Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 1 -
PROCESS FOR THE PREPARATION OF SULFAMIDES
This invention relates to a process for producing
sulfamides, and to novel intermediates used in the
process.
Sulfamides are conventionally prepared by the use of
strongly electrophilic reagents such as sulfamoyl
chloride, sulfonyl dichloride, phosphorus oxychloride or
phosphorus pentachloride. Belgian patent 667.311
discloses a method of making sulfamides employing an
N-alkyl sulfamoyl chloride. However, all such reagents
involve aggressive synthetic methods, and indeed can be
inconvenient or dangerous in their practical,
industrial, application.
The invention provides a process for the production of
aryl sulfamides that avoids the use of the above
hazardous materials and conditions, and gives a high
yield.
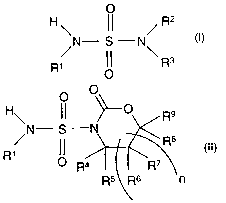
The process of the invention is for the production of an
aryl sulfamide having the formula
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 2 -
O
R
N- S -N
~ \
~ ~ R3
R
O
in which R1, R2 and R3 are each hydrogen, alkyl,
cycloalkyl or aryl, provided that at least one of R1, R2
and R3 is aryl,
which comprises reacting a compound of the formula
O O
R9
N- S -N
~~ ~R$
1
R ~ R4 ~ ~ R'
R5 Rs n
where R4, R5, R6, R~, R8 and R9 are each hydrogen, alkyl
or aryl, and n is 0 or 1, with an amine of the formula
R2R3NH (III), in the presence of a strong base.
The reaction can be carried out at ambient temperature
or at the reflux temperature of the solvent in which the
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 3 -
reaction is performed, and generally the temperature of
the reaction is chosen in the range of from 0~ C. to
100~ C. A polar, aprotic, solvent is preferred, as, for
example, acetonitrile.
A strong base is required for the reaction to proceed,
and examples include triethylamine,
1,8-diazabicyclo[5,4,0]under-7-ene(DBU),
1,5-diazabicyclo[4,3,0]non-5-ene(DBN) or
1,4-diazabicyclo[2,2,2]octane(TED). Preferably from one
to three equivalents of base are employed.
In the above formulae, an alkyl group can be substituted
or unsubstituted, and is preferably C1_6 alkyl, being
branched or unbranched. A cycloalkyl group preferably
containing from 3 to 9 carbon atoms, and may, for
example, be substituted by one to three alkyl groups
such as methyl. When substituted, the alkyl group can
be substituted by halo, C1-6 alkoxy, C3_9 cycloalkyl,
optionally substituted phenyl or optionally substituted
heteroaryl. An aryl group can be, for example, naphthyl
or, preferably, phenyl, and can be substituted or
unsubstituted. A substituted aryl group is substituted
with one or more, preferably one to three, substituents
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 4 -
selected from, for example, an electron-donating
substituent such as, for example, C1_4 alkyl, C1_4
alkoxy, C1_4 alkylthio, hydroxy, amino, or an electron-
withdrawing substituent such as, for example, carboxy,
nitro, cyano, trifluoromethyl, halo, C1_4 alkyl-SO- and
C1_4 alkyl-S02-.
Preferably, R1, R2 and R3 are selected from hydrogen,
C1_6 alkyl and optionally substituted phenyl. In
formula (II) above, R4, R5, R6, R~, R8 and R9 are
preferably hydrogen, and n is preferably 0. It may,
nevertheless, be desirable to employ a terminal moiety
in which one or more of R4 to R9 is alkyl or aryl, for
instance, in the preparation of stereoisomers.
