Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02352451 2001-07-24
3040-CA
HIGH THROUGHPUT METHOD FOR DISCOVERY OF GENE CLUSTERS
FIELD OF INVENTION:
[0001 ] The invention relates to the fields of microbiology and genomics, and
more
particularly to a high-throughput method for discovery of gene clusters.
BACKGROUND:
[0002] The method of the present invention allows rapid discovery of gene
clusters
involved in metabolic pathways or other processes without having to sequence
the
entire genome.
[0003] Microbial genes whose products act in a coordinated fashion, for
example a
biosynthetic pathway, are often arranged in close physical proximity to one
another in
the organism's genome. Such genes are said to form a gene cluster. Gene
clusters
are associated with a variety of metabolic pathways, notably the biosynthesis
of
microbial natural products. Gene clusters may also provide resistance to
therapeutic
drugs. (See for example, Schouten et al., Molecular analysis of Tn1546-like
elements
in vancomycin-resistant enterococci isolated from patients in Europe shows
geographic
transposon type clustering, Antimicrob Agents Chemother, 45(3):986-9). Gene
clusters
are also associated with pathogenicity islands from various organisms. (See
for
example, Kuroda et al., Whole genome sequencing of methicillin-resistant
Staphylococcus aureus, Lancet, 357(9264):1225-40; Carniel E., The Yersinia
high-
pathogenicity island: an iron-uptake island, Microbes Infect. 3(7):561-9;
Nicholls etal.,
Identification of a novel genetic locus that is required for in vitro adhesion
of a clinical
isolate of enterohaemorrhagic Escherichia coli to epithelial cells, Mol.
Microbiol
35(2):275-88). Gene clusters are also responsible for catabolic pathways. (See
for
example, Velasco et al., Genetic and functional analysis of the styrene
catabolic cluster
of Pseudomonas sp. strain Y2, J. of Bacteriology, 180(5):1063-1071; Buchan et
al., Key
aromatic-ring-cleaving enzyme, protocatechuate 3,4-dioxygenase, in the
ecologically
important marine Roseobacter lineage, Appl. and Env. Microbiol., 66(11 ): 4662-
4672;
Masai et al., Genetic and biochemical characterization of a 2-Pyrone-4,6-
dicarboxylic
acid hydrolase involved in the protocatechuate 4,5-cleavage pathway of
Sphingomonas
paucimobilis SYK-6, J. of Bacteriology, 181 (1 ):55-62; Ferrandez et al.,
Catabolism of
phenylacetic acid in Escherichia coli, J. of Biological Chemistry, 273(40),
25974-25986).
[0004] Bioactive small molecules or natural products produced by microbial
secondary metabolism are a prime example of compounds produced by gene
clusters.
1
CA 02352451 2002-O1-17
3040-CA
The genes encoding natural product biosynthetic pathways in both prokaryotes
and
eukaryotes are clustered together in gene clusters. These gene clusters, which
typically range in size from 50 kilobase pairs (kbp) to 200 kbp, also usually
contain
resistance genes and pathway-specific regulatory genes. See, for example, Cole
S.T.
and Saint Girons I., Bacterial genomics, FEMS Microbial Rev., 14(2):139-60.
[0005] Gene clusters in general are of significant interest in various fields.
For
example, gene clusters such as the Tn1546-like elements that are responsible
for the
spread of vancomycin resistance in clinical isolates of enterococci are of
great interest
to the medical field. The rapid identification of such clusters allows a
better
understanding of the spread and mechanisms of action of such gene clusters.
Gene
clusters for catabolic pathways are of interest in the field of bioremediation
for the
breakdown of toxic agents from contaminated environments and in the field of
chemical
engineering for the generation of economically valuable molecules from common,
inexpensive materials. Gene clusters known as pathogenicity islands render
otherwise
harmless bacteria to highly pathogenic threats. For example, E.coli 0157 is a
clinically
important and often lethal pathogen that differs in part from the non-
pathogenic E. coli
K12 in that the former contains pathogenicity islands. Identification of such
pathogenicity islands are of great importance to the medical field.
[0006] Natural product biosynthetic gene clusters are of significant interest
in the field
of combinatorial biosynthesis and metabolic engineering. It is now commonplace
to
make novel molecules by genetic engineering of natural product biosynthetic
genes and
there are many different approaches to generating novel metabolites. Novel
pathways
may be created by rearranging the genes in the gene cluster or by combining
one or
more of the genes from the ger7e cluster with other genes. For example, genes
encoding enzymes catalyzing decorating reactions such as hydroxylation,
methylation,
acetylation, oxidation, reduction etc. of known natural products can be
inserted into or
deleted from a pathway gene cluster to effect production of unnatural
metabolites.
Alternatively, novel products may be generated by gene addition, gene
knockout, gene
substitution, or site-specific modification of genes encoding the enzymes that
catalyze
decorating reactions. Novel products can also be generated in surrogate host
organisms from heterologous libraries created from prokaryotic or eukaryotic
gene
clusters. Individual genes drawn from microbial gene clusters may be used as
biocatalysts either in cell-free systems such as a purified or partially
purified enzymatic
activities or in an appropriate heterologous expression host. Improved methods
to
2
CA 02352451 2001-07-24
3040-CA
rapidly discover gene clusters involved in the biosynthesis of microbial
natural products
expands the repertoire of genes available for use in combinatorial
biosynthesis and as
biocatalysts.
[0007] The emergence of bacteria resistant to multiple antibiotics has led to
renewed
interest in isolating variants of known antibiotics and novel antibiotics, and
also in
identifying new genes and gene products that could serve as new targets for
new or
existing antibiotics. However, methods for natural product discovery have
faced many
challenges. Discovery efforts that focus on plant derived natural products are
hampered by limited source material, typically low concentrations of active
metabolite,
difficulty extracting useful quantities of the natural product produced, and
the fact that
many secondary metabolic biosynthetic loci are expressed only under particular
growth
conditions (for example, pathogen infestation) that are poorly understood and
may be
difficult to reproduce experimentally. Discovery efforts that focus on
microbial derived
natural products are hampered by difficulties in cultivating the microbes;
indeed most
microbes cannot be cultivated. In addition, many cultivated microorganisms are
not
amenable to fermentation. Furthermore many secondary biosynthetic loci are not
expressed to detectable levels under in vitro conditions. Furtf iermore,
natural products
produced under in vitro conditions often vary according to the growth
conditions, e.g.
nutrients provided, and may not be representative of the full biosynthetic
potential of the
microorganism. Thus, there is a need for improved methods for discovery of
gene
clusters involved in the biosynthesis of natural products.
[0008] There also exist limitations in current methods to clone natural
product
biosynthetic loci from known producer microorganisms. Many known methods are
time
consuming and require genetic tools that that are not available for most
microorganisms, for example mutagenesis followed by complementation with
genomic
libraries, or transposon-tagging. Other known methods are time consuming and
have
limited chance of success, for example heterologous expression, and
heterologous
expression of resistance in order to clone linked genes. Other known methods
require
prior knowledge of the structural class of the natural product and DNA
sequence
information from a related biosynthetic locus, for example PCR amplification
or cloning
by hybridization analysis. Thus, it is desirable to obtain method of rapidly
identifying
and cloning from microbial genomes the complete genetic locus responsible for
the
biosynthesis of natural products having antibiotic activity.
3
CA 02352451 2001-07-24
3040-CA
[0009] The clustering together of biosynthetic and resistance genes encoding
natural
product biosynthetic pathways has allowed for several methods for searching
for
natural product biosynthesis gene clusters. For example, resistance has been
used as
a selective probe for clones (Walczak et al., Nonactin biosynthesis: the
potential
nonactin biosynthesis gene cluster contains type II polyketide synthase-like
genes,
FEMS Microbiol. Lett. 183(2000), 171-175), or selection based on the activity
of a
single enzyme within the cluster (Jones and Hopwood, 1984, Molecular cloning
and
expression of the phenoxazinone synthase gene from Streptamyces antibioticus,
J.
Biol. Chem. 259, 14151-14157).
