10-~ M
(left number) and 10-~ M (right number) of inhibitor. The manner in
which the stimulation is determined is described in " III_
Androgenic/Antiandrogenic Activity ". Lower numbers are desirable.
~~5 ND means that a determination was not done.
In column 9, the antiandrogenic activity of preferred type 3
3a-hydroxysteroid deriydrogenase inhibitors expressed by the
concentration which produce .50% of inhibition (ICSO in nM) of DHT-
30 induced proliferation of Shionogi cells is reported (bracketed centered
numbers). The percentage of inhibition of DHT-induced proliferation of
Shionogi cells at concentrations of 10-~IvI (left number) and 10-~M (right
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
_5~
number) of inhibitor is also reported. The manner in which the inhibition
is determined is described in " III- Androgenic/ Antiandrogenic Activity".
Lower numbers are desirable. ND means that a determination was not
done.
In column 10, the estrogenic activity of preferred type 3
3a-hydroxysteroid dehydrogenase inhibitors expressed as the percentage
of stimulation of the proliferation of ZR-75-1 cells at concentrations of 10-
~1VI (left number) and 10~M (right number) of inhibitor. The manner in
which the stimulation is determined is described in "IV-
Estrogenic/Antiestrogenic Activity" Lower numbers are desirable. ND
means that a determination was not done.
In column 11, the antiestrogeruc activity of preferred type 3
3a-hydroxysteroid dehydrogenase inhibitors expressed as percentage of
inhibition of Erinduced proliferation of ZR-75-1 cells at a concentrations
of 10-~1VI (left number) and 10-~ (right number) of inhibitor is reported.
The manner in which the inhibition is determined is described in "IV-
Estrogenic/Antiestrogenic Activity". Lower numbers are desirable. ND
means that a determination was not done.
In column 12, the binding on androgen receptor expressed as percentage
of inhibition of the binding of [3H]R1881 at the concentration of 10-8M
(stared number at 10-~ M) (left number) and 10-~M (stared number at
10-5 M) (right number) of inhibitor is reported. The manner in which the
percentage of inhibition is determined is described in "V-Androgen
Receptor (AR) Assays". Lower numbers are desirable.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-54-
In column 13, the binding on progesterone receptor expressed as
percentage of inhibition of the binding of [3H]R5020 at the concentration
of 10-~M (stared number at 10-~ M) (left number} and 10-~M (stared
number at 10-5 M) (right number) of inhibitor is reported. The manner in
which the percentage of inhibition is determined is described in "VI-
Progesterone Receptor Assay". Lower numbers are desirable.
In column 14, the binding on glucocorticoid receptor expressed as
percentage of inhibition of the binding of [6,7-~H*(I~j-dexamethasone at
the concentration of 'I0$M (stared number at 10-~ M) (left number) and
10~ M (stared number at 10-~ M) (right number) of inhibitor is reported.
The manner in which the percentage of inhibition is determined is
described in " VII- Glucocorticoid Receptor Assay ".
In column 15, the binding on estrogen receptor expressed as percentage
of inhibition of the binding of [3H]EZ at the concentration of 10-~M (stared
number 10-~ (left number) and 10-~M (stared number 10-s) (right number)
of inhibition is reported. The manner in which the percentage of
inhibition is determined is described in " VIII-Estrogen Receptor (ER)
Assay ".
EFFICACY OF THE PREFERRED INHIBITORS
I- In Viyo Assays of Bioavailability of Human t~.pe 3
3a-hydro_xysteroid dehydro~enase inhibitors
1) Princiyle
The assays of the bioavailability of type 3 3a-hydroxysteroid
dehydrogenase inhibitors were performed in male Sprague Dawley rats
CA 02339368 2001-02-02
Wn 00/07576 PCT/CA99/00724
-55-
by measuring the plasma concentrations of the compounds after single
oral administration of the compounds. The measurements at various
time intervals were for values greater than or equal to 1.0 ng/ mL and
less than or equal to 50 ng/ mL.
a) Animals and treatment
Male Sprague-Dawley rats [CrI:CD(SD)Br] weighing 275-350 g were
obtained from Charles-River Canada Inc. and housed 2 per cage during
the acclimation period and individually during the study period. The
animals were maintained under a regimen of 12 hours light: 12 hours
dark (lights on at 08:x). Animals received certified Rodent feed (Lab
Diet # 5002, pellets) and tap water ad libifum. Rats were fasted (access to
water only) starting on the evening prior to dosing.
Each compound to be tested was administered to three animals as a
suspension in 0.4°~ nnethylcellulose by oral gavage at a dose of 0.5
mg/rat (1.0 ml/rat). Four to eight new compounds were tested each day
and one group of animals received megestrol acetate (MGA} under the
same conditions on each dosing day as a reference. One blood sample of
~0.7 ml was collected from the jugular vein of rats under
Isoflurane-induced anesthesia at 1, 2, 3, 4, and 7 hours post-gavage.
Blood samples were immediately transferred into a refrigerated 0.75 ml
Microtainer containing EDTA and kept in an ice-water bath until
centrifugation at 3000 rpm for 10 minutes. Plasma separation was
performed rapidly (less than 50 minutes) after blood collection. One
aliquot of 0.25 ml of plasma was then transferred into a borosilicate tube
(13 x 100) and was rapidly frozen on dry-ice. Plasma samples were kept
CA 02339368 2001-02-02
WO 00/07576 PCTICA99/00724
-56-
at -80°C until measurement of plasma concentration of the inhibitors)
by
LCMS/ MS.
2) LCMS measurements
a) Apparatus
1. Vacuum manifold
2. Turbo Vap LV evaporator
3. Mass spectrometer API III or API-300 (PE/Sciex) with
associated peripherals
4. Automatic Injector
5. HPLC pump
6. Infusion pump
7. Calibrated pipets
b) Reagents and Solutions
1. Methanol, HI'LC grade
2. Water, '~tJltrapure (Super Q)
3. Ethanol, reagent grade
4. N-butyl chloride, HPLC grade
5. Acetone, HPLC grade
6. Male rat plasma (EDTA)
7. type 3 3a-hydroxysteroid dehydrogenase inhibitors in
reference standard ethanol solution approximately 100
~g/mL
8. EM 248 Internal Standard reference standard (solution of
50 ng/mL)
9. Mass calibrator solution Polypropylene Glycol (PE/Sciex)
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-57
c) Mass Spectrometer Conditions
Detector: Mass spectrometer API-300 (PE/Sciex)
Interface: Turbo Ion spray inlet (split 1/5)
Auxiliary flow: 4.5L/ minute (nitrogen)
Nebulizer Flow: 11
Curtain Gas Flow: 11
Probe Temperature: 460 °C
Pressure: Approximately 3 x 10-5 Torr
CAD gas thickness: 3
Count Control: 1
Mobile Phase: Gradient of Methanol with 1 mm Ammonium
formate and 'Water with 1 mrn Ammonium formate
Flow Rate: 1 mL/ minute
d) Mass SpectrometerAnalysis Parameters for EM-1128
Dwell time: 150 cosec
Pause time: 30 cosec
Duration: 4 minute
MRM mode for
' EM-1118 analysis : 444.2 and 398.3
Injection: 10 ~L
Data handling: "API Standard Software" update version.
e) Preparation of Standard Solutions
Stock solutions for each type 3 3a-hydroxysteroid
dehydrogenase inhibitors were prepared in methanol and,
when not in use, the methanol solutions were stored at -20°C.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99100724
-58
Calibration curve standard solutions for each compound were
prepared in male rat plasma as illustrated in Table 1.
A solution of internal standard in methanol containing EM-248
at 50 ng/mL, was prepared from stock standard solutions of
EM-248 stored at -20°C.
Concentration of Volume of solution Volume of plasma
inhibitor 3a,-HSD
Std 50 ng/mL 90 ~1 of 1 ug/mL 1.71 mL
Std 20 ng/mL 0.8 mL of 50 ng/mL 1.2 mL
Std 10 ng/ 0.9 mL of 20 ng/ mL 0.9 mL
mL
Std 5 ng/ mL 0.8 mL of 10 ng/ mL 0.8 mL
Std 2 ng/ mL 0.6 mL of 5 ng/ mL 0.9 mL
Std 1 ng/mL 0.5 mL of 2 ng/mL 0.5 mL
Std 0 N/A 0.5 mL
Blank N/ A 0.5 mL
Extraction Procedure for Type 3 Inhibitors From Rat Plasma
Aliquots of rat plasma (0.250 mL) were transferred to 13 x 100
mm borosilicate tubes. Water ('1.0 mL) and internal standard
solution (0.1 mL) were added to each sample and vortexed for
2 min. A mixture of N-butyl chloride and acetone (v:v, 7:3) (3
mL) was added to each sample and vortexed for 2 min. This
step was repeated and the combined organic phases were
evaporated to dryness under nitrogen in a Turbo Vap
evaporator at 35° C. The residue was reconstituted with 1 mL
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-59
of methanol and evaporated in a Turbo Vap evaporator at
35°C. The final extract was reconstituted into 0.1 mL of
methanol/water (v:v, 75:25) and then transferred into a conical
vial for injection into the mass spectrometer.
g) Assay
The assay :procedure was performed by analyzing, in
duplicate, rat plasma samples spiked at six different Type 5
inhibitor concentrations (1, 2, 5, 10, 20 and 50 ng/mL). The
lower limit of quantitation (LOQ) was established at 1.0
ng/ mL. Values lower than 1.0 ng/ mL were expressed as
below limit of quantification (BLQ).
h) Linearity
The assay procedures for EM-1118 were found to be linear
over the 1.0 to 50 ng/mL range. Weighted (1/X) linear
regression analysis gave a correlation (r2) of 0.991.
i) Calculation of ALIC Values
For all compounds studied, the area under the plasma
concentration versus time curve (AUC~ from time 0 to 7 hours
post-dosing was determined. AUC~~ values were calculated
by the linear trapezoidal method (model-independent) for
each rat and data were expressed as mean AUC0.TtSEM (n=3).
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-60-
II. Enzymatic assay far types 1, 2, 3 and 517(3-HSD and types 1 and 3
3a-HSD
Enzyme sources. 293 cells transiently transfected with expression vectors
encoding types 1, 2 and 3 17~i-HSD (Luu-The et al., J. Steroid Biochem.
Molec. Biol., 55: 58I-5~87, 1995) type 5 17~i-HSD (described in WO
97/11162), and types 1 and 3 3a-HSD (Dufort et al. Biochem. Biophys.
Res. Commun. 228: 474-479, 1996), using the calcium phosphate
procedure (Kingston et al., In: Current Protocols in Molecular Biology.
Edited by E.M. AusbE~l, R. Brent, R.E. Kingston, D.D. Moore, J.G.
Seidman, J.A. Smith, K. Struhl. John Wiley & Sons, New York, pp. 9.1.1-
~15 9.1.9, 1991; Luu-The et al., J. Invest. Dermatol., 102: 221-226, 1994).
For
assays using cell subfractions, cells were sonicated in 50 mM sodium
phosphate buffer (pH 7.4), containing 20°/ glycerol and 1 mM EDTA and
centrifuged at 10 000 xg for 30 min before centrifugation for 100 000 x g
for 1 h to separate the mitochondrial and microsomal fractions,
:?0 respectively. The cytosoa fractions (100 000 x g supernatant) was used to
determine type 1 activit~r while the microsomal fraction (pellet at 100 000
x g) was used for measurement of types 2 and 317(3-HSD activities.
Incubation. The enzymatic reaction was carried out at 37°C in 1 ml
of 50
:~5 rnM sodium phosphate buffer, pH 7.4, containing 20°6 glycerol, 1 mM
EDTA, and 2 mM cofactors (NADPH or NAD+) for 1 h in the presence of
0.1 ~M 14C-labeled substrate: estrone for types 1 17~i-HSD, DHEA and 4-
androstene-3,17-dione (L14), for type 3 and 5 17(i-HSD, testosterone for
type 2 17~-HSD as well as,androstanedione and DHT for types 1 and 3
:.0 3a-HSD activities, in ab:>ence or presence of increasing concentration of
preferred inhibitor of t:he invention, was added to freshly changed
culture medium in a 6-well culture plate.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-61-
After incubation for 1 h, the steroids were extracted twice with 2 ml of
ether. The organic phase were pooled and evaporated to dryness. The
steroids were solubilized in 50 ~l of dichloromethane, applied to Silica
gel 60 thin layer chromatography (TLC) plate (Merck, Darmstad,
'l0 Germany) then separated by migration in the toluene-acetone (4:1)
solvent system. Substrates and metabolites were identified by
comparison with reference steroids and revealed by autoradiography
and quantitated using the Phosphoimager System (Molecular Dynamics,
Sunnyval, CA). Transfection could be also performed with HeLa, SW-13,
293, CUS-1 cells, the preferred cell line is 293 cells.
