Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
DEGRADABLE POLYVINYL ALCOHOL) HYDROGELS
Related Application
This application claims priority to U.S. Serial No. 60/165,531, filed on
November 15,
1999.
Field of the Invention
The present invention relates generally to degradable hydrogels and more
specifically
to degradable polyvinyl alcohol) (PVA) hydrogels that are suitable for use as
biomaterials.
Background of the Invention
Biocompatible hydrogels have become a favored material for many biomedical
to applications. In many cases, it is preferable to employ a hydrogel that is
biodegradable so that
the body can rid itself of the foreign material over a period of time. One
biodegradable
hydrogel is disclosed in U.S. Patent No. 5,410,016 to Hubbell et al. .This
material is made
from biodegradable, polymerizable macromers having a water soluble region, at
feast one
degradable region which is hydrolyzable under in vivo conditions, and free
radical
t5 polymerizable end groups having the capacity to form additional covalent
bonds resulting in
macromer interlinking, wherein the polymerizable end groups are separated from
each other
by at least one degradable region. In preferred embodiments, the macromers
include a
central backbone of polyethylene glycol flanked by degradable regions of
polycaprolactone
or polylactide, which are in turn flanked by polymerizable vinyl groups.
2o A primary disadvantage of the macromers and hydrogels disclosed by Hubbell
is that
they are inflexible in design. PEG has only two groups which are easily
modified, the
terminal hydroxyl groups, and those groups are modified with the biodegradable
and
polymerizable groups. Moreover, Hubbell does not disclose any ways in which
the
macromers can be modified or in which the hydrogel can be modified after its
formation.
25 Also, the degradable PEG material developed by Hubbell et al. exhibits a
large degree of
swelling in aqueous solutions, which is disadvantageous in many applications.
PVA based hydrogels are disclosed in U.S. Patent Nos. 5,508,317 and 5,932,674
to
Muller. However, these hydrogels are not degradable.
PVA hydrogels offer many advantages over PEG based hydrogels. For example, the
3o availability of pendant OH groups along a PVA backbone adds versatility in
terms of the
various modifications that could be made to the macromer (e.g. attachment of
degradable
segments, active agents, hydrophobic groups, etc).
Moreover, a PVA system with its pendant OH groups allows for variations in
loading
(density) of the attached groups, and this is an important feature to have in
a macromer.
35 Third, PEG hydrogels are noted for their superior swelling in aqueous
environments. This
swelling property could be undesirable for certain applications. With a PVA
hydrogel, the
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
choice of a suitable PVA (with appropriate attached groups if desired) can
yield a non-
swellable, minimally swellable, or even shrinkable system.
Fourth, PVA possesses greater adhesive properties than PEG. This might be
desirable
for certain applications. Furthermore, PVA due to its hydrocarbon backbone has
greater
oxidative stability than PEG and.it can be stored as aqueous solutions as
opposed to PEG that
has to be stored as a freeze-dried powder.
Lastly, the preparation of a PVA macromer can be done in aqueous medium with a
final ultrafiltration step for purification. As opposed to this, PEG-based
acrylates/
methacrylates are prepared in organic solvents, and if not purified well can
have toxic
to residuals such as triethylamine hydrochloride.
A disadvantage of the PVA hydrogels that have been developed is that they are
not
degradable. Accordingly, it would be advantageous to have a PVA hydrogel that
is
degradable and methods for making such hydrogels. Moreover, it would be
advantageous to
have a degradable hydrogel having multiple pendant groups that allow for the
attachment of
is various modifiers.
Summary of the Invention
The invention is directed to biodegradable biocompatible hydrogels based on
polyvinyl alcohol) and methods for their preparation. The degradable hydrogels
can be
formed in vivo or ex vivo. The hydrogels can be used for a number of
biomedical
2o applications, including, but not limited to, implants, embolic agents,
wound healing dressings,
adhesion prevention, sealants, bulking agents. coatings for biomaterials, and
delivery of
biologically active compounds such as drugs, genes, proteins, and enzymes. The
hydrogels
are advantageous in that the PVA backbone can be easily modified and can
provide hydrogels
having very different properties.
25 The methods for preparation of the hydrogels involve the use of
prepolymers. The
prepolymers have a PVA backbone and pendant chains that include a
polymerizable group.
In one embodiment, the pendant chains also include a biodegradable region. In
another
embodiment, biodegradable regions are incorporated into the hydrogel during
its formation.
In a first general embodiment, a PVA prepolymer having crosslinkable groups
and a
3o second component having a degradable region flanked by crosslinkable groups
are combined
under conditions suitable for crosslinking the groups. The resulting hydrogel
that is formed
contains PVA chains linked by degradable regions.
In a second general embodiment, PVA prepolymers are formed having pendant
crosslinkable groups separated from the PVA backbone by a biodegradable
region.
35 Hydrogels are formed by exposing the prepolymers to conditions that
initiate crosslinking of
the crosslinkable groups.
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
The many pendant hydroxyl groups of PVA allow great versatility in design of
hydrogels with desired characteristics. Suitable hydrogels can be formed
without having to
employ each of the hydroxyl groups for attaching crosslinkable groups. Certain
hydroxyl
groups can be modified before the prepolymer is formed, after the prepolymer
is formed, or
even after the hydrogel is formed.
Brief Description of the Drawings
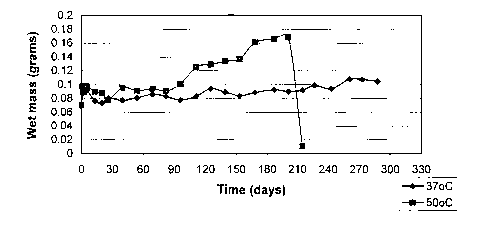
Figure 1 illustrates the mass loss over time in pH 7.4 buffer for a hydrogel
made from
a 3-ester acrylate modified PVA at 1 meq/g crosslinker density. ~ indicates
the degradation
at 37C; ~ indicates the degradation at SOC.
Figure 2 illustrates the mass loss for a hydrogel made from 3-ester acrylate
modified
PVA in pH 9.0 buffer. ~ indicates the degradation at 0.~ meq/g crosslinker
density and SOC;
0 indicates the degradation at 0.~ meq/g crosslinker density and 70C; x
indicates the
degradation at 1.0 meq/g crosslinker density and SOC; . indicates the
degradation at 1.0 meq/g
crosslinker density and 70C; ~ indicates the degradation at 1.8 meq/g
crosslinker density and
~5 HOC: and I indicates the degradation at 1.8 meq/g crosslinker density and
70C.
Figure 3 illustrates the mass loss for a hydrogel made from 3-ester
methacrylate
modified PVA at 1 meq/g crosslinker density in pH 7.4 buffer. v indicates an
average wet
weight of four samples at 37C (wet); ~ indicates the degradation at SOC (wet);
~ indicates
the degradation at 37C (dry); and x indicates the degradation at SOC (dry).
2o Figure 4 illustrates the mass loss for a hydrogel made from 5-ester
acrylate modified
PVA at 1 meq/g crosslinker density in 10 mM HEPES buffer at pH 7.4 and pH 9Ø
v
indicates the degradation at 37C and pH 7.4; ~ indicates the degradation at
37C and pH 9.0;
~ indicates the degradation at 70C and pH 7.4; and x indicates the degradation
at 70C and
pH 9Ø
z~ Figure ~ illustrates the mass loss for a hydrogel made from 5-ester
acrylate modified
PVA at 1 meq/g crosslinker density in 0.1 M phosphate buffer at pH 7.4 and pH
9Ø ~
indicates the degradation at 37C and pH 7.4; ~ indicates the degradation at
37C and pH 9.0;
~ indicates the degradation at 70C and pH 7.4; and x indicates the degradation
at 70C and
pH 9Ø
30 Detailed Description of the Invention
Biodegradable hydrogels based on polyvinyl alcohol) have been developed which
can be rapidly formed in an aqueous surrounding, e.g., in vivo. The PVA based
hydrogels
can be designed to degrade as fast as a few hours to more than 1 year.
Degradation rates are
determined in one respect by selection of an appropriate degradable region.
For example, use
35 of hydroxyethyl methacrylate (HEMA)-lactate as the biodegradable/
crosslinkable groups
will likely provide faster degradation than the use of a 3-ester methacrylate
such as mono-2-
3
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
(methacryoyloxy)ethyl succinate. Other factors that will affect the
degradation rate are the
density of the pendant chain bearing the degradable region, the length of the
degradable
region, the hydrophobicity of the network, and the crosslinking density.
Two Component Embodiment
In one embodiment, the hydrogels are formed by crosslinking two components.
Component A is a prepolymer having a PVA backbone having pendant chains with
crosslinkable groups, and component B is a molecule having a degradable region
flanked by
crosslinkable groups.
Component A: Prepolymer Backbone with Polymerizable Groins
to P VA
The prepolymers have a backbone of a polyhydroxy polymer, such as PVA or
copolymers of vinyl alcohol that contain, for example, a 1,3-diol skeleton.
