Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02360826 2001-10-31
-1-
CONJUGATED POLYCARBAZOLE DERIVATIVES AND PROCESS FOR
THE PREPARATION THEREOF
The present invention pertains to improvements in the field of
conjugated polymers. More particularly, the invention relates to conjugated
polycarbazole derivatives and to a process for the preparation thereof.
A conjugated polymer is a polymer which possesses a delocalized pi-
electron system along its backbone as described, for example, by D.J. Sandman
in
"Trends in Polymer Science", Vol. 2, p. 44 (1994). Conjugated polymers are
considered as a very important class of electroactive and photoactive
materials by
both academic and industrial laboratories. The synthesis over the last twenty
years of
highly pure polyacetylene, polythiophenes, polyphenylenes, polyfluorenes,
ladder
polymers and other conjugated polymers optimized physical properties has led
to a
significant improvement in the performance of these polymeric materials and to
a
better understanding of their structure-property relationships. However, up to
now,
only poorly conjugated poly(N-alkyl-3,6-carbazole) derivatives are available
so that
these are not optimized for the development of light-emitting diodes,
electrochromic
windows, electrochemical sensors, photovoltaic cells, photoconductors,
photorefractive materials, transistors, etc.
It is therefore an object of the present invention to provide conjugated
polycarbazole derivatives having improved optical and electrochemical
properties.
According to one aspect of the invention, there is provided a conjugated
poly(N-alkyl-2,7-carbazole) of formula (I):
/ /
~ O)
n
N
R
CA 02360826 2005-08-O1
-2-
wherein R represents a linear or branched alkyl group having 1 to 22 carbon
atoms, and n is an integer of about 3 to about 100, with the proviso that when
R
is a decyl radical, n cannot represents an integer from 2 to 6.
The present invention also provides, in another aspect thereof, a
process for preparing a conjugated poly(N-alkyl-2,7-carbazole) of the formula
(I) defined above, which comprises treating a N-alkyl-2,7-difunctionnalized
carbazole of formula (II):
x ~ / \ / x O>
I
R
wherein R is as defined above and X represents a trifluoromethanesulfonyl
1 o group or a halogen atom selected from the group consisting of bromine,
chlorine and iodine atoms, with triphenylphosphine and 2,2'-bipyridine in the
presence of zinc and nickel chloride, to cause polymerization of the compound
of formula (II).
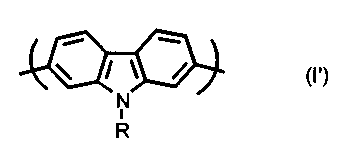
According to a further aspect of the invention, there is provided a
conjugated polymer comprising alternated units of formula (I'):
/ \ /
N
R
wherein R is as defined above.
The conjugated polycarbazole derivatives according to the
invention, comprising units of formula (I'), have interesting optical and
2 o electrochemical properties which render them suitable for use in the
manufacture of light-emitting diodes, electrochromic windows, electrochemical
sensors, photovoltaic cells, photoconductors, photorefractive materials and
the
like.
CA 02360826 2001-10-31
-3-
The following non-limiting examples illustrate the invention, reference
being made to the accompanying drawings, in which:
Figure 1 is the absorption (Abs.) and emission (PL) spectra of poly(N-
octyl-2,7-carbazole) in choroform and in the solid state; and
Figure 2 is the cyclic voltammogram of poly(N-octyl-2,7-carbazole)
cast on a platinum electrode, in acetonitrile containing O.1M N Bu4NBF4, at a
scan
rate of 10 mV/s.
EXAMPLE 1: Preparation of poly(N-octyl-2,7-carbazole)
Following the procedure developed by Smith and Brown and reported
in J. Am. Chem. Soc., Vol. 73, p. 2438 (1951), 4,4'-dinitro-2-biphenylamine
(Aldrich
Co.) was treated with NaNOz and NaN3 to give the corresponding azide via the
transformation of the amino group into a diazonium salt. A ring closure
reaction,
assured by a nitrene intermediate, was carried out to give 2,7-
dinitrocarbazole in a
66 % yield. This compound was then reduced using SnCl2 in a mixture of acetic
acid/HCl (5 :1) to give 2,7-diaminocarbazole in a 78 % yield. Then, the amino
groups
of the resulting product were transformed to iodine atoms; the reaction was
carried
out in a 3M HCI solution using NaN02 and KI. N-octyl-2,7-diiodocarbazole was
prepared in a 93% yield from 2,7-diiodocarbazole upon reaction with KZC03 and
1-
bromooctane in anhydrous DMF at 80 °C. All monomers were characterized
by NMR
and mass spectrometry. Homopolymerization was achieved by a Yamamoto reaction
described in Macromolecules, Vol. 25, p. 1214 (1992), using N-octyl-2,7-
diiodocarbazole as the starting material and triphenylphosphine, 2,2'-
bipyridine, and
zinc and NiClz as catalysts. Poly(N-octyl-2,7-carbazole) was obtained in a 78
% yield.
The synthetic scheme is summarized as follows:
CA 02360826 2001-10-31
-4-
1 ) NaN02, H2S04
02N ~ ~ ~ ~ N02 AcOH OZN ~ ~ ~ ~ N02
2) NaN3
NH2 Y=72 % N3
(1 ) (2)
SnCl2
0, -N2 ' ~ AcOH/HCl (5:1 ) '
Y=66 % 02N \ / \ / N02 L-12[V \ / \ / NH2
H (3) Y= 78
(4)
1 ) HCI ~aq.~, NaN02 . . 1 ) K2C03, DMF
I \ / \ / I 80 C ~ I \ / \ / I
N
2) KI ~aq.~, 24 h, r.t. H 2) C8H»Br
(5) Global yield for step (6)
4 and 5 = 38%
PPh3, Zn, NiCl2
2,2'-bipyridine
N DMAc, 80 °C, 3 d N n
Y=78%
C$H» CBH~~
It is also possible to use, instead of N-alkyl-2,7-diiodocarbazole, N-
alkyl-2,7-dichlorocarbazole which can be obtained from a different synthetic
pathway, according to the following scheme:
CA 02360826 2001-10-31
- 5 -
B(OH)2 CI Pd(PPh3)a
Benzene/NaZC03 aq.
