Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
2-PHENYLCARBAMOYL-BENZIMIDAZOLES
"Cross Reference to Related Application"
This application claims the benefit of U.S. Provisional Application Serial No.
60/181,236, 7946P, filed on February 9, 2000.
FIELD OF THE INVENTION
The subject invention relates to 2-phenyl-carbamoylbenzimidazoles useful for
treatment or prevention of ischemic reperfusion injury of myocardial and other
tissue and
other cardiovascular and inflammatory diseases and disorders.
SUMMARY OF THE INVENTION
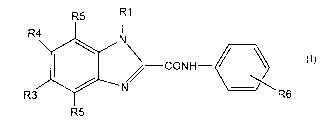
The subject invention includes compounds having the structure:
R5 11
R' V
-CONH- ~ ~ ~t~
R~ R6
R5
wherein:
(a) R1 is selected from alkyl, aryl, alkoxy, and aryloxy;
(b) R3 and R4 are independently selected from hydrogen, halo, alkyl, alkoxy,
alkylthio, and mono-or dialkylamino; except that R3 and R4 are not both
hydrogen;
(c) each RS is independently selected from hydrogen, halo, cyano, alkyl,
hydroxy,
alkoxy, thio, alkylthio, amino and mono- or dialkylamino;
(d) each R6 is independently selected from hydrogen, halo, nitro, cyano,
alkyl,
aryl, heterocyclyl, hydroxy, alkoxy,' aryloxy, thio, alkylthio, arylthio,
amino,
alkylamino, arylamino, acyl, alkylacyl, arylacyl, amido, alkylamido,
arylamido, sulfonyl, alkylsulfonyl, arylsulfonyl, phosphonyl, alkylphosphonyl,
arylphosphonyl, carboxy and its alkyl and aryl esters;
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
an optical isomer, diastereomer, or enantiomer or mixture thereof; a
pharmaceutically-
acceptable salt, hydrate, or biohydrolyzable ester, amide or imide thereof;
pharmaceutical
compositions containing such compounds; and methods of using such compounds
for
treating or preventing reperfusion injuries~to tissues.
DETAILED DESCRIPTION OF THE INVENTION
There are about 1.1 million myocardial infarctions in the United States each
year
and 350,000 people die. Myocardial infarction (MI), or heart attack, occurs
when the
blood vessels supplying the heart become completely or partially occluded.
Treatment
with thrombolytic agents to restore blood flow (reperfusion) is first line
treatment in
many cases. However, the benefit of reperfusion is compromised by the acute
inflammatory response associated with it, resulting in a syndrome called
reperfusion
injury.
Inflammation generally serves a protective role. For example, at sites of
bacterial
infection, bacterial endotoxins induce the production of inflammatory
cytokines which
recruit circulating leukocytes, including neutrophils and monocytes, to
destroy the
bacteria. Once the infection is cleared, the inflammation subsides. However,
there are
conditions where the inflammatory signal is sustained (rheumatoid arthritis)
or is
unnecessarily severe (ischemia-reperfusion injury).
An essential feature of inflammation is the migration of neutrophils (PMNs)
from
blood into tissues. This migration is preceded by a cascade of events mediated
by
adhesion molecules. The adherence of PMNs to vascular endothelial cells
requires the
interaction of adhesion molecules on the surface of both cell types. These
molecules
belong to three distinct families: the selectins, the integrins and the
immunoglobulin
superfamily. Neutrophils first roll along endothelial cells, a process
mediated by the
selectins. At sites of inflammation, firm adherence is mediated by the
interaction of [32
integrins on PMNs and ICAM-1 (intercellular adhesion molecule-1) expressed on
the
endothelial cells. Finally, transendothelial migration of PMNs into tissues
leads to tissue
damage. Compounds able to block the adhesion of neutrophils to endothelium
would be
useful in the treatment of a variety of conditions involving, ischemia-
reperfusion injury
including, but not restricted to, myocardial infarction, coronary artery
bypass grafting,
2
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
angioplasty, angina, stroke, peripheral vascular disease, inflammatory bowel
disease,
ulcerative colitis, burns, frostbite, adult respiratory distress syndrome,
asthma, tissue and
organ transplants, general surgery, replantation, acute renal failure,
rheumatoid arthritis,
psoriasis, hepatitis, pancreatitis, sunburn, radiation, ulcer, and shock. (For
recent reviews
see: C. Cornejo, J. Harlan, R. Winn, in Adhesion Molecules in Health &
Disease, L. Paul
and T. Issekutz, Eds., Marcel Dekker, 1997, Chapter 18; J. Prince, C.
Ballantyne,
Emerging Therapeutic Targets, 1999, 263-277.)
Glossary of Terms
Unless otherwise specified, the following terms have the indicated meanings
when
used in this application.
The term "alkyl" means a hydrocarbon chain which is linear, branched or
cyclic,
saturated or unsaturated (but not aromatic), substituted or unsubstituted. The
term may be
used alone or as part of another word where it may be shortened to "alk"
(e.g., in alkoxy,
alkylamino). Preferred linear alkyl have from 1 to about 20 carbon atoms, more
preferably from 1 to about 8 carbon atoms, more preferably still from 1 to
about 4 carbon
atoms; most preferred are methyl or ethyl. Preferred cyclic and branched alkyl
have from
3 to about 20 carbon atoms, more preferably from 3 to about 10 carbon atoms,
more
preferably still from 3 to about 6 carbon atoms: Preferred cyclic alkyl have
one
hydrocarbon ring, but may have two, three, or more, fused or spirocyclic
hydrocarbon
rings. Alkyl may be unsaturated only with one or more double bonds ("alkenyl")
(no
triple bonds), preferably with one, two, or three double bonds, more
preferably with one
double bond. Alkyl may be unsaturated with one or more triple bonds
("alkynyl"),
preferably with one triple bond. More preferred alkyl are saturated
("alkanyl"). The term
"alkylene" means an alkyl which is attached to 2 or more moieties. Preferred
substituents
of alkyl include alkyl, aryl, halo, hydroxy, alkoxy, aryloxy, amino,
alkylamino,
arylamino, thio, alkylthio, arylthio, acyl, alkylacyl, arylacyl, carboxy,
alkylester, arylester,
amino, alkylamino, arylamino, sulfonyl, alkylsulfonyl, arylsulfonyl, nitro,
cyano,
heterocycle. Preferred alkyl are unsubstituted.
The term "aryl" means an aromatic hydrocarbon ring which is substituted or
unsubstituted. The term may be used alone or as part of another word (e.g., in
aryloxy,
arylamino). Preferred aryl have from 6 to about 14 carbon atoms in the
aromatic ring(s),
3
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
and a total of from about 6 to about 20, preferably to about 12, carbon atoms.
