Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
METHOD AND DISPOSABLE DEVICES FOR MICRO EXTRACTION
The present invention relates generally to extraction technology.
More specifically the invention relates, in a first aspect, to an apparatus
having
disposable elements for carrying out liquid-liquid naicro extraction and
liquid-liquid-
liquid micro extraction.
The invention further relates to methods for liquid-liquid micro extraction
and liquid-
liquid-liquid micro extraction whereby there is obtained a high enrichment of
analyte in
the acceptor solution.
Finally, the invention relates to a special disposable device for use in
liquid-liquid micro
extraction.
With regard to liquid-liquid micro extraction, this relates especially to
extraction of an
analyte from an aqueous sample solution to an organic solvent where the
analyte is
enriched in the organic solvent.
Where the aforementioned liquid-liquid-liquid micro extraction is concerned,
an analyte
is extracted from an aqueous sample solution through a water immiscible liquid
to an
aqueous acceptor solution.
Introduction
In capillary separation methods such as gas chromatography (GC), capillary
electro-
phoresis (CE), capillary electrochromatography (CEC) and micro high
performance
liquid chromatography (HPLC) injection volumes ,are in the nl to l range.
Sample pre-
treatment is necessary when these methods are used to determine analytes in
complex
matrices such as biological fluids. The principal objectives of sample pre-
treatment
involves concentration of the analytes to a concentration suitable for
detection and
removal of as many interfering compounds as possible. The use of an extraction
technique is common in the pre-treatment of most -types of samples. Sample
extraction
is the most tedious and time consuming step in the analysis of drugs present
in the
pg/ml to g/ml range of biological fluids such as blood, serum, plasma, or
urine.
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
2
No sample preparation technique is able to trap analytes in 1 - 50 l of
solvent for direct
injection into the analytical instrument. Ideally the analytes should be
trapped in an
organic solvent for injection into a GC instrument and in an aqueous solvent
for
injection into a CE instrument or a micro HPLC instrument. The most frequently
used
extraction techniques are liquid-liquid extraction (LLE) and solid-phase
extraction
(SPE). The primary goal of these extraction techniques is to extract the
analytes quanti-
tatively from the analytical matrix. When these techniques are used the
analytes are
nonnally collected in 0.2 - 10 ml of extraction solvent.
Quantitative extraction in LLE can only be achieved by using large volumes of
extraction solvent relative to the sample volume. For the extraction of a 1 ml
sample of
a biological fluid 0.5 - 10 ml of extraction solvent is used. In order to
obtain enrichment
the extract is evaporated and the analytes reconstituted in a smaller amount
of solvent.
In SPE the final extraction volume is governed by the bed volume. The bed
volume is
the amount of solvent required to fill all the internal pores and interstitial
spaces of the
particles. For a 40 micron, 60 Angstrom sorbents, bed volumes are in the order
of 120
l per 100 mg of sorbent. A 1 ml sample of a biological fluid is normally
extracted on
100 mg sorbent. The minimum elution volume required is 2 bed volumes or 0.24
ml of
solvent. The consequence is that a maximum of 4 times enrichment is obtainable
in SPE
of a 1 ml sample. In order to achieve higher enrichments the elution solvent
must be
evaporated and the analyte reconstituted in a smaller volume of solvent.
Due to practical limitations it is difficult to reconstitute a sample in
solvent volumes
smaller than 100 l. When the analyte is reconstituted in 100 l of solvent,
analyte
concentration will not exceed 10 times the analyte concentration in a 1 mi
sample.
Higher enrichments are often necessary to detect trace amounts of analytes by
the
capillary separation methods. This fact greatly reduces the applicability of
the capillary
separation methods in bioanalysis.
Micro extraction
A solution to this problem is to apply a micro extraction (ME) technique, in
which the
analytes are extracted from a large volume of sarnple solution into a small
volume of an
acceptor phase. The acceptor phase can either be a solid such as in solid-
phase micro
extraction (SPME), or an organic solvent such as in liquid-liquid micro
extraction
(LLME). A large sample volume is used in order to collect quantifiable amounts
of the
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
3
analytes and enrichment is facilitated by trapping the analytes in the small
volume of the
acceptor, phase. The extraction is carried out until equilibrium.