The backbone can
also contain hydroxyl groups in the form of 1,2-glycols, such as copolymer
units of 1.2-
dihydroxyethylene. These can be obtained, for example, by alkaline hydrolysis
of vinyl
15 acetate-vinylene carbonate copolymers. Other polymeric diols can be used,
such as
saccharides.
In addition, the prepolymers can also contain small proportions, for example
of up to
20%, preferably of up to 5%, of comonomer units of ethylene, propylene,
acrylamide,
methacrylamide, dimethacrylamide, hydroxyethyl methacrylate, alkyl
(meth)acrylates, alkyl
20 (meth)acrylates which are substituted by hydrophilic groups, such as
hydroxyl, carboxyl or
amino groups, methyl acrylate, ethyl acrylate, vinylpyrrolidone, hydroxyethyl
acrylate, allyl
alcohol, styrene, polyalkylene glycols, or similar comonomers usually used.
Polyvinyl alcohols which can be used as prepolymer backbones are commercially
available PVAs, for example Vinol° 107 from Air Products (MW=22,000 to
31.000, 98-98.8
2s % hydrolyzed), Polysciences 4397 (MW=25,000, 98.5 % hydrolyzed), BF 14 from
Chan
Chun, Elvanol° 90-50 from DuPont and UF- 120 from Unitika. Other
producers are, for
example, Nippon Gohsei (Gohsenol°), Monsanto (Gelvatol°),
blacker (Polyviol°) or the
Japanese producers Kuraray, Deriki, and Shin-Etsu. In some cases it is
advantageous to use
Mowiol~ products from Hoechst, in particular those of the 3-83, 4-88, 4-98, 6-
88. 6-98. 8-88,
8-98. 10-98, 20-98, 26-88, and 40-88 types.
It is also possible to use copolymers of hydrolyzed or partially hydrolyzed
vinyl
acetate, which are obtainable, for example, as hydrolyzed ethylene-vinyl
acetate (EVA), or
vinyl chloride-vinyl acetate, N-vinylpyrrolidone-vinyl acetate, and malefic
anhydride-vinyl
acetate. If the prepolymer backbones are, for example, copolymers of vinyl
acetate and
35 vinylpyrrolidone, it is again possible to use commercially available
copolymers, for example
the commercial products available under the name Luviskol° from BASF.
Particular
4
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
examples are Luviskol VA 37 HM, Luviskol VA 37 E and Luviskol VA 28. If the
prepolymer
backbones are polyvinyl acetates. Mowilith 30 from Hoechst is particularly
suitable.
Polyvinyl alcohols that can be derivatized in accordance with the invention
preferably
have a molecular weight of at least 10.000. As an upper limit, the polyvinyl
alcohols may
have a molecular weight of up to 1,000,000. Preferably, the polyvinyl alcohols
have a
molecular weight of up to 300,000, especially up to approximately 100,000 and
especially
preferably up to approximately 30,000.
The polyvinyl alcohols usually have a poly(2-hydroxy)ethylene structure. The
polyvinyl alcohols derivatized in accordance with the disclosure may, however,
also comprise
to hydroxy groups in the form of 1,2-glycols.
The PVA system can be a fully hydrolyzed PVA, with all repeating groups being -
CH~-CH(OH), or a partially hydrolyzed PVA with varying proportions (25% to 1%)
of
pendant ester groups. PVA with pendant ester groups have repeating groups of
the structure
CH~-CH(OR) where R is COCH; group or longer alkyls, as long as the water
solubility of the
~5 PVA is preserved. The ester groups can also be substituted by acetaldehyde
or butyraldehyde
acetals that impart a certain degree of hydrophobicity and strength to the
PVA. For an
application that requires an oxidatively stable PVA, the commercially
available PVA can be
broken down by NaI04-KMn04 oxidation to yield a small molecular weight (3-4K)
PVA.
The PVAs are prepared by basic or acidic, partial or virtually complete
hydrolysis of
2o polyvinyl acetate. In a preferred embodiment, the polyvinyl alcohol
derivatized in accordance
with the invention comprises less than 50% of vinyl acetate units, especially
less than about
25% of vinyl acetate units. Preferred amounts of residual acetate units in the
polyvinyl
alcohol derivatized in accordance with the invention, based on the sum of
vinyl alcohol units
and acetate units. are approximately from 3 to 25%.
25 The prepolymers contain pendant groups that can be crosslinked to one end
of the
component B molecules. Various crosslinkable groups can be used.
Crosslinkable Groups
Crosslinking of components may be via any of a number of means, such as
physical
crosslinking or chemical crosslinking. Physical crosslinking includes, but is
not limited to,
3o complexation, hydrogen bonding, desolvation, Van der wals interactions, and
ionic bonding.
Chemical crosslinking can be accomplished by a number of means including, but
not limited
to, chain reaction (addition) polymerization, step reaction (condensation)
polymerization and
other methods of increasing the moiecular weight of polymers/oligomers to very
high
molecular weights. Chain reaction polymerization includes but is not exclusive
to free radical
35 polymerization (thermal, photo, redox, atom transfer polymerization, etc.),
cationic
polymerization (including onium), anionic polymerization (including group
transfer
CA 02391618 2002-05-14
WO 01!44307 PCT/US00/42190
polymerization), certain types of coordination polymerization. certain types
of ring opening
and metathesis polymerizations, etc. Step reaction polymerizations include all
polymerizations which follow step growth kinetics including but not limited to
reactions of
nucleophiles with electrophiles, certain types of coordination polymerization,
certain types of
ring opening and metathesis polymerizations, etc. Other methods of increasing
molecular
weight of polymers/oligomers include but are not limited to polyelectrolvte,
formation,
grafting, ionic crosslinking, etc.
In one embodiment, a two part redox system is employed. One pan of the system
contains a reducing agent such as ferrous salt. Various ferrous salts can be
used, such as
ferrous gluconate dehydrate, ferrous lactate dehydrate, or ferrous acetate.
The other half of the
solution contains an oxidizing agent such as hydrogen peroxide. Either or both
of the redox
solutions can contain macromer, or it may be in a third solution. The two
solutions are
combined and the agents react to initiate the crosslinking.
Other reducing agents can be used, such as, but not limited to cuprous salts,
cerous
~ 5 salts. cobaltous salts, permanganate, and manganous salts. Other oxidizing
agents that can be
used include, but are not limited to. t-butyl hydroperoxide, t-butyl peroxide.
benzoyl
peroxide, cumyl peroxide, etc.
The crosslinkable groups consist preferably of the following groups:
(meth)acrylamide, (meth)acrylate, styryl, vinyl ester, vinyl ketone, vinyl
ethers, etc.
2o Specific Prepolymers
Prepolymers suitable for use are disclosed in U.S. Patent Nos. x,932,674,
x,508.317,
~,66~.840, 5,849,841, 6,011,077, x,939,489, and 5,807,927.
In one embodiment, units containing a crosslinkable group conform, in
particular. to
the formula I
CH CH
R~
R N R3
in which R is a linear or branched C,-Cg alkylene or a linear or branched C,-
C,
alkane. Suitable alkylene examples include octylene, hexylene, pentylene,
butylene,
propylene, ethylene, methylene, 2-propylene, 2-butylene and 3-pentylene.
Preferably lower
alkylene R has up to 6 and especially preferably up to 4 carbon atoms. The
groups ethylene
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
and butylene are especially prefen-ed. Alkanes include, in particular,
methane, ethane, n- or
isopropane, n-, sec- or tert-butane. n- or isopentane, hexane, heptane, or
octane. Preferred
groups contain one to four carbon atoms. in particular one carbon atom.
Ri is hydrogen, a C,-C6 alkyl. or a cycloalkyl, for example, methyl, ethyl,
propyl or
butyl and RZ is hydrogen or a C,-C6 alkyl, for example, methyl, ethyl, propyl
or butyl. R, and
R~ are preferably each hydrogen.
R3 is an olefinically unsaturated electron attracting copolymerizable radical
having up
to 2~ carbon atoms. In one embodiment, R3 has the structure
C R4~N CO C CH2
n
to where R.~ is the
Rs
group if n=zero, or the
C CH2
Rs
C-
R~
bridge if n=1;
RS is hydrogen or C,-C.~ alkyl, for example, n-butyl, n- or isopropyl, ethyl,
or methyl;
n is zero or I , preferably zero: and
R6 and R~; independently of one another, are hydrogen, a linear or branched C,-
Cg
alkyl, aryl or cyclohexyl, for example one of the following: octyl, hexyl,
pentyl, butyl, propyl,
ethyl, methyl, 2-propyl, 2-butyl or 3-pentyl. R6 is preferably hydrogen or the
CH3 group, and
R~ is preferably a C,-C4 alkyl group. R6 and R~ as aryl are preferably phenyl.