Preferred
aryl is phenyl or naphthyl; most preferred is phenyl. The term "arylene" means
an aryl
which is attached to two or more other moieties. Preferred substituents of
aryl include
alkyl, aryl, halo, hydroxy, alkoxy, aryloxy, amino, alkylamino, arylamino,
thio, alkylthio,
arylthio, acyl, alkylacyl, arylacyl, carboxy, alkylester, arylester, amino,
alkylamino,
arylamino, sulfonyl, alkylsulfonyl, arylsulfonyl, nitro, cyano, heterocycle.
More preferred
aryl are unsubstituted.
The term "heteroatom "means a nitrogen, oxygen, or sulfur atom.
The term "heterocycle" or "heterocyclyl" means a cyclic alkyl or aryl with one
or
more heteroatoms substituted for carbon atoms in the ring(s), preferably 1, 2
or 3
heteroatoms in the ring(s). Preferred heterocycle substituents are the same as
for alkyl.
The term "heteroaryl" refers to the subset of heterocycele which comprise an
aromatic
ring. Preferred heteroaryl have from 5 to about 14, more preferably to about
10, more
preferably still 5 or 6, carbon plus heteroatoms in the ring(s), and a total
of from 5 to
about 20, more preferably to about 12, carbon plus heteroatoms.
The term "safe and effective amount" means an amount of a pharmacologically
active compound sufficient to significantly induce a positive modification in
the condition
to be treated, but low enough to avoid serious side effects (at a reasonable
benefit/risk
ratio), within the scope of sound medical judgment. A safe and effective
amount of a
compound will vary with the particular condition being treated, the size and
age and
physical condition of the patient, the severity of condition, the duration of
the treatment,
the nature of concurrent therapy, the particular pharmaceutically-acceptable
carrier
utilized, and like factors within the knowledge and expertise of the attending
physician.
The term "pharmaceutically-acceptable carrier" or "pharmaceutically-acceptable
excipients" means one or more compatible solid or liquid excipients which are
suitable
for administration to a human or lower animal. The term "compatible" means
that the
excipients are capable of being commingled with the pharmacologically active
compound
or compounds, and with each other, in a manner such that there is no
interaction which
substantially reduces the pharmaceutical efficacy of the composition under
ordinary use
situations. The excipients do not have substantial pharmacological activity
themselves,
but may function, for example, as diluents, lubricants, disintegration
enhancers,
4
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
dissolution enhancers, encapsulating materials, preservatives, colorants,
flavorants, and
the like. Pharmaceutical-acceptable excipients must be of sufficiently high
purity and
sufficiently low toxicity to render them suitable for administration to the
human or lower
animal being treated.
The term "unit dosage form" means a composition comprising an amount of a
pharmacologically active compound that is suitable for administration to a
human or a
lower animal subject in a single dose, according to good medical practice.
A "biohydrolyzable ester" is an ester of a carboxylic acid containing 2-
phenylcarbamoyl-benzimidazole of the present invention that does not interfere
with the
activity of the present compounds or that is readily converted by an animal to
yield an
active phenylcarbamoyl-benzimidazole. Such esters include lower alkyl esters,
lower
acyloxy-alkyl esters (such as acetoxymethyl, acetoxyethyl,
aminocarbonyloxymethyl,
pivaloyloxymethyl and pivaloyloxyethyl esters), lactonyl esters (such as
phthalidyl and
thiophthalidyl esters), lower alkoxyacyloxyalkyl esters (such as
methoxycarbonyloxymethyl, ethoxycarbonyloxyethyl and
isopropoxycarbonyloxyethyl
esters), alkoxyalkyl esters, choline esters and alkyl acylamino alkyl esters
(such as
acetamidomethyl esters).
The Compounds
The subject invention involves 2-phenyl-carbamoylbenzimidazoles compounds
having the structure:
R5 11
R' V
\ I
-CONH
R~ R6
R5
In structure (I), R1 is selected from the group consisting of alkyl, aryl,
alkoxy, and
aryloxy. The alkyl and aryl portions of preferred R1 moieties have from 1 to
about 14
carbon atoms.
More preferred R1 is selected from unsubstituted or substituted alkyl having
from 1
to about 12 carbons atoms, and unsubstituted or substituted phenyl or
naphthyl.
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
More preferred alkyl R~ include unsubstituted or substituted linear alkyl
having
from about 2 to about 8 carbon atoms, more preferably still from about 3 to
about 6
carbon atoms; still more preferred is n-propyl or n-butyl or n-pentyl. More
preferred R1
include branched alkyl having from about 3 to about 8 carbon atoms, more
preferably still
from about 3 to about 6 carbon atoms; still more preferred is isobutyl or
isopentyl. More
preferred R1 include cyclic alkyl having from 3 to about 8 carbon atoms, more
preferably
still from 3 to about 6 carbon atoms. Preferred substituents for such linear,
branched or
cyclic alkyl include halo, hydroxy, alkoxy, amino, mono- and dialkylamino,
thio,
alkylthio, aryl (especially phenyl), and heterocycle; more preferred is such
alkyl being
unsubstituted. Preferred R1 which are linear, branched or cyclic alkyl are
saturated or
unsaturated with one or more double bonds; more preferred are saturated.
More preferred aryl R1 include unsubstituted or substituted phenyl. Preferred
substituents for such phenyl include halo, hydroxy, alkoxy, amino, mono- and
dialkylamino; also preferred is for such phenyl being unsubstituted.
More preferred aralkyl R1 include unsubstituted or substituted benzyl.
Preferred
substituents for such benzyl include halo, hydroxy, alkoxy, amino, mono- and
dialkylamino; also preferred is for such benzyl being unsubstituted.
In structure (I), R3 and R4 are independently selected from the group
consisting of
hydrogen, halo, alkyl, alkoxy, alkylthio, and mono- or dialkylamino, except
that R3 and
R4 are not both hydrogen. The alkyl portions of preferred R3 and R4 moieties
have from
1 to about 8 carbon atoms.
Preferred R3 and R4 include hydrogen, alkoxy having from 1 to about 6,
preferably
to about 3, carbon atoms; alkylthio having from 1 to about 6, preferably to
about 3, carbon
atoms; monoalkylamino or dialkylamino each alkyl having from 1 to about 6,
preferably
to about 3, carbon atoms; and alkyl having from 1 to about 6, preferably to
about 3,
carbon atoms. Preferred substituents on the alkyl of such moieties include
halo, hydroxy,
alkoxy, amino, mono- or dialkylamino, thio, alkythio; more preferred is for
such R3 and
R4 moieties to be unsubstituted. More preferred still is for at least one of
R3 and R4, to
be ethoxy or especially methoxy. Preferably one of R3 and R4 is hydrogen; more
preferably R3 is hydrogen.
