Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02400657 2002-08-16
WO 01/64645 1 PCT/FI01/00203
DERIVATIVES OF QUINOLINE AS ALPHA-2 ANTAGONISTS
FIELD OF THE INVENTION
The present invention relates to therapeutically active derivatives of
quinoline, including the pharmaceutically acceptable salts and esters thereof,
and their use as alpha-2 antagonists.
BACKGROUND
Some compounds exhibiting alpha adrenergic activity are well known in
the art. Those compounds may be used for the treatment of a wide variety of
diseases and conditions of the peripheric system and the central nervous
system (CNS).
The alpha adrenergic receptors are divided into alpha-1 and alpha-2
adrenoceptors, each of which are further divided into subtypes. Accordingly,
alpha-2 adrenoceptors in humans have been subdivided into three
pharmacological subtypes known as alpha-2A, alpha-2B and alpha-2C
~s adrenoceptors. A fourth subtype, alpha-2D, is known in rat, bovine and
porcine
and it corresponds to alpha-2A in man. These subtypes have a distinct
distribution in human and animal tissues. For instance, alpha-2C adrenoceptors
are concentrated in the CNS and they appear to play a role in the modulation
of
various CNS-mediated behavioural and physiological responses.
2o Compounds that are non-specific to any of the above-mentioned alpha-2
subtypes, and compounds that are specific to a certain alpha-2 subtypes are
already known. For example, atipamezole is a non-specific alpha-2
antagonists. Atipamezole has been described in, for example, EP-A-183 492
(cf. p.13, compound XV) and A. Haapalinna et al., Naunyn-Schimiedeberg's
25 Arch. Pharmacol. Vol. 356, 1997, p.570-582. U.S. Patent No. 5,902,807
describes compounds that are selective antagonists for the alpha-2C subtype
and may be used in the treatment of mental illnesses, e.g. mental disturbances
induced by stress. Such compounds include, for example, MK-912 and BAM-
1303. Furthermore, WO-A-99 28300 discloses substituted imidazole derivatives
3o having agonist-like activity for alpha-2B or 2B/2C adrenoceptors. The
disclosures of all documents cited above in this paragraph are incorporated by
reference herein.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
2
As to the derivatives of quinoline, Medicinskaja parazitologija I
parazitarnye bolezni, vol.5, 1991, 55-7 (Mikhailitsyn F.S. et al.) and J. Med.
Chem., vo1.20(8), 1977, 987-996 (Cain F.C. et al.) describe, for example,
acridine derivatives as anticancer and/or antiparasitic agents. In addition, a
publication by Adams et al in 1985 (Mol. Pharm. 27, 480-491 ) reports on the
binding of diquinolines, diacridines and a number of monoacridines on rat
brain
alpha-1-, alpha-2- and beta-adrenoceptors.
SUMMARY OF THE INVENTION
An object of the present invention is to provide further antagonists of
alpha-2 adrenoceptors that can be used for the treatment of diseases or
conditions of the pheripheric or central nervous system where alpha-2
antagonists are indicated to be useful. Accordingly, an object of the present
invention is to provide further compounds to be used as alpha-2 antagonist
agents in the treatment of mammals, including humans and animals.
Another object of the present invention is to provide further compounds
useful as selective alpha-2C antagonist agents for the treatment of various
disorders or conditions of the central nervous system where alpha-2C
antagonists are indicated to be useful.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 shows the effects of atipamezole and compound 1 on
dexmedetomidine-induced hypolocomotion.
Figure 2 shows the effects of atipamezole and compound 1 in a forced
swimming test in Balb/c mice.
DETAILED DESCRIPTION OF THE INVENTION
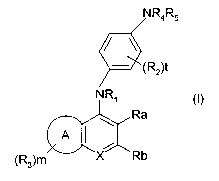
One embodiment of the present invention covers compounds of formula
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
3
NR4R5
'(R2)t
NR~
Ra
A
(R3)m N Rb
wherein,
R~ is H or (C~-Cg)alkyl;
each R2 is independently OH, halogen, (C~-C6)alkyl, (C2-C6)alkenyl,
(C~-Cg)alkoxy, halo(C~-C6)alkyl, N02, NH2, mono- or di(C~-Cg)alkylamino,
(C~-Cg)alkyl-S- or hydroxy(C~-C6)alkyl;
A is a benzene ring or (C5-C7)cycloalkyl;
when A is a benzene ring each Rg is independently OH, halogen, (C~-
C6)alkyl, (C2-C6)alkenyl, (C~-C6)alkoxy, halo-(C~-Cg)alkyl, N02, NH2, mono-
or di(C~-C6)alkylamino, (C~-Cg)alkyl-CO-, mono- or di(C~-Cg)-alkylcarbamoyl,
(C~-Cg)alkyl-S-, hydroxy(C~-Cg)alkyl or NH2-CO-;
when A is (C5-C7)cycloalkyl each R3 is independently OH, halogen,
(C~-Cg)alkyl, (C~-Cg)alkoxy, mono- or di(C~-C6)alkylamino or hydroxy(C~
2o Cg)alkyl;
R4 and R5 form, together with the N-atom to which they are attached,
-N X
wherein X is O or =NRg; R6 is H, OH, NH2, (C~-C6)alkyl, (C2-
Cg)alkenyl, CN-(C~-Cg)alkyl, (C~-Cg)alkoxy-CO-(C~-Cg)alkyl, (C~-C6)alkyl-
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
4
CO-, NH2-CO-, mono- or di(C1-Cg)alkylcarbamoyl, hydroxy(C1-Cg)alkyl, (C3-
C6)cycloalkyl, phenyl, naphthyl or benzyl, wherein the said phenyl, naphthyl
or
benzyl is optionally substitued with one to three substituent(s) each
independently being OH, halogen, N02, NH2, (C1-Cg)alkyl, (C1-Cg)alkoxy,
mono- or di(C1-Cg)alkylamino or halo-(C1-C6)alkyl;
or R4 and R5 form, together with the N-atom to which they are attached,
/' (CH2)n
-N
(R6)r
wherein n = 1 or 2; Rg is as defined above; and r = 0 to 3;
or R4 and R5 form, together with the N-atom to which they are attached,
1-imidazolyl, 1-imidazolinyl or 1-triazolyl, each of which can optionally be
substituted with one to three substituent(s) R7 each independently being (C1-
15 Cg)alkyl or NH2;
or one of R4 and R5 is -S02Rg and the other of R4 and R5 is H or (C1-
Cg)alkyl; Rg is (C1-Cg)alkyl, phenyl, naphthyl or benzyl, wherein the said
phenyl, naphthyl or benzyl is optionally substituted with one to three
2o substituent(s) Rg each independently being OH, halogen, N02, NH2, (C1-
C6)alkyl, (C1-C6)alkoxy or mono- or di(C1-Cg)alkylamino;
Ra and Rb are independently H, OH, halogen, (C1-Cg)alkyl, (C2-
C6)alkenyl, (C2-Cg)alkynyl, (C1-Cg)alkoxy, halo(C1-C6)alkyl, N02, NH2,
25 mono- or di(C1-C6)alkylamino, (C1-C6)alkyl-S- or CN;
or Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed benzene ring optionally substituted with one to
three substituent(s) R'3 each independently being OH, halogen, (C1-C6)alkyl,
so (C2-C6)alkenyl, (C1-Cg)alkoxy, halo-(C1-C6)alkyl, N02, NH2, mono- or di(C1-
C6)alkylamino, (C1-Cg)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, (C1-
Cg)alkyl-S-, hydroxy(C1-Cg)alkyl or NH2-CO-;
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
or Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed five to seven membered carbocyclic ring optionally
substituted with one to four substituent(s) R10 each independently being OH,
halogen, (C1-Cg)alkyl, (C1-Cg)alkoxy, mono- or di(C1-Cg)alkylamino or
hydroxy(C1-C6)alkyl;
or Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed bicyclo[2.2.1]-heptane ring optionally substituted
with one to four substituent(s) each independently being OH, halogen, (C1-
C6)alkyl or (C1-C6)alkoxy;
or Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed five or six membered heterocyclic ring with one ring
heteroatom =NR11, which heterocyclic ring is optionally substituted with one
to
three substituent(s) R10 as defined above; R11 is H or (C1-Cg)alkyl, or R11 is
phenyl optionally substituted with one to three substituents R12 each
independently being OH, halogen, N02, NH2, (C1-C6)alkyl, (C1-C6)alkoxy or
mono- or di(C1-Cg)alkylamino;
2o m is 0 to 3; and
tisOto3,
or pharmaceutically acceptable salts and esters thereof.