III- Shionogi Activity
Androgenic/antiandrogenic activity of some preferred compounds has
been measured using the Shionogi mouse mammary carcinoma cells.
,>
..0
Materials. Minimal essential culture medium (MEM), non-essential
amino acids, and fetal calf serum were purchased from Flow
Laboratories. In order to remove endogenous steroids, serum was
incubated overnight at 4 °C with 1 °~ activated charcoal (Norit
A, Fisher)
~!5 and 0.1 °~ Dextran T-70 (Pharmacia). A 2-h supplementary adsorption
was performed at 25°C, in order to further remove protein-bound
steroids. Serum was also inactivated by a 20-min incubation at 56°C.
5a-dihydrotestosterone (DHT) was obtained from Steraloids. The
antiandrogen hydroxyflutamide (OH-FLU) was kindly supplied by Drs.
a,0 T.L. Nagabuschan and R. Neri (Schering Corporation, Kenilworth;
U.S.A.).
CA 02339368 2001-02-02
- WO 00/0?576 PCT/CA99/00724
-62-
Cell dispersion, culture and cloning. Shionogi male mice bearing
androgen-sensitive mammary tumors were obtained from Drs. Keishi
Matsumoto, Osaka, Japan, and Yvonrte Lefebvre, Ottawa, Canada. For
primary culture, tumors were excised and washed in ice-cold sterile 25
mM Hepes buffer (137 m.M NaCI; 5 mM KCI; 0.7 mM Na2HP04; 10 mM
glucose, pH 7.2). After mincing with scissors, the tumor minces were
digested for 2 h at 37°C in Hepes buffer containing 3.8 mg/ml
collagenase (Clostridium, Boehringer), 1.5 mg/ml hyaluronidase II
(Sigma), and 3% bovine serum albumin fraction V (Schwartz-Mann).
Dispersed cells were collected by centrifugation (500 x g for 10 min),
7.5 washed twice by suspension in minimal essential medium (MEM)
containing 5 ~6 dextran-coated charcoal-treated fetal calf serum
(DCC-FCS),1°/ non-essential amino acids,10 IU/ml penicillin, 50 ~.g/ml
streptomycin, and 100 nM dihydrotestosterone (DHT) (Steraloids).
2:0 Cells were plated in the same medium at a density of 75 000 cells/ml in
75 cm2 flasks under an atmosphere of 5 ~ carbon dioxide in air at 37°C.
The medium was changed weekly. The steroids and antisteroids were
dissolved in ethanol and kept in stock solutions chosen to yield final
ethanol concentrations less than 0.01 °~ in the culture medium. Such a
25 concentration of ethanol does not affect cell growth.
Cells were subcultured at near-confidence by gentle digestion in a
solution of 0.1 % pancreatin (Flow Laboratories) in Hepes buffer
containing 3 mM ethylenediaminetetraacetic acid (EDTA) (pH 7.2). Cells
30 were pelleted by centrifugation, resuspended in culture medium,
counted in a Coulter counter, and replated as described above. Soft agar
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-63-
cloning was performed as described (Stanley et al., Cell 10: 35-44,1977) in
the presence of I00 nM DHT.
Measurement of Cell Growth and Sensitivity to Steroids and
Antisteroids. Cells were plated in 24-well plates at a density of 20 000
'10 cells/well. The indicated increasing concentrations of agents were added
to triplicate dishes, and cells were grown for 10-12 days with changes of
medium every 3-4 days. Cell number was measured by direct counting
in a Coulter counter.
:!5 Calculations and Statistical Analysis. EDSO values of action of DI-iT and
glucocorticoids were calculated according to a least-square regression as
described (Rodbard, Endocrinology 94: 1427-1431, 1974). Statistical
significance was calculated according to a multiple-range test (Kramer,
Biometrics 12: 307-310,1956).
~!0
IV- Estrogeruc/Antiestrogenic Activity
Estrogenic/antiestrogenic activity of some preferred compounds has
been measured using the ZR-?1-1 human breast cancer cell Iine as
described in more detail below.
t:5
Maintenance of Stock Cultures. ZR-75-1 cells (83rd passage) were
obtained from the American Type Culture Collection (Rockville, MD)
and routinely cultured in phenol red free RPMI 1640 supplemented with
1 nM estradiol (EZ), 2 mM L glutamine, 1 mM sodium pyruvate, 15 mM
30 N-2-hydroxyethyl piperazine-N'-2-ethanesulfonic acid, 100 IU
penicillin/ml, 100 ~tg streptomycin/ml, and 10°~ (v/v) fetal bovine
serum (Hyclone, Logan, UT) under a humidified atmosphere of 95% air,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
5% CC~, at 37 °C. All media and medium supplments were purchased
from Sigma. Cells were subcultured weekly by treatment with a
pancreatic solution containing EDTA (0.2 g/L). The cell cultures used for
the experiments herein describied were between passages 89 and 94.
Measurements of Cell :Proliferation. Cells in their logarithmic growth
phase were harvested, briefly centrifuged, and resuspended in RPMI
1640. Cells were then plated in triplicate in LIMBRO 24-well plastic
culture plates (2 cm2/well). Since plating density influences the effect of
hormones on ZR-75-1 cell growth, cells were plated at a density of 1 x 104
cells/ well. After 72 h, medium was replaced with fresh medium
containing the inhibitor at the concentration of 3.10- and 10-~ M in
absence or presence of 0.1 M estradiol (E2). Control cultures received the
ethanol vehicle only. Cells were then allowed to grow at 37 °C for 10
days with medium changes (of identical composition) every 2 days. In
absence of inhibitors, in 0.1M estradiol (E2)-containing medium, ZR-75-1
cells have doubling time of about 48 h.
After E2 and/or antiestrogen treatment, cells were harvested by addition
of 0.5 ml of a pancreatin solution (Sigma) for 5-10 min at 37 °C before
addition of 0.5 ml of RPMI 1640 containing 5 % dextran coated
charcoal-free bovine serum in order to block enzymatic action. Cell
number (0.10 ml aliquot) was determined by measurement of DNA
content as previously described (Simard et al., Endocrinology 126:
3223-3231,1990).
;30
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-65-
V- Androgen Receptor (AR) Assays
Tissue Preparation. Male Sprague-Dawley rats (Crl: CD(SD)Br) weighing
200-300g were obtained from Charles-River Inc. (St-Constant, Quebec,
Canada). The rats were gonadectomized under general anesthesia
(Isoflurane) and killed by cervical dislocation 24 hours later. The ventral
7.0 prostates were rapidly removed, dissected free from adhering tissue and
frozen on dry-ice. Prostates were kept at -80~C until assay.
All subsequent steps were performed at 0-4°C. Prostates were
homogenized in 5 vol (wt/vol) of buffer A (25 mM Tris-HCI, 1.5 mM
EDTA disodium salt, 10 mM a-monothioglycerol, 10°~ glycerol, and
10
mM sodium molybdate, pH 7.4), using a Polytron PT-10 homogenizer
(Brir~lanan Instruments, Canada) at a setting of 5 for three periods of 10
sec, with intervals of 10 sec for cooling. The homogenate was then
centrifuged at 105,000 x g for 60 min in a Beckman L5-65 ultracentrifuge
~ (Fullerton, CA). The protein concentration of the cytosol fraction was
measured according to the method of Bradford (Anal. Biochem. 72:
248-254,1976), using bovine serum albumin as standard.
Androgen Receptor Assay. Androgen binding was measured using the
hydroxylapatite assay (HAP). In brief, the radioactive steroid [3HJR1881
solubilized in ethanol was diluted into buffer A. Aliquots of prostate
cytosol preparation (0.1 ml) were then incubated with 8 nM [3H]R1881
(0.1 ml, 200,000 cpm) in the presence or absence of the indicated
concentrations of unlabeled compounds (0.1 ml, prepared in buffer A
' 30 containing 10% ethanol) for 16-18 h at 0-4°C. Triamcinolone
acetorude
(150 nM) was added in order to mask progesterone receptors. Unbound
steroids were separated by incubation for 40 min at 0-4°C with 0.3 ml
CA 02339368 2001-02-02
- WO 00/07576
PCT/CA99/00724
-66-
HAP prepared in buffer P (50 mM, Tris-HCI, 10 mM ICH2P04, pH 7.4) as
follows: 10 g HAP were washed with buffer P until the supernatant
reached a pH of 7.4 and then following centrifugation and decantatian of
the supernatant, 37.5 mI of buffer P were added. After incubation with
HAP and 10 minutes of centrifugation at 1,000 x g, the pellet was washed
3 times with 1 ml buffer P. Thereafter, the radioactivity was extracted
from the pellet by incubation at room temperature for 60 minutes with 1
ml EtOH. After centrifugation, the supernatant was decanted into a
scintillation vial and the pellet was extracted again with ethanol.
Thereafter, 10 ml Formula-989 scintillation liquid was added to pooled
supernatant and the radioactivity was measured in a Beckman counter.
Calculations. The results were reported as the percentage of inhibition of
the binding of [3H]R1881 at the concentrations of 10-~ and 10-~ M of the
inhibitor.
:?0
VI- Progesterone Receptor Assay
Chemicals. [17a-methyl-3H]-promegestone (R5020) (84 Ci/mmol} and
the corresponding unlabeled compound were purchased from New
England Nuclear (Lachine, Quebec, Canada) . All other chemicals were of
:?5 analytical grade.
Stock solutions of the unlabeled steroids were kept at 4°C in
ethanol. The
desired steroid solutions were then prepared by appropriate dilution in
buffer B (10 mM Tris-HCI, 1.5 mM EDTA, 10 mM a-monothioglycerol,
~~0 pH 7.4) containing 30% ethanol.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-67-
Tissue preparation. Female Sprague-Dawley rats weighing 200-300g
were obtained from Charles-River Inc. (St-Constant, Quebec, Canada).
The rats were gonadectomized under general anesthesia (Isoflurane) and
killed by cervical dislocation 24 hours later. The uteri are rapidly
removed, dissected free from adhering tissue and frozen on dry-ice.
Tissues were kept at -80°C until use.
Cytosol preparation. All procedures were performed at 4°C. Tissues
were pulverized frozen in dry ice with a Thermovac pulverizes. 'The
samples were then homogenized in 10 vol (w/v) of buffer A (25 mM
:l5 Tris-HCI, 1.5 mM EDTA, 10 mM a-monothioglycerol, 10°~ glycerol, 10
mM sodium molybdate, pH 7.4) using a Polytron PT-10 homogenizes
(Brinkmann Instruments, Canada) at a setting of 5 for two periods of 10
sec, with intervals of 10 sec for cooling. The homogenate was then
centrifuged at 105,000 x g for 90 min. The supernatant was used
~!0 immediately for assay.
Bindin Assays. Progesterone binding was measured using the
dextran-coated charcoal adsorption technique. Incubations were
performed at 0-4°C. for 16-18 h using 100 ~1 of cytosol, 100 ~.1 of
~'.5 [3H]-85020 (5 nM final, which contained 1,000 nM of dexamethasone in
order to mask the glucocorticoid receptors) and 100 ul of unlabeled
compounds at the indicated concentrations. Each concentration was done
in triplicate. Assay was ended with 300 ~.1 of DCC (1 % Norit A and 0.1
Dextran T-70 in Buffer B). After 10 min of incubation, tubes were
' 30 centrifuged at 2,000 x g for 10 min. and decanted in vials with 6 ml of
BCS liquid scintillation (New England Nuclear, Dupont). The
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-68-
radioactivity was measured in a Beckman counter at a counting efficiency
of 35~.
Calculations. The results were reported as the percentage of the
inhibition of the binding of [3H]R5020 at the concentrations of 10-~ and
10-~M of the inhibitors.
VII- Glucocorticoid Receptor Assay
Chemicals. [6,7-3H(N)]-Dexamethasone (39 Ci/mmol) was purchased
from New England Nuclear (Lachine, Quebec, Canada) while unlabeled
dexametasone was obtained from Steraloids (Wilton, NH). All other
chemicals were of analytical grade.