[0010] Natural product biosynthetic gene clusters have also been identified
using
hybridization to highly-conserved heterologous genes. Hybridization-based
approaches
have been used in relation to Type I polyketide ketosynthase domains (Beyer et
a1.,1999, Metabolic diversity in myxobacteria: Identification of the myxalamid
and the
stigmatellin biosynthetic gene cluster of Stigmatella aurantiaca Sg a15 and a
combined
polyketide-(poly)peptide gene cluster from the epothilone producing strain
Sorangium
cellulosum So ce90, Biochim. Biophys. Acta, 1445, 185-195; Suwa et al., 2000,
Identification of two polyketide synthase gene clusters on the linear plasmid
pSLA2-L in
Streptomyces rochei. Gene 246, 123-131 ) or Type I I polyketide ketosynthase
domains
(Malpartida et a1.,1987, Homology between Streptomyces genes coding for
synthesis of
different polyketides used to clone antibiotic biosynthetic genes, Nature,
325, 818-821;
Lombo et al., 1996, Characterization of Streptomyces argillaceus genes
encoding a
polyketide synthase involved in the biosynthesis of the antitumor mithramycin,
Gene,
172, 87-91 ), non-ribosomal peptide synthetase (NRPS) domains (Beyer et
al.,supra), or
other highly conserved natural product biosynthesis genes (Steffensky et al,
2000,
Identification of the novobiocin biosynthetic gene cluster of Streptomyces
spheroids
NCIB 11891, Antimicrob. Agents Chemother. 44, 1214-1222). However, these and
other hybridization methods are often cluster-specific, and may not have broad
application to smaller gene clusters or non-modular gene clusters. In
addition, many of
these methods are labor-intensive, and involve sequencing significant amounts
of DNA
encoding genes that are not involved in the biosynthesis of a natural product.
Because
probes or primers are often imperfect, natural product gene clusters may be
missed.
Furthermore, probes or primers may not reveal the natural product biosynthetic
loci
sought as organisms often contain multiple natural product biosynthetic loci.
4
CA 02352451 2001-07-24
3040-CA
[0011] Advances in gene detection and sequencing methods have improved
accuracy of hybridization-based approaches that involve sequence comparison to
databases of known sequences. As of April 1999, the sequences for more than
150
different gene clusters encoding natural product biosynthesis pathways have
been
placed into the public databases (Strohl W.R., Biochemical Engineering of
Natural
Product Biosynthesis Pathways, Metabolic Engineering 3(2000), 4-14). With
recent
advances in sequencing brought about by automatic sequencers, such as the ABI
Prism 3700 Genetic Analyzer, which is capable of handling a throughput of over
half a
billion bases per day, the number of natural product gene clusters available
in public
databases is expected to grow exponentially. This wealth of information
facilitates
increasingly accurate and informative bioinformatic analyses.
[0012] There is a continuing need for high throughput methods for
identification of all
gene clusters in a microbial genome. There is also a need for methods for
detecting
natural product loci in a genome with minimal DNA sequencing, and in
particular
minimal sequencing of DNA encoding genes for primary metabolism. There is also
a
need for improved methods for detecting the biosynthetic loci for secondary
metabolic
pathways in an organism without having to sequence the entire microbial genome
of
the organism. There is also a need for improved genomics-based methods for
detecting gene clusters responsible for the biosynthesis of natural products
in microbial
organisms, which methods are rapid, use less reagents, and are less labor-
intensive.
SUMMARY OF THE INVENTION:
[0013] A genomics based method to rapidly search through the genome of a
microorganism in order to discover genes clusters without having to sequence
entire
genome has been developed. The method can be used to detect any cluster of
genes
that act together in a coordinated manner and are clustered together on a
chromosome.
In one embodiment, the method may be used to detect a gene cluster involved in
the
synthesis of a natural product. In another embodiment, the method may be used
to
detect a gene cluster involved in a catabolic pathway such as the degradation
of
phenolic compounds. In yet another embodiment, the method may be used to
detect a
gene cluster for a pathogenicity island from an organism. In yet another
embodiment,
the method may be used to detect a gene cluster that confers resistance to a
natural
product.
[0014] The invention is not limited to gene clusters having a particular
structure or
sequence pattern, for example modular Type I polyketide synthase genes, but
rather
CA 02352451 2002-09-24
3W0-CA
rnsy be used to identify a wlds vattety of structurally divarss gene dunbsrs,
inducting but
not lim>~d to those responsible for the biosynthesis of natural products such
as
orthosomyclns, glyooaylated Ilpodepeipeptides, and benzodlazepine antibiotics.
The
invention may also be used to Identify pdyketide synthase genes.
(00951 Discovery of flene dusters using the present inv~antion is not
dependent upon
expressafion of the natural product. Thus, It Is possible be discover new
ruatural products
that a~ not expressed at a level sufficient for detedian using more
traditional
approaches. In one embodiment, the organism Is a known producer of a natural
product, although the gene dustsr rosponsibts for pmdudion of the Imawn
natural
p,flducx is unknown. in another smbodiment, ~e or>ganiam b knahm So pnoduCS a
patifarlar nsturial product or multiple natural products but also contains a
further Qene
cluster far the biasyrdhesis of nahual produce undetected by traditional
methods. in
another embodiment the orgaMam Is not !mown to produce a rwtural product. The
genvme of many microorganisms contains multiple natural product blosyrtthetlc
led and
the present invention may be used to detect all natural produvt biosynthetic
led present
In an organism's genome while mfNmt~ng the amount of DNA saquendrrp required.
In
addition, the methods of the present invention do not npulro the arlthretlon,
growth or
fermentation of organiama.
[001 The Invention inrohres the Boning of germ caustars by 8 method chat
oombinss
random DNA sequendng fdlowad by cornputsr analysis of the DNA sequence. The
method further invdve$ the use of spot or shotgun DNA Soqussncing ar a genoma
uair~
a library of small Bones with o~oomitant mapping of them to cosmld NM'arles.
In one gaped, the invsndon provides a method fa deLectlng genes whidt ad
together
in a coordinated manner and era clustered together in a genome, sold method
comprising the atepe of: (s) pnpartng, tram isolated genomfo DIVA, a nndorn
large
inasrt Dbrary of DNA fragments of about 3t) Idlo6ese pairs (kbp) be about X00
kbp: (b)
determining the bNA sequence of at least part of a plurality of the fragments
to tam
random C3enc Sequence Tags (GETS); (c) comparing, under computer controi~ the
DNA
sequence or the plurality of GSTa with esquenoea in a database oontatning
glnea,
gene fra~pmsnts, DNA sequences or amino acid ssquenaes known to be part of a
duster of asnes that act tr~ether In a coordinated manner and that ana
clustered
together on a chromosome to iderdity a C38T that has airnilar structure to a
gene. gene
fnsgment, DNA ssquenoe or amino add aequerroe Io7own to be part or a duster of
genes that eat together in a voordineted manner, and (d) using the 08T having
almllar
8
CA 02352451 2002-09-24
3040-CA
abuv6ure to a gene, gene fragment, DNA saquencs or amine acid seqwnoe known to
be part of a duster of gauss that ad together in a coordinated manner to
detect a DNA
fragment from the large insert library, which DNA fraOmsnt from the large
Insert library
contains the OSr end genes which act together in a oQOrdirwted manner end are
dustend to0ether on a chromosome.
[Q01T] In anothsr rped, the irtvanlion prwidss a method for detecting genes
which
ad froge~er in a coordinated manner and aro duetend together in a genome
comprising: (a) preparing, tl~om Isolated genomic ONA, a random small trtaert
library of
DNA fragments of the penomic ONA and a rmdom large insert litxary of DNA
hagmsnts of the genomio DNA: (b) determining the DNA sequsnoe of et least part
d a
pluralNy at the fiagrnenb in the amali Insert Iltxery to form random t3sne
Sequarxe
Taps (aSTa): (c) ~i~n9. under computer control, the DNA eequenoe of the G8Ts
or the amino acid sequence ocrroaponding to the DNA seqwnos of 1ha r38Te wkh
sequences in ril database ooMaining genes, gene fragments, DNA, or amino acid
sequenaee known to be pad of a dusbr d genes that act together in a
ooordirtarted
manner and arse clustered together on a chromosome to identify a CiST that has
similar
structure tn o g~, gene fragment. DNA or amino add sequtrnoe known to be part
of a
duster of genes that ad together in a coordinated manner, and (d) usir~ the
C~9T
having aimllar.stnlctun to a gene, gene fra~nent, 17NA or amino acid sequence
known
to be pert of a duster of genes that act together in a coordinated manner to
detect a
DNA tragnwnt from the large insert library, which DNA frngmsnt from the large
Invert
library oantains the osT end genes which act topNh~ in a coordinated manner
and are
dusterod together on a chrornosomc.
