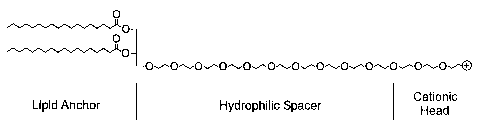Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
1
CATIONIC PEG-LIPIDS AND METHODS OF USE
RELATED APPLICATIONS
This application claims priority to U.S. Provisional Patent Application
Serial No. 60/130,151 filed April 20, 1999, the teachings of which are
incorporated
herein by reference in their entirety for all purposes.
FIELD OF THE INVENTION
This invention relates to cationic lipid conjugates, and more particularly,
to cationic polymer lipid conjugates and lipid-based drug formulations
thereof,
containing one or more bioactive agents.
BACKGROUND OF THE INVENTION
Current vectors for gene delivery and gene therapy are comprised of viral
based and non-viral based systems. Lipid-based non-viral systems include
cationic lipid
plasmid DNA complexes. Limitations of these systems include large sizes,
toxicity and
instability of the complexes in the serum. Unfortunately, the foregoing
drawbacks limit
the applications for these complexes.
Researchers have devoted tremendous effort to the design of long
circulation stealth liposomes that can be used for systemic delivery (see,
Papahadjopoulos, D. et al., Proc. Natl. Acad. Sci. 88:11460-11464 (1991);
Klibanov,
A.L. et al., .I. Liposome Res., 2:321 ( 1992); Woodle, M.C. et al., Biochim.
Biophys. Acta.,
1113:171 (1992); Torchilin, V.P. et al., In: Stealth Liposornes. Ed. By D.
Lasic, F.
Martin. CRC Press, Boca Raton, FL, pp. 51-62 (1995); Allen, T.M. et al.,
Biochim.
Biophys. Acta., 1237:99-108 (1995) and Zalipsky, S. et al., J. Controlled
Release,
39:153-161 (1996)). In certain instances, and depending on the formulation,
stealth
liposomes are often comparatively inefficient at facilitating cellular uptake
and therefore
the therapeutic efficacy is reduced.
In general, the molecular mechanism of liposomal longevity in vivo can be
attributed to steric hindrance resulting from hydrophilic polymer surface
barriers. The
hydrophilic polymer barriers prevent or reduce the rate of the adsorption of
macromolecules from the blood and sterically inhibit both electrostatic and
hydrophobic
interactions between liposomes and blood components. Thus, although the
longevity of
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
stealth liposomes has been increased by the insertion of hydrophilic polymers,
the
cellular uptake of the stealth liposomes often is inefficient.
Over the past decade, it has also become clear that liposomal systems
possessing cationic lipids are highly effective transfection agents in vitro
(Felgner, P.L. et
al., Nature 337:387-388 (1989); Felgner, P.L. et al., Proceedings of the
National
Acadentv of Sciences of the United States of America 84:7413-7417 ( 1987)).
The
addition of cationic liposomes to plasmid DNA gives rise to large DNA-lipid
complexes
that possess excellent transfection properties irz vitro, but which are
ineffective in vivo
due to their rapid clearance from the circulation by cells of the
reticuloendothelial system
(RES). The need for a non-viral lipid-based system capable of systemic
delivery of genes
to cells led to the recent development of stabilized plasmid-lipid particles
(SPLPs)
(Wheeler, J.J. et al., Gene Then-apy 6:271-281 (1999)).. These particles are
small (about
70 nm), contain a single copy of a plasmid vector, possess stealth properties
resulting
from a surface coating,of p9ly(ethyleneglycol) (PEG)., and protect DNA from
degradation
by serum nucleases.
Enhancing intracellular delivery of liposomes and/or their contents
represents one of the major remaining problems in the development of the next
generation of drug delivery systems.. In,order to optimize the delivery of
drugs
(conventional or genetic) in vivo, general methods for increasing the
interactions of
liposomes with cells need to be developed. To date, attempts include the use
of specific
targeting information on the liposome surface, such as an antibody (see,
Meyer, O. et al.,
Journal ofBiological Chemistry 273:15621-15627 (1998); Kao, G.Y. et al.,
Cancer Gene
Therapy 3:250-256 ( 1996,);. Hansen, C.B. et al., Biochimica et Biophysi.ca
Acta 1239:133-
144 (1995)), vitamin- (see, Gabizon, A. et al., Bioconjzcgate Chemistry 10:289-
298
(1999); Lee, R.J. et al., Journal ofBiological Chemistry 269:3198-3204 (1994);
Reddy,
J.A. et al., Critical Reviews in Therapezctic Drug Carrier Systems 15:587-627
(1998);
Holladay, S.R. et al., Biochimica_ et Biophysica Acta 1426:195-204 ( 1999);
Wang, S. et
al., Journal of Controlled Release 53:39-48 (1998)) , oligopeptide- (see,
Zalipsky, S. et
al., Bioconjugate Chemistry 6:705-708 (1995); Zalipsky, S. et al.,
Bioconjugate
Chemistry 8:111-118 (1997)), or the use of oligosaccharide constructs specific
for a
particular membrane protein or receptor. Unfortunately, these methods have not
been
successful in achieving this goal, despite promising in vitro results. While
specific
targeting of liposomes to tissues remains an important area of research, other
approaches
may also provide significant improvements in the effectiveness of liposomal
carriers.
SI1BSTIT'IJT'E SHEET (RULE 26)
22-06-2001 ~ CA0000451
' CA 02370690 2001-10-19
JUN GG G001 4:11 PM FR W.GEORGIR URNCOUUER 682 Oc74 TO 011438923994465 P.06
In view of the foregoing, what is needed in the art is a lipid-based drug
formulation with increased longevity coupled with increased cellular uptake.
The present
invention satisbes this and other needs.
SUMMARY OF THE INVENTION
In certain aspects, the present invention relates to new conjugates that can
be ~incoxporated or inserted into stabilized plasmid lipid particles to
enhance tsansfection
efficiencies. The conjugates of the present invention possess a lipid aachor
for anchoring
the conjugate into the bilsycr lipid particle, wherein the lipid anchor is
attached to a non-
immunogenic polymer, such as a PEG moiety, and wherein the non-immunogenic
polymer is, in turn, attached tv a polycationic moiety, such as a positively
charged
moiety. As such, the present invention provides a compound of Formula I: . _
A w Y
In Formula I, "A" is a lipid moiety attached to a non-immunogeaic
polymer. "W," in Fvnmula I, is a non-immunogenic polymer, and "Y", in Formula
I, is a
polycationic moiety. In one embodiment, Y is about 1 tv about 10 basic amino
acids or
derivatives thereof. In another embodiment, Y is a member selected from the
group
consisting of lysine, arginine, asparagine, glutamine, derivatives thereof and
combinations thereof. In yet another embodiment, Y is a cationic group having
four
lysine residues or derivatives thereof.
In certain preferred embodiments, the compounds of Formula I contain
groups that.give rise to compounds having the general structure of Formula II:
A X-(CH= CH2 O)p Z Y
II
In Formula II, "A" is a lipid, such as a hydrophobic lipid. In Formula II,
~ ~~ "X" is a single bond or a fu etianal group th i~ cvvalently attaches the
lipid to at least one
ethylene oxide unit, i.e., (-CHZ- CHZ-O-). In Formula II, "Z" is a single bond
or a
functional group that cvvalently attaches the at least one ethylene oxide unit
to a cationic
group. In Formula il,~"Y" is a polycativnic moiety. In Formula II, the index
'h" is an ..
integer ranging in value from about 6 to about 160.
In other aspects, the present invention relates tv a lipid-based drug
formulation comprising: (a) a compound having Formula I
A w Y I
3
AMENDED SHEET
Fmof ~o; +~~?~mFmnnt nt ~na c.~~t ~.. .o~z v nn ~
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
wherein: A, W and Y have been defined;
(b) a bioactive agent; and
(c) a second lipid.
In certain embodiments, the lipid-based drug formulation is in the form of
a liposome, a micelle, a virosome, a lipid-nucleic acid particle, a nucleic
acid aggregate
and mixtures thereof. In certain other embodiments, the bioactive agent is a
therapeutic
nucleic acid or other drugs.
In yet other aspects, the present invention relates to a method for
increasing intracellular delivery of a lipid-based drug delivery system,
comprising:
incorporating into the lipid-based drug delivery system a compound of Formulae
I or II,
thereby increasing the intracellular delivery of the lipid-based drug delivery
system.
Additional aspects and advantages of the present invention will be
apparent when read with the following detailed description and the
accompanying
drawings.
DEFINITIONS
The term "lipid" refers to a group of organic compounds that include, but
are not limited to, esters of fatty acids and are characterized by being
insoluble in water,
but soluble in many organic solvents. They are usually divided into at least
three classes:
(1) "simple lipids" which include fats and oils as well as waxes; (2)
"compound lipids"
which include phospholipids and glycolipids; (3) "derived lipids" such as
steroids.
The term "vesicle-forming lipid" is intended to include any amphipathic
lipid having a hydrophobic moiety and a polar head group, and which by itself
can form
spontaneously into bilayer vesicles in water, as exemplified by most
phospholipids.
The term "vesicle-adopting lipid" is intended to include any amphipathic
lipid which is stably incorporated into lipid bilayers in combination with
other
amphipathic lipids, with its hydrophobic moiety in contact with the interior,
hydrophobic
region of the bilayer membrane, and its polar head group moiety oriented
toward the
exterior, polar surface of the membrane. Vesicle-adopting lipids include
lipids that on
their own tend to adopt a non-lamellar phase, yet which are capable of
assuming a bilayer
structure in the presence of a bilayer-stabilizing component. A typical
example is DOPE
(dioleoylphosphatidylethanolamine). Bilayer stabilizing components include,
but are not
limited to, polyamide oligomers, peptides, proteins, detergents, lipid-
derivatives, PEG-
lipid derivatives such as PEG coupled to phosphatidylethanolamines, and PEG
SUBST>;Z'UTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
conjugated to ceramides (see, U.S. Application Serial No. 08/485,608, now U.S.
Patent
No. x,885,613, which is incorporated herein by reference).
The term "amphipathic lipid" refers, in part, to any suitable material
wherein the hydrophobic portion of the lipid material orients into a
hydrophobic phase,
while the hydrophilic portion orients toward the aqueous phase. Amphipathic
lipids are
usually the major component of a lipid vesicle. Hydrophilic characteristics
derive from
the presence of polar ar charged groups such as carbohydrates, phosphato,
carboxylic,
sulfato, amino, sulfhydryl, nitro, hydroxy and other like groups.
Hydrophobicity can be
conferred by the inclusion of apolar groups that include, but are not limited
to, long chain
saturated and unsaturated aliphatic hydrocarbon groups and such groups
substituted by
one or more aromatic, cycloaliphatic or heterocyclic group(s). Examples of
amphipathic
compounds include, but are not_limited to, phospholipids, aminolipids and
sphingolipids.
Representative examples of phospholipids include, but are not limited to,
phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine,
phosphatidylinositol, phosphatidic acid, palmitoyloleoyl phosphatidylcholine,
lysophosphatidylcholine, lysophosphatidylethanolamine,
dipalmitoylphosphatidylcholine,
dioleoylphosphatidylcholine, distearoylphosphatidylcholine or
dilinoleoylphosphatidylcholine. Other compounds lacking in phosphorus, such as
sphingolipid, glycosphingolipid families, diacylglycerols and ~i-acyloxyacids,
are also
within the group designated as, amphipathic lipids. Additionally, the
amphipathic lipid
described above can be mixed; with other lipids including triglycerides and
sterols.
The term "neutral lipid" refers to any of a number of lipid species that
exist either in an uncharged or neutral zwitterionic form at a selected pH. At
physiological pH, such lipids include, for example, diacylphosphatidylcholine,
diacylphosphatidylethanolamine, ceramide, sphingomyelin, cephalin,
cholesterol,
cerebrosides and diacylglycerols.
The term "hydrophopic lipid" refers to compounds having apolar groups
that include, but are not limited to, long chain saturated and unsaturated
aliphatic
hydrocarbon groups and such groups optionally substituted by one or more
aromatic,
cycloaliphatic or heterocyclic group(s). Suitable examples include, but are
not limited to,
diacylglycerol, dialkylglycerol, N-N-dialkylamino, 1,2-diacyloxy-3-
aminopropane and
1,2-dialkyl-3-aminopropane.
The term "diacylglycerolyl" denotes 2-fatty acyl chains, R' and RZ having
independently between 2 and 30 carbons bonded, to the l- and 2-position of
glycerol by
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
6
ester linkages. The acyl groups can be saturated or have varying degrees of
unsaturation.
Diacylglycerol groups have the following general formula:
0
CH,O ~ R~
O
CH-O R-
CH,O-
The term "dialkylglycerolyl" denotes two C,-C3o alkyl chains bonded to
the 1- and 2-position of glycerol by ether linkages. Dialkylglycerol groups
have the
following general formula:
CH,ORi
CH-ORz
CH,O
The term "N-N-dialkylamino" denotes
~C i C3o alkyl
\Ci C3o alkyl
The term "1,2-diacyloxy-3-aminopropane" denotes 2-fatty acyl chains C~-
C3o bonded to the 1- and 2-position of propane by an ester linkage. The acyl
groups can
be saturated or have varying degrees of unsaturation. The 3-position of the
propane
molecule has a -NH- group attached. 1,2-diacyloxy-3-aminopropanes have the
following
general formula:
SUBS?'ITUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
O
CH,O \ Ri
O
CH-O~ R'
CH,N
The term "1,2-dialkyl-3-aminopropane" denotes 2-alkyl chains (C~-C3O)
bonded to the 1- and 2-position of propane by an ether linkage. The 3-position
of the
propane molecule has a -NH- group attached. 1,2-dialkyl-3-aminopropanes have
the
following general formula:
CH=O C,-C3o-Alkyl
CH-O Ci-C3o-Alkyl
CH,N
The term "non-cationic lipid" refers to any neutral lipid as described above
as well as anionic lipids. Examples of anionic lipids include, but are not
limited to,
phosphatidylglycerol, cardiolipin, diacylphosphatidylserine,
diacylphosphatidic acid, N-
dodecanoyl phosphatidylethanolamines, N-succinyl phosphatidylethanolamines, N-
glutarylphosphatidylethanolamines, lysophosphatidylglycerols, and other
anionic
modifying groups joined to neutral lipids.
The term "cationic lipid" refers to any of a number of lipid species that
carry a net positive charge at a selected pH, such as physiological pH. Such
lipids
include, but are not limited to, N,N-dioleyl-N,N-dimethylammonium chloride
("DODAC"); N-(2,3-dioleyloxy)propyl)-N,N,N-trimethylammonium chloride
("DOTMA"); N,N-distearyl-N,N-dimethylammonium bromide ("DDAB"); N-(2,3-
dioleoyloxy)propyl)-N,N,N-trimethylammonium chloride ("DOTAP"); 3 -(N-(N',N'-
dimethylaminoethane)-carbamoyl)cholesterol ("DC-Chol") and N-(1,2-
dimyristyloxyprop-3-yl)-N,N-dimethyl-N-hydroxyethyl ammonium bromide ("DMRIE")
Additionally, a number of commercial preparations of cationic lipids are
available which
can be used in the present invention. These include, for example, LIPOFECTIN~
SUBSTTTLTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
(commercially available cationic liposomes comprising DOTMA and 1,2-dioleoyl-
sn-3-
phosphoethanolamine ("DOPE"), from GIBCO/BRL, Grand Island, New York, USA);
LIPOFECTAMINE~ (commercially available cationic liposomes comprising N-(1-(2,3-
dioleyloxy)propyl)-N-(2-(sperminecarboxamido)ethyl)-N,N-dimethylammonium
trifluoroacetate ("DOSPA") and("DOPE"), from GIBCOBRL); and TRANSFECTAM~
(commercially available cationic lipids comprising dioctadecylamidoglycyl
carboxyspermine ("DOGS") in ethanol from Promega Corp., Madison, Wisconsin,
USA).
The following lipids are cationic and have a positive charge at below
physiological pH:
DODAP, DODMA, DMDMA and the like.
The term "fusogenic" refers to the ability of a liposome or other drug
delivery system to fuse with membranes of a cell. The membranes can be either
the
plasma membrane or membranes surrounding organelles, e.g., endosome, nucleus,
etc.
Fusogenesis is the fusion of a, liposome to such a membrane.
The term "dendrimer" includes reference to branched polymers that
possess multiple generations. In dendrimers, each generation creates multiple
branch
points. . '
The term "ligand" includes any molecule, compound or device with a
reactive functional group and includes lipids, amphipathic lipids, carrier
compounds,
bioaffinity compounds, biomaterials, biopolymers, biomedical devices,
analytically
detectable compounds, therapeutically active compounds, enzymes, peptides,
proteins,
antibodies, immune stimulators, radiolabels, fluorogens, biotin, drugs,
haptens, DNA,
RNA, polysaccharides, liposomes, virosomes, micelles, immunoglobulins,
functional
groups, targeting agents, or toxins. The foregoing list is illustrative and
not intended to
be exhaustive.
The term "ATTA" or "polyamide" refers to, but is not limited to,
compounds disclosed in U.S. Patent Application Serial No. 09/218,988, filed
December
22, 1998. These compounds include a compound having the formula
R' O Rz
R N (CH~CH,O)m (CHZ)p C-(~1H-CSI-C)q R3
O n
wherein: R is a. member selected from the group consisting of hydrogen,
alkyl and acyl; R~ is a member. selected from the group consisting of hydrogen
and alkyl;
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
or optionally, R and R1 and the nitrogen to which they are bound form an azido
moiety;
Rz is a member of the group selected from hydrogen, optionally substituted
alkyl,
optionally substituted aryl and a side chain of an amino acid; R3 is a member
selected
from the group consisting of hydrogen, halogen, hydroxy, alkoxy, mercapto,
hydrazino,
amino and NR'~R', wherein R'~ and R' are independently hydrogen or alkyl; n is
4 to 80;
m is 2 to 6; p is 1 to 4; and q is 0 or 1. It will be apparent to those of
skill in the art that
other polyamides can be used in the compounds of the present invention.
As used herein, the term "alkyl" denotes branched or unbranched
hydrocarbon chains, such as, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-
butyl, iso-
butyl, tertbutyl, octa-decyl and 2-methylpentyl. These groups can be
optionally
substituted with one or more functional groups which are attached commonly to
such
chains, such as, hydroxyl, bromo, fluoro, chloro, iodo, mercapto or thio,
cyano, alkylthio,
heterocyclyl, aryl, heteroaryl, carboxyl, carbalkoyl, alkyl, alkenyl, nitro,
amino, alkoxyl,
amido, and the like to form alkyl groups such as trifluoromethyl, 3-
hydroxyhexyl, 2-
carboxypropyl, 2-fluoroethyl, carboxymethyl, cyanobutyl and the like.
The term "alkylene" refers to a divalent alkyl as defined above, such as
methylene (-CHZ-), propylene (-GHzCHZCH2-), chloroethylene (-CHCICHz-), 2-
thiobutene (-CHZCH(SH)CHZCHZ-), 1-bromo-3-hydroxyl-4-methylpentene (-
CHBrCHaCH(OH)CH(CH3)CHZ-), and the like.
The term "alkenyl" denotes branched or unbranched hydrocarbon chains
containing one or more carbon-carbon double bonds.
The term "alkynyl" refers to branched or unbranched hydrocarbon chains
containing one or more carbon-carbon triple bonds.
The term "aryl"denotes a chain of carbon atoms which form at least one
aromatic ring having preferably between about 6-14 carbon atoms, such as
phenyl,
naphthyl, indenyl, and the like, and which maybe substituted with one or more
functional
groups which are attached commonly to such chains, such as hydroxyl, bromo,
fluoro,
chloro, iodo, mercapto or thio;:cyano, cyanoamido, alkylthio, heterocycle,
aryl,
heteroaryl, carboxyl, carbalkoyl, alkyl, alkenyl, nitro, amino, alkoxyl,
amido, and the like
to form aryl groups such as biphenyl, iodobiphenyl, methoxybiphenyl, anthryl,
bromophenyl, iodophenyl, chlorophenyl, hydroxyphenyl, methoxyphenyl,
formylphenyl,
acetylphenyl, trifluoromethylthiophenyl, trifluoromethoxyphenyl,
alkylthiophenyl,
trialkylammoniumphenyl, amidophenyl, thiazolylphenyl, oxazolylphenyl,
imidazolylphenyl, imidazolylmethylphenyl, and the like.
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
The term "acyl" denotes the -C(O)R group, wherein R is alkyl or aryl as
defined above, such as formyl, acetyl, propionyl, or butyryl.
The term "alkoxy" denotes -OR-, wherein R is alkyl.
The term "amido" denotes an amide linkage: -C(O)NR- (wherein R is
5 hydrogen or alkyl).
The term "amino" denotes an amine linkage: -NR-, wherein R is
hydrogen or alkyl or a terminal NH~_
The term "carboxyl" denotes the group -C(O)O-, and the term "carbonyl"
denotes the group -C(O)-.
10 The term "carbonate" indicates the group -OC(O)O-.
The term "carbamate" denotes the group -iVHC(O)O-, and the term "urea"
denotes the group -NHC(O)NH-.
The term "phosphoro" denotes the group -OP(O)(OH)O-.
The term "basic .amino acid" refers to naturally-occurring amino acids as
well as synthetic amino acids and/or or amino acid mimetics having a net
positive charge
at a selected pH, such as physiological, pH. This group includes, but is not
limited to,
lysine, arginine, asparagine, glutamine, histidine and the like.
The term "phosphorylethanolamino" denotes the group
-OP(O)(OH)OCH~CHZNH-.
The term "phosphorylethanolamido" denotes the group -
OP(O)(OH)OCH~CHZNHC(O)-.
The term "phospho" denotes a pentavalent phosphorous moiety -
P(O)(OH)O-~
The term "phosphoethanolamino" denotes the group -
P(O)(OH)OCHZCH~NH-.
The term "phosphoethanolamido" denotes,the group-
P(O)(OH)OCHzCHzNHC(O)-.
The term "ethylene oxide unit" denotes the group -OCH~CH~-.
The term.''CPL"refers to a cationic-polymer-lipid e.g., cationic-PEG-
lipid. Preferred CPLs are comppunds of Formulae I and II.
The term."d-DSPE-CPL-M" is encompassed by the term "CPL1" which
refers to a DSPE-CPL having,one positive charge. The "d-" in d-DSPE-CPL-M
indicates
that the CPL contains a fluorescent dansyl group. It will be apparent to those
of skill in
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
11
the art that a CPL can be synthesized without the dansyl moiety, and thus the
term
"DSPE-CPL-M" is encompassed by in the term "CPLI" as defined above.
The term "d-DSPE-CPL-D" is encompassed by the term "CPL2" which
refers to DSPE-CPL having two positive charges.
The term "d-DSPE-CPL-Tl" is encompassed by the term "CPL3" which
refers to DSPE-CPL having three positive charges.
The term "d-DSPE-CPL-Q1" is encompassed by the term "CPL4a" which
refers to DSPE-CPL having four positive charges.
The term "d-DSPE-CPL-Q5," or, alternatively, DSPE-PEGQuadS, or,
alternatively, DSPE-CPL-4, are all encompassed by the term "CPL4 (or CPL4b)"
which
refer to a DSPE-CPL having four positive charges. By modifying the headgroup
region,
CPLs were synthesized which contained 1 (mono, or M), 2 (di, or D), 3 (tri, or
T), and 4
(quad, or Q) positive charges:.- Various Quad CPLs were synthesized, hence
these are
numbered Q 1 through Q5.
The abbreviations "HBS". refers to Hepes-buffered saline, "Rho-PE" refers
to rhodamine-phosphatidylethanolamine, and "LIJVs" refers to "large
unilamellar
vesicles."
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 illustrates a structural design of a cationic-polymer lipid (CPL)
conjugate.
Figure 2 illustrates a synthetic scheme for the preparation of cationic-
PEG-lipid conjugates having varying amount of charged head groups (a.)
EtzN/CHC13;
(b.) TFA /CHC13; c. Et3N / CHCI3 Na, Ns-di-t-Boc-L-Lysine N-hydroxysuccinide
ester.
Figure 3 illustrates a CPL incorporated liposome. The large unilamellar
vesicles (LUV) have incorporated different examples of CPLs (CPL1, CPL2, CPL4,
and
CPLB, respectively).
Figure 4 illustrates a distribution of DSPE-CPL-4 between the inner/outer
leaflets of a liposomal membrane. CPL-4-LUVs (DSPC/Chol/DSPE-CPL-4, 55:40:5
mole %) were prepared by extrusion method as described herein. The
distribution of
outer leaflet CPLs was quantified by a fluorescamine assay. For the outer
leaflet CPLs,
the following assay was used. An appropriate amount of CPL-4-LUVs was diluted
with
1M Borate buffer (pH 8.5)an d cooled in ice-water. 20 pl of 10% Triton X-100
was
SUBSTIZ'LIT'E SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
12
added to the above sample solution to solubilize the membrane, and then an
additional 20
q.l of a cooled fluorescamine ethanol solution (10 mg/ml) was added and then
measured.
Figure 5 illustrates a cellular uptake study of CPL-4 LUVs in BHK cells
in PBS-CMG. The controls were LUVs (DSPC/Chol, 60:40) and CPL-4-LUVs
(DSPC/Ch/DSPE-CPL-4, 5~:40:~) were prepared by extrusion as described herein.
Figure 6 illustrates cellular uptake of CPL-4-LUVs in BHK cells in
DMEM (with 10% FBS). The control used LUVs (DSPC/Chol, 60:40). CPL-4-LUVs
(DSPC/Ch/DSPE-CPL-4, 5:40:5), which were prepared by extrusion as described
herein.
