Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
PROCESS FOR MAKING FUSED-RING IMIDAZO-CONT AIMING COMPOUNDS
FIELD OF THE INVENTION
The subject invention relates to processes for making certain substituted
fused-ring
imidazo compounds.
BACKGROUND
Some fused-ring imidazo compounds have pharmacological activity in processes
known to be associated with one or more of cardiovascular activity,
inflammatory
mechanisms, oncology, and regulation of protein transport from cells. The
subject
invention processes are useful for making such compounds.
SUMMARY OF THE INVENTION
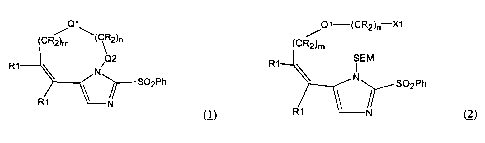
The subject invention involves processes for making compounds having the
structure:
(CR2 \( ~ 2~n
Q2
R1
N
~S02Ph
R 1 ~~N
wherein:
(a) m is an integer from 0 to about 6;
(b) n is an integer from 0 to about 6;
(c) -Q1- is selected from nil, -CR=CR-, -O-, -S-, -NR-, -C(O)-, -NR-C(O)-, -
OC(O)-
(d) -Q2- is nil or -C(O)-;
(e) each -R is independently selected from hydrogen, alkyl, aryl, and
heterocycle;
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
(f) each -R1 is independently selected from hydrogen, alkyl, aryl,
heterocycle,
or the two Rl's are attached to form a cycloalkenyl, aryl or heterocyclic
ring;
the process comprising the following Steps:
(A) treating a compound having the structure:
/Q1-(CR2)n-X1
R1 SEM
N SOZPh
R1 N (2)
wherein m, n, -Q1- and -R1 are the same as for compound (1); and -X1 is
selected from -Cl, -Br, -I, -OH and -COOH;
(i) if -Xl is -OH, treating compound (2) with MsOCI or TsOCI and
Et3N in solvent, whereby -X1 is converted to -X2, -X2 being -
OMs or -OTs, respectively; or treating compound (2) with a
halogenating reactant in solvent, whereby -Xl is converted to -X2,
-X2 being -C1 or -Br or -I;
(ii) if -Xl is -COON, treating compound (2) with phosgene or oxalyl
chloride in solvent, whereby -X 1 is converted to -X2, -X2 being -
C(O)Cl;
whereby some or all of the intermediate thus formed in this Step (A) may
further spontaneously react to form compound (1);
(B) if -X1 is -C1 or -Br or -I, or if the conversion to compound (1) in Step
(A) is
insufficient, treating compound (2) or the reaction product of Step (A),
respectively, with nBu4NF in solvent, whereby conversion to compound (1)
occurs.
2
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
The subject invention also involves processes having additional Steps before
and/or
after Steps (A) and/or (B). The subject invention also involves combinatorial
libraries of
compounds made according to subject processes.
DESCRIPTION OF THE INVENTION
Glossary of Terms
As used herein unless specified otherwise, "alkyl" means a hydrocarbon chain
which is branched, linear or cyclic, saturated or unsaturated (but not
aromatic),
substituted or unsubstituted. The term "alkyl" may be used alone or as part of
another
word where it may be shortened to "alk" (e.g., in alkoxy, alkylacyl).
Preferred linear
alkyl have from one to about twenty carbon atoms, more preferably from one to
about ten
carbon atoms, more preferably still from one to about six carbon atoms, still
more
preferably from one to about four carbon atoms; most preferred are methyl or
ethyl.
Preferred cyclic and branched alkyl have from three to about twenty carbon
atoms, more
preferably from three to about ten carbon atoms, more preferably still from
three to about
seven carbon atoms, still more preferably from three to about five carbon
atoms.
Preferred cyclic alkyl have one hydrocarbon ring, but may have two, three, or
more,
fused or spirocycle hydrocarbon rings. Preferred alkyl are unsaturated with
from one to
about three double or triple bonds, preferably double bonds: more preferably
they are
mono-unsaturated with one double bond. Still more preferred alkyl are
saturated.
Saturated alkyl are referred to herein as "alkanyl". Alkyl unsaturated only
with one or
more double bonds (no triple bonds) are referred to herein as "alkenyl".
Preferred
substituents of alkyl include halo, alkyl, aryl, heterocycle, hydroxy, alkoxy,
aryloxy, thio,
alkylthio, arylthio, amino, alkylamino, arylamino, amide, alkylamide,
arylamide, formyl,
alkylacyl, arylacyl, carboxy and its alkyl and aryl esters and amides, sulfo,
alkylsulfo,
arylsulfo, sulfino, alkylsulfino, arylsulfino, phospho, alkylphospho,
arylphospho,
phosphino, alkylphosphino, arylphosphino, nitro, and cyano. Substituents of
cycloalkyl
also include cycloalkyl, aryl and heterocycle rings which are fused or
spirocycle with the
initial cycloalkyl. Also, unsubstituted alkyl are preferred.
As used herein, "heteroatom" means a nitrogen, oxygen, or sulfur atom.
3
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
As used herein, "alkylene" means an alkyl which connects two other moieties,
"heteroalkylene" means an alkylene having one or more heteroatoms in the
connecting
chain.
As used herein unless specified otherwise, "aryl" means an aromatic
hydrocarbon
ring (or fused rings) which is substituted or unsubstituted. The term "aryl"
may be used
alone or as part of another word (e.g., in aryloxy, arylacyl). Preferred aryl
have from six
to about fourteen, preferably to about ten, carbon atoms in the aromatic
ring(s), and a
total of from about six to about twenty, preferably to about twelve, carbon
atoms.