CI / \ / \ CI
reflux, 18 h
CI Br Y = 93% N02
P(OEt)3
reflux, 24 h
Y=60%
K2CO3 .
CBHI~Br
CI \ ~ ~ / CI - CI \ ~ ~CI
DMF, 80°C
CgH~7 (9) 24 h H
Y = 86 % K2COg,TBAH
PPh3, Zn, NiCl2 C18H3~Br
2,2'-bipyridine Acetone, reflux, 24 h
DMAc, 100°C, 3 d Y = 86
Y = 75 % Ni(COD)2
COD, 2,2'-bipyridine _ _
DMF, 100°C, 2 d
/ CI \ ~ ~ / CI
Y=75 % [V
C~sH37 (1
R = C8H1~ or ClgH3~
This scheme involves a coupling between 4-chlorophenylboronic acid
(Aldrich Co.) and 1-bromo-4-chloro-nitrobenzene (Aldrich Co.), followed by a
ring
closure using P(OEt)3 and an alkylation of the nitrogen atom in DMF and KZC03
using octylbromide or 1-bromooctadecane.
EXAMPLE 2 : Preparation of poly(N-octyl-2,7-carbazole-alt-9,9-dioctyl-2,7-
fluorene) and poly[N-(2-ethylhexyl)-2,7-carbazole-alt-5,5'-(2,2'-
bithiophene)].
CA 02360826 2005-08-O1
- 6 -
Alternated copolymers were prepared from Suzuki couplings
(described by Ranger, M. et al. in Macromolecules, Vol. 30, p. 768 (1997))
between di-boronic functionalized aromatic units and N-alkyl-2,7-
diiodocarbazole derivatives. Poly(N-octyl-2,7-carbazole-alt-9,9-dioctyl-2,7-
fluorene) was prepared from a reaction between 2,7-bis(4,4,5,5-tetramethyl-
1,3,2-dioxaborolan-2y1)-9,9-dioctylfluorene and N-octyl-2,7-diiodocarbazole
using (PPh3)4Pd(0) as catalyst in a mixture of THF and 2 M KZC03 aqueous
solution. Moreover, Stille couplings (described by Yu, L. et al. in Acc. Chem.
Res., Vol. 29, p. 13 (1996)) between distannyl aromatic derivatives and N-
1 o alkyl-2,7-diiodocarbazole derivatives are possible. As an exemple, poly[N-
(2-
ethylhexyl)-2,7-carbazole-alt-5,5'-(2,2'-bithiophene)] was obtained with a
good
yield from N-(2-ethylhexyl)-2,7-diiodocarbazole and of 5,5'
bis(trimethylstannyl)-2,2'-bithiophene, in presence of Cl2(PPh3)2Pd(0) in THF.
These polymerization reactions are summarized in the following
scheme:
O ~ . O
I \ / \ / I + ~g \ / \ / g
N '
O O
I CaH~~ CsH~~
C8H17
Pd(PPh3)4
K2COs i ~ ~ / ~ /
THF, 3 d ~ N~ ~ n
Reflux I C8H~~ C8H~~
Y = 75 % C$H 17
I~ \ ~ I + Me3Sn S /S\ SnMe3
N ~ /
I
CgH 17
Pd C12(PPh 3)2 ~ ~ / \
THF \ / \ / ~S / S
Reflux, 3 d N
Y=52%
CaH~~
CA 02360826 2001-10-31
-
EXAMPLE 3 : Preparation of poly (N-octyl-2,7-carbazole)
In order to obtain a reactive monomer in better global yield than
obtained with 2,7-diiodocarbazole derivatives, N octyl-2,7-
bis(trifluoromethanesulfonyl)carbazole was synthesized as follows:
r (0H)2
Pd(PPhg)4
N02 ~ Benzene, KZCOg 2M - -
+ ~ H3C ~ ~ ~ ~ OCH3
/ / Reflux, 2h
OCH3 OCH3 y = 90 % 02N (11)
P(OEt)3
Reflux, Sh
Y=70%
H3C0 \ ~ ~ / OCH3
N (12)
H
NaOH,TBAH
1-bromooctane
Acetone, reflux, 24 h
Y=91
N Y = 68 % (13)
(14)
CBH~~ CBH~~
TfZO, DMAP
Pyridine
0°C - r.t., 24 h
Y=76%
Pinacolborane* -'
PdCl2(dPP~ 0.
Tf0 \ ~ ~ / OTf NEt3, CHZCIz ~p N
(15) EO°C, 24 h
Y=59% C8H17 (16)
C8Ht7
(15), Pd(PPh3)4>
PPhg, Zn, NiCl2 KzC03 (aq.)/THF
2,2'-bipyiridine reflux 3 d
DMF, 80°C, 3 d Y = 86 %
Y=83%
\ ~ ~ / \
N n ~ n
C8H17
CaH~7
~ BBrg, CHzCl2
HO \ ~ ~ / OH p°C _ r.t., 24h H3C0 \ / \ / OCHg
N
CA 02360826 2001-10-31
- g -
The biphenyl unit (11) was obtained using a Suzuki coupling between
4-methoxyphenylboronic acid and 4-bromo-3-nitroanisole in standard conditions.