The compounds of formula I can be used for the manufacture of a
medicament for the treatment of diseases or conditions where alpha-2
antagonists are indicated to be effective.
The following subgroups (1 ) to (18) of compounds of formula I taken
alone or in any combination with each other are possible:
(1 ) A is a benzene ring;
(2) A is a (C5-C7)cycloalkyl;
(3) Ra and Rb are independently H, OH, halogen, (C1-Cg)alkyl, (C2-
C6)alkenyl, (C2-C6)alkynyl, (C1-C6)alkoxy, halo(C1-C6)alkyl, N02,
NH2, mono- or di(C1-Cg)alkylamino, (C1-Cg)alkyl-S- or CN; e.g. H,
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
6
halogen, (C1-C6)alkyl, (C2-C6)alkenyl, (C2-Cg)alkynyl, (C1-
C6)alkoxy or halo(C1-Cg)alkyl; such as H, halogen, (C1-Cg)alkyl or
(C1-Cg)alkoxy; e.g. H or (C1-C3)alkyl, wherein the said (C1-Cg)alkyl
includes both straight and branched chain radicals of up to 3 carbon
atoms;
(4) Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed benzene ring unsubstituted or substituted
with one to three, e.g one or two, such as one, substituent(s) R'g
each independently being OH, halogen, (C1-Cg)alkyl, (C2-
Cg)alkenyl, (C1-Cg)alkoxy, halo-(C1-C6)alkyl, N02, NH2, mono- or
di(C1-Cg)alkylamino, (C1-Cg)alkyl-CO-, NH2-CO-, mono- or di(C1-
C6)alkylcarbamoyl, (C1-Cg)alkyl-S or hydroxy(C1-C6)alkyl; e.g. OH,
halogen, (C1-Cg)alkyl, (C2-Cg)alkenyl or (C1-Cg)alkoxy; such as
halogen, (C1-C6)alkyl or (C1-C6)alkoxy; e.g. (C1-Cg)alkyl or (C1-
C6)alkoxy;
(5) Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed five to seven membered carbocyclic ring
optionally substituted with one to four, e.g. one or two, such as one,
substituent(s) R1 p each independently being OH, halogen, (C1-
2o Cg)alkyl (C1-C6)alkoxy, mono- or di(C1-Cg)alkylamino or
hydroxy(C1-C6)alkyl; such as (C1-C6)alkyl or (C1-C6)alkoxy; e.g.
(C1-C6)alkyl;
(6) Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed bicyclo[2.2.1 ]-heptane ring optionally
substituted with one to four, e.g. one or two, such as one,
substituent(s) each independently being OH, halogen, (C1-Cg)alkyl
or (C1-Cg)alkoxy; e.g. (C1-C6)alkyl
(7) Ra and Rb form, together with the carbon ring atoms to which they
are attached, a condensed five or six membered heterocyclic ring
3o with one ring heteroatom =NR11, which heterocyclic ring is optionally
substituted with one to three, e.g. one or two, such as one
substituent(s) R1 p as defined above; and R11 is H or (C1-Cg)alkyl, or
R11 is phenyl optionally substituted with one to three, e.g. one or two,
such as one, substituents R12 each independently being OH,
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
7
halogen, N02, NH2, (C1-C6)alkyl, (C1-Cg)alkoxy or mono- or di(C1_
C6)alkylamino;
(8) R4 and R5 form, together with the N-atom, 1-piperazinyl which is 4-
substituted with Rg, wherein Rg is as defined above; e.g. (C1-
Cg)alkyl, (C2-Cg)alkenyl, CN-(C1-C6)alkyl, (C1-C6)alkoxy-CO-(C1-
C6)alkyl, (C1-C6)alkyl-CO-, NH2-CO-, mono- or di(C1-
Cg)alkylcarbamoyl, hydroxy(C1-C6)alkyl, (C3-Cg)cycloalkyl or
optionally substituted benzyl; such as (C1-Cg)alkyl, (C2-C6)alkenyl,
hydroxy(C1-Cg)alkyl, (C3-C6)cycloalkyl; e.g. (C1-Cg)alkyl;
(9) R4 and R5 form, together with the N-atom, a morpholino ring;
(10) R4 and R5 form, together with the N-atom, a 1-piperidinyl or 1-
pyrrolidinyl optionally substituted with Rg, wherein Rg is as defined
above, possibly (C1-Cg)alkyl, (C2-C6)alkenyl, CN-(C1-Cg)alkyl, (C1-
C6)alkoxy-CO-(C1-C6)alkyl, (C1-C6)alkyl-CO-, NH2-CO-, mono- or
di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl or (C3-C6)cycloalkyl;
such as (C1-Cg)alkyl, (C2-C6)alkenyl, hydroxy(C1-Cg)alkyl, (C3-
Cg)cycloalkyl; e.g. (C1-C6)alkyl;
(11 ) R4 and R5 form, together with the N-atom, 1-imidazolyl, 1-
imidazolinyl or 1-triazolyl, each of which can optionally be substituted
2o with one to three, e.g. one or two, such as one, substituent(s) R7
each independently being (C1-C6)alkyl or NH2; e.g. R4 and R5 form,
together with the N-atom, 2-amino-imidazol-1-yl or 2-amino-
imidazolin-1-yl;
(12) one of R4 and R5 is -S02Rg and the other of R4 and R5 is H or
(C1-Cg)alkyl; Rg is independently (C1-Cg)alkyl, phenyl, naphthyl and
benzyl, wherein the said phenyl, naphthyl or benzyl is optionally
substituted with one to three, e.g. one or two, such as one,
substituent(s) Rg each independently being OH, halogen, N02, NH2,
(C1-C6)alkyl, (C1-Cg)alkoxy or mono- or di(C1-Cg)alkylamino;
(13) m is 0 to 3; e.g. 0, 1 or 2; e.g. 0 or 1; such as 0;
(14) t is 0 to 3; e.g. 0, 1 or 2; e.g. 0 or 1; such as 0; and/or
(15) R1 is H;
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
8
(16) R1 is (C1-C6)alkyl;
(17) R2 is independently OH, halogen, (C1-C6)alkyl, (C2-C6)alkenyl,
(C1-Cg)alkoxy, halo(C1-C6)alkyl, N02, NH2, mono- or di(C1-
C6)alkylamino, (C1-C6)alkyl-S- or hydroxy(C1-Cg)alkyl; such as OH,
halogen, (C1-C6)alkyl, (C1-Cg)alkoxy or hydroxy(C1-Cg)alkyl; e.g.
(C1-C6)alkyl or (C1-Cg)alkoxy; and/or
(18) R3 is independently OH, halogen, (C1-Cg)alkyl, (C2-C6)alkenyl,
(C1-Cg)alkoxy, halo-(C1-Cg)alkyl, N02, NH2, mono- or di(C1-
Cg)alkylamino, (C1-C6)alkyl-CO-, mono- or di(C1-Cg)-
alkylcarbamoyl, (C1-Cg)alkyl-S-, hydroxy(C1-C6)alkyl or NH2-CO-;
such as OH, halogen, (C1-Cg)alkyl, (C2-C6)alkenyl, (C1-Cg)alkoxy,
halo-(C1-Cg)alkyl; e.g. halogen, (C1-Cg)alkyl or (C1-Cg)alkoxy.