Preferred aryl is phenyl or naphthyl; most preferred is phenyl (Ph). Preferred
substituents
of aryl include halo, alkyl, aryl, heterocycle, hydroxy, alkoxy, aryloxy,
thio, alkylthio,
arylthio, amino, alkylamino, arylamino, amide, alkylamide, arylamide, formyl,
alkylacyl,
arylacyl, carboxy and its alkyl and aryl esters and amides, sulfo, alkylsulfo,
arylsulfo,
sulfino, alkylsulfino, arylsulfino, phospho, alkylphospho, arylphospho,
phosphino,
alkylphosphino, arylphosphino, nitro, and cyano. Substituents of aryl also
include
cycloalkyl and heterocycle rings which are fused with the aryl ring or rings.
Also,
unsubstituted aryl are preferred.
As used herein unless specified otherwise, "heterocycle" or "heterocyclic"
means
a saturated, unsaturated or aromatic cyclic hydrocarbon ring (or fused rings)
with one or
more heteroatoms in the hydrocarbon ring(s). Preferred heterocycles have from
one to
about six heteroatoms in the ring(s), more preferably one or two or three
heteroatoms in
the ring(s). Preferred heterocycles have from three to about fourteen,
preferably to about
ten, carbon plus heteroatoms in the ring(s), more preferably from three to
about seven,
more preferably still five or six, carbon plus heteroatoms in the rings(s);
and a total of
from three to about twenty carbon plus heteroatoms, more preferably from three
to about
ten, more preferably still five or six, carbon plus heteroatoms. Preferred
heterocycles
have one ring, but may have two, three, or more, fused rings. More preferred
heterocycle
rings include those which are one ring with 5 or 6 carbon plus heteroatoms in
the ring
with no more than three ring heteroatoms, no more than two of which are O and
S. Still
more preferred are such 5- or 6-ring atom heterocycles with one or two ring
atoms being
O or S and the others being C; or with one, two or three ring atoms being N
and the
4
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
others being C. Such preferred 5- or 6-ring atom heterocycles are preferably
saturated,
unsaturated with one or two double bonds, or aromatic. Such preferred 5- or 6-
ring atom
heterocycles are preferably a single ring; or fused with a 3- to 6-ring atom
hydrocarbon
ring which is saturated, unsaturated with one double bond, or aromatic
(phenyl); or fused
with another such 5- or 6-ring atom heterocyclic ring. Heterocycles are
unsubstituted or
substituted. Preferred heterocycle substituents are the same as for alkyl.
Processes of the Invention
Scheme I
The subject invention processes include those depicted in Scheme I:
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
(A_):
(1) if-X1 =-OH:
SOCI2 or TsOCI or MsOCI,
,01-(CR2)n-X1
EtgN in CH2CI2; or NBS,
(CR2)m PhgP or PBrg, pyridine
in
R1 SEM DMF; or Nal or KI, acid
in
I DMF
N
S02Ph (2) if -XI = -COOH:
CI2C0
~
R1 or (CICO)2 in CH2CI2
\
N (3) if -X1 = CI or -Br
or -I:
(?)
skip to Step (B)
/Q 1-(CR2)n-X2
(CR2)m (B) if needed:
nBu4NF
R1~ SEM in THF
S02Ph
(3)
~al~
(CR2)m tCR2)n
R 1 /Q2
N S02Ph
R1
N
(1)
In Scheme I and other Schemes herein, m is an integer from 0 to about 6,
preferably from 0 to about 2, more preferably 0 or 1; n is an integer from 0
to about 6,
preferably from 0 to about 2, more preferably 0 or 1; m + n is from 0 to 12,
preferably
from 0 to about 4, more preferably from 1 to about 3, more preferably still 2
or 3.
In Scheme I and other Schemes herein, each -R is independently selected from
hydrogen, alkyl, aryl, and heterocycle. Non-hydrogen -R are preferably
selected from
phenyl, heterocycle having 5 or 6 ring atoms including 1 or 2 heteroatoms, and
alkyl
having from 1 to about 6 carbon atoms; such R are unsubstituted or
substituted,
6
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
preferably unsubstituted. Preferably no more than 2 of all the -R's is other
than
hydrogen, more preferably no more than 1; more preferably still all -R's are
hydrogen.
In Scheme I and other Schemes herein, each -R1 is independently selected from
hydrogen, alkyl, aryl and heterocycle, or both -R1's are attached to form a
cycloalkenyl,
aryl or heterocyclic ring. Preferably the two -R1's are attached to form a
cycloalkenyl,
aryl, or heterocyclic ring; more preferably a cycloalkenyl or aryl ring; more
preferably
still an aryl ring, especially phenyl. When the two -R1's are attached forming
a phenyl
ring, one or preferably both of the positions on the phenyl ring para to the
positions of
attachment of the -R1's shown in Scheme I have non-hydrogen substituents
attached
thereto. Such substituents are preferably attached to the phenyl ring by a
heteroatom, the
heteroatom preferably being oxygen; such substituents are preferably alkoxy,
especially
methoxy.
In Scheme I and other Schemes herein, -Ql- is selected from nil, -CR=CR-, -O-,
-S-, -NR-, -C(O)-, -NR-C(O)-, and -OC(O)-; preferably from nil, -O-, -S-, -NR-
,
and -C(O)-. More preferred -Q1- is nil. When -Q1- is nil, m + n is preferably
from 1 to
about 4, more preferably from 1 to about 3.
In Scheme I and other Schemes herein, -X1 is selected from -Cl, -Br, -I, -OH
and
-COOH. If -X1 is -OH, then -X2 can be -OMs or OTs, and -Q2- is nil.