Then, a Cadogan ring closure reaction was carried out in hot triethylphosphite
to give
2,7-dimethoxycarbazole (12) as reported in Macromolecules, Vol. 18, p. 1388
(1985).
This compound was alkylated using finely powdered NaOH, phase transfer agent
and
primary alkyl bromide in anhydrous acetone. From compound (13), a standard
deprotection reaction using BBr3 in methylene chloride was achieved to give
2,7-
dihydroxycarbazole (14), in relatively good yields. Finally, monomer (14) was
treated
with DMAP and trifluoromethanesulfonic anhydride in cold pyridine to give
monomers (15) that can undergo Yamamoto, Stifle or Suzuki polycondensation
reaction. When necessary, boronic esters can be introduced at the 2,7-
positions with
pinacolborane and Pd(II) using similar procedure published in J. Org. Chem.,
Vol. 65,
p. 164 (2000) to obtain compound (16) to perform Suzuki polycondensation
reaction.
Materials. All chemicals were purchased from Aldrich Co. and were
used without further purification.
4,4'-dinitro-2-azidobiphenyl (2): To a solution of 10.0 g (38.6 mmol)
of 4,4'-dinitro-2-biphenylamine in a mixture of 200 mL of acetic acid and 40
mL of
sulfuric acid at 0°C was added dropwise 5.32 g (77.2 mmol, Aldrich Co.)
of sodium
nitrite. The mixture was stirred at 5-10°C for 2h after 5.00 g of urea
(to destroy the
excess nitrous acid), S00 mL of ice-water and 5 g of activated carbon was
added. The
cold suspension was stirred again for 20 min and filtrated rapidly through a
biichner
funnel into a flask immersed in an ice bath. A solution of 5.07 g (77.2 mmol,
Aldrich
Co.) of sodium azide in 100 mL of water was added dropwise to the yellow clear
filtrate. The resulting solution was stirred at 0°C for 1 h and at room
temperature for
24 h. The mixture was quenched with 500 mL of a solution of NaHC03 in water
and
extracted three times with ethyl acetate. The organic layer was dried over
magnesium
sulfate and the solvent was removed under vacuum. Recristallization in ethanol
afforded 7.37 g of the title product as a yellow solid. M.P. 171-172°C
(Yield: 72%).
CA 02360826 2001-10-31
-9-
1H NMR (300 MHz, CDC13, ppm): 8.37 (d, 2H, J= 8.8 Hz); 8.20 (d, 1 H, J= 2.2
Hz);
8.16 (dd, 1 H, J = 10.3 and 2.2 Hz); 7.89 (dd, 2H, J = 8.8 and 2.8Hz); 7.79
(d, 1 H, J =
8.1 Hz).
'3C NMR (75 MHz, CDC13, ppm): 143.84; 140.35; 137.98; 133.20; 131.81; 124.29;
120.71; 115.44.
HRMS: Calculated for C12H~N5O4: 285.0498 Found: 285.0505
2,7-dinitrocarbazole (3): To 600 mL of boiling kerosene (first washed
with concentrated sulfuric acid) was added very slowly 6.00 g (21.0 mmol) of
compound 2. The solution was maintained to reflux for 1 h. After cooling, the
solution was kept at 4°C for 24 h. The precipitate was filtered through
a biichner
funnel and the solid was washed with petroleum ether. Very pure material was
obtained by recristallization in ethanol to afforded 3.38 g of the title
product. M.P.
>300°C. (Yield: 66 %).
1H NMR (300 MHz, Acetone-d6, ppm): 11.41 (s, 1H); 8.55 (d, 2H, J= 2.2 Hz);
8.49
(d, 2H, J= 8.8 Hz); 8.15 (dd, 2H, J= 8.8 and 2.2 Hz).
'3C NMR (75 MHz, Acetone-d6, ppm): 141.07; 126.38; 122.09; 114.76.
HRMS: Calculated for C12H~N3O4: 257.0436 Found: 257.0431
2,7-diaminocarbazole (4). To a solution of compound 3 (6.00g, 23.3
mmol) in a mixture of acetic acid (200 mL) and hydrochloric acid (35 mL) was
added
44.3 g (0.24 mol, Aldrich Co.) of tin(II) chloride. The mixture was refluxed
for 24 h
under argon. After cooling, the precipitate was separated from the solvent by
filtration
and washed several times with cold acetic acid. The resulting diammonium salt
was
dissolved in water followed by addition of an aqueous solution of sodium
hydroxide
until the pH was around 10. The precipitate was collect by filtration and
dried under
vacuum. Recristallization in ethanol afforded 3.60 g of the title product as a
shiny
gray solid. M.P. 248°C (dec.). (Yield: 78 %).
1H NMR (300 MHz, Acetone-d6, ppm): 9.45(s, 1H); 7.53(d, 2H, J= 8.1 Hz) ; 6.62
(d,
2H, J= 1.5 Hz); 6.47 (dd, 2H, J= 17.0 and 2.2 Hz); 4.45 (s, 4H).
isC NMR (75 MHz, Acetone-d6, ppm) : 146.53; 142.53; 119.72; 116.58; 108.70;
96.42.
