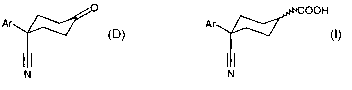Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
Process and Intermediates for Preparing a Cyclohexylnitrile
Area of the Invention
This invention relates to a method and intermediates for preparing certain
nitrites
which are useful for making cyclohexanoic acids. The latter are
pharmaceutically active
agents.
Background of the Invention
The process and intermediates of this invention provide a means for making
certain
4-substituted-4-(3,4-disubstituted phenyl)cyclohexanoic acids which are useful
for treating
asthma and other diseases which can be moderated by affecting the PDE 4
enzyme. The
final products of particular interest are fully described in U.S. patent
5,552,438 issued 03
September 1996. The information and representations disclosed therein, in so
far as that
information and those representations are necessary to the understanding of
this invention
and its practice, are incorporated herein by reference, in total.
This invention discloses a method for preparing a cyclohexanoic acid by
cyanohydrin homologation of a cyclohexanone precursor.
Summary of the Invention
A process for preparing a compound of formula (I)
COOH
Ar
CT N ~' (I)
where Ar is an aromatic group,
wherein the process comprises reducing a a,(3-unsaturated cyclohexene
carboxylic
acid of formula (A).
COON
Ar
STN ~' (A)
In as second aspect, this invention relates to a process for preparing an a,(3-
unsaturated cyclohexene carboxylic acid of formula (A)
1
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1101083
COOH
Ar
CAN ~' (A)
which process comprises hydrolysing a compound of formula (B).
CN
Ar
cTN '' (B)
In yet a further aspect, this invention relates to a process for preparing a
compound
of formula (I) by the cyanohydrin homologation of cyclohexanone (X) as
described herein
O
Ar
N (X)
wherein Ar is an aromatic group.
Description of the Invention
This invention can be used to homologate any cyclohexanone. In this invention
the
cyclohexanone has a 4-position nitrite group with or without another group at
that 4
position. Since the homologation is believed to be independent of the 4-
position
substitutent(s), the scope of the compounds which are homologated herein are
simply
examples of the chemistries being done on the 1-position carbon. Herein the
invention is
exemplified by compounds which have an aromatic group at the 4 position in
addition to the
nitrite group.
A preferred group of compounds which can be prepared by this homologation
route
are those of formula (Ia)
X / R~
R..
R~X2
Ra
(Ia)
wherein
2
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
Rl is -(CR4R5)rR6 wherein the alkyl moieties may be optionally substituted
with
one or more halogens;
R2 is -CH3 or -CH2CH3 optionally substituted by 1 or more halogens;
R3 is -CN;
risOto6;
R4 and RS are independently selected from hydrogen or a C1_2 alkyl;
R6 is hydrogen, methyl, hydroxyl, aryl, halo substituted aryl, aryloxyCl_3
alkyl,
halo substituted aryloxyCl_3 alkyl, indanyl, indenyl, C~_11 polycycloalkyl,
tetrahydrofuranyl, furanyl, tetrahydropyranyl, pyranyl, tetrahydrothienyl,
thienyl,
tetrahydrothiopyranyl, thiopyranyl, C3_6 cycloalkyl, or a C4_6 cycloalkyl
containing one or
two unsaturated bonds, wherein the cycloalkyl and heterocyclic moieties may be
optionally
substituted by 1 to 3 methyl groups or one ethyl group;
R~ is hydrogen or Cl-6 alkyl;
X is YR2;
X2 is O or NR~;
Y is O or S(O)m where m is 0, 1 or 2; and
one of R' or R" is hydrogen and the other is COOH or a salt thereof.
Preferred X groups for formula (Ia) are those wherein Y is oxygen. The
preferred
X2 group for formula (Ia) is that wherein X2 is oxygen. Preferred R2 groups
are a C1-2
alkyl unsubstituted or substituted by 1 or more halogens. The halogen atoms
are preferably
fluorine and chlorine, more preferably fluorine. More preferred R2 groups are
those
wherein R2 is methyl, or the fluoro-substituted alkyls, specifically a C 1 _2
alkyl, such as a -
CF3, -CHF2, or -CH2CHF2 moiety. Most preferred are the -CHF2 and -CH3
moieties.