Figure 7 illustrates a cellular uptake of CPL-liposomes in BHK cells in
PBS-CMG after 4 hr incubation. LUVs (DSPC/Chol, 60:40) and CPL-LUVs
(DSPC/Chol/DSPE-CPL, 5:40:5) were prepared by extrusion as described herein.
Figure 8 illustrates the preparation of CPL-LUVs by detergent dialysis.
Lipids were codissolved in chloroform at the indicated ratios, following which
the
solvent was removed by nitrogen gas and high vacuum. The lipid mixture was
dissolved
in detergent/buffer (OGP in HBS) and dialysed against HBS for 2-3 days. The
LUVs,
which formed during dialysis, were then fractionated as shown on Sepharose CL-
4B.
Panel A: Fractionation of DOPE/DODAC/CPL4[3.4K] /PEGCerC20/Rho-PE
(79.5/6/4/10/0.5); Panel B: Fractionation of DOPE/DODAC/CPL4[1K]/
PEGCerC20/Rho-PE (79.5/6/4/10/0.5); and Panel C: Fractionation of
DOPE/DODAC/CPL4[3.4K] /PEGCerC20/Rho-PE (71.5/6/12/10/0.5).
Figure 9 Panel A illustrates the insertion of DSPE-CPL-QS into DOPC
LUVs (100 nm). DOPC LUVs (2.6 ~.mol lipid) were incubated with 0.214 ~mol DSPE-
CPL-QS (total volume 300 uL) at 60°C for 3 hours, following which the
sample was
applied to a column of Sepharose CL-4B equilibrated in HEPES-buffered saline.
1 mL
fractions were collected and assayed for dansyl-labelled CPL and rhodamine-PE
as
described herein. Panel B illustrates the insertion of DSPE-CPL-QS into LUVs
(100 nm)
composed of DOPE/DODAC/PEG-Cer-C20 (84/6/10). LUVs (5 p,mol lipid) were
incubated with 0.43 ~.mol DSPE-CPL-QS (total volume ~ 19 q.l) at 60°C
for 3 hours,
following which the sample was applied to a column of Sepharose CL-4B
equilibrated in
HEPES-buffered saline. The qlution of free CPL is also shown, demonstrating a
straightforward method for isolation of the CPL-LUV. 1 mL fractions were
collected and
assayed for dansyl-labelled CPL and rhodamine-PE as described herein. Panel C
illustrates retention of DSPE-CPL-QS in LUVs (100 nm) composed of
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
13
DOPE/DODAC/PEG-Cer-C20 (84/6/10). The main LLTV fraction from Fig. 9 Panel B
was re-applied to a column of Sepharose CL-4B equilibrated in HEPES-buffered
saline.
1 mL fractions were collected and assayed for dansyl-labelled CPL and
rhodamine-PE as
described.
Figure 10 illustrates the effect of time and temperature on the insertion of
d-DSPE-CPL-Q1 into DOPE/DODAC%PEG-Cer-C20 LLTVs. For each of the 3
temperatures, 3 ~.mol lipid was combined with 0.17 umol CPL (total volume 240
ql). At
l, 3. and 6 hours, 1 pmol of lipid was withdrawn and cooled in ice to halt
insertion of
CPL. The samples were passed down a column of Sepharose CL-4B to remove excess
CPL, and assayed for CPL insertion.
Figure 11 illustrates the effect of initial CPL/lipid ratio on final CPL
insertion levels. Initial CPL/lipid molar ratios were 0.011, 0.024, 0.047,
0.071, 0.095,
and 0.14. Final mol % inserted were 0.8, 1.8, 3.4, ~.0, 6.~, and 7Ø Right-
hand axis is
-insertion.
Figure 12 illustrates the insertion of DSPE-CPL-Q1 and DSPE-CPL-QS
into neutral vesicles. The initial CPL/lipid molar ratio was 0.065 for Q 1
(2.~ pmol lipid
and 0.21 umol CPL) and 0.034 for Q5. Samples were incubated at 60°C for
3 hours. The
DOPC and DOPC/Chol LL'Vs were prepared by extrusion, while the others were
prepared by detergent dialysis. As described herein, the presence of 4%
methanol in the
QS samples appear to account for the higher insertion observed for this
sample. Sample
compositions were as follows: DOPC/Chol (55/45), DOPC/PEG-Cer-C20 (90/10),
DOPC/Chol/PEG-Cer-C20 (45/45/10).
Figure 13 illustrates the effect of chain length of PEG-Cer on mol-% CPL
inserted. LUVs composed of DOPE/DODAC/PEG-Cer-C20 (84/6/10),
DOPE/DODAC/PEG-Cer-C14 (84/6/10), and DOPE/DODAC/PEG-Cer-C8 (79/6/15)
were incubated in the presence of between 2-8.6 mol % d-DSPE-CPL-Q1 at
60°C for 3
hrs.
Figure 14 illustrates the effect of PEG-Cer-C20 content on insertion of d-
DSPE-CPL-QS. Vesicles composed of DOPC/DODAC/PEGCerC20, with the latter lipid
ranging from 4-10 mol %, were incubated in the presence of CPL-QS (initial
CPL/lipid
molar ratio = 0.071).
Figure 15 illustrates the uptake of CPL-LLIVs incubated in PBS/CMG on
BHK cells. Approximately 105 BHK cells were incubated with 20 nmol of
DOPE/DODAC/PEGCerC20 (84/6/10) LUVs containing (1) no CPL, (2) 8% DSPE-
iz
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
14
CPL-D, (3) 7% DSPE-CPL-T1, and (4) 4% DSPE-CPL-Q~. Incubations were performed
at 4°C and 37°C, the former giving an estimate of cell binding,
and the latter of binding
and uptake. By taking the difference of the two values, an estimate of lipid
uptake at
37°C was obtained.
Figure 16 Panel A illustrates a structure of the CPL4. Panel B illustrates a
protocol for the insertion of CPL4 into the SPLP system.
Figure 17 Panel A illustrates a model for DOPE/DODAC/PEG-Cer-C20
LUVs, i.e., a standard liposome containing a PEG-lipid (or "stealth" lipid).;
Panel B
illustrates the same LLJVs with CPL (i.e. long chain) inserted. "Long chain"
refers to the
polymer W being the same length or greater length than the polymer component
of the
PEG-lipid. Thus, the charged group of the CPLI is immediately exposed to the
outside
environment.; and Panel C illustrates the same LWs with CPL, with a short
chain
inserted. A "short chain" CPL, wherein polymer W is shorter than the
corresponding
polymer of the PEG-lipid.
Figure 18 Panel A illustrates a time-course for the uptake of SPLP system
(~) compared to DOPE:DODAC (l:l) liposomes complexed to pLuc (~) on BHK cells.
Lipid concentration was 20 ~,M. Panel B illustrates transfection efficiencies
of 1.~
pg/mL pLuc obtained using the SPLP system compared to those obtained using
complexes after 4 hour (~) or 8 hour (~) incubations.
Figure 19 illustrates a column profile, following insertion of 3.5 mol
%initial(~ mol % final) CPLa into SPLP, for the separation of CPL-SPLP from
free CPL.
Profiles for lipid (~), CPL (a), and DNA (0) with respect to the total amount
applied to a
Sepharose CL-4B column are shown. Panel B shows the column profile for
Fraction #9
from Panel A.
Figure 20 illustrates a time course for the insertion of CPL, (15 nmol) into
SPLP (200 nmol).
Figure 21 illustrates a time course for the uptake of 20 p.M of SPLP
possessing 0% (~), 3% (0), or 4% (~) CPL4 in BHK cells.
Figure 22 illustrates transfection of BHK cells by SPLP (2.5 pg/mL pLuc)
following insertion of various mol % of the CPL4 compared to SPLP alone (0%
CPL).
Transfections were carried out by incubating the samples on top of the cells
for 4 or 9
hours and replacing with complete media for a complete 24 hours incubation
(see also
Figure 33).
Figure 23 tabulates CPL insertion results.
SUBSTTTUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
Figure 24 also tabulates CPL insertion results.
Figure 25 illustrates the post-insertion method for preparation of CPL-
containing liposomes. The preformed liposomes were made of DSPC/Chol (5:45,
mol:mol). The CPL was incubated with the preformed liposomes at 60°C
for 2 hour.
Panel A illustrates separation of free CPL and CPL-LLJVs by gel filtration
after post-
insertion. Panel B illustrates elution of fraction 10 (Panel A) on a Sepharose
CL-4B
column.
Figure 26 illustrates cellular uptake of the stealth liposomes containing
DSPE-CPLs in BHK cells in DMEM (10% FBS). Control LLJVs (DSPC/Chol/PEG-PE,
10 56:40:4) and CPL-LUVs (DSPC/Chol/PEG-PE/CPL, ».5:40:2:2) were prepared by
extrusion as described herein.
Figure 27 illustrates cellular uptake of stealth liposomes containing
DSPE-CPLs in BHK cells in PBS-CMG. Control LUVs (DSPC/Chol/PEG-PE, X6:40:4)
and CPL-LUVs (DSPC/Chol/PEG-PE/CPL, ».x:40:2:2) were prepared by extrusion as
15 described herein.
Figure 28 Panel A: Chemical structures of various CPLs; Panel B:
Chemical structures of various CPLs. Note that CPL: (Panel A) is identical to
CPL.~b
(Panel B); and Panel C: Chemical structures of various CPLs.
Figure 29 illustrates a synthetic embodiment to generate compounds of
the present invention.
Figure 30 illustrates a structure of dansylated CPL4. CPL. possesses four
positive charges at the end of a PEG3aoo molecule which is attached to a DSPE
molecule.
The CPLa is dansylated by incorporation of a dansylated lysine.
Figure 31 illustrates an effect of cation concentration on the
deaggregation of SPLP-CPL.. The mean diameter and standard deviation of the
particles
in the presence of increasing [Cation], Caz+ (~) and M~z+ (~), from 0 mM to 70
mllil,
was measured using quasi-elastic light scattering (QELS). To 180 nmol of SPLP-
CPLa
in 400 mL in a Nicomp tube was added small quantities of either CaCh or M~CIZ
(500
mM stock solutions). Measurement of the mean diameter ~ standard deviation of
the
particles in the presence of differing amounts of the cation were made using a
Nicomp
Model 270 Submicron Particle Sizer. The diameters of the particles do not
dramatically
change, however, the Gaussian distributions do get broader. Thus, the standard
SUBSTTI'LTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
16
deviations were used as a measure of deaggregation with smaller deviations
indicating
less aggregation.
Figure 32 illustrates uptake of SPLP containing various percentages of
CPL.. Panel A. Time course for the uptake of 20 ~iVI SPLP possessing 0 mol %
(~), 2
mol % (~), 3 mol % (1), or 4 mol % (~) CPL. and DOPE:DODAC complexes (~) by
BHK cells. The insertion of the CPL: into SPLP and the preparation of
complexes was
performed as described herein The mol % of CPL. in the SPLP-CPL4 was also
determined, as described herein. BHK cells were plated in 12-well plates at
1x10'
cells/well. To 200 ~.L of sample (containing SPLP-CPL. or complex + CaCI~) was
added
800 ~L of DMEM + 10% FBS. The resulting CaCI~ concentration was diluted to 20%
of
the original. Following incubation periods of 2, 4, 6 and 8 hours, the cells
were lysed with
600 mL of lysis buffer and the rhodamine fluorescence and BCA assays were
measured
for the lysate, as described herein (see Figure 21).
Figure 33 illustrates tansfection of BHK cells by SPLP (~.0 ~g/mL pLuc)
following insertion of various mole percentages of CPL (2, 3, and 4 mol %).
The CPL.
was inserted into SPLPs using the procedure described herein. As a comparison,
SPLP (0
mol % CPL) and DOPE:DODAC (1:1) complex transfections were also performed.
BHK cells were plated at 1x10' in 96-well plates. Transfections were carried
out by
incubating the samples [20 ~,L (SPLP-CPLa + CaCl2) + 80 ~L of complete media]
on the
cells for 4 hours followed by a 24 hour complete incubation. The CaCh
concentration
again is diluted to 20% of the original concentration. Following the 24 hour
incubation,
the cells were lysed with lysing buffer and the luciferase and BCA assays were
performed
(see Figure 22).
Figure 34 illustrates the effect of [Cation], Ca2+ (~) and Mg~T (~), on the
transfection of SPLP-CLP.~ (5.0 ug/mL pLuc) on BHK cells. SPLP-CPL4 + CaCl2 or
MgCl2 was mixed with DMEM +10% FBS and the mixtures were applied to 1x10' BHK
cells plated in a 96-well plate. Following a complete 48 hour incubation, the
transfection
media was removed and the cells were lysed with lysing buffer and the
luciferase activity
and protein content were measured as described earlier.
Figure 35 illustrates the effect of [Cation], Ca2+ (~) and Mg'- (~), on the
lipid binding and uptake of 80 ~M SPLP-CPLa on BHK cells. The samples
possessing
varying concentrations of the cation (0-14 mM final concentration) were
incubated on
1x105 BHK cells for 4 hours at which time the cells were lysed and the
rhodamine
fluorescence and protein content were measured.
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
17
Figure 36 illustrates transfection of SPLP-CPL., SPLP and complexes
(each containing ~.0 pg/mL pCMVLuc) at longer time points. Transfection of
SPLP-
CPLa (4 mol % CPL) + 40 mM;";~;a, CaCh (~), SPLP (1), DOPE:DODAC complexes
(~), and Lipofectin complexes (~) was performed on 1x10' BHK cells. The
transfection
media was incubated on the cells for 4, 8 or 24 hours, after which the
transfection media
was replaced by complete media for the 4 and 8 hour timepoints. Then at a
total
incubation time of 24 hours (20, 16, and 0 hours, respectively, after removal
of the
transfection media), the cells were lysed and the luciferase activity and
protein content
were measured.
Figure 37 illustrates transfection potency and toxicity of SPLP-CPL.
compared to Lipofectin complexes. A. Transfection activity for SPLP-CPL. +
CaCh (~),
SPLP (~), and Lipofectin (~) on 1x10' BHK cells incubated for 24 and 48 hours
followed by immediate cell lysis, and measurement of luciferase activity and
protein
content. B. Measurement of the cellular survival following 24 and 48 hour
incubations of
the SPLP-CPL. + CaCh (~), Lipofectin (~), and DOPE/DODAC (l:l) complexes on
1x10' BHK cells. Following incubation, the cells were lysed and the protein
content
from the BCA assay was used as a measure of protein survival.
Figure 38 illustrates the transfection of BHK cells using both long and
short chained CPLs. The presence of the short chained PEG in the CPL results
in a
decrease by a factor of about 4 compared to the transfection by the long
chained CPL.
Figure 39 illustrates the transfection of Neuro-2a cells. SPLP + 4 mol
CPL4-lk produces 4 orders of magnitude of gene expression more than SPLP alone
in
Neuro-2a cells.
Figure 40 illustrates in vivo pharmacokinetics of SPLP containing a short
chain CPL,.
DETAILED DESCRIPTION OF THE INVENTION
AND PREFERRED E1VIBODIMENTS
A. Compounds And Synthesis
In certain aspects, the present invention provides cationic-polymer-lipid
conjugates (CPLs), such as distal cationic-polyethylene glycol)-lipid
conjugates that can
be incorporated into conventional and stealth liposomes for enhancing, inter
alia, cellular
SUBSTI'TL1TE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
18
uptake. The CPLs of the present invention have the following architectural
features: ( 1 )
a lipid anchor, such as a hydrophobic lipid, for incorporating the CPLs into
the lipid
bilayer; (2) a hydrophilic spacer, such as a polyethylene glycol, for linking
the lipid
anchor to a cationic head group; and (3) a polycationic moiety, such as a
naturally
occurring amino acid, to produce a protonizable cationic head group. As such,
the
present invention provides a compound of Formula I:
A W Y
wherein A, W and Y are as previously defined.
With reference to Formula I. "A" is a lipid moiety such as an amphipathic
lipid, a neutral lipid or a hydrophobic lipid that acts as a lipid anchor.
Suitable lipid
examples include vesicle-forming lipids or vesicle adopting lipids and
include, but are
not limited to, diacylglycerolyls, dialkylglycerolyls, N-N-dialkylaminos, 1,2-
diacyloxy-3-
aminopropanes and 1,2-dialkyl-3-aminopropanes.
"W" is a polymer or an oligomer, such as a hydrophilic polymer or
oligomer. Preferably, the hydrophilic polymer is a biocompatable polymer that
is non-
immunogenic or possesses low inherent immunogenicity. Alternatively, the
hydrophilic
polymer can be weakly antigenic if used with appropriate adjuvants. Suitable
non-
immunogenic polymers include, but are not limited to, PEG, polyamides,
polylactic acid,
polyglycolic acid, polylactic acid/polyglycolic acid copolymers and
combinations
thereof. In a preferred embodiment, the polymer has a molecular weight of
about 250 to
about 7000 daltons.
"Y" is a polycationic moiety. The term polycationic moiety refers to a
compound, derivative, or functional group having a positive charge, preferably
at least 2
positive charges at a selected pH, preferably physiological pH. Suitable
polycationic
moieties include basic amino acids and their derivatives such as arginine,
asparagine,
glutamine, lysine and histidine; spermine; spermidine; cationic dendrimers;
polyamines;
polyamine sugars; and amino polysaccharides. The polycationic moieties can be
linear,
such as linear tetralysine, branched or dendrimeric in structure. Polycationic
moieties
have between about 2 to about 15 positive charges, preferably between about 2
to about
12 positive charges, and more preferably between about 2 to about 8 positive
charges at
selected pH values. The selection of which polycationic moiety to employ may
be
determined by the type of liposome application which is desired.
SUBSTTTUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
19
The charges on the polycationic moieties can be either distributed around
the entire liposome moiety, or alternatively, they can be a discrete
concentration of
charge density in one particular area of the liposome moiety e. J., a charge
spike. If the
charge density is distributed on the liposome, the charge density can be
equally
distributed or unequally distributed. All variations of charge distribution of
the
polycationic moiety are encompassed by the present invention.
The lipid "A", and the non-immunogenic polymer "W", can be attached
by various methods and preferably, by covalent attachment. Methods known to
those of
skill in the art can be used for the covalent attachment of "A" and "W".
Suitable
linkages include, but are not limited to, amide, amine, carboxyl, carbonate,
carbamate,
ester and hydrazone linkages. It will be apparent to those skilled in the art
that "A" and
"W" must have complementary functional groups to effectuate the linkage. The
reaction
of these two groups, one on the lipid and the other on the polymer, will
provide the
desired linkage. For example, when the lipid is a diacylglycerol and the
terminal
hydroxyl is activated, for instance with NHS and DCC, to form an active ester,
and is
then reacted with a polymer which contains an amino group, such as with a
polyamide
(see, U.S. patent Application No. 09/218,988, filed December 22, 1998), an
amide bond
will form between the two groups.
In certain embodiments, "W" is bound, preferably covalently bound, to
"Y". As with "A" and "W", a covalent attachment of "W" to "Y" can be generated
by
complementary reactivity of functional groups, one on the polymer and the
other on the
polycationic moiety. For example, an amine functional group on "W" can be
reacted
with an activated carboxyl group, such as an acyl chloride or NHS ester, to
form an
amide. By suitable choice of reactive groups, the desired coupling can be
obtained.
Other activated carboxyl groups include, but are not limited to, a carboxylic
acid, a
carboxylate ester, a carboxylic acid halide and other activated forms of
carboxylic acids,
such as a reactive anhydride. Reactive acid halides include for example, acid
chlorides,
acid bromides, and acid fluorides.
In certain instances, the polycationic moiety can have a ligand attached,
such as a targeting ligand. Preferably, after the ligand is attached, the
cationic moiety
maintains a positive charge. In certain instances, the ligand that is attached
has a positive
charge. Suitable ligands include, but are not limited to, a compound or device
with a
reactive functional group and includes lipids, amphipathic lipids, carrier
compounds,
bioaffinity compounds, biomaterials, biopolymers, biomedical devices,
analytically
SUBSTTTUTE SHEET (RULE 26)
WO00/62813 ~ 02370690 2001-10-19 pCT/CA00/00451
detectable compounds, therapeutically active compounds, enzymes, peptides,
proteins,
antibodies, immune stimulators, radiolabels, fluorogens, biotin, drugs,
haptens, DNA,
RNA, polysaccharides, liposomes, virosomes, micelles, immunoglobulins,
functional
Groups, other targeting moieties, or toxins.
In certain preferred embodiments, other moieties are incorporated into the
compounds of Formula I to form the compounds of Formula II:
A X (CH, CH, O)" Z Y
II
In Formula II, "A" is a lipid moiety such as, an amphipathic lipid, a neutral
lipid or a
hydrophobic lipid moiety. Suitable lipid examples include. but are. not
limited to,
10 diacylglycerolyl, dialkylglycerolyl, N-N-dialkylamino, 1,2-diacyloxy-3-
aminopropane
and 1,2-dialkyl-3-aminopropane.
In Formula II, "X" is a single bond or a functional group that covalently
attaches the lipid to at least one ethylene oxide unit. Suitable functional
groups include,
but are not limited to, phosphatidylethanolamino, phosphatidylethanolamido,
phosphoro,
15 phospho, phosphoethanolamino, phosphoethanolamido, carbonyl, carbamate,
carboxyl,
carbonate, amido, thioamido, oxygen, NR wherein R is a hydrogen or alkyl group
and
sulfur. In certain instances, the lipid "A" is directly attached to the
ethylene oxide unit by
a single bond. The number of ethylene oxide units can range from about 1 to
about 160
and preferably from about 6 to about ~0.
20 In Formula II, "Z" is a single bound or a functional croup that covalently
attaches the ethylene oxide unit to the polycationic moiety. Suitable
functional groups
include, but are not limited to, phospho, phosphoethanolamino,
phosphoethanolamido,
carbonyl, carbamate, carboxyl, amido, thioamido, NR wherein R is a member
selected
from the group consisting of hydrogen atom or alkyl group. In certain
embodiments, the
terminal ethylene oxide unit is directly attached to the polycationic moiety.
In Formula II, "Y" is a polycationic moiety as described above in
connection with Formula I. In Formula II, the index "n" is an integer ranging
in value
from about 6 to about 160.
In an illustrative embodiment, compounds of Formula II can be
synthesized using a generalized procedure as outlined in Figure 2. Figure 2
illustrates
one particular embodiment of the present invention and thus, is merely an
example that
SUBSTIT'IJTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
21
should not limit the scope of the claims herein. Clearly, one of ordinary
skill in the art
will recognize many other variations, alternatives, and modifications that can
be made to
the reaction scheme illustrated in Figure 2. With reference to Figure 2, a
solution of a
lipid, such as DSPE, and a base, such as triethylamine in a chloroform
solution is added
to (t-Boc-NH-PEG3.~oo-C42NHS), and the solution is stirred at ambient
temperature. The
solution is then concentrated under a nitrogen stream to dryness. The residue
is then
purified by repeated precipitation of the chloroform mixture solution with
diethyl ether
until disappearance of the lipid using chromatography. The purified CPL
conjugate is
dissolved in a solvent, followed by addition of TFA, and the solution is
stirred at room
temperature. The solution can again be concentrated under a nitrogen stream.
The
residue is then purified by repeated precipitation of the mixture with diethyl
ether to offer
a lipid-PEG-NHS, such as a DSPE-PEG-NHS or, alternatively, DSPE-CPL-1 with one
protonizable cationic head group. The ratio of the phosphoryl-lipid anchor and
the distal
primary amine can then be measured by phosphate and flourescamine assays as
described
herein.
In this illustrative embodiment, the number of protonizable amino groups
can be increased to create a polycationic moiety. By incrementally adding
stoichiometric amounts of, for example, a Na,Ns-di-t-Boc-L-Lysine N-
hydroxysuccinide
ester, the polycationic moiety can be increase from about 2 to about 16
positive charges.
As describe previously, the positive charges can be incorporated using any
number of
suitable polycationic moieties such as lysine, arginine, asparagine,
glutamine, histidine,
polyamines and derivatives or combinations thereof. Using the synthesis
methods of the
present invention, the number of cationic groups, such as amino groups, can be
readily
controlled during the CPL synthesis.
B. Lipid-Based Drug Formulations
In certain aspects, the present invention provides a lipid-based drug
formulation comprising:
(a) a compound having the general structure of Formula I:
A W Y
wherein A, W and Y are as previously defined; (b) a bioactive agent; and
optionally, (c) a second lipid. In preferred embodiments, the lipid-based drug
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
22
formulation of the present invention comprises the second lipid, such as a PEG-
lipid
derivative.
In certain preferred embodiments, the lipid-based drug formulations of
the present invention comprise
(a) a compound of Formula II:
A X (CHI CH,-0)~ Z Y
II
wherein A, X, Z, Y and n have been previously defined; (b) a bioactive agent;
and
optionally, (c) a second lipid. In preferred embodiments, the lipid-based drug
formulation of the present invention comprises the second lipid, such as a PEG-
lipid
derivative.
After the CPLs have been prepared, they can be utilized in a variety of
ways including, for example, in lipid-based drug formulations. In this aspect,
the lipid-
based formulations can be in the form of a liposome, a micelle, a virosome, a
lipid-
nucleic acid particle, a nucleic acid aggregate and other forms which can
incorporate or
entrap one or more bioactive agents. In certain aspects, the lipid-based drug
formulations
of the present invention comprise a second lipid.