CA 02360826 2001-10-31
- 10-
HRMS: Calculated for Cl2HnN3: 197.0953 Found: 197.0948
2,7-diiodocarbazole (5). To a solution of 1.50 g (7.56 mmol) of
compound 4 in 100 mL of 3 M HCl solution at 0°C was added very slowly
1.10 g
S (15.9 mmol) of sodium nitrite in 5 mL of water. The mixture was stirred at
0°C for 2 h
and then added to 100 mL of a solution of potassium iodide in distillated
water. The
stirring was kept for 24 h at room temperature. The precipitate was collect by
filtration and washed with aqueous solution of NaHC03. The solid was dried
under
vacuum for 24 h and use directly in the next reaction without further
purification.
However, the crude material could have been purified by column chromatography
(silica gel, 10 % ethyl acetate in hexanes as eluent) but the reaction yield
would be
greatly affected, probably due to the degradation of the product on silica
gel.
1H NMR (300 MHz, Acetone-d6, ppm): 10.54(s, 1H); 7.93 (m, 4H) ; 7.53 (dd, 2H,
J=
7.4 and 1.5 Hz).
1 S 13C NMR (75 MHz, Acetone-d6, ppm) : 141.77; 128.88; 122.69; 120.79;
120.74;
90.89.
HRMS: Calculated for C~2H~IzN: 418.8668 Found: 418.8675
N octyl-2,7-diiodocarbazole (6). To a solution of compound 5 (3.00 g)
in 30 mL of anhydrous DMF was added 660.mg (4.78 mmol, Aldrich Co.) of
anhydrous K2C03. The solution was stirred at 80°C for 2 h under argon
after 0.93 g
(4.82 mmol) of bromooctane was added. The mixture was stirred at 80°C
for 24 h
and then quenched with 30 mL of water. The aqueous layer was extracted three
times
with 50 mL of diethyl ether. The organic layer was dried over magnesium
sulfate and
the solvent was removed under vacuum. The residue was purified by column
chromatography (silica gel, hexanes as eluent) followed by recristallization
in
methanol to give 1.55 g of the title product as a white solid. M.P. 82-
84°C. (Global
yield for the last two steps: 38 %).
1H NMR (300 MHz, CDCl3, ppm): 7.78 (d, 2H, J = 8.1 Hz); 7.73 (s, 1H); 7.52
(dd,
2H, J= 8.8 and 1.5 Hz); 4.17 (t, 2H, J= 7.4 Hz); 1.82 (m, 2H); 1.30 (m, 10 H);
0.88
(t, 3H, J= 5.9 Hz).
CA 02360826 2001-10-31
-11-
i3C NMR (75 MHz, CDCl3, ppm) : 141.25; 128.20; 121.84; 121.81; 117.96; 90.80;
43.25; 31.81; 29.28; 29.17; 28.80; 27.15; 22.64; 14.11.
HRMS: Calculated for C2oH23IZN: 530.9920 Found: 530.9906
1-chloro-4-(4'-chlorobenzene)-2-nitrobenzene (7): In a 100 mL flask,
4-chlorophenylboronic acid (2.00g, 12.8 mmol, Aldrich Co.), 1-bromo-4-chloro-2-
nitrobenzene (2.72 g, 11.5 mmol, Aldrich Co.), 18 mL of benzene and 12 mL of
aqueous KZC03 2M were mixed. The resulting solution was degassed with a
vigorous
flow of argon. Tetrakis(triphenylphosphine)Pd(0) (0.5-1.0 mol %) was then
added
under argon and the mixture was refluxed for 2h. The mixture was filtered
through a
Buchner funnel and the filtrate was extracted three times with diethyl ether.
The
combine organic layer was washed with brine and dried over magnesium sulfate.
The
solvent was removed and the residue was purified by column chromatography
(silica
gel, hexanes as eluent) to provide 2.87 g of the title product as a yellow
solid. M.P.
88-89 °C (Yield: 93%).
1H NMR (300 MHz, Acetone-d6, ppm): 8.06 (d, 1H, J= 2.2 Hz); 7.82 (dd, 1H, J=
5.9
and 2.2 Hz); 7.61 (d, 1H, J= 8.8 Hz); 7.52 (dd, 2H, J= 8.8 and 2.2 Hz); 7.40
(dd, 2H,
J= 8.1 and 2.2 Hz).
i3C NMR (75 MHz, Acetone-d6, ppm) : 136.08; 134.97; 134.67; 134.15; 133.43;
130.43 (2C); 129.61; 124.88 (2C).
2,7-dichlorocarbazole (8): A 25 mL flask was charged with 2.00 g of
compound 2 and 10 mL of triethylphosphite. The resulting mixture was refluxed
under argon for 5 h. The excess of triethylphosphite was distillated under
vacuum
(30°C, 0.25 mm Hg) and the crude product was purified by column
chromatography
(silica gel, 10 % ethyl acetate in hexanes) to provides 1.05 g of the title
product as a
white solid. M.P. 188-189°C (Yield: 60 %).
1H NMR (300 MHz, CDC13, ppm): 8.02 (s, 2H); 7.91 (d, 2H, J= 8.1 Hz); 7.38 (d,
2H,
J= 1.5 Hz); 7.22 (dd, 2H, J= 8.8 and 1.5 Hz).
13C NMR (75 MHz, CDC13, ppm): 140.18; 131.86; 121.43; 121.10; 120.62; 110.87.
CA 02360826 2001-10-31
-12-
N ((2-ethylhexyl)-2,7-dichlorocarbazole) (9): To a solution of 900 mg
compound 8 (3.81 mmol) in 20 mL of anhydrous DMF was added 1.06 g (7.67 mmol,
Aldrich Co.) of anhydrous KZCO3. The solution was stirred at 80°C for 2
h under
argon after 1.47 g (7.61 mmol) of 2-ethylhexylbromide was added. The mixture
was
stirred at 80°C for 24 h and then quenched with 30 mL of water. The
aqueous layer
was extracted three times with 50 mL of diethyl ether. The organic layer was
dried
over magnesium sulfate and the solvent was removed under vacuum. The residue
was
purified by column chromatography (silica gel, hexanes as eluent) to give 1.15
g of
the title product as a colorless oil (Yield = 86 %).