Stock solutions of the unlabeled steroids were kept at 4°C in
ethanol. The
desired steroid solutions were then prepared by appropriate dilution in
:ZO buffer B (10 mM Tris-HCl, 1.5 mM EDTA, 10 mM a-monothioglycerol,
pH 7.4) containing 30% ethanol.
Tissue ~~aration. Male Sprague-Dawley rats weighing 200-3008 were
obtained from Charles-River Inc. (St-Constant, Quebec, Canada). The rats
were killed by cervical dislocation and the liver were rapidly removed,
dissected free from adhering tissue and frozen on dry-ice. Tissues were
kept at -80°C until use.
C t~l .preparation. All procedures were performed at 4°C. Tissues
~~0 were eminced and homogenized in 10 vol (w/v) of buffer A (25 mM
Tris-HCI, 1.5 mM EDTA, 10 mM a-monothioglycerol, 10% glycerol, 10
mM sodium molybdate, pH 7.4) using a Polytron PT-10 homogenizer
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-69-
(Brinkmann Instruments, Canada) at a setting of 5 for two periods of 10
sec, with intervals of 10 sec for cooling. The homogenate was then
centrifuged at 105,000 x g for 90 min. 'The supernatant was used
immediately for assay.
CA 02339368 2001-02-02
- WO 00/07576
PCT/CA99/00724
-70-
Binding Assays. Glucocorticoid binding was measured using the
dextran-coated charcoal adsorption technique. Incubations were
performed at 0-4°C. for 16-18 h using 100 ~1 of cytosol, 100 ul of
[3HJ-Dexamethasone (5 nM final) and 100 ~1 of unlabeled compounds at
the indicated concentrations. Each concentration was done in triplicate.
'10 Assay was ended with 300 ~1 of DCC (2.5°~ Norit A and 0.25% Dextran
T-70 in Buffer B). After 10 min of incubation, tubes were centrifuged at
2,000 x g for 10 min. and decanted in vials with 6 ml of BCS liquid
scintillation (New England Nuclear, Dupont). The radioactivity was
measured in a Beckman counter at a counting efficiency of 35%.
'15
Calculations. The results were reported as the percentage of the
inhibition of the binding of [3H]-dexamethasone at the concentrations of
10$ and 10-~M of the inhibitor.
:?0 VIII-Estrogen Receptor (ER Assay_
Tissue Preparation. Female Sprague-Dawley rats (CrI: CD(SD)Br)
weighing 200-300g were obtained from Charles-River Inc. (St-Constant,
Quebec, Canada). The rats were gonadectomized under general
anesthesia (Isoflurane) and killed by cervical dislocation 24 hours later.
The uteri were rapidly removed, dissected free from adhering tissue and
frozen on dry-ice. Uteri were kept at -80~C until assay.
All subsequent steps were performed at 0-4°C. Uteri were
homogenized
in 10 vol (wt/vol) of buffer A (25 mM Tris-HCI,1.5 mM EDTA disodium
a0 salt, 10 mM a-monothioglycerol, 10% glycerol, and 10 mM sodium
molybdate, pH 7.4), using a Polytron PT-10 homogenizer (Brinkman
Instruments, Canada) at a setting of 5 for three periods of 10 sec, with
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-71-
intervals of 10 sec for cooling. The homogenate was then centrifuged at
105,000 x g for 60 min in a Beckman L5-65 ultracentrifuge (Fullerton, CA).
The protein concentration of the cytosol fraction was measured according
to the method of Bradford (Anal. Biochem. 72: 248-254, 1976), using
bovine serum albumin as standard.
:l0
Estrogen binding was measured using the dextran-coated charcoal
adsorption technique as described previously (Asselin et al.,
Endocrinology, 101: 666-671, 1977; Asselin and Labrie, J. Steroid
Biochem., 9: 1079-1082, 1978). Briefly, [3HJE2 solubilized in ethanol were
9l5 diluted into buffer A. Aliquots of uterine cytosol preparation (0.1 ml)
were incubated with 5 nM [3H]E2 {200,000 cpm, 0.1 ml) in the presence
or absence of the indicated concentrations of unlabeled compounds
(0.1 ml, prepared in buffer A containing 10% ethanol) for 3 h at room
temperature. Unbound steroids were then separated by incubation for 15
:!0 min at 0-4°C with 0.3 ml 0.5~ Norit-A and 0.05°6 Dextran T-
70 in buffer B
(1.5 mM EDTA disodium salt, 10 mM monothioglycerol, and 10 mM
Tris-HCI, pH 7.4) and centrifuged at 3,000 x g for 15 min. Aliquots of the
supernatant (0.3 ml) were removed for radioactivity measurement. After
the addition of 10 ml Formula-989 scintillation liquid (New England
~!5 Nuclear-DuPont), the radioactivity was measured in a Beckman counter
at a counting efficiency of 62%.
Calculations. The results were reported as the percentage of inhibition of
the binding of Ez at the concentrations of 10-~ and 10-~ M of the inhibitor.
' ~' 0
Primary criteria in selecfiing preferred inhibitors include bioavailability ,
desirable inhibition of type 3 3a.-hydroxysteroid dehydrogenase and type
CA 02339368 2001-02-02
WO 00/07576
PCT/CA99/00724
-72-
517(3-hydroxysteroid dehydrogenase, extent of undesirable inhibition on
type 2 17~i-hydroxysteroid dehydrogenase and androgenicity. It is
believed that the methyl groups in 5' position in EM 01645 and EM 01667
and analogous compounds promote selectivity of type 5 17j3-HSD
inhibition (versus undesirable type 2 inhibition). It is also believed that
free hydroxy group in 3-position has a benific effect as well as the
substitution in position 2.
Applicants have tested a wide range of compounds for effectiveness as
inhibitors of type 3 3a-HSD. It is believed, based on this laboratory
work, that certain characteristics of molecular structure discussed herein
provide favorable characteristics to the steroidal compounds of the
invention. For example, it is believed that an aromatic A-ring and a
moiety in position 3 that is either hydroxyl or a common pro-drug group
that is converted to hydroxyl in vivo are characteristics which favor good
2n inhibition of type 3 3a-HSD when combined with appropriate
substitution at position 2 or 4. Substituents at positions 2 and/or 4 are
preferably independently selected from the group consisting of hydrogen
cyano, fluoro, chloro, bromo, and vitro (provided that the substituents at
2 and 4 are not simultaneously hydrogen). The substituents that have
2;i worked well at 2 position have tended to work well at 4 position and vice
versa. Placing the same substitution at both 2 and 4 may be easier from
manufacturing standpoint, and compounds of that type that we have
tested have tended to be good inhibitors of type 3 3a-HSD.
3CI Applicants have also found that inhibitors of type 3 3a-HSD have better
selectivity when provided with D-ring subsHtuents such as those
described herein at the 16 or 17 position of a steroidal nucleus. By
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-73-
"selectivity", it is meant that these preferred D-ring substituents,
especially those set forth at 17 position, tend to suppress undesirable
interactions between the inhibitors of the invention and, for example,
enzymes whose inhibition is not desired or receptors whose activation is
not desired. Some of the parameters tested (both desirable and
undesirable) are set forth in tables herein a prime (') after their table
number. (See also the detailed explanations following the tables). As
may be seen from these tables, preferred compounds of the invention
effectively inhibit activity of type 3 3a-HSD while substantially avoiding
numerous undesirable activities for which Applicants tested the same
T.5 compounds. For example, appropriate D-ring substituents tend to
reduce undesirable androgenic or estrogenic activities. We have found
that 17-spiro-lactone and 17a-benzyl substituents give a good selectivity
for type 3 3a-HSD. Not all of the type 3 3a-I-ISD compounds discussed
herein are claimed because some of the compounds also have good
0 activity against type 5 17/3-HSD and are claimed in a separate patent
application by Applicants directed to this separate activity.
"In vitro" inhibition of the transformation of 4-androstenedione
(4-dione) to testosterone ('I~ by type 3 3a-HSD.
25 The inhibition of type 3 3a-HSD was preliminarily determined using the
inhibition of the transformation of DHT to androstane-3a,17~i-diol as
described above in "II Enzymatic assay for types 1,2,3 and 517(3-HSD and
types 1 and 3 3a-HSD" and reported in column 2 of Tables 1 and 2. To
complete this data, in Tables 3 and 4, the inhibition of the transformation
30 of 4-androstenedione (4-dione) to testosterone (T) by type 3. 3a-HSD. by
some preferred inhibitors is reported.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-74-
Enzymatic assay of the transformation of 4-androstenedione (4-dione)
to testosterone (T) by type 3 3a-HSD
Enzyme source:
Purified human type 3 3a-HSD expressed over in E. coli.
T'he coding region of human type 3 3a-HSD was amplified by PCR and
inserted in a pGEX-1~,T(Amersham Pharmacia Biotech, Inc., Quebec ,
Canada) vector in order to produce a fusion protein with glutathion~-
transferase. Expression over of the type 3 3a-HSD in E. coli, purification
of the protein on the glutathione-Sepharose 4B affinity column
(Amersham Pharmacia Biotech), and cleavage of the fusion protein by
thrombin were performed as described by the manufacturer.
Incubation:
The purified enzyme was incubated in a final volume of 2 ml of 50 mM
sodium phosphate buffer (pH 7.5), 20~ glycerol,1 mM EDTA and O.lpM
of [14C]-labeled steroid and 1 mM of NADPH. After 2 h incubation, the
steroids were extracted twice with 1 ml of ether. The organic phases were
pooled and evaporated to dryness. The steroids were solubilized in 50 ~1
of dichloromethane, applied to Silica gel 60 TLC plates (Merck,
2;5 Darmstad, Germany), before separation by migration in the toluene-
acetone (4:1) solvent system. Substrates and metabolites were identified
by comparison with reference steroids and revealed by autoradiography
and quantified using the Phosphoimager System (Molecular Dynamics,
Surulyval, CA).
31J
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-75-
Table 3
Laboratory Name %
Inhibition
of
the
transformation
of
4-
dione
to
T
by
type
3
3a-I-iSD
E-7
E-6
EM-1124 98 99
EM-1125 72 gg
EM-1126 82 95
EM-1131 80
EM-1667c 39 91
EM-1645 65 91
See the structures in table 1
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
- 6
Table 4
R17~ .
7a,
R16a
R2
16(3
R~'
vo
NAME RZ R4 R16' Ri~ Rl~ Rl~ ~ Inhibition of
the
transformation
of 4-
dione to T by
type 3
3a-HSD
Ea E-~
EM-1926NC- H CH3 CH3 H -OH 94 g7
EM-2060NC- H 13 H H -OC~s 88 99
EM-2200H -CN H H =O 80 97
EM-2132NC- H H H -C~-h -0H 92 99
EM-2318NC- H H H -Ph -0H 90 97
EM-2150NC- H -CF-IzPhH =O 83 99
EM-2330NC- H H H H -CONH2 90 98
EM-2359NC- H H H CHz~(p--OH 79 98
t-Bu)
CA 02339368 2001-02-02
WO 00/07576 PCTlCA99/00?24
_ ~7
Of the compounds in the foregoing tables, the most preferred and their
molecular structures are set forth below
O
C
EM-1125
EM-OI667-C
EM-01645
EM-1834
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
EM-2359
EM-1926
EM-2132
EM-2318
EM-2330
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-79-
EXAMPLES OF SYNTHESIS OF PREFERRED INHIBITORS
The IR spectra herein were taken on a Perkin-Elmer 1600 Series FT-IR
spectrophotometer. Proton NMR spectra were recorded on a Brucker
AC-F 300 instrument. The following abbreviations have been used: s,
:LO singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quadruplet;
and m,
multiplet. The chemical shifts (8) were referenced to chloroform (7.26
ppm for 1H and 77.00 ppm for 13C) and were expressed in ppm. Optical
rotations were measured at room temperature on a Jasco DIP 360
polarimeter. Mass spectra (MS) were obtained on a V.G. Micromass 16F
:l5 machine. Thin-layer chromatography (TLC) was performed on 0.25 mm
Kieselgel 60F254 plates (E. Merck, Darmstadt, FRG). For flash
chromatography, Merck-Kieselgel 60 (230-400 mesh A.S.T.M.) was used.
Unless otherwise noted,, starting material and reactant were obtained
commercially and were used as such or purified by standard means. All
:'0 solvents and reactants purified and dried were stored under argon.