It has been found that the nature of the substituent on
an aryl group, for example a substituted phenyl, can
surprisingly affect the reaction. Electron-donating
substituents assist the reaction. Thus it is preferred
that the substituent R1 is optionally substituted alkyl
or phenyl optionally substituted with an electron-
donating substituent, and a preferred process is one for
the preparation of a compound of the formula
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 5 -
O
R2
~
N- S -N
/ \
( ) R3
R
O
in which R1 is alkyl or phenyl optionally substituted
with an electron-donating substituent, and R2 and R3 are
each hydrogen, alkyl or optionally substituted phenyl,
provided that R1 is phenyl optionally substituted with
an electron-donating substituent and/or R2 is optionally
substituted phenyl,
which comprises reacting a compound of the formula
O O
~o
N- S -N
R O
(IV)
with an amine of the formula R2R3NH, in the presence of
a strong base. A particularly preferred process is one
for the production of a compound of the above formula in
CA 02391978 2002-05-15
WO 01/36383 PCT/LTS00/28877
- 6 -
which R1 is C1-6 alkyl or phenyl optionally substituted
with an electron-donating substituent, R2 is C1-6 alkyl
or optionally substituted phenyl, and R3 is hydrogen,
provided that R1 is phenyl optionally substituted with
an electron-donating substituent and/or R2 is optionally
substituted phenyl.
Compounds of formula (IV) where R1 is phenyl optionally
substituted with an electron-donating substituent are
novel, with the exception of compounds in which R1 is
3-methylbutyl or phenyl, and these novel compounds are
included as an aspect of the present invention. They
are stable, mainly crystalline solids, which can be
readily isolated from the reaction medium.
Compounds of formula R2R3NH (III) employed in the above
reactions are well known chemical compounds. As
indicated above, some of the reactants of formula (II)
are novel, but they can nevertheless be readily prepared
by methods well known in the art. For example,
compounds of formula (II) can be prepared by the
reaction of chlorosulfonylisocyanate with an alcohol of
formula
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
Ra Rs R7
Hal C C C OH
r~
R5 R7 Rs
n
where Hal is chloro or bromo,
to give
O
II ~-~ R9
CI S - N
R
O Ra ~ ~ ~ R7
R5 Rs n
which, in turn, when reacted with an amine of formula
R1NH2, yields the desired compound of formula (II). The
use of an appropriate optically pure alcohol can enable
the
production of diastereoisomers from which pure chiral
sulfamides can be derived.
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
g
Examples of reactions according to the invention are as
follows:
The sulfamides of formula (I) can be put to many uses.
One such is disclosed in EP-A 0 897921, in which a
sulfamide is cyclised to produce a benzothiadiazine
dioxide intermediate employed in the preparation of
pharmaceutically active compounds.
0
O~ ~ Et3N(2eqya-t3CN
~O
/ \ H . '~- Refluc
Q /\
H-~-N~ -~ -~ Refl~nc
VO
O
(l_ ~ ~/ \ ~''N(2e9Y~~
~I- -S~NHz
RaB~nc
O
O ~ \ /
~1 y O
O~'O + ~ / \ ~~ ~ \ H~O,
O
O O OW-l
/ \ ~ ~ / \
~O
H_~ N V -1- ~~ O
O O
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 9 -
The following Examples illustrate the invention.
T7CTT1~?TT TT 1
1,1-Dimethylpropylamino-1-sulfonic acid (4-
methylphenyl)-amide
2-Oxo-oxazolidine-3-sulfonic acid (4-methylphenyl)-amide
To a 1 L reactor, charged with dichloromethane (176 ml)
under an inert atmosphere (N2) was added chlorosulfonyl
isocyanate (CSI) (34.8 ml, 56.6 g, 0.40 mol) and the
solution was cooled to 5 ~C.
A solution of 2-bromoethanol(28.4 ml, 50.0 g, 0.40 mol,
1.0 equiv) in dichloromethane (176 ml) was added to the
reaction mixture over 30 minutes under cooling to keep
the temperature reaction mixture between 5-7 ~C.