1H NMR (300 MHz, CDC13, ppm): 7.90 (d, 2H, J= 8.1 Hz); 7.32 (d, 2H, J= 1.5
Hz);
7.19 (dd, 2H, J= 8.8 and 2.2 Hz); 4.01 (m, 2H); 2.01 (m, 1H); 1.33 (m, 8H);
0.90 (m,
6H).
i3C NMR (75 MHz, CDC13, ppm): 141.74; 131.71; 121.01; 120.89; 119.81; 109.29;
47.64; 39.16; 30.87; 28.64; 24.40; 23.06; 14.03; 10.92.
N octadecane-2,7-dichlorocarbazole (10): To a solution of 3.00 g of
compound 8 (12.7 mmol) in SO mL of anhydrous acetone was added 3.51 g (25.4
mmol, Aldrich Co.) of KzC03 and 8.47 g (25.4 mmol, Aldrich Co.) of 1-
bromooctadecane. The solution was refluxed 24 h under argon. The mixture was
poured into 100 mL of distillated water and the aqueous layer was extracted
with
three portions of diethyl ether. The combined organic fractions were washed
with
brine and dried over magnesium sulfate. The solvent was removed under vacuum.
Recristallization from methanol followed by a recristallization in hexanes
afforded
4.77 g of the title product as a white solid. M. P. 72-74 °C (Yield =
86 %).
1H NMR (300 MHz, CDC13, ppm): 7.90 (d, 2H, J= 8.1 Hz); 7.33 (d, 2H, J= 1.5
Hz);
7.19 (dd, 2H, J = 8.8 and 2.2 Hz); 4.05 (t, 2H, J = 7.3 Hz); 1.82 (m, 2H);
1.32 (m,
30H); 0.90 (t, 3H, J= 5.9 Hz).
13C NMR (75 MHz, CDCl3, ppm): 141.29; 131.75; 121.09; 120.94; 119.83; 109.03;
43.34; 31.97; 29.74 (7C); 29.64; 29.59; 29.51; 29.40; 29.35; 28.76; 27.20;
22.73;
14.15.
CA 02360826 2001-10-31
-13-
1-methoxy-4-(4'-methoxybenzene)-2-nitrobenzene (11): In a 100 mL
flask, 4-methoxyphenylboronic acid (4.00g, 26.3 mmol, Aldrich Co.), 4-bromo-3-
nitroanisole (5.50g, 25.0 mmol, Aldrich Co.), 30 mL of benzene and 20 mL of
KZC03
2M in water were mixed. The resulting solution was degassed with a vigorous
flow of
argon. Pd(PPh3)4 (0.5-1.0 mol %) was then added under argon and the mixture
was
refluxed for 2 h. The mixture was filtered through a Buchner funnel and the
filtrate
was extracted with three portions of diethyl ether. The combined organic
fractions
were washed with brine and dried over magnesium sulfate. The solvent was
removed
under reduced pressure and the residue was purified by column chromatography
(silica gel, 10 % ethyl acetate in hexanes as eluent) to provide 5.50 g of the
title
product as a yellow solid. M.P. 123-125 °C (Yield: 90 %).
1H NMR (300 MHz, CDCl3, ppm): 7.32 (m, 2H); 7.21 (d, 2H, J = 8.8 Hz); 7.13
(dd,
1H, J= 5.9 and 2.2 Hz); 6.94 (d, 2H, J= 8.8 Hz); 3.88 (s, 3H); 3.83 (s, 3H).
i3C NMR (75 MHz, CDCl3, ppm): 159.41; 158.84; 149.74; 132.80; 129.47; 129.22;
128.22; 118.60; 114.15; 108.96; 55.92; 55.30.
HRMS: Calculated for C14Hi3NOa: 259.0844 Found: 259.0851
2,7-dimethoxycarbazole (12): A 25 mL flask was charged with 5.30 g
(20.4 mmol) of compound 11 and 15 mL of triethylphosphite. The resulting
mixture
was refluxed under argon for 5 h. After cooling at room temperature, the
precipitate
was filtrated, washed with a large amount of cold methanol and dried under
reduced
pressure to provides 3.25 g of the title product as a white solid. M.P. 273-
274°C
(Yield: 70 %).
1H NMR (300 MHz, DMSO-db, ppm): 10.99 (s, 1H); 7.85 (d, 2H, J= 8.5 Hz); 6.95
(d,
2H, J= 1.8 Hz); 6.74 (dd, 2H, J= 6.3 and 2.0 Hz).
i3C NMR (75 MHz, DMSO-db, ppm): 157.60; 141.08; 119.95; 116.53; 107.34; 94.73;
55.26.
N-octyl-2,7-dimethoxycarbazole (13): A 50 mL flask was charged
with 3.00 g (13.2 mmol) of compound 12, 5.10 g (26.4 mmol, Aldrich Co.) of 1-
bromooctane, 1.06 g (26.4 mmol) of sodium hydroxide reagent grade, 134 mg
(0.39
mmol) of tetrabutylamonium hydrogensulfate (TBAH) and 25 mL of anhydrous
CA 02360826 2001-10-31
-14-
acetone. The resulting mixture was refluxed under argon for 24 h and then
poured into
100 mL of distillated water. The aqueous layer was extracted three times with
diethyl
ether. The combined organic fractions was dried over magnesium sulfate and the
solvent was removed under reduced pressure. The residue was purified by column
chromatography (silica gel, 5 % ethyl acetate in hexanes as eluent) to provide
4.06 g
of the title product as white fluffy solid. M.P. 63-64°C (Yield: 91 %).