2o In another embodiment, R3 is an olefinically unsaturated acyl group of
formula R8-
CO-, in which R8 is an olefinically unsaturated copolymerizable group having
from 2 to 24
carbon atoms, preferably from 2 to 8 carbon atoms, especially preferably from
2 to 4 carbon
atoms. The olefinically unsaturated copolymerizable radical Rg having from 2
to 24 carbon
atoms is preferably alkenyl having from 2 to 24 carbon atoms, especially
alkenyl having from
25 2 to 8 carbon atoms and especially preferably alkenyl having from 2 to 4
carbon atoms, for
example ethenyl, 2-propenyl, 3-propenyl, 2-butenyh hexenyl, octenyl or
dodecenyl. The
7
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
groups ethenyl and 2-propenyl are preferred, so that the group -CO-Rg is the
acyl radical of
acrylic or methacrylic acid.
In another embodiment, the group R3 is a radical of formula
-[CO-NH-(R9-NH-CO-O)q-R, o-O]P-CO-Rg
wherein q and q are zero or one and
R9 and Rio are each independently lower alkylene having from 2 to 8 carbon
atoms,
arylene having from 6 to 12 carbon atoms, a saturated divalent cycloaliphatic
group having
from 6 to 10 carbon atoms, arylenealkylene or alkylenearylene having from 7 to
14 carbon
atoms or arylenealkylenearylene having from 13 to 16 carbon atoms, and
Rg is as defined above.
Lower alkylene R9 or R,o preferably has from 2 to 6 carbon atoms and is
especially
straight-chained. Suitable examples include propylene, butylene, hexylene,
dimethylethylene
and, especially preferably, ethylene.
Arylene R9 or Rio is preferably phenylene that is unsubstituted or is
substituted by
~5 lower alkyl or lower alkoxy, especially 1,3-phenylene or 1,4-phenylene or
methyl-1,4-
phenylene.
A saturated divalent cycloaliphatic group R9 or R,o is preferably
cyclohexylene or
cyclohexylene-lower alkylene, for example cyclohexylenemethylene, that is
unsubstituted or
is substituted by one or more methyl groups, such as, for example,
2o trimethylcyclohexylenemethylene, for example the divalent isophorone
radical.
The arylene unit of alkylenearylene or arylenealkylene R9 or Rio is preferably
phenylene, unsubstituted or substituted by lower alkyl or lower alkoxy, and
the alkylene unit
thereof is preferably lower alkylene, such as methylene or ethylene,
especially methylene.
Such radicals R9 or R,o are therefore preferably phenylenemethylene or
methylenephenylene.
25 Arylenealkylenearylene R9 or R,o is preferably phenylene-lower alkylene-
phenylene
having up to 4 carbon atoms in the alkylene unit, for example
phenyleneethylenephenylene.
The radicals R9 and R,o are each independently preferably lower alkylene
having from
2 to 6 carbon atoms, phenylene, unsubstituted or substituted by lower alkyl,
cyclohexylene or
cyclohexylene-lower alkylene, unsubstituted or substituted by lower alkyl,
phenylene-lower
3o alkylene, lower alkylene-phenylene or phenylene-lower alkylene-phenylene.
The divalent group -R9-NH-CO-O- is present when q is one and absent when q is
zero. Prepolymers in which q is zero are preferred.
The divalent group -CO-NH-(R9-NH-CO-O)q-R,o-O-- is present when p is one and
absent when p is zero. Prepolymers in which p is zero are preferred.
35 In prepolymers in which p is one, q is preferably zero. Prepolymers in
which p is one,
q is zero, and R,o is lower alkylene are especially preferred.
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
All of the above groups can be monosubstituted or polysubstituted, examples of
suitable substituents being the following: C~-C~ alkyl, such as methyl, ethyl
or propyl, -
COOH, -OH, -SH, C,-C4 alkoxy (such as methoxy, ethoxy, propoxy, butoxy. or
isobutoxy), -
NO~, -NHZ, -NH(C,-C~), -NH-CO-NHS, -N(C,-Ca alkyl)2, phenyl (unsubstituted or
substituted by, for example, -OH or halogen, such as Cl. Br or especially I), -
S(Ci-C.~ alkyl), a
5- or 6-membered heterocyclic ring, such as, in particular, indole or
imidazole, -NH-C(NH)-
NH~, phenoxyphenyl (unsubstituted or substituted by, for example, -OH or
halogen. such as
Cl. Br or especially I), an olefinic group, such as ethylene or vinyl, and CO-
NH-C(NH)-NHS.
Preferred substituents are lower alkyl, which here, as elsewhere in this
description, is
to preferably C,-C~ allyl, C,-C4 alkoxy, COOH, SH, -NHS, -NH(C,-C.~ alkyl), -
N(C,-C,~ alkyl)
or halogen. Particular preference is given to C,-Ca alkyl, C,-C4 alkoxy, COON
and SH.
For the purposes of this invention. cycloalkyl is, in particular, cycloalkyl,
and aryl is,
in particular, phenyl, unsubstituted or substituted as described above.
Modifiers
15 The prepolymers can include further modifier groups and crosslinkable
groups such as
those described in U.S. Patent No. 5,932,674. Crosslinkable groups and the
optional further
modifier groups can be bonded to the prepolymer skeleton in various ways, for
example
through a certain percentage of the 1,3-diol units being modified to give a
1,3-dioxane, which
contains a crosslinkable radical. or a further modifier, in the 2-position.
Modifiers that might
2o be attached to the hydroxyls include those to modify the hydrophobicity,
active agents or
groups to allow attachment of active agents, photoinitiators, modifiers such
as polymers or
molecules to enhance or reduce adhesiveness, polymers to impart
thermoresponsiveness,
polymers to impart other types of responsiveness, and additional crosslinking
Qroups.
Component B
25 Component B is a molecule having a degradable region flanked by
crosslinkable
groups. Component B contains at least 1 group capable of crosslinking with the
crosslinkable
groups of component A. Component B also includes at least one group capable of
crosslinking with other components B after they have been attached to
component A. The
mechanism by which component A crosslinks with component B can be different
than the
3o mechanism by which component B crosslinks with other component B's after
they are
attached to component A's. When component A is a specific prepolymer as
described above,
component B includes a group capable of crosslinking with the olefinically
unsaturated group
of component A. Component B can include other copolymers in addition to the
degradable
region and crosslinkable group.
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
Crosslinkable Grozrps
Any of the crosslinkable groups described above with respect to component A
can
also be used on component B. Different types of crosslinking may be employed
for
crosslinking of A to one end of B and of B with other B's.
Degradable Regions
The degradable region is preferably degradable under in vivo conditions by
hydrolysis. The degradable region can be enzymatically degradable. For
example, the
degradable region may be polymers and oligomers of glycolide, lactide, E-
caprolactone, other
hydroxy acids, and other biologically degradable polymers that yield materials
that are non-
1o toxic or present as normal metabolites in the body. Preferred poly(a-
hydroxy acids are
poly(glycolic acid), poly(DL-lactic acid) and poly(L-lactic acid). Other
useful materials
include poly(amino acids), poly(anhydrides), poly(orthoesters),
poly(phosphazines), and
poly(phosphoesters). Polylactones such as poly(E-caprolactone), poly(s-
caprolactone),
poly(8-valerolactone) and poly(y-butyrolactone), for example, are also useful.
Enzymatically
~5 degradable linkages include poly(amino acids), gelatin, chitosan, and
carbohydrates. The
biodegradable regions may have a degree of polymerization ranging from one up
to values
that would yield a product that was not substantially water soluble. Thus,
monomeric,
dimeric, trimeric, oligomeric, and polymeric regions may be used. The
biodegradable region
could, for example, be a single methacrylate group.
2o Biodegradable regions can be constructed from polymers or monomers using
linkages
susceptible to biodegradation, such as ester, acetal, carbonate, peptide,
anhydride, orthoester,
phosphazine, and phosphoester bonds.
Methods of Making Biodegradable PVA Hydrogels from Components A and B
Methods for Making the Prepolvmers
35 Methods for making the prepolymers are taught in the U.S. Patents to Muller
cited
above.
The prepolymers of formulae I are extraordinarily stable. Spontaneous
crosslinking
by homopolymerization does not typically occur. The prepolymers of formula I
can
furthermore be purified in a manner known per se, for example by precipitation
with acetone,
3o dialysis, or ultrafiltration. Ultrafiltration is especially preferred. By
means of that
purification process the prepolymers of formula I can be obtained in extremely
pure form, for
example in the form of concentrated aqueous solutions that are free, or at
least substantially
free, from reaction products, such as salts, and from starting materials.
The preferred purification process for the prepolymers of the invention,
ultrafiltration,
35 can be carried out in a manner known per se. It is possible for the
ultrafiltration to be carried
out repeatedly, for example from two to ten times. Alternatively, the
ultrafiltration can be
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
carried out continuously until the selected degree of purity is attained. The
selected degree of
purity can in principle be as high as desired. A suitable measure for the
degree of purity is,
for example, the sodium chloride content of the solution, which can be
determined simply in
known manner.