Most preferred are those compounds wherein R1 is -CHI-cyclopropyl,
cyclopentyl,
3-hydroxycyclopentyl, methyl or CF2H; X is YR2; Y is oxygen; X2 is oxygen; and
R2 is
CF2H or methyl. The most preferred compounds are cis and traps 4-cyano-4-(3
cyclopentyloxy-4-methoxyphenyl)cyclohexanoic acid, in particular the cis
isomer, which is
the equitorial form the acid.
As regards the preferred embodiments for formula (A), they are those wherein:
R1 is -(CR4R5)rR6 wherein the alkyl moieties may be optionally substituted
with
one or more halogens;
R2 is -CH3 or -CH~CH3 optionally substituted by 1 or more halogens;
R3 is -CN;
risOto6;
R4 and RS are independently selected from hydrogen or a C 1 _2 alkyl;
3
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
R6 is hydrogen, methyl, hydroxyl, aryl, halo substituted aryl, aryloxyC I _3
alkyl,
halo substituted aryloxyCl_3 alkyl, indanyl, indenyl, C~_I I polycycloalkyl,
tetrahydrofuranyl, furanyl, tetrahydropyranyl, pyranyl, tetrahydrothienyl,
thienyl,
tetrahydrothiopyranyl, thiopyranyl, C3_6 cycloalkyl, or a C4_6 cycloalkyl
containing one or
two unsaturated bonds, wherein the cycloalkyl and heterocyclic moieties may be
optionally
substituted by 1 to 3 methyl groups or one ethyl group;
R~ is hydrogen or CI-6 alkyl;
X is YR2; and
X2 is O or NR~;
Y is O or S(O)m where m is 0, I or 2.
Preferred X groups for formula (A) are those wherein Y is oxygen. The
preferred
X2 group for formula (A) is that wherein X2 is oxygen. Preferred R2 groups are
a C I-2
alkyl unsubstituted or substituted by 1 or more halogens. The halogen atoms
are preferably
fluorine and chlorine, more preferably fluorine. More preferred R2 groups are
those
wherein R2 is methyl, or the fluoro-substituted alkyls, specifically a C I _2
alkyl, such as a -
CF3, -CHF2, or -CH2CHF2 moiety. Most preferred are the -CHF2 and -CH3
moieties.
Most preferred are those compounds wherein RI is -CH2-cyclopropyl,
cyclopentyl,
3-hydroxycyclopentyl, methyl or CF2H; X is YR2; Y is oxygen; X2 is oxygen; and
R2 is
CF2H or methyl.
The homologation of substituted cyclohexan-1-ones to the corresponding
cyclohexanoic acids are useful in making the compounds of U.S. patent
5,552,483. With
reference to Scheme 1, one route is to convert 4-cyano-4-(3-cyclopentyloxy-4-
methoxyphenyl)cyclohexan-1-one (the ketone) to the cyanohydrin of formula 2,
then to the
a,(3-unsaturated nitrile 3, thereafter to the saturated nitrile, and finally
to the saturated acid 5
or 6, i.e., sequence I ~~~~ 2 ~~~» 3 ~~~w 7 ~~~ 5 ~~~~ 6. The key step in this
sequence is hydrolysis of
the saturated nitrite 7 to the saturated acid 5 and 6.
Scheme 1
Ketone TMS~ , OH CN
(X) (or NaCN) Ar~CN + Ar~OH
RCN CT ~'N
2a 2b
4
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
SO~ Ar~CN
~CT/~~~'N
3
Reduction
Hydrolysis
Mg (O)
H or
Ar~ CN Hz/Pd
CI ~N
COOH
7 Ar
(axial nitrite also RCN 4
present in mixture)
Reduction
Hydrolysis
~ ~rCOOH
H Ar
Arm
C(O)OH MeOH, H+ CN
CN t-BuOK, heat
Saponification
An alternative novel reaction sequence is the one in Scheme 1 illustrated by
steps 1
~~~~ 2 ~~~~ 3 ~~~ 4 ~~~ 5 ~~~ 6. Intermediate 4 in this sequence, the a,(3-
unsaturated carboxylate is
reduced with Pd/C/cyclohexene and ammonium bicarbonate or via catalytic
hydrogenation
to yield a mixture of cis and traps isomers.
This invention can be applied generally to the preparation and reduction of an
a,(3-
unsaturated cyclohexene carboxylate as illustrated in Scheme 2.