Alternatively, if -
X1 is -OH, -X2 can be -CI or -Br or -I, and -Q2- is nil. If -Xl is -COOH, then
-X2 is -
C(O)Cl, and -Q2- is -C(O)-. If -X1 is -CI or -Br or -I, Step (A) is skipped,
and -X2 is the
same as -X1 (compound (2) and compound (3) are the same).
In Scheme I and other Schemes herein, -SEM has the structure: -
CH20CH2CH2-Si(CH3)3.
St A
In Step (A) of Scheme I, the reactants are dependent on -X1.
If -Xl is -OH, compound (2) can be treated with either methylsulphonyl
chloride
(MsOCI) or p-toluenesulfonyl chloride (TsOCI), preferably in the presence of
base (e.g.
triethylamine (Et3N)), in solvent, preferably dichlorolmethane. This reaction
is
preferably carried out under an inert atmosphere, more preferably under an
argon
atmosphere. The MsOCI or TsOCI is preferably added slowly, preferably over a
period
7
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
of up to about 2 h, more preferably from about 1/4 h about 1 h. More
preferably still
over about 1/2 h. During addition of the MsOCI or TsOCI, the temperature of
the
reaction mixture is preferably from about -20 °C to about 25 °C,
more preferably about 0
°C. After addition of MsOCI or TsOCI is complete, the reaction mixture
is preferably
warmed to a temperature of from about 0 °C to about 40 °C, more
preferably to about
room temperature; at this temperature, the reaction mixture is stirred
preferably for from
about 1/2 h to about 6 h, more preferably for about 1 h.
Alternatively, if -X1 is -OH, compound (2) is reacted with a halogenating
reactant
such that the -OH is converted to -Cl, -Br or -I (-X2), thus forming compound
(3).
Known halogenating reaction conditions that are compatible with compounds of
structure
(2), (3), and (1) are suitable. Preferred halogenation reactions are selected
from the
following:
(a) Compound '2) can be treated with thionyl chloride (SOCl2), preferably in
the presence of base (e.g., triethylamine), in solvent, preferably
dichloromethane,
to produce compound (3) wherein -X2 is Cl. This reaction is preferably carried
out under an inert atmosphere, more preferably under an argon atmosphere. The
thionyl chloride is preferably added slowly, preferably over a period of up to
about 2 h, more preferably from about 1/4 h about 1 h. More preferably still
over
about 1/2 h. During addition of the thionyl chloride, the temperature of the
reaction mixture is preferably from about -20 °C to about 25 °C,
more preferably
about 0 °C. After addition of thionyl chloride is complete, the
reaction mixture is
preferably warmed to a temperature of from about 0 °C to about 40
°C, more
preferably to about room temperature; at this temperature, the reaction
mixture is
stirred preferably for from about 1/2 h to about 6 h, more preferably for
about 1 h.
(b) Compound ~2) can be treated with NBS, preferably in the presence of
triphenylphosphine (Ph3P), in solvent, preferably dimethyl formamide (DMF), or
with tribromophosphine (PBr3), preferably in the presence of base (e.g.,
pyridine), in solvent, preferably dichloromethane, to produce compound (3)
wherein -X2 is -Br. This reaction is preferably carried out under an inert
atmosphere, more preferably under a nitrogen atmosphere. The temperature of
8
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
the reaction mixture is preferably from about 0 °C to about 40
°C, more preferably
about 20 °C. The reaction mixture is stirred preferably for from about
1/2 h to
about 12 h, more preferably for about 2 h.
(c) Compound (2) can be treated with sodium iodide (NaI) or potassium
iodide (KT), preferably in the presence of strong acid (e.g., phosphoric acid,
sulfuric acid), in solvent, preferably DMF, to produce compound '3) wherein -
X2
is -I. The temperature of the reaction mixture is preferably from about 0
°C to
about 90 °C, more preferably about 20 °C. The reaction mixture
is stirred
preferably for from about 1 h to about 12 h, more preferably for about 2 h.
If -X1 is -COOH, compound ~2) is treated with phosgene (C12C0) or oxalyl
dichloride ((C1C0)2) in solvent, preferably dichloromethane. This reaction is
preferably
carried out under an inert atmosphere, more preferably under an argon
atmosphere. The
phosgene or oxalyl dichloride is preferably added slowly, preferably over a
period of up
to about 2 h, more preferably from about 1/4 h about 1 h. More preferably
still over
about 1/2 h. During addition of the phosgene or oxalyl dichloride, the
temperature of the
reaction mixture is preferably from about -20 °C to about 25 °C,
more preferably about
0 °C. After addition of phosgene or oxalyl dichloride is complete, the
reaction mixture is
preferably warmed to a temperature of from about 0 °C to about 40
°C, more preferably
to about room temperature; at this temperature, the reaction mixture is
stirred preferably
for from about 1/2 h to about 6 h, more preferably for about 1 h.
In Step (A), -X1 is converted to -X2. If -Xl is -OH, then -X2 is selected from
-
OMs, -OTs, -Cl, -Br and -I. If -X1 is -COOH, then -X2 is -C(O)Cl.
If -X is -Cl or -Br or -I, Step (A) is not needed, and -X2 is the same as -X1,
i.e.,
compound (2) is also compound (3). In this case, only Step (B) is needed to
produce
compound ( 1 ).
Depending on the reaction conditions in Step (A), the -SEM protective group on
the imidazo nitrogen may be split off and the reactive -X2 moiety may
spontaneously
react at that position to close the ring, thus forming compound ( 1 ). If this
ring closure is
sufficient in Step (A), then Step (B) is not needed; otherwise Step (B) is
used to achieve
sufficient ring closure.