6
08-03-2002 . US0104212
CA 02399799 2002-08-09
MRR-08-2902 14 : 43 f 8~C, HC PRTENT D I ~ . 513 622 ~e10 P . ~0~r
1n stnictiue (1), RS denotes moietias at positions 4 and 7 of the
benzimidaZOle clng5. Each RS is
independently selected from the group consisting of hydrogen, halo, cyano,
alkyl, hydroxy,
alkoxy, thin, alkyithio, amino, and mono- or dialkylamino. Ths allyl portions
of preferred R5
moieties have from 1 to about 8 carbon atoms. .
Pn:frr<ed R5 include hydroEen, halo, alkyl having from 1 to about 6,
preferably to about 3.
carbon atoms, alkoxy having from 1 to about 6, preferably about 3, carbon
atoms, monoatkyl- or
dialkylamino each alkyl having from 1 to about 6, prefernbly to about 3,
carbon atoms, and alkylthio
having from 1 to about 6, preferably to about 3, catbon atoms. Preferred
substituettts on the alkyl of
such RS moieties include alkoxy, amino, and alkyl; mere preFerrcd is for the
alkyl of such moieties to
be unsubstituted.
More preferred is for each RS to be independet>tty selected from hy~ogerf,
halo, and
unsubstitutcd alkyl having from 1 to about 3 carbon atoms. Morc preferred is
for no more than
one R5 being other than hydrogen. Most preferred is both RS being hydrogen.
In Structure (I), each R6 is independently 5elccbed from the group consisting
of hydrogen,
halo, vitro, cyano, alkyl aryl. heterocyclyl, hydroxy, alkoxy, aryloxy, thin,
alkylthio, arylthio,
amino, alkylatnino, arylamino, aryl, alkylacyl, arylacyl, amide, ~alkylarnido,
arylamido, sulfonyl,
alkylsulfonyl, arylsulfonyl, phosphonyl, alkylphosphonyl, arylphosphonyl,
carboxy and its alkyl
and aryl esters. Preferred is each R6 being selected from hydrogen, halo,
vitro, about C 1-C~
alkyl, phenyl, hydroxy, about Cl-C4 alkoxy, thin, about C1-C4 alkylthio,
amino, abourt CI-C4
mono. or dialkylamino. More preferred is each R6 being selected from hydrogen,
fluoro, chlom,
vitro, methyl, ethyl, triflummethyl, hydroxy, methoxy, ethmcy,
trifluoromethoxy. Prefrrred is for
no more titan three, mute preferably no mots than twa, more preferably still
no more than one, R6
being other than hydrogen.
Prefemd compounds of the subject invention include the following examples
having
structure (I) and the indicated substituents:
Eza a ~ R3 R4 RS ~6
1 isoburyl H -OCH3 both H H
2 isobutyl H -OCH, both H p-F
3 isobutyl H -OCH3 both H -o-OCHg
7
AMENDED SHEET
~m~~ ,~;+~n~m~mnnn ~n~~~ ~.-.,~ ..,. ~~io n nnn
08-03-2002 US0104212
CA 02399799 2002-08-09
MRR-~-~ 14 : 43 FAG HC PfaTENT D I V . S 13 6c2 X00 F . 0507
4 isobucyl H -OCHa bath H -a-F
isvbutyl H -OCHa both H ~m-OCH3
6 isobutyl H -OCH3 both H -p-OCH3
isobutyl H -OCH3 both H p-NOZ
8 isobutyl H -0CH3 both H -o-0CF3
Subject invention compounds iaclude all active optical isomers, diasteroomers
and
enantiomers, and mixtures thereof. of the above compounds. Subject invention
compounds
include pharmaceutically-acceptable salts, hydrates, and biohydmlirable
esters, amides. and
imides of such compounds.
Synthesis of Compotmds
' The following provides goneral schemes for making subject invention
eompauads, and
specific methods for synthesizing p~rfemd subject invention compounds. Unless
otherwise
stated, all commercially available reagerts are used without futtttet
purification. Reactions are
genccally run under an inert atmosphere (argon or nitrogen). Brine refers to
sanrrated aqueous
sodium chloride. Residual solvent is removed under vacuum ca. 0.03 mm H at mo
( g) m
temperature (rt).
Structures of the compounds synthesised are confirm! ping the following
analytical
tools. Proton NMR spectra ere taken on a GE QE-300 (300 MI~z) spectrometer, a
Bniker AC-
304 (380 MHz) quad-nuclei probe system or on a Varian Unityplus (300 MHz). All
chemical
i shifts are reported in b scale as parts per million (ppm) dowrrfsled from
(CH3)4Si. Spectra taken
' in CDCl3 are referenced either to (CH3)4Si or to a residual CHCl3 (7.24
ppm). Spectra taken in
D20 are referenced to HOD (4.80 ppm), those is (C03)2C0 are referenced to
residual
(CH3)ZCO (2.04 ppm), those in CD30D ere referenced to residual CH30H (3.30
ppm), and
those in (CD3)ZSO are referenced to residual (CH3)2S0 (2.49 ppm). Carbon-13
spectra are
taken on a GE QE-300 (75 MHz) speeuometa or a Bniker AC-300 (75 MHz) quad-
nuclei probe
system. Spectra taken in CDCI3 are referenced to solvent (78 ppm). those in
CD30D are
referenced to solvent (49 ppm), those in (CD3)ZSO are referenced to solvent
(39.7 ppm). arid
those in (CD3yZC0 are refe~tced to solvent (206_5, 29.8 ppm). Mass spectra are
determined on
a Fision's Trio 2000 equipped with a robotic probe or a Fisons Platform II
Mass Spectrometer.
Chemical ionization spectra are obtained using methane and/or
8
AMENDED SHEET
Fmflf tea;+~flplfl'-eJ~?ftfl~ ~?f1~~~ ~.~~~ ~,. ~~io ~ nn~
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
ammonia as a reagent gas. ESI compound introduction is via Hewlett Packard
1050
HPLC autosampler using methanol, 0.2% formic acid, and 0.2 mM ammonium acetate
as
the eluting solvent. Thin layer chromatography is performed on silica gel 60-
F254
precoated plates. Flash chromatography is performed using silica gel 60
(Merck, 230-400
mesh). Melting points are obtained with an Electrothermal 1A9200 or a MelTemp
II
capillary melting point apparatus and are uncorrected.
Scheme I is a general scheme useful for synthesizing many subject invention
compounds:
Scheme I
R5
R~
a b
R
K5 R5
A
R5 R5
R~
R
R5 R5
B
9
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
R5 R5 ,R1
R4 \ NHR1 R ~4
N f
R3 / 02 R3 ~ N
R5 R5
C D
R5 ,R1
R /4
N
CONH
N ~ R6
R3
R5
E
a: NaN02, propionic acid; b: HN03; c: Tf20/Et3N, toluene; d: R1-NH2; e: HCOOH,
10% Pd/C; f n-BuLi,THF, R6-PhNCO.