Anhydrous reactions were performed under an inert atmosphere, the set-
up assembled and cooled under argon. Organic solutions were dried
over magnesium sulfate, evaporated on a rotatory evaporator and under
reduced pressure. Starting materials and reagents were available from
:'5 Aldrich Chemical Company, Inc. (Milwaukee, Wisconsin)
CA 02339368 2001-02-02
WO 00/07576
PCT/CA99/00724
LIST OF ABBREVIATIONS
DHP 3,4-dihydra-2H-pyran
EDTA Ethylenediaminetetraacetic acid
HPLC High pressure liquid chromatography
P'TSA p-toluenesulfonic acid
THF Tetrahydrofuran
THP Tetrahydropyranyl
TMS Tetramethylsilyl
CA 02339368 2001-02-02
- WO 00/0?576 PCT/CA99/00724
_81 _
Example 1
Synthesis of 2-nitro-1,3,5(10)-estratrien-
17-spiro-S-lactone derivatives
:l0 These syntheses are described in Scheme 1.
Scheme 1
O O THPO
W= OZN ~ '_~ HO.,v
HO I ~ 1 RO~ 02N
3
br- 2. R-H TBDMSO
THPO ~.. 26 R-78DMS HO
O
,,J Ho 0
,,J
f
02N \ -- O2N \ ~. _. _
TBDMSO I ~ ~ ~ 5 OZN
HO HO , EM-1121
O R
g ~,~ R2
_ __~ 02N ~
HO ~
EM -1121 R~ ~ Me, RZ - H
EM-1131 R~ - ~ Ry~ Ma (epima of EM-I 126)
g EM-1125 R~-R~-Me
~. NaNO~. HNO~, AcOH b. TBDMSCI, imidazole c HCC(CH~OT1~, Meh d. Hs, Pd/GCO~ a
5!4 HCJ, MeOH
f. )ones rageot g. LDA Mel
»5
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
82
Example 1A
3-hydroxy-2-vitro-1,3,5(10)-estratrien-17-one (2a). The titled compound
was prepared as described by Stubenrauch and ICnuppen The procedure
is described below.
7.0 Estrone (1,18.004 g, 66.6 mmol) was dissolved in boiling acetic acid (540
mL) and allowed to cool down to 50°C. The nitrating mixture was
prepared from 70°~6 nitric acid (4.5 mL, 70 mmol), water (10 mL) and a
few crystal of sodium nitrite, warmed up to 50 °C and added dropwise to
the solution of estrone with stirring. After stirnng overnight at room
7.5 temperature, the yellow precipitate was filtered by suction and
recrystallized from 92~ aqueous acetic acid. 4-vitro derivative (6.800 g,
32~) was thus obtained as a pale yellow solid. IR (v) 3227 (OH), 2931,
2864,1723 (C=0),1626,1584,1523,1458, 2404,1374,1295,1264,1245,
1211,1169,1085,1062,1027, 954, 930, 908, 881, 823, 796, 719, 654, 588, 556,
2.0 530, 494 cm-1; 1H NMR (Pyridine-ds) 8 0.85 (3H, s,18'-CH3), 2.85 (2H, d,
6'-CH2), 5.00 (1H, s, OH), 7.11 (1H, d, J=8.7 Hz, 2'-CH), 7.26 (1H, d, J=8.7
Hz,1'~; 13C NMR (Pyridine-ds) S 13.8 '(C-18), 21.6 (C-15), 24.4 (C-11),
25.7 (C-7), 26.2 (C-12), 32.0 (C-6), 35.9 (C-16), 37.7 (C-8), 44.0 (C-14),
47.9
(C-13), 50.1 (C-9),115.4 (C-2),128.4 (C-1 ),129.0 (C-10),131.8 (C-5),148.4
25 (C-3), 219.2 (C-17).
The reaction filtrate from above was evaporated under reduced pressure
and the residue was recrystallized from EtOH/ Hz0 8.5:1.5. A brown
solid (7.854 g) was obtained which was further purified by flash
30 chromatography on SiOz column (EtOAc/hexanes, gradient 8-20%) to
give pure compound 2a (6.284 g, 30%) as a yellow solid. IR (n): 3300
(OH), 2933, 2864,1737 (C =O),1630,1562,1522,1480,1431,1372,1311,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
1252,1216,1146,1084,10154,1035,1008, 905, 832, 762, 722, 662, 600, 520
cm-1.1H NMR (Pyridine-~ds) b 0.85 (3H, s,18'-CI-~), 2.76 (2H, d, 6'-CH2),
4.99 (1H, s, OH), 6.98 (11-I, s, 4'-CH), 7.96 (1H, s, l'-CH). 130 NMR
(Pyridine-ds) 813.8 (C-18), 21.7 (C-15), 25.8 (C-11), 26.1 (C-7), 29.6 (C-12),
31.9 (C-6), 35.9 (C-16), 37.8 (C-8), 43.5 (C-14), 47.9 (C-13), 50.3 (C-
9),119.8
DLO (C-4),122.2 (C-1),132.8 (C-IO),147.8 (C-2),152.6 (C-3), 219.1 (C-17).
Example 1B
3-(tert-butyldimethylsilyloxy)-2-vitro-1,3,5(10)-estratrien-17-one (2b). A
~! 5 solution of 2-vitro-estrone (2a,1.118g, 3.55 mmole), imidazole (0.6708,
9.84 mmole) and TBDMS~CI (0.7818, 5.18 mmole) in dry DMF (50 mL) was
stirred under Ar(g) ovenught. The mixture was then poured onto
ice/water (80) mL. The white precipitate was filtered, washed with
water and then dried in 'racuo to give (2b) as a yellowish powder (1.447
a!0 g, 95~). [a]~D+123.9° (c L03, CHC13); IR (NaCI) 2933, 2860,1736
(s,C=O),
1617,1561,1518,1492,1408,1351,1291,1256,1054, 909, 832, 790, 697 cm-1;
iH NMR S 0.24 (6H, s, Si(CH3)2), 0.92 (3H, s,18-CHs),1.01 (9H, s,
SiC(CHa)3),1.40-1.78 (6H,, m),1.90-2.35 (5H, m). 2.37-2.60 (2H, m), 2.90
(2H, m, 6-CHZ), 6.67 (1H, s, 4-CH), 7.76 (lH,s,1-CH);13C NMR 8 220.2,
~'.5 147.2,143.8,139.6,133.3,122.6,122.1, 50.3, 47.9, 43.6, 37.8, 35.8, 31.3,
29.4,
26.1, 25.7, 25.6, 21.5,18.2,13.8, -4.4.
Example 1C
3.0 3-(tent-butyldimethylsilyloxy)-17(3-hydroxy-2-rutro-17a-(4'-(2"-
tetrahydro-2"H pyranyloxy)-butynyl)-1,3,5(10)-estratriene (3). To a
stirred solution of tetrahydro-2-(butynyloxy)-2H-pyran (1.71 mL,10.91
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
_84
mmole) in dry THF (75 m~L) under Ar (g) at -35 °C was dropwise added
(MeLi 1.4M in ether 7.80 mL,10.92 mmole). The solution was stirred for
45 mdn. after which was added at -35 °C a solution of ketone 2b (1.294
g,
3.01 mmole) in dry THF (20 mL). After 75 min. ice (20 g) and saturated
aqueous NaHCOa (70 mL) were added to the reaction mixture and the
1~~ aqueous phase was extracaed with EtOAc. The combined organic layers
were washed with brine, dried with magnesium sulfate, filtered and then
concentrated in vacuo. T7he crude yellow oil was purified on SiOz (40 g,
2:8 EtOAc/hexanes) to give compound 3 as a yellow foam (1.617 g, 92%).
[a]~-57.5° (c 0.72, CHCI<<);1R (NaCI) 3423 (broad, OH), 2936, 2870,
2366,
1;5 1654,1630,1578,1560,1527,1481,1458,1438,1313,1268,1121,1080,1032,
899, 869, 761, 669 cm-I; ~H NMR 8 0.23 (6H, s, Si(CHs)2), 0.87 (3H, s,18-
Cl~),1.00 (9H, s, SiC(CH;s)3),1.20-2.35 (20H, m), 2.55 (2H, t, J=6.9Hz,
CCCHZ), 2.84 (2H, m, 6-~H2), 3.55 (2H, m, CH20 of chain), 3.85 (2H, m,
CH20 of THP), 4.65 (1H, m, CH of THP), 6.65 (1H, s, 4-Cl-n, 7.76 (1H, s,1-
2il CH); 13C NMR 8147.0,194.2,139.5,133.9,122.6,122.0, 98.8, 84.5, 83.4,
79.8, 65.8, 62.2, 49.4, 47.1, 43.2, 38.9, 32.6, 30.6, 29.6, 26.7, 26.2, 25.6,
25.4,
22.8, 20.4,19.4,18.2,12.7, ~-4.4.
2;i Example 1D
3-(tent-butyldimethylsilyloxy)-17~i-hydroxy-2-vitro-17a-(4'-(2"-
tetrahydro-2"H-pyranyloacy)-butyl)-1,3,5(10)-estratriene (4). A solution
of compound 3 (2.OOg, 3.42 mmol) and 5% Pd/CaCOa (400 mg) in dry
MeOH (400 mL) was stirred under H2(g) atmosphere (balloon) for 1 h.
30 The mixture was then filtered through celite and the filtrate rotary
evaporated. The residue ~Nas purified on silica gel (2:8 EtOAc/hexanes)
to give compound 4 as a ~rhite foamy solid (1.483 g, 74%). [a]D~ +31,3°
(c
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-85
0.90, CHC13); IR (NaCI) 3458 (broad, OH), 2935, 2860,1616,1563,1518
(NOz),1491,1408,1348 (rJ02),1291,1256,1119,1070,1023, 925, 893, 832,
784, 672 cm-1; ~H NMR 8 0.23 (6H, s, Si{CHa)2), 0.91 (3H, s,18-CI-0,1.00
(9H, s, SiC(CHa)s),1.20-2.38 (26H, m), (2.85 (2H, m, 6-CH2), 3.48 (2H, m,
CH20 of chain), 3.83 (2H, m, CH20 of THP), 4.59 (1H, m, CH of THP),
6.65 (1H, s, 4-CH), 7.75 (1:H, s,1-CH); 13C NMR 8146.9,144.1,139.4,
134.0,122.4,121.9, 98.9, 83.2, 67.6, 67.6, 62.4, 62.4, 49.3, 46.6, 36.3, 34.1,
3L3, 30.7, 30.3, 29.5, 26.9, 26.0, 25.5, 25.4, 23.3, 20.4,19.6,18.1,14.3, -
4.4.
Example 1E
17a-(4'-hydroxybutyl)-3;U7(3-dihydroxy-2-vitro-1,3,5(10)-estratriene (5).