1H NMR (300 MHz, CDC13, ppm): 7.88 (dd, 2H, J = 5.9 and 1.5 Hz); 6.84 (m, 4H);
4.17 (t, 2H, J = 7.4 Hz); 3.95 (s, 6H); 1.86 (m, 2H); 1.35 (m, l OH); 0.92 (t,
3H, J =
4.4 Hz).
~3C NMR (75 MHz, CDCl3, ppm): 158.22; 141.98; 120.13; 117.12; 106.65; 93.65;
55.77; 43.07; 31.87; 29.44; 29.23; 28.70; 27.33; 22.67; 14.12.
HRMS: Calculated for CZZH29N02: 339.2198 Found: 339.2193
N-octyl-2,7-hydroxycarbazole (14): A 250 mL flame dried flask was
charged with 3.30 g (9.72 mmol) of compound 13 and 100 mL of anhydrous
methylene chloride. The solution was cooled at -78°C and 24.3 mL (48.6
mmol) of
boron tribromide (1M in methylene chloride, Aldrich Co.) was added over 0.5 h.
The
resulting mixture was stirred under argon at -78°C for 3 h and at room
temperature for
12 h. The mixture was quenched slowly with 50 mL of HCl 10 % (v/v) to destroy
the
excess of boron tribromide and extracted with 3 portions of 50 mL methylene
chloride. The combined organic fractions was dried over magnesium sulfate and
the
solvent was removed under reduced pressure. Recristallization from
toluene/hexanes
afforded 1.86 g of the title product as a slightly gray solid. M.P. 144-
145°C (Yield: 68
%).
1H NMR (300 MHz, Acetone-d6, ppm): 8.22 (s, 2H); 7.76 (d, 2H, J= 8.8 Hz); 6.88
(d,
2H, J = 2.2 Hz) ; 6.70 (dd, 2H J = 6.6 and 2.2 Hz); 4.19 (t, 2H, J = 7.4 Hz);
1.82 (m,
2H); 1.31 (m, 10 H); 0.86 (t, 3H, J= 6.6 Hz).
i3C NMR (75 MHz, Acetone-d6, ppm): 156.30; 142.88; 120.37; 117.15; 108.49;
95.78; 43.24; 32.39; 29.85; 29.20; 28.95; 27.24; 23.14; 14.21.
HRMS: Calculated for CZOH25N02: 311.1885 Found: 311.1891
CA 02360826 2001-10-31
-15-
N-octyl-2,7-bis(trifluoromethanesulfonyl)carbazole (15): A 25 mL
flask was charged with 2.00 g (6.46 mmol) of compound 14, 790 mg (6.46 mmol,
Aldrich Co.) of dimethylaminopyridine (DMAP) and 16 mL of anhydrous pyridine.
The mixture was cooled at 0°C and 5.47 g (19.4 mmol, Aldrich Co.)
of
trifluoromethanesulfonic anhydride was added dropwise. After 10 min., 4 mL of
pyridine was added to dissolve the white precipitate formed during the
addition of
anhydride. The mixture was stirred at 0°C for 1 h and at room
temperature for 24 h.
The excess of anhydride was destroy with slow addition of 25 mL of distillated
water.
The mixture was extracted three times with 25 mL of diethyl ether. The
combined
organic fractions were washed successively with five 50 mL portions of
distillated
water, five 50 mL portions of aqueous CuS04 O.1M, three 50 mL portions of
brine
and again with SO mL portion of distillated water. The mixture was dried over
magnesium sulfate and the solvent was removed under reduced pressure. The
crude
product was purified by column chromatography (silica gel, 5 % ethyl acetate
in
hexanes as eluent) to provide 2.83 g of the title product as a red oil.
(Yield: 76 %).
1H NMR (300 MHz, CDC13, ppm): 8.00 (d, 2H, J= 8.8 Hz); 7.31 (d, 2H, J= 2.2
Hz);
7.17 (dd, 2H, J = 5.9 and 2.2 Hz); 4.17 (t, 2H, J = 7.4 Hz); 1.83 (m, 2H);
1.30 (m,
l OH); 0.89 (t, 3H, J= 6.6 Hz).
i3C NMR (75 MHz, CDC13, ppm): 148.23; 141.31; 125.34; 121.73; 121.63; 121.09;
116.83; 112.84; 112.58; 102.53; 43.60; 31.68; 29.22; 29.04; 28.68; 27.15;
22.57;
13.90.
HRMS: Calculated for CZZH23F6NO6S2: 575.0871 Found: 575.0877
N-octyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-
yl)carbazole (16): A flame dried 25 mL flask was charged successively with
1.50 g
(2.60 mmol) of compound 15, 11 mL of 1,2-dichloroethane, 38 mg (0.05 mmol) of
PdCl2(dppfJ, 2.2 mL of triethylamine and 1.14 mL (7.86 mmol) of 4,4,5,5-
tetramethyl-1,3,2-dioxaborolane. The mixture was stirred under argon for 4 h
at 80°C
and then poured in 50 mL of distillated water. The aqueous layer was extracted
with
three portions of CHC13. The combined organic layers were dried over magnesium
sulfate and the solvent was removed under reduced pressure. The crude dark red
oil
was puriifed by column chromatography (NEt3 pretreated silica gel, 5 % ethyl
acetate
CA 02360826 2001-10-31
-16-
in hexanes) to provide 837 mg of the title product as a white solid. M.P. 168-
169°C
(Yield: 59 %).