Figure 1 illustrates the difference between traditional LLE and LLME. A 1 ml
sample
containing I g/ml of the analyte is extracted into I ml of solvent in LLE and
into 10 l
of solvent in LLME. The analyte partition coefficient between the aqueous
matrix and
the organic solvent is 100. Figure 1 shows that 0.99 gg of the analyte is
extracted into
the organic extract by LLE and that 0.0099 g remains in the sample. Analyte
concen-
tration in the extract is 0.99 g/ml. In -LLME 0.5 g of the analyte is
extracted into the
organic acceptor phase while 0.5 gg remains in the sample. Sample
concentration in the
acceptor phase is 50 g/ml. This fact demonstrates that very high enrichments
are
obtainable in LLME without solvent evaporation and reconstitution.
SPME is a well established solvent free sample preparation technique. In SPME
the
acceptor phase is a solid polymer coated on a fibre. The polymeric acceptor
phase is
non-volatile and acts as a sorbent for partitioning of the analytes. The
volume of the
polymeric acceptor phase is less than 1 l. SPME was originally developed for
the
analysis of organic compounds in water samples and the method is particularly
useful
for trace analysis of volatile organic compounds prior to GC analysis. When
SPME is
applied to bioanalytical samples such as plasma or urine several difficulties
are
observed. In drug analysis enrichment from a biological matrix is greatly
reduced as
compared to enrichment from a pure water sample. This is due to reduced
capacity of
the acceptor phase. In addition the polymeric acceptor phase is easily
contaminated
when SPME is applied to bioanalysis and cross contamination between samples
may
easily occur. These facts greatly limit the applicability of SPME in
bioanalysis. A
solution to this problem is to apply a LLME technique.
Basic principles of LLME
The basic theory of SPME is well documented and is applicable to LLME. If we
define
the acceptor/sample partition coefficient as K. , the volume of acceptor
solution as Va,
the volume of sample solution as VS and the initial sample concentration as
Co, the
amount of analyte trapped in the acceptor phase, n, is shown in equation 1:
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
4
Kw Va VS Ca
1. n =
Kas Va-l- Vs
The concentration of analyte trapped in the acceptor phase, C,,, is:
2. Ca = n/Va
When Kas Va VS the amount of analyte collected in the acceptor phase is:
3. n=K,,, VaCoandCa=KaSCo=
Table I shows the equilibrium concentration of the; analyte trapped in 0.001
ml - 1 ml of
acceptor phase after extraction of a I ml sample solution containing 1 g/ml
of the
analyte. The partition coefficients are 10,100 ,1000 and Go.
Table 1. Concentration of analyte trapped in variable volumes of acceptor
solution
with partition coefficients ranging from 10 - oo.
Volume of acceptor Concentration of analyte in the acceptor phase ( g/ml)
phase (ml) K=10 K= 100 K= 1000 00
0.001 9.9 91 500 1000
0.01 9 50 91 100
0.025 8 28.6 38.5 40
0.05 6.7 16.7 19.6 20
0.1 5 9.1 9.9 10
0.5 1.7 1.9 2.0 2.0
1 0.91 0.99 1 1
The concentrations shown in Table 1 demonstrate that as long as the partition
coefficients are high, the enrichments obtained by LLME into 0.001 - 0.05m1 of
acceptor phase are superior to the enrichments obtained by traditional
extraction
methods.
CA 02353130 2004-09-10
There are, however, practical problems related to extraction of biological
fluids with
small volumes of organic solvents. In biological fluids an emulsion is readily
formed
during liquid-liquid extraction and small volumes of solvent are easily
emulsified. Even
after centrifugation small solvent volumes are difficult to collect.
Extraction of bio-
logical fluids with solvent volumes less than 50 l is therefore not performed
in
traditional analytical procedures.
One possible way to solve these problems is to use so-called disposable
sponges or to
use the devices described in the attached Figures 3 and 4.
Disposable extraction sponges
The problems mentioned above can be solved by disposable extraction sponges.
Disposable extraction sponges are used to immobilise 10 - 50 l of extraction
solvent.
LLME with extraction sponges is particularly suited for sample preparation of
biological fluids prior to GC, analysis. Solvent immobilised into extraction
sponges
eliminates the handling problems encountered with small solvent volumes since
immobilised solvents are not emulsified and are easily collected after
extraction.
Materials used in the manufacture of extraction sponges should be solvent
resistant,
porous and compressible. In addition the materials should be sufficiently
hydrophobic to
immobilise water immiscible solvents. The pore size may range from a few
micrometers
up to nzillimeters. Expanded polymers and polymeric foams are particularly
suited.
Examples of solvent resistant polymers are Teflon*, Tefzel*, Halar*,
polyethylene and
polypropylene. The size of the polymeric material is cut to fit immobilisation
of a
predetermined volume of solvent.