The prepolymers of formulae I are crosslinkable in an extremely effective and
controlled manner.
Methods for Making H ~~dro~els
The methods of making a hydrogel from components A and B involves combining
the
components under conditions suitable for crosslinking of components A and B
and,
optionally in a second step, crosslinking of components B after they have been
attached to
components A.
The crosslinking is suitably carried out in a solvent. A suitable solvent is
in principle
any solvent that dissolves components A and B. for example water, alcohols,
such as lower
alkanols, for example ethanol or methanol, also carboxylic acid amides. such
as
t5 dimethylformamide, or dimethyl sulfoxide, and also a mixture of suitable
solvents, such as,
for example, a mixture of water with an alcohol, such as, for example, a
water/ethanol or a
water/methanol mixture. The combination of A and B is preferably carried out
in a
substantially aqueous solution. In accordance with the invention, the
criterion that the
prepolymer is soluble in water denotes in particular that the prepolymer is
soluble in a
2o concentration of approximately from 3 to 90% by weight, preferably
approximately from 5 to
60% by weight, in a substantially aqueous solution. Insofar as it is possible
in an individual
case, prepolymer concentrations of more than 90% are also included in
accordance with the
invention.
Within the scope of this invention, substantially aqueous solutions of the
prepolymer
35 comprise especially solutions of the prepolymer in water. in aqueous salt
solutions, especially
in aqueous same solutions that have an osmolarit~~ of approximately from 200
to 450
milliosmol per 1000 ml (unit: mOsm/1), preferably an osmolarity of
approximately from 250
to 350 mOsm/l, especially approximately 300 mOsm/l, or in mixtures of water or
aqueous salt
solutions with physiologically tolerable polar organic solvents, such as, for
example, glycerol.
30 Solutions of the prepolymer in water or in aqueous salt solutions are
preferred.
The viscosity of the solution of the prepolymer in the substantially aqueous
solution
is, within wide limits, not critical, but the solution should preferably be a
f7owable solution
that can be deformed strain-free.
The molecular weight of the prepolymer is also. within wide limits, not
critical.
35 Preferably, however, the prepolymer has a molecular weight of from
approximately 3,000 to
approximately 200.000, most preferably from about 3,000 to 30,000.
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
Components A and B are preferably combined such that a hydrogel is formed
having
crosslinking in an amount of from approximately 0.2~ to 10 milliequivalents of
crosslinker
per gram of PVA (meq/g), more desirably about 0.25 to 1,5 meq/g.
In order to encourage inter crosslinking between A and B prior to intra
crosslinking
of A with A or B with B, a large excess of B can be used, such as a ten fold
increase. It is
possible that a partially degradable hydrogel will result from this system.
Such a partially
degradable hydrogel may be desirable for some applications.
Preferably, the prepolymers used in the process according to the invention can
be
purified in a manner known per se, for example by precipitation with organic
solvents. such
1o as acetone, filtration and washing, extraction in a suitable solvent,
dialysis or ultrafiltration,
ultrafiltration being especially preferred. By means of that purification
process the
prepolymers can be obtained in extremely pure form, for example in the form of
concentrated
aqueous solutions that are free, or at least substantially free, from reaction
products, such as
salts. and from starting materials, such as, for example, non-polymeric
constituents.
The preferred purification process for the prepolymers used in the process
according
to the invention, uitrafiltration, can be carried out in a manner known per
se. It is possible for
the ultrafiltration to be carried out repeatedly, for example from two to ten
times.
Alternatively, the ultrafiltration can be carried out continuously until the
selected degree of
purity is attained. The selected degree of purity can in principle be as high
as desired. A
2o suitable measure for the degree of purity is, for example, the sodium
chloride content of the
solution, which can be determined simply in a known manner, such as by
conductivity
measurements.
One additive that is added, where appropriate, to the solution of the
prepolymer is an
initiator for the crosslinking, should an initiator be required for
crosslinking the crosslinkable
groups. Moreover, it may be desirable to employ different crosslinking means
for
crosslinking component A to component B and for crosslinking component B to
other
component B's after they are attached to component A's. For example, it may be
desirable to
employ salt crosslinking for crosslinking component A to component B but to
employ redox
initiated free radical crosslinking for crosslinking components B.
3o Single Component Embodiment
In the single component embodiment, a prepolymer is used to produce the
hydrogel.
The prepolymer includes a PVA backbone having pendant chains having a
degradable region
and crosslinkable group. In this embodiment, as opposed to the two component
embodiment,
the biodegradable regions are attached to the PVA backbone to form a
prepolymer prior to
crosslinking of the prepolymer into a hydrogel. It is desired that at least a
majority of the
crosslinkable groups are attached to the polymer via the degradable region.
The prepolymer
12
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
when polymerized results in a crosslinked network that is permeable to water
but water
insoluble. This crosslinked PVA network has degradable segments in between the
crosslinked
moieties. As such, hydrolytic/enzymatic degradation of these degradable
segments would
result in the degradation of the entire network, or a degradable PVA system.
The degradable segment may be attached to the backbone in two ways. One method
involves the preparation of a degradable crosslinker containing both the
polymerizable
segment and the degradable segment, followed by its attachment to the PVA
backbone. The
attachment of the degradable crosslinker to the backbone can be done by
suitable
functionalization of pendant hydroxyl groups on the PVA backbone and/or the
degradable
crosslinker.
A second method involves the attachment of the degradable segment first to the
PVA
backbone, following which the crosslinkable moiety is attached to the
degradable segment.
The ease of synthesis as well as the type of degradable segment and
crosslinker used
determines the type of method used. The choice of a suitable degradable
segment, its length
o and loading dictate the final degradation profile achieved, although this is
also a function of
certain environmental factors such as pH, temperature and buffer
concentrations.
The PVA Backbone
Generally, the requirements for the PVA in this embodiment are the same as for
the
two component embodiment. Polyvinyl alcohols) which can be used as prepolymer
?o backbones are commercially available PVAs, for example Vinol° 107
from Air Products
(MW=22,000 to 31.000, 98-98.8 °io hydrolyzed), Polysciences 4397
(MW=25,000, 98.5
hydrolyzed), BF 14 from Chan Chun, Elvanol° 90-50 from DuPont and UF-
120 from
Unitika. Other producers are, for example. Nippon Gohsei (Gohsenol°),
Monsanto
(Gelvatol~). W'acker (Polyviol°) or the Japanese producers Kuraray,
Deriki, and Shin-Etsu.
?s In some cases it is advantageous to use Mowiol° products from
Hoechst, in particular those
of the 3-83. 4-88. 4-98, 6-88, 6-98, 8-88, 8-98, 10-98, 20-98, 26-88, and 40-
88 types.
It is also possible to use copolymers of hydrolyzed or partially hydrolyzed
vinyl
acetate, which are obtainable, for example, as hydrolyzed ethylene-vinyl
acetate (EVA), or
vinyl chloride-vinyl acetate, N-vinylpyrrolidone-vinyl acetate, and malefic
anhydride-vinyl
3o acetate. If the prepolymer backbones are, for example, copolymers of vinyl
acetate and
vinylpyrrolidone, it is again possible to use commercially available
copolymers, for example
the commercial products available under the name Luviskol° from BASF.
Particular
examples are Luviskol VA 37 HM, Luviskol VA 37 E and Luviskol VA 28. If the
prepolymer
backbones are polyvinyl acetates, Mowilith 30 from Hoechst is particularly
suitable.
35 The PVA should preferably have a molecular weight of at least 10,000. As an
upper
limit, the polyvinyl alcohols may have a molecular weight of up to 1,000,000.
Preferably. the
13
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
polyvinyl alcohols have a molecular weight of up to 300.000, especially up to
approximately
100,000 and especially preferably up to approximately 30,000.
PVAs usually have a poly(2-hydroxy)ethylene structure. The PVA may, however,
also comprise hydroxy groups in the form of 1,2-glycols.
The PVA system can be a fully hydrolyzed PVA, with all repeating groups being -
CH2-CH(OH), or a partially hydrolyzed PVA with varying proportions (25% to 1%)
of
pendant ester groups. PVA with pendant ester groups have repeating groups of
the structure
CHZ-CH(OR) where R is COCH3 group or longer alkyls, as long as the water
solubility of the
PVA is preserved. The ester groups can also be substituted by acetaldehyde or
butyraldehyde
1 o acetals that impart a certain degree of hydrophobicity and strength to the
PVA. For an
application that requires an oxidatively stable PVA, the commercially
available PVA can be
broken down by NaIO~-KMnO:, oxidation to yield a small molecular weight (3-4K)
PVA.
The PVA is prepared by basic or acidic, partial or virtually complete
hydrolysis of
polyvinyl acetate. In a preferred embodiment, the polyvinyl alcohol
derivatized in accordance
with the invention comprises less than 50% of vinyl acetate units, especially
less than 20% of
vinyl acetate units. Preferred amounts of residual acetate units in the
polyvinyl alcohol
derivatized in accordance with the invention, based on the sum of vinyl
alcohol units and
acetate units, are approximately from 3 to 20%, preferably approximately from
5 to 16%.