9
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
Ste B
If the solvent for Step (B) is different from that used in Step (A), the Step
(A)
solvent is removed from the final Step (A) reaction mixture, preferably by
evaporation
under vacuum at room temperature.
Compound (3) or the reaction product from Step (A) is treated with
tetrabutylammonium fluoride (nBu4NF) in solvent, preferably tetrahydrofurane
(THF).
This treatment is preferably carried out at a temperature of from about 0
°C to about 60
°C, more preferably at about room temperature, preferably for a period
of from about 1/2
h to about 12 h, more preferably for about 4 h.
Compound (1) is preferably isolated and purified from the reaction mixture of
Step (A) or/and Step (B) by evaporating off organic solvent, washing the
product with
water and/or aqueous solutions, separating the organic layer from the aqueous
layer,
drying off the organic layer by evaporation, and purifying by chromatography.
Scheme II
The subject invention processes disclosed in Scheme I above optionally include
additional Step (C) shown in Scheme II:
Q1-(CR2)~-X3
SEM
R1 (CR2)m JgSn
\' S02Ph
R1 'Z NN
L) L)
(C): Q1-(CR2)n-X3
Pd(PPh3)4 R1 (CR2)m
DTBMP SEM
if Z = OTf: LiCI I
N S02Ph
Dioxane R1
N
(2)
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
In Scheme II, m, n, -R, -Ql-, and -Rl are the same as defined for Scheme I
above.
-X3 is a subset of -X1, and is -OH or -COOH.
In Scheme II and other Schemes herein, -Z is -Br, -I, or -OTf, preferably -Br.
In Scheme II and other Schemes herein, -J is alkanyl having from 1 to about 4
carbon atoms; preferred J is methyl or n-butyl.
Ste C
In Step (C), compound (4) and compound (5) are reacted in solvent, preferably
dioxane, preferably in the presence of Pd(PPh3)4 catalyst, to produce compound
(2). A
small amount of a radical scavenger, preferably 2,6-di-tert-butyl-4-
methylphenol
(DTBMP) is included in the reaction mixture for Step (C). If -Z is
trifluoromethanesulfonate (-OTfJ, lithium chloride (LiCI) is also preferably
included in
the reaction mixture. The components of the reaction mixture are preferably
combined at
about room temperature; then the reaction mixture is heated to about reflux
temperature.
The reaction mixture is held at about reflux temperature for a period of from
about 2 h to
about 24 h, preferably for about 5 h. The reaction mixture is preferably
retained under an
inert atmosphere, preferably under an argon atmosphere during Step (C). The
reaction
mixture is preferably cooled to about room temperature. The cooled reaction
mixture is
preferably treated with a mixture of ether and saturated aqueous potassium
fluoride
solution, preferably about a 1:1 mixture, preferably for from about 1 h to
about 24 h,
more preferably for about 15 h.
Purified compound ~2) is obtained from the reaction mixture, preferably by
filtration, ether washing, water and aqueous solution washing, drying, and
purifying by
chromatography.
Scheme III
For the processes of the subject invention, the Steps shown in Scheme III are
an
alternative to those of Scheme II, and are an optional addition to the Steps
of Scheme I.
Scheme III below is used to produce compound (2a) which is a subset of
compound (2):
11
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
(D):
SEM Pd(PPhg)4
DTBMP
(CR2)m + ~3Sn N S02Ph if Z = OTf:LiCl
T
N Dioxane
Z
(5)
(li)
Y
I
(CR2)m
SEM W-(CR2)n-X1 (8_)
T N EtgN
\/ S02 Ph
/r CH2CI2
N
L)
Q3-(CR2)n-X1
(CR2)m
T ~ SEM
N S02Ph
N
L)
In Scheme III, m, n, -R, -J, and -X1 are the same as for Schemes I and II.
Ring T
represents all ring moieties that the two -R1's of compound (2) can form when
they are
attached. -Q3- is a subset of the moieties of -Ql-; -Q3- is selected from -O-,
-S-, -NR-, -
NR-C(O)-, and -OC(O)-; preferably from -O-, -S-, and -NR-.
In Scheme III, -Y is -NHR, -OH, or -SH; preferably -NHR or -OH; more
preferably -OH.
In Scheme III, -W is -I, -Br, or -C(O)V, preferably -Br or -C(O)V, more
preferably -Br. -V is -OH, -Cl, or -Br, preferably Cl.
Ste D
12
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
In Step (D) of Scheme III, compound (6) and compound (5) are reacted in
solvent, preferably dioxane, preferably in the presence of Pd(PPh3)4 catalyst
to produce
compound (7). The preferred conditions for Step (D) of Scheme III are the same
as those
for Step (C) of Scheme II.
Ste E
In Step (E), compound (7), and compound (8) are reacted, preferably in the
presence of a base, in solvent, preferably dichlorolmethane, to produce
compound (2a).
Preferred bases useful for this step include sodium carbonate and
triethylamine; more
preferred is triethylamine. This step is preferably carried out at a
temperature of from
about 0 °C to about 45 °C, more preferably at about room
temperature, preferably for a
period of from about 2 h to about 14 h, more preferably for about 6 h.
Scheme IV
The subject invention processes include the preparation of compound (9) from
compound ( 1 ) or compound ( 11 ) as depicted Scheme IV
13
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
Q1 D1
(CR2 \(CR2)n (G)~ / \(CR2)n
NBS ,Q2
R1 /Q2 CCI4 R
N S02Ph S02Ph
R1
N N
L)
Q1~
CR2)n
R2-SnJg (13) I
or
R2-B(OH)2 (14)p2
Pd(PPh3)4
\'S02Ph
Tolun /r
e
N
11)
(F): /Q1 \
R3-NH2 (12)
nBuLi (CR2)m iCR2)n
THF R1 ~ N j 2
NH-R3
R1
N
(9)
In Scheme N, m, n, -R, -Ql-, -Q2-, -J, and -R1 are the same as specified for
Schemes I and II above.