5-Methoxy-2-nitrophenol (B from Scheme I with R4 = methoxy and R3 and both
RS = H): To a 2 liter round bottom flask, fit with a mechanical stirrer and an
additional
funnel, add propionic acid (300 mL) and 3-methoxyphenol (37.2 g, 0.3 mol). The
resulting mixture is cooled to 0°C and a solution of sodium nitrite (21
g, 0.304 mol) in
water (50 ml) is slowly added. After stirring for 1 hr at 0°C, fuming
nitric acid (40 mL) is
slowly added. The resulting slurry is stirred at 0°C for 1 hour and
then warmed to room
temperature over 2 hours. Water (250 mL) is added dropwise at room
temperature, and
the resulting solid is filtered and washed with 300 mL 50% aqueous propionic
acid to
provide, after drying, 5-methoxy-2-nitrophenol as a tan solid.
N-Alkyl-5-methoxy-2-nitroaniline (C from Scheme I with R1 = alkyl, R4=
methoxy, and R3 and both RS = H): To a one-liter round bottom flask, toluene
(300 mL),
S-methoxy-2-nitrophenol (5.0 g., 0.03 mol), and triethylamine (6.68 g, 0.066
mol) are
added. The resulting solution is then cooled to 0°C and triflic
anhydride (Tf20) is slowly
added via syringe (9.3g, 0.033 mol). The reaction mixture is stirred at
0°C for 5 minutes;
the amine (R1-NH2) (0.12 mol) is added and the reaction mixture is heated to
reflux for
5.5 hrs. After cooling to room temperature, the reaction content is filtered
through a plug
of silica gel (eluted with 90:10 hexane:ethyl acetate) and concentrated via
rotary
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
evaporation to provide crude N-alkyl-5-methoxy-2-nitroaniline. The material is
used as is
in the next synthetic step.
N1-Alkyl-6-methoxybenzimidazole (D from Scheme I with R1 = alkyl, R4 =
methoxy, and R3 and both RS = H): To a 250 mL round bottom flask is added 88%
formic acid (50 .mL) and N-alkyl-5-methoxy-2-nitroaniline (0.02 mol). To this
homogeneous mixture is added an ethyl acetate slurry of 10% Pd-C (600 mg). The
resulting heterogeneous reaction mixture is heated to 100°C for one
hour, cooled to room
temperature, and filtered through Celite (elute with water). The filtrate is
then made basic
with the addition of 28% NH40H and then washed with ethyl acetate (3x 100 mL).
The
combined organics are dried (MgS04), filtered, and concentrated via rotary
evaporation
to give a brown residue. The residue is chromatographed (Si02, 50:50
hexane:ethyl
acetate) to provide N1-alkyl-6-methoxybenzimidazole.
Alkyl 6-methoxy-N-(R6-Ph)benzimidazole-2-carboxamides (E from Scheme I
with R1 = alkyl, R4 = methoxy, and R3 and both RS = H): To a 50 mL round
bottom
flask under Ar is added 1-alkyl-6-methoxybenzimidazole (1.0 equiv., 0.98 mmol)
and
anhydrous THF (10 mL). The solution is cooled to -78°C, n-butyl lithium
(1.4 equiv.,
1.37 mmol) is added dropwise and the resulting mixture is stirred at -
78°C for 30 minutes.
Neat R6-phenylisocyanate is then added via syringe. The mixture is then
stirred at -78°C
for 10 minutes, then warmed to room temperature and stirred for an additional
15
minutes. Saturated sodium bicarbonate is then added (20 mL), followed by the
addition
of water (20 mL). The resulting mixture is extracted with ethyl acetate (3x 50
mL). The
combined are washed with brine (1 x 100 mL), dried (MgS04), filtered and
concentrated
to give crude benzamide. This residue is then chromatographed (hexane:ethyl
acetate) to
give pure N-1-alkyl-6-methoxybenzimidazole-2-benzamide.
Scheme II is another general scheme useful for synthesizing many subject
invention compounds.
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Scheme II
R5 R5
R4 N02 h R4 ~ NOZ
\ I
N ~CN
R3 NH2 R3
R5 R5
G H
R4 R5 ~H k
\ /~ CN
N
R3
R5
J
R5 O_R2 R5 O-R2
R4 \ N m R4 \ N
I ~>-CN --~ I i~CONH-Ar
N ~ N
R3 R3
R5 R5
K L
h: (CH20)n, KCN, HOAc, ZnCl2; j: EtOH, KOH; k: R2-OH, DEAD, Ph3P, THF;
m: ArOH, H+; or ArNH2 H20, H2Ru(PPh3)4~
Scheme III is another general scheme useful for synthesizing many subject
invention compounds:
12
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Scheme III
R5 R5
HZN ~ F n H2N ~ F
P
R3 I / R3 I / NOZ
R5 R5
M N
R5 R5 R1
BocHN ~ F q BocHN ~ NH
R3 ~ N02 R I ~ N02
R5
R5
P
R5 R1 R5 R1
BocHN I ~ NH BocHN ~ N_ OEt
g I ~-.-~ t
O
R3 ~ NHz R3 R5 N
R5
R S
R5 R1 R5 R1
H2N I j N\ /OEt a HZN I j N~NH-Ar
'NCO R3 N O
R3 5 5
T U
n: PhCHO, HN03, H2S04; p: (Boc)2C0, CH2Cl2; q: Rl-NH2, CH3CN; r: 10% Pd/C,
EtOH; s: (CHO-COOEt)n/toluene, I2/EtOH; t: CF3COOH, CH2Cl2; u: Ar-NH2,
Scheme IV is another general scheme useful for synthesizing many subject
compounds:
13
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Scheme IV
R5 R50~ R6 R5 ~ R6
R4 ~ NH2 ~ R4 ~ NH W R4 ~ NH
HgCO I ~ N02 HgCO I ~ N02 H3C I ~ NOZ
R5 R5
R5
V W X
R5 ~ R6 R5 / R6
R4~NH y _R4~N O
I I
HgCO ~ NH2 HgCO ~ N OEt
R5 R5
Y Z
R5 ~ R6
z R4 ~ N O
HgCO I ~ N NH-Ar
R5
AA
v: R6-COC1, CH2C12; w: B2H6, THF; x: 10% Pd/C, EtOH; y: (CHO-COOEt)n, I2/EtOH,
z: NH2-Ar.
The Compositions
The subject invention includes pharmaceutical compositions comprising a safe
and
effective amount of a 2-phenyl-carbamoylbenzimidazoles compound described
hereinabove and pharmaceutically-acceptable excipients. The compositions may
also
optionally include other pharmacologically active compounds, particularly
those having
activity as thrombolytics (e.g., abciximab, reteplase), streptokinase or
tissue plasminogen
activators (e.g., streptokinase), anticoagulents (e.g., heparin, aspirin),
beta-blockers (e.g.,
carvedilol, propanalol), and calcium channel Mockers (e.g., verapamil,
nifedipine).