A possible subgroup of the compound of formula I is a compound of
formula IA:
N R4R5
'(R2)t
NR~
IA
/~~CH2)i
(R3)m N (Rio)J
wherein R1, R2, R3, R4, R5, R1 p; m and t are as defined above; i is 1 to
3; and j is 0 to 4.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
9
Another possible subgroup of the compound of formula I is a compound
of formula IB:
NR4R5
(R2)t
NR~
Ra IB
A
(R3)m N Rb
wherein A, R~ , R2, R3, R4, R5, Ra, Rb, m and t are as defined above;
and Ra and Rb are independently H, OH, halogen, (C~-Cg)alkyl, (C2-
C6)alkenyl, (C2-C6)alkynyl, (C~-Cg)alkoxy, halo(C~-C6)alkyl, N02, NH2,
mono- or di(C~-Cg)alkylamino, (C~-C6)alkyl-S- or CN; or Ra and Rb form,
together with the carbon ring atoms to which they are attached, a condensed
five to seven membered carbocyclic ring optionally substituted with one to
three
1o substituent(s) Rip ; each independently being OH, halogen, (C~-Cg)alkyl,
(C~-
Cg)alkoxy, mono- or di(C~-Cg)alkylamino or hydroxy(C~-C6)alkyl.
Another possible subgroup of the compound of formula I is a compound
of formula IC:
Ra R5
'(R2)t
NR~
IC
\ \ (R~o)J
(CH2)i
N
(R3)m N i
R11
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
wherein R1, R2, Rg, R4, R5, R10, R11, m and t are as defined above; i
is 1 or 2; and j is 0 to 3.
Another possible subgroup of the compound of formula I is a compound
5 of formula ID:
NRQRS
~(R2)t
NR~ ID
\ \ \
i
(R3)m N (R'3)P
wherein R1, R2, R3, R'3, R4, R5, m and t are as defined above and p is
0 to 3; for example wherein m is 1 and R3 is (C1-Cg)alkoxy.
In a possible subgroup of the compound of formula I, IA, IB, IC or ID, R4
and R5 form, together with the N-atom to which they are attached,
-N X
U
wherein X is =NRg; Rg is as defined above under formula I; such as (C1-
Cg)alkyl, (C2-C6)alkenyl, hydroxy(C1-C6)alkyl or (C3-Cg)cycloalkyl ; e.g. (C1-
C6)alkyl.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
11
Another embodiment of the invention provides new compounds of
formula II:
N R4R5
'(R2)t
NR~ II
Ra
A
(R3)m N Rb
wherein,
R1 is H or (C1-Cg)alkyl;
each R2 is independently OH, halogen, (C1-C6)alkyl, (C2-C6)alkenyl, (C1-
Cg)alkoxy, halo(C1-C6)alkyl, N02, NH2, mono- or di(C1-Cg)alkylamino, (C1-
Cg)alkyl-S- or hydroxy(C1-Cg)alkyl;
A is a benzene ring or (C5-C7)cycloalkyl;
when A is a benzene ring each R3 is independently OH, halogen, (C1-
C6)alkyl, (C2-C6)alkenyl, (C1-C6)alkoxy, halo-(C1-Cg)alkyl, N02, NH2, mono-
or di(C1-C6)alkylamino, (C1-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl,
(C1-C6)alkyl-S-, hydroxy(C1-C6)alkyl or NH2-CO-;
when A is (C5-C7)cycloalkyl each Rg is independently OH, halogen, (C1-
2o Cg)alkyl, (C1-Cg)alkoxy, mono- or di(C1-Cg)alkylamino or hydroxy(C1-
C6)alkyl;
R4 and R5 form, together with the N-atom to which they are attached,
-N X
U
wherein X is O or =NRg; Rg is H, OH, NH2, (C1-Cg)alkyl, (C2-Cg)alkenyl,
CN-(C1-C6)alkyl, (C1-Cg)alkoxy-CO-(C1-C6)alkyl, (C1-C6)alkyl-CO-, NH2-CO-,
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
12
mono- or di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, (Cg-Cg)cycloalkyl or
benzyl, wherein the said benzyl is optionally substitued with one to three
substituent(s) each independently being OH, halogen, N02, NH2, (C1-C6)alkyl,
(C1-Cg)alkoxy, mono- or di(C1-Cg)alkylamino or halo-(C1-Cg)alkyl;
or Rq. and R5 form, together with the N-atom to which they are attached,
/-(CH2)n
N
~(R6)r
wherein n = 1 or 2; Rg is as defined above; and r = 0 to 3;
or R4 and R5 form, together with the N-atom to which they are attached, 1-
imidazolyl, 1-imidazolinyl or 1-triazolyl, each of which can optionally be
substituted with one to three substituent(s) R7 each independently being (C1-
Cg)alkyl or NH2;
Ra and Rb are independently H, OH, halogen, (C1-Cg)alkyl, (C2-
C6)alkenyl, (C2-Cg)alkynyl, (C1-Cg)alkoxy, halo(C1-Cg)alkyl, N02, NH2,
mono- or di(C1-Cg)alkylamino, (C1-Cg)alkyl-S- or CN;
or Ra and Rb form, together with the carbon ring atoms to which they are
2o attached, a condensed benzene ring optionally substituted with one to three
substituent(s) R'3 each independently being OH, halogen, (C1-C6)alkyl, (C2-
C6)alkenyl, (C1-C6)alkoxy, halo-(C1-C6)alkyl, N02, NH2, mono- or di(C1-
Cg)alkylamino, (C1-Cg)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, (C1-
Cg)alkyl-S-, hydroxy(C1-Cg)alkyl or NH2-CO-;
or Ra and Rb form, together with the carbon ring atoms to which they are
attached, a condensed five to seven membered carbocyclic ring optionally
substituted with one to four substituent(s) R10 each independently being OH,
halogen, (C1-Cg)alkyl, (C1-Cg)alkoxy, mono- or di(C1-Cg)alkylamino or
3o hydroxy(C1-Cg)alkyl;
or Ra and Rb form, together with the carbon ring atoms to which they are
attached, a condensed bicyclo[2.2.1]-heptane ring optionally substituted with
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
13
one to four substituent(s) each independently being OH, halogen, (C1-Cg)alkyl
or (C1-C6)alkoxy;
or Ra and Rb form, together with the carbon ring atoms to which they are
attached, a condensed five or six membered heterocyclic ring with one ring
heteroatom =NR11, which heterocyclic ring is optionally substituted with one
to
three substituent(s) R10 as defined above; R11 is H or (C1-C6)alkyl, or R11 is
phenyl optionally substituted with one to three substituents R12 each
independently being OH, halogen, N02, NH2, (C1-Cg)alkyl, (C1-Cg)alkoxy or
mono- or di(C1-Cg)alkylamino;
m is 0 to 3; and
tisOto3,
or of a pharmaceutically acceptable salt or ester thereof, with the provisos,
that
a) when A is a benzene ring, m is 0 or 1, t is 0, R1 is H, R3 is CI or N02,
and Ra and Rb form, together with the carbon ring atoms to which they are
2o attached, a condensed benzene ring, and X is NR6, then R6 is not H, -CH3, -
CH2CH3, -COCH3, or -CO-NH2;
b) when A is a benzene ring, then Ra and Rb are not at the same time H;
c) when A is a benzene ring, m is 1, t is 0, R1 is H, and Ra and Rb form,
together with the carbon ring atoms to which they are attached, a condensed