The compounds of Formulae I and II can be used in lipid-based
formulations such as those described in for example, the following copending
U.S. Patent
Applications Serial Numbers 08/454,641, 08/485,458, 08/660,025, 08/484,282,
60/055,094, 08/856,374, 60/053,813 and 60/063,473, entitled "Methods for
Encapsulating Nucleic Acids in Lipid Bilayers," filed on October 10, 1997 and
bearing
Attorney Docket No. 016303-004800, U.S. Patent No. 5,703,055, U.S. Patent
Application
No. 09/218,988, filed December 22, 1998, the teachings all of which are
incorporated
herein by reference in their entirety for all purposes. This specification
sets out a variety
of liposome types and a variety of methods for incorporating CPLs into
liposomes, all of
which are examples of the broad methods and compositions claimed herein.
The lipid components and CPLs used in forming the various lipid-based
drug formulations will depend, in part, on the type of delivery system
employed. For
instance, if a liposome is employed, the lipids used in the CPL will generally
be selected
from a variety of vesicle-forming or vesicle-adopting lipids, typically
including
SUBSTITUTE SKEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
23
phospholipids and sterols, such as phosphatidylenthanolamine (PE),
phosphatidylserine
(PS), phosphatidylinositol (PI), phosphatidylglycerol (PG), phosphatidic acid
(PA),
which have been suitably functionalized, and the like. In contrast, if a
micelle is
employed, the lipids used in the CPL will generally be selected from
sterylamines,
alkylamines, Cg-C22 alkanoic acids, lysophospholipids, detergents and the
like. It will
be readily apparent to those of skill in the art that the acyl chains can be
varied in length
and can be saturated or possess varying degrees of unsaturation. The more
saturated the
acyl chains the more rigid the membrane. Higher degrees of unsaturation impart
more
fluidity into the vesicle's membrane. Similarly, the other lipid components
(e.g., lipids,
cationic lipids, neutral lipids, non-cationic lipids, etc.) making up the drug
delivery
systems of the present invention will vary depending on the drug delivery
system
employed. Suitable lipids for the various drug delivery systems will be
readily apparent
to those of skill in the art.
When the lipid-based drug formulations are used to deliver therapeutic
genes or oligonucleotides intended to induce or to block production of some
protein
within the cell, cationic lipids can be included in the formulation, e.g.,
liposome, micelle,
lipid-nucleic acid particle, etc. Nucleic acid is negatively charged and can
be combined
with a positively charged entity to form a lipid complex suitable for
formulation and
cellular delivery.
As used in this specification, "cationic lipid" generally refers to a lipid
with a cationic head group situated at or near the liposome membrane (when
incorporated
in a liposome). CPLs are distinguished from cationic lipids by the polymer "W"
which in
certain instances, has the effect of placing the cationic charge at a
significant distance
from the membrane.
Examples of suitable cationic lipids include, but are not limited to, the
following: DC-Chol, (see, Gao, et al., Biochem. Biophys. Res. Comm., 179:280-
285
(1991); DDAB; DMRIE; DODAC (see, United States Patent Application Serial
Number
08/316,399, filed September 30, 1994, which is incorporated herein by
reference);
DOGS; DOSPA; DOTAP; and DOTMA. In a presently preferred embodiment, N ,N
dioleoyl-N,N dimethylammonium chloride is used in combination with a
phosphatidylethanolamine.
In addition, other cationic lipids useful in producing lipid-based carriers
for gene and oligonucleotide delivery are LIPOFECTIN (U.S. Patents Nos.
4,897,35;
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
24
4,946,787; and 5,208,036 issued to Eppstein, et al.) and LIPOFECTACE (U.S.
Patent
No. x,279,883 issued to Rose). Both agents, as well as other transfecting
cationic lipids,
are available from Life Technologies, Inc. in Gaithersburg, Maryland.
In one preferred embodiment, the CPL-liposomes of the present invention
are optimized for systemic delivery applications. In certain applications, the
polymer
length in the CPL is shorter than the normal neutral PEG chains (M.W. 2000-
5000
Daltons) used for stealth liposomes. In this instance, the shorter polymer in
the CPL is
about 250 to about 3000 Daltons and more preferably, about 1000 to about 2000
Daltons.
In this embodiment, the second lipid is for example. a PEG;.~oo-lipid and the
compound of
Formula I is, for example, A- PEG~ooo-1'. (see, Figure 17C).
Without being bound by any particular theory, when the shorter polymer is
used, it is believed that the distal charges) of the CPL is hidden within the
normal PEG
exclusion barrier, thus allowing retention of long circulation lifetimes while
at the same
time, extending the positive charges away from the liposomal surface. This
embodiment
enhances interactions between liposomes and a target cell. The use of
different sized
polymers, such as PEG chains, in the CPLs and the neutral PEG-lipids used to
modulate
vesicle circulation and cellular uptake, allows for a new generation of
stealth liposomes
as drug carriers. It is believed that the optimized polymer length can vary
with the
specific conditions such as in vitro or in vivo applications, local or
systemic
administration, and different lipid formulations.
In another embodiment, the polymer length in the CPL has a larger MW
than the normal neutral PEG chains used for stealth liposomes. In this
instance, the
second lipid is for example, a PEG~ooo-lipid and the compound of Formula I has
a
formula of, for example, A- PEG3.~oo-Y. (see, Figure 17B).
In certain formulations and applications, the type of CPL i.e. the length of
the polymer chain, and the amount of cationic charge per molecule, and the
amount of
such CPL in a formulation e.g., SPLP, can be optimized to obtain the best
balancing of
clearance properties. In certain instances, long chain CPLs and higher levels
of such
CPLs are to be preferred to increase transfection. In other instances, short
chain CPLs
incorporated in the formulations are optimized for longer circulation
lifetimes in animals.
In one embodiment of the present invention, a fusogenic liposome or
virosome is provided. It will be readily apparent to those of skill in the art
that the CPLs
of the present invention can advantageously be incorporated into various types
of
fusogenic liposomes and virosomes. Such fusogenic liposomes and virosomes can
be
SUBSTTTUTE SKEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
designed to become fusogenic at the disease or target site. Those of skill in
the art will
readily appreciate that a number of variables can be used to control when the
liposome or
virosome becomes fusogenic. Such variables include, for example, the
composition of
the liposome or virosome, pH, temperature, enzymes, cofactors, ions, etc.
In one embodiment, the fusogenic liposome comprises: a lipid capable of
adopting a non-lamellar phase, yet capable of assuming a bilayer structure in
the presence
of a bilayer-stabilizing component (such as a PEG-lipid derivative); and a
bilayer-
stabilizing component reversibly associated with the lipid to stabilize the
lipid in a bilayer
structure. Such fusogenic liposomes are advantageous because the rate at which
they
10 become fusogenic can be not only predetermined, but varied as required over
a time scale
of a few minutes to several tens of hours. It has been found, for example,
that by
controlling the composition and concentration of the bilayer-stabilizing
component, one
can control the rate at which the BSC exchanges out of the liposome i~t vivo
and, in turn.
the rate at which the liposome becomes fusogenic (see, U.S. Patent No.
5,88,613). For
15 instance, it has been found that by controlling the length of the lipid
acyl chain(s), one
can control the rate at which the BSC exchanges out of the liposome in vivo
and, in turn,
the rate at which the liposome becomes fusogenic. In particular, it has been
discovered
that shorter acyl chains (e.g., C-8) exchange out of the liposome more rapidly
than longer
acyl chains (e.g., C-20). Alternatively, by controlling the composition and
concentration
20 of the BSC, one can control the rate at which the BSC is degraded, i.e.,
broken down, by
endogenous systems, e.g., endogenous enzymes in the serum, and, in turn, the
rate at
which the liposome becomes fusogenic.
The polymorphic behavior of lipids in organized assemblies can be
explained qualitatively in terms of the dynamic molecular shape concept (see,
Cullis, et
25 al., in "Membrane Fusion" (Wilschut, J. and D. Hoekstra (eds.), Marcel
Dekker, Inc.,
New York, (1991)). When the effective cross-sectional areas of the polar head
group and
the hydrophobic region buried within the membrane are similar then the lipids
have a
cylindrical shape and tend to adopt a bilayer conformation. Cone-shaped lipids
which
have polar head groups that are small relative to the hydrophobic component,
such as
unsaturated phosphatidylethanolamines, prefer non-bilayer phases such as
inverted
micelles or inverse hexagonal phase (H ). Lipids with head groups that are
large relative
to their hydrophobic domain, such as lysophospholipids, have an inverted cone
shape and
tend to form micelles in aqueous solution. The phase preference of a mixed
lipid system
depends, therefore, on the contributions of all the components to the net
dynamic
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
26
molecular shape. As such, a combination of cone-shaped and inverted cone-
shaped lipids
can adopt a bilayer conformation under conditions where either lipid in
isolation cannot
(see, Madden and Cullis, Biochinz. Biophys. Acta, 684:149-153 (1982)).
A more formalized model is based on the intrinsic curvature hypothesis
(see, e.g., Kirk, et al., Biochemistry, 23:1093-1102 (1984)). This model
explains
phospholipid polymorphism in terms of two opposing forces. The natural
tendency of a
lipid monolayer to curl and adopt its intrinsic or equilibrium radius of
curvature (Ro)
which results in an elastically relaxed monolayer is opposed by the
hydrocarbon packing
constraints that result. Factors that decrease the intrinsic radius of
curvature, such as
increased volume occupied by the hydrocarbon chains when double bonds are
introduced,
tend to promote H phase formation. Conversely, an increase in the size of the
headgroup
increases Ro and promotes bilayer formation or stabilization. Introduction of
apolar
lipids that can fill the voids between inverted lipid cylinders also promotes
H phase
formation (see, Gruner, et al., Proc. Natl. Acad. Sci. USA, 82:3665-3669
(1989); Sjoland,
et al., Biochemistn~, 28:1323-1329 (1989)).
As such, in one embodiment, the lipids which can be used to form the
fusogenic liposomes of the present invention are those which adopt a non-
lamellar phase
under physiological conditions or under specific physiological conditions,
e.g., in the
presence of calcium ions, but which are capable of assuming a bilayer
structure in the
presence of a BSC. Such lipids include, but are not limited to,
phosphatidylenthanolamines, ceramides, glycolipids, or mixtures thereof. Other
lipids
known to those of skill in the art to adopt a non-lamellar phase under
physiological
conditions can also be used. Moreover, it will be readily apparent to those of
skill in the
art that other lipids can be induced to adopt a non-lamellar phase by various
non-
physiological changes including, for example, changes in pH or ion
concentration (e.g.,
in the presence of calcium ions) and, thus, they can also be used to form the
fusogenic
liposomes of the present invention. In a presently preferred embodiment, the
fusogenic
liposome is prepared from a phosphatidylethanolamine. The
phosphatidylethanolamine
can be saturated or unsaturated. In a presently preferred embodiment, the
phosphatidylyethanolamine is unsaturated. In an equally preferred embodiment,
the
fusogenic liposome is prepared from a mixture of a phosphatidylethanolamine
(saturated
or unsaturated) and a phosphatidylserine. In another equally preferred
embodiment, the
SUBSTIT'LTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
27
fusogenic liposome is prepared from a mixture of a phosphatidylethanolamine
(saturated
or unsaturated) and a cationic lipid.
In one embodiment, the lipid-based drug formulations of the present
invention comprise a bilayer stabilizing component (BSC). Suitable BSCs
include, but
are not limited to, polyamide oligomers, peptides, proteins, detergents, lipid-
derivatives,
PEG-lipids such as PEG coupled to phosphatidylethanolamine, and PEG conjugated
to
ceramides (see, U.S. Patent No. 5,885,613, which is incorporated herein by
reference).
Preferably, the bilayer stabilizing component is a PEG-lipid, or an ATTA-
lipid. As
discussed herein, in certain preferred instances, the PEG or the ATTA of the
BSC has a
greater molecular weight compared to the polymer "W" of the CPL. In other
instances,
the BSC has a smaller molecular weight compared to the "W" of the polymer. The
present invention encompasses all such variations.
In accordance with the present invention, lipids adopting a non-lamellar
phase under physiological conditions can be stabilized in a bilayer structure
by BSCs
which are either bilayer forming themselves, or which are of a complementary
dynamic
shape. The non-bilayer forming lipid is stabilized in the bilayer structure
only when it is
associated with, i.e., in the presence of, the BSC. In selecting an
appropriate BSC, it is
preferable that the BSC be capable of transferring out of the liposome, or of
being
chemically modified by endogenous systems such that, with time, it loses its
ability to
stabilize the lipid in a bilayer structure. Only when liposomal stability is
lost or
decreased can fusion of the liposome with the plasma membrane of the target
cell occur.
The BSC-lipid, therefore, is "reversibly associated" with the lipid and only
when it is
associated with the lipid is the lipid constrained to adopt the bilayer
structure under
conditions where it would otherwise adopt a non-lamellar phase. As such, the
BSC-lipids
of the present invention are capable of stabilizing the lipid in a bilayer
structure, yet they
are capable of exchanging out of the liposome, or of being chemically modified
by
endogenous systems so that, with time, they lose their ability to stabilize
the lipid in a
bilayer structure, thereby allowing the liposome to become fusogenic.
Typically, the CPL is present in the lipid-based formulation of the present
invention at a concentration ranging from about 0.05 mole percent to about 50
mole
percent. In a presently preferred embodiment, the CPL is present at a
concentration
ranging from 0.05 mole percent to about 25 mole percent. In an even more
preferred
embodiment, the CPL is present at a concentration ranging from 0.05 mole
percent to
about 15 mole percent. One of ordinary skill in the art will appreciate that
the
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
28
concentration of the CPL can be varied depending on the CPL employed and the
rate at
which the liposome is to become fusogenic.
In one embodiment of the present invention, the liposomes contain
cholesterol. It has been determined that when cholesterol-free liposomes are
used in vivo,
they have a tendency to absorb cholesterol from the plasma lipoproteins and
cell
membranes. Cholesterol, if included, is generally present at a concentration
ranging from
0.2 mole percent to about 50 mole percent and, more preferably, at a
concentration
ranging from about 35 mole percent to about 4~ mole percent.
C. Preparation of CPL-Liposomes
A variety of general methods for making CPL-containing liposomes (or
"CPL-liposomes") are discussed herein.
Two general techniques include "post-insertion," that is, insertion of a
CPL into for exam;~ie, a pre-formed liposome vesicle, and "standard"
techniques,
wherein the CPL is included in the lipid mixture during for example, the
liposome
formation steps. The post-insertion technique results in liposomes having CPLs
mainly
in the external face of the liposome bilayer membrane, whereas standard
techniques
provide liposomes having CPLs on both internal and external faces.
In particular, "post-insertion" involves forming vesicles (by any method),
and incubating the pre-formed vesicles in the presence of CPL under
appropriate
conditions (usually 2-3 hours at 60°C). Between 60-80% of the CPL can
be inserted into
the external leaflet of the recipient vesicle, giving final concentrations up
to 7 mol
(relative to total lipid). The method is especially useful for vesicles made
from
phospholipids (which can contain cholesterol) and also for vesicles containing
PEG-lipids
(such as PEG-Ceramide).
In an example of a "standard" technique, the CPL-LUVs of the present
invention can be formed by extrusion. In this embodiment, all of the lipids
including
CPL, are co-dissolved in chloroform, which is then removed under nitrogen
followed by
high vacuum. The lipid mixture is hydrated in an appropriate buffer, and
extruded
through two polycarbonate filters with a pore size of 100 nm. The resulting
vesicles
contain CPL on both internal and external faces. In yet another standard
technique, the
formation of CPL-LUVs can be accomplished using a detergent dialysis or
ethanol
dialysis method, for example, as discussed in U.S. Patent Nos. 5,976,567 and
5,981,501,
SUBSTITUTE SKEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
29
both of which are incorporated herein by reference. The extrusion method and
the
detergent dialysis method are explained in detail in the Example section.
D. Liposome Preparation and Sizing
A variety of methods are available for preparing and sizing Iiposomes as
described in, e.~., Szoka, et al., Ann. Rev. Biophys. Bioeng., 9:467 (1980),
U.S. Pat. Nos.
4,186,183, 4,217,344, 4,235,871, 4,261,975, 4,485,054, 4,501,728, 4,774,085,
4,837,028.
4,946,787, PCT Publication No. WO 91/17424, Deamer and Bangham, Biochim.
Biophys. Acta, 443:629-634 (1976); Fraley, et al., Proc. Natl. Acad. Sci. USA,
76:3348-3352 (1979); Hope, et al., Biochim. BiopJtvs. Acta, 812:55-65 (1985);
Mayer, et
al., BioclTim. Biophys. Acta, 858:161-168 (1986); Williams, et al., Proc.
Natl. Acad. Sci.,
85:242-246 (1988), the text Liposomes, Marc J. Ostro, ed., Marcel Dekker,
Inc., New
York, 1983, Chapter 1, and Hope, et al., Client. Phys. Lip., 40:89 (1986), all
of which are
incorporated herein by reference. Suitable methods include, but are not
limited to,
sonication, extrusion, high pressure/homogenization, microfluidization,
detergent
dialysis, calcium-induced fusion of small liposome vesicles, and ether-
infusion methods.
all of which are well known in the art. One method produces multilamellar
vesicles of
heterogeneous sizes. In this method, the vesicle-forming lipids are dissolved
in a suitable
organic solvent or solvent system and dried under vacuum or an inert gas to
form a thin
lipid film. If desired, the film may be redissolved in a suitable solvent,
such as tertiary
butanol, and then lyophilized to form a more homogeneous lipid mixture which
is in a
more easily hydrated powder-like form. This film is covered with an aqueous
buffered
solution and allowed to hydrate, typically over a 15-60 minute period with
agitation. The
size distribution of the resulting multilamellar vesicles can be shifted
toward smaller sizes
by hydrating the lipids under more vigorous agitation conditions or by adding
solubilizing detergents, such as deoxycholate.
Extrusion of liposome through a small-pore polycarbonate membrane or
an asymmetric ceramic membrane is an effective method for reducing liposome
sizes to a
relatively well-defined size distribution. Typically, the suspension is cycled
through the
membrane one or more times until the desired liposome size distribution is
achieved.
The liposomes may be extruded through successively smaller-pore membranes, to
achieve gradual reduction in liposome size. For use in the present invention,
liposomes
having a size ranging from about 0.05 microns to about 0.40 microns are
preferred.
SUBSTITUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
E. Use Of Liposomes As Drug Delivery Vehicles
The lipid-based drug formulations and compositions of the present
invention (e.g., liposomes, micelles, lipid-nucleic acid particles, virosomes,
etc.) are
useful for the systemic or local delivery of bioactive agents such as
therapeutic agents,
prophylactic agents and diagnostic agents. Such delivery systems are described
in greater
detail in, for example, the following copending U.S. Patent Applications
Serial Numbers
08/454,641, 08/485,458, 08/660,025, 08/484,282, 60/055,094, 08/856,374,
60/053,813
and 60/063,473, the teachings of all of which are incorporated herein by
reference.
The following discussion refers generally to liposomes; however, it will be
10 readily apparent to those of skill in the art that this same discussion is
fully applicable to'
the other drug delivery systems of the present invention (e.g., micelles,
virosomes, lipid-
nucleic acid particles, etc.).
For the delivery of therapeutic agents, the compositions can be loaded
with a therapeutic agent and administered to the subject requiring treatment.
The
15 therapeutic agents which are administered using the present invention can
be any of a
variety of drugs which are selected to be an appropriate treatment for the
disease to be
treated or prevented. Often the drug will be an antineoplastic agent, such as
vincristine,
doxorubicin, mitoxantrone, camptothecin, cisplatin, bleomycin,
cyclophosphamide,
methotrexate, streptozotocin, and the like. Especially preferred antitumor
agents include,
20 for example, actinomycin D, vincristine, vinblastine, cystine arabinoside,
anthracyclines,
alkylative agents, platinum compounds, antimetabolites, and nucleoside
analogs, such as
methotrexate and purine and pyrimidine analogs. It may also be desirable to
deliver anti-
infective agents to specific tissues by the present methods. The compositions
of the
present invention can also be used for the selective delivery of other drugs
including, but
25 not limited to, local anesthetics, e.g., dibucaine and chlorpromazine; beta-
adrenergic
blockers, e.g., propranolol, timolol and labetolol; antihypertensive agents,
e.g., clonidine
and hydralazine; anti-depressants, e.g., imipramine, amitriptyline and
doxepim; anti-
conversants, e.g., phenytoin; antihistamines, e.g., diphenhydramine,
chlorphenirimine and
promethazine; antibiotic/antibacterial agents, e.g., gentamycin,
ciprofloxacin, and
30 cefoxitin; antifungal agents, e.g., miconazole, terconazole, econazole,
isoconazole,
butaconazole, clotrimazole, itraconazole, nystatin, naftifine and amphotericin
B;
antiparasitic agents, hormones, hormone antagonists, immunomodulators,
neurotransmitter antagonists, antiglaucoma agents, vitamins, narcotics, and
imaging
agents.
SUBSTTTTJTE SKEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
31
As mentioned above, cationic lipids can be used in the delivery of
therapeutic genes or oligonucleotides intended to induce or to block
production of some
protein within the cell. Nucleic acid is negatively charged and may be
combined with a
positively charged entity to form a lipid complex or a fully encapsulated
stable plasmid-
lipid particle.
Particularly useful antisense oligonucleotides are directed to targets such
as c-myc, bcr-abl, c-myb, ICAM-1, C-erb B-2 and BCL-2.
The CPLs of the present invention are also useful in the delivery of
peptides, nucleic acids, plasmid DNA, minichromosomes and ribozymes.
Another clinical application of CPLs of this invention is as an adjuvant for
immunization of both animals and humans. Protein antigens, such as diphtheria
toxoid,
cholera toxin, parasitic antigens, viral antigens, immunoglobulins, enzymes
and
histocompatibility antigens, can be incorporated into or attached onto the
liposomes
containing the CPLs of the present invention for immunization purposes.
1 S Liposomes containing the CPLs of the present invention are also
particularly useful as Garners for vaccines that will be targeted to the
appropriate
lymphoid organs to stimulate an immune response.
Liposomes containing the CPLs of the present invention can also be used
as a vector to deliver immunosuppressive or immunostimulatory agents
selectively to
macrophages. In particular, glucocorticoids useful to suppress macrophage
activity and
lymphokines that activate macrophages can be delivered using the liposomes of
the
present invention.
Liposomes containing the CPLs of the present invention and containing
targeting molecules can be used to stimulate or suppress a cell. For example,
liposomes
incorporating a particular antigen can be employed to stimulate the B cell
population
displaying surface antibody that specifically binds that antigen. Liposomes
incorporating
growth factors or lymphokines on the liposome surface can be directed to
stimulate cells
expressing the appropriate receptors for these factors. Using this approach,
bone marrow
cells can be stimulated to proliferate as part of the treatment of cancer
patients.
Liposome-encapsulated antibodies can be used to treat drug overdoses.
The tendency of liposomes having encapsulated antibodies to be delivered to
the liver has
a therapeutic advantage in clearing substances, such as toxic agents, from the
blood
circulation. It has been demonstrated that whereas unencapsulated antibodies
to digoxin
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
32
caused intravascular retention of the drug, encapsulated antibodies caused
increased
splenic and hepatic uptake and an increased excretion rate of digoxin.
Liposomes containing the CPLs of this invention also find utility as
carriers for introducing lipid or protein antigens into the plasma membrane of
cells that
lack the antigens. For example, histocompatibility antigens or viral antigens
can be
introduced into the surface of viral infected or tumor cells to promote
recognition and
killing of these cells by the immune system.
In addition, liposomes containing the CPLs of the present invention can be
used to deliver any product (e.g., therapeutic agents, diagnostic agents,
labels or other
compounds) including those currently formulated in PEG-derivatized liposomes.
In certain embodiments, it is desirable to target the liposomes of this
invention using targeting moieties that are specific to a cell type or tissue.
Targeting of
liposomes using a variety of targeting moieties, such as ligands, cell surface
receptors,
glycoproteins, vita;xzins (e.g., riboflavin) and monoclonal antibodies, has
been previously
described (see, e.g., U.S. Patent Nos. 4,957,773 and 4,603,044, the teachings
of which are
incorporated herein by reference). The targeting moieties can comprise the
entire protein
or fragments thereof.
In some cases, the diagnostic targeting of the liposome can subsequently
be used to treat the targeted cell or tissue. For example, when a toxin is
coupled to a
targeted liposome, the toxin can then be effective in destroying the targeted
cell, such as a
neoplasmic cell.
In another aspect, the present invention provides a method for increasing
intracellular delivery of a lipid-based drug formulation, comprising:
incorporating into
the lipid-based drug formulation, a compound of Formulae I or II, thereby
increasing the
intracellular delivery of the lipid based drug formulation compared to a
formulation
without a compound of Formulae I or II. The compounds of Formulae I or II
increase
intracellular delivery about 10 fold to about 1000 fold and preferably, about
10 fold to
about 100000 fold.
In another aspect, the present invention provides a method of increasing
the blood-circulation time of a parenterally administered lipid-based drug
formulation,
the method comprising: incorporating into the lipid-based drug formulation
about 0.1 to
20 mole percent of a compound of Formulae I or II.
In other aspects, the present invention provides a method for transfection
of a cell with a lipid-based drug formulation, comprising: contacting the cell
with a lipid-
SUBSTTTITTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
33
based drug formulation having about 0.1 to 20 mole percent of a compound of
Formulae I
or II. Moreover, a method for increasing the transfection of a cell with a
lipid-based
drug formulation, comprising: contacting the cell with a lipid-based drug
formulation
having about 0.1 to 20 mole percent of a compound of Formulae I or II, whereby
the
transfection efficiency of the lipid-based drug formulation is increased
compared to the
transfection efficiency of a lipid-based drug formulation without the compound
of
Formulae I or II.