5
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
Scheme 2
CN COOH COON
Ar~ ~ Ar~ Reduction Ar
CI N ~' CI N ~' CI ~'N
3 4 5
equilibration
equatorial carboxylic acid (cis isomer)
Compound 4 is prepared by the hydrolysis of the a,~3 unsaturated nitrite 3
with base
(for example 2 equivalents of Ba(OH)~). This produces the unsaturated
carboxylic acids
which are stable and can be characterized. Although the catalytic
hydrogenation of 3 is
difficult to achieve, hydrogenation of 4 occurs readily and produces the
saturated
carboxylate as a mixture of cis and traps acid. This mixture can be converted
to the cis
isomer by equilibration of the methyl esters.
Reaction Scheme 3 illustrates how a by-product of the base hydrolysis of
compound
7 in Scheme 1 can be converted to the 4-cyanocyclohexanoic acid.
Scheme 3
COOH
7 KOH/EtOH ~ Ar
CT N ~' S
SOCIz
COOH
Ar
ICON'H'2
8
The following examples are set out to illustrate the invention. They are not
intended to limit it in any way or fashion. Reference is made to the claims
for what is
reserved to the inventors hereunder.
6
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
Specific Exemplification
Example I
Preparation of 4-f3-(Cyclopentyloxy)-4-methoxyphenyll-1,4-dicarbonitrile
cyclohexan-1-ol:
Trimethylsilyl Cyanide Method
To a 100 ml round bottom flask equipped with magnetic stirrer and a nitrogen
inlet
was charged 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-I-one
(prepared as
illustrated in, for example, U.S. patent 5,552,438) ( 12.50 g, 40 mmol), zinc
iodide (0.35 g,
I .l mmol) and methylene chloride (50 mL). Stirring produced a clear solution.
This
solution was charged with trimethylsilyl cyanide (TMSCN).(8 mL, 5.952 g, 59.9
mmol).
After stirring for 1 hour under nitrogen 5 drops of the solution was quenched
into dilute acid
and examined by reverse phase HPLC.
The reaction was cooled to 15 °C and treated with a stream of HCl (gas)
for 15 min.
The reaction was evaporated on the rotovap and the resulting thick oil was
suspended in
ethyl acetate (150 mL) and washed with 2 x 50 mL 3N HCI, brine (25 mL) and
finally
evaporated to a residue. The residue was treated with ethanol (95%) (15 mL)
and the
solution cooled to 0 °C overnight. No crystallization had occurred. The
solution was
refluxed and treated sequentially with acetone (5 mL) and water (deionized, 15
mL). The
solution was cooled to room temperature and treated with 5 mg and seeds of
authentic
cyanohydrin. The reaction was cooled to 0 °C and stirred for 1.5 hours.
The resultant solids
were filtered and washed with cold H20-EtOH ( 1: I ) ( 10 mL).
The product was dried at 45 °C at 20 inches of mercury. The elemental
analysis was
satisfactory for CZOH,41V~0~ Theory: C, 70.58; H, 7.11; N, 8.23. Found: C,
70.66; H, 7.03;
N, 8.47. . The NMR in CDC 13 (400 MHz) was in agreement with the assigned
structure
(see below).
400 MHz NMR data for 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-1,4-dicarbonitrile
cyclohexan-1-of
ABSORPTION No. of Multiplicity/ Coupling
protons constant
1.59-1.62 2 singlet, broad
1.75-2.0 6 multiplet, broad
2.1-2.48 (incl 8 multiplet,
2.20 s)
2.90 I singlet O-H
3.85 3 singlet O-CH3
4.80 1 Multiplet (broad) cyclopentyl
7
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
6.85 1 doublet , J = Arom
8 Hz
7.0 2 Multiplet (narrow) Arom
Mass spectral data
(Parent Conditions Assignment Relative
340) Intensity
Mass
Absorption
375 Neg ion/ DCI [m+Cl - 88.5
methane
348 Neg ion/ DCI [375-HCN] ~ 100
methane
339 Neg ion/ DCI [M-H] - 25.75
methane
334 Neg ion/ DCI 17.1
methane
244 Neg ion/ DCI [375 - CSH9 36.9
methane -HCN] -
341 DCI/methane [M+H]+ 19.9
314 DCI/methane [M+H-HCN] + 74.3
287 DCI/methane [M - 2 HCN] 100.0
272 DCI/methane [M+245 + C2H5] 14.84
245 DCI/methane [M -CSH9-HCN]H+36.9
Example 2
Alternative Preparation of 4-f3-(Cyclopentyloxy)-4-methoxyphenyll-1,4-
dicarbonitrile
cyclohexan-1-ol: Sodium Cyanide Method
To a 20 mL round bottom flask equipped with magnetic stirrer and internal
thermometer was added 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-1-
one
(1.00 g, 3.19 mmol) and sodium cyanide (0.325 g, 6.6 mmol) in water (5 mL).