25 benzene ring, which is optionally substituted with Br, and X is O, then R3
is not
N02 or -OCH3;
d) when A is a benzene ring, m is 0, t is 0, R1 is H, and Ra and Rb form,
together with the carbon ring atoms to which they are attached, a condensed
nonsubstituted benzene ring, then X is not O;
so e) the compound is not 4-[4-[(7-Chloro-2-methyl-4-quinolinyl)amino]phenyl]-
1-diethylcarbamoylpiperazine, 4-[4-[(6-Chloro-2-methoxy-9-
acridinyl)amino]phenyl]-1-diethylcarbamoylpiperazine, 6-amino-4-[[3-chloro-4-
(1 H-imidazol-1-yl)phenyl]amino]-7-metoxy-3-quinolinecarbonitrile or 4-[[3-
chloro-4-(1 H-imidazol-1-yl)phenyl]amino]-7-methoxy-6-vitro-3-
35 quinolinecarbonitrile.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
14
A possible subgroup of the compound of formula II is a compound of
formula IIA,
N R4R5
'(R2)t
NR~
IIA
\ \
(CH2)i
(R3)m N (Rio)J
wherein R1, R2, R3, R4, R5, R10, m and t are as defined in formula II; i is 1
to 3; and j is 0 to 4; for example
wherein i is 2, j is 0 or 1 and R10 is (C1-C3)alkyl, wherein the (C1-C3)alkyl
includes both straight and branched chain radicals of up to 3 carbon atoms; or
wherein m is 0 or 1 and R3 is (C1-C3)alkyl or halogen, wherein the (C1-
C3)alkyl includes both straight and branched chain radicals of up to 3 carbon
atoms; or
wherein the compound is [4-(4-Methylpiperazin-1-yl)phenyl]-(1,2,3,4-
tetrahydroacridin-9-yl)amine, 2-{4-[4-(1,2,3,4-Tetrahydroacridin-9-
yl)aminophenyl]piperazin-1-yl}ethanol, [4-(4-Methylpiperazin-1-yl)phenyl]-(2-
methyl-1,2,3,4-tetrahydro-acridin-9-yl)amine, (8-Fluoro-1,2,3,4-
tetrahydroacridin-9-yl)-[4-(4-methylpiperazin-1-yl)phenyl]amine, [4-(4-
2o Methylpiperazin-1-yl)phenyl]-(2,7-dimethyl-1,2,3,4-tetrahydroacridin-9-
yl)amine,
[4-(4-Methylpiperazin-1-yl)phenyl]-(7,8,9,10-tetrahyd ro-6H-
cyclohepta[b]quinolin-11-yl)amine or [4-(4-Methylpiperazin-1-yl)phenyl]-
(1,1,3,3-
tetramethyl-1,2,3,4-tetrahydroacridin-9-yl)amine.
2s Another possible subgroup of the compound of formula II is a compound of
formula IIB,
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
NR4R5
(R2)t
NR~
IIB
Ra
A
i
(R3)m N Rb
wherein A, R1, R2, R3, Rq., R5, m and t are as defined in formula II; and
Ra and Rb are independently H, OH, halogen, (C1-Cg)alkyl, (C2-
5 C6)alkenyl, (C2-C6)alkynyl, (C1-C6)alkoxy, halo(C1-C6)alkyl, N02, NH2,
mono- or di(C1-C6)alkylamino, (C1-C6)alkyl-S- or CN; or Ra and Rb form,
together with the carbon ring atoms to which they are attached, a condensed
five to seven membered carbocyclic ring optionally substituted with one to
three
substituent(s) R10; each independently being OH, halogen, (C1-C6)alkyl, (C1-
1o Cg)alkoxy, mono- or di(C1-Cg)alkylamino or hydroxy(C1-Cg)alkyl; for example
wherein A is a benzene ring; or
wherein m is 0 or 1; or
wherein R3 is (C1-C6)alkyl or (C1-Cg)alkoxy; or
wherein Ra and Rb are independently H or (C1-C3)alkyl, wherein the (C1-
C3)alkyl includes both straight and branched chain radicals of up to 3 carbon
atoms; or
wherein A is a six membered carbocyclic ring and Ra and Rb form,
together with the carbon ring atoms to which they are attached, a condensed
five to seven membered carbocyclic ring, wherein the carbocyclic ring can
optionally be substituted with one to three substituent(s) each independently
being OH, halogen, (C1-Cg)alkyl, (C1-Cg)alkoxy or hydroxy(C1-Cg)alkyl; or
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
16
wherein the compound is (3-Ethyl-2,8-dimethylquinolin-4-yl)-[4-(4-
methylpiperizin-1-yl)phenyl]amine, (2-Methylquinolin-4-yl)-[4-(4-
methylpiperazin-1-yl)phenyl]amine, (3-Ethyl-2,6-dimethylquinolin-4-yl)-[4-(4-
methylpiperazin-1-yl)phenyl]amine, (3-Ethyl-6-methoxy-2-methylquinolin-4-yl)-
[4-(4-methylpiperazin-1-yl)phenyl]amine, (3-Ethyl-2-methylquinolin-4-yl)-[4-(4-
methylpiperazin-1-yl)phenyl]amine, 4-[4-(4-Methylpiperazin-1-
yl)phenylamino]quinoline-3-carbonitrile, 3-Isopropyl-2-methylquinolin-4-yl)-[4-
(4-
methylpiperazin-1-yl)phenyl]amine, (2,3-Dimethylquinolin-4-yl)-[4-(4-
methylpiperazin-1-yl)phenyl]amine, [4-(4-Methylpiperazin-1-yl)phenyl]-
(1,2,3,4,5,6,7,8-octahydroacridin-9-yl)amine or (3-Ethyl-2-methylquinolin-4-
yl)-
methyl-[4-(4-methylpiperazin-1-yl)phenyl]amine.
Another possible subgroup of the compound of formula II is a compound of
formula IIC,
NR4R5
,(R2)t
NR~
\ \ (R~o)j IIC
H2)i
(R3)m N N
R»
wherein R1, R2, R3, R4, R5, R10, R11, m and t are as defined in formula
II; i is 1 or 2; and j is 0 to 3.
Another possible subgroup of the compound of formula II is a compound of
formula IID,
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
17
N R4R5
'(R2)t
NR~ IID
\ \ \
(R3)m N (R'3)p
wherein R1, R2, R3, R'g, R4,R5, m and t are as defined in formula II; and p
is 0 to 3; for example
wherein m is 1 and R3 is (C1-C6)alkoxy; or
wherein the compound is 2-{4-[4-(Acridin-9-yl)aminophenyl]piperazin-1-yl}-
ethanol, (4-Methoxyacridin-9-yl)-[4-(4-methylpiperazin-1-yl)-phenyl]amine,
Acridin-9-yl-[4-(piperidin-1-yl)phenyl]amine, Acridin-9-yl-[4-(4-
benzylpiperazin-1-
yl)phenyl]amine, Acridin-9-yl-[4-(4-methylpiperidin-1-yl)phenyl]amine, Acridin-
9-
1o yl-[4-(3-hydroxymethylpiperidin-1-yl)-phenyl]amine, Acridin-9-yl-[4-
pyrrolidin-1-
yl)phenyl]amine, Acridin-9-yl-[4-(4-cyclopropylpiperazin-1-yl)phenyl]amine,
Acridin-9-yl-[4-(4-isopropylpiperazine-1-yl)phenyl]amine, (Acridin-9-yl)-
methyl-
[4-(4-methylpiperazin-1-yl)phenyl]amine or Acridin-9-yl-[2,5-diethoxy-4-
(morpholin-4-yl)phenyl]amine.
In a possible subgroup of the compound of Formula II, IIA, IIB, IIC, or IID,
R4 and R5 form, together with the N-atom to which they are attached,
- ~N R6
2o wherein Rg is (C1-Cg)alkyl, (C1-C6)alkenyl, (Cg-C6)cycloalkyl or
hydroxy(C1-Cg)alkyl, for example, R6 is (C1-Cg)alkyl.
The compounds of formulae I and II and the subgroups IA, IB, IC, ID,
IIA, IIB, IIC, and IID thereof, as well as the pharmaceutically acceptable
esters
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
18
and salts thereof, are referred to below as the compounds of the invention,
unless otherwise indicated.