A solution of compound 4 (300 mg, 0.510 mmol) in 5 % HCi in MeOH (10
mL) was stirred at room i:emperature and under argon atmosphere for
12 h. The reaction mixtw~e was then poured into NaHCOa/ice and
MeOH was evaporated under reduced pressure. The aqueous phase was
extracted with EtOAc and the combined organic layers were washed
with brine, dried over M~,.~SO~, filtered and evaporated to dryness. This
gave a crude yellow foams (198 mg,100 ~). Purification by flash
chromatography (coiumuk loaded with CHzCiz and then eluted with
EtOAc/ CHZC1Z 2:8, 4:6,1:1, 6:4, 9:1) gave compound 82 as a yellow solid
(127.0 mg, 64°~). Rf 0.21 {8:2 EtOAc/Hexanes); M.p.184-6 °C;
[a]~o
+58.8° (c.1.00, CHCi); IR (v) 3335, 2934,
2865,1735,1719,1654,1630,1576,
1522,1479,1434,1373,1305,1266,1169,1112,1067,1033,1000, 896, 874,
762, 659 cm-1;1H NMR 8 0.90 (3H, s), 3.69 (2H, d, J=5.7 Hz), 6.84 (1H, s),
7.98 (1H, s),10.42 (1H, s);1sC NMR 814.3,19.8, 23.3, 26.1, 26.8, 29.8, 31.2,
33.3, 34.3, 36.1, 39.0, 43.2, 46.5, 49.4, 61.7, 62.8, 83.4,118.8,121.4,131.6,
133.7,149.2,152.8.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-86-
.5 Example 1F
2-Nitro-1,3,5(10)-estraetriene-3-ol-17(R)-spiro-2'-(6'-oxo)tetrahydropyran
(EM-1124). To a stirred solution of compound 5 (128 mg, 0.33mmole) in
dry acetone (25 mL) at U °C was slowly added a first 1.1 equivalent of
Jones' reagent {1.25 M, 0.29 mL, 0.80 mmol). The orange solution was
11) then stirred for 0.5 h then a second equivalent was added. The dark
solution was stirred for a further 0.5 h then quenched with isopropanol
(green precipitate formed). the mixture was stirred for 10 min. then
filtered through celite and. the filtrate rotary evaporated. The residue
was taken in EtOAc then ~Nashed with aq. sat. NaHCOs, HzO, brine,
1;i dried (MgS04), filtered, rotovaped. The crude solid was purified by flash
chromatography on Si02 {3:7 EtOAc/Hexanes) to give EM-1124 (108 mg,
85%) as a yellow solid. M.p. 213 °C; [aJ~p +90,0' {c 0.70, CHC13); IR
{NaC1) 3198, 2934, 2876, 2245,1720 (s,C=O, Iactone),1630,1577,1522,
1480,1434,1378,1314,126'7,1234,1199,1169,1151,1120,1070,1036,1024,
2(I 992, 914, 851, 759, 732, 662,, 585 cm-1; 1H NMR 81.02 (3H, s,18-CH3),1.20-
2.23 (16H, m), 2.25-265 {3H, m), x.90 (2H, m, 6-CHZ), 6.85 (1H, s, 4-CH),
7.97 (1H, s,1-Cl~,10.41 (1:H, s, OH phenol); I3C NMR S 171.9,152.8,
148.9,133.3,131.7,121.5,1:L8.9, 93.0, 48.8, 47.1, 43.1, 38.4, 33.9, 31.6,
29.7,
29.4, 27.9, 26.8, 25.8, 23.4,15.8,14.2.
Example 1G
2-Nitro-1,3,5(10)-estratrien-3-ol-17(R)-spiro-2'-(5'-methyl-6'-
oxo)tetrahydropyran (EM-1126, EM-1131). LDA was prepared as
follows: To a stirred solution of diisopropylamine (92 ~L, 71 mg, 0.70
30 mmol) in dry THF {5 mL) at -78 °C under Ar(g) was added n-BuLi (1.2
M/Hexane, 580 ~L, 0.68 mmol) and the solution was then stirred at 0
°C
for 25 min. then cooled down to -78 °C. A solution of EM-1124 (66 mg,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-87
0.17 mmol) in dry THF (5 mL) was added and the resulting dark orange
solution was then stirred for 30 min. Dry HMPA (2 mL) was added and
after 15 min., MeI (107 ~I,, 243 mg,1.71 mmol). The solution was then
stirred for a further 4 h. rfhe reaction was quenched with aqueous
saturated NH4Cl and ext~~acted with EtOAc. The organic phase was
washed with 1 M aqueous CuS04 (4X), H20, aqueous 1M Na2SOa, brine,
dried (MgS04), filtered then rotary evaporated to give a crude solid (103
mg). Purification by flash chromatography on Si02 (1:9 -> 2:8
EtOAc/Hexanes) providE~d first EM-1126 (11 mg,16~) closely followed
by EM-1131 {34 mg, 34%) both as yellow solids. EM-1126: M.p. 204-6 °C;
[a]~D +73.4 ° (c 1.67, CDC:I~); IR v 3422 (br, OH), 2937, 2874,1725
(vs, CO),
1630,1577,1525,1479,14;i8,1432,1378,1311,1269,1249,1205,1188,1150,
1118,1088,1071,1007, 990, 934, 896, 760, 731, 668, 585, 495 cm-1; 1H NMR
81.03 (3H, s),1.30 (3H, d, J=7.1 Hz),1.31-1.77 (10H, m),1.89-2.03 (5H, m),
2.15 (1H, td, J=7.1 Hz, ]'=;i.0 Hz), 2.30-2.50 (2H, m), 2.90 (2H, dd, J=8.3
Hz,
J'=4.9 Hz), 6.85, (1H, s), 7.'98 (1H, s),10.43 (1H, s, OH);13C NMR 8174.8,
152.9,149.0,133.4,131.7,121.5,118.9, 93.4, 48.7, 47.1, 43.I, 38.5, 36.2, 34.6,
31.6, 29.7, 28.6, 26.9, 25.9, :25.2, 23.4,17.4,14.4.
EM-1131 (5'-epimer of EM-1126, real configuration not determined):
M.p. 206-8 °C; (a~~n +62.6 ° (c 0.68, CDC13); IR v 3422
(br, OH), 3192,
2934, 2876, 2858, 2824,1721 (vs, CO),1631,1578,1522,1482,1458,1436,
1377,1314,1271,1237,12CI4,1173,1120,1103,1082,1051,1019,1002, 933,
901, 877, 860, 759, 663, 63E~, 600, 495 cm-~; 1H NMR 81.01 (3H, s),1.24 (3H,
d, J=7.0 Hz),1.31-1.80 (101, m),1.89-2.20 (6H, m), 2.35 (1H, br s), 2.55
(1H, sextuplet, J=7.5 Hz), 2.89 (2H, t, J=5.2 Hz), 6.84 (1H, s), 7.96 (1H, s),
10.41 (H, s, OH) ;13C NM1C~ ~ 175.8,152.8,148.9,133.3,131.6,121.4,118.8,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
92.5, 77.4, 77.0, 76.6, 48.5, 47.0, 43.0, 38.4, 33.8, 33.4, 31.6, 29.7, 27.16,
26.7,
25.8, 24.3, 23.6,17.2,14.3.
Example 1H
2-Nitro-1,3,5(10)-estratrien-3-ol-17(R)-spiro-2'-(5',5'-dimethyI-6'-
UO oxo)tetrahydropyran (El'vi-1125). LDA was prepared as follows: To a
stirred solution of diisopropylamine (206 ~L,159 mg,1.57 mmol) in dry
THF (12 mL) at -78 °C under Ar(g) was added n-BuLi (1.2 M/Hexane,
1,28 mL,1.53 mrnol) and the solution was then stirred at 0 °C for 20
min.
then cooled down to -78 '°C. A solution of a mixture of EM-1126 and EM-
7.5 1131 (153 mg, 0.38 mmol',I in dry THF {10 mL) was added and the
resulting dark orange solution was then stirred for 20 min. Dry HMPA
(4.7 mL) was added and after 15 min., MeI (238 ~,L, 544 mg, 3.83 mmoI).
The solution was then stirred for 5 min. then was warmed up to -30
°C
and stirred for a further 1 h. The reaction was quenched with aqueous
2.0 saturated NH4Cl and exh~acted with EtOAc. The organic phase was
washed with brine {6X), aqueous 1M Na2SOs, brine, dried (MgS04),
filtered then rotary evaporated to give a crude liquid. Purification by
flash chromatography on Si02 (1:9 -> 2:8 EtOAc/Hexanes) provided
EM-1125 (82 mg, 52°~) as a yellow solid. M.p.195-7 °C;
[«]gin +72.8 ° (c
25 1.61, CDCIs); IR v 3421 (br, OH), 3194, 2954, 2927, 2873,1718 (vs, CO),
1631,1578,1523,1476,1458,1438,1386,1312,1298,1271,1204,1151,1118,
2059,1032,1016, 931, 898, 872, 855, 758, 663, 595 cm-1; 1H NMR b 1.02
(3H, s),1.28 (6H, s),1.32-1.77 (10H, m),1.85-2.15 (6H, m), 2.36 (1H, br s),
2.89 (2H, dd, J=8.2 Hz, J'=4.9 Hz), 6.85 (1H, s), 7.97 (1H, s),10.42 (H, s,
31) OH);13C NMR b 177.7,152.8,149.0,133.3,131.7,121.5,118.9, 93.4, 48.6,
47.1, 43.1, 38.5, 37.8, 34.7, 31.6, 31.5, 29.7, 27.7(4), 27.6(8), 26.7, 25.9,
25.5,
23.3,14.4.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
Example 2
Synthesis of 2-cy~ano-1,3,5(10)-estratrien-30' -17(R)-spiro-2'
(5',5'-dim~ethyl-6'-oxo) tetrahydropyran (13)
'LO Scheme 2
0 0
/ " TBDMSCI / LiC:C(CH~OTHP
H \ ~ Imidazole/DMF~ TBDMSO \ I THE
6
OH
--C:C~CH~OTHP
--(CH~~OTHP
v Hz, 5!iG P~ / TsOH, MeOH
TBDMSO \ EtOAc
TBDMSO
9
OH
--(CIiZ)~OH
Jones (1.5 eq) iHMDS, Mel
TBDMS \ Acetone .~F -'''
11
IfBAF, THE
12
13
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-90
3-t-butyldimethylsilyloxy-1,3,5(10)-Estratrien-17-one (7). The ether was
prepared horn estrone (6) following the method described Pelletier et al.
(Steroids 59: 536-547,1994).
3-t-butyldimethylsilyloxy-17}i-hydroxy-27a-{4'-(2"-tetrahydro-2"H-
pyranyl)butyn-1'-yl}-1,3,5(10)-estratriene (8). To a solution of
HC=-C(CH2)20THP (18.3 mL,117 mmol) in dry THF (600 mL) at 0 °C,
was
added dropwise n-butyllithium (43.7 mL,109 mmol) and the mixture was
stirred for 90 min. The rnixture was cooled to -78 °C and a solution of
TBDMS-estrone 7 (15 g, 39 mmol) in THF (500 mL) was added dropwise.
Then, the reaction mixhire was allowed to come to room ternperature and
left stirring for a period of 15 h. Solvents were evaporated to the half
volume and 200 mL of water was added. The mixture was extracted with
EtOAc (3 x 200 mL), the organic layer was washed with brine, dried
(MgS04~ and evaporated to dryness. The residue was purified over silica
gel column chromatography with hexanes/EtOAc (9/1) as an eluent to
furnish 15.1 g (72~) of the product; IR (NaCI cm-1) 3432, 2934, 2858,1607,
1495,1287,1256,1033, 9:i8, 839;1H NMR (300 MHz, CDCl3) 8 7.12 (d,1H,
=8.4 Hz), 6.62 (dd,1H, J=2.4, 8.4 Hz), 6.54 (d,1H, J=2.2 Hz), 4.66 (br.s.,
1H), 3.89-3.79 (m, 2H), 3.56-3.50 (m, 2H), 2.79 (br.s., 2H), 2.56 (t, 2H,
J=7.0
:25 Hz), 2.35-2.17 (m, 3H), 2.07-1.23 (m,17H), 0.98 (s, 9H), 0.87 (s, 3H,18-
Me),
0.19 (s, 6H);13C NMR (75 MHz, CDCl3) b 153.3,137.8,133.0,126.1,119.9,
117.1, 98.7, 84.7, 83.2, 80.0, 65.8, 62.1, 49.5, 47.2, 43.7, 39.4, 39.0, 32.9,
30.6,
29.7, 27.3, 26.4, 25.7, 25.4, 22.8, 20.3,19.3,18.1,12.8, -4.4.
3-t-butyldimethylsilylo~y 17(3-Hydroxy-17a-(4'-(2"-tetrahydro-2"H-
pyranyl)butan-1'-yl}-1,3,.5(10)-estratriene (9). 5% Palladium on activated
carbon (1.5 g,10°~ wt) was added to a solutian of the alkyne 8 (15.1 g,
28
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-91
mmol) in EtOAc (500 mL) at room temperature. The flask was purged with
Hi three times (vacuum followed by H2) and left stirring under 1 atm
pressure of H2. The reaction was followed by TLC. After a period of 3 h,
the mixture was filtered over a plug of celite and the solvent was removed
under reduced pressure. The crude product was used in the next step
IO without further purification; IR (NaCI, cm-1) 3474, 2935, 2858, 2607, x570,
1496,1471,1286,12.57,1156,1137,1119,1033, 954, 839, 780;1H NMR (300
MHz, CDC13) 8 7.12 (d, l.H, J=8.4 Hz), 6.62 (dd,1H, J=2.1, 8.4 Hz), 6.55 (s,
1H), 4.59 {br.s.,1H), 3.92:-3.73 (m, 2H), 3.55-3.38 (m, 2H), 2.82-2.77 (m,
2H),
2.30-1.33 (m, 26H), 0.97 {s, 9H), 0.90 {s, 3H,18-Me), 0.18 (s, 6H);13C NMR
(75 MHz, CDC13) 8153.27,137.81,133.08,126.02,119.87,117.06, (98.90,
98.84), 83.38, 67.61, 62.33, 49.50, 46.67, 43.81, 39.58, 36.35, 34.28, 31.60,
30.75,
30.36, 29.62, 27.51, 26.26, 25.67, 25.47, 23.37, 20.45,19.66,18.12,14.35, -
4.43.