The reaction
was stirred and cooled to 0-5 °C and an aqueous solution of sodium
bisulfite (0.625 g, in 2.5
mL water) as the temperature reached 8 °C. The reaction was cooled and
after 30 min. the
reaction had set up as a solid. The reaction was warmed to 8-10 °C, and
acetone (2 mL) was
added. The reaction was continued at 20 °C for 90 min. The reaction was
partitioned
8
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
between water (5 mL) and ethyl acetate ( 10 mL). The organic layer was washed
with water
(3 x) and brine, and dried with anhydrous MgS04. Evaporation of the ethyl
acetate gave a
solid which was dried in a vacuum oven at 25 °C for 15 hours.
300 MHz NMR data for compound 2:
ABSORPTION IVo of protonsMultiplicity/
Coupling
constant
1.60 4 (include singlet, broad
solvent)
1.75-2.0 6 multiplet, broad
2.1-2.48 (incl 8 multiplet,
2.20 s)
2.96 1 unresolved doubletO-H
3.85 3 singlet O-CH3
4.80 1 Multiplet (broad)cyclopentyl
6.88 1 doublet , J = Arom
7.4 Hz
7.0 2 Multiplet (narrow)Arom
Example 3
Dehydration of 4-(3-(Cyclopentyloxy)-4-methoxyphenyll-1 4-dicarbonitrile
cyclohexan-1
ol
To a 10 mL 3-necked round bottom flask equipped with magnetic stirring, a
thermometer, and reflux condenser was charged with 4-[3-(cyclopentyloxy)-4-
methoxyphenyl]-1,4-dicarbonitrile cyclohexan-1-of (2 in Scheme 1) (0.50 g,
1.46 mmol),
toluene (1.5 mL) and pyridine (0.60 mL). The resulting solution was cooled to
0 °C and
treated with thionyl chloride (0.35 g, 2.94 mmol) in toluene (0.5 mL). A
precipitate formed
after 1-2 min. The reaction was heated to 80 °C and refluxed for an
additional 2.0 hours.
The solution was cooled and poured into a mixture of HCl and ice. The reaction
was
extracted with 2 x 15 mL of ethyl acetate. The organic phase was washed with
0.6 N HCI,
5% sodium carbonate and with brine. The organic layer was dried (MgS04) and
evaporated
to a solid, i.e., compound 3. The NMR in CDC13 was clean and compatible with
that of the
structure 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexene-1,4-
dicarbonitrile, i.e.,
nitrite 3 in Scheme 1.
Elemental analysis: theoretical C~oHZ2N~0~: C, 74.51; H, 6.88; N, 8.69. Found:
C,
74.23; H, 6.98; N, 8.64.
NMR of the unsaturated nitrite in CDC13 showed a diagnostic vinyl C-H at 6.67
ppm.
300 MHz NMR data
9
CA 02397296 2002-07-12
WO 01/51455 PCT/LTSO1/01083
ABSORPTION No of Multiplicity/ Coupling
protons constant
1.55-1.67 3-4 broad overlapping
1.80-2.0 3-4 multiplet, broad
2.05-2.18 I-2 multiplet,
2.2-2.5 2 2 groups of protons
2.6-2.9 2-3 2 groups of protons
3.84 3 singlet O-CH3
4.80 1 Multiplet (broad) cyclopentyl
6.67 1 triplet (broad) vinyl
proton
6.83-6.95 3 six overlapping bandsArom
Example 4
Alternative Method for Dehydrating 4-f3-(Cyclopentyloxy)-4-methoxyphenyl]-I 4
dicarbonitrile cyclohexan-1-of
To a 1000 mL 3-necked round bottom flask equipped with overhead stirring, a
pressure equalizing dropping funnel and a nitrogen inlet was charged crude 4-
[3
(cyclopentyloxy)-4-methoxyphenyl]-1,4-dicarbonitrile cyclohexan-1-of (2) along
with
toluene ( 140 mL) and pyridine (40 mL). A clear solution was produce with
stirring. The
reaction was cooled to 10 °C and thionyl chloride (about 20 mL) was
added at a rate to keep
the reaction < 15° C. The reaction was refluxed and monitored by HPLC
(4.6 x 250 mm
Beckman Ultrasphere; wavelength at 230 nm, flow rate 1.0 mL/min) until
essentially all of
the starting material had disappeared, approximately 2.5 hours.