[001] In a further aspect, the invention provides a high throughput method for
identifying a gene or gone clustor Involved In the biosyntheaia ar a~ miaobiel
natural
product comprising: (e) proparing. from isolated genomic DNA, a random large
insert
library of DNA fragments of about 30 kilabase pelts (kbp) to about 30o kbp;
(b)
determining the DNA sequence of at least part of a plurality of the fragments
to form
random Gene Sequence Tsge (GBTa); (o) vompering, under computer control, the
DNA
Gequenoe of the G6Ts or the amino add sequence oomesponding to the DNA
ssquertcs of the G8Ts with sequences In ~ databeae containing genes, pane
fragments, DNA sequences or emin4 sad sequences known to be involved tn the
biosynthosle of microbial natural product to identify a t3$T tlyat has a
similar structure
to a gene, gene fragment, DNA sequences or emirio add sequence known to be
7
CA 02352451 2002-09-24
3040-tJA
Invoh~d (n the biosynthesis of mlarobial natural produaEs; and (d) using the
~3ST having
similar structure to a gene, gene fragment, DNA or amino add sequence known to
be
involved in the b'rosynlheais of microbial natural products, or portions
thereof. to identity
a DNA fragment from tho large insert library, which DNA fra0ment contains the
GST
and a gene or gstle cluster Involved In the btosyntneris of a microbial
natural product
In a further aspect, the irnentlori provklsa a high throughput method for
Identifying a gene or gene duster involved In the biotsynthesis of a microbial
natural
product comprising: e) preparing, from isolated genomlc DNA, a random :tmoll
Insert
library of DNA fn~ments of tho genomlo DNA and a random large insert library
of DNA
fragments of the genornle DNA; b) determining the DNA sequence of at least
part of a
plurality of fragments in the small Insert library to form Oene Sequence Te~ge
(OSTs~
c) oomparktg, under computer oontrd. the ONA sequence of the t3STs or the
amino
add sequence corresponding to ttw DNA sequence of the GSTs with eequenaee in a
detatsfl» Dining genes, gene fragments. DNA or amino add sequences known M
be involved in the biottyntheela of microbial natural products to identify a
OST that has
sim3lsr structure to a gene, gene fragment, DNA or amino acid sequence known M
be
involved in tho biosynthesis of microbial natural products; and d) using the
48T having
similar structure to a gene, gene fragment, DNA or amino add sequence known to
be
Irnolved in the biosynthesis of microbial neturwi products, or portions
theroot, td identify
a DNA fragment from the large insert library, which DNA fragment aontelns the
a9T
end a Gene or gene eluabsr involved in the biosynthesis of a microbial nabual
product.
(0019] In a lurthor asplc~, the Invlrtdon prnvldes a method for scanning the
genorne
of a miaoorgaNsm in identifjr a gene duster Involved in the blosyrthseis of s
llpopeptide, said mefhad comprising: (a) Providing genomic DNA from a
microorganism:
(b) preparing a randomly generated smell insert librar~r of DNA fragments of
about 1.5
kilobase pairs (ktip) to about 10 kbp of the genomio DNA, and a randomly
generadsd
large insert library of DNA fragment of the genomic DNA of about 10 kbp to
about 300
kbp; (c) ssquendng at tasst pert a plurality of the fragments In the small
insert library ~
form random Gene 8equenCe Tags (OSTb) of about 300 base pelts (bp) to about
700
bp, translating the DNA sequences of the GSTs Into the oornsapondirfa amino
void ,
sequence and providing the amino aad sequence of the QSTa In computer readable
form; (d) comparing, under camputar control, the amino sold slquencea of the
6STa
with asqwnoas in n dstebeae oontainins amino add sequsr~css Imovm to be
Involved
in the biosynthesis of llpopeptldsa to identify a GST that hss a similar
atnrcture to an
7a
CA 02352451 2002-09-24
3040-C~A
amino add sequence known to be inv~ONAd in the blosynthssla of 4popePtides:
and (e)
using the G6T of step d) as a hydridhatlat probe to screen tlx large Insert
library of
Aenomic DNA to detect a DNA fraernent containing a gene duster involved In the
biosynlhosis of a lipopepffde; wheroln the 9enom~ DNA la obtained from a
microorparlism sele~'ted from the group consisting of (a) actinomyootor of tho
genera
Nocardhr, Qeodermetophllua, Acflnoplanea, Mlvromonopors. Nocardloldes,
Saccherothrhc, Amycolatopsis, KuiisnsHa, Saccheromorrospora.
Siccharopolyapora,
Kiteepto9porla, StnptnmYcsa, AMoro~blepaa~ v~rPt~~Grum and Actinomadrrra;
and (b) myxoooocelea of the genera Myxoooocu~, StJpmrstelJa and PolyangJum.
in a further gaped, the invention provides a method for scanning the genome of
a mic~oor~genlsm to identify a gene duster involved in the biosyntheses of am
enediyne,
satd method vomprising: a) providing penomic DNA tram a rnicroorpanism: b)
prsparlna
a randomly generated smell insert Hbrary of DNA fragments of about t .4
kiiobase pairs
(ld~P) to' about 10 kbp of the gsr>arnio DNA, artd randomly ganeratsd Isrge
Nleert ~'Y
of DNA fra9mertb of the genomlc DNA of about 10 kDp to about 300 kbp; c)
sequencing et least part of s plurality of the fragments in the small enssrt
1(brary to form
random Gene Sequence Taps (GSTs) of about 300 hasp pain (bp) to about T00 bp,
translating the DNA sequence at the GSTs into the caorrasponding amino aad
sequsrtar
and providing the amino add sequences of the GSTs in aomputar readaDte form;
d)
comparinfl, under computer oontral, the amino acid sequences of the GSTs with
sequences tn a database contaINnQ arnlno skid sequences known to de Involved
In the
biosynthesis of enediynas to Ident~y a GST that has a sinNler structure to an
amino sdd
sequence (mown to be involved In the biosynthssts of er~diynes; and e) using
the GST
of step d) as a hybridization probe to screen the IuBa Insert IiDrary of
genomic DNA roc
detect a DNA ha~ment oor>blninp a gene olustor Invalved In the biosyntheses of
an
enediyne; wherein the sertomic DNA is obtained frvrn a miaoorganism ssleelsd
from
the group oonaisbng of (a) acdnomycebee o? the genera Nocardfa,
Gaodarmatoiph~ua.
Adlnoplane$, Miorvmonaspwa. Noa~iaMes~ rothrsr, Amyrroraiopsls,
Kutzrwrfa, Saccharor»orwapore. Si~~lrer~opo~pO~s, ~~'°p°"ie,
mtr~lphomyvea.
Microbiospore. S~p~Pa~Brum end Aatrnomeduro: and (b) rrryo~cococcaies of the
genes bus, 5riyn»rdrlls and Pdyangium.
In a further aspect, the, invention provides a method for xannlng the gerroms
of
a mieroorpaniam to identify a gone cluster involved In the biowynthesia of an
orthoaumydn. said method comprising: (a) providing Qanom~ DNA from a
7b
CA 02352451 2002-09-24
rnicroafrgsnism; (b) preparing a randomly penersAsd amaN inxrt library of DNA
fragmsMa ad about 1.6 kitobass pairs (kbp) to about 1 D kbp of true genomlc
DNA. and a
randomly generated large Insert library of ONA fragmer>ts of the genomie DNA
of about .
lcbp bo about 300 Id~p: (c) asquenGnp at least part of a piurallty of the
fragments in
the small Insert Ilbrary to form random Gene Sequenoa Tags (GSTs) of aoout 300
by to
about 700 bp. translailng the DNA eaquenoe of the 08Ts into the aorreepondlng
amino
add sequenpe and providing the amino acid sequences of the f3STe in computer
readable form: (d) oompsring, under computer control, the ammo add sequences
of the
GSTs ~ sequences In a database containinfl amino add sequences ltnown io be
involved In the bi~yMhe:ls of orthosomydns to identify a C38T that has a
aimlier
strucbtra to an smirro add sequence knavm to 6a irnotved in tho biosyMltssb of
orthosamyclne; end (e) using the 08T of step d) as a hybridfzadan Probe to ~n
~
large insert library of pmornic DNA to detect a DNA fragment containing a gene
cluster
involved In the biosynthesis of an ortfl~nmydn: Wherein the gertomic DNW Is
obtained
from a microor~genixn selected from the group consisting v~f (a) adinomycstss
of the
Genera Nocardja. Oeodermatap~hifus, Aa~op~oa~ ~'~'°r'~P°~~
N°~~~~s
SaccharothrGc, Amyicofatop~ie. Ktazrwrle. Sweehanorrronoeporn,
SaooiwraporyapoR.
Kitaeatosporla. SYnptomY~es~ : ~~~ and Aclirromadura:
arxf (b) myxococcales of the generra Myxoooocut. ma~~ and Potyanglum.