Some examples of pharmaceutically-acceptable carriers or components thereof
are
sugars, such as lactose, glucose, and sucrose; starches, such as cornstarch
and potato
starch; cellulose and its derivatives, such as sodium carboxymethyl cellulose,
ethyl
cellulose, cellulose acetate; powdered tragacanth; malt; gelatin; talc; solid
lubricants,
such as stearic acid, magnesium stearate; or calcium sulfate; vegetable oils,
such as
peanut oil, cottonseed oil, sesame oil, olive oil, corn oil, and oil of
theobroma; polyols
14
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
such as propylene glycol, glycerin, sorbitol, mannitol, and polyethylene
glycol; alginic
acid; emulsifiers, such as the Tweens~; wetting agents such as sodium lauryl
sulfate;
coloring agents; flavoring agents; excipients; tableting agents; stabilizers;
antioxidants;
preservatives; pyrogen-free water; isotonic saline; and phosphate buffer
solutions.
The choice of a pharmaceutically-acceptable carrier to be used in conjunction
with a compound is basically determined by the way the compound is to be
administered.
The compounds and compositions of the present invention may be administered
systemically. Routes of administration include topical or transdermal (patch,
ointment,
cream, powder, etc.); oral; parenteral, including subcutaneous, intramuscular,
or
intravenous injection; topical; rectal; colonic; intraperitoneal;
intraoccular; sublingual;
buccal; inhalation; and/or intranasal. The preferred route of administration
is parenteral,
especially intravenous injection on a daily or as needed basis.
The appropriate amount of the compound to be used may be determined by
routine experimentation with animal models. Such models include, but are not
limited to
the ferret, canine, and non human primate models. Generally, an amount between
0.01
pg/kg to 100 mg/kg of body weight per day is administered dependent on the
potency of
the compound or compositions used.
Preferred unit dosage forms for injection include sterile solutions of water,
physiological saline, or mixtures thereof. Parenteral unit dosage form
compositions may
be in the form of solutions ready for injection or dry (e.g. lyophilized)
compositions
which are reconstituted with water or saline solutions prior to injection. The
pH of said
solutions should be adjusted to about 7.4. Suitable carriers for injection or
surgical
implants include hydrogels, controlled- or sustained release devises,
polylactic acid, and
collagen matrices. Other suitable carriers for injection include dextrose,
mannitol, lactose,
lecithin, albumin, sodium glutamate, and the like.
Compositions of the subject invention are also preferably provided in unit
dosage
form. A unit dosage form composition preferably contains from about SOmg, more
preferably from about 200mg, also preferably from about SOOmg, preferably to
about
2000mg, more preferably to about 1000mg, also preferably to about SOOmg, of a
2-
phenylcarbamoyl benzimidazole compound disclosed above.
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
The subject compositions may be in a variety of forms suitable (for example)
for
peroral, topical, or parenteral administration. Depending upon the particular
route of
administration desired, a variety of pharmaceutically-acceptable carriers well-
known in
the art may be used. These include solid or liquid fillers, diluents,
hydrotropes, surface-
active agents, and encapsulating substances. The amount of carrier components
employed
in conjunction with the active compound is sufficient to provide a practical
quantity of the
material for administration per unit dose of the active compound. Techniques
and
compositions for making the subject unit dosage forms are described in the
following
references: Modern Pharmaceutics, vol. 7, chapters 9 & 10, Banker and Rhodes,
editors,
1979; Lieberman et al., Pharmaceutical Dosage Forms: Tablets, 1981; and Ansel,
Introduction to Pharmaceutical Dosage Forms, 2d edition, 1976.
As indicated above preferred dosage form of the subject invention is intended
for
parenteral administration. Preferred pharmaceutically-acceptable excipients
for such
compositions include sterile, pyrogen-free water and physiological saline
solution.
Parenteral unit dosage form compositions may be in the form of solutions ready
for
injection or dry (e.g., lyophilized) compositions which are reconstituted with
water or
saline solution prior to injection.
Preferred compositions of the subject invention also include those intended
for
peroral administration, such as tablets, capsules, powders and liquids.
Suitable
pharmaceutically-acceptable excipients for such compositions include sugars,
starches,
cellulose and its derivatives, malt, gelatin, talc, calcium sulfate, magnesium
sulfate,
vegetable oils, synthetic oils, polyols, algenic acid, phosphate buffers,
emulsifiers,
alcohols, and water.
Methods of Using the Compounds
The 2-phenyl-carbamoylbenzimidazoles compounds of the subject invention are
useful for the treatment of ischemia-reperfusion injury. Although not limited
to any
specific mechanism, it is believed that the compounds act via modulation of
adhesion
molecule metabolism. Therefore, the subject compounds are potentially useful
for the
treatment of ischemia-reperfusion injury including: cardiovascular disease
(myocardial
ischemia, angina, cardiac arrhythmia, heart failure, hypertension); treatment
to reduce
neurotoxic injury associated with anoxia or ischemia which typically follows
stroke,
16
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
cardiac arrest, or perinatal asphyxia; for treatment to reduce reperfusion
injury following
organ transplantation; for treatment of frostbite, inflammatory valve disease,
psoriasis,
asthma, adult respiratory distress syndrome; for treatment of chronic
inflammatory lung
diseases including emphysema, bronchitis; and for treatment of fibrosis,
urticaria,
angioedema, vasculitis, migarine, rheumatoid arthritis, gout, and allergy.
It is known that a variety of processes are involved in reperfusion injury,
inflammation and related processes. Not being bound by theory, the following
mechanism is of interest regarding the subject invention. A key event in the
reperfusion
injury damage process is the up-regulation, expression, activation of
intracellular
adhesion molecule-1 (ICAM-1) on endothelial cells. ICAM-1 can then interact
with
neutrophils resulting in the transmigration of the neutrophils into the tissue
with
subsequent release of deleterious enzymes and destructive reactive oxygen
molecules.
Thus, compounds which can interfere with the up-regulation, expression, or
activation of
ICAM-1 are likely to have a beneficial effect for ischemic reperfusion events.
These
compounds can be administered via oral, intra-vascular, subcutaneous, intra-
musclar,
intra-nasal, intra-rectal, intra-occular, sublingual/buccal, inhalation, and
topical (patch,
ointment, powder, or cream) routes, as long as an effective dose is delivered
to the source
of the ICAM-1.
Although not intended to be bound by theory, it is believed that the subject
invention 2-phenylcarbamoyl benzimidazoles, significantly reduce ICAM-1 up-
regulation, expression, or activation. In other assays, subject compounds
demonstrate
activity which correlates with protection to the heart. The following are test
methods
useful for determining such activities of compounds.
Assay For The Inhibition of Expression of ICAM-1
Tissue: The expression of adhesion of molecules (ICAM-1 in particular) is
performed on Human Umbilical Vein Endothelial Cells (HUVEC) obtained from
Clonetics Corp. (Cat# CC2519), San Diego, CA.