3-t-butyIdimethylsilylo,~cy 17(3-hydroxy-17a-(4'-hydroxybutan-1'-ylj-
1,3,5(10)-estratriene (10),. To a solution of the THP ether 9 (15.1 g, 2:8
mmol)
in MeOH (400 mL), was added p-toluenesulfonic acid monohydrate (150
mg, 0.8 mmol) and the reaction was stirred over a period of 5 h. A
saturated solution of NaHC03 (100 mL) was added and volume of solvent
was reduced to half on a rotary evaporator. The mixture was extracted
:25 with CH2Ch, the organic phase was washed with brine, dried (MgS04~ and
evaporated to dryness. The crude product was used in the next step
without pufification; IR (NaCI, cm-1) 3356, 2931, 2858,1608,1496,1471,
1286,1256, 954, 839, 780;1H NMR (300 MHz, CDCl3) 8 7.12 (d,1H, j=$.5
Hz), 6.61 {dd,1H, J=2.5, 8.5 Hz), 6.55 (s,1H), 3.69 {br.d, 2H, J=5.2 Hz), 2.82-
2.78 (m, 2H), 2.35-2.26 (m~,1H), 2.20-1.94 (m, 2H),1.90-1.81 (m,1H),1.62-
1.22 (m,17H), 0.98 (s, 9H), 0.90 (s, 3H,18-Me), 0.19 (s, 6H);13C NMR (75
MHz, CDCIs) b 153.27, 137.81,133.05,126.02,119.89,117.09, 83.59, 62.56,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-92
49.50, 46.69, 48.81, 39.58, 35.98, 34.32, 33.20, 31.61, 29.62, 27.51, 26.26,
25.68,
23.37,19.74,18.14,14.36, -4.40.
3-t-butyIdimethylsilyloxy-1,3,5(10)-estratrien-17(R)-spiro-2'-(6'-oxo)
tetrahydropyran (11). To a solution of the diol 10 (12.5 g, 27 mmol) in
acetone (500 mL) at 0°C, was added dropwise of a 2.7 M solution of
Jones
reagent (15.1 mL, 41 mrnol). The reaction was stirred for 30 min. 2-
Propanol (100 mL) was added, followed by of a saturated solution of
NaHC03 (200 mL). The volume of solvents was reduced to half by
evaporation and the mixaure was extracted with EtOAc. The organic phase
was washed with brine, dried (MgS04~ and concentrated under reduced
pressure. The residue was purified a silica gel column chromatography
with hexanes/acetone {6~/1) to afford 8.6 g of the Iactone. (68% yield for 3
steps); IR {NaCI, cm-1): 2!x60, 2930, 2857,1732,1607,1496,1284,1264,1244,
1037, 958, $40;1H NMR (;300 MHz, CDC13) 8 7.11 (d,1H, J=8.4 Hz), 6.61
:?0 (dd,1H, J= 2.3, 8.4Hz), 6.;56 (s,1H), 2.85-2.79 (m, 2H), 2.58-2.39 (m, 2I-
~,
2.38-2.25 {m,1H), 2.21-2.'.LO (m,1H), 2.03-1.27 (m,15H),1.02 (s, 3H,18-Me),
0.97 (s, 9H), 0.18 (s, 6H); »3C NMR (75 MHz, CDCl3) 8172.00,153.36,137.63,
132.62,126.02,119.92,117.19, 93.25, 48.88, 47.26, 43.68, 39.05, 33.98, 31.96,
29.50, 29.48, 27.94, 27.46, 25.98, 25.67, 23.48,18.12,15.87,14.30, -4.43.
~!5
3-t-butyldimethylsilyloxy-1,3,5(10)-estratrien-17(R)-spiro-2'-(5'-5'-
dimethyl-6'-oxo) tetrahydropyran(12). In a dry 1L flask under argon, was
dissolved the lactone 11 (8.6 g,19 mmol) in dry THF (300 mL), and cooled
to 0°C. A 1M solution of L iHMDS (47.3 mL, 47.3 mmol) was added
30 dropwise. The mixture was stirred 15 min at 0°C and cooled to -
78°C and
then methyl iodide (5.9 rr~L, 79 mmol) was added. The reaction was stirred
1 h at this temperature and then allowed to warm to room temperature
CA 02339368 2001-02-02
WO 00/0?576 PCT/CA99/00724
-93-
over a period of 2 h. A saturated solution of NH4Cl (200 mL) was added
and the mixture was extracted with EtOAc. The organic layer was washed
with a saturated solution of Na2S20s, brine, dried (MgS04> and
concentrated under reduced pressure. The residue was purified by column
chromatography with hexanes/acetone (5/1) as an eluent to afford 7.4 g
(81 %) of the dimethyl compound; IR (NaCI, cm-1) 2954, 2930, 2858,1725,
1496,1287,1258,1150, 7.137, 956, 840; ~H NMR (300 MHz, CDCI3) 8 7.11 (d,
1H, J=8.5Hz), 6.62 (dd, '1H, J=2.4, 8.5Hz), 6.55 (d,1H, J=2.IHz), 2.81-2.78
(m, 2H), 2.36-2.28 (m,1lH), 2.20-1.38 (m,16H),1.28 {s, 3H),1.27 (s, 3H),1.02
(s, 3H,18-Me}, 0.97 (s, 9H), 0.18 (s, 6H);'3C NMR (75 MHz, CDC13) 8
177.79,153.33,137.62,132.62,125.99,119.90,117.14, 93.66, 48.67, 47.24,
43.65, 39.06, 37.74, 34.79, 31.96, 31.56, 29.50, 27.73, 27.61, 27.42, 26.01,
25.65,
25.55, 23.26,18.11,14.4:?, -4.43.
1,3,5 (10)-estratrien-3-ol-17(R)-spiro-2'-(5',5'-dimethyl-6'-oxo)
tetrahydropyran (13): 7'o a solution of the silyl ether 12 (7.1 g,14.7 mmol)
in THF (300 mL) at 0°C, was added dropwise a 1M solution of TBAF (17.6
mL,17.6 mmol) and the reaction was stirred for 15 min. Ice water (200 mL)
was added to precipitate the compound. The flask was placed on a rotary
evaporator to reduce the volume of THF, and then placed on an ice bath.
The precipitate was collected by filtration, washed with cold water and
dried in an oven (30°C) over a period of 24 h to furnish 5.4 g
(100°~) of the
3-OH compound; IR (NaCI, cm-1): 3357, 2932, 2871,1695,1287,1158;1H
NMR (300 MHz, CDCIa) 8 7.14 (d,1H, J=8.4Hz), 6.63 (dd,1H, J=2.6, 8.4
Hz), 6.55 (d,1H, J=2.6Hz), 4.62 (br.s,1H, OH), 2.81-2.79 (m, 2H), 2.38-2.29
(m,1H), 2.20-1.81 {m, 5H),1.76-1.31 (m,11H),1.29 {s, 3H),1.28 (s, 3H),1.01
(s, 3H,18-Me};13C NM:R (75 MHz, CDCIs) 8178.06,153.52,138.08,132.19,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-94
126.42,115.26,112.74, 93.80, 48.69, 47.29, 43.65, 39.14, 37.81, 34.84, 31.98,
31.61, 29.53, 27.76, 27.64, 27.39, 26.12, 25.59, 23.29,14.43.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-95-
Example 3
Synthesis of EM-01667
Scheme 3
° ° o
o J ' o'~s
a ~ ~ ~ se ~ b ~ ci
I I i 1a I w
HO ~ H HO
13 + 4-isomer
EM-01667
a. PhSeCI, CHG3 b. NCS, CHCI3
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-96-
3-Hydroxy-2-phenylselenenyI-estra-1,3,5(10)-triene-17(R)-spiro-2'-(5',5'-
dimethyl-6'-oxo) tetrahydropyran (14). A solution of 3-hydroxy-estra-
1,3,5(10)-triene-17(R)-spiro-2'-(5',5'-dimethyl-6'-oxo)tetrahydropyran (406
mg,1.10 mmol) (13) and phenylselenenyl chloride (253 mg,1.32 mmol) in
dry CHC13 {24 mL) under Ar (g) was stirred at 0 °C for one hour then at
r.t. overnight. The resulting yellow solution was poured onto ice/H20
then extracted with CHzCl2 (3x). The combined organic phase was dried
(cotton plug) then rotary evaporated to give a crude foamy solid.
Purification by flash chromatography (Si02) using 1:9 EtOAc/Hexane as
eluent gave 7 (353 mg, 61 %) with the 4-isomer (86 mg,15%). Compound
7: [a]~p +77.7 ° (c 1.14, CHC13); IR v 3366, 3050, 2965, 2928,
2869,1709,
1603,1576,1550,1458,1438,1384,1349,1310,1294,1262,1202,1157,1141,
1114,1065,1017, 984, 89:2, 845, 736, 689, 665, 593, 555, 498, 460 cm-1; 1H
NMR(CDCl3) (8) 1.02 (3H, s),1.27(9) (s, 3H),1.28(4) (s, 3H),1.27-1.80
(11H, m),1.88-2.28 (6H, m), 2.87 (2H, t, J= 4.8 Hz), 6.24 (1H, s, OH), 6.80
(1H, s), 7.21 (5H, br s), 7.52 (1H, s) ppm; 13C NMR(CDCl3) (b) 14.4, 23.3,
25.5, 26.1, 27.2, 27.6, 27.7, 29.5, 31.5, 31.8, 34.7, 37.7, 38.9, 43.4, 47.2,
48.6,
93.6,111.6,114.7,126.5, 129.2(6),129.3(4),131.2,133.3,134.7,141.4,154.4,
177.8 ppm.
2-Chloro-3-hydroxy-estra-1,3,5(10)-triene-17(R)-spiro-2'-(5',5'-dimethyl-
6'-oxo) tetrahydropyran (EM-01667). A solution of 14 (116 mg, 0.22
mmol) and N-chlorosuccinimide (44 mg, 0.33 mmol) in dry CHCl3 (15
mL) under Ar(g) at 0 °C were stirred for 30 min. The solution was
poured onto ice/ H20 then was extracted with CH2Cl2 (3x). The
combined organic phase was dried (cotton plug) then rotary evaporated
to give a crude solid. Purification by flash chromatography (Si02) using
1:29->1:19 EtOAc/Toluene as eluent gave EM-01667 (51 mg, 57%) as a
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-97-
white solid. Rf 0.28 (3:T EtOAc/Hexane); m.p. 241 °C; [a]~D +62.b
° (c
1.09, CDCl3); IR (v) 3253 (br, OH), 2936, 2873,1685 (CO),1608,1501,1458,
1418,1388,1314,1300,1.263,1223,1160,1016, 987, 884, 844, 737, 673, 598
crn-'; 1H NMR (CDCI3) (8) T.Ol (3H, s),1.28 (6H, s),1.25-1.75 (11H, m),
1.85-2.28 (6H, m), 2.80 (2H, dd, J'=8.7 Hz, J"=3.9 Hz), 5.33 (1H, br s, OH),
6.73 (1H, s), 7.19 (1H, s) ppm; 13C NMR(CDC13) (8) 14.4, 23.3, 25.6, 26.1,
27.2, 27.7, 27.8, 29.0, 31.6, 31.8, 34.8, 37.8, 38.8, 43.4, 47.2, 48.6,
93.6,116.0,
117.1,125.6,133.5,137.1, 249.0,177.8 ppm.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-98-
Example 4
Synthesis of EM-01728
Scheme 4
O OH
a ~ N, I ~ b / \
Se
HO I ~ HO~ ~ HO I
16
OH
a CI
~b
Ho ' -
EM-01T28
a. PhCHyMgCI, THF b. PhSeCI t. NCS
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
_9~
17-benzyl-1, 3, 5, (10)estratriene-3,17(3-diol (15)
To a solution of estrone (5.0 g,18.5 mrnol) in dry THF (200 mL) at
0°C:
under Ar(g) was added benzyI magnesium chloride (2M in THF, 65 mL)
and the solution was stirred overnight. Aqueous saturated NHaCI
solution was added at 0°C and the solution was extracted with CH2Cl2 (3
times), washed with brine, dried with MgS04, filtered and then
evaporated. The product was purified by flash chromatography (RP-
C18, 30:30:40 -10:30:60, Hz0/ MeOH/ CH3CN) to give 1 as a white solid
(4.0 g, 60~) and estrone: (1.5g, 30% recovery). Compound 1: Rf: 0.25
(2:98, MeOH/ CH2C12); IR (v) 3284, 2926, 2856,1725,1686,1655,1606,
1561,1499,1439,1378,1343,1321,1286,1252,1221,1156,1082,1029,1016,
930, 915, 888, 872, 820, 786, 732, 700, 646, 586 cm-1;1H NMR (CDaOD) (b)
0.94 (3H, s, H18),1.32-2.35 (13H, m), 2.67 (1H, d, J=13.5 Hz, CH-Ph), 2.78
(2H, m, H6), 2.87 (1H, d, J=13.5 Hz, CH-Ph), 6.49 (1H, d, J=2.4 Hz, H4),
6.56 (1H, dd, J=8.3 Hz, J''=2.5 Hz, H2), 7.10 (1H, d, J=8.3 Hz, H1), 7.18-7.30
(5H, rn, Ph) ppm ;13C NMR {CDaOD) (b) 15.3, 24.1, 27.7, 28.8, 30.8, 32.4,
32.8, 41.4, 43.6, 45.2, 50.9, 84.5,113.7,116.1,126.9,127.2,128.7,132.3,136.6,
138.8,140.3,155.9 ppm.