Degradable Region
2o Generally the same degradable regions can be used as are described above
for the two
component embodiment. The PVA based hydrogels can be designed to degrade as
fast as an
hour to a day (with cross linkers containing rapidly degradable segments such
as HEMA-
glycolate and HEA-glycolate), to a few days or more than 1 year. with
degradable segments
such as 3-ester or ~-ester methacrylates or acrylates (for example, using mono-
2-
(methacryloyloxy)ethyl succinate (a 3-ester methacrylate), or mono-2-
(Acryloyloxy)ethyl
succinate (AOES, a 3-ester acrylate). The desired degradability can be
achieved using an
appropriate degradable region, an appropriate region length, by varying the
hydrophobicity of
the network with pendant groups, and by varying the density or loading of the
degradable/
crosslinking chains.
3o Crosslinkable Groups
The crosslinkable groups that can be used in the single component embodiment
are
the same as those described above with respect to the two component
embodiment.
Modifiers
The prepolymers can include further modifier groups and crosslinkable groups
such as
those described in U.S. Patent No. 5,932,674. Crosslinkable groups and the
optional further
modifier groups can be bonded to the prepolymer skeleton in various ways, for
example
14
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
through a certain percentage of the 1.3-diol units being modified to give a
1,3-dioxane, which
contains a crosslinkable radical, or a further modifier, in the 2-position.
Modifiers that might
be attached to the hydroxyls include those to modify the hydrophobicity,
active agents or
groups to allow attachment of active agents, photoinitiators, modifiers such
as polymers or
molecules to enhance or reduce adhesiveness, polymers to impart
thermoresponsiveness,
polymers to impart other types of responsiveness, and additional crosslinking
groups.
Methods of Making Biodegradable PVA Hydrogels from a Single Component
The prepolymers can be made in at least two ways. In one method, a degradable
crosslinker containing both the polymerizable group and the degradable region
is prepared,
followed by its attachment to the PVA backbone. The attachment of the
degradable cross
linker to the backbone can be done by suitable functionalization of pendant
hydroxyl groups
on the PVA backbone and/or groups on the degradable cross linker.. In a second
method, the
degradable segment is first attached to the PVA backbone, following which the
cross linkable
moiety is attached to the degradable segment.
The methods for attaching the crosslinkable group to the degradable region and
for
attaching the degradable region to the PVA will vary according to the type of
crosslinkable
group and degradable region used. One skilled in the art will be capable of
designing a
synthetic scheme.
Degradable glycolide or lactide based crosslinkers can be prepared according
to the
2o reference Furch, M. et al., Polymer, 39(10):1977-1982 (1998). It should be
noted that this
experimental procedure offers several advantages. First, a variety of
degradable segments
derived from glycolide, lactide, caprolactone, cyclic anhydrides, etc. can be
prepared each of
which provides a specific degradation profile. Second, this technique also
allows for a "I-
step" synthesis of a degradable cross linker by simple control of the initial
''feed ratio" of
35 monomer to initiator. Third, by supplying a feed of different monomer types
and monomer
ratios, one could prepare a variety of random or block copolymers to achieve a
specific
degradation pattern. Fourth, one could incorporate end cappers such as
succinic anhydride in
the initial monomer feed to prepare acid-terminated segments, thereby
eliminating an
additional synthetic step.
3o Methods for Using the Prepolymers and Hydrogels
The hydrogels can be used for a number of biomedical applications, including,
but not
limited to, implants, embolic agents, wound healing dressings, adhesion
prevention, sealants,
bulking agents, coatings for biomaterials, and delivery of biologically active
compounds such
as drugs, genes, proteins, and enzymes.
35 In one embodiment, a hydrogel is formed from the prepolymers prior to
implantation
in or application or administration to a patient. In another embodiment, a
hydrogel is formed
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
in situ at the intended site of use. By way of example, a soft tissue implant
can be formed by
injecting a solution of prepolymers into the site where the implant is to be
formed, along with
the second component if the two component embodiment is used. Crosslinking of
the
prepolymers is initiated to form the hydrogel. Bulking agents and embolic
agents can be
s similarly formed. The compositions can also be used to create tissue
supports by forming
shaped articles within the body to serve a mechanical function. Such supports
include, for
example, sealants for bleeding organs, sealants for bone defects and space-
fillers for vascular
aneurysms. Further, such supports include strictures to hold organs, vessels
or tubes in a
particular position for a controlled period of time.
The compositions can be applied in a number of ways, such as injection, via
catheter,
by spray, by pouring or spreading on a surface.
The composition can be used for encapsulation of various agents, such as
therapeutic,
diagnostic and prophylactic agents. For example, the compositions can be used
to deliver an
active agent which can be any of a variety of materials, including proteins,
carbohydrates,
tJ nucleic acids. and inorganic and organic biologically active molecules.
Specific examples
include enzymes, antibiotics, antineoplastic agents, local anesthetics,
hormones,
antiangiogenic agents, antibodies, neurotransmitters, psychoactive drugs,
drugs affecting
reproductive organs, and oligonucleotides such as antisense oligonucleotides.
Cells, tissues,
and organelles can also be encapsulated.
2o In a variation of the method for controlled drug delivery, the prepolymers
are
polymerized with the biologically active materials to form microspheres or
nanoparticles
containing the biologically active material. The macromer, photoinitiator, and
agent to be
encapsulated are mixed in an aqueous mixture. Particles of the mixture are
formed using
standard techniques, for example, by mixing in oil to form an emulsion,
forming droplets in
25 oil using a nozzle, or forming droplets in air using a nozzle. The
suspension or droplets are
irradiated with a light suitable for photopolymerization of the macromer.
The present invention will be further understood by reference to the following
non-
limiting examples.
Examples
3o Moderate to Slow De~radin~ Systems
Hydrogels having moderate to slow degradation times, ranging from a few days.
to
months, to a year were prepared. Degradation profiles are shown in Tables 1
and 2.
Example l: Preparation of Degradable PvA containing 3-ester acrylate cross
linker.
35 3-ester acrylate modified PVA was prepared having 0.5 to 1.8
milliequivalents of the
side chain per gram of PVA (meq/g). The following recipe is for 1.0 meq/g. The
proportion
16
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
of crosslinker chain to PVA was adjusted to prepare prepolymers with other
meq/g. Dried
mono-2-(acryloyloxy)ethyl succinate (AOES, 2.3 g, 10.6 mmoles) in
dichloromethane (DCM,
35 mL) was maintained under nitrogen at about 5°C. One glass stopper
was removed and
replaced with a rubber septum. Dicyclohexylcarbodiimide (DCC) solution (5 mL,
5 mmoles)
was removed from the sure seal bottle by syringe under nitrogen flow. The DCC
solution
was added to the AOES solution slowly over about 2 minutes. A precipitate
formed almost
immediately. After complete addition of the DCC solution, the flask was
removed from the
ice bath and the solution was stirred at room temperature. The reaction was
followed by the
disappearance of the DCC peak at about 2116 em-~ . The reaction was done in
less than 2
hours.
When the reaction was complete, the dicyclohexylurea (DCU) byproduct was
removed by filtration through a glass frit. The precipitate was rinsed with a
little DCM and
the DCM was removed from the filtrate using the rotary evaporator. About 5 mL
or more of
DMSO was added as needed in order to dissolve the anhydride. At this time,
29.4 g of an
t5 18% PVA solution in DMSO was added. The PVA used was Mowiol 3-83. Upon
addition
of the PVA solution, some polymer may precipitate. If this does occur, stir
the solution until
the solution is homogenous. Heating the solution to 60°C may be
required. Vl~'hen the
solution was homogeneous. 5 drops of triethylamine was added. The solution was
stirred at
room temperature overnight. The following day, the solution was heated to
60°C for I hour.
?o The solution was precipitated into a 10-fold excess of acetone (vs. volume
of DMSO).
Additional DMSO may be needed to dilute the solution so the polymer is able to
precipitate
adequately. About 12-15% solids is appropriate.
Formatlation, casting, and curing of degradable PV.A
A 20% solution of the degradable PVA prepolymers in water was prepared with
0.3%
25 Irgacure. The mixture was warmed to 60°C for about 15-30 minutes
until the polymer
completely dissolved. To prepare flats, 2 drops of the polymer solution were
transferred into
a polypropylene mold, the mold was closed, and the solution was irradiated for
20 seconds
(2.0-2.5 mW/cm2 at 310 nm, and intensity of 65-75 mW/cm' at 365 nm). Plugs
were
prepared by adding 35 drops of the formulation to the open male end of the
polypropylene
3o mold and irradiating under the same conditions. The cross linked gels were
analyzed for
degradation profiles.
17
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
Protocol for In vitro Degradation Experiments
A 10 mM HEPES solution at pH 7.4 was prepared containing 0.200 g/L sodium
azide.