Endpoint: Concentration of material that inhibits 50% of the expression of
ICAM-1 on the surface of HUVECs upregulated with 300U/ml of TNF-alpha (ICSp).
Method: Thaw 1 vial of frozen HUVEC rapidly at 37°C for ~2 min (5 x
105-1 x
106 cells in 1 ml medium), then transfer cells to 45 ml pre-warmed growth
medium (EGM
17
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
for HUVEC) in a 225 cm2 flask (seeded at 2500 - 5000 cells/cm2) and place in a
humidified 37°C incubator with 5% C02. Change medium after 24-30h (to
remove dead
cells and cytopreservatives), and change every 2-3 days thereafter - cells
should be
confluent after 5-7 days of growth. Trypsinize cells to remove from flask -
spin (200g, 5
min) to pellet cells - resuspend cells in 50 ml medium (want ~l-2 x 105
cells/ml) and
plate 100 ml cell suspension in 96-well, collagen-coated plates (use ~1-2 x
104 cells/well)
- grow to confluence (1-2 days). Remove old culture medium (discard) and add
90 ml
fresh medium containing TNF-alpha (or other ICAM-1 stimulator at desired
concentration) or 90 ml medium alone (for unstimulated control wells). Add 10
ml
medium (or PBS or PBS, 1% DMSO) containing compound to be tested ( at lOx
desired
concentration) or additional 10 ml medium alone (or PBS or PBS, 1% DMSO) for
control
wells - (final compound concentration = lx; final DMSO concentration = 0.1%,
if used) -
incubate 4h at 37°C. Remove medium (discard) and fix cells in 200 ml
80% acetone:20%
H20 for 20 min at (-20°C). Remove acetone:H20 (discard) and allow
plates to air dry -
and store at (-20°C) overnight in desiccator. Wash plates with PBS (5 x
250 ml), then add
200 ml BLOTTO solution to block non-specific binding - incubate 1 h at room
temperature. Remove BLOTTO (discard) and wash plates with PBS (2 x 250 ml),
then
add 100 ml ICAM-1 antiserum (in BLOTTO) - incubate 1h at room temperature and
remove ICAM-1 antiserum (discard) and wash plates with PBS (5 x 250 ml), then
add
100 ml goat anti-mouse-HRP antiserum conjugate (in BLOTTO) - incubate 1h at
room
temperature. Remove antiserum-HRP conjugate (discard) and wash plates with PBS
(5 x
250 ml), then wash with citrate buffer (1 x 300 p.1; discard). Add 100 ml HRP
substrate
and incubate for 5-20 min at room temperature (time may vary; watch color
development), add 50 ml 1N H2S04 to wells to stop reaction. Read plates at 490
nm on
plate reader.
Solutions for ICAM-1 ELISA:
(1) 80% acetone:20% H20 (v:v) - store at (-20°C).
(2) (lx) Dulbecco's phosphate buffer solution (DPBS), w/o Ca++ or Mg++, pH
7.5 - (Sigma D-5652, lx powder or Sigma D-1408, lOx liquid, dilute 1:10
before use).
(3) BLOTTO - 5% (w:v) non-fat dry milk (Carnation or other) in DPBS.
~8
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
(4) Mouse anti-human ICAM-1 monoclonal antiserum (Research Diagnostics;
catalog # RDI-CBL450-lx; anti CD54-clone 15.2) - stock solution (1 mg/ml) -
dilute 1:1000 in BLOTTO just prior to use (1 mg/ml final antiserum
concentration).
(5) Goat anti-mouse IgG-horseradish peroxidase conjugate (IgG-HRP, DAKO
Corp; catalog # P0447) - stock solution ( 1 mg/ml) - dilute 1:1000 in BLOTTO
just prior to use (1 mg/ml final concentration).
(6) Citrate buffer, pH 5.0 - 65.3 mM sodium phosphate (dibasic, 12-hydrate; MW
= 358.4; 23.4 g/1) and 34.7 mM citric acid (anhydrous, free acid; MW = 192.1;
6.67 g/1) - check pH (5.0), store at 4°C.
(7) HRP substrate - o-phenylenediamine dihydrochloride (OPD; Sigma, P6912; 5
mg OPD/tablet) - add 1 tablet/10 ml citrate buffer (at room temperature), then
add 4 ml 30% H202 (Sigma, H1009)/10 ml substrate solution just prior to use
- final concentration = 0.5 mg OPD/ml and 0.012% H202.
(8) Human tumor necrosis factor alpha (TNF-alpha, Boehringer-Manheim; catalog
# 1371843) - 10 mg/vial (in 1 ml) - 108U activity/mg = 106 U/10 mg - diluted
to 20 ml endothelia cells basal media (EBM) (50000 U/ml; 500 ng/ml) -
aliquot 150 ml (7500U; 75 ng) into eppendorf tubes (x133) - store at
20°C - for
each experiment, add one aliquot to 25 ml EBM - final concentration = 300
U/ml or 3 ng/ml TNF-alpha.
Assay For The Inhibition of Human Umbilical Vein Endothelial Cell
(HUVEC)/Neutrophil Adhesion
Tissue: Adhesion is performed on Human Umbilical Vein Endothelial Cells
(HUVEC) obtained from Clonetics (Cat# CC2519).
Endpoint: Concentration of material that inhibits 20% of the PMN adhesion to
HUVECs upregulated with 300 U/ml of TNFalpha (IC20)
Method:
I. Cells:
A. Endothelia Cells:
19
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Human umbilical vein endothelial cells (HUVEC) are purchased as frozen cells
in
1 ml aliquots (Clonetics Corporation, San Diego, CA). Endothelial Growth
Media-Umbilical Vein (EGM-UV), bullet kit additives, trypsinization reagents
(trypsin neutralizing solution and HEPES buffer) are also purchased from
Clonetics. The flask is placed at 37°C in a 5% C02 + 95% air, 100%
humidity
incubator. One vial of liquid N2 frozen cell is thawed in the 37°C
water bath, the
whole vial placed in a T-275 flask with 50 mls of fresh media and placed in
the
C02 incubator. The media is replaced 24-48 hrs later. Confluency should occur
within 4-5 days. Media is changed at least once during that period. The
monolayer in the flask is detached using the trypsin solution, after the
monolayer
is washed with Hanks balanced salt solution (HBSS). The trypsinized cells are
centrifuged at 200xGs for 5 minutes and resuspended in approximately 150 mls
of
media. 100 u1 aliquots are placed in each well of 96-well plate that had been
previously coated with collagen. The monolayer in the plate should be
confluent
within 48 hrs.