G. Use of the Liposomes as Diagnostic Agents
The lipid-based drug formulations or compositions, e.g., liposomes,
prepared using the CPLs of this invention can be labeled with markers that
will facilitate
diagnostic imaging of various disease states including tumors, inflamed
joints, lesions,
etc. Typically, these labels will be radioactive markers, although fluorescent
labels can
also be used. The use of gamma-emitting radioisotopes is particularly
advantageous as
they can easily be counted in a scintillation well counter, do not require
tissue
homogenization prior to counting and can be imaged with Gamma cameras.
Gamma- or positron-emitting radioisotopes are typically used, such as
99Tc~ 24Na~ SICr~ 59Fe~ 67Ga, 86Rb, 111In~ 125h and ~95Pt as gamma-emitting;
and
such as 68Ga, 82Rb, 22Na 75Br, 122I and 18F as positron-emitting.
The liposomes can also be labelled with a paramagnetic isotope for
purposes of in vivo diagnosis, as through the use of magnetic resonance
imaging (MRI)
or electron spin resonance (ESR). See, for example, U.S. Pat. No. 4,728,575,
the
teachings of which are incorporated herein by reference.
H. Loading and Administerine the Lit~osomes
The following discussion refers generally to liposomes; however, it will be
readily apparent to those of skill in the art that this same discussion is
fully applicable to
the other drug delivery systems of the present invention (e.g., micelles,
virosomes, lipid-
nucleic acid particles, etc.). Methods of loading conventional drugs into
liposomes
include, for example, an encapsulation technique, loading into the bilayer and
a
transmembrane potential loading method.
In one encapsulation technique, the drug and liposome components are
dissolved in an organic solvent in which all species are miscible and
concentrated to a dry
film. A buffer is then added to the dried film and liposomes are formed having
the~drug
SUBSTTTL1TE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
34
incorporated into the vesicle walls. Alternatively, the drug can be placed
into a buffer
and added to a dried film of only lipid components. In this manner, the drug
will become
encapsulated in the aqueous interior of the liposome. The buffer which is used
in the
formation of the liposomes can be any biologically compatible buffer solution
of, for
example, isotonic saline, phosphate buffered saline, or other low ionic
strength buffers.
Generally, the drug will be present in an amount of from about 0.01 ng/mL to
about 50
mg/mL. The resulting liposomes with the drug incorporated in the aqueous
interior or in
the membrane are then optionally sized as described above.
Transmembrane potential loading has been described in detail in U.S.
Patent No. 4,885,172, U.S. Patent No. 5,059,421, and U.S. Patent No.
5,171,578, the
contents of which are incorporated herein by reference. Briefly, the
transmembrane
potential loading method can be used with essentially any conventional drug
which can
exist in a charged state when dissolved in an appropriate aqueous medium.
Preferably,
the drug will be relatively lipophilic so that it will partition into the
liposome membranes.
A transmembrane potential is created across the bilayers of the liposomes or
protein-liposome complexes and the drug is loaded into the liposome by means
of the
transmembrane potential. The transmembrane potential is generated by creating
a
concentration gradient for one or more charged species (e.g., Na+, K+ and/or
H+) across
the membranes. This concentration gradient is generated by producing liposomes
having
different internal and external media and has an associated proton gradient.
Drug
accumulation can than occur in a manner predicted by the Henderson-Hasselbach
equation.
The liposome compositions of the present invention can by administered
to a subject according to standard techniques. Preferably, pharmaceutical
compositions
of the liposome compositions are administered parenterally, i.e.,
intraperitoneally,
intravenously, subcutaneously or intramuscularly. More preferably, the
pharmaceutical
compositions are administered intravenously by steady infusion. Suitable
formulations
for use in the present invention are found in Remington's Pharmaceutical
Sciences, Mack
Publishing Company, Philadelphia, PA, 17th ed. (1985). The pharmaceutical
compositions can be used, for example, to diagnose a variety of conditions, or
treat a
diseased state. The diseases include, but are not limited to, inflammation
associated with
rheumatoid arthritis, post-ischemic leukocyte-mediated tissue damage
(reperfusion
injury), acute leukocyte-mediated lung injury (e.g., adult respiratory
distress syndrome),
SUBSTIZ'LTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
septic shock, and acute and chronic inflammation, including atopic dermatitis
and
psoriasis. In addition, various neoplasms and tumor metastases can be treated.
Preferably, the pharmaceutical compositions are administered
intravenously. Thus, this invention provides compositions for intravenous
administration
5 which comprise a solution of the liposomes suspended in an acceptable
carrier, preferably
an aqueous carrier. A variety of aqueous carriers can be used, e.g., water,
buffered water,
0.9% isotonic saline, and the like. These compositions can be sterilized by
conventional.
well known sterilization techniques, or may be sterile filtered. The resulting
aqueous
solutions may be packaged for use as is or lyophilized, the lyophilized
preparation being
10 combined with a sterile aqueous solution prior to administration. The
compositions may
contain pharmaceutically acceptable auxiliary substances as required to
approximate
physiological conditions, such as pH adjusting and buffering agents, tonicity
adjusting
agents, wetting agents and the like, for example, sodium acetate, sodium
lactate, sodium
chloride, potassium chloride, calcium chloride, sorbitan monolaurate,
triethanolamine
15 oleate, etc.
The concentration of active ingredient in the pharmaceutical formulations
can vary widely, i.e., from less than about 0.05%, usually at or at least
about 3-5% to as
much as 10 to 30% by weight and will be selected primarily by fluid volumes,
viscosities,
etc., in accordance with the particular mode of administration selected. For
diagnosis, the
20 amount of composition administered will depend upon the particular label
used (i.e.,
radiolabel, fluorescence label, and the like), the disease state being
diagnosed and the
judgment of the clinician.
The following examples serve to illustrate, but not to limit the invention.
EXAMPLES
25 I. EXAMPLE I
A. General Overview
Distal cationic-polyethylene glycol)-lipid conjugates (CPL) were
designed, synthesized and incorporated into conventional and stealth liposomes
for
enhancing cellular uptake. The present approach uses either inert, nontoxic or
naturally
30 occurred compounds as components for the CPL synthesis. CPLs were
synthesized with
the following architectural features: 1) a hydrophobic lipid anchor of DSPE
for
incorporating CPLs into liposomal bilayer; 2) a hydrophilic spacer of
polyethylene glycol
SUBSTIT'L1TE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
36
for linking the lipid anchor to the cationic head group; and 3) a naturally
occurring amino
acid (L-lysine) was used to produce a protonizable cationic head group. The
number of
charged amino groups can be controlled during the CPL synthesis. It has been
demonstrated that DSPE-CPLs were almost quantitatively incorporated into
liposomal
bilayer by a hydration-extrusion method. Quite surprisingly, in an in vitro
model., it was
confirmed for the first time that liposomes possessing distal positively
charged polymer
conjugates with preferably four or more charges efficiently bind to host cell
surfaces and
enhance cellular uptake in mammalian cells.
B. Materials and Methods
I. Abbreviatio~ts: DSPE, Distearoyl-sn-glycero-3-
phosphoethanolamine; DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine; DSPE-
PEGZOOO, 1,2-distearoyl-3-phosphatidylethanolamine-PEG~ooo; TFA,
trifluoroacetic acid;
CPL, cationic-polv (ethylene glycol)-lipid conjugate; DSPE-CPL, (cationic-
polyethylene
glycol)-DSPE conjugate; DSPE-CPL-1, DSPE-CPL with one positive charge; DSPE-
CPL-2, DSPE-CPL with two positive charges; DSPE-CPL-4, DSPE-CPL with four
positive charges; DSPE-CPL-8, DSPE-CPL with eight positive charges; Rh-PE, (or
Rho-
PE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine Rhodamine B
sulfonyl).
2. Chemical: t-Boc-NH-PEG3aoo-COzNHS was obtained from
Shearwater polymers, Inc (Huntsville, AL). Na, Ns-di-t-Boc-L-Lysine N-
Hydroxysuccinide Ester, triethylamine and cholesterol were obtained from Sigma-
Aldrich Canada Ltd (Oakville, ON). Trifluoroacetic acid, ethyl ether and
chloroform
were obtained from Fisher Scientific (Fair Lawn, NJ). 1,2-Distearoyl-sn-
glycero-3-
phosphoethanolamine and 1,2-distearoyl-sn-glycero-3-phosphocholine were
obtained
from Avanti Polar Lipids, Inc (Alabaster, Al). 1,2-distearoyl-3-
phosphatidylethanolamine-PEGZOOO was obtained from Genzyme (Cambridge, MA).
3. Synthesis of DSPE-CPL-1: To a solution of DSPE ( 121 mg, 161
mmol) and Et3N (200 ~L) in CHC1; (2 mL) at 45 °C was added t-Boc-NH-
PEG3.~oo-
COzNHS (500 mg, 147 pmol in 2 mL dry CHC13), and the solution was stirred for
3 hr at
ambient temperature. The solution was concentrated under a nitrogen stream to
dryness.
The residue was purified by repeat precipitation of the chloroform mixture
solution with
diethyl ether until disappearance of DSPE spot on TLC. The purified DSPE-PEG
SUBSTITUTE SHEET (RULE 16)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
37
conjugate was dissolved in 2 mL CHC13 followed by addition of 2 mL TFA, and
the
reaction solution was stirred at room temperature for :~ hr. The solution was
again
concentrated under a nitrogen stream to dryness. The residue was purified by
repeat
precipitation of the chloroform mixture solution with diethyl ether to offer
DSPE-PEG-
NH? as DSPE-CPL-1 with one protonable cationic head group: yield 500 mg (120
p,mol,
80%); R; 0.4 (CHCIz/MeOH, 9/1, v/v); The ratio of phosphoryl-lipid anchor and
the
distal primary amine was measured by phosphate and flourescamine assays and ~H
NMR.
4. General procedure for the synthesis of DSPE-CPL-2, DSPE-CPL-
4 and DSPE-CPL-8 (see Fig. 2 for schematic): To a solution of DSPE-CPL-1 (250
mg,
60 ~mol) and Et;N (200 ~L) in CHC13 (2 mL) was added Na, NE-di-t-Boc-L-Lysine
N-
Hydroxysuccinide Ester (50 mg, 113 ~mol in 2 mL dry CHCl3), and the solution
was
stirred for 3 hr at ambient temperature. Disappearance of positive amine-
active spot on
TLC by nihydrin visualization indicated that the reaction was completed. The
solution
was concentrated under a nitrogen stream to dryness. The residue was purified
by repeat
precipitation of the chloroform mixture solution with diethyl ether until
disappearance of
t-Boc-Lysine spot on TLC. The purified DSPE-PEG-conjugates were dissolved in 2
mL
CHC13 followed by addition of 2 mL TFA, and the reaction solution was stirred
at room
temperature for 4 hr. The solution was again concentrated under a nitrogen
stream to
dryness. The residue was purified by repeat precipitation of the chloroform
mixture
solution with diethyl ether to offer DSPE-CPL2: yield 250 mg (57 pmol, 95%);
R~ 0.4
(CHC13/MeOH, 9/1, v/v); The ratio of phosphoryl-lipid anchor and the distal
primary
amine was 1 measured by phosphate and flourescamine assays. DSPE-CPL4 and DSPE-
CPL8 were synthesized in a similar manner.
S. Preparation of large zcnilanzellar vesicles: Large unilamellar
vesicles (LUV) were prepared by extrusion as described by Hope et al. (see
Hope, M.J.,
et al., (1985) Production of large unilamellar vesicles by a rapid extrusion
procedure.
Characterization of size distribution, trapped volume and ability to maintain
a membrane
potential. Biochinz. Biophys. Acta. 812, 55-65). Appropriate amounts of lipid
mixtures
(DSPC/Chol, 60:40 mol/mol) with or without DSPE-CPLs (as set out in Table 3)
containing trace amounts of Rh-PE in chloroform, were dried under a stream of
nitrogen
gas to form a homogeneous lipid film. The trace amount of solvent was then
removed
under a vacuum overnight. The lipid film was hydrated in HBS buffer (pH 7.5)
with or
without HPTS (50 mM) by vortex mixing. The resulting multilamellar vesicles
(MLVs)
SUBSTTrUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
38
were extruded 10 times through two stacked 100 nm polycarbonate filters
(Nuclepore)
employing an extrusion device (Lipex Biomembranes, Inc., Vancouver, BC,
Canada) at
65°C. Unincorporated DSPE-CPLs and in some cases untrapped free HPTS
were
removed by chromatography using a 1.1 x 20 cm Sepharose CL-6B column (Sigma
Chemical Co., St. Louis, MO, USA) equilibrated with HBS buffer.
6. Determination of liposome si=e: Liposome size was determined
by quasi-elastic light scattering (QELS) using a Nicomp 370 submicron particle
sizer
(Santa Barbara, CA).
C. Results and Discussion
This example was carried out to synthesize and assess the efficacy of the
distal positively charged cationic polymer lipid conjugates (CPL) to enhance
the cellular
uptake of CPL-incorporated liposomes. The present approach uses inert,
nontoxic and
naturally occurring compounds, e.g., amino acids, as components for the CPL
synthesis.
Several CPLs were designed with the following architectural features: 1 ) a
hydrophobic
lipid anchor for incorporating the CPLs into the liposomal bilayer; 2) a
hydrophilic
spacer for linking the lipid anchor to the cationic head group; and 3) a
cationic head
group. Moreover, the amount and nature of the cationic group can be changed
according
to the final application. In this example, a naturally occurring amino acid, L-
lysine, was
used to produce a protonizable amino group. The number of amino group can be
controlled during the CPL synthesis.
In analyzing these compounds, structure-function relationships in these
cellular uptake enhancers may be identified. As an initial step, a variety of
these CPLs
with differing amounts of charge were screened for their ability to enhance
uptake (see,
Figures 5-7). In addition, the physico-chemical properties of the synthesized
CPLs and
the ability of these CPLs to incorporate into the liposome bilayers were also
studied (see,
Tables 1-3 and Figures 5-7). In an in vitro model, it was confirmed that these
distal
charged polymer conjugates significantly enhance liposome uptake in mammalian
cells.
Table 1. Physicochemical properties of cationic CPL
Sample NHZ / P ratio
DSPE-CPL-1 0.98
DSPE-CPL-2 2.05
DSPE-CPL-4 3.96
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
39
DSPE-CPL-8 7.88
Table 2. pH gradients for CPL-liposomes
Sample Lipid composition D pH
DSPC/Ch (60:40) 1.84
2 DSPC/Ch/CPL-4 (57.x:40:2.5) 1.11
DSPC/Ch/CPL-8 (57.~:40:2.~) 0.85
4 DSPC/Ch/PEG-PE (54:40:6) 1.59
DSPC/Ch/PEG-PE/CPL-4 (54:40:2:4) 1.01
6 DSPC/Ch/PEG-PE/CPL-8 04:40:2:4) 1.13
Table 3. CPL incorporated lip osomes
and their
properties.
Lipid composition Size (nm)CPL incorp.(%)
1, DSPC/Ch(60:40) 110 -
2, DSPC/Ch/CPL-1(57.5:40:2.5) 120 98.~
3, DSPC/Ch/CPL-2(57.5:40:2.5) 122 94.~
4, DSPC/Ch/CPL-4(57.5:40:2.5) 122 98.1
5, DSPC/Ch/CPL-8(57.5:40:2.5) 122 97.6
6, DSPC/Ch/CPL-1(55:40:5) 120 98.5
7, DSPC/Ch/CPL-2(55:40:5) 122 . 94.5
8, DSPC/Ch/CPL-4(55:40:5) 122 98.1
9, DSPC/Ch/CPL-8(55:40:5) 122 97.6
10, DSPC/Ch/PEG-PE(54:40:6) 128 -
11, DSPC/Ch/PEG-PE/CPL-1(54:40:2:4)130 96.7
12, DSPC/Ch/PEG-PE/CPL-2(54:40:2:4)130 101
13, DSPC/Ch/PEG-PE/CPL-4(54:40:2:4)130 104
14, DSPC/Ch/PEG-PE/CPL-8(54:40:2:4)130 110
15, DSPC/Ch(60:40) 110
16, DSPC/Ch/CPL-4(57.5:40:2.5)120 98.5
17, DSPC/Ch/CPL-8(57.5:40:2.5)122 94.5
18, DSPC/Ch/PEG-PE(54:40:6) 128 -
19, DSPC/Ch/PEG-PE/CPL-4(54:40:2:4)130 96.7
20, DSPC/Ch/PEG-PE/CPL-8(54:40:2:4)130 101
II. EXAMPLE II
This example illustrates that LUVs containing CPL4 can be formed by a
5 detergent dialysis method.
The LLJVs contain DOPE, DODAC, PEG-Cer-C20, and CPL.[3.4K] (or
CPLa[1K]). Two preparations were made with the CPL comprising 4 mol % of the
original lipids:
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
TABLE 4
Li id mol-
DODAC 6
DOPE 79.5
CPL, 4
PC-C20 10
_
-.
Rho-PE ~ 0.5
The lipids indicated above were co-dissolved in chloroform, which was
then removed under nitrogen followed by 2 hours under high vacuum. The dry
lipid
mixture (10 p.mol total) was then hydrated in 83 ~L of 1 M OGP and 1 mL Hepes-
5 buffered saline (20 muI Hepes 150 rWI NaCI pH 7.~) at 60°C with
vortexing until all the
lipid was dissolved in the detergent solution.
The lipid-detergent mixture was transferred to Slide-A-Lyzer dialysis
cassettes, and dialysed against at least 2 L HBS for 48 hours, with a least
two changes of
buffer in that time. Removal of detergent by dialysis results in formation of
LIlVs. To
10 determine whether all of the CPL was incorporated into the LUVs following
dialysis, the
lipid samples were fractionated on a column of Sepharose CL-4B (see Figures 8A
and
8B). The fractionation profiles show LUVs formed with either CPL:[3.4K] or
CPL.[1K].
The final concentration of CPL in the LLJV fraction (fractions 7 - 10) was
estimated from initial and final dansyl/rhodamine ratios, and from estimating
the
15 proportion of total dansyl and rhodamine fluorescence present in the LIJV
peak.
Essentially identical results were obtained.
In order to examine the effect of increasing the initial CPL concentration,
a sample was made with the following proportions:
TABLE 5
Li mol-
id -
DODAC 6
DOPE 71.5
CPL4 12
PC-C20 10
- Rho-PE 0.5
20 The column profile for fractionation of this sample is shown in Figure 8C.
The results for all 3 samples are given below:
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
41
TABLE 6
Sam original mol %-insertedfinal mol
le %CPL %CPL
Figure 4 64.6 2.6
8A
Figure 4 82.8 3.3
8B
Figure 12 50.9 6.1
8C
Conclusion: LLTVs containing CPLa can be formed by detergent dialysis.
Not all of the CPL. is incorporated into the vesicle, and the proportion that
is
incorporated falls as the initial CPL/lipid molar ratio is increased. In the
present case,
beginning with 4 mol % CPL, about 3 mol % was incorporated into the LUV. For
an
initial CPL content of 12 mol %, a final content of 6 mol % was achieved. It
is also
worth noting that the behavior of the CPL,[1K] is very similar to that of the
CPL.[3.4K].
This is also true in post-insertion studies. In certain instances, the ideal
length of the
hydrophilic spacer will allow the cationic groups to extend out from the
liposomal
surface at a distance shorter than the normal neutral PEG that is typically
being used to
provide stealth properties for increased liposomal circulation lifetimes.
III. EXAMPLE III
A. Overview
In this example, a non-specific targeting approach is described that
involves increasing the electrostatic attraction between liposomes and cells
by
incorporation of positively-charged lipid molecules into preformed vesicles.
This
approach leads to dramatic increases in cell binding/uptake in vitro in BHK
cells. The
methodology is demonstrated to work for neutral vesicles and for vesicles
composed of
lipids used in the construct of lipid-based gene carriers. The approach
outlined herein
thus has numerous applications ranging from delivery of conventional drugs to
gene
therapy.
B. Materials and Methods
1. Materials: 1,2-dioleoylphosphatidylcholine (DOPC), 1,2-
dioleoylphosphatidylethanolamine (DOPE), and 1,2-dioleoyl-sn-glycero-3-
phosphoethanolamine-N-(Lissamine Rhodamine B Sulfonyl) (Rhodamine-PE) were
obtained from Avanti Polar Lipids. Cholesterol was obtained from Sigma
Chemical Co.
DODAC and PEGCerC20, PEGCerCl4, and PEGCerC8 were generous gifts from Inex
Pharmaceuticals.
SUBSTTTUT'E SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
42
?. Synthesis of cationic-PEG lipids: The details of the synthesis of
the CPLs is described herein. Two types of CPLs were synthesized which
differed in the
lipid anchor portion of the molecule. In one, the anchor was
distearoylglycerol (DSG),
while the other contained distearoylphosphatidylethanolamine (DSPE). The
molecule
consists of the anchor portion, to which is attached a PEG;~oo chain. At the
end of the
PEG chain, a charged "headgroup" is attached, often made up of lysine residues
linked
together. By modifying the headgroup region, CPLs were synthesized which
contained 1
(mono, or M), 2 (di, or D), 3 (tri, or T), and 4 (quad, or Q) positive
charges. Several
different Quad CPLs were synthesized, hence these are numbered Q 1 through Q5.
The
nomenclature chosen to describe these compounds specifies the type of lipid
anchor and
the identity of the headgroup (e.g., d-DSPE-CPL-QS). The lower case "d"
indicates a
dansylated derivative.
3. Preparation of Vesicles by Detergent Dialysis. In general, vesicles
were formed using a detergent dialysis method (see, Wheeler, J.J., et al.
(1999) Stabilized
plasmid-lipid particles: construction and characterization. Gene Therapy 6,
271-281, the
teachings of which are incorporated herein by reference). The lipids, as
described in
Example II, were co-dissolved in chloroform in the appropriate ratios,
following which
the chloroform was removed under a stream of nitrogen and placed under high
vacuum
for 2 hours. An aliquot of the non-ionic detergent octylglucopyranoside (1 M
in water)
(OGP) was then added to the dry lipid film, which was incubated for 10-20
minutes at 60
°C with frequent vortexing. This was followed by addition of 20 muI
HEPES 150 rrWl
NaCI pH 7.5, with further warming and vortexing until all the lipid was
dispersed and a
clear solution was obtained. For 20 mg of lipid, 0.125 mL of OGP and 1 mL of
HBS
were used. The lipid-detergent solutions (1-2 mL) were then transferred to
Slide-A-
Lyzer dialysis membranes (3 mL volume) and exhaustively dialysed at room
temperature
against HBS over a period of 48 hours. In general, a total volume of 8 - 10 L
of HBS
was used (4 - 5 changes of 2L) for sample volumes of 1 - 8 mL.
Vesicles of DOPC and DOPC/Chol (55:45) were prepared by extrusion as
previously described (Hope, M.J., et al., (1985) Production of large
unilamellar vesicles
by a rapid extrusion procedure. Characterization of size distribution, trapped
volume and
ability to maintain a membrane potential. Biochim. Biophys. Acta 812, 5~-6~).
4. Insertion of Cationic PEG-lipids into preformed vesicles. The
cationic PEG-lipids (CPLs) were stored as micellar solutions in HBS or, in a
few cases,
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
43
in methanol. The CPL and the vesicles were combined to Qive the desired molar
ratio
(up to 11.6 mol % CPL relative to vesicle lipid), and incubated for a given
time at the
desired temperature. For most insertions, the standard conditions involved a 3
hour
incubation at 60 °C. Following insertion, the samples were cooled on
ice, and the CPL-
LUV was separated from free CPL by passage down a column ( 1.5 x 15 cm) of
Sepharose CL-4B equilibrated in HBS. Figure 16 illustrates the insertion
protocol for
SPLPs (an analogous procedure).
The insertion levels of CPL were measured by fluorescence. In all cases,
the vesicles contained either 0.25 mol % or 0.~ mol % rhodamine-PE, and the
CPL
contained a dansyl group. After combining the CPL and lipids, a 15 p,L aliquot
(initial
fraction) was set aside for analysis. The amount of CPL inserted into the
vesicles could
then be quantified by measuring the initial dansyl/rhodamine (D/R)
fluorescence ratio,
and the D/R ratio of the isolated CPL-LUVs. Fluorescence parameters: for the
rhodamine assay, the excitation wavelength was 560 nm, and the emission
wavelength
was 590 nm. For the dansyl assay, the excitation wavelength was 340 nm, and
the
emission wavelength was 510 nm. In general, the excitation and emission slit
widths
were 10 and 20 nm, respectively. The assay was performed as follows: to an
aliquot of
the initial sample (?-3 p.L) or the CPL-LUV (20-40 ~L) was added 30 p.L of 10%
Triton
X-100 followed by 2 mL of HBS. The fluorescence levels of both the dansyl and
rhodamine labels were read consecutively using a wavelength program as per the
above
parameters with an emission filter of 410 nm. The %-insertion was calculated
as follows:
-insertion = (~D/R~CpL-LW)*lOO/~D/R~~N~TIAL
5. Measurement of Lipid Concentrations: Following insertion, it is
necessary to know the lipid concentration of each sample for cell binding
studies. This
can be done quickly by fluorescence. Following detergent dialysis, the lipid
concentration of each sample was measured using the standard phosphate assay
(Fiske,
C.H., and Subbarow, Y. (1995) The colorimetric determination of phosphorus. J.
Biol.
Chem. 66, 375-400). An aliquot was then diluted to approximately 3 rnul. By
comparing
the rhodamine fluorescence of this sample, whose lipid concentration is known,
with the
CPL-LUVs prepared from that stock, allows determination of CPL-LUV
concentrations.
Lipid concentrations of LUVs were measured using the standard phosphate assay.