In yet another arnbodlment, the Invention pravide9 a method for seanning the
psnome of a mlaroorQanism to Identify a polyketida Qynthew one or a gene
duller
Indudlna a potyketide synthase gene. said method comprtslnp: (e) piovidinp
qenamic
DNA from o mtcrooreanfsm; (b) preparing.a randomly generated emaA Insert
Ilbrevy of
DNA fragrrfarr~ of about 1.5 kUobaee pairs (kbp) to about 10 kbp of the
genomio DNA,
and a randomly generated large insert library ~ DNA fragments of the Qenomlc
DNA of ,
about 10 kbp to about 300 I~p; (c) sequendng at least part of a plunltty of
the
fragmar'tb In the small insert Ubrary to form random Owns Sequence Taps (G8Ts)
of
about 300 brass pelrs (bp) to about 700 bp, translating the DNA sequences of
the GSTs
into the .corresponding amino add sequence pnd Providing the amine ec~id
sequences
of the C33'Ts in oorrtpuber raadseis form: (d) comparing. undo' muter contrd,
the
amino add sequences of the GSTs with sequences in a database containing amino
add spusnces known to be assodated wtth a polyketide eynthase be Wsnttfy a
OST'
tiler has a simeler struCturs to en ornino add sequence knoNm to be aaeodatvd
with a
polyketfde ayntnsse; (s) using th~ G5T of step d~ as a hybridlsstlon probe to
screen the
7c
CA 02352451 2002-09-24
3040~CJv
iar~e Insert pbrary of panomic DNA to detect a DNA fragment oontatnlrro a
polyketlde
synthaaa pang or a gene duster Inducting poiykedde synthaae pane; wherein the
ger>amlc DNA Is obeslned from a mla~oorpanlam selected from the group
ooneiatlng of
(a) actlnomyostes of the genera Nocerrdie, Gealsm~atqph~us, Adlnophnee,
AMcromortospora. Noavdl0ldi~s.Sar'xheroif~roc. AmycaJai~qpria, Kutrrtar~a.
Sac~haromono~poie. SecchawPdYspora. Kite9arosporia, 5'f~ptamyYovs. A~o~~.
Sbyptoepo~lanpium and ActlnOmsdt~a~ arid (b) myxooac~ies of the genera
~y~CCOCUa, Stl~ntatalJe end Polyar~ium.
IOp~pl Although the Inwa~tion requires th~ aequandnq of hundnrAde of fragments
of
genomk DNA, the Invsatmsnt Is smell when aompatad to asquendr~ the ire
penome of a miaoor~snlam~
~OOi21] Other objedx. Naatuns and advaraa~es of the ptr>~nt Invenflon will
become
apparsnt fnxn the fiallowires detwtted dascrlption. tt ohould t~s underatood~
however that
1hs detatisd description and the apectflc examples, whllo indlaetlng preferred
embodlmsrtta of the Invention, aro given by way of IAustratjon only. sings
various
chan~a and modHlc~tlons within the spirit end poops of the Invention will
become
apparent to those tikilled In the art from this detailed dssoriptrjon.
7d
CA 02352451 2002-O1-17
3040-CA
BRIEF DESCRIPTION OF THE DRAWINGS:
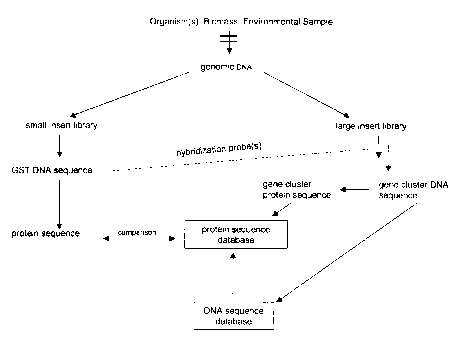
[0022] Figure 1 is a schematic view of a method for discovery of a gene
cluster
according to one embodiment of the invention.
[0023] Figure 2 illustrates construction of a small insert library and a large
insert
library according to the method of Figure 1.
[0024] Figure 3 illustrates selection of Gene Sequence Tags (GSTs) from the
small
insert library for use of probes for screening the large insert library
according to the
method of Figure 1.
[0025] Figure 4 illustrates identification and cloning of the gene cluster
from the large-
insert library according to the method of Figure 1.
DETAILED DESCRIPTION OF THE INVENTION:
[0026] The present invention provides an efficient strategy for identifying
genomic
sequences harboring a target gene cluster.
[0027] The meaning of gene cluster, as used in the specification, refers to
any group
of two or more genes that act together in a coordinated manner and that are
clustered
together on a chromosome. The meaning of gene cluster is not restricted to or
associated with any particular type of metabolic pathway. Rather, the target
gene
cluster of the invention may be associated with a wide range of metabolic
pathways or
cellular processes including, but not limited to, the biosynthesis of natural
products, the
degradation of a compound, conferring resistance to therapeutic drugs, or
pathogenicity
islands from various organisms.
[0028] The meaning of genome extends to all DNA contained within an organism,
including naturally occurring plasmids or other episomal DNA, or in the case
of
eukaryotes, compartmentalized DNA.
[0029] The approach involves the generation of two random DNA libraries from
genomic DNA of a microorganism of interest, namely a small insert library and
a large
insert library. The small insert library serves as a genomic sampling library
and is
sometimes referred to herein as the GST library. Probes derived from sequences
obtained from the small insert library are used to identify and isolate from
the large
insert library (sometimes referred to herein as the gene identification
library or GIL)
significantly larger genomic DNA fragments that include the probe together
with its
flanking sequences, and genes of the target gene cluster. The small insert
library is
formed of a population of randomly generated fragments so as to provide an
adequate
8
CA 02352451 2002-O1-17
3040-CA
sampling of the entire DNA contained within a microorganism. Advantageously,
the
population includes fragments of all biosynthetic
8 (a)
CA 02352451 2002-09-24
304D-CA
bd in the genome. Frepmsnta fhom the smelt Insert library are raequenced to
provide
Gene Sequence Tags {OSTs). The aSTs that correspond to fragments of the target
gene Guater by homology oornparwan with a database are used as probes to
identffy
the large insert done(s) oorttalning the genes that form the earget gene
dusbsr.
(0430] The penomic DNA may be der9ved from any protcaryotlo or eukaryotla
microoreenism known or suspected to contain a flene cJueter, The aenomic DNA
may
be drawn from a population of uncultured microor~anisma found in their natural
habitat
or environment or from blomo$a, thereby avoiding problems aasodated with
cultivation
and tertnentation of microbes. The gsnamic DNA may also be dsdved from
aulturod
microorganisms, either mixed or puftlled. A pnafarrod souroe of the qsnomb bNA
ie
mkxvorpan(sms. ouch as bedsria rnd ltrnpi. Bectrrlat spades suitable for use
in the
method indude subetantlelly ell badertet spades, both animat~snd plant-
pathogenic
end nonpethopenic. Prehrrod micnno~anism~ far the purposes o1 identifying
net<x~el
product blosynthesls dusters Include but are not limited to bacteria of the
order
Acdrromyoetelee and Myxocoocales. Prelemed genera of Actinomyosbs Indude
Nocsrdla, Goaderrr~rEOphilus, Adinoplanee, Mlorornonosporri. Nooerrlioides,
Saed~srvthd~c. AmycototoPsic, Kut~tarie. arnmonoeporo, Seocharopolyspor'a.
K~satospo,~, Stnsptomyoes, Mkxabispore, Stroptosporan9lum, Actfnomadura.
Preferred Qenera of the order Myxocoacales include Stipmatelle, Myxo~e and
Potyangiu m. The taxonomy of Iidinomycetss iri complex end reference is rnede
to
OoodfWicw {1886) t3uprapanerlo olrsslticatbn of actinomycer~aa. B~Y'a ~~
°f
~rnaElc 9acterlology, Vol. 4, William: end Wilklns. amore, pp 2322-2339, end
to
Embtay end 8ts~ckebrsndt, (1684), The molecular phylogeny and sysaematics of
the
aetinomyoetes, Annu. Rw Mkn~blo~ . 48. 25?-289 for species than may else be
used
with the present Invention. One skilled in the art would understand that the
profaned
source of DNA wiu depend on the Gatnet gene duster, s.g" actinomycetss and
badlli for
natural products. pseudamoneds for cataballc pathways, etc_
(pp31] The gErrwmic DNA can be isolated fnxn samples using venous teahrfiquea
wall
known In the art (NUO>oic AoMa In the Env~N~Orvr~erri' A~letHods S
ApplJbadbne. J.T.