LLME with disposable extraction sponges
The extraction sponges are filled into a container with the solvent to be
immobilised.
The sponges are compressed to remove air trapped in the pores and are
thereafter soaked
in the solvent. The sponges are then ready for extraction.
The sample solutions are filled into extraction vials. Typical volumes of
sample solution
are 0.5 - 5 ml. Quantitative analysis is always performed by adding an
internal standard
to the sample solution. The intennal standard is added to the sample prior to
the
extraction and follows the analyte through all the analytical steps. The
internal standard
compensates for all fluctuations in the procedure. The chemical nature of the
sample is
*TRADEMARK
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
6
altered prior to extraction to facilitate analyte extraction into the organic
solvent. This
involves optimisation of pH and addition of salt.
One solvent sponge with immobilised solvent is removed from the container and
added
to the extraction vial. Extraction is performed by stirring. Any kind of
stirring, for
example, a magnetic stir bar, can be used. The extraction is continued until
equilibrium
(10 - 30 min). The solvent sponge is then removed from the sample vial.
Immobilised
solvent with the enriched analyte is liberated by compression. Compression can
be
facilitated in any device suitable for squeezing. For example the sponge can
be
compressed in a disposable medical syringe equipped with a needle and the
liberated
solvent is filled into micro sampling vials made to fit into a GC
autoinjector.
Sponges able to immobilise 25 l of solvent are suitable in many applications.
As
shown in Table I enrichments of 30 are obtained for analytes having a
partition
coefficient of 100. A solvent volume of 25 l is suji'ficiently large to allow
easy
handling. This solvent volume is also large enough to avoid overloading and
reduced
enrichment during extraction. One sponge is used for each sample and used
sponges can
be stored safely in a container prior to destruction. Compared to traditional
methods for
sample preparation, LLME with solvent sponges greatly reduces solvent
consumption
and hazards to workers and the environment.
The present invention aims to solve the problem introduced above by utilising
so-called
micro back extraction, referred to above and hereinafter as liquid-liquid-
liquid micro
extraction (LLLME), to obtain a sufficiently high concentration of the
material to be
analysed in the acceptor solution.
The principles of LLLME will be explained in more detail below.
Liquid-liquid-liquid micro extraction (LLLME)
Separation techniques used in capillary electrophoresis such as capillary zone
electrophoresis, micellar electrokinetic chromatography and capillary
electrochromatography favour injection of low ionic strength aqueous samples.
Due to
the injection of nl volumes of samples, high enrichments are required in
bioanalysis of
drugs present in trace amounts in biological fluids.
Most drugs are ionic. Ionic organic substances can be enriched by LLLME. The
principle of LLLME for isolation of ionic organic inolecules is illustrated in
Figure 2.
CA 02353130 2004-09-10
7
Clean-up and concentration of analytes are based on partitioning of the
analytes from a
large volume of the aqueous sample matrix through a membrane and into a small
volume of an aqueous acceptor phase. The membrane acts as a clean-up barrier
between
two aqueous phases. Both basic and acid compounds can be enriched with LLLME.
The
pH of the matrix is adjusted so that the analytes are uncharged. This permits
them to
pass through the membrane into the aqueous acceptor solution on the other
side. The pH
of the acceptor solution is adjusted to a pH where the analytes are ionised,
thus
preventing them from re-entering the membrane. Only small uncharged molecules
can
pass through the membrane and only molecules which are soluble in the membrane
and
in the acceptor solution can be enriched. Water soluble neutral substances
remain in the
matrix. Neutral hydrophobic substances partition into the membrane and not
into the
acceptor phase. Substances with the opposite charge as the analytes remain in
the
matrix. LLLME is thus a powerful clean-up technique.
The driving force for the extraction is dependant on the product of the
analyte partition
coefficients between the membrane and the sample solution and between the
acceptor
phase and the membrane which is equivalent to the analyte partition
coefficient between
the acceptor phase and the sample matrix. Compounds having a large partition
coefficient between the two aqueous phases 'will be enriched. This partition
coefficient
will be large for many drugs. LLLME therefore has the potential to act as both
a
powerful enrichment and clean-up technique for many ionic drugs.