After stirring for about 30 minutes, a solution of
p-toluidine (48.0 g, 0.45 mol, 1.1 equiv) and
triethylamine (125 ml, 90.5 g, 0.90 mol, 2.2 equiv) in
dichloromethane(358 ml) was added to the reaction
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 10 -
mixture over 30 minutes under cooling to keep the
temperature reaction mixture around 5-7 ~C.
After a stirring period of about 30 minutes 0.2N HCl
(0.4 L) was added. Additional concentrated HC1 (37o w/w)
was added until the pH of the water layer was ~2. After
decantation and separation of the aqueous layer, the
organic layer was washed with 0.05 N HCl (0.4 L) and
water (0.4 L).
To the washed and separated organic layer, water (0.4 L)
was added followed by the removal of dichloromethane
under vacuum. The resulting suspension was stirred for
an additional 30 minutes.
The reaction mixture was filtered and the filter cake
washed with water (0.2 L) and dried at 50 ~C under
reduced pressure to yield 90.82 g (0.355 mol) of crude
2-oxo-oxazolidine-3-sulfonic acid (4-methylphenyl)-
amide.
Crude 2-oxo-oxazolidine-3-sulfonic acid
(4-methylphenyl)-amide (50g) was suspended in
dichloromethane (50 ml) and stirred for one hour at room
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 11 -
temperature. The suspension was filtered, washed with
dichloromethane (40 ml) and dried under vacuum at 50 ~C
to yield pure 2-oxo-oxazolidine-3-sulfonic acid
(4-methylphenyl)-amide (34.3 g). mp 159-160 ~C.
1,1-Dimethylpropylamino-1-sulfonic acid (4-
methylphenyl)-amide
Triethylamine (3.50 ml, 2.55 g, 25.2 mmol, 2.5 equiv)
and tert-amylamine (1.50 ml, 1.12 g, 12.8 mmol,
1.3 equiv) were added to a solution of 2-oxo-
oxazolidine-3-sulfonic acid (4-methylphenyl)-amide
(2.56 g, 10 mmol, 1.0 equiv) in acetonitrile (12.5 ml).
This mixture was heated at reflux for 8 h.
After cooling, water (40 ml) was added and the
acetonitrile was removed by distillation under vacuum.
Dichloromethane (25 ml) was added to the resulting water
emulsion and acidified with 1 ml HCl (37o w/w). After
decantation and separation the organic layer was washed
with 25 ml 0.05 N HCl and water (25 ml).
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 12 -
The organic layer was concentrated at room temperature
under vacuum yielding the crude 1,1-Dimethylpropylamino-
1-sulfonic acid (4-methylphenyl)-amide (1.802 g,
7.9 mmol) as a viscous yellow oil which slowly
crystallised.
Crude 1,1-Dimethylpropylamino-1-sulfonic acid (4-
methylphenyl)-amide (1.40 g, 6.13 mmol) was suspended in
hexane (25 ml) and stirred at room temperature during
4 h.
The suspension was filtered, and the solid washed with
hexane (10 ml). After drying the solid under vacuum at
50 ~C, pure 1,1-Dimethylpropylamino-1-sulfonic acid (4-
methylphenyl)-amide (609 mg, 2.67 mmol) was obtained. mp
92.5-93 ~C.
4-Methvlphenvlylamino-1-sulfonic acid
(4-methanesulfonylphenyl)-amide
Triethylamine (7.0 ml, 5.10 g, 50 mmol, 2.5 equiv) and
4-methanesulfonyl-phenylamine(4.28 g, 25 mmol, 1.25
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 13 -
equiv) were added to a solution of 2-oxo-oxazolidine-3-
sulfonic acid (4-methylphenyl)-amide (5.12 g, 20 mmol)
in acetonitrile (25 ml). This reaction mixture was
heated at reflux for 8 hours.
After cooling, water (50 ml) was added and the
acetonitrile was removed by distillation under vacuum.
'I'o the obtained water emulsion were added
dichloromethane (40 ml) and HC1 (0.6 ml, 37o w/w).