The phosphate buffer used was a 100 mM solution at pH 9.0 containing 0.200g/L
sodium
azide.
Determination of Initial Wet Weight
Prior to the degradation experiment, the gels were stored in USP water at
4°C. A gel
sample was removed from solution using small forceps. The excess water was
removed from
the gel by touching the sample to the side of the beaker. The sample was then
placed in a
vial, the lid placed on the vial and weighed on a 4-place balance.
t o Determination of the initial dry weight
After the wet weight of the samples has been determined the lid was removed
and the
samples were exposed in order for it to start to dry. A few pieces of dry ice
were placed in a
small Dewar flask. Ethanol was slowly added to the dry ice. Wait until the
evolution of COZ
has slowed before beginning. The vial was held without the lid using crucible
tongs and
t 5 about 1 cm of the bottom of the vial was submerged into the cold ethanol
for 1 minute. At
this time the vial was removed from the cold ethanol, the excess ethanol was
dried from the
bottom of the vial using a paper towel and the vial was placed in the freezer
and kept there for
at least 1 hour. The samples were placed into the freeze-drier overnight. The
next day, the
dried samples were weighed with the lids on.
20 General Procedure
After the weights were determined, 5 mL of the appropriate buffer was
immediately
added using a 10 mL pipet. The lid was replaced and the vial was placed into
the appropriate
oven.
When a mass measurement was taken, the vial was removed from the oven and
25 allowed to cool for a few minutes, then a small plastic pipet was used to
remove the excess
water without touching the sample. After all the excess water was removed, all
the excess
water from the lid and inside the vial was removed without touching the
sample. The lid was
replaced and the weight of the sample was determined as described above for
the wet weight
determination. After this was done, either some fresh HEPES solution was added
to the vial
30 or the sample was freeze-dried and the dry weight of the sample was
determined.
Figure 1 illustrates the mass loss over time in pH 7.4 buffer for a hydrogel
made from
a 3-ester acrylate modified PVA at I meq/g crosslinker density. v indicates
the degradation
at 37C; ~ indicates the degradation at SOC.
Figure 2 illustrates the mass loss for a hydrogel made from 3-ester acrylate
modified
35 PVA in pH 9.0 buffer. ~ indicates the degradation at 0.5 meq/g crosslinker
density and SOC;
4 indicates the degradation at 0.5 meq/g crosslinker density and 70C; x
indicates the
18
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
degradation at 1.0 meq/g crosslinker density and SOC; ~ indicates the
degradation at I .0 meq/g
crosslinker density and 70C; ~ indicates the degradation at 1.8 meq/g
crosslinker density and
SOC: and I indicates the degradation at 1.8 meq/g crosslinker density and 70C.
Example 2: Preparation of Degradable PVA containing 3-ester methacrylate cross
linker.
The same procedure as Example 1 was followed, using mono-2-
(methacryloyloxy)ethyl succinate.
Figure 3 illustrates the mass loss for a hydrogel made from 3-ester
methacrylate
modified PVA at 1 meq/g crosslinker density in pH 7.4 buffer. v indicates an
average wet
to weight of four samples at 37C (wet); ~ indicates the degradation at HOC
(wet); ~ indicates
the degradation at 37C (dry); and x indicates the degradation at SOC (dry).
Example 3: Preparation of Degradable PVA containing 5-ester acrylate cross
linker.
AOES (3 g, 13.88 mmoles) and ethylene glycol (4.3088, 69.4 mmoles) were dried.
A
few milligrams of dimethylaminopyridine (DMAP) was added to the solution. The
solution
was cooled to 4°C, and 1 M DCC solution ( I 3.88 mL, I 3.88 mmoles) was
added over 25
minutes. At this time the solution was removed from the ice bath and stirred
at room
temperature for 4 hours. The DCU precipitate was removed by filtration and the
filtrate was
extracted with ~% HCI (2 x 75 mL), I M NaHC03 (2 x 75 mL) and deionized water
(2 x 5
2o mL). The organic phase was dried with MgS04, the MgSO~ was filtered, and
the filtrate
concentrated under reduced pressure. The product was dried overnight in a
vacuum oven at
room temperature.
The product of the above reaction (0.915 g. 3.52 mmoles) was dissolved in 10
mL of
dichloroethane (DCE). Succinic anhydride (0.352 g, 3.~2 mmoles) and 1-
methylimidazole
(72 pL) was added to the solution. The solution was heated to 60 ~C for 3
hours, then cooled
to room temperature, and extracted with 10% HC1 (2 x 50 mL) and deionized
water (2 x 50
mL). The organic layer was dried using MgSO~, the MgSO~ was filtered, and the
filtrate
concentrated under reduced pressure. The product was dried in a vacuum oven
overnight at
room temperature. 'H NMR CDC13 (vinyl, 6.4 ppm (d), 6.2 ppm (q), 5.8 ppm (d);
CHZ of
3o ethylene glycol, 4.2-4.4 ppm; CHI of succinic acid, 2.6-2.8 ppm). FT-IR
(COOH, 2300-3600
cm-'; ester, 1732 cm-'; vinyl, 2957, 1636 cm-').
5-ester acrylate having 3 meq/g acetal was prepared similarly.
Figure 4 illustrates the mass loss for a hydrogel made from ~-ester acrylate
modified
PVA at 1 meq/g crosslinker density in 10 mM HEPES buffer at pH 7.4 and pH 9Ø
v
3s indicates the degradation at 37C and pH 7.4; ~ indicates the degradation at
37C and pH 9.0;
19
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
~ indicates the degradation at 70C and pH 7.4; and x indicates the degradation
at 70C and
pH 9Ø
Figure 5 illustrates the mass loss for a hydrogel made from 5-ester acrylate
modified
PVA at 1 meq/g crosslinker density in 0.1 M phosphate buffer at pH 7.4 and pH
9Ø v
indicates the degradation at 37C and pH 7.4; s indicates the degradation at
37C and pH 9.0;
~ indicates the degradation at 70C and pH 7.4; and x indicates the degradation
at 70C and
pH 9Ø
Example 4: Preparation of Degradable PVA containing carboxyethylacrylate cross
linker.
The same procedure as Example I was followed using carboxyethylacrvlate and 3-
83
PVA at 1.0 meq/g.
Example 5: Preparation of Degradable PVA containing vinyl azlactone cross
linker.
The same procedure as Example 1 was followed using vinylazlactone and 4-88 PVA
is at 1.0 meq/g. Azlactone-modified PVA was prepared according to the
literature reference
Muhlebach, A. et. al., J. Polynz Sci., Polvm. Chenz Ed. 1997. 3.5, 3603-3611.
Example 6: Preparation of Degradable PVA containing lOK PEG diazlactone
cross linker.
PEG lOK diol (10 g) was dissolved in 52 mL DCM. The material was dried by
reflux
20 of DCM through molecular sieves for an hour. Vinyl azlactone (0.5105 g,
0.0037 moles) was
added to the PEG solution, followed by 20 p1 of 1,8-diazabicyclo{5,4,0}undec-7-
ene (DBU).
The solution was heated under reflux for 24 hours. After the solution cooled,
it was poured
into 500 mL of hexane. The precipitate was filtered and dried in a vacuum oven
at room
temperature overnight.
~5
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
Table 1: Days to Complete Degradation in 0.1 M Phosphate Buffer
Crosslinker days days days days days Diameter
at at at at in j
pH pH pH pH 1N in PBS
7.4 9.0 7.4 9.0
37C 37C 70C 70C NaOH (37C)
j
i
3-ester acrylate105 12-14 4 2 -
flat
( 1.0 me /a)
3-ester acrylate>76 - - - -
plug
(1.2~ meq/g)
3-ester methacrylatei > 33-35 8-8.6 3 2-3
178
( 1.0 me /Q) minutes
~-ester acrylateI 36-382 1.7 0.5 -
flat
( 1.O me / )
J-ester acrylate>86 - - - - 19 mm
plug
( 1.~ meq/g)
~-ester acrylate69 - - - - 18 mm
plug
( 1.3 me /a) (hazv)
-ester acrylate46 15-17 - - - -
plug
(0.7~ meq/g)
~-ester acrylate25-26 - 1.3 - - I 21 mm
plug '.
(0.5 meq/g)
5-ester acrylate13-14 - - - - 22 mm
plug ',
(0.4 me / )
~-ester acrylate8-13 - - - - 30 mm
plug
(0.34 me /~) i
~-ester acrylate>38 - - - - 18 mm
plug
(0.4 meq/g +
3
me / acetal) , I
I
~-ester acrylate>3~ - 4 - - - 20 mm
plug i ',
(0.3 meq/g +3 j
meq/g acetal)
Carboxyethvlacrv_> 188 > 188 > 188 >
late 'I ! 188
Vinyl azlactone> 18~ > 185 > 185 > 7 hours
' 193
PEG lOK diazlactone57, >145 7 3.5 ~38
6~
minutes
PEG 35K diacrvlate- 16 - 1-5 seconds
hours hours
Note 1: gels not completely degraded, but unable to remove buffer solution
without removing
the pieces of gel.