The reaction was cooled to 40 °C in an ice bath and quenched with 6N
HCl ( 100
mL) followed by addition of ethyl acetate (400 mL). The organic layer was
separated and
washed with two 100 mL portions of 3N HCI, 10% aqueous sodium bicarbonate (
100 mL),
and finally brine ( 100 mL). The organic layer was evaporated to constant
weight and
dissolved in warm isopropanol (50 mL). The solution was cooled to 0 °C
and the solid was
stirred at that temperature for 2 hours, and filtered and washed with cold
isopropanol ( 10
mL). Drying in a vacuum oven at 40 °C for 16 h (10 mmol Hg) gave a
solid, i.e., compound
3. The NMR in CDC 13 was the same as the spectrum of the product prepared in
Example 3.
The melting point was 133.5-134.5 °C. Elemental Analysis of a
recrystallized sample:
Calculated: C, 74.51; H, 6.88; N, 8.69. Found: C, 74.23; H, 6.98; N, 8.64.
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
Example 5
Reduction of 4-f3-(Cyclopentyloxy)-4-methoxyphenyll-1-cyclohexene-1 4-
dicarbonitrile to
Form 4-[3-(Cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-1 4-dicarbonitrile
This example illustrates the preparation of compounds 7 in Scheme 1.
To a 1000 mL 3-necked round bottom flask equipped with magnetic stirring, an
internal thermometer and a reflux condenser was charged with 4-[3-
(cyclopentyloxy)-4-
methoxyphenyl]-I-cyclohexene-1,4-dicarbonitrile, (10.00 g, 31.0 mmol) and
methanol (200
mL). The solution was stirred and heated to 60 °C, at which time a fine
suspension resulted.
The reaction was charged with magnesium turnings (4.02 g, 165 mmol, 5
equivalents)
which had been activated by drying in a vacuum oven at 40-50 °C. The
reaction was
refluxed for 3 h. The reaction mixture was cooled to < 30 °C and
treated with 6N HCI. The
reaction solvent was evaporated on the rotovap (40 °C) to constant
weight and the residue
treated with ethyl acetate (200 mL). The isolated organic layer after
filtration was washed
with water (2 x 50 mL) and brine (2 x 15 mL). The organic layer was
concentrated on a
rotovap to an oil, i.e. 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-
1,4-
dicarbonitrile, compound 7 in Scheme 1. The product was diluted with absolute
ethanol and
used directly in the hydrolysis of the nitrile with potassium hydroxide.
In the same manner as above the unsaturated nitrile (2.53 g,7.80 mmol) was
reduced with Mg(0) ( 0.98 g, 40.3 mmol, 5 eq) in methanol (50 mL) at 55
°C. After
refluxing for 3 h, the product was isolate as an oil. this oil was used in the
hydrolysis
without further purification.
Example 6
Base hydrolysis of 4-f3-(Cyclopent~y)-4-methoxyphenyll-1-~clohexane-1 4-
dicarbonitrile
4-[3-(Cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-1,4-dicarbonitrile (7)
(2.120 g, 6.53 mmol) and ethanol ( 10 mL) were charged to a 100 mL 3-necked
round
bottom flask equipped with magnetic stirring, an internal thermometer and a
reflux
condenser. The solution was stirred and refluxed until clear. After removing
heat the
reaction was charged dropwise with a solution of KOH in water (3.4 g, 60.6
mmol in 10 mL
water). This solution was then refluxed for 4 h. The reaction mixture was
reduced to one-
half volume on the rotovap and partitioned between ethyl acetate (50 mL) and 6
N HCl (12
mL). The organic layer was separated and washed with deionized water (2 x 15
mL), brine
( I x 15 mL), and evaporated to give a mixture of cis and traps isomers of 4-
cyano-4-[3-
(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-1-carboxylic acid as an oil.