The compounds of the invention may have chiral carbon atoms) in their
structure. The invention includes within its scope all the possible
stereoisomers
of the compounds, including geometric isomers, e.g. Z and E isomers (cis and
trans isomers), and optical isomers, e.g. diastereomers and enantiomers.
Furthermore, the invention includes in its scope both the individual isomers
and
any mixtures thereof, e.g. racemic mixtures. The individual isomers may be
obtained using the corresponding isomeric forms of the starting material or
they
may be separated after the preparation of the end compound according to
conventional separation methods. For the separation of, for example, optical
isomers, e.g. enantiomers, from the mixture thereof the conventional
resolution
methods, e.g. fractional crystallisation, may be used.
Physiologically acceptable salts, e.g. acid addition salts with both organic
and inorganic acids are well known in the field of pharmaceuticals. Non-
limiting
examples of these salts include chlorides, bromides, sulfates, nitrates,
phosphates, sulfonates, formates, tartrates, maleates, citrates, benzoates,
salicylates and ascorbates. Pharmaceutically acceptable esters, when
2o applicable, may be prepared by known methods using pharmaceutically
acceptable acids that are conventional in the field of pharmaceuticals and
that
retain the pharmacological properties of the free form. Non-limiting examples
of
those esters include esters of aliphatic or aromatic alcohols, e.g. lower
alkyl
esters, e.g. methyl, ethyl and propyl esters.
Terms employed herein have the following meanings: A halogen or halo
refers to fluorine, chlorine, bromine or iodine, e.g. fluorine or chlorine.
The term
(C1-C6)alkyl as employed herein as such or as part of another group includes
both straight, and branched chain radicals of up to 6 carbon atoms, for
example
of 1 to 4 carbon atoms, e.g. methyl, ethyl, n-propyl, i-propyl, n-butyl, i-
butyl or s-
3o butyl. The term (C1-C6)alkoxy as such or as part of another group refers to
-O-
(C1-Cg)alkyl, wherein (C1-Cg)alkyl is as defined above. The term (C2-
Cg)alkenyl includes both straight and branched chain radicals of up to 6
carbon
atoms, for example of 2 to 4 carbon atoms, containing (a) double bond(s). The
term (C2-Cg)alkynyl includes both straight and branched chain radicals of up
to
6 carbon atoms, for example of 2 to 4 carbon atoms, containing (a) triple
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
19
bond(s). The term halo-(C1-Cg)alkyl refers to (C1-Cg)alkyl radical, as defined
above, that is substituted by one or more halo radicals as defined above, for
example trifluoromethyl, difluoromethyl etc. The term mono-or di(C1-
Cg)alkylcarbamoyl refers to a carbamoyl radical which is N-substituted with
one
or two (C1-C6)alkyl radical(s), as defined above.
The compounds of the invention can be prepared analogously or
according to a variety of synthetic routes known in the literature using
suitable
starting materials, for example, according to or analogous to methods
described by B.F. Cain et al. in J.Med.Chem., vo1.20(8), 1977, pp. 987-996,
1o and by W.A. Denny et al. in J.Med.Chem., vo1.25, 1982, p.276-315, the
contents of which are hereby incorporated by reference.
In general, the compounds of the invention can be prepared e.g.
analogously or according to the following reaction scheme 1:
Scheme 1
(R2)t NRaRs
NR4Rs R N
CI
1
/ \ Ra (R2)t (R3)m
\ \ Ra
i
(R3)m \ ~ ~ + I /
N Rb /
N Rb
NHR1
III IV I'
wherein Ra, Rb, R1, R2, R3, R4, R5, m and t are as defined above.
The above reaction is a conventional acid-catalyzed coupling of the
chloro-compound of formula II I with a substituted aromatic amine of formula
IV.
The reaction is carried out at room or elevated temperature in a suitable
2o solvent, e.g. alcohol, such as methanol, in acidic conditions, to obtain
the
compound of formula I' which is then isolated from the reaction mixture in a
usual manner.
The starting compounds III and IV are commercially available or can be
prepared according to or analogously to the methods described in the
literature
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
(see e.g. J.Med.Chem., vo1.20(8), 1977, pp. 987-996, and J.Med.Chem., vo1.25,
1982, p.276-315, as mentioned above).
Accordingly, a substituted aromatic NR1-amine IV can e.g. be prepared
starting from the corresponding nitro compound which is reduced with a
reducing
agent, e.g. in the presence of SnC12~H20, in a suitable solvent, e.g. DMF, and
optionally alkylated with R1 in a manner known in the art, when R1 =
(C1_6)alkyl is
desired.
The starting material III can be prepared e.g. according to following scheme
2:
Scheme 2
O CI
R m ~R3)m ~ ~ Ra
Ra gOCl2
N/\Rb DMF / N~Rb
V III
wherein Ra, Rb, R3, and m are as defined above.
In scheme 2 a compound V is reacted e.g. with thionyl chloride in the
presence of a small amount of DMF, to obtain the 9-chlorinated reactant III.
The reaction is carried out at room or elevated temperature.
It is obvious for a skilled person that, in the above reactions, any starting
material or intermediate can be protected, if necessary, in a manner well
known
in the chemical field. Any protected functionality can subsequently be
deprotected in a manner known in the art.
2o The above disclosed synthetic routes are meant to illustrate the
preparation of the compounds of the invention and the preparation is by no
means limited thereto, i.e. other synthetic methods that are within the
general
knowledge of a skilled person are also possible.
The compounds of the invention may be converted, if desired, into their
pharmaceutically acceptable salt or ester form using methods well known in the
art.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
21
The following examples are meant only for illustrating purposes and
does not limit the scope of the invention defined in claims.
EXAMPLE 1
Acridin-9-yl-[4-(4-methylpiperazin-1-yl)-phenyl]amine
Step 1
1.04 g (5.0 mmol) of N-(4-nitrophenyl)piperazine was dissolved in 5 ml of
dimethylformamide. Sodium hydride (0.24 g, 6.0 mmol) was added to the
reaction mixture in three portions under nitrogen atmosphere and cooling over
a period of 10 min. After 30 min of stirring, 0.31 ml (6.0 mmol) of methyl
iodide
was added dropwise to the reaction mixture at 0 °C. Stirring was
continued for
1 h at room temperature, the reaction mixture was then evaporated to dryness,
and purified by silica gel chromatography (methylene chloride : methanol
triethylamine 94 : 5 : 1 ) to yield 1.0 g (90 %) of 1-methyl-4-(4-
~5 nitrophenyl)piperazine.
Step 2
0.999 g (4.5 mmol) of 1-methyl-4-(4-nitrophenyl)piperazine, 10.15 g (45
mmol) of tin(II)chloride dihydrate and 20 ml of dimethylformamide were mixed
and stirred overnight at 80 °C. Most of dimethylformamide was
evaporated
2o under vacuum. The remaining slurry was poured into ice water, neutralized
with a sodium bicarbonate (sat.) solution, and filtered. The filtrate was
extracted
several times with ethylacetate and chloroform to provide 0.636 g (74 %) of 4-
(4-methylpiperazin-1-yl)aniline.
Step 3
25 0.488 g (2.5 mmol) of 9-(10H)acridone, 2.5 ml of thionyl chloride and a
catalytic amount (a few drops) of dimethylformamide were mixed at 80
°C. After
30 min of stirring, the reaction mixture was evaporated, the residue was
dissolved in chloroform and poured into cold aqueous ammonia. The ammonia
solution was extracted several times with chloroform. The combined organic
3o phases were washed with 2M ammonia solution, dried over sodium sulfate and
evaporated to obtain 0.517 g (97 %) of 9-chloroacridine.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
22
Step 4
0.191 g (1.0 mmol) of 4-(4-methylpiperazin-1-yl)aniline, 2.5 ml of
methanol and a few drops of concentrated hydrochloric acid were mixed and
heated under reflux. The 9-chloroacridine (1-1,5 equivalents) and 2.5 ml
s methanol were mixed separately and added to the reaction mixture in small
portions. After 30 min of stirring, two drops of concentrated hydrochloric
acid
were added, and heating was continued for 2 h. The reaction mixture was then
evaporated to dryness and purified by chromatography (silica gel column;
gradient from 100 % methylene chloride to 90 % methylene chloride and 10
~o methanol; when 4-(4-methylpiperazin-1-yl)aniline eluted from the column,
the
eluent was changed to methylene chloride : methanol : triethylamine 94 : 5 : 1
).