Trovore, D.D. van Elasas, Pringer Laboratory, 1885). Preferably, the genomic
DNA
obtained will be of high molecular w~ight and free of enzyme inhibitors and
other
contaminsnb. In a preferred embodiment, the sire of the genomic DNA Is of a
molQcufar weight higher than 80 kb. The 9enomlc DNA is employed to pfoduCa
t,WO
random rooembinant DNA Ilbmrles. The rycombinant DNA library may be prepe~red
.
9
CA 02352451 2002-O1-17
3040-CA
without prescreening the organism or population of organisms, cultured or not,
for the
presence of the target gene cluster. The genomic DNA fragments may be
generated
and subcloned into an appropriate cloning vector by a variety of procedures.
Ideally,
the genomic DNA fragments will be as random as possible. Mechanical shearing
methods such as sonication, nebulization and the like, or passage through a
fine needle
with manual pressure are preferred methods, however enzymatic methods such as
partial digestion with a frequently cutting restriction enzyme (for example
Sau3Al or
Taql) and other methods can also be employed. When a mechanical shearing
method
is employed, the ends of such fragments may be "repaired" or blunted to
generate
uniform ends that can be enzymatically ligated to the appropriate restriction
sites) of
the vector either directly or with the use of DNA linkers. Smaller inserts are
preferentially cloned.
[0032] Any conventional cloning vector, suitable for genomic DNA libraries,
may be
used including phage-derived, plasmids, cosmids, phosmids, Bacterial
Artificial
Chromosomes (BACs), and Yeast Artificial Chromosomes (YACs). One skilled in
the
art will select an appropriate cloning vector based on the circumstances, e.g.
typical
plasmid cloning range of 0.1 to 10 kbp, typical cosmid cloning range 30 to 50
kbp,
typical BAC cloning range 75-300 kbp etc. In general, the DNA sequence is
inserted
into an appropriate restriction endonuclease sites) on the cloning vector by
procedures
known in the art. Such procedures and others are deemed to be within the scope
of
those skilled in the art.
[0033] A small insert library of relatively short genomic DNA fragments is
constructed.
DNA fragments forming the small insert library are cloned into an appropriate
vector
and serve as a source for genetic sampling. One suitable vector that may be
used to
prepare the small insert library is the pBluescriptT"~ II cloning vector
(Stratagene). Other
suitable vectors include but are not limited to pUC19 and related vectors,
Lambda
vectors, M13 cloning vectors, pBR322 and related vectors.
[0034] Advantageously, the small insert library size, i.e. the number of
individual
clones, is as random as possible and is large enough to provide an adequate
representation of the DNA contained within the microorganism of interest. By
estimating size of the target gene cluster and the size of the genome, a
preferred small
library size may be determined. For example, the frequency of sequences
containing
genes from secondary metabolic pathways producing natural products in the
small
insert library reflects their occurrence in the genome. If the microorganism
has one or
CA 02352451 2001-07-24
3040-CA
more naturally occurring plasmids of moderate to high copy number, or is a
microorganism whose genome is segmented in a non proportional fashion, the
resulting
small insert library will reflect this disproportionality. To overcome any
bias that may
arise due to such genetic disproportionality, a larger number of small insert
clones may
have to be processes and the size of the large insert library may likewise
have to be
increased under such circumstances. Alternatively, the chromosomal DNA may be
purified by methods known in the art to overcome problems due to a high copy
number
of plasmids. In any event, the number of cloned DNA fragments in the short
insert
library or the library size must provide a reasonable probability that genes
from the
target gene cluster will be found in the representative fragments forming the
short insert
library. If it is known what experimental conditions allow for the expression
of a gene or
enzymatic activity believed to belong to a target gene cluster, the small
insert library
may be enriched for clones containing DNA fragments thereof by activity
selection or
screening.
[0035] Preferably, the size of the DNA fragments forming the short insert
sampling
library will be substantially uniform. The actual size of the DN,A fragments
in the short
insert library may vary, but the size must be of a length to provide
sufficient sequence
data to identify a fragment as part of the target gene cluster. In one
embodiment of the
invention, the size of the DNA fragments in the short insert library is about
1.5 kpb to
about 10 kbp, in a preferred embodiment the size of the DNA fragments is about
1.5
kbp to about 5 kbp, in a more preferred embodiment the size of the DNA
fragments is
about 1.5 kpb to about 3 kbp. Since the current sequencing technology can
routinely
provide sequence information, referred to herein as a "read", of up to 700 bp,
and
sequencing can be carried out with primers flanking both sides of the insert,
it is
advantageous that the insert be at least the length of two reads so that each
read yields
different sequence data. The use of larger inserts increases the probability
of obtaining
intact genes together with required regulatory sequences that may be expressed
in the
cloning host, especially if the cloning host is closely related to the
organism from which
the genomic DNA was isolated. This may not be desirable as this may skew the
population towards non-toxic or non-detrimental DNA fragments or beneficial
DNA
fragments.
[0036] A large insert library of relatively long DNA fragments is also
constructed. The
DNA fragments forming the large insert library are cloned into an appropriate
vector
and serve as a screening library from which the target gene clusters) is/are
obtained.
11
CA 02352451 2002-O1-17
3040-CA
Suitable vector systems for use in preparing the large insert library include
but are not
restricted to Lambda vectors such as Lambda DASH II, cosmid vectors such as
pWE15
or SuperCosT""-1, P1 cloning vectors such as pAd10sacBll, fosmid vectors such
as
pFos1, Bacterial Artificial Chromosome (BAC) vectors such as pBeIoBAC11, and
Yeast
Artificial Chromosomes (YAC) vectors such as pYAC4. The vector is selected to
be
stably propagated in an appropriate host. It is noted that the short insert
library and the
large insert library need not be done in the same host organism, i.e., E.
coli, Bacillus,
Saccharomyces cerevisiae, human cell lines, etc.
[0037] Preferably, the size of the genomic DNA fragments in the large insert
library
will be substantially uniform. The size of the genomic DNA fragments in the
large insert
library will vary widely depending on the vector system used. In the case
where a
cosmid vector system is employed, the size of the DNA fragment in the large-
insert
library is about 30 kbp to about 50 kbp.
[0038] Where the genomic DNA is isolated from a purified organism, an
appropriate
number of the large insert clones is one that allows several-fold coverage of
the
genome of interest. Where the DNA is isolated from a mixed population of
organisms,
the number of large insert clones should preferably be larger so as to
maximize the
probability to find overlapping clones.
[0039] Short lengths of DNA from either end of cloned inserts in the short
insert
library are sequenced using a forward primer (F) or a reverse primer (R) to
provide a
plurality of Gene Sequence Tags (GSTs). However, if the large insert library
includes a
significant number of clones and the DNA sequencing technology reproducibly
yields
adequate sequence information from the ends of such inserts, the GSTs can be
generated from the large insert library. In such a case, the ability to
sequence a
significant number of ends from a large insert library can substitute for
sequencing of
the small insert library. In a preferred embodiment a GST is produced from
each of the
cloned inserts in the short insert library. The length of the GST sequence
will depend
on the sequencing technology used but typically ranges from about 300 by with
a
traditional (manual) DNA sequencing apparatus up to about 700 by or more with
an
automated DNA sequencer such as an a 3700 ABI capillary electrophoresis DNA
sequencer (Applied Biosystems). In one embodiment the GSTs are about 700 base
pairs in length.
[0040] The sequence of each GST is provided in computer readable form for in
silico
screening of a database containing genes, gene fragments or DNA known to be
12
CA 02352451 2002-O1-17
3040-CA
involved in the target gene cluster. In one embodiment the in silico screening
is based
on the nucleic acid sequence of the GSTs. In a preferred embodiment, the
nucleic acid
sequence of the GST is translated to its corresponding amino acid sequence,
and the in
silico screening is based on the amino acid sequence of the GSTs against a
database
containing proteins or protein fragments known to be involved in the target
gene cluster.
Advantageously, translation of the nucleic acid sequence of the GSTs to their
corresponding amino acid sequence or of a database of genes, gene fragments
and
DNA to the corresponding amino acid sequences is computer-assisted.
[0041] The nucleic acid sequence or the amino acid sequence of the GSTs, in
computer readable form, is compared under computer control using publicly
available
bioinformatics tools such as BLASTT"~, ProdornT"", ClustalT"~, etc. to a DNA
or protein
database containing genes, gene fragments, or clusters of genes, or their
corresponding protein products known to be involved in the target gene
cluster. The
database may be a public gene database such as GenBank, EMBL, or a private
database. A preferred database for the identification of natural product
biosynthetic
genes is the DecipherT"~ database of microbial genes, available on a
subscription basis
from Ecopia BioSciences Inc., St.-Laurent, Quebec.