If we define the acceptor/membrane partition coefficient as Kal, the
membrane/sample
partition coefficient as Kls, the acceptor/sample partition as Kas, the
acceptor volume as
Va, the membrane volume as VI, the sample volume as Vs, and the initial sample
concentration as Co, the amount of analyte extracted by LLLME, n, is:
Kai K1s Va Vs Co Kas Va Vs Co
4. n - -
Kal Kis Va + Kls Vl + Vs Kas Va + Kls Vl + Vs
In LLLME the membrane volume should be as small as possible. Then Kis Vi will
be
negligible and equation 4 can be reduced to :
Kas Va Vs Co
5. n =
KasVa+Vs
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
8
Equation 5 can be used to estimate analyte enrichrxient from a 1 ml sample
solution
containing I g/ml of the analyte as a function of acceptor phase volume and
the
partition coefficient. The results obtained are equivalent to the results
shown in Table I
showing that enrichments from a 1 ml sample into 0.001-0.05 ml of acceptor
solution
are superior to the enrichments obtained by traditional extraction methods.
Many ionic drugs have partition coefficients larger than 100 between two
aqueous
phases: one having a pH where the drugs are charged and the other a pH where
the
drugs are uncharged. When an analyte with a partition coefficient of 100 is
trapped in 10
l acceptor solution from 1 ml sample with a concentration of 1 g/ml, analyte
concen-
tration in the acceptor phase is 50 g/ml. This fact demonstrates that LLLME
is able to
provide enrichments not obtainable by any other extraction method. LLLME is
therefore particularly useful as an extraction technique for modern capillary
separation
methods such as CE.
The chemical nature of the membrane is important in obtaining short analysis
times.
Extractions should be continued until equilibrium between the three phases is
established. If the membrane/sample partition coefficient is low, equilibrium
times will
be long and will approach infinity for analytes which are very poorly soluble
in the
membrane. The solvent forming the membrane should therefore be a good solvent
for
the target analyte. The chemical nature of the membrane is also important for
tuning of
the selectivity.
The present invention aims to improve the known art and to utilise the above
suggested
possibilities and therefore relates, in a first aspect, to an apparatus for
carrying out
liquid-liquid micro extraction or liquid-liquid-liquid micro extraction with
high
enrichment, and the apparatus is characterised in that it comprises
a) a container for a sample solution having volume Vs with dissolved
substance,
analyte, to be analysed,
b) a second container arranged in the first container, preferably a disposable
container, having permeable membrane wal.ls, for an acceptor solution, having
volume Va, wherein
1) Vs:Va _ 50 and
2) about 1 l _ Va <_ 50 l,
c) stirring means, preferably a magnetic bar.
CA 02353130 2001-05-31
WO 00/33050 PCT/N099100359
9
In one embodiment the container for the acceptor solution is a microporous
hollow
fibre, optionally of an active polymer.
As mentioned in the introduction, the invention also relates to methods for
extraction
and thereby relates, in a first extraction aspect, to a method for liquid-
liquid micro
extraction with high enrichment by using the above described apparatus, and
this
method is characterised in that
a) the container for acceptor solution is lowered into an acceptor solution so
that
the membrane wall is impregnated with, and the container is filled with, a
defined volume of the acceptor solution,
b) the container filled under a) is transferred to the container having a
defined
volume of the sample solution with the analyte that is sought,
c) the sample solution with analyte is stirred until extraction equilibrium is
established for the analyte in the two solutions, and
d) the acceptor solution containing enriched analyte is removed from its
container
for analysis of the analyte.
In a second extraction aspect, the invention relates to a method for liquid-
liquid-liquid
micro extraction with high enrichment by the use of the apparatus according to
claim 1,
and this method is characterised in that
a) the walls of the container for the acceptor solution are impregnated with,
for
immobilisation of, a liquid that is immiscible with the sample solution and
the
acceptor solution,
b) the container for acceptor solution is filled with a defined volume thereof
and
c) is lowered into the container having a defined volume of the sample
solution
with the analyte that is sought,
d) the sample solution with analyte is stirred uintil extraction equilibrium
is
established between
i) the sample solution and the immobilised liquid, and
ii) the immobilised liquid and the acceptor solution, and
e) the acceptor solution with enriched analyte is removed from its container
for
analysis of the analyte.
The latter method is particularly suited for enrichment of acidic or basic
analytes. For
example, basic analytes can be enriched from basic, aqueous, biological
samples by
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
utilising an acceptor liquid in the formof an acidified, aqueous liquid and an
organic
liquid immobilised in the membrane that is immiscible with the aqueous
liquids.
Here, as mentioned previously, it may be advantageous to use a microporous
hollow
fibre, but it is also possible to use an active polymer.