A final amount of 0.126 g (34 %, overall yield 12 %) of the title compound in
pure form was obtained.
~H NMR (DMSO-ds, 500 MHz): 8.05 (2H, m), 7.73 (2H, m), 7.66 (2H, m),
15 7.13 (2H, m), 6.97 (4H, m), 3.35 (4H, m), 2.98 (4H, m), 2.58 (3H, s); MS
(E1+):
368 (M+)
EXAMPLE 2
Acridin-9-yl-[2,5-diethoxy-4-(morpholin-4-yl)phenyl]amine
2o Following the procedure outlined in Step 4 of Example 1, but substituting
2,5-diethoxy-4-morpholinoaniline dihydrochloride for 4-(4-methylpiperazin-1-
yl)aniline, afforded the title compound with an overall yield of 23 %.
'H NMR (CDC13, 500 MHz): 8.19 (2H, m), 8.13 (2H, m), 7.56 (1 H, s),
7.42 (2H, m), 7.04 (2H, m), 6.42 (1 H, s), 3.91 (4H, m), 3.15 (4H, m), 3,10
(4H,
2s q, J = 7.27 Hz), 1.41 (6H, t, J = 7.27 Hz); MS (ESI+ TOF): 444 (M+)
EXAMPLE 3
Acridin-9-yl-[4-(morpholin-4-yl)phenyl]amine
Following the procedure outlined in Step 4 of Example 1, but substituting
30 4-(morpholin-1-yl)aniline for 4-(4-methylpiperazin-1-yl)aniline, afforded
the title
compound with an overall yield of 64 %.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
23
~H NMR (CDC13, 500 MHz): 7.99 (4H, m), 7.55 (2H, m), 7.17 (2H, m),
7.04 (2H, m), 6.88 (2H, m), 3.88 (4H, m), 3.15 (4H, m); MS (ESI+ TOF): 356
(M+)
EXAMPLE 4
(3-Ethyl-2,8-dimethylquinolin-4-yl)-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure outlined in Step 4 of Example 1, but substituting
4-chloro-2,8-dimethyl-3-ethylquinoline for 9-chloroacridine, afforded the
title
compound with 27 % overall yield.
'H NMR (CDC13, 500 MHz): 7.60 (1 H, m), 7.41 (1 H, m), 7.16 (1 H, m),
6.79 (2H, m), 6.62 (2H, m), 5.62 (1 H, s), 3.12 (4H, m), 2.79 (3H, s), 2.79
(2H, q,
J = 7.63 Hz), 2.78 (3H, s), 2.59 (4H, m), 2.36 (3H, s), 1.15 (3H, t, J = 7.63
Hz);
MS (ESI+ TOF): 375 (M+)
EXAMPLE 5
(2-Methylquinolin-4-yl)-(4-(4-methylpiperazin-1-yl)phenyl]amine
Following the procedure outlined in Step 4 of Example 1, but substituting
4-chloro-2-methylquinoline for 9-chloroacridine, afforded the title compound
2o with 24 % overall yield.
'H NMR (CDC13, 500 MHz): 8.02 (1 H, m), 7.91 (1 H, m), 7.64 (1 H, m),
7.44 (1 H, m), 7.23 (2H, m), 7.00 (2H, m), 6.59 (1 H, m), 3.26 (4H, m), 2.61
(4H,
m), 2.55 (3H, s), 2.38 (3H, s); MS (ESI+ TOF): 333 (M+)
EXAMPLE 6
[4-(4-Methylpiperazin-1-yl)phenyl]-(quinolin-4-yl)amine
Following the procedure outlined in Step 4 of Example 1, but substituting
4-chloroquinoline for 9-chloroacridine, afforded the title compound with 11
overall yield.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
24
'H NMR (CDC13, 500 MHz): 8.28 (1 H, m), 8.25 (1 H, m), 8.06 (1 H, m),
7.66 (1 H, m), 7.50 (1 H, m), 7.25 (2H, m), 6.97 (2H, m), 3.25 (4H, m), 2.64
(4H,
m), 2.40 (3H, s); MS (ESI+ TOF): 319 (M+)
EXAMPLE 7
(3-Ethyl-2,6-dimethylquinolin-4-yl)-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure outlined in Step 4 of Example 1, but substituting
4-chloro-1,6-dimethyl-3-ethylquinofine for 9-chloroacridine, afforded the
title
1o compound with 18 % overall yield.
~H NMR (CDC13, 500 MHz): 7.90 (1 H, m), 7.49 (1 H, m), 7.40 (1 H, m),
6.81 (2H, m), 6.67 (2H, m), 3.12 (4H, m), 2.77 (2H, q, J = 7.56 Hz), 2.78 (3H,
s), 2.58 (4H, m), 2.36 (3H, s), 2.35 (3H, s), 1.15 (3H, t, J = 7.56 Hz); MS
(ESI+
TOF): 375 (M+)
EXAMPLE 8
(3-Ethyl-6-methoxy-2-methylquinolin-4-yl)-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure outlined in Step 4 of Example 1, but substituting
4-chloro-3-ethyl-6-methoxy-2-methylquinoline for 9-chloroacridine, afforded
the
title compound with 5 % overall yield.
MS (ESI+ TOF): 391 (M+)
EXAMPLE 9
(3-Ethyl-2-methylquinolin-4-yl)-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure outlined in Step 4 of Example 1, but substituting
4-chloro-3-ethyl-2-methylquinoline for 9-chloroacridine, afforded the title
compound with 17 % overall yield.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
~H NMR (CDC13, 500 MHz): 7.91 (1 H, m), 7.72 (1 H, m), 7.55 (1 H, m),
7.25 (1 H, m), 6.80 (2H, m), 6.67 (2H, m), 5.72 (1 H, s), 3.11 (4H, m), 2.79
(2H,
q, J = 7.59 Hz), 2.77 (3H, s), 2.57 (4H, m), 2.34 (3H, s), 1.17 (3H, t, J =
7.59
Hz); MS (ESI+ TOF): 361 (M+)
5
EXAMPLE 10
(4-Methoxyacridin-9-yl)-[4-(4-methylpiperazin-1-yl)phenyl]amine
Following the procedure outlined in Step 3 of Example 1, but substituting
9-hydroxy-4-methoxyacridine for 9-(10H)acridone, afforded 9-chloro-4-
methoxyacridine, which was reacted with 4-(4-methylpiperazin-1-yl)aniline
(according to the Step 4 in the procedure of Example 1 ) to obtain the title
compound with 24 % overall yield.
MS (ESI+ TOF): 399 (M+)
~5 EXAMPLE 11
[4-(4-Methylpi perazin-1-yl)phenyl]-(1,2,3,4-tetrahydroacridin-9-
yl)amine
Following the procedure outlined in Step 3 of Example 1, but substituting
1,2,3,4-tetrahydro-9-(10H)acridone for 9-(10H)acridone, afforded 9-chloro-
20 1,2,3,4-tetrahydroacridine, which was reacted with 4-(4-methylpiperazin-1-
yl)aniline (according to Step 4 in the procedure of Example 1 ) to obtain the
title
compound with 46 % overall yield.