1H NMR (300 MHz, CDC13, ppm): 8.13 (d, 2H, J= 7.4 Hz); 7.89 (s, 2H); 7.69 (d,
2H,
J = 7.4 Hz); 4.39 (t, 2H, J = 7.4 Hz); 1.90 (m, 2H); 1.41 (m, 12 H); 1.30 (m,
l OH);
0.88 (t, 3H, J= 6.6 Hz).
isC NMR (75 MHz, CDC13, ppm): 140.47; 125.09; 124.86; 120.01; 115.28; 83.80;
42.91; 31.84; 29.37; 29.20; 29.17; 27.12; 24.95; 22.64; 14.11. (The missing
peak is
due to the carbon linked to the boronic function which shows no signal in'3C
NMR).
HRMS: Calculated for C32H4~BZNOq: 531.3691 Found: 531.3700
EXAMPLE 4: Preparation of poly (N-octyl-2,7-carbazole)
In a 10 mL flask, 1.00 g (1.88 mmol) of compound 6, 296 mg (1.13
mmol) of triphenylphosphine, 405 mg (6.20 mmol) of zinc powder 99.998% 100
mesh, 15.0 mg (0.09 mmol) of 2,2'-bipyridine 12.0 mg (0.09 mmol) of anhydrous
nickel (II) chloride and 3 mL of anhydrous DMAc were stirred under argon for 3
days
at 100°C. The whole mixture was then poured into a cold mixture of
methanol/HCl
(5:1 v/v). The precipitated material was recovered by filtration through a
Buchner
funnel and washed with dilute HCI. The solid material was washed for 24 h in a
Soxhlet apparatus using acetone to remove oligomers and catalyst residues. The
resulting solid was dilute again in chloroform and filtrated on 0.2 pm
filtering paper to
remove all traces of nickel. The resulting solid was dried under reduced
pressure for
24 h. (Yield: 78 %).
EXAMPLE 5: Preparation of poly (N-octadecane-2,7-carbazole)
In a 25 mL flask, 384 mg (2.46 mmol, Aldrich Co.) of 2,2'-bipyridine,
676 mg (2.46 mmol, Aldrich Co.) of Ni(COD)2 and 0.25 mL (2.05 mmol, Aldrich
Co.) of 1,5-cyclooctadiene (COD) 7 mL of degassed anhydrous DMF were stirred
under argon for 1h at 80°C. 1.00 g (2.05 mmol) of compound 10 in 7 mL
of degassed
anhydrous DMF was added and the mixture was stirred for 48 h under argon at
100°C. The whole mixture was then poured into a cold of methanol. The
precipitated
material was recovered by filtration through a Buchner funnel. The solid
material was
washed for 24 h in a Soxhlet apparatus using acetone to remove oligomers and
CA 02360826 2001-10-31
-17-
catalyst residues. The resulting solid was dilute again in chloroform and
filtrated on
0.2 ~m filtering paper to remove all traces of nickel. The resulting solid was
dried
under reduced pressure for 24 h. (Yield: 75 %).
EXAMPLE 6: Preparation of poly (N-octyl-2,7-carbazole-alt-9,9-dioctyl-2,7-
fluorene)
In a 10 mL flask, 225 mg (0.42 mmol) of compound 6, 271 mg (0.42
mmol) of 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2y1)-9,9-
dioctylfluorene and
mg of (PPh3)4Pd(0) were dissolved in a degassed mixture of THF (2.5 mL) and
10 aqueous 2 M KZC03. The solution was refluxed under argon for 3 days. The
whole
mixture was then poured into cold methanol (100 mL). The precipitated material
was
recovered by filtration through a Buchner funnel and washed with dilute HCI.
The
solid material was washed for 24 h in a Soxhlet apparatus using acetone to
remove
oligomers and catalyst residues. The resulting polymers were soluble in THF
and
1 S CHC13. (Yield: 78 %).
EXAMPLE 7: Preparation of poly [N-2-ethylhexyl-2,7-carbazole-alt-S,5'-(2,2'-
bithiophene)]
In a 50 mL flask, 541 mg (1.10 mmol) of 5,5'-bis(trimethylstannyl)-
2,2'-bithiophene, 531 mg (1.00 mmol) of N 2-ethylhexyl-2,7-diiodocarbazole and
25
p.g of C12(PPh3)ZPd(0) were dissolved in 30 mL of degassed THF. The solution
was
refluxed under argon for 3 days. The whole mixture was then poured into cold
methanol (300 mL). The precipitated material was recovered by filtration
through a
Buchner funnel and washed with dilute HCI. The solid material was washed for
24 h
in a Soxhlet apparatus using acetone to remove oligomers and catalyst
residues. The
resulting polymers were soluble in THF and CHC13. (Yield: 52 %).
EXAMPLE 8: Preparation of poly (N-octyl-2,7-carbazole-alt-2,5-thiophene) (PTC)
In a 25 mL flask was added 350 mg (0.66 mmol) of compound (3), 276
mg (0.67 mmol) of compound (8), 9 mg (13 ~mol, Aldrich Co.) of PdCl2(PPh3)2
and 8
mL of anhydrous THF. The mixture was refluxed under argon for 72 h. The yellow
suspension was poured in cold methanol. The precipitate was collected by
flitration
CA 02360826 2001-10-31
-18-
and washed with acetone in a Soxhlet apparatus for 48 h. After being dried
under
reduced pressure, an orange solid was obtained. (Yield: 68 %).