In Scheme IV, -R2 may be hydrogen in which case Steps (G) and (H) are not
used. -R2 is selected from hydrogen, halo, alkyl, aryl, heterocycle, carboxy
and its alkyl
esters and amides. Preferred -R2 is selected from hydrogen, halo, C1-C4 alkyl,
and
phenyl. More preferred -R2 is selected from hydrogen and unsubstituted and
substituted
phenyl; substituents on such phenyl are preferably selected from hydroxy,
alkoxy, thio
and alkylthio. Most preferred -R2 is hydrogen.
In Scheme IV, -R3 is selected from hydrogen, alkyl, aryl, and heterocycle.
Preferred -R3 is selected from alkyl, aryl, and heterocycle. More preferred -
R3 is
unsubstituted and substituted phenyl and benzyl. Preferred substituents for
such phenyl
14
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
and benzyl are selected from halo, C1-C4 alkyl, aryl, hydroxy, alkoxy,
aryloxy, thio,
alkylthio, arylthio, amino, alkylamino, arylamino, formyl, alkylacyl,
arylacyl, carboxy
and its alkyl and aryl esters, amides, thioesters and thioamides. More
preferred -R3 is
benzyl, wherein the alpha carbon of the benzyl is unsubstituted or
substituted; preferred
substituents are selected from alkyl (preferably C1-C4), aryl and heterocycle.
Ste F
In Step (F), compound ( 1 ) or compound ( 11 ) is combined with compound ( 12)
and n-butyllithium (nBuLi) in solvent, preferably tetrahydrofuran, to produce
compound
(9). Compound (12) is preferably dissolved in solvent first, and the resulting
solution is
cooled to a temperature of from about -30 °C to about 5 °C,
preferably about 0 °C. The
solution is preferably under an inert atmosphere, more preferably under an
argon
atmosphere. nBuLi is preferably added slowly to the solution over a period of
from
about 0.2 h to about 1 h, more preferably over a period of about 0.5 h. The
resulting
mixture being stirred for a period of from about 1J2 h to about 1 h, more
preferably about
3/4 h. Compound (1) or compound (11) dissolved in solvent, preferably THF, is
then
added. The reaction mixture is preferably warmed to room temperature, and then
preferably heated to about reflux temperature for a period of from about 2 h
to about 24
h, more preferably for about 12 h. When the reaction is complete, the reaction
mixture is
preferably quenched with methanol.
Compound (9) is preferably purified from the reaction mixture by evaporating
the
solvent, redesolving in solvent, preferably dichloromethane, washing with
water and
aqueous solutions, drying, and purifying by chromatography.
Steps (G1 and (H)
A non-hydrogen -R2 is optionally obtained on compound (9) by performing
optional Steps (G) and (H) of Scheme IV.
In Step (G), compound (1) and N-bromosuccinimide (NBS) are combined in
solvent, preferably carbon tetrachloride. A radical initiator, preferably
benzoyl peroxide,
is preferably added. The reaction mixture is heated to a temperature,
preferably from
about 0 °C to about 100 °C, more preferably about 90 °C.
The reaction mixture is held at
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
this elevated temperature for a period of from about 5 min to about 120 min,
more
preferably for about 10 min.
Purified compound (10) is preferably obtained by filtration, evaporation, and
purification by chromatography.
In Step (H), compound (10) is combined with compound 13) or compound 14)
in the presence of Pd(PPh3)4 catalyst in solvent, preferably toluene, to
produce
compound 11 ). A small amount of radical scavenger, preferably DTBMP, is
preferably
added to the reaction mixture of Step (H). The reaction mixture is heated,
preferably to
reflux, under an inert atmosphere, preferably a nitrogen atmosphere,
preferably for a
period of from about 3 h to about 24 h, more preferably for about 6 h. After
the reaction
is complete, the reaction mixture is preferably cooled to about room
temperature.
Purified compound 11) is obtained from the reaction mixture, preferably by
extraction, treating with aqueous KF, filtration, washing with water and
aqueous
solutions, extracting, drying, and purification by chromatography.
Scheme V
The subject invention processes optionally includes one or more additional
steps
to produce compound (5), as depicted in Scheme V:
16
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
SEM
N SEM-CI N PBSSPh
DMF THF
N N
(15) (16)
SEM SEM
N S~ MCPBA N
S02Ph
Ph
CH2C12
N N
17) 18)
SEM
nBuLi JgSn
JgSnCI ~ ~SOZPh
THF //,/N
(5)
In Scheme V, -J and -SEM are the same as specified in Schemes II and III
above.
The steps of Scheme V needed as additions to the subject invention processes
depends on which of compounds (15), (16), (17), and (18) is available as a
starting raw
material.
Compound 15), imidazole, is reacted with SEM-Cl, preferably in the presence of
sodium hydride, in solvent, preferably dimethylformamide (DMF), preferably
under an
inert atmosphere, preferably at a temperature of from about -20 °C to
about 60 °C,
more preferably at about room temperature, preferably for a period of from
about 1/2 h to
about 12 h, more preferably for about 2 h, to produce compound (16).