11
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
Example 7
Conversion of cis-4-(Aminocarbonyl)-4-f3-Lyclopentyloxy)-4-
methoxyphenyllcyclohexanecarboxylic acid to cis-4-C~no-4-(3-cyclopent~y-4-
methoxyphenyl)-r-cyclohexanecarboxylic acid
To a 5 mL 1-necked round bottom flask equipped with a magnetic stirrer, a
nitrogen
inlet, and an oil bath was charged the cis-4-(aminocarbonyl)-4-[3-
(cyclopentyloxy)-4-
methoxyphenyl]cyclohexanecarboxylic acid (0.050 g, 0.138 mmol) and toluene
(0.50 mL).
The flask was heated to 70 °C and the suspension treated with thionyl
chloride (0.25 mL,
0.408 g, 3.43 mm, 25 eq) added in a single portion. A clear yellow solution
was produced.
The reaction was heated a total of 4 h at bath temperature 75 °C. The
reaction was cooled to
10 °C and a small aliquot was evaporated with nitrogen and examined by
HPLC. The
HPLC showed complete disappearance of the acid-amide and clean conversion to a
mixture
of the cis and traps isomers of 4-cyano-4-[3-(cyclopentyloxy)-4-methoxyphenyl]-
1-
cyclohexane-1-carboxylic acid.
Example 8
Hydrolysis of 4-f3-(Cyclopentyloxy)-4-methoxyphenyll-1-cyclohexane-1 4-
dicarbonitrile
To a 50 mL 3-necked round bottom flask equipped with magnetic stirrer, an
internal
thermometer, and a reflux condenser was charged equal amounts of cis saturated
nitrile
(compound 7 in Scheme 1, 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-
1,4
dicarbonitrile) (0.5016 g, 1.54 mmol) and the corresponding traps
dicarbonitrile (0.5016 g,
1.54 mmol) (total 3.08 mmol) and absolute ethanol (10 mL). The reaction was
heated to
reflux for 5 min, to obtain a solution. The reaction was treated slowly with a
solution of
NaOH ( 1.16 g, 29 mmol, 9.4 eq) in deionized water ( 10 mL). The rate of
addition was
adjusted to avoid precipitation of the starting nitriles. The solution was
stirred and refluxed
for 4-5 h. At that time the HPLC showed (80% PAR) of the desired product a
cis/trans
mixture of acids and a side product identified as an acid-amide, that is, the
4-position nitrile
had converted to -CONH2. The solvent was evaporated to give an oil which was
treated
with 6 N HCl ( 10 mL) and ethyl acetate (35 mL). The layers were separated and
the organic
layer washed with water (2 x 10 mL) and brine (1 x 5 mL) and dried (magnesium
sulfate).
The product (compound 5 and its 4-position -CONH2 analog cis-4-(aminocarbonyl)-
4-[3-
(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-1-carboxylic acid) was
concentrated by
evaporation at reduced pressure.
12
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
This crude product was placed in a 50 mL, 3-neck flask equipped with a
magnetic
stirrer, nitrogen inlet, and a thermometer. Toluene (9.0 mL) was added and
then thionyl
chloride ( 1.0 mL, I .6 g) and the reaction heated at 70-75 °C. The
reaction was judged to be
complete after 3 h. The reaction was evaporated on the rotovap to 4 mL, and
transferred to
a separatory funned with ethyl acetate (30 mL). The top layer was washed with
water (2 x
5.5 mL), 3 N HCl ( 1 x 5 mL), water ( 1 x 5 mL) and brine ( I x 5 mL) before
drying with
magnesium sulfate. The resulting organic layer was evaporated to constant
weight ( 1.15 g)
and dissolved in boiling ethyl acetate and treated with hexane. The product
was further
treated by using a base-acid treatment of the combined aqueous layers,
followed by
extraction into ethyl acetate. Evaporating the ethyl acetate gave a residue
which was
dissolved then re-dissolved in ethyl acetate (5 mL) and hexane (4 mL).
Product, a mixture
of cis and traps 4-cyanocyclohexanoic acid, was crystallized from this solvent
system.