[0042] Advantageously, the reference database used for homology comparison
contains at least one or preferably multiple homologues of one or more genes
of the
target gene cluster. A homologous amino acid sequence is one that differs from
an
amino acid sequence by one or more conservative amino acid substitutions. Such
a
sequence encompasses allelic variants, as well as sequences containing
deletions or
insertions that retain the functional characteristics of the polypeptide.
Homologous
amino acid sequences include sequences that are identical or substantially
identical to
the amino acid sequence. By amino acid sequence substantially identical is
meant a
sequence that differs from the sequence of reference by a majority of
conservative
amino acid substitutions. Conservative amino acid substitutions are
substitutions
among amino acids of the same class. These classes include, for example, amino
acids having uncharged polar side chains, such as asparagine, glutamine,
serine,
threonine, and tyrosine; amino acids having basic side chains, such as lysine,
arginine,
and histidine; amino acids having acidic side chains, such as aspartic acid
and glutamic
acid; and amino acids having nonpolar side chains, such as glycine, alanine,
valine,
leucine, isoleucine, proline, phenylalanine, methionine, trytophan, and
cysteine.
13
CA 02352451 2002-O1-17
3040-CA
[0043] Homology comparison of the GSTs and the sequences in the database may
be assessed by % identity or by E value. The E value relates the expected
number of
chance alignments with an alignment score at least equal to the observed
alignment
score. An E value of 0.00 indicates a perfect homolog. The E values are
calculated as
described in Altschul et al. J. Mol. Biol., October 5; 215(3) 403-10. The E
value assists
in the determination of whether two sequences display sufficient similarity to
justify an
inference of homology. An E value of 10-x° will generally be indicative
of two proteins
that are significantly related to one another, an E value of 10-'5 being
especially
significant. However the length and accuracy of the sequenced being compared
with
the database will strongly influence the value of E considered significant.
The use of a
filter to mask stretches of low complexity or highly biased amino acid
sequences can be
used to increase the specificity of homology comparisons.
[0044] Comparison of sequences may also be assessed by ClustalT"" alignments
showing conserved positions between the GSTs and the sequences of the
database.
In this manner, GSTs likely to belong to genes involved in the target gene
cluster are
identified. Amino acid sequences are aligned to maximize identity. Gaps may be
artificially introduced into the sequence to attain proper alignment. Once the
optimal
alignment has been set up, the degree of homology is established by recording
all of
the positions in which the amino acid of both sequences are identical,
relative to the
total number of positions.
[0045] Clones that contain a GST that have a similar primary amino acid
sequence
based on homology comparison with gene fragments known to be involved in the
target
gene cluster are sequenced from the short insert library. In a preferred
embodiment,
the DNA clone from the small insert library that corresponds to a GST of
interest can be
sequenced from the other end using a universal reverse primer and analyzed for
homology to the reference database. Sequencing at the opposite end of the
insert from
which the GST was derived identifies clones whose inserts contain GSTs that
correspond to gene fragments known to be involved in the target gene cluster
at both
ends of a single short insert. Insert clones that display homology to the
target gene
cluster at both ends are likely to contain sequences from the target gene
cluster.
Identification of clones having homology to the target gene cluster requires
the
presence of characterized homologues in the reference database.
14
CA 02352451 2001-07-24
3040-CA
[0046] The GSTs that correspond to genes or gene fragments known to be
involved
in the target gene cluster are used as or used to derive hybridization probes
to isolate
the corresponding DNA fragments in the large insert library by standard
hybridization
procedures on high density array matrices such as nylon membranes or DNA
microchips. Such hybridization probes can be nucleic acids, DNA or RNA,
containing a
sequence from the cloned DNA fragment, in full or in part, that is labeled
either with a
radioisotope such as 32P or with a non-radioactive detection system such as
digoxygenin (Roche). With organisms whose genome is highly biased in that it
is highly
GC-rich or AT-rich, larger probes can lead to non-specific hybridization or
background.
Therefore, for GC-rich organisms such as actinomycetes relatively short
oligonucleotide
probes of approximately 20 nucleotides are preferred over longer PCR-amplified
fragments. In the event that the desired gene cluster extends beyond the
boundaries of
a large insert clone or a series of overlapping large insert clones, the DNA
sequence at
these boundaries can be used to design additional probes which can be used in
another round of hybridization to identify other overlapping large insert
clones. This
second round of hybridization can be performed at any stage of detection and
cloning
of the target gene cluster from the large insert library or final assembly of
the target
gene cluster.
[0047) The insert of the large insert clone is entirely sequenced by any
method known
in the art. In one embodiment, the insert of the large insert clone is
sequenced by a
shotgun DNA sequencing technique. In other embodiments, the insert of the
large
insert clone is sequenced by a technique selected from a subcloning technique,
a
primer walking technique, and a nested deletion technique.
[0048] The cloned sequences are then assembled and the open reading frames are
identified using appropriate methods known to one skilled in the art. These
methods or
criteria for gene identification can vary depending on the nature of the
organism from
which the genomic DNA was isolated. Overlapping large insert clones can be
assembled together using computer algorithms to generate a (large, contiguous
DNA
contig sequence or multiple DNA contig sequences that are sE;parated by
relatively
small gaps. One skilled in the art can then analyze these contigs of DNA
sequence
using bioinformatics tools to identify the open reading frames and regulatory
sequences. The sequences are assembled into the target gene cluster by
additional
computer analysis. The sequences of the DNA contigs and the proteins which
they are
predicted to encode can then be submitted to appropriate databases.
CA 02352451 2002-O1-17
3040-CA
[0049] Reviewing the method by reference to the figures, high molecular weight
genomic DNA of interest is isolated from a cell mass or biomass (Figure 1 ). A
small
insert library and a large insert library are constructed by a shotgun cloning
approach
so as to contain randomly generated fragments of DNA (Figure 2). The small
insert
library is composed of about 500 individual clones each containing a piece of
genomic
DNA insert in the range of 1.5-3 kb carried on a cloning vector that can be
propagated
in a suitable host organism. The large insert library is composed of an
appropriate
number of individual clones each containing a piece of genomic DNA of interest
that is
at least 30 kb carried on a cloning vector that can be propagated in a
suitable host
organism. The small insert library serves as a genomic DNA sampling that is
sequenced to generate Gene Sequence Tags (GSTs) as illustrated in Figure 3.
Computer-assisted analysis of the GSTs identifies those GSTs likely to reside
within the
target gene cluster (GSTs of interest). Molecular probes) are then designed
from the
GSTs of interest and are used to identify, by nucleic acid hybridization, the
clones in the
large insert library that contain the probe(s). Once identified, the large
insert clones) of
interest are sequenced by a shotgun method similar to that employed on genomic
DNA
for the generation of the small insert library (Figure 4). A sufficient number
of shotgun
sequences are done so as to allow for computer-assisted reconstruction or
assembly of
the entire sequence of the large insert clone(s).
[0050] It is to be understood that the embodiments described herein are for
illustrative
purposes only and that various modifications or changes in light thereof will
be
suggested to persons skilled in the art and are to be included within the
spirit and
purview of this application and scope of the appended claims.
[0051] The following examples use many techniques well known and accessible to
those skilled in the art. Enzymes are obtained from commercial sources and are
used
according to the vendors' recommendations or other variations known to the
art.
Abbreviations and nomenclature are employed as commonly used in professional
journals such as those referred to herein.
EXAMPLES:
[005] Example 1' Identification of the ramoplanin biosynthetic locus in
Actinoplanes
sa. ATCC 33076.
16
CA 02352451 2001-07-24
3040-CA
[0053] Actinoplanes sp. ATCC 33076 was previously shown to naturally produce
ramoplanin, a biologically active lipodepsipeptide (U.S. Patent No.
4,303,646). The
genetic locus involved in the production of this compound was not previously
identified.
[0054] Actinoplanes sp. strain ATCC 33076 was obtained from the American
Tissue
Culture Collection (ATCC) and cultured according to standard microbiological
techniques (Kieser et al., supra). Confluent mycelia from oatrneal agar plates
were
used for the extraction of genomic DNA as previously described (Kieser et al.,
supra)
and the size range of the DNA obtained was assessed on agarose gels by
electrical
field inversion techniques as described by the manufacturer (FIGE, BioRad).
The DNA
serves for the preparation of a small size fragment genomic sampling library
(GSL),i.e.
the small insert library, as well as a large size fragment cluster
identification library
(CIL), i.e. the large insert library. Both libraries contained DNA fragments
generated
randomly from genomic DNA and, therefore, they represented the entire genome
of
Actinoplanes sp.