Compound (16) is reacted with phenyldisulfide, preferably in the presence of n-
butyllithium, in solvent, preferably THF, preferably at a temperature of from
about -80 °C
to about 25 °C, more preferably starting at a temperature of about -80
°C and ending at
about room temperature, preferably for a period of from about 1/2 h to about 6
h, more
preferably for a period of about 1/2 h after addition of the n-butyllithium at
17
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
about -80 °C, for about 1 h after addition of the phenyldisulfide at
about 0 °C, and for
about 1 h at about room temperature, to produce compound 17).
Compound 17) is reacted with an oxidizing agent, preferably MCPBA, in
solvent, preferably dichloromethane, preferably under an inert atmosphere,
preferably at
a temperature of from about 0 °C to about 40 °C, more preferably
at about room
temperature, preferably for a period of from about 2 h to about 24 h, more
preferably for
about 15 h, to produce compound 18).
Compound (18) is reacted with trialkyltin chloride, preferably tributyltin
chloride,
in solvent, preferably THF, preferably in the presence of n-butyllithium,
preferably under
inert atmosphere, preferably at a temperature of from about -80 °C to
about 40 °C, more
preferably at about room temperature, preferably for a period of from about
1/2 h to
about 6 h, more preferably for a period of about 1/2 h after the addition of n-
butyllithium
at about -80 °C, for about 1 h after addition of tributyltin chloride
at about 0 °C, and for
about 4 h at room temperature, to produce compound ~5).
Examples
The following examples provide further information regarding the subject
invention processes. They are simply exemplary and do not limit the scope of
the subject
invention.
Step Example 1
H SEM
N N
- ~ ~ SEM = ~~~~SiMe3
N ~ N
A
A suspended solution of NaH (6.5 g, 0.162 mol, washed with hexane twice) in
anhydrous
DMF (300 ml) is cooled in an ice/acetone bath (bath temp. -15 °C).
Solid imidazole (10
g, 0.145 mol) is added in small portions and the mixture is stirred at room
temperature
(r.t.) for 0.5 h; the solution becomes clear. SEM-Cl (25 g, 0.150 mol) is
added dropwise
by a syringe pump at r.t. over 1 h; NaCI precipitates during the addition. The
mixture is
18
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
stirred at r.t. for about 1 h. Progress of the reaction is monitored by TLC
(CH2Cl2/MeOH, 9:1 ). H20 ( 10 ml) is added with caution to quench the
reaction. The
solvent is evaporated in vacuo. The residue is dissolved in Et20 (200 ml) and
washed
with H20 (4 x SOmI), brine (50 ml), dried (MgS04), filtered, and evaporated in
vacuo to
give compound A as an orange liquid.
Step Example 2
IEM IEM
N N
\ ~ _~. \ ~-S w
N N
A B
To a solution of SEM-protected imidazole A (1.48 g, 7.50 mmol) in dry THF (75
ml)
under argon at -78 °C, n-BuLi (1.6 M in hexane) (6 ml, 9.60 mmol) is
added dropwise
and the mixture is stirred at -78 °C for 30 min. Phenyl disulfide (2.1
g, 9.60 mmol) in
THF (2 ml) is then added dropwise. The dry ice/acetone bath is replaced with
an ice bath
after this addition. The mixture is stirred at 0 °C for 1 h, then at
r.t. for 1 h. Progress is
monitored by TLC (CH2C12/MeOH, 9:1 ). H20 (5 ml) is added to quench the
reaction.
The solvent is evaporated in vacuo, and the residue is dissolved in Et20,
washed with
S% NaHC03 (3 x 20m1), brine (20 ml), dried (MgS04), evaporated in vacuo, and
purified by chromatography (silica gel, hexane/EtOAc 3:1) to give B as a
yellow oil.
Step Example 3
SEM SEM
N S
w \~ w
N ~ ~ N
g C
19
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
3-chloroperoxybenzoic acid (MCPBA, 80-85%) (17.03 g, 78.9 mol) is added to a
solution of SEM-protected 2-phenylsulfide imidazole B (9.71 g, 31.6 mmol) in
anhydrous CH2Cl2 (160mL), and the reaction is stirred under argon at room
temperature
for 1 S hours. Progress monitored by TLC (hexane/EtOAc, 3:1 ). Sodium
thiosulfate (3.9
g) is added to remove excess MCPBA. The mixture is filtered. The filtrate is
washed
with S% Na2C03 (3 x 150 mL), brine (150 mL), dried (MgS04), filtered,
evaporated in
vacuo, and purified by chromatography (silica gel, hexane/EtOAc 3:1 ) to give
C as a
light yellow oil.
Step Example 4
SEM
p O SEM
S~ \ nBu3Sn
N
C D
To a solution of SEM-protected 2-phenylsulfone imidazole C (8.61 g, 25.4 mmol)
in
anhydrous THF (250 mL) under argon at -78 °C, n-BuLi (1.6 M in hexane)
(19.0 mL,
30.0 mmol) is added dropwise by a syringe pump; the solution is stirred at -78
°C
for 30 minutes. Tributyltin chloride (6.9 mL, 25.4 mmol) is added dropwise by
a syringe
pump. The mixture is stirred at room temperature for one hour. Progress is
monitored
by TLC (hexane/EtOAc, 9:1 ). H20 (30 mL) is added to quench the reaction. The
solvent is evaporated in vacuo. The residue is dissolved in ether (S50 mL) and
washed
with saturated NH4C1 (3 x 150 mL), brine (150 mL), dried (MgS04), filtered,
evaporated
in vacuo, and purified by chromatography (silica gel, gradient: hexane (500
mL),
hexane/EtOAc, 50:1; hexane/EtOAc, 12:1) to give D as a clear oil.