Note 2: The sample held its shape for 4 days at 70°C, but lost its
shape when cooled to room
temperature
21
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
Table 2: Days to Complete Degradation in 10 mM HEPES Buffer
Crosslinkerdays days days days Comments
at at at at
pH pH pH pH
7.4 9.0 7.4 9.0
37C 37C 70C 70C
3-ester >281 - 94-104- 33% mass increase
in
methacrylate 274 days 'I
( 1 meq/g)
3-ester >273 - - 11 73% mass increase
acrylate in
(1 me / 272 da s
)
5-ester 178 1 13 2.5 Some sample
acrylate ~-17 pieces
(1 meq/g) left
Fast degrading systems
Hydrogels having fast degradation times, ranging from hours to a day were
prepared.
Degradable glycolide cross linkers were prepared according to the reference
Furch, M. et al.,
Polymer, 39( 10):1977-1982 ( 1998).
Materials:
Glycolide obtained from PolySciences was used as received. Hydroxyethyl
acrylate
(HEA) and Hydroxyethyl methacrylate (HEMA) were dried over molecular sieves (4
A) and
distilled under reduced pressure before use. Triethyl aluminum (Et;AI. 1 M
solution in
hexane) was obtained from Aldrich and used as received. Triethyl aluminum
(Et3Al, 2M
solution in toluene) was obtained from Aldrich and used as received. Anhydrous
methylene
chloride (DCM, >99.9%, Aldrich), anhydrous DMSO (Aldrich), and diethyl ether
were used
15 as received. All glassware was dried in the oven, and flame dried under
nitrogen flow before
use. All transfers were done under strictly anhydrous conditions via syringe
or cannula.
Example 7: Preparation of Degradable PVA containing HEMA Glycolate-COOH
cross linker.
Preparation ofHEMA-Glvcolate-COOHcross linker
2o HEMA-glycolate-OH (F.W. 246g/mol, Sg, 20 mmol) was taken in a 3-necked
flask
fitted with a water condenser. Succinic anhydride (3.1 S g, 30.5 mmol), a
pinch of 4-
methoxyphenol, and 200 mL of anhydrous dichloroethane (DCE) were added to the
above
reaction flask and the contents were stirred. 1-methyl imidazole(3.~ mL, 44
mmol) was
added to the above reaction flask and the contents were stirred overnight
(~18h) at 70 ~C
25 using an oil bath. The mixture was cooled to room temperature and
transferred to a
separatory funnel. The contents were washed with 10% HCI (2x100 mL), followed
by DI
water (2x100 mL). Saturated NaCI was used to break up any emulsions in the
course of the
work up. The organic layer (DCE) was separated, dried over MgSO~, and the
MgS04 filtered
off. The filtrate was concentrated on a rotary evaporator to yield the HEMA-
glycolide-
3o COOH product as yellow oil. The product was characterized by IR, Proton NMR
and Carbon
22
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
NMR spectroscopy. ' H NMR (CDC13) 6.3 ppm (d, 1 H) of vinyl, 5.6 ppm (d, 1 H)
of vinyl,
4.6-4.8 ppm (m, 4H) of CHZ of glycolate unit, 4.4 ppm (m, broad, 4H) includes
CHz groups
of HEMA, 2.6-2.8 ppm (m. 4H) of succinic anhydride end-capper, 2.0 ppm (s, 3H)
of CH3
group of HEMA.
Preparation ofdeQradable PY:A containing HEI'tTA-Glvcolate-COOHcross linker
The HEMA-glycolate-COOH cross linker was attached to PVA by the following
procedure. 60 mL of DCM was taken in a flame dried 3-necked flask fitted with
a Soxhlet
and a condenser (attached to the Soxhlet). The Soxhlet was pre-filled with dry
molecular
sieves. The contents were refluxed for 2.~h. The HEMA-glycolate-COON (F.W. 346
g/mol,
2.0 g, ~.8 mmol) in 20 mL of anhydrous DCM was added to the reaction flask by
syringe and
the contents were gently refluxed for an additional O.Sh. The contents were
cooled to room
temperature and dicyclohexylcarbodiimide ( 1 M solution in DCM, 2.9 mL, 2.9
mmol) was
added in drops to the reaction flask by syringe. The mixture remained clear
but turned turbid
in a few minutes due to the precipitation of the dicyclohexyl urea byproduct.
The contents
were stirred at room temperature for 3h, and the reaction was followed for
completion by IR
analysis. The contents were filtered to remove the urea byproduct and the
filtrate was
concentrated on a rotary evaporator. The residue obtained after concentration
is the
anhydride of HEMA-glycolate-COOH. This product was taken in about 100 mL of
anhydrous DMSO and the contents were stirred.
2o Polyvinyl alcohol) [PVA 4-88, 14.2g, 20% solution in anhydrous DMSO] was
taken
in a dry flask and 80 mL of DMSO was added to the flask by cannula. Triethyl
amine (2 mL)
was added to the PVA solution and the contents were stirred for ~~ minutes.
Following this,
the anhydride product in DMSO (prepared above) was cannulated in drops to the
PVA
solution with rapid stirring. The contents were stirred overnight at room
temperature. and
2s then for 2h at 60 °C using an oil bath. The contents were cooled to
room temperature and the
DMSO solution of PVA was precipitated into acetone (DMSO:acetone was 1:10,
v/v) in
drops with rapid stirring. A fine white precipitate was obtained that was
isolated by filtration
or centrifuging process (dependent on the type of precipitate obtained). The
precipitate was
then dried under vacuum to yield a dry white fibrous solid which is the
degradable PVA
3o containing the HEMA-glycolate cross linker (loading of degradable segment
on PVA is 1
meq/g).
Formulation, casting, and curing of degradable PVA
A 30% solution of the degradable PVA prepolymer in water was prepared with 1
Irgacure. The mixture was warmed to 60°C for about 15-30 minutes until
the polymer
35 completely dissolved. The polymer solution was then transferred into
polypropylene molds
and UV irradiated for 30 seconds (2.0-2.5 mW/cm' at 310 nm, and intensity of
65-75
23
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
mW/cm' at 365 nm) to yield a cross linked gel that was analyzed for
degradation profiles.
The degradation profiles were determined as described below. After UV
irradiation. the flats
were placed in a suitable buffer at a specific temperature and the time to
dissolution was
determined. A static set up was used.
s The degradation times of this hydrogel in various buffers at various
temperatures is
shown in Table 3. The phosphate buffer used was a 100 mM solution at pH 9.0
(with 0.2%
NaN;). The HEPES buffer used was a 10 mM solution at pH 9.0 (without 0.2%
NaN3).
Materials used were flats that were cured by UV light for 30 seconds. in
polypropylene molds
of 246 um thickness. Loading of cross linker groups (XL) on PVA was 1 meq/g
Table 3. Degradation of [PVA-glycolate-XL) as a function of buffer type and
temperature
Buffer ! TemperatureBuffer Time to DegradationGel Form
I (~C) Volume (mL)(hours)
i
I Phosphate37 10 5.50 ~ Dissolved
i
I
I Phosphate37 10 6.50 Dissolved
Phosphate60 10 3.50 Dissolved
I I
i Phosphate60 10 3.60 Dissolved
I
Phosphate70 10 3.35 Dissolved
'
Phos hate_ 70 10 3.35 Dissolved
HEPES 37 10 29.0 Dissolved
HEPES 37 10 29.0 Dissolved
HEPES 50 10 20.0 Dissolved
HEPES 50 10 20.0 Dissolved
HEPES 70 10 3.33 Dissolved
~
HEPES 70 ~ 10 ~ 3.33 ~ Dissolved
Example 8: Preparation of Degradable PVA containing HEA-Glycolate cross
15 linker.
Synthesis ofHEA-Glycolate-OH
60 mL of DCM and Et3Al solution in toluene (2 M solution, 1.6 mL, 3 mmol) were
placed in a flask that had been flame dried and purged with nitrogen, and
equipped with a
rubber septum. The contents in the flask were cooled to 0 ~C for about 16
minutes. Under
2o vigorous stirring, freshly distilled hydroxyethyl acrylate (HEA) (0.4 mL, 3
mmol) in 16 mL
of DCM was added by cannula to the flask containing the Et3Al solution. The
color of the
solution turned yellow on addition of the HEA and staved yellow for a few
seconds. The
contents were stirred at 0 ~C for 10 minutes and then at room temperature for
1h. The flask
was then transferred to an oil bath and stirred at 40 ~C for an additional 30
minutes.