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
44
Following CPL insertion, lipid concentrations were estimated for cell binding
studies
from the rhodamine fluorescence.
6. Uptake of CPL-containing L UVs by BHK cells. Approximately
10' BHK cells were incubated in PBS/CMG medium with 20 nmol of
DOPE/DODAC/PEGCerC20 (84/6/10) LUVs containing either (1) no CPL, (2) 8%
DSPE-CPL-D, (3) 7% DSPE-CPL-T1, or (4) 4% DSPE-CPL-Q5. Incubations were
performed for 1, 2, 4, and 6 hours at 4 °C and 37 °C, the former
giving an estimate of cell
binding, and the latter of binding and uptake. By taking the difference of the
two values,
an estimate of lipid uptake at 37 °C was obtained. For each timepoint,
the cells were
ruptured and assayed for lipid and protein. Lipid concentrations were measured
from
rhodamine fluorescence, while protein was determined using the BCA assay.
Lipid
concentrations were measured using rhodamine fluorescence, while protein was
determined using the BCA assay kit obtained from Pierce.
C. Results and Discussion
1. Development of Insertion Protocol. The transfer of pegylated
lipids from micellar aggregates to vesicles has been previously described
(see, Uster,
P.S., et al., (1996) Insertion of polyethylene glycol) derivatized
phospholipid into pre-
formed liposomes results in prolonged in vivo circulation time. FEBS Letters
386, 243-
246; Zalipsky, S., et al., (1997) Polyethylene glycol)-grafted liposomes with
oligopeptide or oligosaccharide ligands appended to the termini of the polymer
chains.
Bioconjugate Chem. 8, 111-118). This idea was tested with DSPE-CPUs. This is
demonstrated in Figure 9(A) for DOPC LUVs. The co-elution of the dansyl and
rhodamine labels demonstrates incorporation of the CPL in the LUVs. In this
case, 84%
of the CPL was incorporated into the LUVs, and thus only a trace of free CPL
is observed
trailing the CPL-LUV fractions. This is more clearly seen in Figure 8(B),
where the
DSPE-CPL-QS has been inserted into a more complex positively charged vesicles
composed of DOPE/DODAC/PEGCerC20 (84/6/10). Here, the co-elution of the two
fluorescent labels at approx. 9 mLs demonstrates 70% insertion of the CPL into
the
vesicles. The free CPL elutes in a broad peak centered at 16 mLs, which is
separate from
the vesicle peak, allowing for easy isolation of the CPL-LUV. Once inserted,
the DSPE-
CPL-QS is retained and does not exchange out of the vesicles. The CPL-LUV
fraction
from Figure 9(B) was re-eluted on the column of Sepharose CL-4B. As shown in
Figure
9(C), all of the CPL remains with the LUVs.
SUBSTTTUTE SKEET (RULE Z6)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
The effects of incubation temperature and time on the insertion process are
shown in Figure 10. DSPE-CPL-Q 1 was incubated in the presence of
DOPE/DODAC/PEGCerC20 (84/6/10) at room temperature, 40°C, and
60°C, with
aliquots withdrawn at 1, 3, and 6 hours. The highest insertion levels were
achieved at 60
3 °C, which was therefore used in subsequent insertions. Although
slightly higher insertion
was obtained at 6 hr, we chose 3 hr to minimize sample degradation.
Aside from time and temperature, the parameter that will have the greatest
influence on final CPL insertion levels is the initial CPL/lipid ratio.
Assuming about
70% insertion, a series of incubations were performed with CPL/lipid molar
ratios
10 varying between 0.011 to 0.14, with the aim to achieve CPL-LLJVs containing
1, 2, 4, 6.
8, and 12 mol % CPL. These results are shown in Figure 1 l, where it is seen
that the
insertion level remains close to 70% up to an initial CPL/lipid ratio of .095,
above which
it drops to 50% for CPL/lipid = 0.14.
Similar results were obtained for other vesicle systems, including
15 DOPE/DODAC/PEGCerCl4 and DOPE/DODAC/PEGCerCB. In general, the insertion
levels obtained with DODAC-containing samples fell in the range of 70-80% for
initial
CPL/lipid < 0.1. In order to see whether the insertion levels were effected by
the
presence of cationic lipid, several experiments were performed on neutral
vesicles
containing DOPC. The compositions examined were: (1)DOPC, (2) DOPC/Chol, (3)
20 DOPC/PEGCerC20, and (4) DOPC/Chol/PEGCerC20. The results, shown in Figure
12,
reveal that for the DSPE-CPL-Q1, somewhat less insertion was achieved in the
neutral
systems: between 45 - 65%. This may be due to reduced attraction benveen the
negatively-charged DSPE anchor and the membrane surface. Regardless, the
results
demonstrate that significant insertion can be achieved for both neutral and
positive
25 vesicles.
It should be noted that the insertion levels for the DSPE-CPL-QS also
shown in Figure 13 are much higher than for the Q 1 (70 - 84%). There is a
reason for
the differential behavior of the Ql and QS CPLs in these systems, when in
prior
experiments they behaved very similar. This particular batch of QS was
prepared in
30 methanol, a solvent in which the lipids may exhibit greater storage
stability. As
explained herein, it has been found that the presence of methanol in the
incubation
mixture leads to higher insertion.
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
46
A large number of insertions have been performed using other CPLs in
addition to the QS and Q1. These results are summarized in Tables 7 and 8,
(Figures 23
and 24) where some composition-dependent trends can be ascertained. First, the
same
trend seen above in Figure 11 with the QS hold for several CPLs with differing
charge.
As the initial ratio of CPL/lipid is increased, the percentage of CPL inserted
decreases. If
we look at the T1, Q1, and QS incubations where CPL/lipid = 0.022 - 0.024, the
%-
insertion ranges from 76 - 80%. However, for CPL/lipid = 0.086 - 0.095, the %-
insertion range decreases to 62 - 68%.
Another trend is illustrated in Figure 13. For initial CPL/lipid ratios >
0.04, slightly less CPL-Q1 is inserted into LUVs containing PEG-Cer-C20 than
into those
containing either of the shorter chain PEGs. In addition to the type of PEG
anchor
present, the quantity of PEG-Cer also has an effect on insertion, as seen in
Figure 14. As
the PEG-Cer-C20 content is increased from 4 to 10 mol %, the insertion levels
of CPL-
QS fall from 71 to 62%.
As those of skill in the art will readily appreciate, the lipid anchor can be
varied and the insertion levels may vary depending on the lipid used as the
lipid anchor.
For instance, some experiments were performed with CPL containing a DSG
(distearoylglycerol) anchor: in all cases the insertion levels were much
lower, from 17 -
40%, than in CPL containing a DSPE anchor. Using the methods and assays of the
present invention, those of skill in the art can readily identify suitable
lipid anchors.
In order to check for possible aggregation following CPL insertion, quasi-
elastic light scattering (QELS) was used to examine the effect of insertion on
particle
diameter. DOPE/DODAC/PEG-Cer-C20 vesicles were found to have a diameter of 119
X39 nm. Following insertion of 1.8 mol % CPL4b, a slight increase in diameter
to
approx. 135 X42 nm was observed, but both the mean diameter and standard
deviation
remained constant up to 7 mol % CPL. The increase from 120 nm to 135 nm could
reflect a slightly larger diameter resulting from the presence of the longer
CPL PEG
chains or it could indicate a small amount of vesicle aggregation. To
differentiate
between these two possibilities, CPL-LUVs were examined by fluorescence
microscopy,
using a rhodamine filter. While control LUVs exhibited no signs of
aggregation,
significant levels were observed for CPL-LUVs. However, it was found that
addition of
mM CaCl2 completely prevented this effect.
As described in Materials & Methods, estimates were obtained for the
uptake of various CPL-LUVs on BHK cells incubated on PBS/CMG. The data, shown
in
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
47
Figure 1 S, reveals that the presence of positive charge on the CPLs can lead
to significant
enhancement in uptake by BHK cells. LUVs composed of DOPE/DODAC/PEGCerC20
(and thus exhibiting a net positive charge) showed little uptake on the BHK
cells. LUVs
containing 8 mol % of DSPE-CPL-D showed similar low uptake values. Uptake was
only slightly increased by the presence of 7 mol % of DSPE-CPL-Tl. However, a
significant increase in uptake was realized for DSPE-CPL-QS present at only
4.1 mol %.
Several points can be surmised from this data. While it is clear that an
increase in the
positive charge present at some distance from the LUV surface leads to an
increase in
uptake, it is not total charge alone that plays a role in enhanced cell
binding. The
quantity of positive charge present for the DSPE-CPL-D and DSPE-CPL-QS samples
is
approximately equal, and yet the former shows little binding compared to the
latter. The
DSPE-CPL-T1 sample has a greater positive charge than the DSPE-CPL-QS sample,
and
yet exhibits only 1/3 the uptake. It would appear that localization of a
sufficient positive
charge density at the distal end of the CPL molecule is an important parameter
in
ensuring interaction with cells. In a preferred embodiment, at least four
charges are used
to achieve efficient cell binding.
The dramatic effect of CPL insertion on LUV binding to BHK cells is
most clearly visualized using fluorescence microscopy. In the absence of CPL,
vesicles
composed of DOPE/DODAC/PEG-Cer-C20 and containing a trace of rhodamine-PE
exhibit little binding to cells. Incorporation of 3 mol % CPL4b leads to high
levels of
vesicle binding and uptake. Although much of the lipid appears to be binding
to the cell
surface, some small punctate structures can be seen, indicating that uptake of
vesicles is
also occurring. An important point to note is that the cells appear healthy
following
incubation in the presence of the CPL-LUVs. In contrast, DNA-cationic lipid
complexes
are known to display significant toxicity.
One of the major remaining hurdles in liposomal drug delivery is the
problem of how to ensure that the contents of a carrier system are taken up
and utilized
by a specific target cell. It is now believed that the cellular uptake of
liposomes involves
adsorption or binding at the cell surface, followed by endocytosis. Thus
factors which
interfere with cellular binding will lead to low levels of intracellular
delivery. This is of
particular importance for 'stealth' or long-circulating liposomes that are
coated with a
surface layer of a hydrophilic polymer such as PEG. The very characteristic of
the PEG
coating which imparts long-circulation lifetimes - the formation of a steric
barrier that
prevents interaction with serum proteins, will also minimize interactions with
cells. On
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
48
the other hand, factors that enhance surface binding may be expected to lead
to increased
cellular uptake. One approach involves attaching molecules specific for
membrane
receptors to liposomal surfaces. Possible candidates include oligopeptides
(see, Zalipsky
et al., Bioconjugate Chemistry 6, 705-708 (1995); Zalipsky et al.,
Bioconjugate
Chemistry 8, 111-118 (1997)) oligosaccharides (see, Zalipsky et al.,
Bioconjugate
Chemistry 8, 111-118 (1997)), folate (see, Gabizon et al., Bioconja~gate
Chemistry 10,
289-298 (1999); Lee et al., Journal ofBiological Chemistry 269, 3198-3204
(1994);
Reddy et al., Critical Reviews in Therapezctic Drug Carrier- Systems 15, 587-
627 (1998);
Wang et al., Joz~rnal of Controlled Release, 53, 39-48 ( 1998)), riboflavin
(see, Holladay
et al., Biochimica et Biophysica Acta 1426, 195-204 (1999)), or antibodies
(see, Meyer et
al., .Journal of Biological Chemistry 273, 15621-15627 ( 1998); Kao et al.,
Cancer Gene
Therapy 3 250-256 (1996); Hansen et al., Biochimica et Biophysica Acta 1239,
133-144
(1995)]. An alternate approach is to modify the charge characteristics of the
liposome.
It is well known that inclusion of either negative (see, Miller et al.,
Biochemistry 37,
12875-12883 (1998); Allen et al., Biochimica et Biophysica Acta 1061, 56-64
(1991);
Lee et al., Biochemistry 32, 889-899 (1993); Lee et al., Biochimica et
Biophysica Acta
1103, 185-197 (1992)) or positive (see, Miller et al., Biochemistry 37, 12875-
12883
(1998)) charges in liposomes can lead to enhanced cellular uptake. Cationic
DNA-lipid
complexes, which are efficient in vitro transfection agents (see, Felgner et
al. Nature 337,
387-388 (1989); Felgner et al., Proceedings of the National Academy of
Sciences of the
United States ofAmerica 84, 7413-7417 (1987); Kao et al., Cancer Gene Therapy
3 250-
256 (1996); Felgner et al., Annals of the New York Academy of Sciences 772,
126-139
(1995); Jarnagin et al., Nucleic Acids Research 20, 4205-4211 (1992)), are
taken up via
endocytosis.
This example describes a new approach for enhancing the interaction of
liposomes with cells, a necessary step in the development of non-viral systems
capable of
intracellular delivery. The approach involves the insertion of novel cationic-
PEG-lipids
into pre-formed liposomes, leading to a cationic vesicle in which the positive
charge
involved in cell interaction is located some distance away from the vesicle
surface. The
process is illustrated in Figure 16 for the insertion of a CPL into sterically-
stabilized
LUVs composed of DOPE, the cationic lipid DODAC, and PEG-Cer-C20. This lipid
composition was chosen for study for two reasons: first, it allows for
efficient entrapment
of plasmid DNA within small vesicular structures by virtue of the presence of
positively
charged DODAC (see, Wheeler et al. Gene Therapy 6, 271-281 (1999)), and thus
has
SUBSTTTLTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
49
potential as a gene delivery system (see below). Secondly, this composition is
representative of the many sterically-stabilized drug delivery systems which
contain
PEG-lipids. Insertion of CPLs leads to localization of positive charge above
the surface
PEG layer, thereby allowing electrostatic interactions between the CPLs and
cell
surfaces. This should lead to increased cellular interactions for both
conventional- and
PEG-containing liposomes.
The CPLs are conjugates of DSPE, a dansyl-lysine moiety, the hydrophilic
polymer PEG3aoo~ and a mono- or multivalent cationic headgroup. The PEG
functions as
a spacer, separating the charged headgroup from the lipid anchor and vesicle
surface.
Incubation of a wide variety of neutral and cationic LLTVs with micellar CPLs
resulted in
the incorporation of up to 6-7 mol % (relative to total vesicle lipid) of CPL
in the outer
vesicle monolayer (see tables in Figures 23 and 24). The insertion efficiency
was quite
high, with approximately 70 - 80% of added CPL incorporating into the LUVs
(see
tables in Figures 23 and 24). The most important factors influencing the CPL
insertion
levels were the incubation temperature (Figure 10) and initial CPL/lipid ratio
(Figure 11).
The composition of the liposome was found to affect the final CPL levels to a
lesser
degree (see tables in Figures 23 and 24). Following insertion, the CPL-LUV
could be
efficiently separated from free CPL by gel exclusion chromatography. Similar
insertion
levels were obtained for all CPLs, with headgroup charges ranging from one to
four
charges per molecule . With this knowledge, vesicles could be prepared
containing a
desired level of CPL with reasonable accuracy.
High insertion levels (up to 7 mol %) could be achieved for vesicles
containing as much as 10 mol % PEG-Cer-C20. It is possible that a portion of
the PEG-
Cer's are lost during the insertion process, as PEG-Cer's will exchange from
vesicles
during circulation. This may explain why the highest insertion levels are
achieved with
PEG-Cer-C8, which has the greatest propensity to exchange. However, analysis
of LUVs
and SPLPs containing PEG-Cer-C20 by HPLC before and after insertion of CPL4
reveal
only a slight loss of PEG-Cer-C20 (from about 10 mol% to 8 mol%).
As shown in Figure 15, cationic LLTVs composed of
DOPE/DODAC/PEG-Cer-C20 exhibit little uptake when incubated on BHK cells.
Although positively charged vesicles exhibit enhanced binding to some cell
lines, this can
be attenuated by the presence of PEG on the liposome surface (see, Miller et
al.,
Biochemistry 37, 12875-12883 (1998)). Clearly, for these systems, the presence
of 6 mol
of positively charged DODAC leads to only low uptake levels after 6 hours.
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
Incorporation of approximately 7 mol % of dicationic-CPL has little effect on
uptake,
which was only slightly improved in the presence of approx. 7 mol %
tricationic-CPL.
The best results were obtained with the CPL4b (at 4 mol %), which possessed 4
positive
charges. At 6 hours incubation, a ten-fold increase in uptake was observed
relative to the
starting vesicles. Several points can be surmised from this data. The first is
that the
presence of positively charged groups at some distance from the LUV surface
can lead to
significant increases in cellular uptake. In this case, the positive charges
of the CPL
(PEG MW = 3400) are located above the surface coating of PEG (MW = 2000), and
thus
are available for interactions with cells. However, it is not total charge
alone that plays a
10 role in enhanced cell binding. The quantity of positive charge present for
the CPL and
CPL.~b samples is approximately equal, and yet the former shows little uptake
compared
to the latter. The CPL3 sample has a greater positive charge than the CPL:~b
sample, and
yet exhibits only 1/3 the uptake. It would appear that localization of a
sufficient positive
charge density at the distal end of the CPL molecule is an important parameter
in
15 ensuring interaction with cells. At least four charges seem to be required
for efficient cell
binding to occur.
The protocol described for insertion of CPL into conventional and
sterically-stabilized CPL is ideal for demonstrating the methodology using in
vitro
applications. In both cases, the added positive charge is physically distant
from the
20 surface, and is available for interactions with cells. This is particularly
important for
polymer-coated vesicles that are designed for minimal interactions with serum
proteins
and cells such as macrophages. However, this system may not be ideal for in
vivo
applications, where it may be desirable to initially hide or screen the CPL
charge to
reduce clearance and allow accumulation of the vesicles at the tissue of
choice. Thus,
25 alternative embodiments employ shorter PEG spacer chains in the CPL, or
longer PEG
chains in the PEG-Cer molecules. The PEG-Cer molecules are known to exchange
out of
the particle during circulation see, Webb et al., Biochimica et Biophysica
Acta 1372, 272-
282 (1998)], which would leave the CPL exposed for cellular interactions.
As mentioned above, the cationic liposomes employed in the present study
30 are composed of a fusogenic lipid (DOPE), a cationic lipid (DODAC), and a
stabilizing
lipid (PEG-Cer-C20), the latter of which imparts long-circulating properties
to the
vesicles. This lipid composition was modeled after a new class of lipid-based
DNA
carrier systems known as stabilized plasmid-lipid particles (SPLPs) see,
Wheeler et al.
Gene Therapy 6, 271-281 (1999)). SPLPs are small (70 nm) particles that
encapsulate a
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
51
single plasmid molecule. The presence of a PEG coating on the liposome surface
imparts
long circulation properties as well as protecting the plasmid from degradation
by serum
nucleases. SPLPs thus represent the first carrier systems with real potential
for systemic
in vivo gene therapy applications. The approach described here greatly
enhances the
tranfection potency of these particles by increasing cellular binding and
uptake, which
leads to increased intracellular delivery of plasmids. The inclusion of CPL in
conventional formulations (e.g., anticancer drugs) also leads to increased
efficacy.
IV. EXAMPLE IV
A. Overview
This example employs CPLs incorporated into stable plasmid-lipid articles
(SPLPs) for in vitro transfection of cells.
Incubation of these particles on BHK cells for up to 8 hours resulted in an
increase in uptake as the amount of inserted CPL increased from 2-4 mol %.
Transfection of the SPLP system increased with the addition of CPL with lSmNI
CaCh in
the transfection media. The SPLP alone showed very low transfection at both a
4 and a 9
hour transfection followed by 24 hour complete incubation in fresh media. The
addition
of 15 mM CaCh final concentration in the media to the SPLPs, increased
transfection on
BHK cells by 10-fold at both time points. In the presence of 15 mM CaCla, SPLP
+ 2%,
3% and 4% CPL transfect 2000- to 5000-times higher than that of the SPLP alone
at both
time points. The 4 mol % CPL shows the greatest increase in transfection:
approximately
4500 times higher, followed by the 3% and then the 2% CPL samples. Therefore,
the
presence of the CPL, DSPE-Quads in the SPLP increased in both uptake and
transfection
to levels comparable to or above those achieved with the complexes.
B. Materials and Methods
1. Svnthesis of the DSPE-Quad.S: The dansylated DSPE-Quads
(CPL) was prepared in our laboratory as described by Chen et al (2000).
2. Incorporation of DSPE-Quads Into SPLP: Inex Pharmaceuticals,
Inc. supplied the SPLP. The incorporation of the CPL into the SPLP was
performed by
incubation of the CPL with the SPLP at 60°C for 2-3 hours in HBS. The
resulting
mixture was then passed down a Sepharose CL-4B column equilibrated with HBS,
75
mM CaCl2, pH 7.5 to remove the unincorporated CPL from the SPLP with the
incorporated CPL. Fractions (1 mL) were collected and assayed for CPL (dansyl
assay),
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
52
phospholipid, and DNA (PicoGreen assay). The final samples were prepared to
contain
2, 3, or 4 mol % of the CPL. The dansyl assay involved preparing a standard
curve of 0.5
to 2.5 mol % of dansylated CPL in BBS and determining the concentration of the
CPL in
the sample. The phospholipid was extracted from the SPLP by extracting the
lipid using
the Bligh-Dyer extraction technique (Bligh & Dyer, 1952) and then performing a
Fiske-
Subarrow assay on the organic phase of the extraction. The PicoGreen assay was
performed by comparing the sample in the presence of PicoGreen and Triton X-
100 using
a DNA standard curve. The final % insertion of the CPL was determined by
dividing the
CPL concentration by the lipid concentration.
The optimal time for insertion of the CPL into the SPLP was determined
using SPLP prepared with 0.5 mol % Rh-DSPE. 15 nmol of the dansylated CPL
(DSPE-
QuadS) was mixed with 200 nmol of the labeled SPLP and the sample was
incubated at
60°C for various time points (0.5, 1, 2, 3, and 4 hours). At these time
points the sample
was removed fromz the water bath and was passed down a Sepharose CL-4B column.
The
major fraction was collected from the column and the dansyl to rhodamine
fluorescence
ratios were measured. The parameters used for the rhodamine fluorescence were
a 7~er of
560 nm and a ~.~m of 600 nm and for the dansyl fluorescence were a n.e~ of 340
m and a
~.em of 510 nm. The excitation and emission slit widths for both of these were
10 nm and
nm, respectively. By comparison of the dansyl/rhodamine ratio for the sample
before
20 the column to that after the column, the % insertion was determined at each
time point.
3. QELS of CPL-SPLP: The diameter of these particles was
determined using a Nicomp Particle Sizer.
4. Freeze-Fracture EM: Freeze-fracture EM was performed on the
2%, 3%, and 4% CPL samples by methods which will be described by K. Wong
~. Serum Stability of Particles: The stability of the DNA within these
CPL-SPLP was determined by incubating the samples (25~L), containing 6~g of
plasmid
DNA (pLuc) for various time periods (0, 1, 2, and 4 hours) in 50% mouse serum
(25~.L)
at 37°C. At each time point, other than the zero time point, 11 ~L of
the mixture was
removed, the volume was made up to 45~,L using water and the samples were
placed on
ice. The DNA was then extracted from the lipid using one volume of
phenol:chloroform
(1:1). Following a 20 min centrifugation in a microfuge, the top aqueous phase
was
removed. The zero time point was obtained by removing 5.5~,L of the sample
prior to
SUBSTTT'UTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
53
serum addition and performing the extraction. Twenty microliters of the
aqueous phase
was then mixed with 2~L of loading buffer and the sample was run on a 1%
agarose gel
in TAE buffer. Following one hour, the gel was placed on a transilluminator
and a
photograph was taken.
6. Lipid Uptake Stacdies: For the uptake studies, 1x10' BHK cells
were grown on 12 well plates overnight in 2 mL of complete media (DMEM + 10%
FBS)
at 37°C in 5% CO~. Then 20 nmol of the 2, 3, and 4 mol % CPL-SPLP
samples
containing 0.5% rhodamine-DSPE were mixed with HBS + 75mM CaCl~ to a final
volume of 200qL and this was added to the top of the cells followed by the
addition of
800~.L of complete media. This was allowed to incubate on top of the cells for
2, 4, 6,
and 8 hours at which time the cells were washed three times with PBS and were
lysed
with 600~L of 0.1% Triton X-100 in PBS, pH 8Ø The rhodamine fluorescence of
the
lysate was then measured on a fluorometer using a 7~e,~ of 560 nm and a n,em
of 600 nm
using slit widths of 10 and 20 nm, respectively. An emission filter of 430 nm
was also
used. A 1.0 mL microcuvette was used. The lipid uptake was determined by
comparison
of the fluorescence to that of a lipid standard (5 nmol). This value was then
normalized
to the amount of cells present by measuring the protein in 50~L of the lysate
using the
BCA assay.
Fluorescence micrographs were taken on a Zeiss fluorescence microscope.
7. Transfection Studies: For the in vitro transfection studies, 5x10'
BHK cells were plated in 24-well plates in complete media. These were
incubated
overnight at 37°C in 5% COZ. SPLP, SPLP + 75mM CaCl2, DOPE:DODAC
(1:1)/DNA
complexes, and CPL-SPLP systems (2, 3, and 4 mol % CPL) containing 2.5p.g of
DNA
were made up to 100~L using HBS or HBS + 75mM CaCIZ and were placed on the
cells.
Then 400p,L of complete media was added to this. At 4 and 9 hours, the
transfection
media was removed and replaced with complete media containing penicillin and
streptomycin for a complete 24 hour transfection. At the end of the
transfection period,
the cells were lysed with lysis buffer containing Triton X-100. Following this
lysis, 10-
20p,L of the lysated was transferred to a 96-well luminescence plate. The
luminescence
of the samples on the plate were measured using a Luciferase reaction kit and
a plate
luminometer. The luciferase activity was determined by using a luciferase
standard curve
SUBSTITUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
54
and was normalized for the number of cells by measuring the protein with the
BCA assay
on 10-20 ~L of the lysated.