Step Example 5
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
SEM
O
nBugSn ~ S~~O Me0
OH
~N I +
Me0 / Br
D E_
OH
\ SEM
O
-~ / N SI
Me0
F N I
Pd(PPh3)4 (0.0177g, 0.015 mmol) is added to a solution of stannylimidazole D
(0.51 g,
0.80 mmol), 4,5-dimethoxy-2-(2-hydroxyethyl)phenyl bromide E (0.33 g, 1.1
mmol), and
LiCI (0.087 g, 2.1 mmol) in anhydrous dioxane (4.0 mL) at room temperature. A
few
crystals (~ 2 mg) of a radical scavenger, 2,6-di-tert-butyl-4-methylphenol, is
added and
the reaction is heated to reflux under argon for 5 hours. The reaction is
cooled to room
temperature and treated with a 1:1 mixture of ether and saturated aqueous KF
solution
(10 mL) for 15 hours. Progress is monitored by TLC (hexane/EtOAc, 3:1). The
mixture
is filtered through a pad of Celite with ether rinses. The filtrate is washed
with water (3 x
12 mL), brine (3 x 12 mL), dried (MgS04), filtered, evaporated in vacuo, and
purified by
chromatography (silica gel, hexane/EtOAc, 2:3) to give F as an orange oil.
Step Example 6
21
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
M
O
S~~O
\
F
Me0 \
O
Me0 \
G
To a solution of F (2.2 g, 4.24 mmol) and Et3N (886 mL ) in dichloromethane
(200 mL)
under argon atmosphere at 0 °C, methylsulfonyl chloride (MsCI, 492 mL)
is added over a
period of 0.5 h. The reaction is then warmed to room temperature and stirred
for 1 h.
TLC (EtOAc:Hexane, 1:1 ) is used to monitor the reaction; it indicates that
Ms0-ester
formation and the SEM-cleavage followed by ring closure occur in one pot. The
mixture
is diluted with CH2Cl2 (25 mL), and washed with cold HCl aqueous (0.5N),
NaHC03
aqueous, H20, brine and dried over MgS04. Filtration and evaporation of
solvent gives
a yellow solid. Purification using preparative HPLC (EtOAc:hexane, a gradient
from 1:1
to 1:0) provides product G ( 1.3 g).
Step Example 7
22
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
Me0 Me Me
Ii/ O H2N
Me0 / S/ ~ +
N
_H
G
Me0
N I ~
Me0
N ~
\\' Me
Me
J
In a 25 mL single neck round bottom flask, equipped with a magnetic stir bar,
argon
inlet, and rubber septum, 1-methyl-1-phenyl-ethylamine (H~ (164 mg, 1.2 mmol)
in
anhydrous THF under argon atmosphere is cooled to 0 °C. nBuLi in hexane
(0.5 mL, 2.4
M) is added to the solution slowly. The reaction turns light yellow. After
stirnng for 45
minutes, compound G (150 mg, 0.405 mmol) in THF (1 mL) is added. The solution
is
warmed to room temperature, and is further heated to reflux for 12 h. TLC
(CH2C12:CH30H, 99:1) indicates completion of the reaction. The solution is
quenched
with MeOH and evaporated to give a residue which is redissolved in CH2C12,
washed
with aqueous NaHC03 (5%), H20, brine, and dried over Na2S04. The crude
product,
after filtration and evaporation in high vacuum in order to remove any excess
amine H, is
put3fied by chromatography (CH2C12:CH30H, 99:1) to provide subject invention
compound J.
Step Example 8
23
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
Me0 \
\
/ I "
Me0 ~ N
N Me
Me
J
Me0 \
Me \
Me0 ~ ~N
N ~Me
Me
P
1n a 25 mL single neck round bottom flask, compound J (181 mg, 0.5 mmol) in
anhydrous THF (10 mL) under argon atmosphere is cooled to 0 °C, then
NaH (18 mg)
pre-washed with hexane is added as a suspension in hexane to the solution.
After stirring
for 15 minutes, methyl iodide (71 mg, 0.5 mmol) in THF (0.5 mL) is added. The
solution is warmed to room temperature, and further heated to reflux for 2 h
to complete
the reaction. The solution is quenched with MeOH and evaporated to give a
residue
which is redissolved in CH2C12, washed with aqueous NaHC03 (5%), H20, brine
and
dried over Na2S04. The crude product is purified by chromatography
(CH2C12:CH30H
from 99:1 to 95:5) to provide subject invention compound P.
Step Example 9
24
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
Me0
O
SI/O
Me0
~N
G
Me0
O
8~~0
Me0
N
Br
N-bromosuccinimide (NBS) solid (98 mg, 0.26 mmol) is added to a solution of
compound G (0.5 mmol) in 15 mL of CCl4. Radical initiator benzoyl peroxide (2
mol%) is subsequently added. The flask is placed into a 90 °C oil bath.
After 10 min
stirnng, the reaction is complete. Filtration of the mixture through a celite
pad, and
evaporation of the filtrate gives a residue. Purification by chromatography
(EtOAc:hexane, 1:3 to 1:1 ) affords compound K.
Step Example 10
S~o
N
Br
Me0
O
N S~~O
Me0
N
L
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
To a solution of K (0.22mmo1) and Pd(PPh3)4 (l3mg, 0.056mmol) in 7 mL of
anhydrous
toluene are added phenyltributyltin (0.26mmo1) and a few crystals (~ 2 mg) of
2,6-di-tert-
butyl-4-methylphenol. The reaction mixture is allowed to reflux at 110
°C under
nitrogen for 6 hours to complete the reaction. The reaction mixture is allowed
to cool,
and is then diluted with 1-2 mL of ethyl acetate (EtOAc). The resultant
mixture is
washed with water, then brine, extracted with CH2Cl2, dried over Na2S04, and
filtered.