'H NMR (CDC13, 500 MHz): 7.96 (1 H, m), 7.72 (1 H, m), 7.56 (1 H, m),
7.27 (1 H, m), 6.83 (2H, m), 6.73 (2H, m), 5.82 (1 H, s), 3.14 (6H, m), 2.67
(2H,
25 m), 2.58 (4H, m), 2.35 (3H, s), 1.94 (2H, m), 1.84 (2H, m); MS (ESI+ TOF):
373
(M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
26
EXAMPLE 12
Acridin-9-yl-[4-(piperidin-1-yl)phenyl]amine
0.202 g (1.0 mmol) of 1-bromo-4-nitrobenzene and 0.20 ml of piperidine
(2.0 mmol) were dissolved in 3 ml of dimethylsulfoxide. 0.207 g (1.5 mmol) of
potassium carbonate was added and the reaction mixture was heated to
100°C. After 2 h 100 ml of water was added, and the mixture was
extracted a
few times with dichloromethane. The combined organic layers were dried over
sodium sulfate and evaporated to obtain crude 4-(piperidin-1-yl)-1-
nitrobenzene. Following the procedure outlined in Step 2 of Example 1, but
substituting 4-piperidin-1-yl-1-nitrobenzene for 1-methyl-4-(4-
nitrophenyl)piperazine, afforded 4-piperidin-1-ylaniline, which was reacted
with
9-chloroacridine (according to Step 4 in the procedure of Example 1 ) to
obtain
the title compound with 6 % overall yield.
~H NMR (CDC13, 500 MHz): 8.05 (2H, m), 8.00 (2H, m), 7.48 (2H, m),
7.09 (4H, m), 6.86 (2H, m), 3.13 (4H, m), 1.71 (4H, m), 1.58 (2H, m); MS (ESI+
TOF): 354 (M+)
EXAMPLE 13
Acridin-9-yl-[4-(4-methylpiperidin-1-yl)phenyl]amine
2o Following the procedure of Example 12, but substituting 4-
methylpiperidine for piperidine, afforded the title compound with 6 % overall
yield.
MS (ESI+ TOF): 368 (M+)
EXAMPLE 14
Acridin-9-yl-[4-(3-hydroxymethylpiperidin-1-yl)phenyl]amine
Following the procedure of Example 12, but substituting 3-
hydroxymethylpiperidine for piperidine, afforded the title compound with 3
overall yield.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
27
'H NMR (CDC13, 500 MHz): 7.93 (2H, m), 7.81 (2H, m), 7.47 (2H, m),
7.04 (4H, m), 6.92 (2H, m), 3.68 (2H, m), 3.59 (2H, m), 2.78 (1 H, m), 2.61 (1
H,
m), 1.95 (1 H, br s), 1.83 (2H, m), 1.72 (1 H, m), 1.23 (2H, m); MS (ESI+
TOF):
384 (M+)
EXAMPLE 15
Acridin-9-yl-[4-(pyrrolidin-1-yl)phenyl]amine
Following the procedure of Example 12, but substituting pyrrolidine for
piperidine, afforded the title compound with 50 % overall yield.
'H NMR (CDC13, 500 MHz): 8.00 (4H, m), 7.56 (2H, m), 7.18 (2H, m),
7.00 (2H, m), 6.50 (2H, m), 3.27 (4H, m), 2.02 (4H, m); MS (ESI+ TOF): 340
(M+)
EXAMPLE 16
~5 Acridin-9-yl-[4-(piperazine-1-yl)phenyl]amine
Following the procedure outlined in Step 2 of Example 1, but substituting
N-(4-nitrophenyl)piperazine for 1-methyl-4-(4-nitrophenyl)piperazine, afforded
4-(piperazin-1-yl)aniline, which was reacted with 9-chloroacridine (according
to
Step 4 in the procedure of Example 1 ) to obtain the title compound with 16
20 overall yield.
'H NMR (CD30D, 500 MHz): 8.21 (2H, m), 7.94 (4H, m), 7.42 (2H, m),
7.38 (2H, m), 7.20 (2H, m), 3.56 (4H, m), 3.43 (4H, m); MS (ESI+ TOF): 355
(M+)
25 EXAMPLE 17
Acridin-9-yl-[4-(4-acetylpiperazine-1-yl)phenyl]amine
35 mg (0.10 mmol) of 9-[4-(piperazin-1-yl)phenyl]aminoacridine
(Example 16) was dissolved in 2 ml of chloroform, 7.1 ~I (0.10 mmol) of acetyl
chloride and catalytical amounts of pyridine and triethylamine were added.
After
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
28
2 h of stirring at room temperature, the solvents were removed under vacuum,
and the residue was purified by silica gel chromatography (eluent chloroform
methanol 6:1 ). The fractions with the title compound were combined,
evaporated, taken up to water and extracted with chloroform. After drying over
sodium sulfate, the organic layer was evaporated to obtain the title compound
(6 %, overall yield 1 %).
MS (ESI+ TOF): 397 (M+)
EXAMPLE 18
Acridin-9-yl-[4-(4-benzylpiperazine-1-yl)phenyl]amine
Following the procedure outlined in Step 1 of Example 1, but substituting
9-[4-(piperazin-1-yl)phenyl]aminoacridine (Example 16) for N-(4-
nitrophenyl)piperazine, and benzyl bromide for methyl iodide, afforded the
title
compound (15 %, overall yield 2 %).
~5 'H NMR (CDC13, 500 MHz): 8.00 (4H, m), 7.48 (2H, m), 7.34 (4H, m),
7.28 (1 H, m), 7.12 (4H, m), 6.88 (2H, m), 3.59 (2H, s), 3.21 (4H, m), 2.63
(4H,
m); MS (ESI+ TOF): 445 (M+)
EXAMPLE 19
2o Acridin-9-yl-[4-(4-isopropylpiperazine-1-yl)phenyl]amine
Following the procedure outlined in Step 1 of Example 1, but substituting
9-[4-(piperazin-1-yl)phenyl]aminoacridine (Example 16) for N-(4-
nitrophenyl)piperazine, and isopropyl iodide for methyl iodide, afforded the
title
compound (11 %, overall yield 2 %).
25 MS (ESI+ TOF): 397 (M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
29
EXAMPLE 20
2-{4-[4-(1,2,3,4-Tetrahyd roacridi n-9-yl)am i nophenyl] pi perazi n-1-
yl}ethanol
0.14 ml (2.0 mmol) of 2-bromoethanol and 0.17 ml (2.4 mmol) of acetyl
chloride were dissolved in 2 ml of dichloromethane. 0.28 ml (2.0 mmol) of
triethylamine was added, and the reaction mixture was stirred at room
temperature. After 1 h 50 ml of dichloromethane was added to the reaction
mixture, which was then washed with a sodium bicarbonate (sat.) solution, a 10
citric acid solution and water. The organic phase was dried over sodium
sulfate and evaporated to obtain 0.241 g (72 %) of 2-bromoethylacetate.
Following the procedure outlined in Step 1 of Example 1, but substituting 2-
bromoethylacetate for methyl iodide, afforded 2-[4-(4-nitrophenyl)piperazin-1-
yl]ethylacetate (64 %), which was reduced according to the procedure of Step 2
in Example 1 to give the corresponding amine 2-[4-(4-aminophenyl)piperazin-1-
yl]ethylacetate in quantitative yield. 2-[4-(4-aminophenyl)piperazin-1-
yl]ethylacetate was reacted with 9-chloro-1,2,3,4-tetrahydroacridine
(according
to the procedure of Step 4 in Example 1) to obtain 2-{4-[4-amino-(1,2,3,4-
tetrahydroacridin-9-yl)phenyl]piperazin-1-yl)ethyl acetate with 47 % yield.
Treating the ester with 4 equivalents of lithium hydroxide in a dioxane /water
2o solution overnight, afforded the title compound with 60 % yield (overall
yield 20
%).