In a final aspect, as mentioned above, the invention relates to a disposable
device for use
in liquid-liquid micro extraction, which is characterised in that it has the
form of a
sponge-like body having a defined pore volume foir absorption of an
immobilised
acceptor solution for an analyte from a volume of a sample solution.
The invention will now be illustrated in more detail with reference to the
accompanying
drawings where:
- Figure 1 shows a comparison between liquid-liquid extraction and liquid-
liquid
micro extraction,
- Figure 2 shows the principle for liquid-liquid-liquid micro extraction,
- Figure 3 shows a possible device for utilisation in LLLME, or LLME,
- Figure 4 shows another possible device for LLLME, or LLME,
- Figure 5 shows chromatograms obtained in connection with Example 1, and
- Figure 6 shows electropherograms obtained in connection with Example 2.
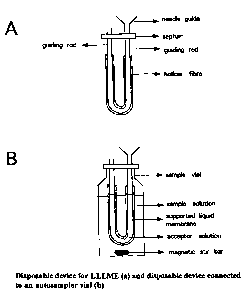
Disposable devices for LLLME
Devices for LLLME should accomplish extraction from a large sample volume
through
a negligible volume of a membrane into a small volume of an aqueous acceptor
solution. The membrane should be a thin film with. a large surface area. The
membrane
can either be a solid (liquid-solid-liquid micro extraction, LSLME) or a
liquid (liquid-
liquid-liquid micro extraction, LLLME).
In LLLME the solvent forming the liquid membrane should be immobilised. Any
material able to immobilise a water immiscible solvent can be used.
Hydrophobic
hollow fibres are particularly useful. The fibres can be made of a polymeric
materials
such as Teflon, polypropylene or polyethylene. The inner diameter of the
hollow fibre is
in the range of 0.05 -1 mm, the wall thickness is typically in the range of
0.01 - 0.3
mm and the average pore size is in the range of 0.01 - 10 m. The length of
the fibre is
typically 2 - 10 cm to allow fixed volumes of acceptor solution in the range
of 5- 50 l
to be filled into the hollow fibre.
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
11.
In LSLME the membrane may be a polymeric film made of a materiale able to sorb
uncharged compounds. Examples of such materials are polydimethylsiloxane
(PDMS),
polyacrylate or polystyrene divinyl benzene. These; materials are also used as
well
known sorbents in SPME. The membrane thickness is preferably 0.5 - 50 m. The
membrane may be supported by a rigid framework and formed into tubes with the
same
dimensions as the hollow fibres described above.
Devices for LLLME should be disposable. Impurities from one sample may be
trapped
in the membrane and these impurities may contaminate other samples. One sample
pre-
treatment device should therefore be used for each sample. Disposable devices
should
also be connected to commercially available sample preparation vials.
Disposable devices for LLLME are shown in Figures 3a and 4a. Figures 3b and 4b
show
the devices connected to sample vials filled with sample solution. The guiding
rod can
be a stainless steel rod or a rod made of a polymeric material. The top of the
guiding rod
can be connected to a needle guide to allow filling of the hollow fibre with
acceptor
solution from a syringe. The hollow fibre in Fig. 3b is connected at both ends
to guiding
rods.
Sample preparation with disposable LLLME devices
In LSLME the membrane can be used as it is. In LLLME the liquid membrane is
formed by dipping the hollow fibre into the organic solvent for 5-30 sec. to
allow the
solvent to penetrate into the pores of the fibre. Acceptor solution is then
filled into the
fibre by a syringe. Normally, fibres with an inner tube volume of 10 1 are
preferred,
since 10 l of acceptor solution gives high analyte enrichments and 10 l
volumes can
be handled with commercially available syringes. 'Che acceptor solution has a
pH where
the target analytes are charged. Extraction is performed by connecting the
LLLME
device to the sample vial. The sample filled into the sample vial is buffered
to a pH
where the analytes are neutral. Typical sample volumes are 0.5 - 5 ml of a
biological
fluid. An internal standard is always added to the sample solution before
extraction to
compensate for fluctuations in the procedure. Extraction is performed by
stirring, for
example with a magnetic stir bar placed in the sample vial. Extraction is
continued until
equilibrium between the three phases is established. When equilibrium is
reached (15-
60 min) the acceptor solution is collected with a syringe and filled into
autosampler
vials for automated injection into the analytical instrument.
CA 02353130 2003-05-01
WO 00/33050 PCT/N099/00359
12
The invention will now be illustrated in more detail with the aid of the
following
examples.