The filtrate is treated with 3 mL of 30% aqueous KF at room temperature for
2h. The
solid is filtered off. The filtrate is diluted with CH2C12 and washed with
water, 30%
aqueous NH40H (3X), brine, extracted with EtOAc, dried (Na2S04), and
concentrated
in vacuo to yield crude product. Chromatography purification (silica gel,
EtOAc:hexane,
1:l to 1:0) yields compound L.
Step Example 11
O Me Me
O
H2N
L M
- Me Me
N
In a 25 mL single neck round bottom flask, equipped with a magnetic stir bar,
argon
inlet, and rubber septum, (1-methyl-1-phenyl-ethylamine) (~ (1.2 mmol) in
anhydrous
THF under argon atmosphere is cooled to 0 °C. nBuLi in hexane (0.5 mL,
2.4 M) is
added slowly to the solution. The reaction turns light yellow. After stirring
for 45
minutes, compound L (180 mg, 0.405 mmol) in THF (1 mL) is added. The solution
is
26
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
warmed to room temperature, and is further heated to reflux for 12 h to
complete the
reaction. The solution is quenched with MeOH and evaporated to give a residue
which is
redissolved in CH2C12, washed with aqueous NaHC03 (5%), H20, brine and dried
over
Na2S04. The crude product, after filtration and evaporation in high vacuum in
order to
remove excess amine M, is purified by chromatography to provide subject
invention
compound N.
Step Example 12
SEM
nBugSn ~ I~O Me0 ~ N02
N ( / ~ OTf
D O
P
Pd(PPh3)4 (0.0177g, 0.015 mmol) is added to a solution of stannylimidazole D
(0.51 g,
0.80 mmol), 2-vitro-4-methoxyphenol triflate O (0.33 g, 1.1 mmol), and LiCI
(0.087 g,
2.1 mmol) in anhydrous dioxane (4.0 mL) at room temperature. A spatula tipful
of
radical scavenger, 2,6-di-tert-butyl-4-methylphenol, is added and the reaction
is heated to
reflux under argon for 5 hours. The reaction then is cooled to room
temperature and
treated with a 1:1 mixture of ether and saturated aqueous KF solution ( 10 mL)
for 15
hours (monitored by TLC, hexane/EtOAc 3:1 ). The mixture is filtered through a
pad of
Celite with ether rinses, and the filtrate is washed with water (3 x 12 mL),
brine (3 x 12
mL), dried (MgS04), filtered, evaporated in vacuo, and purified by
chromatography
(silica gel, hexane/EtOAc 3:1 ) to give compound P as an orange oil.
27
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
Step Example 13
Me0 ~ N02 Me0 NH2
EM ~ IEM
N SI//O ~ N SI//O
w - ~ ~- w
N I ~ N
9
Pd (0.20 g, 0.19 mmol, 10% on activated carbon) is added to a solution of
compound P
(0.91 g, 1.85 mmol) in ethyl acetate (37 mL). After the reaction mixture is
hydrogenated
under H2 at 40 psi for 2 hours, methanol (10 mL) is added to the mixture and
hydrogenation continues for 3 additional hours. The mixture is filtered
through silica,
and the filtrate is evaporated in vacuo to give compound Q as an orange oil.
Step Example 14
0'\ ~
Me0 ~ NH2 O I CI
SEM
O II
N S~O CI ~CI
N
R
S~O
I/~
S
28
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
A solution of 4-dimethylaminopyridine (0.18 g, 1.5 mmol) in anhydrous CH2Cl2
(3 mL)
is added to a solution of the compound Q (0.69 g, 1.5 mmol) in anhydrous
CH2Cl2 (15.7
mL) and the solution is stirred under argon at room temperature. After 10
minutes,
chloroacetyl chloride (C1C(O)CH2Cl) (0.12 mL, 1.5 mmol) is added dropwise. The
solution is stirred at room temperature for 20 hours (monitored by TLC,
hexane/EtOAc
3:1). The solution is diluted with CH2Cl2, washed with H20 (2 x 20 mL) and
brine (25
mL), dried (MgS04), filtered, evaporated in vacuo, and purified by
chromatography
(silica gel, gradient: hexane/EtOAc 10:1, 5:1, 1:1) to provide compound S as a
white
solid (53.3 mg).
Step Example 15
0
H
Me0 N
O Me Me
O
N ~~ \ + H2N
N
S H
T
1-Methyl-1-phenyl-ethylamine (68 mg, 0.5 mmol) in anhydrous THF under argon
atmosphere is cooled to 0 °C. nBuLi in hexane (0.2 mL, 2.4 M) is added
to this solution
slowly. The reaction mixture turns light yellow. After stirring for 45
minutes, compound
S (39 mg, 0.1 mmol) in THF (1 mL) is added. The solution is warmed to room
temperature, and is further heated to reflux for 12 h. The solution is
quenched with
29
CA 02372469 2001-11-16
WO 00/69857 PCT/US00/13417
MeOH and evaporated to give a residue which is redissolved in CH2C12, washed
with
aqueous NaHC03 (5%), H20, brine, and dried over Na2S04. The crude product,
after
filtration and evaporation in high vacuum in order to remove any excess amine
H, is
purified by chromatography (CH2CI2:CH30H, 99:1) to provide compound T.
While particular embodiments of the subject invention have been described, it
will be obvious to those skilled in the arts that various changes and
modifications of the
subject invention can be made without departing from the spirit and scope of
the
invention. It is intended to cover, in the appended claims, all such
modifications that
are within the scope of this invention.