[0055] For the generation of the GSL library, genomic DNA was randomly sheared
by
sonication. DNA fragments having a size range between 1.5 and 3 kb were
fractionated on a agarose gel and isolated using standard molecular biology
techniques
(Sambrook et al., supra). The ends of the obtained DNA fragments were repaired
using
T4 DNA polymerase (Roche) as described by the supplier. This enzyme creates
DNA
fragments with blunt ends that can be subsequently cloned into an appropriate
vector.
The repaired DNA fragments were subcloned into a derivative of pBluescript SK+
vector (Stratagene) which does not allow transcription of cloned DNA
fragments. This
vector was selected as it contains a convenient polylinker region surrounded
by
sequences corresponding to universal sequencing primers such as T3, T7, SK,
and KS
(Stratagene). The unique~EcoRV restriction site found in the polylinker region
was used
as it allows insertion of blunt-end DNA fragments. Ligation of the inserts,
use of the
ligation products to transform E. coli DH10B host and selection for
recombinant clones
were performed as previously described (Sambrook et al., supra). Plasmid DNA
carrying the Actinoplanes sp. genomic DNA fragments was extracted and the
insert
size of 1.5 to 3 kb was confirmed by electrophoresis on agarose gels. Using
this
procedure a library of small size random genomic DNA fragments is generated
that
covers the entire genome of the studied microorganism. The number of
individual
clones that can be generated is infinite but only a small number is further
analyzed to
sample the microorganism's genome.
17
CA 02352451 2002-O1-17
3040-CA
[0056] To generate the CIL library, high molecular weight genomic DNA was
partially
digested with a frequent cutting restriction enzyme, Sau3A (G~ATC). This
enzyme
generates random fragments of DNA ranging from the initial undigested size of
the
DNA to short fragments of which the length is dependent upon the frequency of
the
enzyme DNA recognition site in the genome and the extent of the DNA digestion.
Conditions generating DNA fragments having an average length of ~40 kb were
chosen
(Sambrook et al., supra). The Sau3A restricted DNA was ligated into the BamHl
site of
the SuperCos-1 cosmid cloning vector (Stratagene) and packaged into phage
particles
(GigapackT"" III XL, Stratagenej as specified by the supplier. E. coli strain
DH10B was
used as host and 864 recombinant clones carrying cosmids were selected and
propagated to generate the CIL library. Considering an average size of 8 Mb
for a
streptomyces genome and an average size of 35 kb of genomic insert in the CIL
library,
this library represents a 4-fold coverage of the microorganism's entire
genome.
Subsequently, the Actinoplanes sp. CIL library was transferred onto membrane
filters
(Schleicher & Schnell) as specified by the manufacturer.
[0057] The GSL library was analyzed by sequence determination of the cloned
genomic DNA inserts. The universal primers KS or T7, referred to as forward
(F)
primer, were used to initiate polymerization of labeled DNA. Extension of at
least 700
by from the priming site can be routinely achieved using the TF, BDT v2.0
sequencing
kit as specified by the supplier (Applied Biosystems). Sequence analysis of
the
generated fragments (Genomic Sequence Tags, GSTs) was performed using a 3700
ABI capillary electrophoresis DNA sequencer (Applied Biosystems). The average
length of the DNA sequence reads was --700 bp. Further analysis of the
obtained
GSTs was performed by sequence homology comparison to various protein sequence
databases. The DNA sequences of the obtained GSTs were translated into amino
acid
sequences and compared to the National Center for Biotechnology Information
(NCBI)
nonredundant protein database and the proprietary Ecopia natural product
biosynthetic
gene DecipherT"' database using previously described algorithms (Altschul et
al.,
supra). Sequence similarity with known proteins of defined function in the
database
enables one to make predictions on the function of the partial protein that is
encoded by
the translated GST.
(0058] A total of 882 Actinoplanes sp. GSTs were generated and analyzed by
sequence comparison. Sequence alignments displaying an E value of at least e-5
were
considered as significantly homologous and retained for further evaluation.
GSTs
18
CA 02352451 2002-O1-17
3040-CA
showing similarity to a gene of interest can be at this point selected and
used to identify
larger segments of genomic DNA including the gene of interest. Ramoplanins
produced by Actinoplanes sp. belong to the family of polypeptide antibiotics.
Polypeptides are synthesized by nonribosomal peptide synthetase (NRPS) enzymes
that perform a series of condensations and modifications of aminoacids. Many
members of this enzymatic class are found in protein databases rendering
possible the
identification of an unknown NRPS by sequence similarity. Analysis of the
Actinoplanes sp. GSTs revealed the presence of 3 GSTs having similarity to
known
NRPS proteins in the NCBI non redundant protein database (Table 1 ). The
obtained E
values confirm that these GSTs encode partial NRPS sequences. The 3 NRPS GSTs
were selected for the generation of oligonucleotide probes which were then
used to
identify gene clusters harboring the specific NRPS genes in the CIL library.
[0059] Oligonucleotide probes were designed from the nucleotide sequence of
the
selected GSTs, radioactively labeled, and hybridized to the CIL library using
standard
molecular biology techniques (Sambrook et al., supra, Schleicher & Schnell).
Positive
clones were identified, cosmid DNA was extracted (Sambrook et al., supra) and
entirely
sequenced using a shotgun sequencing approach (Fleischmann et al., Science,
269:496-512 ). Identification of the original GSTs, used to generate the
oligonucleotide
probes, within the DNA sequence of the obtained cosmids proved that these
cosmids
indeed carried the gene cluster of interest.
[0060] Generated sequences were assembled using the Phred-PhrapT"" algorithm
(University of Washington, Seattle, USA) recreating the entire DNA sequence of
the
cosmid insert. Reiterations of hybridizations of the CIL library with probes
derived from
the ends of the original cosmid allow indefinite extension of sequence
information on
both sides of the original cosmid sequence until the complete sought-after
gene cluster
is obtained. Application of this method on Actinoplanes sp. and use of the
above-
described NRPS GST probes yielded <j cosmids. Complete sequence of these
cosmids
and analysis of the proteins encoded by them undoubtedly demonstrated that the
gene
cluster obtained was indeed responsible for the production of ramoplanin.
Subsequent
inspection of the ramoplanin biosynthetic cluster sequence (~80 kb) revealed
the
presence of 3 additional GSTs from the GSL library, bringing the total number
of
ramoplanin locus GSTs to 6. Thus, the genetic locus responsible for the
biosynthesis
of ramoplanin was identified by the present invention.
19
CA 02352451 2002-O1-17
3040-CA
[0061] Table 1
LengthProposed Homology ProbabilityProposed function
(bp) function of
protein match
GST1 632 NRPS PIR T362483.0 2 CDA peptide synthetase
I in Streptomyces
coelicolor
GST2 592 NRPS PIR T36248 5.00 -~
28 CDA
peptide
synthetase
I in Streptomyces
i coelicolor
GST3 502 NRPS '~rPIR 7.00 -31
T36180 CDA peptide
synthetase
i III in
Streptomyces
coelicolor
[0062] Example 2: Identification of a new natural product biosynthetic locus
in
Strectomyces mobaraensis.
[0063] Streptomyces mobaraensis was previously shown to naturally produce a
variety of biologically active compounds including piericidins, pactamycin,
and detoxins
(Tamura et al., 1963, Agr. Biol. Chem., Vol. 27, No. 8, pp. 576-582). The
genetic loci
responsible for the production of these compounds and, hence, the enzymatic
mechanisms involved in their biosynthesis was not previously characterized.
[0064] Streptomyces mobaraensis strain NRRL B-3729 was obtained from the
Agricultural Research Service collection (ARS) and cultured according to
standard
microbiological techniques (Kieser et al., supra). All subsequent experimental
procedures were performed as described in Example 1.
[0065] A total of 450 GSTs were generated and analyzed by sequence comparison.
Among these GSTs, two showed similarity to enzymes involved in deoxysugar
biosynthesis (Table 2). There are several classes of natural compounds such as
macrolides, polypeptides, anthracyclines, enediynes, polyenes that are
glycosylated
with typical and/or unusual glycosyl groups. Other metabolites such as
orthosomycins
and aminoglycosides are mainly composed of modified deoxysugar moieties
(Weymouth-Wilson, The role of carbohydrates in biologically active natural
products,
Nat. Prod. Rep.,1997, 99-110). Specific enzymes are required for the
biosynthesis of
unusual sugars from natural sugar precursors as well as glycosyltransferase
enzymes
CA 02352451 2002-O1-17
3040-CA
that catalyze the transfer of the sugar to a specific backbone structure (Liu
and
Thorson, Pathways and mechanisms in the biogenesis of novel deoxysugars by
bacteria, Annu. Rev. Microbiol., 48: 223-256). The presence of two sugar
biosynthetic
genes in Streptomyces mobaraensis is of interest as the natural products shown
to be
produced by this microorganism do not contain any sugar residue. GST1 was used
to
probe the S. mobaraensis CIL library as above-described (Example 1 ).