The device shown in Figure 4a was used to demonstate the potential of liquid-
liquid
micro extraction and liquid-liquid-liquid micro extr*ction. The hollow fibre
used was a
polypropylene fibre with pore size 0.2 m (Accurel PP Q _31/2) and was
purchased from
Akzo Nobel (Wuppertal, Germany). The inner diameter was 600 }im, the vvall
thickness
was 200 m and the length was 5.5 cm.
Example 1: Liquid - liquid micro extraction (LLME)
LLME is demonstrated by the extraction of 5 nmol/mi sample solutions of
diazepam
(D) and prazepam (P) prepared in 1.0 M acetate buffer pH 5.5, in urine and in
human
plasma. A standard solution in octanol (5 nmol/ml) was prepared as a reference
solution
for direct injection into the gas chromatograph. The pH of'the standard
solution in urine
was adjusted to pH 5.5 before extraction. To an aliquote of plasma (1080 l)
was added
120 l methanol to reduce the protein binding of the benzodiazepines prior to
extraction
and the mixture was agitated for 1 min. LLME was acconiplished by placing 1.2
ml of
*
the sample solutions in 2 ml autosampler vials (Chromacol, Trumball, CT.,
USA). The
hollow fibre was filled with 10 l of 1-octanol. After 1 miii, tc- ensure that
the solvent
would completely penetrate the pores, the hollow fibre was inimersed into the
autosampler vials. The sample solution was stirred wit.h ainagnetic stir bar
during
extraction. After 30 min 1 l of octanol was withdrawn from the hollow fibre
with a GC
syringe and injected into the gas chromatograph. The gas chromatographic
separation
was achieved on a poly-(dimethylsiloxane) column (30 x 0.25 rrun i.D., 0.25 mm
film
thickness) and the compounds were detected with a nitrogen-phosphorous
detector
(NPD). Helium (1 ml/min) was used a carrier gas. The chromatographic
separation was
achieved by temperature programming. The temperature was held at 180 C for 1
min
and increased at 20 C /min to 300 C. Figure 5 shows chromatograms of the
reference
solution in octanol (5 mnol/ml) and chromatograms of th-e sample solutions (5
nmol/ml)
of diazepam and prazepam in acetate buffer, in urine and plasma after
enrichment by
LLME. The chromatograms demonstrate preconcentration by a factor of 100 and
70,
respectively, for diazepam and prazepam from the acetate buffer, urine and the
plasma
sample.
Example 2: Liquid-liquid-liquid micro extraction (LLLME)
LLLME is performed with 1-octanol as the immobilised liquid. The hollow fibre
was
immersed for 5 sec in 1-octanol which is sufficient for 1-octanoI to penetrate
and fill the
*TRADEMARK
CA 02353130 2001-05-31
WO 00/33050 PCT/N099/00359
13
pores of the fibre. 10 gl of 0.1 M HCI was used as acceptor solution and was
filled into
the impregated fibre with a syringe. A standard solution of 4 g/ml of
diphenhydramine
in 0.1 M HCl was prepared as a reference for direct injection into the CE
instrument. In
addition, sample solutions of diphenydramine (4 g/ml) were prepared in 0.1 M
NaOH,
in urine and plasma. Before extraction the pH in the urine and plasma sample
solutions
were adjusted to a pH 12-13 with NaOH. 1.5 ml of the sample solutions were
placed in
2 ml autosampler vials. LLLME was accomplished by stirring with a magnetic
stir bar
for 30 min. The acceptor solution was removed after extraction and analysed by
CE.
Separations were perfomed inside a 10 cm effective length (52 cm total length)
x 50 m
internal diameter fused silica capillary. A 20 mM sodium acetate buffer
adjusted to pH
4.5 with acetic acid was utilised as separation buffer. Sample introduction
was
accomplished by hydrodynamic injection with a pressure of 0.5 psi for 5 sec.
Separations were performed at 25 kV, while detection was accomplished at 215
nm.
Electropherograms are shown in Figure 6. The electropherograms show that
diphenhydramine (DH) was preconcentrated by a factor of 90 from the sample
solution
prepared in 0.1 M NaOH and in urine. A preconcentration of 50 was achieved
from
plasma. The lower enrichment from plasma is due to protein binding of the
analyte. For
both of the biological samples, excellent sample clean-up was observed in
addition to
analyte enrichment. In spite of the high sample complexity, almost no matrix
components were observed in the electropherograms obtained by capillary zone
electrophoresis.