Example 9
Alternative Process for Hydrolyzina 4-f3-(Cyclopentyloxy)-4-methoxyphenyll-1-
cyclohexene-1,4-dicarbonitrile
To a 100 mL 3-necked round bottom flask equipped with magnetic stirrer, an
internal thermometer, and a reflux condenser was charged the cisltrans
saturated nitrites
(compound 7, 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexene-1,4-
dicarbonitrite)
(2.12 g, 6.54 mmol) and absolute ethanol ( 10 mL). The reaction was heated to
reflux; a
solution was obtained. The reaction was treated slowly with a solution of KOH
(3.4 g, 60.6
mmol, 9.3 eq) in deionized water (10 mL). The addition was done dropwise over
10 min..
The solution was stirred and refluxed for 4-5 h. At that time the HPLC showed
the absence
of starting material and the presence of 4-cyano-4-[3-(cyclopentyloxy)-4-
methoxyphenyl]-
1-cyclohexane-I-carboxylic acid and the side product, the 4-carboxamide 8 (15
% PAR).
The reaction was evaporated to 1/2 volume and treated with 6 N HCl (12 mL) and
ethyl
acetate (50 mL). The layers were separated and the organic layer was washed
with water (2
x 15 mL) and brine ( 1 x 10 mL) and dried (magnesium sulfate). A crude product
was
obtained by evaporation at reduced pressure.
The crude product obtained as per the previous paragraph was placed in a 100
mL
3-neck flask equipped with a magnetic stirrer, distillation head, and a
thermometer. Toluene
( 15.0 mL) was added and then distilled to a volume of 6 mL. Then fresh
toluene ( 15 mL)
and thionyl chloride (2.0 mL, 3.2 g) was added and the reaction heated at 70-
75 °C. After
1.5 h some unreacted starting material had not gone into solution, so the
reaction was
charged with thionyl chloride ( 1 mL) and toluene (5 mL). After 4 h more the
reaction was
13
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
judged to be complete (HPLC assay). The mixture was filtered and the filtrate
evaporated
to constant weight.
The filtrate (2.71 g) from the preceding paragraph was charged to a 100 mL
round
bottom flask equipped with a magnetic stirrer along with anhydrous
tetrahydrofuran (15
mL). To this well stirred solution was added 20% aqueous NaOH (3 mL) after
which the
mixture was stirred at room temperature for 1 h. The resulting reaction was
concentrated on
the rotovap, treated with ethyl acetate (50 mL) and 6 N HCl ( 10 mL) and the
layers
separated. The organic layer was isolated and washed with water (2 x 10 mL)
brine (lx 5
mL) and dried over magnesium sulfate. Removal of the volatiles left a thick
oil which was
dried at 50 °C for 16 h at 20 mm of Hg. The resulting semisolid (2.00
g) was dissolved in
boiling ethyl acetate ( 10 mL) and then hot hexane (8 mL). The reaction was
slowly cooled
to -3 °C and stirred for 2 h. A thick solid was present. The solid was
collected and washed
with a cold ethyl acetate/hexane mixture. After drying in a vacuum oven at 50
°C ( 1 mm of
Hg), a white solid was obtained (compound 5 in Scheme 1).
300 MHz NMR data: c-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)-r-
cyclohexanecarboxylic acid in CDC13
ABSORPTION No of protonsMultiplicity/ Coupling
constant
1.64-1.7 2 singlet, broad
1.86-1.98 9-10 multiplet, broad
2.28 4 triplet (distorted)
2.45 1 multiplet
3.89 3 singlet O-CH3
4.84 1 septuplet cyclopentyl
6.89 1 doublet , J = 8.4 Arom
Hz
7.01, 7.02, 7.041,2 Multiplet (narrow) Arom
7.048
Carbon 13 NMR (90 MHz, CDC13): 180.22, 149.10, 147.84, 132.78, 122.15, 117.34,
112.97, 111.96, 80.74, (solvent 76 t), 56.07, 42.98, 41.63, 36.44, 32.77,
25.88, 24.10.
[Italicized signals are C-H's and C-H3's].