17-benzyl-2-Phenylselenyl-1, 3, 5, (10)estratriene-3,17(i-diol (16)
To a stirred solution of 1 (508 mg,1.40 mmol) in MeOH/CHC13 (1:10, 33
mL) at 0°C was added PhSeC1 (322 mg,1.68 mmol). After two hours, the
orange solution had turned yellow and was poured onto ice/H20,
extracted with CH2CI2, dried (MgS04), filtered and then evaporated. The
crude solid was purified by flash chromatography (Si02,1:29
EtOAc/Toluene) to give 2 as a beige solid (456 mg, 63%) and the 4-
phenylselenyl isomer (71 mg,10%). Compound 2: 1H NMR (8) 1.00 (3H,
s, H18),1.30-2.38 (14H, m), 2.82 (2H, dd, J=78.1 Hz, J'=13.2 Hz, CH2-Ph),
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
- -100-
2.90-2.93 (2H, m. H6), 6.26 {1H, br s, OH), 6.85 {1H, s, Hl), 7.2I-7.38 (10H,
m), 7.59 (1H, s, H4),13C :f~IMR (b) 14.5, 23.4, 26.4, 27.4, 29.7, 31.3, 33.7,
39.4,
42.4, 43.7, 46.7, 49.5, 82.9, 111.5,114.7,126.3,126.5,128.1,129.2,129.3,
131.0,131.3,133.7,134.8,138.2,141.6,154.4 ppm.
17-Benzyl-2-chloro-1, 3, 5, (10)estratriene-3,17[i-diol (EM-01728)
To a stirred solution of 2 (155 mg, 0.30 mmol) in CHC13 (25 mL) at
0°C
under Ar (g) was added N-chlorosuccinimide (48 mg, 0.36 mmol). After
one hour, the reaction was worked up as in 2 and purification by flash
chromatography (SiOz,1:29 -> 1:19 EtOAc/Toluene) gave EM-01728 as a
white solid (74 mg, 62%). Rf 0.18 (1:19 EtOAc/Toluene); M.p. 220°C;
[a]~D +74.8° (c 0.96, acetone-d~; IR (v) 3544 {OH), 3284 (br, OH),
3023,
2931, 2849,1601,1497,1454,1338,1285,1257,1201,1084,1015, 979, 918,
885, 796, 755, 702, 675, 642, 560, 532, 504 cm-1; 1H NMR (CDaOD) (8) 0.95
(3H, s, H18),1.33-1.75 (15H, m), 2.68 (1H, d, J=15.5 Hz, CH-Ph), 2.76 (1H,
:?0 dd, J=8.3 Hz, J'=3.6 Hz, H6), 2.88 (1H, d, J=15.5 Hz, CH-Ph), 6.60 (1H, s,
H4), 7.16 (IH, s, Hl), 7.I7-7.31 (5H, m, Ph) ppm; 13C NMR (acetone-d6)
{8) 15.2, 24.0, 27.3, 28.3, 29.9, 32.1, 33.5, 40.7, 43.5, 44.6, 48.0, 50.3,
83.6,
117.5,118.3,126.6,127.5,128.5,132.2,134.2,137.8,140.6,151.3 ppm.
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-101 -
Example 5
Synthesis of EM-01831 and EM-01832
Scheme 5
° ° o
~ w ~ ~ ~ ~ G
~o ~ Ho
18
° O OH
Se ~ d CI ~ a CI
" .
i
HO 1 g HO HO
EM-01831 EM-01832
a. NaH, Mel b. TMSI c. PhSeCI d. NCS e. LiAIH,,
CA 02339368 2001-02-02
WO 00/07576 PCTICAy9/00724
-102
16,16-Dimethyl-3- methoxy-1,3,5(10)-estratrien-17-one (17J
To a solution of 3-methoxy-estra-1,3,5{10)triene-17-one (10.00 g, 35 mmol)
in anhydrous THF (500 mL) under Ar(g) at room temperature was added
NaH (60% in oil, 2.55 g, '105 mmol) and iodomethane ( 22 mL, 350 mmol).
The solution was refluxe~d overnight. More NaH (2.55 8,105 mmol) and
1.0 iodomethane (22 mL, 35!) mol) was then added and the solution was
refluxed for another 24 hours. The resulting solution was quenched with
ethanol (100 mL) and then by water onto ice (300 mL). This solution was
extracted using ethyl acetate (2 x 300 mL), washed with brine (2 x 300
mL), dried with MgS04 and evaporated under reduce pressure to give a
yellow solid. Purification by flash chromatography on silica gel using
ethyl acetate/hexane (1:19) as eluent gave 3 (10.19 g, 93%) as a white
solid. IR {v) 2933, 2869,1731 (CO),1609,1502,1467,1382,1315,1258,
1240,1153,1037,1020, 90:2, 850, 816, 781 cm-1; 1H NMR (8) 0.94 {3H, s,
H18),1.09 (3H, s, I6-Me),1.22 (3H, s,16-Me),1.40-2.42 (11H, m), 2.90 (2H,
dd, J=8.0 Hz, J'=3.3 Hz, H6), 3.79 (3H, s, OMe), 6.65 (1H, d, J=2.7 Hz, H4),
6.73 (1H, dd, j=8.4 Hz, j'=-2.7 Hz, H2), 7.21 (1H, d, j=8.5 Hz, H1) ppm; 13C
NMR (8) 14.4, 25.8, 26.0, 26.7, 27.3, 29.7, 32.3, 37.6, 37.9, 44.2, 45.3,
47.2,
49.0, 55.2,111.5,113.9,126.3,132.2,137.7,157.6, 225.1 ppm.
2~i 16,16-Dimethyl-1,3,5(10)-estratrien-3-ol-17-one (18)
A solution of 3-methoxy-estra-1,3,5(10)triene-17-one-16-dimethyl,17 (6.00
g,19 mmol) and iodotrimethylsilane (27 mL,190 mmol) in anhydrous
CHZCl2 (1000 mL) under Ar(g) was refluxed overnight. The resulting
solution was poured onto ice/ H20 (600 mL), extracted with CH2Cl2 (3 x
600 mL), dried with MgSO4, filtered and then evaporated under reduce
pressure. The crude brown solid was purified by flash chromatography
on silica gel using ethyl acetate/toluene (1:9) as eluent to give 4 as a
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-103
white solid (3.50 g, 62°~). IR (v) 3361 (br,s, OH), 3027, 2923,
2876,1860
(w),1717 (CO),1620,1584,1499,1460,1355,1286,1248,1152,1099,102,.'x,
909, 875, 816, 787, 735, 647, 571, 516 cm-~; 1H NMR (8) 0.91 (3H, s, H18),,
1.06 (3H, s,16-Me),1.18 (3H, s,16-Me),1.23-2.35 (11H, m), 2.81 (2H, t,
J=4.5 Hz, H6), 6.56 (1H, d, J=2.3 Hz, H4), 6.62 (1H, dd, J=8.5 Hz, J'=2.6 Hz,
H2), 7.09 (1H, d, J=8.4 Hz;, Hl) ppm;13C NMR (8) 14.4, 25.7, 25.8, 26.6,
27.1, 29.4, 32.2, 37.5, 37.8, 44.0, 45.3, 47.1, 49.1,112.7,115.2,126.2,131.3,
137.7,154.2, 226.3 ppm.
16,16-Dimethyl-2-phenylselenyl-1,3,5(10)-estratrien-3-ol-17-one (19)
To a stirred solution of 18 (2.00 g, 6.70 mmol) in MeOH/CHCI~ (1:10, 550
mL) at 0°C was added PhSeCI (1.56 g, 8.15 mmol). The reaction mixture
was stirred overnight at room temperature. The resulting yellow
solution was then poured onto ice/H20 (500 mL), extracted with CHzC;l2
(2 x 500 mL), dried with MgS04, filtered and then evaporated under
reduced pressure. The product was purified by flash chromatography on
silica gel using ethyl acetate/toluene (0.3: 9.7) to give 5 as a white foam
(2.44 g, 80°~6) and the 2-ch:loro isomer (100 mg, 5%). Compound 5: 1H
NMR (8) 0.94 (3H, s, H18),1.08 (3H, s,16-Me),1.21 (3H, s,16-Me),1.24-
2.36 (11H, m), 2.92 (2H, dd, J=8.4 Hz, J'=3.6 Hz, H6), 6.21 (1H, s, OH), 6.82
2~i (1H, s, H4), 7.17-7.23 (5H, m, Ph), 7.54 (1H, s, H1) ppm.