B. NeutrophilIsolation:
Peripheral blood polymorphonuclear neutrophils (PMNs) are isolated by
established methodology (1). Human blood is obtained from the cubital vein by
conventional venipucture performed by qualified phlebotomist. The blood is
collected in heparinized vacutainers (Vacutainer #6489, green cap, 15 ml draw,
VWR). Thirty ml of blood is used for each assay. The heparinized blood is
diluted with approximately '/2 volume of phosphate buffered saline containing
0.2% glucose (PBS-G). A discontinuous gradient of Histopaque (3 ml of
Histopaque-1119 in the bottom and 3 ml of Histopaque-1077 on top) (Sigma
Chemical Co., St. Louis, MO) is prepared in 6, 15-ml conical centrifuge tubes.
The diluted blood is carefully layered on top of the Histopaque-1077. The
tubes
are centrifuged at 800 x G for 30 min at room temperature. After the
centrifugation step the PMNs are removed by aspiration from the area between
Histopaque-1077/Histopaque-1119 interface and the top of the pelleted red
blood
cells. The PMNs are collected from all the tubes, further diluted to a total
volume
of 30 ml and centrifuged at 600 x G for 15 min. The supernatant is discarded,
and
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
the pellet (containing PMNs and some red blood cells) is treated with 6 ml of
cold
water for 30 seconds to lyse contaminating RBC. Normal osmolarity is
reestablished by adding 3 ml of 2.7% saline. The PMNs are washed an additional
2 times with PBS-G. The viability and number of the PMNs is determined using
the trypan blue exclusion test in a hemocytometer counting chamber.
Occasionally, a small aliquot of the PMNs suspension is used for a Cytofuge
preparation. The Cytofuge slide is stained with Wright's blood stain (Sigma
Chemical Co.) and a differential court performed to evaluate the percent of
PMNs
in the preparation.
II. Upregulation of Endothelia Cells:
The monolayers of endothelial cells in the 96-well plates are upregulated with
300
U/ml of Tumor Necrosis Factor (TNF, Boehinger-Manheim Catalog # 1371-843). The
TNF and compound are added to each well 4 hours prior to the addition of the
PMNs.
III. Fluorescent Labeling of Neutrophils:
After the last wash the neutrophil pellet is resuspended in S mls of PBS-G
(approximately 1-3 x 106/m1). 5 (and 6) carboxyfluorescein diacetate
succinimidyl ester
(CFSE, Molecular Probes, Eugene, OR). A 20 mM stock of the CFSE is prepared by
dissolving 25 mg into 2.24 mls (MW557.5) of DMSO. 5 p1 of the stock is added
to the 5
ml suspension of PMNs for a final concentration of 2 uM. The mixture is
incubated in the
refrigerator for 20 minutes. At the end of this period the PMNs are washed 4
times with
PBS-G. After the final wash the PMNs are resuspended in complete EGM-UV media
to
the desired concentration (usually each well of a 96-well plate receives 0.6-
1.2 x 105
PMNs).
IV. Addition of Test Compounds:
One hundred p1 of media containing the compounds with TNF is used to replace
the media in the wells containing the monolayers of endothelial cells 4 hours
prior to the
addition of the PMNs.
21
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
V. Adhesion Assay:
A. Collection of Data:
CFSE-labeled neutrophils (.7 to 1.5 x 105) in 10 p1 volumes (see III) are
added to
the HUVEC monolayers. The plates are incubated at 37°C in a 5% C02 +
95% air,
100% humidity incubator for 30 min. Non-adherent cells are removed by
centrifugation according to the following protocol:.
(1) A reading is taken in the Cytofluor 2400 after the incubation at
37°C. This
reading is considered as 100% of all the cells added.
(2) The wells are filled with warmed (incubator) media to the top of the well
with a slight convex meniscus (usually 260 u1 (in addition to the 100 u1 or so
already in the wells)).
(3) The wells are sealed using Adhesive Sealing Films for Micro Plates
(Rainin cat# 96-SP-100).
(4) The lid is replaced on the plate and the size and location of any bubbles
recorded on the lids with a marker.
(5) The lids are removed and a folded (4 plies) and cut piece of paper towel
placed on top of the Sealing film. The plate lid is then replaced on top of
the
paper towel and the plate inverted.
(6) The plates are placed on the plate holders of the centrifuge (Sorvall
Model
RT 6000D) and spun by turning the speed down to around 500 RPMs, then
turning the centrifuge on with the timing knob. The speed control is adjusted
until the tachometer read 1100 RPM (this is the equivalent of 200 Gs). At the
moment that the speed of the 1100 RPM is achieved a separate timer was
started. At exactly 2 minutes the timer in the centrifuge is brought to zero
to
stop the motor. The plates are allowed to come to rest without any braking.
(7) The plates are removed and any empty wells recorded. The lid and the
folded paper towel are removed (maintaining the plate upside-down). The
Sealing Film is then removed over the biological material disposal bin and the
media shaken out. The plate is then blotted on the paper towel and any excess
fluid aspirated.
22
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
(8) The plate is returned to the upright position and studied under the
microscope.
(9) A second reading is taken on the Cytofluor. This second reading is used to
determine the percent of PMNs that remained adhered to the monolayer.
(10) The information from the CSV files of the Cytofluor are downloaded and
the data processed in a prepared EXCEL spreadsheet where the background
(PMNs adherent to non-upregulated endothelium) is subtracted and the percent
adhesion inhibition is determined as follows:
Percent
adhesion (Individual adherent PMNs with background subtracted X 100)
inhibition = 100 -
Mean adherent PMNs to upregulated endothelium minus background
(11) The EXCEL spreadsheet calculates a) the percent of PMNs adhering to
the monolayer, b) the percent PMNs adhering to the monolayer minus
background (PMNs adherent to unstimulated endothelial cells) and c) the
percent adhesion inhibition considering the wells receiving TNF alone to the
0% inhibition (negative numbers indicate increase in adhesion).
B. Statistics, Data Handling, and Storage:
Statistics are performed using the double sided t-test with equal variance in
EXCEL and the results recorded as the Pvalue.
VI. Additional Information:
A. To Coat Wells With Collagen:
Dissolve 25 mg of acid soluble rat tail collagen 446 mls of water acidified
with a
few drops of HCI. Sterilize by filtration. Add 50 u1 to each well in a 96-well
plate
(.28 cm2) for 10 ug/well. Incubate overnight in the 37°C incubator.
Aspirate all
the liquid and store in refrigerator until use.
B. To Prepare PBS+0.2% Glucose (Dextrose):
Prepare 2 liters of PBS (Sigma) and add 2 grams of glucose. Sterile filter and
save.
Rat Myocardial Infarct/Renerfusion Iniury Model
23
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Surgical Preparation of Rats:
Male, Sprague-Dawley rats are anesthetized with urethane, 1.25 g/kg ip.
Acarotid
artery and jugular vein are exteriorized and cannulated with PE-50 tubing for
recording
blood pressure and to facilitate intravenous administration of dye or drug. A
Tracheotomy is performed. The animals are connected to a Harvard Rodent
Ventilator
(Model 683, Harvard Apparatus, South Natick, MA) and ventilated at 1.5 m1/100
g body
weight at SO strokes/min. Needle electrodes are placed for a lead II
electrocardiogram.