Alternate synthesis
Example A
Hydrolysis of 4-f3-(Cyclopentyloxy)-4-methoxyphenyll-1-cyclohexene-I 4-
dicarbonitrile
14
CA 02397296 2002-07-12
WO 01/51455 PCT/USO1/01083
To a 100 mL 3-necked round bottom flask equipped with a magnetic stirrer, a
reflux
condenser, and a nitrogen inlet was charged with 4-[3-(cyclopentyloxy)-4-
methoxyphenyl]-
I-cyclohexene-1,4-dicarbonitrile nitrile, compound 3 in Scheme 1, (2.00 g,
6.16 mmol) and
absolute ethanol (25 mL). The reaction was refluxed for 5 minutes; a clear
solution
resulted. To this was added a suspension of Ba(OH)2~8H20 in water (6.00 g, 19
mmol, in
25 mL of distilled water). The solution was refluxed for 3.5 hrs and cooled to
room
temperature. The reaction was acidified with 3 N HCl and extracted with ethyl
acetate ( 1 x)
and t-butylmethyl ether ( 1 x). The organic layers were combined and washed
with water (2
x 15 mL) and brine. After drying with magnesium sulfate the unsaturated acid 4-
cyano-4-
[3-(cyclopentyloxy)-4-methoxyphenyl)-1-cyclohexene-1-carboxylic acid (compound
5 in
Scheme 1) was isolated as an oil by evaporation of the solvents on the
rotovap. This oil also
contained an impurity identified as the unsaturated acid with a 4-carboxamide
(formed by
concomitant hydrolyis of the 4-position nitrile). The product was purified by
preparative
reverse phase liquid chromatography. The product, compound 4 in Scheme 1,
weighed
0.450 g. The unsaturated acid with a 4-carboxamide group was isolated and
identified as
indicated below.
IR FT-IR (KBr): 3300 (s, O-H st), 2235 (CN stretch), 1689 (C=O stretch of
unsat
carboxylic acid); 1650 (C=C stretch), 1517 (C=C stretch), 1434, 1419, (C-H
deform), 1258
and 1145 (C-O stretch).
The NMR (360 mHz) had a signal diagnostic for the vinyl proton at 7.0 ppm
(C6D6) and 7.16 ppm (CDC13).
Proton NMR (360 MHz) c-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)-r-
cyclohexanecarboxylic acid taken in C6D6.
ABSORPTION No of protonsMultiplicity/ Coupling
constant
1.41-2.09 10 4 clusters (broad)
2.15-2.27 1.5 doublet allylic protons
2.38-2.52 I .5 doublet allylic protons
2.80 1 Very broad allylic proton
3.50 3 singlet O-CH3
4.72 1 septuplet cyclopentyl
6.63 1 doublet , J = 8.4 arom (a to
Hz OCH3)
6.82 1 doublet of doublets, arom
(J = 8.4 +
meta coupling, J =
2)
7.0 1 broad singlet vinyl proton
CA 02397296 2002-07-12
WO 01/51455 PCT/USOI/01083
7.1 1 doublet (meta, J= 2) Isolated aryl C-H
C-13 NMR of unsaturated carboxylic acid (CHCL3, 90 HZ). ): 170.7, 149.9,
147.9,
137.36, 131.5, 129.7, 122.3, 117.5, 112.7, 111.9, 80.69, 56.1, 39.7, 33.0,
32.8, 32.6, 24.1,
22.25.
Example B
Transfer hydrogenation of 4-Cyano-4-f3-(c~pentyloxy)-4-methoxyphenyl]-1-
cyclohexene-1-carboxylic acid (4) to c-4-Cyano-4-(3-cyclopentyloxy-4-
methoxyphen~)-r-
cyclohexanecarboxylic acid (5)
To a 5 mL 2-necked round bottom flask equipped with a magnetic stirrer and a
nitrogen inlet was charged 4-cyano-4-[3-(cyclopentyloxy)-4-methoxyphenyl]-1-
IO cyclohexene-1-carboxylic acid (4 in Scheme 1) ( 0.021 g, 0.06 mmol) and
dimethyl
formamide (DMF) (0.5 mL). Ammonium formate (0.050 g) was added to the pot and
hydrogenation catalyst ( 10% Pd/C; Aldrich 20569-9). The reaction was stirred
at 20 °C for
20 h. The reaction was filtered and the dimethyl formamide removed under high
vacuum.
The product was extracted into ethyl acetate (2 x 4 ml). The organic layers
were washed
with water (2 x 4 mL) and brine (I x 5 mL). Evaporation gave an oil which had
an NMR
the same as reference material of the cis form of the desired acid, 4-cyano-4-
[3-
(cyclopentyloxy)-4-methoxyphenyl]-1-cyclohexane-I-carboxylic acid, 5 in Scheme
1.
I6