2-Chloro-16,16-dimethyl-1,3,5(10)-estratrien-3-ol-17-one (EM-01831)
To a stirred solution of 5 (680 mg,1.50 mmol) in anhydrous CHC13 (200
mL) at room temperature and under Ar(g) was added N-
Chlorosuccinimide (246 rr~g,1.84 mmol). The mixture was then stirred at
-30°C for one hour. The reaction was quenched with aqueous saturated
NH4Cl (300 mL), extracted with CH2Cl2 (2 x 300 mL), dried with Na2SOn
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-104
filtered and then evaporated under reduced pressure. The crude product
was purified by flash chromatography using methanol/ethyl
acetate/hexane (0.5:1: 9) as the eluent to give EM-01831 as a yellow solid
(178 mg, 33%). IR (v) 3321 (br, s, OH), 2922, 2853,1724 (CO),1606,1502,
1468,1414,1380,1340,1260,1215,1019, 885, 830, 738, 673, 626 cm-~; 1H
NMR (8) 0.93 (3H, s, H18),1.08 (3H, s,16-Me),1.21 (3H, s,16-Me),1.26-
1.35 (11H, m), 2.84 (2H, dd, J=9.0, j'=4.3 Hz, H6), 5.33 (IH, s, OH), 6.75
(1H, s, H4), 7.20 (1H, s, Hl) ppm; 13C NMR (8) 14.4, 25.7, 26.0, 26.5, 27.3,
29.0, 32.2, 37.5, 37.6, 43.9, 45.3, 47.1, 49.0,116.0,117.2,125.7,133.4,137.0,
149.1, 225.0 ppm
2-Chloro-16,16-dimethyl-1,3,5(10)-estratrien-3,1713-diol (EM-01832)
To a stirred solution of EM-01831 (200 mg, 0.60 mmol) in anhydrous THF
(20 mL) at -78°C under Ar (g) was added LiAlH4 (33 mg, 0.86 mmol). The
temperature of the reaction was allowed to slowly return to rt over 24
hours. The reaction was cooled down to 0 °C, more LiAlH4 (23 rng, 0.60
mmol) was added and the mixture was stirred for another 2 hours. The
reaction mixture was quenched with 1M aqueous Rochelle salt (50 mL)
then extracted with ethyl acetate (3 x 50 mL). The organic layer was
washed with brine (50 mL), dried with MgS04, filtered and then
2.5 evaporated under reduced pressure. The product was purified by flash
chromatography on silica gel using ethyl acetate/ toluene (1:19) as eluent
to give EM-01832 as whitE~ solid (145 mg, 72%). IR (v) 3557 (s, OH), 3388
(s, OH), 3186 (br, s, OH), ~>_921, 2861,1602,1576,1486,1454,1430,1409,
1382,1344,1256,1207,1128,1069,1029, 981, 880, 798, 733, 677, 584, 534
cm-1; 1H NMR (8) 0.81 (31-3, s, H18),1.01 (3H, s,16-Me),1.06 (3H, s, I6-
Me),1.24-2.20 (11H, m), 2.74 (2H, dd, J=8.4 Hz, J'=3.7 Hz), 3.23 (1H, s,
H17), 6.59 (1H, s, H4), 7.13 (1H, s, Hl) ppm; 1'3C NMR (b) 11.5, 25.3, 26.2,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-105
27.2, 29.1, 32.3, 37.7, 37.9,, 39.0, 41.2, 43.8, 45.4, 46.8, 89.8,115.9,117.0,
125.7,134.0,137.3,148.9 ppm.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-106
Example 5
3-hydroxy derivatives of 2-cyano-1,3,5(10)-estratrien
17-spiro-(dimethyl-S-lactone)
Scheme 5
o" ,
o~a
0
is <cx,°~,s~.,c~~H a
HO
O_
O~J~ ~~1J1~
NHiOH HQ
~x° N
Pytidiue. ethanol
RO
Z1a R-Ac ~ K:CO~
21
2I6 R-H ~W Hi0
22e R-a~y~ ac
1;7
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-107-
Example 5A
2-formyl-1,3,5(10)-estratrien-3-ol-17(R)-spiro-2'-(5',5'-dimethyl-6'-
oxo)tetrahydropyran (20). Lactone 13 (1.0 g, 2.72 mmol) was dissolved in
dry 1,2-dichloroethane (9 mL) under argon atmosphere. SnCl4 (0.16 mL,
1.37 mmol) and Bush (0.52 mL, 2.18 mmol) were added successively. The
mixture was stirred at room temperature for 20 min. Formaldehyde (0.23
g, 7.84 mmol) was added and the mixture was stirred at reflux for 6h. The
reaction mixture was poured into aq acid (pH=2) and, was extracted with
CHZCl2. The organic layers were washed with brine solution, dried
(Na2S04) filtered and concentrated in vacuo. The crude product was
7.5 purified by flash chromatography on silica gel, eluting with (95:5 to
80:20)
hexanes-acetone to yield 0.74 g (69 %) of the product; IR (NaCI cm-1): 3164,
2937, 2872,1716,1652, 1571,1487, 1466,1386, 1298,1152, 1017, 914, 731;1H
NMR (CDC>3) 1.00 (s, 3H),1.26 (s, 6H) 1.23-2.40 (m,17H), 2.80-2.90 (m, 2H),
6.66 (s,1H), 7.39 (s,1H}, 9.79 (s,1H),10.77 (s,1H);13C NMR (CDC>3) 814.3,
2.0 23.2, 25.4, 25.9, 26.8, 27.E~, 27.7, 30.0, 31.4, 31.6, 34.6, 37.7, 38.6,
42.9, 47.0,
48.5, 93.4,116.9,118.9,130.3,132.2,147.8,159.2,177.7,196Ø
Example 5B
2-oximino-1,3,5(10)-estratrien-3-ol-17(R)-spiro-2'-(5',5'-dimethyl-6'-
25 oxo)tetrahydropyran (21.). Under argon atmosphere, a solution of
compound 20 (215 mg, 0.54 mmol) in anhydrous ethanol-pyridine 1-1 (4
mL) was treated with hydroxylamine hydrochloride (56.6 mg, 0.814
mmol) and stirred at roorn temperature for 25 min. The reaction mixture
was evaporated, diluted with water, and extracted 3 times with
30 dichloromethane. The combined organic phase was washed with brine,
dried over sodium sulfate, filtered, and evaporated to provide the oxime
113 (217 mg, 98%): ~H NMR (300 MHz, CDCIs) s 1.02 (s, 3H),1.29 (s, 6H),
CA 02339368 2001-02-02
- WO 00/07576 PCT/CAy9/00724
-108-
1.43-1.70 (m,10H), 1.89 :?.12 (m, 6H), 2.22-2.37 (m, 1H), 2.80-2.87 (m, 2H),
6.69 (s,1H), 7.05 (s,1H), ;8.15 (broad s,1H), 8.20 (s,1H), 9.61 (s,1H).
Example 5C
3-acetoxy-2-cyano-1,3,5(10)-estratrien-17(R)-spiro-2'-(5',5'-dimethyI-6'-
1.0 oxo)tetrahydropyran (22a). A solution of compound 21 (180 mg, 0.44
mmol) and acetic anhydride {125 ~L,1.32 mmol) in pyridine (3.5 mL) was
refluxed for 1 h. The reaction mixture was evaporated, diluted with
dichloromethane, and washed 3 times with water, 1 time with saturated
sodium bicarbonate and I time with brine. The organic phase was dried
over magnesium sulfate,, filtered, and evaporated. The crude mixture
was purified by fl;~sh chromatography (dichloromethane to
dichloromethane-ethyl acetate I9-1) to provide acetate 22a (145 mg,
76°~):
IR {CHCls) 2933, 2872, 2229, 1773, 1718, 1613, 1494, 1183 cm-1; iH NMR
(300 MHz, CDCl3) b 1.02 (s, 3H),1.28 (s, 6H),1.34-1.89 (m,11H),1.94-2.33
(m, 6H), 2.37 (s, 3H), 2.89-2.94 (m, 2H), 6.95 (s,1H), 7.55 (s,1H).
Example 5D
2-cyano-1,3,5(10)-estratrien-3-ol-17(R)-spiro-2'-(5',5'-dimethyl-b'-
oxo)tetrahydropyran (22b). A solution of compound 22a (60 mg, 0.14
2;5 mmol) in methanol (5 mL) was treated with 10% potassium carbonate
(0.5 mL) and stirred 30 min. The reaction mixture was acidified to pH 2
with 1 N hydrochloric acid and extracted 3 times with dichloromethane.
The combined organic phase was washed with water, saturated sodium
bicarbonate, and brine, dried over magnesium sulfate, filtered, and
evaporated. The crude mixture was recrystallized in aqueous ethanol to
afford the phenol 22b (28. mg, 52%). IR (CDCIs) 3334, 2932, 2868, 1692,
1312, 1206, 1159 cm-1; II-~l NMR (300 MHz, CDCIs) 8 0.97 (s, 3H), 1.29 (s,
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00?24
-109-
6H), 1.26-2.14 (m, 16H), 2.21-2.28 (m, 1H), 2.82-2.86 (m, 2H), 6.69 (s, IH),
6.91 (s, 1H), 7.35 (s, 1H),; 13C NMR (75 MHz, CDCl3) 8 14.41, 23.30, 25.62,
26.92, 27.69, 27.78, 29.84, 31.62, 31.79, 34.82, 37.84, 38.70, 43.18, 47.23,
48.67, 93.52, 97.01,116.29,116.69,129.44,133.69,144.86,155.73,177.88.
:l0 Example 5E
3-aIkyloxy-2-ryano-1,3,5(10}-estratrien-17(R)-spiro-2'-(5',5'-dimethyl-6'-
oxo)tetrahydropyran (2'.?c). Under argon atmosphere, a suspension of
compound 22b, alkyl iodide (5 equiv) and cesium carbonate (1.5 equiv) in
anhydrous acetonitrile (1 % W/~ was stirred for 16 h with refluxing
condition if necessary. 'The reaction mixture was quenched with brine
and extracted 3 times with dichloromethane. The combined organic
phase was washed with brine, dried over magnesium sulfate, filtered,
and evaporated. The crude mixture was purified by flash
chromatography (dichloromethane to dichloromethane-ethyl acetate 10-
1) and recrystallizaHon (:methanol) to provide compound 22c (e.g., EM-
1396, R=(CH2)20CHs, 75°'6): IR (CHC13) 3013, 2941, 2881,
2229,1710,1610,
1500, 1304, 1136 cm-1; IH NMR (300 MHz, CDC13) 8 1.01 (s, 3H), 1.28 (s,
6H),1.20-1.75 (m,10H), 1.80-2.20 (m, 6H), 2.20-2.35 (m, 1H), 2.80-2.95 (m,
2H), 3.47 (s, 3H), 3.79 (t, J=4.8 Hz, 2H), 4.17 (t, J=4.8 Hz, ZH), 6.67 (s,
1H),
7.44 (s, 1H); 13C NMR (75 MHz, CDC13) 8 14.41, 23.25, 25.58, 25.90, 26.96,
27.66, 27.74, 30.24, 31.59,. 31.78, 34.79, 37.80, 38.68, 43.16, 47.20, 48.60,
59.55, 68.78, 70.71, 93.43, 99.55, 112.80, 116.98, 130.72, 133.31, 144.09,
158.29,177.70.
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-lI0
Example 6
0 0
MOMCI
(Eth~PrN
h. ns, Coy, cF,
a aopsn
ssX
HO M~ 90%
P~OAcy:, PPh,
Zn, KCN
I
CH,CN
D,Ah
64X
MOO MOMO
O
A LiHMDS, RI R R
THF N ~R SOX AcOH N
_7B'C~ 90'C, ANON R
R=CH,,26X
MOMO MOMO
O
O
(CHI" C
NaH. Br(CH=~,Br N
--~ ' .SOX AcOH ~'
THF
D. ON 90'C, ON
n=~.73X MOMO
HO
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-111 -
PHARMACEUTICAL COMPOSITION EXAMPLES
Set forth below, by way of example and not of limitation, are several
pharmaceutical compositions utilizing a preferred active compound
EM-2330 (an inhibitor of type 3 3a-HSD). Other compounds of the
invention or combination thereof may be used in place of (or in addition
to) EM-02318 and EM-02200. The concentration of active ingredient may
be varied over a wide range compatible with the preferred dosages
discussed herein, and depending on preferred frequency of
administration. The amounts and types of other ingredients that may be
7.5 included are well known in the art.
EXAMPLE A
Composition suitable for injection
Ingredient Weight
(by weight of total composition)
EM-2330 0.4
Ethanol 6.4
NaCI 0.8
Water 91.5
Benzyl alcohol 0.9
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-112 -
EXAMPLE B
Composition suitable for use as topical lotion
Ingredient Weight
(by weight of total composition)
EM-2330 1.0
Ethanol 70.0
Propylene glycol 29.0
EXAMPLE C
1.0 Composition suitable for use as topical gel
Ingredient Weight
(by weight of total composition)
EM-2330 1.0
Hydroxypropylcellulose 1.5
Ethanol 70.0
Propylene glycol 27.5
EXAMPLE D
Tablet
1 ~i
Ingredient Weight
(be weight of total composition)
EM-2330 1.0
Gelatin 5.0
Lactose 67.5
Starch 26.5
CA 02339368 2001-02-02
WO 00/07576 PCT/CA99/00724
-I13-
EXAMPLE E
Gelatin capsule
Ingredient Weight
(be weight of total composition
EM-2330 2.0
Lactose hydrous 80.0
Starch 4.8
Cellulose microcrystalline 12.8
Magnesium atearate 0.4
EXAMPLE F
Composition suitable for use as topical gel
Ingredient Weight
(be weight of total composition)
EM-2330 2.0
Ethanol 4.0
Polyethylene glycol 4.0
Gelatin 1.0
NaCI 0.9
Benzyi alcohol 1.0
Water USP gg.l
Other inhibitors of type ;3 3a-hydroxysteroid dehydrogenase may be
7! 5 substituted for EM-2330 in the above formulations. In some
embodiments, two or more type 3 3a-hydroxysteroid inhibitors may be
CA 02339368 2001-02-02
- WO 00/07576 PCT/CA99/00724
-114-
included together, (or one inhibitor of type 3 3a-HSD plus on inhibitor of
type 5173-HSD) in which case the combined weight percent of the two is
preferably double what is shown in the above examples for EM-2330
alone, with a corresponding reduction in the weight percent of the most
prevalent excipient (e.g.,, water, lactose, ethanol or the like). Other active
ingredients of preferred combinations herein may be added in like
manner.
The invention has been described in terms of preferred embodiments and
examples, but is not limited thereby. Those of skill in the art will readily
recognize the broader applicability and scope of the invention which is
limited only by the patent claims herein.