25 Glyeolide (3.6g, 30 mmol) was quickly weighed out into a clean dry flask
with a stir bar and
the flask was sealed with a rubber septum. About 90 mL of DCM was cannulated
into the
flask containing the glycolide and the contents were stirred to effect
dissolution. The
24
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
alycolide solution was then cannulated into the flask containing the HEA-
Et;AI solution at
40 ~C. The contents of the flask remained clear during this addition but
became turbid with
progress of the reaction. The contents were allowed to stir for 20 h at room
temperature. At
the end of the reaction, trifluoroacetic acid (in this case, 120 mL) was added
to the reaction
mixture with vigorous stirring until most of the prepolymer dissolved
(external cooling may
be employed at this stage of the reaction if necessary). On addition of the
trifluoroacetic acid,
the turbid mixture became increasingly translucent. The mixture was then
filtered through a
coarse frit funnel and precipitation of the filtrate was checked in diethyl
ether. The entire
filtrate (about 230 mL) was added in drops to diethyl ether (800 mL) with
rapid stirring to
yield a white precipitate. The precipitate was filtered using a frit funnel
under aspirator
pressure and the product was dried in a vacuum oven overnight at room
temperature. Carbon
NMR and Proton NMR characterization confirms the formation of the product i.e.
HEA
attached to 20 glycolate units (from 10 glycolide units). ~H NMR (CDCI; /
CF;COOD
mixture) 6.6 ppm (d) of vinyl, 6.2 ppm (q) of vinyl, 6.0 ppm (d) of vinyl, ~.0
ppm (m, broad)
1s of CHI of glycolate unit, 4.4-4.6 ppm (m, broad) includes CH~ groups of HEA
and CHzOH of
last glycolate segment of macromer.
This degradable cross linker could be attached to the PVA backbone by
isocyanate
coupling reaction between the hydroxyl groups. A suitable diiisocyanate such
as HMDI
(hexamethylene diisocyanate) or IPDI (isophorone diisocyanate) can be employed
for this
2o reaction.
Example 9: Preparation of Degradable PVA containing HEMA-Glvcolate cross
linker.
Synthesis ofHEMA-Glvcolate-OH
120 mL of DCM and Et;AI solution in hexane ( 1 M solution. 28.8 mL. 29 mmol)
25 were placed in a flask that had been flame dried and purged with nitrogen,
and equipped with
a rubber septum. The contents in the flask were cooled to 0 ~C for about 1 ~
minutes. Under
vigorous stirring, freshly distilled HEMA (3.~ mL, 29 mmol) in 30 mL of DCM
was added
by cannula to the flask containing the Et;AI solution. The color of the
solution turned yellow
on addition of the HEMA and stayed yellow for a few seconds. The contents were
stirred at 0
~C for 10 minutes and then at room temperature for 1 h. The flask was then
transferred to an
oil bath and stirred at 40 ~C for an additional 30 minutes. Glycolide (3.4 g,
29 mmol) was
quickly weighed out into a clean dry flask with a stir bar and the flask was
sealed with a
rubber septum. About 120 mL of DCM was cannulated into the flask containing
the
glycolide and the contents were stirred to effect dissolution. The glycolide
solution was then
35 cannulated into the flask containing the HEMA- Et3Al solution at 40 ~C. The
contents of the
flask remained clear during this addition but became turbid with progress of
the reaction. The
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
contents were allowed to stir for 20 h at room temperature. At the end of the
reaction the
mixture was cooled to 0 ~C in an ice bath and trifluoroacetic acid ( 100 mL)
was added to the
reaction mixture with vigorous stirring until most of the prepolymer
dissolved. The mixture
was then stirred at room temperature for an additional 10 minutes. On addition
of the
trifluoroacetic acid. the turbid mixture became increasingly translucent. The
mixture was then
filtered through a coarse frit funnel and the filtrate was concentrated in a
rotary evaporator to
remove all solvents (DCM, TFA, Hexane). The product was a yellow liquid. One
method of
purification is column chromatography using silica gel with DCM:diethyl ether
mixture
(80:20, v/v) as the eluting solvent mixture. As column chromatography can be
time
consuming, a differential solubility technique was developed to separate the
HEMA-glycolate
product from the catalyst byproduct. It should be noted that both purification
methods
resulted in products that had identical characterization results by TLC and
NMR techniques.
For the differential solubility purification method, the HEMA-glycolate
product (yellow
liquid) was added slowly into dichloroethane to precipitate the unwanted
byproduct. The
~ 5 HEMA-glycolate product remains in solution and can be separated from the
solid byproduct
by filtration. The filtrate is then concentrated on a rotary evaporator under
reduced pressure.
The product is a yellow liquid that is dried in a vacuum oven overnight at
room temperature.
Carbon NMR and Proton NMR characterization confirm the formation of the
product i.e.
HEMA attached to 2 glycolate units (from 1 glycolide unit). 'H NMR (CDC13 /
CF3COOD
2o mixture) 6.3 ppm (d, 1 H) of vinyl, 5.8 ppm (d, 1 H) of vinyl, 5.0 ppm (m,
2H) of CHI of
glycolate unit. 4.4-4.6 ppm (m, broad, 6H) includes CHz groups of HEMA and
CHZOH of last
glycolate segment of oligomer, 2.0 ppm (s, 3H) of CH3 group of HEMA. GPC
analysis was
done of the starting material (HEMA), the product HEMA-glycolate-OH, and the
product
spiked with HEMA. Analysis of the plots shows the presence of the product and
the absence
25 of the starting material, HEMA. Also, GC-MS analysis was done of the
starting material
(HEMA), the product HEMA-glycolate-OH, and the product spiked with HEMA. An
evaluation of the plots show 3 peaks for the product, which correspond to the
major
molecular weight fragments (M+ ions) from the HEMA-glycolate-OH product. No
peaks
corresponding to unreacted HEMA are seen.
3o This degradable cross linker could be attached to the PVA polymer as
described
above in Example 8.
Example 10: Insitu syntlzesis of HEMA-glycolate-COOH
70 mL of DCM and Et3Al solution in hexane (1 M solution, 14.~ mL, 14.5 mmol)
were placed in a flask that had been flame dried and purged with nitrogen, and
equipped with
35 a rubber septum. The contents in the flask were cooled to 0 ~C for about 15
minutes. Under
vigorous stirring, freshly distilled HEMA ( 1.75 mL, 14.4 mmol) in 30 mL of
DCM was
26
CA 02391618 2002-05-14
WO 01/44307 PCT/US00/42190
added by cannula to the flask containing the Et3Al solution. The color of the
solution turned
yellow on addition of the HEA and stayed yellow for a few seconds. The
contents were
stirred at 0 ~C for I S minutes and then at room temperature for l h. The
flask was then
transferred to an oil bath and stirred at 40 ~C for an additional 30 minutes.
Glycolide (5.1 g,
43.2 mmol) was quickly weighed out into a clean dry flask with a stir bar and
the flask was
sealed with a rubber septum. About 125 mL of DCM was cannulated into the flask
containing the glycolide and the contents were stirred to effect dissolution.
The glycolide
solution was then cannulated into the flask containing the HEMA- Et3A1
solution at 40 ~C.
Immediately after the cannulation of the glycolide was complete, a solution of
succinic
anhydride ( 1.5 g, 14.4 mmol) in 60 mL of DCM was cannulated into the reaction
flask. The
reaction mixture was homogeneous after the addition of glycolide and succinic
anhydride
solutions. The mixture was stirred for I S minutes at 40 ~C, and then for 20 h
at room
temperature. At the end of the reaction the mixture was homogeneous and yellow
in color.
The mixture was cooled to 0 ~C in an ice bath, and trifluoroacetic acid (30
mL) was added to
~5 the reaction mixture with vigorous stirring. On allowing the reaction
mixture to stand, a
yellowish white precipitate settled to the bottom of the flask and was removed
by filtration
through a coarse frit funnel. The precipitation of the filtrate was checked in
diethyl ether.
The filtrate (about 300 mL) was added in drops to diethyl ether (1200 mL) with
rapid stirring
to yield a white precipitate. The precipitate was filtered using a frit funnel
under aspirator
2o pressure and the product was dried in a vacuum oven overnight at room
temperature. Carbon
NMR and Proton NMR characterization confirms the formation of the product i.e.
HEMA
attached to glycolate units that are end-capped with an acid group from the
succinic
anhydride end capper. ' H NMR (CDC13 / CF~COOD mixture) 6.3 ppm (d, I H) of
vimrl, ~.8
ppm (d, 1 H) of vinyl, 5.0 ppm (m, 1 OH) of CHZ of alycolate unit, 4.4-4.6 ppm
(m, broad, 6H)
25 includes CHI groups of HEMA and CHZ of last glycolate segment, 2.8-3.0 ppm
(m, 4H) of
succinic anhydride end-capper, 2.0 ppm (s, 3H) of CH3 group of HEMA.
This degradable cross linker could be attached to the PVA polymer as described
above in Example 8.
Modifications and variations of the present invention will be apparent to
those skilled
3o in the art from the foregoing detailed description. All modifications and
variations are
intended to be encompassed by the following claims. All publications, patents,
and patent
applications cited herein are hereby incorporated by reference in their
entirety.
27