C. Results and Discussion
Figures 18A and B show that the uptake and transfection of the SPLP
system is on the order of 105 times lower than complexes.
The CPL, DSPE-Quads, will be used in the following studies. Its
structure is shown in Figure 16A. This molecule possesses four positive
charges at the
end of a PEG3.~oo molecule, which has been covalently attached to the lipid
DSPE. The
incorporation of this CPL into empty liposomes of the same composition as the
SPLP has
been described previously in the above examples.
The incorporation of the CPL into the SPLP involves only a few steps.
These steps are shown in Figure 16B.
The DSPE-Quads was incorporated into SPLPs containing DOPE:PEG-
CerC20:DODAC (84:10:6) at various concentrations of the CPL (from 2-4 mol %).
The
incorporation efficiencies for the various CPL percentages were between 70 and
80% of
the initial. In order to separate the SPLPs possessing the CPL from the
unincorporated
CPL, gel filtration chromatography was employed. A typical column profile for
the 3%
DSPE-Quads is shown in Figure 19A. The CPL, lipid, and DNA all eluted from the
column at the same time in a single peak. There was however a small amount of
unincorporated CPL that eluted at a later stage. To show that the incorporated
CPL
remains incorporated, the sample is re-eluted from the column (Figure 19A). As
it can be
seen in Fig. 19B, no CPL is eluted in the later fractions of the column
indicating that the
CPL remains associated with the lipid.
To determine the optimal incubation period for the insertion of the CPL, a
time course at 60°C was performed (Figure 20). From this figure, it can
be determined
that the optimal insertion occurs between 2 and 3 hours.
The diameter of these particles containing the CPL was determined by
QELS to be 125 mn compared to the SPLP, which had a diameter of 109 nm. To
observe
the structure of these particles compared to the SPLP in the absence of the
CPL, freeze-
fracture EM was performed.
The serum stability of the SPLP in the presence and absence of various
amounts of the CPL was assayed (data not shown). Incubating free DNA with 50%
mouse serum for only 1 hour results in its complete degradation. The serum
stability of
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
the CPL-SPLPs was similar to that for the SPLP system. This indicates that the
DNA in
the CPL-SPLP is as protected as that in the SPLP system without CPL.
The major objective of this study is to increase both the uptake and
transfection of the SPLP system using CPLS. Figure 21 shows the time course
for the
5 uptake of rhodamine labeled SPLP in the presence (2, 3, or 4 mol %) and
absence of the
DSPE-Quads (0%). The uptake of the 4% system is higher than the 3% system,
which is
higher than the 2% system, and all three are much higher than the system
without CPL.
Fig. 22 shows 4 h and 9 h time points of the same formulations.
V. EXAMPLE V
10 This example illustrates the incorporation of a CPL into a Stabilized
Antisense-Lipid Particle ("SALP")
A. Materials and Results
Distearoylphosphatidylcholine (DSPC), was purchased from Northern
Lipids (Vancouver, Canada). 1,2-dioleoyloxy-3-dimethylammoniumpropane (DODAP
15 or AL-1) was synthesized by Dr. Steven Ansell (Inex Pharmaceuticals) or,
alternatively,
was purchased from Avanti Polar Lipids. Cholesterol was purchased from Sigma
Chemical Company (St. Louis, Missouri, USA). PEG-ceramides were synthesized by
Dr.
Zhao Wang at Inex Pharmaceuticals Corp. using procedures described in PCT WO
96/40964, incorporated herein by reference. [3H] or [14C]-CHE was purchased
from
20 NEN (Boston, Massachusetts, USA). All lipids were > 99% pure. Ethanol
(95%),
methanol, chloroform, citric acid, HEPES and NaCI were all purchased from
commercial
suppliers. Lipid stock solutions were prepared in 95% ethanol at 20 mg/mL (PEG-
Ceramides were prepared at 50 mg/mL).
SALPs are first prepared according to the methods set out in PCT Patent
25 Application No. WO 98/51278, published 19 November 1998, and incorporated
herein by
reference. See also, J.J. Wheeler et al., (1999), Gene Therapy, 6, 271-281.
Briefly, a
l6mer of [3H]-phosphorothioate oligodeoxynucleotide Inx-6295 (human c-myc)
having
sequence 5' T AAC GTT GAG GGG CAT 3' (SEQ ID. No: 1 ) (in 300 mM citrate
buffer, pH 3.80) was warmed to 65°C and the lipids (in ethanol) were
slowly added,
30 mixing constantly (DSPC:CHOL:DODAP:PEG-CerCl4; 25:45:20:10, molar ratio).
The
resulting volume of the mixture was 1.0 mL and contained 13 mmol total lipid,
2 mg of
antisense oligodeoxynucleotide, and 38% ethanol, vol/vol. The antisense-lipid
mixture
was subjected to 5 cycles of freezing (liquid nitrogen) and thawing
(65°C), and
SUBSTTTLTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
56
subsequently was passed l OX through three stacked 100 nm filters (Poretics)
using a
pressurized extruder apparatus with a thermobarrel attachment (Lipex
Biomembranes).
The temperature and pressure during extrusion were 65°C and 300-400 psi
(nitrogen),
respectively. The extruded preparation was diluted with 1.0 mL of 300 mM
citric acid,
pH 3.8, reducing the ethanol content to 20%. The extruded sample was dialyzed
(12 000-
14 000 MW cutoff; SpectraPor) against several liters of 300 mM citrate buffer,
pH 3.8 for
3-4 hours to remove the excess ethanol. The sample was subsequently dialyzed
against
HEPES-buffered saline (HBS), pH 7.5, for 12-18 hours to neutralize the DODAP
and
release any antisense that was associated with the surface of the vesicles.
Encapsulation
was assessed either by analyzing the pre-column and post-column ratios of ['H]-
antisense
and [~'~C]-lipid or by determining the total pre-column and post-column ['H]-
antisense
and [~'~C]-lipid radioactivity.
CPL is incorporated after the SALPs are prepared. Approximately 5 p.mol
SALP were mixed with 3-10 mol % CPL (i.e., 0.15-0.5 ~mol CPL). CPL were stored
as
micellar solutions in HBS, or in methanol. When CPL was added in methanol, the
final
methanol concentration of 3-4%. The mixtures were incubated overnight at room
temperature or at 40°C. Unincorporated CPL was removed from the SALP
preparation
by column separation (Sepharose CL-4B equilibrated with HBS, 75mM CaCL~ at pH
7.5). Incorporation efficiency was between 34 and 60%. It is anticipated that
other
organic solvents may improve incorporation efficiency.
TAI. EXAMPLE VI
A. General Overview
In the present example, distal positively charged cationic polyethylene
glycol) lipid conjugates (CPL) were synthesized and assessed for their
efficacy at
enhancing the cellular uptake of CPL-incorporated liposomes. It was confirmed
that
distal charged polymer conjugates bound to a liposome surface enhanced
liposome
uptake in mammalian cells in vitro.
B. Methods
Determination of the critical micelle concentration (CMC)
The CMCs of the CPLs were determined using the NPN assay as
previously reported by Brito and Vaz (see, Brito, R.M.M., and Vaz, W.L.C.
(1986)
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
57
Determination of the critical micelle concentration of surfactants using the
fluorescent
probe N-phenyl-1-naphthylamine. Anal. Biochem. 152, 250-255.). A series of
different
concentrations of CPLs were prepared in HBS buffer (25 rniVl Hepes, 150 mM
NaCI, pH
7.4). 5 ~M of NPN (from a stock NPN solution in 95% ethanol) was added into
the
above CPL solutions. After incubation of the mixtures at room temperature for
30 min,
the fluorescence intensities at ~,em 410 nm using ~,e~=356 nm on a Perkin
Elmer LS 50
Luminescence Spectrometer.
C. Results
Uptake Enhancement of CPL-L UVs in vitro. Cellular uptake of
conventional CPL-liposomes.
The in vitro cellular uptake of CPL-containing liposomes was studied on
baby hamster kidney (BHK) cells. The liposome-associated fluorescent lipid
marker
(Rh-PE) was used as a marker for lipid uptake. As shown in Figures 26 and 27,
CPL:
significantly enhances the cellular uptake compared to control samples (no
CPL) using
both PBS-CMG and serum containing medium. The time dependent uptake of CPL-
LLTVs reaches a maximum after 3 hr. Figure 6 summarized the cell uptake of the
different CPL-containing vesicles after a four hour incubation. Compared to a
control,
reduced cell uptake was observed for CPL,, a moderate increase for CPL (2
fold), and a
large increase for both CPL. and CPLg. The similar degree of increase
resulting from
CPLa and CPLB indicates a charge density of four in the CPLs satisfies the
requirement
for maximum enhanced cellular uptake.
VII. EXAMPLE VII
A. General Overview
This experiment describes the synthesis of a new class of cationic lipids
designed to enhance non-specific targeting by increasing the electrostatic
attraction
between liposomes and cells.
B. Materials and Reagents
tBoc-NH-PEG3~oo-COZ-NHS was obtained from Shearwater Polymers
(Huntsville, AL). Na,NE-di-tBoc-L-lysine-N-hydroxysuccinimide ester, N~-dansyl-
L-
lysine, N-hydroxysuccinimide (NHS), and N,N'-dicyclohexyl-carbodiimide (DCC)
were
SUBSTTTLJTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
58
purchased from Sigma-Aldrich Canada (Oakville, ON). 1,2-Distearoyl-sn-glycero-
3-
phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine
(DSPE)
were obtained from Northern Lipids (Vancouver, BC). Fluorescamine and
Rhodamine-
DSPE (Rh-PE) were obtained from Molecular Probes (Eugene, OR). Cholesterol
(Chol)
was obtained from Sigma Aldrich Canada (Oakville, ON). Trifluoroacetic acid,
diethyl
ether, methanol, triethylamine, and chloroform were obtained from Fisher
Scientific
(Vancouver, BC). All reagents were used without further purification.
General Methods
All reactions were performed in 16 x 100 mm glass test tubes. 1 H NMR
spectra were obtained employing a Bruker MSL 200 spectrometer operating at 200
MHz.
Deuterated chloroform (CDC13) was used as the solvent in the NMR experiments.
Proton
chemical shifts (b) were referenced to CHC13 set at 7.24 ppm. When signals
were
reasonably resolved, their intensities were integrated to allow an estimation
of the
number of protons. The chemical shifts of exchangeable amino group protons,
observed
between 7 - 8 ppm, are not given. These peaks were assigned on the basis of
their
removal by a DSO exchange.
Phosphorus and fluorescamine assays were performed to confirm the ratio
of primary amine per phosphate in each CPL as follows.
The phosphate concentration of the CPL was determined using the Fiske-
Subarrow phosphorus assay (see, Fiske, C.H., and Subbarow, Y. (1995) The
colorimetric
determination of phosphorous. J. Biol. Chem. 66, 375-400.). The primary amine
concentration in the CPL was determined using the fluorophore, fluorescamine.
A
fluorescamine solution (0.6mg/mL) in acetone was prepared. An aliquot of CPL
solution
in HBS (2-4 pL) was made up to 250 ~L with 200 mM sodium borate, pH 8Ø To
this
mixture, 50 L of the fluorescamirie solution was added dropwise with
vortexing, followed
by 1700 p.L of water. The fluorescence of this solution was measured using a
Perkin-
Elmer LS50 Luminescence Spectrometer with ~,e,~ of 397 nm and ~,em of 475 nm,
and
excitation and emission slit widths of 10 nm. The primary amine concentration
of the
CPL was determined from a lysine standard curve.
tBoc-NH-PEG3aoo-CGr(NE-dansyl)lysine (1). tBoc-NH-PEG3aoo-COa-
NHS (500 mg, 147 ~mol) in 3mL of dry chloroform was added slowly to a solution
of
NE-dansyl-L-lysine (65 mg, 171 ~mol) in 1 mL of methanol and 200 p,L of
triethylamine.
After the reaction mixture was stirred at room temperature for 3 h, the
solvent was
SUBSTTTLTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
59
removed under a N~ stream and further dried under vacuum. The crude product
was
washed by first dissolving it in a minimum amount of chloroform with warming
and then
precipitating it out with the addition of 10 mL of diethyl ether. The ether
was added
while vortexing. Precipitation of 1 was accelerated by cooling. The
precipitate was then
pelleted by centrifugation and the ether was discarded. This chloroform/ether
wash and
precipitation procedure was repeated. The dry solid was then dissolved in 4 mL
of
chloroform and cooled in an ice bath for 15 min. Methanol (2 mL) was added if
this
cooled solution was clear. If a precipitate (excess dansyl lysine) developed,
it was
filtered off prior to the addition of methanol. The chloroform/methanol
solution was
washed with 1.2 mL of 0.1 M HC1. The chloroform phase was extracted, dried,
and the
solid redissolved in 6 mL of chloroform/methanol (2:1 v/v) and washed with 1.2
mL of
distilled water. The chloroform phase was extracted, dried to a thick paste
and tBoc-NH-
PEG-COZ-{NE-dansyl)lysine (1) was precipitated with 10 mL of ether. After
centrifugation and the removal of ether, the dried product is a light yellow
solid. Yield:
520 mg (93%). TLC (silica gel) chloroform/methanol (85:15 v/v): Rf0.56.'H NMR
(CDC13): S 1.08 (t), 1.40 (s, 11H), 2.66 (s, 1H), 2.86 (s, 8H), 3.27 (q), 3.50
(t), 3.60 (s,
309H), 3.96 (t), 4.19 (m[broad]), 5.03 (s[broad], 1H), 5.22 (t, 1H), 5.43 (d,
1H), 7.16 (d,
1 H), 7.51 (q, 2H), 8.19 (d, 1 H), 8.27 (d), 8.50 (d, 1 H) ppm.
Dansylated CPL,-tBoc (3). First, tBoc-NH-PEG3,~oo-COZ-(NE-
dansyl)lysine-NHS (2) was prepared as follows. A solution of tBoc-NH-PEG3aoo-
COZ-
(Ne-dansyl)lysine (1) (500 mg, 132 p,mol) and NHS (31.5 mg, 274 ~.mol) in 2 mL
of dry
chloroform was added to DCC (42.8 mg, 207 ~mol) dissolved in 1 mL of dry
chloroform.
The reaction mixture was stirred for 2 h at room temperature. The by-product,
dicyclohexyl urea (DCU), was filtered using a Pasteur pipette with a cotton
plug. The
filtrate, containing tBoc-NH-PEGS.goo-COZ-(NE-dansyl)lysine-NHS (2), was
slowly added
to a solution of DSPE (120.6 mg, 161 ~,mol) in 2 mL of dry chloroform and 200
pL of
triethylamine. The dissolution of DSPE in dry chloroform and triethylamine
required
warming to 65 °C. After the reaction mixture was stirred at room
temperature for 3 h, it
was dried, and chloroform/ether washed and precipitated as described earlier
until the
disappearance of DSPE on TLC as visualized with ninhydrin. This removal of
excess
DSPE required at least three washings. The product, dansylated CPL,-tBoc (3),
was
dissolved in chloroform/methanol (2:1), washed with dilute HCl and water, and
precipitated using ether as described for (1). Yield: 575 mg (96%). TLC
(silica gel)
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
chloroformimethanol (85:15) Rf0.58. 1H NMR (CDC1;): a 0.85 (t, 4H), 1.22 (s,
48H),
1.41 (s, lOH), 1.55 (t), 2.27 (m[broad], 6H), 2.90 (m[broad], 6H), 3.04 (s,
8H), 3.27 (q),
3.61 (s, 275H), 4.14 (m[broad]), 4.32 (d), 4.38 (d), 5.05 (s[broad]), 5.23
(s[broad]), 5.58
(m[broad]), 7.37 (d, 1H), 7.49 (s[broad], 1H), 7.59 (t, 2H), 8.24 (d, 1H),
8.50 (d, 1H),
5 8.59 (d, 1H) ppm.
Dansylated CPL, (4). Trifluoroacetic acid (TFA) (2 mL) was added to a
solution of dansylated CPL,-tBoc (3) (550 mg, 121 pmol) in 2 mL of chloroform
and
stirred for 4 h at room temperature. The solution was concentrated to a thick
paste and
chloroform/ether washed three times. After the removal of ether, the solid was
dissolved
10 in 6 mL of chloroform/methanol (2:1) and washed with 1.2 mL of 5% sodium
bicarbonate. The chloroform phase was extracted, dried and redissolved in 6 mL
chloroform/methanol (2:1 ) and washed with 1.2 mL distilled water. The
chloroform
phase was concentrated to a thick paste and the purified CPL1 (4) was obtained
through a
chloroform/ether ~.vash and vacuum dried. Yield: 535 mg (97%). TLC (silica)
15 chloroform/methanol/water (65:25:4) Rf0.76. 'H NMR (CDCI;). b 0.85 (t, 4H),
1.22 (s,
46H), 1.54 (m[broad], 8H), 2.23 (t, 6H), 2.84 (s, 9H), 3.16 (m[broad], 3H),
3.26 (t, 3H),
3.61 (s, 263H), 3.98 (q), 4.17 (t), 4.33 (d), 4.38 (d), 5.19 (s[broad]), 5.93
(d, 1H), 7.13 (d,
1H), 7.46 (t, 1H), 7.52 (t, 1H), 8.15 (d, 1H), 8.43 (t, 2H) ppm.
Dansylated CPLZ-tBoc (5). A solution of Na,NE-di-tBoc-L-lysine-N-
20 hydroxysuccinimide ester (105 mg, 236 ~mol) in 2 mL dry chloroform was
Gradually
added to a solution of dansylated CPL, (4) (510 mg, 112 pmol) in 2 mL
chloroform
containing 200 pL triethylamine and stirred at room temperature for 3 h. The
completion
of the reaction was indicated by the disappearance of primary amine as
visualized by
ninhydrin assay on TLC. The reaction mixture was concentrated to a thick paste
and
25 chloroform/ether washed (~3 times) until the disappearance of excess Na,NE-
di-tBoc-L-
lysine-N-hydroxysuccinimide ester as checked by TLC. The product was dissolved
in 6
mL chloroform/methanol (2:1) and washed with 1.2 mL 0.1 M HCI. The chloroform
phase was extracted, dried, redissolved in 6 mL chloroform/methanol (2:1 ) and
washed
with 1.2 mL distilled water. The chloroform phase was concentrated to a thick
paste and
30 the purified compound was obtained through a chloroform/ether wash and
vacuum dried.
Yield: 510 mg (96%). TLC (silica gel) chloroform/methanol (85:15) Rf0.58. 'H
NMR
(CDCl3). 8 0.85 (t, 3H), 1.22 (s, 44H), 1.41 (s, 20H), 1.56 (m[broad]), 1.78
(m[broad]),
2.27 (m, 5H), 2.88 (s), 2.91 (s), 2.97 (s), 3.06 (s, 7H), 3.26 (t), 3.44 (t),
3.62 (s, 252H),
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
61
3.97 (t), 4.05 (d), 4.13 (m), 4.33 (d), 4.38 (d), 4.68 (s[broad]), 5.22
(s[broad]), 5.51
(s[broad]), 6.57 (t[broad], 1H), 7.39 (d, 1H), 7.51 (s[broad], 1H), 7.60 (t,
2H), 8.26 (d,
1H), 8.53 (d, 1H), 8.61 (d, 1H) ppm.
Dansylated CPL (6). The synthesis of CPLz (6) was the same as that of
CPL, (4) by deprotecting dansylated CPLZ-tBoc (5) (490 mg, 103 ~mol). Yield:
478 ma
(97%). TLC (silica) chloroform/methanol/water (65:25:4) Rf0.63. 'H NMR
(CDCI;). 8
0.85 (t, 3H), 1.22 (s, 42H), 1.55 (m, lOH), 1.93 (s[broad], 4H), 2.24 (t, 5H),
2.85 (s, 8H),
3.26 (t, 3H), 3.61 (s, 271H), 3.95 (q), 4.17 (s), 4.34 (s), 5.18 (s[broad],
1H), 6.31 (d, 1H),
6.89 (s, 1H), 7.10 (d, 1H), 7.49 (m, 1H), 8.15 (d, 1H), 8.34 (d, 1H), 8.47 (d,
2H) ppm.
Dansylated CPL,-tBoc (7). The synthesis of CPLa-tBoc (7) was the same
as that of CPL-tBoc (5) by reacting Na,NE-di-tBoc-L-lysine-N-
hydroxysuccinimide ester
(170 mg, 383 ~mol) with dansylated CPL (6) (455 mg, 95 umol). Yield: 475 mg
(96%).
TLC (silica gel) chloroform/methanol (85:15) Rf0.58. 'H NMR (CDCI;). 8 0.85
(t, 3H),
1.22 (s, 43H), 1.40 (s, 39H), 1.71 (m[broad], 6H), 2.27 (m, 5H), 2.88 (s),
2.90 (s), 2.95
(s), 3.05 (s, lOH), 3.25 (t, 3H), 3.43 (s), 3.61 (s, 262H), 3.97 (t), 4.05
(d), 4.15 (m), 4.32
(d), 4.37 (d), 4.51 (s[broad]), 4.75 (s[broad]), 4.90 (s[broad]), 5.23
(t[broad], 1H), 5.52
(s[broad]), 5.80 (s[broad], 1H), 7.15 (m[broad], 1H), 7.38 (d, 1H), 7.50 (s,
1H), 7.59 (t,
2H), 8.25 (d, 1H), 8.51 (d, 1H), 8.60 (d, 1H) ppm.
Dansylated CPL4 (8). The synthesis of CPL. (8) was the same as that of
CPL, (4) by deprotecting dansylated CPL-tBoc (7) (450 mg, 86 p.mol). Yield:
440 mg
(97%). TLC (silica) chloroform/methanol/water (65:25:4) Rf0.19. 1H NMR
(CDCl3). 8
0.85 (t), 1.22 (s), 1.53 (m[broad]), 2.34 (m[broad]), 2.86 (s), 3.26 (t), 3.62
(s), 3.87
(s[broad]), 3.97 (t), 4.17 (s[broad]), 4.33 (d), 5.18 (s[broad]), 7.15 (d),
7.43 (s), 7.51 (t),
8.15 (d), 8.32 (d), 8.48 (d), 9.05 (s[broad]) ppm.
Dansylated CPLg-tBoc (9). The synthesis of CPLg-tBoc (9) was the same
as that of CPLZ-tBoc (5) by reacting Na,NE-di-tBoc-L-lysine-N-
hydroxysuccinimide ester
(70 mg, 158 ~.mol) with dansylated CPL4 (8) (100 mg, 19 p,mol). Yield: 112 mg
(96%).
TLC (silica gel) chloroform/methanol (85:15) Rf0.58. 1H NMR (CDCl3). 8 0.84
(t, 3H),
1.08 (s), 1.21 (s, 39H), 1.39 (s, 75H), 1.66 (m [broad]), 2.26 (m, 4H), 2.89
(s, 4H), 3.06
(s, 11H), 3.25 (t, 3H), 3.43 (s), 3.49 (s), 3.60 (s, 248H), 3.96 (t), 4.04
(d), 4.12 (t), 4.31
(d), 4.36 (m), 5.19 (m [broad]), 6.77 (m [broad], 1H), 6.91 (s [broad], 1H),
7.24 (CHCl3),
7.41 (d), 7.50 (s [broad]), 7.60 (t), 8.25 (d, 1H), 8.53 (d, 1H), 8.63 (d, 1H)
ppm.
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
62
Dansylated CPLB (10). The synthesis of CPLg (8) was the same as that of
CPL, (4) by deprotecting dansylated CPLg-tBoc (9) (~0 mg, 8 ~mol). Yield: 48
mg
(96%). TLC (silica) chloroform/methanol/water (65:25:4) Rf0.13. 'H NMR
(CDCl3). 8
0.85 (t, 3H), 1.22 (s, 34H), 1.52 (s [broad]), 2.23 (s [broad]), 2.86 (d),
3.27 (d), 3.61 (s,
274H), 3.96 (t), 4.18 (m [broad]), 7.14 (s [broad]), 7.24 (CHC13), 7.50 (m
[broad]), 8.12-
8.27 (s [broad]), 8.47 (m [broad]) ppm.
C. Results and Discussion
The CPL were synthesized by repeated coupling reaction steps involving
amines and NHS-activated carbonate groups as outlined in Figure 29. This
consists of (a)
incorporating the dansyl fluorescent label to the hydrophilic PEG spacer, (b)
coupling of
the DSPE anchor, and (c) attachment of the cationic headgroup to the lipid.
The
heterobifunctional PEG polymer tBoc-NH-PEG3aoo-COZ-NHS (MW 3400), was chosen
for two reasons. Firstly, it was commercially available. Secondly, it is
insoluble in ether
that provided a very convenient means of purifying its derivatives, 1 -10.
Other reagents
were used in excess to ensure the complete conversion of the PEG polymer to
its
derivatives. The excess reagents were soluble in ether and therefore could be
removed by
washing in ether during purification.
Incorporation of the fluorescent label, N~-dansyl lysine, to the PEG
polymer by coupling the a-amino group of dansyl lysine with the NHS activated
carbonate of PEG gave the lysine derivative 1. The DSPE anchor was coupled via
intermediate 2 that was formed by the esterification of 1 using NHS and DCC.