[0066] Positive clones were identified and sequenced. The original GST1 was
identified within the sequenced cosmid. One reiteration of the same method was
applied providing two overlapping cosmids covering the entire biosynthetic
cluster.
Analysis of the proteins encoded by this cluster demonstrated the presence of
a novel
biosynthetic locus (-45 kb) having the potential to produce an avilamycin-like
compound, member of the orthosomycin group of antibiotics composed of a series
of
deoxysugar residues. Thus, the genetic locus responsible for the biosynthesis
of this
avilamycin-like compound was identified by the present invention.
Table 2
LengthProposed Homology ProbabilityProposed function
of
(bp) function protein match
GST1 738 sugar dehydratasePIR T308732.0 -74 dNDP-glucose
dehydratase in
Streptomyces
viridochromogenes
GST2 601 glycosyltransferasePIR F750992.00E-05 rhamnosyl transferase
in
I~ Pyrococcus abyssii
[0067] Example 3: Identification of the anthramycin producing biosynthetic
locus in
Streptomyces refuineus.
[0068] Streptomyces refuineus var. thermotolerans was shown to produce a
benzodiazepine antibiotic, anthramycin, that covalently binds to the minor
groove of
DNA. Anthramycin has been shown to possess various potent biological
activities
including antibiotic, antitumor and antiviral activities. The biosynthetic
locus responsible
for the production of anthramycin was not previously characterized.
21
CA 02352451 2002-O1-17
3040-CA
[0069] Streptomyces refuineus var. fhermotolerans strain NRRL-3143 was
obtained
from the Agricultural Research Service collection (ARS) and cultured using
standard
microbiological techniques (Kieser et al., supra). Subsequent experimental
procedures
for cloning and analyzing the genetic material of this microorganism were as
described
in Example 1. A total of 486 GSTs were analyzed by sequencing and protein
homology
comparison to the NCBI protein database and the Ecopia Decipher'-""
proprietary
protein database. Precursor feeding studies have established two distinct
moieties in
the anthramycin molecule that derive from tryptophan via the kynurenine
pathway and
catabolism of L-tyrosine (Hurley et al., 1975). The two modified amino acids
are linked
together through an amide bond typically catalyzed by nonribosomal peptide
synthetases (NRPS). Analysis of the S.refuineus GSTs revealed the presence of
a
GST showing aminoacid similarity to an alpha-aminoadipate reductase protein in
Candida albicans, an enzyme that has a domain organization similar to those of
NRPSs
(Table 3). This GST was subsequently used to probe the S. refuineus CIL
library as
described in Example 1.
[0070] Cosmids positive by hybridization were obtained and analyzed by
sequence
determination. The presence of the original GST that was used to screen the
CIL library
was determined in the sequenced cosmid confirming that this cosmid carried the
sought-after gene cluster. After one reiteration of the described method, two
overlapping cosmids covering the entire anthramycin biosynthetic locus were
obtained.
Analysis of the genetic information derived from these two cosmids clearly
demonstrated the presence and defined the boundaries of the anthramycin
biosynthetic
locus (-33 kb). Accordingly, the genetic locus responsible for the
biosynthesis of
anthramycin was identified by the present invention.
[0071 )
Table 3
LengthProposed ~ Homology ProbabilityProposed function
of
(bp) function protein match
GST1 426 reductase gb I2.OOE-06 alpha-~aminoadipate
AAC02241.1 ' reductase in Candida
albicans
22
CA 02352451 2002-O1-17
3040-CA
Example 4: Identification of the Micromonos~oora carbonacea everninomicin
biosynthetic pathway.
[0072] Everninomicins are oligosaccharide antibiotics that are members of the
orthosomycin chemical class. This class is characterized by the presence of
orthoester
groups joining, together with glycosidic linkages, various deoxysugar
residues.
Everninomicins are produced by several variants of the microorganism
Micromonospora carbonacea (Weinstein et al., Antimicrobial Agents and
Chemotherapy - 1964, 24-32, 1964; US Patent 3,499,078).
[0073] The Agricultural Research Service collection (ARS) Micromonospora
carbonacea subsp. aurantiaca strain NRRL 2997 was used to determine the
everninomicin biosynthetic locus. All experimental procedures were as
described
(Example 1 ). The presence of several deoxysugar residues in the chemical
structure of
everninomicins is a clear indication that well-described enzymatic activities
involved in
the generation of these unusual sugar residues should participate in the
biosynthesis of
these compounds. Analysis of the M. carbonacea genome sampling library (GSL),
of a
total of 437 GST, revealed the presence of two GSTs having sequence homology
to
enzymes involved in the synthesis of deoxysugar residues from natural sugar
precursors (Table 4).
[0074] Both GSTs were used as probes for screening the Micromonospora
carbonacea cluster identification library (CIL). Overlapping cosmids positive
for both
probes were obtained suggesting a near proximity for the two GSTs in the gene
cluster.
Analysis of sequenced cosmids revealed the presence of the original GSTs
confirming
that the obtained gene cluster was indeed the targeted one. After two
reiterations of
this method, 3 overlapping casmids were obtained.
[0075] DNA sequence determination of these cosmids and analysis of the encoded
proteins by sequence similarity undoubtedly established this locus as the one
responsible for the biosynthesis of everninomicin. Additional DNA sequence
inspection
of the everninomicin locus (-68 kb) showed that a total of 7 GSTs obtained
from the
original screening of the GSL library, including the ones that were used to
probe the CIL
library, were part of the everninomicin locus. Thus, the genetic locus
responsible for
the biosynthesis of everninomicin was identified using the present invention.
23
CA 02352451 2001-07-24
3040-CA
Table 4
LengthProposed Homology ProbabilityProposed function
(bp) function of
protein match
GST1 787 sugar PIR T308736.00t-90 dNDP-glucose
dehydratase dehydratase in
Streptomyces
viridochromogenes
GST2 601 dNTP-sugar PIR T308729.00t-38 dNDP-glucose synthase
synthase in Streptomyces
viridochromogenes
[0076] Example 5: Identification of a phiC31-like prophaae in Streavtomyces
aizunensis NRRL B-11277
[0077] Streptomyces aizunensis NRRL B-11277 was obtained from the Agricultural
Research Service collection (ARS) and cultured according to standard
microbiological
techniques (Hopwood). Unless otherwise stated, all subsequent experimental
procedures were performed as described in Example 1.
[0078] A total of 462 GSTs were generated and analyzed by sequence comparison.
Three GSTs showed similarity to genes from the actinophage phiC31 (Smith et
al., The
complete genome sequence of the Streptomyces temperate phage phiC31:
evolutionary relationships to other viruses) (Table 5). Prophages are
integrated
versions of the genome of bacterial viruses and hence represent a type of gene
cluster;
that is, they include a collection of closely linked genes whose function is
to propagate
progeny virions. Oligonucleotide probes based on the three GSTs and probed a
S.
aizunensis cosmid library were designed.
[0079] Several positive cosmid clones were identified and among these two non-
overlapping clones were selected for further sequencing analysis as described
in
Example 1. Cosmid 1 consisted of a 35 kb insert that included the sequences of
both
GST 1 and GST 2. Interestingly, the GST1 and GST 2 sequences (in the context
of the
insert of cosmid 1) were flanked by sequences encoding several other phiC31-
like
genes, and most notably these include the "late" genes of phiC31. Cosmid 1
also
included a short sequence with significant similarity to the Cos sites of
phage phiC31
and contained tRNA sequences in close proximity to this Cos-site-like element.
Cosmid
24
CA 02352451 2001-07-24
3040-CA
2 consisted of an insert of at least 32 kb that included the sequences of GST
3. As
expected, the GST 3 sequences (in the context of the insert of cosmid 2) were
flanked
by sequences encoding several other phiC31-like genes, and most notably these
include the "early" genes of phiC3l. Thus, a phiC31-like prophage was
identified within
the genome of S. aizunensis using the present invention.
Table 5
Length Proposed Homology ProbabilityProposed function
of
(bp) function protein match
GST1 501 terminase largeCAA07103 2.00t-66 phiC31 gp33 ;
subunit terminase, large
subunit
GST2 501 protease CAA07105 7.00t-41 phiC31 gp35; protease
GST3 501 primase/helicaseCAA07134 I.OOt-58 phiC31 gp9a;
primase/helicase