The animals are maintained at 37°C by means of electric heating pads
adjusted to the
desired temperature and controlled via a rectal thermistor probe and
controller. The heart
is carefully isolated by a left thoracotomy at the fifth intercostal space,
and the left
anterior descending coronary artery (LAD) is located. A ligature of 6-0 silk
is placed
around the LAD, with the ends threaded through a small length of PE-320 tubing
to
facilitate rapid occlusion and reperfusion of the artery. The LAD is occluded
by clamping
the suture and tubing tight against the heart surface sing 25 mm Schwarz
aneurysm clip.
Occlusion lasts for 90 min and is followed by reperfusion for 3.0-4.5 hr.
Animals are
dosed with drug or vehicle 10 min prior to reperfusion of the affected area of
the heart by
intravenous delivery via a jugular vein. Sham-operated rats are not subjected
to ischemia
or reperfusion. At the end of the experiment, the LAD is permanently re-
occluded and a
mg/ml solution of Evans Blue Stain is administered via the jugular cannula to
identify
the area affected by ischemia, i.e., the area-at-risk (AAR). The stained heart
is rapidly
excised and placed into 0.9% saline at 4°C prior to the determination
of creatine
phosphokinase activity (CPK).
Determination of Creatine Phosphokinase Activity:
The left ventricular free wall (LVFW) is dissected free from the heart and
weighed. The AAR, as defined by the absence of stain, is dissected from the
LVFW and
also weighed. The AAR is homogenized for 5 sec in 4 ml of 0.25 M sucrose
containing 1
mM EDTA and 10 mM mercaptoethanol at 4°C. The homogenate is centrifuged
at 3000 x
g for 30 min at 4°C. The supernatant is decanted for determination of
CPK activity and
the pellet is stored frozen for the isolation and assay of myeloperoxidase
activity. CPK
activity is assayed spectrophotometrically with a commercially supplied
substrate, CPK
Assay Vial~ (Sigma Diagnostics), at a wavelength of 340 nm at 24-
26°C.
24
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Determination of Myeloperoxidase Activity:
Myeloperoxidase (MPO) is isolated from the frozen pellet after the preparation
of
CPK. The pellet is suspended in 50 mM phosphate buffer, pH 6, containing 0.5%
hexadecyltrimethylammonium bromide (HTAB) to a concentration of approximately
10%
sonicated for 10 sec and frozen on dry ice. Three freeze-thaw cycles are done
with 10 sec
of sonication between cycles. The samples are chilled on ice for 30 min
followed by
centrifugation at 12,500 x g for 15 min at 4°C. An aliquot of the
supernatant is assayed
spectrophotometrically for MPO activity in 50 mM sodium phosphate buffer, pH
6,
containing 0.167 mg/ml o-dianisidine dihydrochloride and 0.0005% hydrogen
peroxide at
a wavelength of 460 nm at 24-26°C.
Calculations and Statistical Analysis:
The results are reported as the mean ~ SEM. CPK and MPO activity are
expressed as units/g tissue, where 1 unit of CPK activity is defined as the
quantity of CPK
utilizing 1 pmol peroxide per minute. The AAR is quantified as a percentage of
the
LVFW based on weight. Mean arterial blood pressure (MABP) is calculated as one-
third
the difference between systolic and diastolic blood pressure added to
diastolic blood
pressure. Data are analyzed for statistical significance of treatment effects
at the 95%
confidence level by a pooled t-test or by one-way analysis of variance.
The subject invention involves methods of treating or preventing any of the
diseases and disorders provided hereinabove, especially reperfusion injury, by
administering a safe and effective amount of a the compounds disclosed
hereinabove.
Such methods of treatment can involve administering a unit dosage form of such
compounds parenterally, perorally, or topically. Parenteral administration
includes
intravenous, intramuscular, subcutaneous, intraperitoneal, or other injection
of the dosage
form. Peroral administration involves ingestion of the dosage form and
absorption of the
active from the gastrointestinal tract. Topical administration involves
contacting the
dosage form with the surface of the skin or mucosal tissues, including, but
not limited to,
those of the alimentaryl canal and the respiratory system.
For parenteral administration, the amount of the compound typically
administered
is preferably from about 2 mg/kg, more preferably from about 5 mg/kg,
preferably to
about 2- mg/kg, more preferably to about 10 mg/kg. The frequency of such
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
administration is typically once or twice daily. A treatment regimen typically
is a single
dose, or lasts from about 1 day, preferably from about 5 days, to about 30
days, preferably
to about 15 days.
For peroral administration, the amount of 2-phenylcarbamoyl benzimidazole
compound typically administered is preferably from about 5 mg/kg, more
preferably from
about 10 mg/kg, preferably to about 25 mg/kg, more preferably to about 15
mg/kg. The
frequency of such administration is typically from once to about 4 times
daily. A
treatment regimen typically lasts from about 1 day, preferably from about 5
days, to about
30 days, preferably to about 15 days.
Composition and Method Examples
The following non-limiting examples illustrate the subject invention. The
following composition and method examples do not limit the invention, but
provide
guidance to the skilled artisan to prepare and use the compounds,
compositions, and
methods of the invention. In each case other compounds within the invention
may be
substituted for the example compound shown below with similar results.
Example A
Pharmaceutical compositions in the form of an intravenous solution are
prepared
by conventional methods, such as mixing the following:
I~redient (quantity Lmls,~
Compound of Example 11 1 mg.
Sterile water 10 ml
HCL and/or NaOH pH 7.2-7.5
When 1 ml of the above composition is administered intravenously, either
immediately before or immediately after a tissue damage event (aneurysm
repair,
coronary bypass, transplant surgery, traumatic hemorrhage, organ ischemia due
to
hypoperfusion, sepsis, etc.), tissue damage is avoided or reduced.
Example B
Compound of Example 1 can be substituted with any of Examples 2-8.
26
CA 02399799 2002-08-09
WO 01/58879 PCT/USO1/04212
Pharmaceutical compositions in liquid form are prepared by conventional
methods, formulated as follows:
In. reg diem uantit
Compound of Example 1 Z 1 mg
Phosphate buffered physiological saline 10 ml
Methyl Paraben O.OSmI
When 1.0 ml of the above composition is administered subcutaneously, either
immediately before or immediately after a tissue damage event (aneurysm
repair,
coronary bypass, transplant surgery, traumatic hemorrhage, organ ischemia due
to
hypoperfusion, sepsis, etc.), tissue damage is avoided or reduced.
While particular embodiments of the subject invention have been
described, it would be obvious to those skilled in the art that various
changes and
modifications to the compositions disclosed herein can be made without
departing from
the spirit and scope of the invention. It is intended to cover, in the
appended claims, all
such modifications that are within the scope of this invention.
z Compound of Example 1 can be substituted with any of Examples 2-8.
27