The
resulting PEG lipid, 3, was deprotected by removing the tBoc to form CPL1, 4,
with one
positive charge. The positive charges in the other CPL are carried by the
amino groups
of lysine. Here, the NHS activated and di-tBoc protected lysine was attached
to the free
amino function of CPL, to form intermediate 5 which, upon deprotection,
yielded CPL,
6, with two positive charges. The attachment of two lysine residues to the
amino groups
of CPL, via intermediate 7 gave CPL4, 8, with four positive charges. Thus,
CPLB, 10,
with eight positive charges was synthesized with the attachment of four lysine
residues as
the headgroup. As can be seen, this provides a very convenient means of
synthesizing
multivalent CPL that are of particular interest for non-viral drug delivery
applications.
The structures of the purified intermediates and CPL in Figure 29 were
verified by iH NMR spectroscopy and chemical analysis. The'H NMR spectra
showed
well-resolved resonances for the PEG, tBoc and acyl chains of DSPE at
approximately
SUBSTTrLJTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
63
3.61, 1.41 and 1.21 ppm, respectively, and for the resonances of the dansyl
moiety
(aromatic protons at 7.1-8.5 ppm; methyl protons at 2.8-3.0 ppm). From the
integrated
signal intensities of the former three peaks, it was found that the ratio of
tBoc/PEG or
tBoc/DSPE was 1.0, 2.1, 4.0, and 8.1 for CPL,-tBoc, CPL-tBoc, CPL4-tBoc, and
CPLg-
tBoc, respectively. As each tBoc is attached to an amino group, this gives the
number of
amino groups in the headgroup of each CPL relative to the CPL,. That
essentially
identical results were obtained using the ratios of tBoc relative to both PEG
and DSPE
demonstrates the presence of lipid and polymer in correct proportion to the
headgroup.
The complete cleavage of the tBoc protecting groups was verified by the loss
of tBoc
NMR peaks and chemical analysis which determined the ratio of primary amine to
phosphate in each of the CPL by using the fluorescamine and phosphorus assays.
The
amine/phosphate ratios for CPL,, CPL, CPL:, and CPL$ were found to be 1.0,
2.2, 3.7,
and 8.0, respectively. These corresponded well with the expected number of
positive
charge bearing amino groups of the respective CPL.
1 S The CPL described here possess several attributes which may increase
their usefulness relative to other cationic lipids. Firstly, the phospholipid
anchor will
readily allow efficient incorporation of CPL into liposomal systems. Secondly,
the
dansyl label will permit accurate and convenient quantification of the CPL in
the bilayer
using fluorescence techniques. Finally, the valency of the cationic headgroup
in the CPL
can easily be modified using lysine residues.
VIII. EXAMPLE VIII
A. General Overview
The synthesis of a fluorescent cationic polyethylene glycol) (MW 1000)
lipid conjugates (CPL)' is described. The procedure is very similar to that of
PEG 3400
described in detail previously. However the lower molecular weight PEG
derivatives
may not be insoluble in ether, and therefore could not be readily purified by
ether wash as
before. The synthetic procedure is similar to the one outlined in Figure 29.
B. Abbreviations
tBoc, tert-butyloxycarbonyl; tBoc-NH-PEG,ooo-C~z-NHS, tBoc protected
and NHS activated PEGlooo~ CPL, cationic polyethylene glycol) lipid conjugate;
CPLI,
CPL with one positive charge; CPL, CPL with two positive charges; CPL4, CPL
with
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
64
four positive charges; DCC, N,N'-dicyclohexyl-carbodiimide; DCU, dicyclohexyl
urea;
NHS, N-hydroxysuccinimide; di-tBoc-lysine-NHS, Na,NE-di-tBoc-L-lysine-N-
hydroxysuccinimide ester; DSPE, 1,2-distearoyl-sn-glycero-3-
phosphoethanolamine;
PEG~ooo~ polyethylene glycol) with an average MW of 1000; TFA, trifluoroacetic
acid.
C. Materials and Reagents
tBoc-NH-PEG~ooo-COz-NHS was obtained from Shearwater Polymers
(Huntsville, AL). Na,NE-di-tBoc-L-lysine-N-hydroxysuccinimide ester, N~-dansyl-
L-
lysine, N-hydroxysuccinimide (NHS), and N,N'-dicyclohexyl-carbodiimide (DCC)
were
purchased from Sigma-Aldrich Canada (Oakville, ON). 1,2-Distearoyl-sn-glycero-
3-
phosphoethanolamine (DSPE) was obtained from Northern Lipids (Vancouver, BC).
Fluorescamine was obtained from Molecular Probes (Eugene,OR). Trifluoroacetic
acid,
diethyl ether, methanol, triethylamine, and chloroform were obtained from
Fisher
Scientific (Vancouver, BC). All other reagents were used without further
purification.
tBoc-NH-PEGiooo-CO~-(Ne-dansyl)lysine (1). tBoc-NH-PEG,ooo-COz-
NHS (500 mg, 500 ~mol) in 3mL of dry chloroform was added slowly to a solution
of
NE-dansyl-L-lysine (200 mg, 536 p,mol) in 1.5 mL of methanol and 300 uL of
triethylamine. After the reaction mixture was stirred at room temperature for
3 h, the
solvent was removed under a Nz stream and further dried under vacuum. The
crude
product was dissolved in 6 mL of chloroform/methanol (2:1 v/v), washed once
with 1.2
mL of 0.5 M HCl and rivice with 1.2 mL of distilled water. The chloroform
phase was
extracted, dried to a thick paste and tBoc-NH-PEG-COz-(Ne-dansyl)lysine ( 1 )
was
obtained as a light yellow solid. Yield: 600 mg (95%). TLC (silica gel)
chloroform/methanol (85:15 v/v): Rf0.50.
Dansylated CPL-tBoc (3). First, tBoc-NH-PEGiooo-COz-(NE
dansyl)lysine-NHS (2) was prepared as follows. A solution of tBoc-NH-PEGiooo-
COz-
(Ne-dansyl)lysine (1) (600 mg, 474 p.mol) and NHS (113 mg, 982 p,mol) in 2 mL
of dry
chloroform was added to DCC ( 150 mg, 728 ~,mol) dissolved in 1 mL of dry
chloroform.
The reaction mixture was stirred for 5 h at room temperature. The by-product,
dicyclohexyl urea (DCU), was filtered using a Pasteur pipette with a cotton
plug. The
filtrate, containing tBoc-NH-PEGiooo-COz-(NE-dansyl)lysine-NHS (2), was slowly
added
to a solution of DSPE (365 mg, 488 p,mol) in 3 mL of dry chloroform and 300 ~L
of
triethylamine. The dissolution of DSPE in dry chloroform.and triethylamine
required
SUBSTIT'LTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
warming to 65 °C. After the reaction mixture was stirred overnight at
room temperature,
it was filtered to remove some precipitate (unreacted DSPE) and dried to a
viscous paste.
The paste was dissolved in chloroform/methanol (2:1), washed with dilute HC1
and water
as before. The product, dansylated CPL,-tBoc (3), was obtained after the
removal of
5 solvent and precipitated using 10 mL of ether. Yield: 900 mg (96%). TLC
(silica gel)
chloroform/methanol (85:15) Rf0.58.
Dansylated CPL, (4). Trifluoroacetic acid (TFA), 3 mL, was added to a
solution of dansylated CPL,-tBoc (3) (900 mg, 456 umol) in 3 mL of chloroform
and
stirred for 4 h at room temperature. The solution was concentrated to a thick
paste and
10 chloroform/ether washed three times. After the removal of ether, the solid
was dissolved
in 6 mL of chloroform/methanol (2:1) and washed rivice with 1.2 mL of 5%
sodium
bicarbonate and twice with 1.2 mL distilled water. The chloroform phase was
concentrated to a thick paste and the purified CPL, (4) was obtained through a
chloroform/ether wash and vacuum dried. Yield: 7~0 mg (88%). TLC (silica)
15 chloroform/methanol/water (65:25:4) Rf0.72.
Dansylated CPL-tBoc (5). A solution of Na,NE-di-tBoc-L-lysine-N-
hydroxysuccinimide ester (350 mg, 789 ~mol) in 3 mL dry chloroform was
gradually
added to a solution of dansylated CPL, (4) (750 mg, 400 p.mol) in 3 mL
chloroform
containing 300 pL triethylamine and stirred at room temperature for 3 h. The
completion
20 of the reaction was indicated by the disappearance of primary amine as
visualized by
ninhydrin assay on TLC. The reaction mixture was concentrated to a thick
paste,
redissolved in 6 mL chloroform/methanol (2:1) and washed once with 1.2 mL 0.5
M HCl
four times with 1.2 inL distilled water. The chloroform phase was extracted
and dried.
No further purification was performed. Yield: 700 mg (81 %). TLC (silica gel)
25 chloroform/methanol (85:15) Rf0.58.
Dansylated CPL (6). The synthesis of CPLZ (6) was the same as that of
CPL (4) by deprotecting dansylated CPLZ-tBoc (5) (700 mg, 318 ~mol). Yield:
650 mg
(92%). TLC (silica) chloroform/methanol/water (65:25:4) Rf0.63.
Dansylated CPL,-tBoc (7). The synthesis of CPL4-tBoc (7) was the same
30 as that of CPLZ-tBoc (5) by reacting Na,Ne-di-tBoc-L-lysine-N-
hydroxysuccinimide ester
(500 mg, 1127 ~mol) with dansylated CPLZ (6) (650 mg, 292 p,mol). Besides
washing
with dilute HCL and water no further attempts were made to purify CPL-tBoc
before
SUBSTITUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/004s1
66
deblocking to generate CPL. Yield: 800 mg (Crude). TLC (silica gel) chloroform
/methanol (85:15) Rf0.58 (dansyl peak only).
Dansylated CPL. (8). The synthesis of CPL: (8) was the same as that of
CPL, (4) by deprotecting dansylated CPLa-tBoc (7) (800 mg). The final product
was
purified by column chromatography using silica gel 60, 70 - 230 mesh, and
chloroform/methanol/ammonia solution (65:25:4 v/v). Yield: 300 mg (38%). TLC
(silica) chloroforni/methanol/water (65:25:4) Rf0.15.
IX. EXAMPLE IX
A. General Overview
We show here that CPL: can be inserted into preformed SPLP and that the
resulting SPLP-CPL exhibit improved uptake and markedly improved iu vitro
transfection potency in BHK cells. These results establish that the SPLP
system is
intrinsically a hig:~~i potent transfection vector.
B. Materials and Methods
1. Preparation of SPLP, SPLP-CPLQ, a~zd Complexes
(i). SPLP: SPLP composed of DOPE:DODAC:PEG-CerCzo (84:6:10) and
containing the plasmid pLuc, a modified marker gene expressing luciferase, was
supplied
by INEX Pharmaceuticals Inc.
(ii) SPLP-CPL.: Dansylated CPL. was prepared in our laboratory and
incorporated into SPLP as follows: SPLP at a dose of 500 nmol lipid was
incubated with
different amounts of CPLa (12.5, 19, and 30 nmol) at 60°C for 2 to 3
hours in Hepes
Buffered Saline, pH 7.5 (HBS) to achieve a final incorporation of 2, 3, and 4
mol %,
respectively. SPLP-CPL was separated from unincorporated CPL by gel filtration
chromatography on a Sepharose CL-4B column equilibrated in HBS. Fractions
(1mL)
were collected and assayed for CPL, phospholipid and DNA contents. Fractions
containing all three components were pooled and concentrated for use in
transfection and
uptake studies. The samples from the column were greatly aggregated. To
deaggregate
the systems, addition of CaCl2 or MgCl2 was required. Experiments to determine
the
optimal amount of cation for deaggregation will be described later in the
Methods.
CPL Assay.' The presence of CPL was determined by measuring the
fluorescence of the dansyl group in CPL on a Perkin Elmer LS52 Luminescence
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
67
spectrophotometer using ~,e~ = 340 nm and ~.em = 510 nm with excitation and
emission slit
widths of 10 and 20 nm, respectively. Fluorescence of the dansyl was
quantified using a
standard curve of dansylated CPL in HBS.
Phospholipid Assay. Phospholipid was determined by first extracting the
lipids from SPLP using the Bligh-Dyer technique. and then measuring phosphate
in the
organic phase according to the Fiske-Subbarow method (see, Bligh EG, Dyer WJ A
rapid method of total lipid extraction and purification. Can JBiochem Physiol
1959; 37:
911-917; and Fiske CH, Subbarow Y. The colorimetric determination of
phosphorous. J
Biol Chem 1925; 66: 375-400.).
DNA Assay: DNA content was measured using the PicoGreen Assay kit
(Molecular Probes, Eugene, Oregon) as previously described. (see, Mok KWC, Lam
AMI, Cullis PR. Stabilized plasmid-lipid particles: factors influencing
plasmid
entrapment and transfection properties. Biochim Biophys Acta 1999; 1419: 137-
1~0).
(iii) Complexes: The complexes were prepared, at a charge ratio of 1.5:1
+ve/-ve, by mixing 25 ~L of DOPE:DODAC (0.8mM), kindly supplied by Inex, with
25
~L of 88 pg/mL pLuc, also supplied by Inex, followed by incubation for 30 min
before
addition to cells.
?. Preparation of SPLP Containing 0.5 mol % of Rh-PE for Optimal
Insertion Time Determination and Lipid Uptake Experiments
SPLP were prepared as described by Wheeler et al. (see, Wheeler et al.,
Gene Therapy; 6:271-281 (1999)) with a few modifications. The lipids DOPE, PEG-
CerC~o, DODAC, and rhodamine-DOPE (Rh-PE), all stocks in CHC13, were mixed
together in a molar ratio of (83.5:10:6:0.5) and the CHC13 was completely
evaporated.
The resulting lipid film was dissolved in 20 mM octyl glucopyranoside (OGP)
and 200
~g/mL of plasmid DNA was added to a total volume of 1 mL. The OGP was dialysed
from the sample in a dialysis bag with two changes of buffer (HBS) over 48
hours. The
resulting sample was passed down a DEAF Sepharose column and the effluent was
run
on a discontinuous sucrose gradient as described previously. (see, Gabizon A,
Papahadjopoulos D. Liposome formulations with prolonged circulation time in
blood and
enhanced uptake by tumors. Proc Natl Acad Sci USA 1988; 85: 6949-6953.). The
resulting rhodamine-labeled SPLP possessed a DNA/Lipid ratio of ~60 ~,g/~mol.
SUBSTTTLITE S~iEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
68
3. Determination of Optimal Incubation Time for Insertion of CPL4
Into SPLP
To determine the time required for optimal insertion of CPLa into SPLP, ~
mol % of CPL: (0.3 nmol) was mixed with 6 nmol of SPLP (containing 0.5 mol %
Rh-
PE) in a total volume of 1.5 mL and incubated in a 60°C water bath. At
time points (30
min, 1 h, 2 h, 3 h, and 4 h), 250 ~.L of the mixture was run down a Sepharose
CL-4B
column equilibrated with HBS. The fractions possessing fluorescent dansyl were
combined and the dansyl fluorescence was measured using the parameters
described
above while the rhodamine fluorescence was measured using 7~r~=560 nm,
n.~m=590 nm,
and excitation and emission slit widths of 10 and 20 nm, respectively. These
measurements were also made on a small fraction of the original solution
before the
column. The dansyl/rhodamine ratios are calculated for both the initial and
final samples
to determine the percentage of the initial 5 mol % that was inserted.
4. Deaggregation of SPLP-CPL4 Using CaCl2 and MgCl2
As stated above, the preparation of SPLP-CPLa results in aggregation of
the particles. To deaggregate the system an increase in ionic strength is
required. This
was achieved by the addition of increasing amounts of CaCh or MgCl2 (~00 mM
stock
solution) to a solution of SPLP-CPL,. To 60 p.L of SPLP-CPL4 (3mM lipid) was
added
360 ~L of HBS in a Nicomp tube. The mean diameter ~ standard deviation of the
SPLP-
CPL (0 mM Cation) was then determined by QELS using a Nicomp Model 270
Submicron Particle Sizer. Then the salt (CaCh or MgCh) was added to
concentrations
from 20 mVI to 70 mM. At each interval the mean diameter ~ standard deviation
was
determined by QELS. The mean diameter of the particles hardly changes with
increasing
[Cation], however, the QELS Gaussian distribution gets broader. Therefore, the
standard
deviations were used as a measure of deaggregation.
S. Size Determination of SPLP-CPL4 and SPLP
Freeze-fracture EM was performed on the SPLP-CPL, (no CaCh), SPLP-
CPL.~ + 40 mM CaCl2, and SPLP, according to Wheeler et al. (see, Wheeler JJ et
al.
Stabilized plasmid-lipid particles: construction and characterization. Gene
Therapy 1999;
6: 271-281.). The SPLP-CPL4 contained 4 mol % CPL.. The micrographs of SPLP-
CPL4,
in the presence and absence of CaCl2, were compared to show the visual effect
of Caz+ on
the aggregation. Vesicle diameters of the SPLP-CPLa + 40 mM CaClz and SPLP
were
analyzed by QELS using a Nicomp Model 270 Submicron Particle Sizer.
SUBSTTI'LTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
69
6. Serum Stability of SPLP-CPL4 Particles
The serum stability of the SPLP-CPL containing various % of CPL were
determined by mixing the particles with mouse serum to a final serum
concentration of
50%,,. These mixtures were then incubated for 0, 1, 2, or 4 hours at
37°C. At these time
points, a volume of the mixture containing about 1 ~g of plasmid DNA was
removed and
the DNA was extracted from the lipid and protein using a phenol:chloroform
extraction.
The resulting DNA solutions were then run on a 1 % agarose gel following which
the
DNA was transferred to nitrocellulose and a Southern blot was performed.
7. Lipid Analysis of SPLP-CPL.
To determine the loss of PEG-CerCzo from the SPLP during the insertion
of CPLa, lipid was extracted for the SPLP sample and SPLP-CPL. sample by the
Bligh-
Dyer extraction. The mixtures were then passed through an HPLC and were
assayed for
DOPE and PEG-CerC2o by Northern Lipids, Inc (Vancouver, BC). The DOPE:PEG-
CerC2o ratios for the SPLP-CPL was compared to that for the SPLP and the
amount of
1 S PEGylated lipid in the outer monolayer of the SPLP was determined.
8. Uptake Studies
For all in vitro experiments, the cells used were a transformed BHK cell
line (tk-). For the uptake studies, 1x10' BHK cells were grown on 12-well
plates
overnight in 2 mL of complete media (DMEM + 10% FBS) at 37°C in 5% CO2.
SPLP,
SPLP-CPL. + 40 mM CaClz, or DOPE:DODAC complexes (200 ~L), each containing
0.5 mol % Rh-PE as lipid marker were mixed with 800 p.L of complete media and
this
mixture was added to the top of the cells at a lipid dose of 20 p.M. After
incubation at
37°C for 2, 4, 6, or 8 hours, the cells were washed with PBS and lysed
with 600 ~L of
lysis buffer (0.1% Triton X-100 in PBS). The rhodamine fluorescence of the
lysate was
measured in a 1.0 mL microcuvette on a Perkin-Elmer LS52 Luminescence
Spectrophotometer using a 7~eX of 560 nm and a 7~em of 600 nm with slit widths
of 10 and
20 nm, respectively. An emission filter of 430 nm was also used. Lipid uptake
was
determined by comparison of the fluorescence in the lysate to that of a lipid
standard and
normalized to the amount of cells as determined by the BCA protein assay
(Pierce,
Rockford, IL). Where indicated, fluorescence micrographs were taken on an
Axiovert
100 Zeiss Fluorescent microscope (Carl Zeiss Jena GmbH) using a rhodamine
filter from
Omega Opticals (Brattleboro, VT) with the following specifications, ~.e,~
560~20 nm,
600nm LP, and DC 590nm.
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00100451
9. Effect of Type and Concentration of Cation on Lipid Binding and
Uptake
This uptake experiment was performed with the same SPLP-CPLa
(containing 0.5 mol % Rh-PE) as above. 5x10'' BHK cells were plated overnight
in 1 mL
5 of complete media in 24-well plates. The SPLP-CPLa (40 nmol) was mixed with
CaCh
or Mach at various initial concentrations of 20 mM to 70 mM in a total volume
of 100
p.L. To this was added 400 ~.L of complete media resulting in final [Cation]
of 4 rrWl to
14 mM. This mixture was then added to the top of the cells and the cells
incubated for 4
hours. After incubation the cells were washed twice with PBS and 600 ~L of
lysis buffer
10 (0.1% Triton X-100 in PBS) was added. As above, the rhodamine fluorescence
was
measure and the lipid uptake was determined comparing the resulting
fluorescence to that
of a standard sample containing a known amount of lipid. The resulting values
were then
normalized to the number of cells by measuring the protein content using the
BCA
protein assay kit.
15 10. Transfection Stacdies
1x10' BHK cells were plated in 96-well plates in 150 ~L complete media
and incubated overnight at 37°C in S% COZ. SPLP and SPLP-CPL,
containing between 2
and 4 mol % CPL, were prepared to deliver 0.5 ~g of DNA in a total volume of
20 ~L
using HBS (SPLP), or HBS + 40 mM CaCI2 (SPLP-CPL) and were added to 90 p.L of
20 complete media. Samples were incubated with the cells for 4 hours. The
transfection
media was then replaced with complete media for a complete 24 hour incubation.
Cells
were then lysed with 100 p,L of lysis buffer, and 40 ~L of the lysate was
transferred to a
96-well luminescence plate. Luciferase activity was determined using a
Luciferase
reaction kit (Promega, Madison, WI), a luciferase standard (Boehringer-
Manheim), and a
25 ML3200 microtiter plate, luminometer from Molecular Dynamics (Chantilly,
VA).
Activity was normalized to the number of cells as measured by the BCA protein
assay
(Pierce, Rockford, IL). From the uptake and transfection experiments above, it
was
determined that 4 mol % CPL4 in SPLP-CPL4 gave optimal results. Thus, the rest
of the
experiments were performed with SPLP-CPL containing 4 mol % CPL..
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
71
II. Time Course for the transfection of SPLP-CPL versz~s SPLP and
complexes
Samples and cells were prepared as described for the above transfection
study, and incubated together at 37°C. As well, Lipofectin (Gibco BRL,
)complexes
containing pLuc were prepared at a charge ratio of 1.5:1. At 4, 9, and 24
hours, the
transfection media was removed and in the case of the 4 and 9 hour
transfections,
replaced with complete media for a complete 24-hour incubation. At 24 h, all
cells were
lysed and assayed for luciferase activity and protein content (BCA assay), as
above.
12. Transfection Potency and Toxicity of SPLP-CPL4
BHK cells were incubated with SPLP, SPLP-CPLa + 40 mM CaCh, and
Lipofectin complexes for 24 or 48 hours. After the incubation period the cells
were
immediately lysed and the luciferase activity was measured and was normalized
to the
amount of protein present, as above.
As a rough measure of cell survival at the above time points, the protein
concentration after cell lysis at 24 and 48 hours was measured and compared
for the
SPLP-CPL, + 40 mM CaCh and the Lipofectin complexes.
13. Comparison of effect of Ca2+ and Mg?+ on Transfection of BHK
Cells
Cells were plated and used as above. SPLP-CPL4 (5.0 ~.g/mL) with either
CaCh or MgClz at concentrations of 20 mM to 70 rnll~I were combined in a
volume of 20
~L and mixed with complete media, resulting in final [Cation] of 4 mM to 14
mM.
Following incubation on the cells for 48 hours, the cells were washed and
lysed, and the
luciferase activity and protein content were measured as above.
14. Measz~rement of transfection efficiency of SPLP-CPL4
The transfection efficiency of the SPLP-CPL was measured by preparing
SPLP-CPL4 containing encapsulated pEGFP (kindly supplied by Inex), that
expresses
GFP (green fluorescence protein), using the detergent dialysis procedure.
(see, Wheeler et
al. supra). 400 p,g/mL of pEGFP was encapsulated within 10 mM DOPE:PEG-
CerC20:DODAC (84:10:6), followed by the insertion of 4 mol % of CPL..
DOPE:DODAC complexes and Lipofectin complexes containing pEGFP were also
prepared at a charge ratio of 1.5:1. The transfections were performed as
described earlier
at a DNA dose of 5.0 p,g/mL. Following incubation of the samples for 24 and 48
hours,
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
72
the transfection media was removed, the cells were washed. and fresh media was
added
to the cells. The cells were then viewed under the Zeiss fluorescence
microscope. The
total number of cells within the frame were counted; then the number of cells
expressing
the GFP were counted using a fluorescein filter (Omega Opticals) with the
following
specifications, 7~e~=470~20 nm, ~,em 535~?2.5 nm, and DC ~SOOnm. The
efficiency of
transfection is the number of cells expressing the GFP divided by the total
number of
cells.
C. Results and Discussion
1. SPLP-CPL4 aggregate following insertion of CPL4 and de-
aggregate following addition of divalent cations.
LLTV containing CPL tend to aggregate, and that this aggregation can be
inhibited by increasing the ionic strength of the medium. It was found that
SPLP-CPL,
were also susceptible to aggregation, and that this aggregation could be
reversed by
adding NaCI, CaCh or MgCl2 to the SPLP-CPL. formulation. This effect is
illustrated in
Figure 31 which shows the effect of the addition of CaCI~ and MgCh on
aggregation of
SPLP-CPL, as monitored by the change in the standard deviation of the mean
diameter of
the particles measured by quasi-elastic light scattering (QELS). For both
cations the
standard deviation decreases with increasing cation concentration with optimal
de-
aggregation occurring above 30 to 40 mM. This behavior could also be
visualized by
freeze-fracture electron microscopy. Freeze-fracture micrographs of SPLP
reveal small
monodisperse particles, whereas SPLP-CPL: prepared in the absence of CaCh are
highly
aggregated. The addition of 40 mM CaCI? reverses this aggregation to produce
monodisperse particles similar to the SPLP preparation. For details of sample
preparation and electron microscopy, (see, Wheeler et al., Gene Therapy; 6:271-
281
( 1999)).