EXAMPLE 9: Preparation of poly (N-octyl-2,7-carbazole-alt-2,5-
S dioxyethylenethiophene) (PEDOTC)
In a 10 mL flask was added 190 mg (0.36 mmol) of compound (14),
107 mg (0.36 mmol) of compound (7), 8 mg (7 pmol) of Pd(PPh3)4, 3 mL of
anhydrous THF and 2 mL of KZC03 2M in water. The mixture was refluxed under
argon for 72 h. The black suspension was poured in cold methanol. The
precipitate
was collected by filtration and washed with acetone in a Soxhlet apparatus for
48 h.
After being dried under reduced pressure, a black solid was obtained. (Yield:
68 %).
EXAMPLE 10: Preparation of poly (N-(2-ethylhexyl)-2,7-carbazole-alt-4-butyl-
N,N
Bis(p-phenyl)phenylamine) (PPAC)
In a 10 mL flask, 501 mg (0.87 mmol) of N (2-ethylhexyl)-2,7-
bis(trifluoromethanesulfonyl)carbazole, 474 mg (0.87 mmol) of 4-butyl-N,N
Bis(p-
(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)phenylamine) and 10 mg of
(PPh3)4Pd(0) were dissolved in a degassed mixture of THF (3 mL) and aqueous 2
M
KZC03 (2 mL). The solution was refluxed under argon for 3 days. The whole
mixture
was then poured into cold methanol (100 mL). The precipitated material was
recovered by filtration through a Biichner funnel and washed with dilute HCI.
The
solid material was washed for 24 h in a Soxhlet apparatus using acetone to
remove
oligomers and catalyst residues. The resulting polymers were soluble in THF
and
CHCI3. (Yield: 75 %).
EXAMPLE 11: Preparation of poly (N-octyl-2,7-carbazole)
In a 10 mL flask, 500 mg (0.87 mmol) of compound 15, 137 mg (0.52
mmol) of triphenylphosphine, 188 mg (2.88 mmol) of zinc powder 99.998% 100
mesh, 7.0 mg (0.09 mmol) of 2,2'-bipyridine 12.0 mg (0.04 mmol) of anhydrous
nickel (II) chloride and 3 mL of anhydrous DMF were stirred under argon for 3
days
at 100°C. The whole mixture was then poured into a cold mixture of
methanol/HCI
(S:1 v/v). The precipitated material was recovered by filtration through a
Buchner
CA 02360826 2001-10-31
-19-
funnel and washed with dilute HCI. The solid material was washed for 24 h in a
Soxhlet apparatus using acetone to remove oligomers and catalyst residues. The
resulting solid was dilute again in chloroform and filtrated on 0.2 ~m
filtering paper to
remove all traces of nickel. The resulting solid was dried under reduced
pressure for
24 h. (Yield: 83 %).
EXAMPLE 12: Preparation of poly (N-octyl-2,7-carbazole)
In a 10 mL flask, 540 mg (0.94 mmol) of compound 15, 500 mg (0.94
mmol) of compound 16 and 10 mg of (PPh3)4Pd(0) were dissolved in a degassed
mixture of THF (6.6 mL) and aqueous 2 M K2C03 (2.8 mL). The solution was
refluxed under argon for 3 days. The whole mixture was then poured into cold
methanol ( 100 mL). The precipitated material was recovered by filtration
through a
Buchner funnel and washed with dilute HCI. The solid material was washed for
24 h
in a Soxhlet apparatus using acetone to remove oligomers and catalyst
residues. The
resulting polymers were partially soluble in THF and CHC13. (Yield: 86 %).
The resulting conjugated homopolymers and copolymers are soluble in
common organic solvents, such as chloroform and tetrahydrofuran. The number-
average molecular weight (measured by size exclusion chromatography against
monodisperse polystyrene standards) of these polymers is about 10 kDa with a
polydispersity of 2. They can be processed by spin coating or by simple
casting to
yield thin polymer films with good mechanical properties. As reported in
Figure 1,
the solution and solid-state optical properties of poly(N-octyl-2,7-carbazole)
have
been investigated in more details. In dilute solutions or as thin films, this
polymer
exhibits an absorption maximum around 380-390 nm, indicating a pale-yellow
color
in both forms. This absorption maximum is significantly red-shifted compared
to that
previously reported for poly(N-alkyl-3,6-carbazole)s (i.e. 300-320 nm) and can
be
related to a more conjugated structure. Moreover, poly(N-octyl-2,7-carbazole)
exhibits an intense blue emission upon radiative excitation, with a quantum
yield of
about 80% in chloroform, at room temperature. In solution, poly(N-octyl-2,7-
carbazole) shows a maximum of emission at 417 nm followed by two vibronic side-
bands at 439 and 474 nm whereas in the solid state, the polymer is slightly
more
CA 02360826 2005-08-O1
-20-
conjugated with an emission maxium at 437 nm followed by two other maxima
at 453 and 492 nm. These solid-state and solution emission spectra are
slightly
red-shifted compared to poly(9,9-dioctyl-2,7-fluorene) and could be related to
the electron-donating effect of the nitrogen atom in the inner ring. Moreover,
poly(N-octyl-2,7-carbazole) shows a relatively low oxidation potential at 0.75
V vs Ag/AgCI (Figure 2). This oxidation potential is lower than those reported
for poly(N-alkyl-3,6-carbazole)s at 0.85 and 1.2 V vs Ag/AgCI and is an
indirect proof of the more delocalized structure in 2,7-linked polycarbazoles.
This combination of electrical and optical properties is particularly
interesting
1 o for the development of a novel class of blue-light emitting materials.
Moreover, with the possibilities of structural modifications through the
synthesis of various alternating copolymers, it is possible to develop tunable
light-emitting materials.