After decantation and separation of the aqueous layer,
0.05 N HCl (25 ml) was added to the organic layer. At
this stage crystallisation occurred. Dichloromethane
was removed by distillation under vacuum at room
temperature.
The resulting suspension was filtered and the solid
washed with water (40m1) and dichloromethane (1 ml).
After drying under vacuum at 50 ~C, 4-methylphenylamino
1-sulfonic acid (4-methanesulfonylphenyl)-amide (4.64 g,
13.6 mmol) was obtained, mp 165.5-167 ~C.
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 14 -
1-Methvlethvlamino-1-sulfonic acid
(4-methanesulfonylphenyl)-amide
2-Oxo-oxazolidine-3-sulfonic acid isopropyl-amide
To a 250 L glass lined reactor initially charged with
dichloromethane (42 L) was added chlorosulfonyl
isocyanate (4.5 kg, 31.8 mol) at room temperature and
under a nitrogen atmosphere. The reaction mixture was
cooled to about 1 ~C. and a solution of 2-bromoethanol
(4.00 kg, 1 equiv) in dichloromethane (14 L) was slowly
added over 51 minutes in order to keep the reaction
temperature between 0 and 10 ~C. Stirring of the
reaction mixture was continued at the same temperature
for a minimum of 30 minutes. Progress of the reaction
was monitored by 1H-NMR. A mixture of isopropylamine
(2.1 kg, 1.1 equiv) and triethylamine (7.1 kg) in
dichloromethane (28 L) was then added at such an
addition rate that the reaction temperature was
maintained between 0 and 10 ~C. The solution was heated
up to room temperature. Aqueous hydrochloric acid
(~0.2 N, 28.5 kg) was then added and the pH of the
reaction mixture was adjusted to about 2 by addition of
concentrated hydrochloric acid (450 ml in 2 portions).
CA 02391978 2002-05-15
WO 01/36383 PCT/US00/28877
- 15 -
The reaction mixture was decanted and the separated
organic layer washed with aqueous hydrochloric acid
(28.1 kg, 0.05 N). The decanted and separated organic
layer was washed with water (28 kg). To the decanted
and separated organic layer, water (28 kg) was then
added and the reactor was placed under vacuum to distil
the maximum of dichloromethane while controlling the
temperature below 25 ~C. (84.4 kg of distillate). The
resulting suspension was stirred for a minimum of
2 hours at room temperature, filtrated, rinsed twice
wish water (2 x 7 L) and dried under vacuum at about
50 ~C during 16 hours to afford the 2-oxo-oxazolidine-3-
sulfonic acid isopropyl-amide, mp 107.5-108.5 ~C.
1-Methylethylamino-1-sulfonic acid
(4-methanesulfonylphenyl)-amide
A 100 L glass lined reactor was charged with
acetonitrile (17.8 kg) and 4-methylsulfonylaniline
hydrochloride (3.36 kg, 16.2 mol) under stirring at room
temperature. Triethylamine (4.5 kg) and 2-oxo-
oxazolidine-3-sulfonic acid isopropyl-amide (3.70 kg,
CA 02391978 2002-05-15
WO 01/36383 PCT/IJS00/28877
- 16 -
1.1 equiv) were then added at the same temperature. The
reaction mixture was heated to reflux and stirred at the
same temperature for a minimum of 6 hours. The solution
was then slowly cooled to room temperature and kept
agitated over night. Water was slowly added over
40 minutes and the reactor was placed under vacuum to
distil as much as possible of acetonitrile (27.8 kg of
distillate) while maintaining the reaction temperature
below 40 ~C. The suspension was cooled to room
20 temperature and stirred for a minimum of 2 hours before
filtering the product. The cake was rinsed with water
(16.2 kg) and dried under vacuum at about 50 ~C. for a
minimum of 16 hours to yield the 1-methylethylamino-1-
sulfonic acid (4-methanesulfonylphenyl)-amide, mp
164-165 ~C.