The sizes of SPLP and SPLP-CPL4 in the presence of CaCI? were
compared using QELS and freeze-fracture electron microscopy. QELS studies
revealed
the mean diameter of SPLP and SPLP-CPL4 to be 80 ~ 19 nm and 76 ~ 15 nm,
respectively, whereas the freeze-fracture studies indicated to diameters of 68
~ 11 nm and
64 ~ 14 nm. These values for SPLP are in close agreement with previous
studies.
SUBSTTTI1TE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
73
2. Chemical composition and stabilitv of SPLP-CPL4.
The lipid composition of SPLP-CPLa and SPLP are given in Table 9
below:
Table 9
Loss of PEG-CerC2o from SPLP following CPL insertion.
[DOPE] [PEG-CerC~_o] DOPE:PEG-C,% PEG-CerC,o
after
(rrWl ) (mM) insertion
0.786 0.07140.0004 11.010.1
(81.6:7.:1; mol) 79.7=0.9%
SPLP-CPL,, 0.7900.007 0.05720.0003 13.810.1 (x=~.9O.lmol
,')
(81.6:x: mol)
By analysis of the SPLP itself, the molar ratio of DOPE:PEG-CerC~o was
11.0(~0.1 ):1. This corresponds to a system of DOPE:PEG-CerCZO:DODAC of
(81.6:10.9:7.4). From the results, 79.7 ~ 0.9 % of the PEG-CerC2o remains
following
CPL, insertion. This corresponds to a final mol % of PEG-CerCzo of 5.9 ~ 0.1
mol %.
This means that about 1.5 ~ 0.1 mol % of PEG-CerCao was replaced during the
insertion
of CPL.. If we assume that on the inner leaflet and outer leaflet the same
amount of
PEG-CerCzo is initially present at 7.4 mol %, the outer leaflet will possess
4.4 ~ 0.1 mol
°,'o of PEG-CerCZO after insertion. Since we inserted -4.5 mol % CPL4
into SPLP (9.0
mol % in the outer leaflet), resulting in a total of 13.4 ~ 0.1 mol % of total
PEG in the
outer leaflet.
The stability of SPLP and SPLP-CPL in 50% mouse serum for up to 4
hours. In all cases. the DNA was completely protected from serum degradation.
3. SPLP-CPLa exhibit enhanced uptake into BHK cells and
dramatically enhanced transfection potency.
The next set of experiments was aimed at determining the influence of
incorporated CPL, on the uptake of SPLP into BHK cells and the resulting
transfection
potency of the SPLP-CPL4 system. SPLP containing up to 4 mol % CPL, were
prepared
in the presence of 40 mM CaCl2 and were added to BHK cells (final CaClz
concentration
8 mM) and incubated for varying times. The cells were then assayed for
associated
SPLP-CPLa as indicated in Methods. As shown in Figure 32, uptake of SPLP that
contain
SUBSTITUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
74
no CPL, is minimal even after 8 h of incubation, however uptake is
dramatically
improved for SPLP containing 3 mol % or higher levels of CPL.. For example,
SPLP
containing 4 mol % CPL4 exhibit accumulation levels at 8 h that are
approximately 50-
fold higher than achieved for SPLP. This enhanced uptake can be visually
detected using
fluorescence micrographs of BHK cells following incubation with rhodamine-
labeled
SPLP and SPLP-CPL4 for 4 h. The presence of 4 mol % CPL, clearly results in
improved
levels of cell-associated SPLP.
The transfection properties of SPLP, SPLP-CPL. and plasmid DNA-
cationic lipid complexes (DODAC/DOPE; 1:1; 1.5:1 +ve/-ve c.r.) were examined
using
the incubation protocol usually employed for complexes. This consisted of
incubation of
10'~ BHK cells with SPLP, SPLP-CPLa and complexes containing 0.5 ~g pCMVLuc
for 4
h, followed by removal of SPLP, SPLP-CPL4 or complexes that are not associated
with
the cells, replacement of the media, incubation for a further 20 h and then
assaying for
luciferase activity. '~'ne SPLP-CPL4 preparations contained 7 mNI CaCh in the
incubation medium. As shown in Figure 33, the presence of the CPL, resulted in
dramatic
increases in the transfection potencies of the SPLP system. SPLP-CPL4
containing 4 mol
CPL. exhibited luciferase expression levels some 3x103 higher than achieved
with
SPLP. (see,Mok et al., Biochim Biophys Acta, 1419:137-150 (1999)).
4. Ca2+ is required for transfection activity of SPLP-CPLQ:
It was of interest to determine the influence of Ca2+ on the transfection
activity of SPLP-CPL4. SPLP containing 4 mol % CPLa were incubated with BHK
cells
for 48 h in the presence of 0-14 mlVl MgCh and CaCh and the luciferase
activities then
determined. As shown in Figure 34, the transfection activity was influenced by
the
presence of Ca2+ in the transfection medium. At the optimum CaCl2
concentration of 10
mM, SPLP-CPL exhibited transfection potencies that were more than 10~ times
higher
than if MgCh was present.
Uptake of SPLP-CPL4 into BHK cells was monitored following a 4 h
incubation in the presence of 0-14 mM MgCl2 and CaCl2. As shown in Figure 35
the
amount of SPLP-CPL4 taken up by BHK cells is the same for both Mg2+ and Ca2+-
containing media. The uptake of the SPLP-CPL4 decreases as the concentration
of
divalent cations increases, which likely arises due to shielding of the
negatively charged
binding sites for the CPLa on the surface of the BHK cells.
SUBSTTTUTE SI3EET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
SPLP-CPL exhibit tr-ansfection potencies in vitro that are
comparable to or greater than achieved acsing complexes.
The results shown in Figure 33 indicating that complexes give rise to
-100-fold higher levels of transfection than SPLP-CPL. were obtained for a
fixed 4 h
5 incubation time with the BHK cells, followed by a 20 h hold time to achieve
maximum
expression. Given that the SPLP-CPL4 are stable systems it is likely that
uptake into the
BHK cells would continue over extended time periods. The transfection levels
achieved
when the incubation time of the SPLP-CPL, and the complexes with the BHK cells
was
extended to 8 and 24 h, followed by hold times of 16 and 0 h respectively were
10 examined. Two types of plasmid DNA-cationic lipid complexes were used,
namely
DOPE:DODAC (l:l) complexes (1.5:1, c.r.) and complexes obtained using the
commercial transfection reagent Lipofectin (DOPE/DOTMA [1:1] complexes, 1.5:1
c.r.).
As shown in Figure 36, the transfection potency of the SPLP-CPLa increases
markedly
with increased incubation times, suggesting that a limiting factor for
transfection
15 achieved at a 4 h incubation time was the rate of uptake of the SPLP-CPL
system. At the
24 h incubation time transfection levels are achieved that are comparable to
those
achieved by Lipofectin or DOPE/DODAC complexes.
Further experiments were conducted to determine transfection levels after
24 and 48 h incubation times with luciferase activities assayed immediately
following the
20 incubation period. As shown in Figure 37A the activity of Lipofectin
(DOPE/DOTMA;
1:1) complexes leveled off at 2000 ng/mg after 24 h. In contrast, the activity
of SPLP-
CPL~ formulation continued to increase as the incubation time was increased,
achieving
luciferase expression levels corresponding to 4000 ng/mg at 48 h. This
activity is
approximately 106 times higher than observed for SPLP (in the absence of CaZ~
and
25 almost double the levels that can be achieved by Lipofectin complexes.
Similar results
were obtained for the DOPE:DODAC complexes.
6. SPLP-CPL4 are non-toxic and efficient transfection agents.
It is well known that plasmid DNA-cationic lipid complexes can be toxic
to cells. The SPLP-CPL4 contain low levels of cationic lipid and are
potentially less toxic
30 than complexes. The toxicity of SPLP-CPLa and complexes was assayed by
determining
cell viability following a 48 h exposure to levels of SPLP-CPL4 and complexes
corresponding to 0.5 ~g plasmid and ~30 nmol total lipid. As shown in Figure
37B,
SPLP-CPL4 exhibit little if any toxicity. Cell survival was only 30% after a
48 h
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
76
incubation with Lipofectin complexes whereas ~95% of the cells were viable
following a
48 hour incubation with SPLP-CPL4.
The efficiency of transfection, indicated by the proportion of cells
transfected by a vector, is also an important parameter. The proportion of
cells
transfected were estimated using plasmid carrying the green fluorescent
protein (GFP)
gene. Transfection was detected by expression of the fluorescent protein
inside a cell
employing fluorescence microscopy. As shown in Figure 37A and 38B,
approximately
35% of the cells at 24 h and 50% at 48 h were transfected by SPLP-CPL.,, with
no
apparent cell death. In contrast, Lipofectin complexes exhibit maximum
transfection
efficiencies of less than 35% and only ~SO% cell survival after the 24 h
transfection
period. Similar low transfection efficiencies and high toxicities were also
seen with
DOPE:DODAC complexes.
The results of this study demonstrate that the incorporation of CPL into
SPLP results in improved uptake into BHK cells and a dramatically enhanced
transfection potency of SPLP when Ca2+ is present. There are three points of
interest.
The first concerns the chemical composition and structure of the SPLP-CPL.
system and
the generality of the post-insertion procedure for modifying the trophism and
transfection
potency of SPLP. The second concerns the relation between enhanced uptake of
SPLP,
the presence of Ca2+ and the transfection activities observed. Finally, it is
of interest to
compare the properties of the SPLP-CPL system with plasmid DNA-cationic lipid
complexes.
The second point of discussion concerns the mechanism whereby CPL4
increases the transfection potency of the SPLP system. Clearly the presence of
the CPL,
increases the uptake of SPLP into the BHK cells, however the increase in
transfection
potency is almost entirely dependent on the additional presence of Ca'+. It
may be noted
that, following an 8 h incubation, the presence of 4 mol % CPL4 increases the
uptake of
SPLP into BHK cells by approximately 50-fold, whereas the transfection potency
(in the
presence of Caz+) is increased by a factor of 104. Previous work conducted on
SPLP has
shown that the presence of Caz+ results in a maximum increase in transfection
potency of
600 and that this increase in potency results from an ability of Ca2+ to
assist in
destabilizing the endosomal membrane following uptake, rather than an increase
in
uptake itself. In turn, this suggests that the improvement in transfection
potency for the
SPLP-CPL4 system over the SPI;P system arises from the CPL4-dependent'increase
in
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
77
uptake multiplied by the Ca''+-dependent improvement in intracellular delivery
following
uptake.
The final area of discussion concerns the advantages of the SPLP-CPL
system over other non-viral vectors, which include the well-defined modular
nature of the
SPLP-CPL4 system as well as toxicity and potency issues. First, the well-
characterized
nature of the SPLP-CPL. as small, homogeneous, stable systems containing one
plasmid
per particle contrast with non-viral systems such as plasmid DNA-cationic
lipid
complexes which are large, inhomogeneous, unstable systems containing ill-
defined
numbers of plasmids per complex. An important point is that SPLP are basic
components of more sophisticated systems, such as SPLP-CPL:, which can be
constructed in a modular fashion. For example, post-insertion of PEG-lipids
which
contain specific targeting ligands in place of the cationic groups of CPL
should result in
SPLP that are specifically targeted to particular cells and tissues. With
regard to toxicity,
it is clear that SPLP-CPL4 are markedly less toxic to BHK cells in tissue
culture. This is
presumably related to the low proportions of cationic lipid contained in SPLP
as
compared to complexes. The transfection potency and efficiency of SPLP-CPLa is
clearly comparable to the levels that can be achieved with complexes. It
should be noted
that this finding suggests that models of transfection by complexes that
involve.
In the present example, the superiority of SPLP-CPL. compared to
commercially available complex systems (e.g. Lipofectin) has been
demonstrated. Thus,
a synthetic virus has been developed that will have high transfection potency
but none of
the problems associated with viruses. Many points can be made to corroborate
these
statements. The first point revolves around the placement of the charge.
Whereas on
complexes the charges are located on the surface of the lipid bilayer, the
SPLP-CPL
possess charges on the vesicle surface which are localized a good distance
from the
liposomal surface, above the protective PEG coating which surrounds the
liposome. In
the case of the complexes, proteins binding to the liposome surface can lead
to
recognition and clearance by macrophages of the RES. (see, Chonn et al., JBiol
Chem;
267:18759-18765 (1992)) In the SPLP-CPL, the charge on the surface of the
bilayer is
protected by the PEG coating, such that this should not occur. However, the
charge on
the SPLP-CPL4 will allow the association of the liposomes with cells resulting
in
eventual uptake and transfection.
The size and serum stability of the SPLP-CPL4 compared to complexes
are important parameters for effective gene delivery systems, especially if
one wishes to
SUBSTTTZJTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
78
approach the capabilities of viral systems. The SPLP-CPL. have been shown here
to be
of relative small size 0100 nm) compared to complexes, which are frequently on
the
order of microns in diameter. The small size should allow for accumulation at
sites with
larger fenestration (e.g. tumors, and inflammation sites). (see, Kohn et al.,
Lab Invest;
67:596-607 ( 1992)). As stated earlier, DNA in the SPLP-CPL4 was shown to be
protected from the external environment (i.e. inaccessible to degradation by
DNase
within serum), whereas DNA in complexes is susceptible to DNase. (see, Wheeler
et al.,
Gene Therapy; 6:271-281 (1999)).
Viruses (see, Hermonat et al., Proc. Natl. Acad. Sci. USA; 81:6466-6470
(1984); Lebkowski et al., Molec Cell Biol; 8:3988-3996 (1988); Keir et al., J
Neurovirology, 3:322-330 (1997)]and lipid/DNA complexes (see, Felgner et al.,
Proc
Natl Acad Sci USA, 84:7413-7417 (1987); Felgner et al., JBiol Chem; 269:2550-
61
(1994); Hofland et al., Proc Natl Acad Sci USA; 93:7305-7309 (1996); Bebok et
al.,
JPharm Exp Ther; '79:1462-1469 (1996); Gao et al., Gerte Therapy; 2:710-722
(1995))
have been shown to possess high in vitro transfection potencies. It therefore
reasons that
the SPLP-CPL system, if it is to attain viral qualities, should be capable of
attaining
these high transfections. This has actually been achieved by the SPLP-CPL.
system on
BHK cells, with transfection levels reaching a factor of two higher than a
commercially
available complex system (i.e. Lipofectin). This is a huge improvement over
SPLP,
which showed only a small amount of transfection.
Efficient systemic delivery and transfection of genetic drugs are achieved
using this SPLP-CPL system due to the above benefits. Very high transfections
in vitro
with SPLP-CPLa have been achieved. In addition, a system wherein the
positioning of
the positive charges on the CPL, so that the PEG of the PEG-Cer initially
masks it. This
is achieved by the synthesis of DSPE-PEG-CPL4 with a shorter PEG moiety. This
allows
for its accumulation at disease sites followed by the controlled release of
the PEG-Cer,
exposing the positive charges to the surrounding cells.
EXAMPLE X
This example shows transfection rates of BHK cells by long- versus short-
chained CPLs.
Using synthesis methods from above, CPL (PEG 3.4k) and CPL(PEG lk)
were generated and each inserted into a separate SPLP system containing PEG-
ZOOO-Cer
C20 as described above. Figure 38 illustrates transfection rates of the CPLs
having a
SUBSTTTUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
79
PEG 3.4k versus a CPL having a PEG l k. The short-chained PEG in the CPL
results in a
decrease by a factor of about 4 compared to the transfection by the long
chained CPL.
Without being bound by any particular theory, it is believed that the long
chain CPL
(PEG3aoo) sticks out above the surface, whereas the short chain CPL (PEGiooo)
is buried
(masked) in the surface of the SPLP. The reduced in vitro transfection of the
short chain
CPL clearly suggests that it has improved in vivo circulation.
EXAMPLE XI
This example shows that CPL8 behaves similar to CPL4 with respect to
insertion into LUVs, and that transfection can be achieved with CPLB-LIJV
systems.
Table 10
Insertion of CPLg in SPLP and LLJV.
SPLP Initial mol % Insertion Final mol % CPLB
% CPLB
1.11 97% 1.07
1.39 85% 1.19
1.67 95% 1.60
1.94 87% 1.70
LUV Initial mol % Insertion Final mol % CPLB
% CPLB
1.05 79% 0.82
1.39 71 % 0.99
1.74 76% 1.32
2.11 89% 1.88
The insertions of the CPLg into LUV and SPLP is very similar to what was
observed for the insertions of CPL. For the transfection and uptake of these
particles on
BHK cells, variable results are obtained, with the CPLB performing better than
the CPL4
sometimes and vice versa at other times.
EXAMPLE XII
In this in vitro example using mouse neuroblastoma cell line Neuro-2a
(ATCC-CCL-131), the SPLP-CPL4[lk] is used to determine gene expression with
respect to varying Ca2+ concentrations and to compare to gene expression using
a
SUBSTITUTE SFIEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
standard SPLP (PEG-CerC20 10%; CPL4[lk] 4%; and other components; DNA:lipid
ratio =0.05).
5 x 10~ cells/well are plated in 24-well plates in 1mL of complete media
(MEM(Eagle) with non-essential amino acids and Hanks' buffered salt solution
with 10%
5 FBS). Plates are incubated overnight at 37°C with 5.0% COa. To each
group set out
below is added 500 ~L transfection media in triplicate.
TABLE 11
GROUP SPLP or SPLP-CPL[Ca2+] (250 mVI)Complete Media
(pg) (]1L) (ALL)
A (0 mM Ca2+) 2.5 0 1980
B (2 mM Ca2+) 2.5 48 1932
C (4 mM Ca2+) 2.5 96 1884
D (6 mM Ca2+) 2.5 144 1836
E (8 mM Ca2+) 2.5 192 1788
F ( 10 mM Ca2+)2.5 240 1740
G (12 mM Ca2+) 2.5 288 1692
H ( 14 mM Ca2+)2.5 336 1644
10 2.5 ~.g DNA is added per well in fully encapsulated SPLPs (0.5 mL total
solution). Plates are incubated for 8 hrs. Transfection media is removed. 1mL
of
complete media is added back. Cells are incubate for another 24 hrs at
37°C, 5.0% COz.
For analysis, media is removed from cells and they are washed 2x with
PBS then frozen at -70°C. Cells are lysed with 150-200 ~,L lx CCLR;
then shaken 5
15 minutes on plate shaker. 20 ~.L lysate is transferred to a 96-well
luminescence plate.
Plates are read to determine luciferase activity.
The results are shown in Figure 39. As shown therein, SPLP + 4 mol
%CPL4-lk produces 4 orders of magnitude of gene expression more than SPLP
alone in
Neuro-2a cells. Effects of calcium are not considered to be significant in
this experiment.
20 The amount of luciferase produced remains the same from 2-14 mM Ca2+.
SUBSTIT'LTTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
81
EXAMPLE XIII
This in vivo example discloses pharmacokinetics and biodistribution of
CPL.-1-k LLTVs (SPLPs containing short chain CPLs) in C57/b16 mice. Different
SPLP
formulations containing increasing amounts of CPL--~-lk are assayed in vivo to
determine
optimal clearance characteristics.
CPL.-lk SPLPs are prepared according to previous protocols. Before use,
all samples are characterized to determine actual composition prior to
administration. All
samples are filter sterilized prior to dilution to working concentration. All
samples are to
provided in sterile crimp top vials. All vials are labeled with the
formulation date, lipid
composition, and specific activity. 3[H]CHE is incorporated at 1 pCi/mg Lipid.
The
following formulations are made and analyzed:
A: 3[H]CHE-LLTV DOPE:DODAC:PEGC20::84:6:10
B: 3[H]CHE-LUV DOPE:DODAC:PEGC20::84:6:10 + 1 mol % CPL-4-lk
C: 3[H]CHE-LIJV DOPE:DODAC:PEGC20::84:6:10 + 2 mol % CPL-4-lk
D: 3[H]CHE-LLJV DOPE:DODAC:PEGC20::84:6:10 + 3 mol % CPL-4-lk
E: 3[H]CHE-LLJV DOPE:DODAC:PEGC20::84:6:10 + 4 mol % CPL-4-lk
Experiments used 100 C57/b16 mice, female, 18-23 g all ordered from
Harlan Sprague Dawley. All animals housed in cages of 4 animals per group in
25
groups.
SUBSTTI'L1TE S~iEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
82
TABLE 12
Group Mice Treatment TimepointAssay
~
A 4 A:DOPE:DODAC:PEGC20:84:6:10 IS PK
I min
B 4 A: DOPE:DODAC:PEGC20::84:6:10 l hr PK
C ~l A: DOPE:DODAC:PEGC20::84:6:10 4 hr PK
D 4 A: DOPE:DODAC:PEGC20::84:6:10 8 hr PK
~
E 4 A: DOPE:DODAC:PEGC20::84:6:10 24 PK
~ hr
~
F ~ B: DOPE:DODAC:PEGC20::84:6:I0+ 15 PK
1 mol % CPL-.l-lk min
G 4 B: DOPE:DODAC:PEGC20::84:6:10+ 1 hr PK
l mol % CPL-.l-lk I
H l B: DOPE:DODAC:PEGC20::84:6:10 4 hr PK
I + 1 mol % CPL-4-lk
I .l B: DOPE:DODAC:PEGC20::84:6:10 8 hr PK
+ l mol % CPL-4-lk I
-t B: DOPE:DODAC:PEGC20::84:6:10 24 PK
+ l mol % CPL-~l-1 k hr
K d C: D(?PE:DODAC:PEGC20::84:6:10+2IS PK
mol ,'CPL--i-lk min
L ~l C: DOPE:DODAC:PEGC20::84:6:10+21 hr PK
mol %CPL-i-lk
M 4 ~ C: DOPE: DODAC:PEGC20::84:6:10+24 hr PK
mol % CPL-l-lk
N 4 C: DOPE:DODAC:PEGC20::84:6:10+28 hr PK
mol % CPL-l-Ik
O 4 C: DOPE:DODAC:PEGC20::84:6:10 24 PK
+ 2 mol % CPL-1-Ik hr
P 4 I D: DOPE:DODAC:PEGC20::84:6:10IS PK
+ 3 mol % CPL--t-lk min
Q 4 D: DOPE:DODAC:PEGC20::84:6:10 1 hr PK
+ 3 mol % CPL-.i-Ik
R I ~ D: DOPE:DODAC:PEGC20::84:6: 4 hr I PK
4 I O + 3 mol % CPL-l-I k
S 4 D: DOPE:DODAC:PEGC20::84:6:10 8 hr PK
+ 3 mol % CPL-4-lk
T 4 D: DOPE:DODAC:PEGC20::84:6:10 24 PK
+ 3 mol % CPL-.l-1 k hr
U 4 E: DOPE:DODAC:PEGC20::84:6:10 15 PK
+4 mol % CPL-l-lk min
V 4 E: DOPE:DODAC:PEGC20::84:6:10+41 hr PK
mol % CPL-4-lk
W 4 E: DOPE:DODAC:PEGC20::84:6:10 4 hr PK
+4 mol % CPL-lk
X 4 ~ E: DOPE:DODAC:PEGC20::84:6:108 hr PK
+4 mol % CPL-4-lk
Y 4 E: DOPE:DODAC:PEGC20::84:6:I0 24 PK
+4 mol % CPL-.t-lk hr
Mice were treated with 3[H]CHE-LLJV administered by tail vein LV. in a
total volume of 200 p,l . Mice receive one treatment only. At the indicated
time-points
mice are weighed, sacrificed, and blood will be collected by cardiac puncture
then
evaluated for 3[H]CHE. Formulations are expected to be well tolerated. Mice
are treated
SUBSTITUTE SHEET (RULE 26)
CA 02370690 2001-10-19
WO 00/62813 PCT/CA00/00451
83
according to certified animal care protocols. Any mice exhibiting signs of
distress
associated with the treatment are terminated at the discretion of vivarium
staff. All mice
are terminated by COz inhalation followed by cervical dislocation. Measurement
of
3[H]CHE from blood is determined according to standard protocols.
In vivo pharmacokinetics of SPLP containing short chain CPL: are
illustrated in Figure 40. It is observed that that increasing amounts of the
CPL. in the
SPLP tends to increase the rate of clearance from the blood. CPLa incorporated
a 1 mol
gives clearance results which are similar to SPLPs without CPLa. Incorporation
of higher
amounts of CPL4 tends to increase the rate of clearance of the SPLP from the
blood.
SPLP- CPL. [lk] (1%) shows best plasma clearance characteristics with a t,;a
of 6-7
hours. Anything greater than 1 mol % clears more rapidly.
The results disclosed in this specification indicate a further refinement of
SPLP technology. In particular, from these results it is clear that the type
of CPL (i.e. the
length of the polymer chain; and the amount of cationic charge per molecule)
and the
amount of such CPL in an SPLP must be optimized to obtain the best balancing
of
clearance properties in vivo with enhanced transfection ability. In vitro data
has shown
long chain CPLs and higher levels of such CPLs are to be preferred to increase
transfection. However, as seen in previous comparisons of SPLPs versus lipid
complexes, lipid formulations that work best in vitro are not best suited in
vivo. In vivo
results herein demonstrate that short chain CPLs incorporated at approximately
1 % are
optimized for circulation lifetimes in animals.
It is understood that the examples and embodiments described herein are
for illustrative purposes only and that various modifications or changes in
light thereof
will be suggested to persons skilled in the art and are to be included within
the spirit and
purview of this application and scope of the appended claims. All
publications, patents,
and patent applications cited herein are hereby incorporated by reference in
their entirety
for all purposes.
SUBSTITUTE SI3EET (RULE 26)