MS (ESI+ TOF): 403 (M+)
EXAMPLE 21
2-~4-[4-(Acridin-9-yl)aminophenyl]piperazin-1-yl}ethanol
Following the procedure outlined in Step 1 of Example 1, but substituting
9-[4-(piperazin-1-yl)phenyl]aminoacridine (Example 16) for N-(4-
nitrophenyl)piperazine, and 2-bromoethyl-acetate for methyl iodide, afforded 2-
(4-[4-amino-(1,2,3,4-tetrahydroacridin-9-yl)phenyl]piperazin-1-yl)ethyl
acetate
(31 %). Treating the ester with 8 equivalents of lithium hydroxide in dioxane
/water solution overnight, afforded the title compound with 37 % yield
(overall
yield 2 %).
MS (ESI+ TOF): 399 (M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
EXAMPLE 22
Acridin-9-yl-[4-(4-cyclopropylpiperazine-1-yl)phenyl]amine
mg (0.10 mmol) of 9-[4-(piperazin-1-yl)phenyl~aminoacridine
(Example 16) was dissolved in 1 ml of methanol. 571 (1.0 mmol) of acetic acid,
221 (0.11 mmol) of (1-ethoxycyclopropyloxy)-trimethylsilane and a small
amount of 3 ~ molecular sieves were added. The reaction mixture was stirred
at room temperature under nitrogen atmosphere. After 30 min 28 mg (0.45
mmol) of sodium cyanoborohydride was added and the reaction mixture was
heated at 50°C over night. The solvents were removed under vacuum, and
the
residue was purified by chromatography (silica gel column, eluent
chloroform/methanol 6:1 ) to obtain 3.8 mg (10 %, overall yield 2 %) of the
title
compound.
MS (ESI+ TOF): 395 (M+)
15 EXAMPLE 23
4-[4-(4-Methylpiperazin-1-yl)phenylamino]quinoline-3-carbonitrile
0.91 ml (10 mmol) of aniline and 1.69 g (10 mmol of
ethyl(ethoxymethylene)cyanoacetate were dissolved in 10 ml of pyridine and
heated under reflux. After 3 h pyridine was removed under vacuum, and the
2o residue was purified by chromatography (silica gel column, eluent 1
methanol in dichloromethane). 1.08 g (50 %) of
ethyl(anilinomethylene)cyanoacetate were obtained. The compound was
cyclized by heating in a biphenyl/phenyl ether mixture. After cooling a
precipitate was filtered and washed with diethyl ether to give 4-
2s hydroxyquinoline-3-carbonitrile (49 %). Following the procedure outlined in
Step
3 of Example 1, but substituting 4-hydroxyquinoline-3-carbonitrile for 9-
(10H)acridone, 4-chloro-3-cyanoquinoline (90 %) was obtained, which was
reacted with 4-(4-methylpiperazin-1-yl)aniline (according to the procedure of
Step 4 in Example 1 ), to afford the title compound with 12 % yield (overall
yield
30 3 %).
'H NMR (CDC13, 500 MHz): 8.66 (1 H, s), 8.01 (1 H, m), 7.78 (1 H, m),
7.73 (1 H, m), 7.42 (1 H, m), 7.15 (2H, m), 6.94 (2H, m), 3.29 (4H, m), 2.63
(4H,
m), 2.39 (3H, s); MS (ESI+ TOF): 344 (M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
EXAMPLE 24
31
(3-Isopropyl-2-methylquinolin-4-yl)-(4-(4-methylpiperazin-1-
yl)phenyl]amine
4.56 ml (50 mmol) of aniline and 10.7 ml (60 mmol) of ethyl 2-
isopropylacetoacetate were mixed with 50 ml of chloroform. 0.48 g (2.5 mmol)
of para-toluenesulfonic acid was added, and the reaction mixture was refluxed
with continuous removal of the water produced in the reaction. After 2 d
chloroform was removed in vacuum, and the residue was refluxed in 10 ml of
phenyl ether. After cooling, the precipitate was filtered and washed with
diethyl
ether to give 4-hydroxy-3-isopropyl-2-methylquinoline (21 %). Following the
procedure of Step 3 in Example 1, but substituting 4-hydroxy-3-isopropyl-2-
methylquinoline for 9-(10H)acridone, 4-chloro-3-isopropyl-2-methylquinoline
(100 %) was obtained, which was reacted with 4-(4-methylpiperazin-1-
yl)aniline (following the procedure of Step 4 in Example 1 ), to afford the
title
compound with 61 % yield (overall yield 13 %).
'H NMR (CDC13, 500 MHz): 7.95 (1 H, m), 7.73 (1 H, m), 7.54 (1 H, m),
7.24 (1 H, m), 6.79 (2H, m), 6.60 (2H, m), 5.72 (1 H, s), 3.61 (1 H, q, J =
7.28),
3.11 (4H, m), 2.81 (3H, s), 2.57 (4H, m), 2.34 (3H, s), 1.38 (6H, d, J =
7.28);
MS (ESI+ TOF): 375 (M+)
EXAMPLE 25
(2,3-Dimethylquinolin-4-yl)-[4-(4-methylpiperazin-1-yl)phenyl]amine
Following the procedure of Example 24, but substituting ethyl 2-
methylacetoacetate for ethyl 2-isopropylacetoacetate, afforded the title
compound with 11 % yield.
'H NMR (CDC13, 500 MHz): 8.02 (1 H, m), 7.78 (1 H, m), 7.59 (1 H, m),
7.33 (1 H, m), 6.83 (2H, m), 6.69 (2H, m), 5.90 (1 H, s), 3.13 (4H, m), 2.73
(3H,
s), 2.58 (4H, m), 2.35 (3H, s), 2.24 (3H, s); MS (ESI+ TOF): 347 (M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
EXAMPLE 26
32
[4-(4-Methylpiperazin-1-yl)phenyl]-(2-methyl-1,2,3,4-tetrahydro-
acridin-9-yl)amine
1.37 g (10 mmol) of 2-aminobenzoic acid and 1.23 ml (10 mmol) of 4-
s methylcyclohexanone were dissolved in 10 ml of phosphorus oxychloride and
the reaction mixture was heated under reflux in a nitrogen atmosphere. After 3
h most of the phosphorus oxychloride was removed under vacuum. The
remaining brown syrup was poured into a cold sodium bicarbonate (sat.)
solution and washed once with chloroform. The yellow precipitate forming in
the
basic water solution was filtered (2-methyl-1,2,3,4-tetrahydro-9-(10H)-
acridone,
46 %). Following the procedure of Step 3 in Example 1, but substituting 2-
methyl-1,2,3,4-tetrahydro-9-(10H)acridone for 9-(10H)acridone, gave 9-chloro-
2-methyl-1,2,3,4-tetrahydroacridine (40 %), which was reacted with 4-(4-
methylpiperazin-1-yl)aniline (according to the procedure of Step 4 in Example
15 1 ), to afford the title compound with 39 % yield (overall yield 7 %).
~H NMR (CDC13, 500 MHz): 7.97 (1 H, m), 7.70 (1 H, m), 7.55 (1 H, m),
7.24 (1 H, m), 6.83 (2H, m), 6.72 (2H, m), 5.81 (1 H, br s), 3.23 (1 H, m),
3.14
(5H, m), 2.86 (1 H, m), 2.57 (4H, m), 2.35 (3H, s), 2.24 (1 H, m), 2.03 (1 H,
m),
1.93 (1 H, m), 1.57 (1 H, m), 1.10 (3H, d); MS (ESI+ TOF): 387 (M+)
EXAMPLE 27
[4-(4-methylpiperazin-1-yl)phenyl]-(7,8,9,10-tetrahydro-6H-
cyclohepta[b]quinolin-11-yl)amine
Following the procedure of Example 26, but substituting cycloheptanone
for 4-methylcyclo-hexanone, afforded the title compound (0,2 %).
(ESI+ TOF): 387 (M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
33
EXAMPLE 28
[4-(4-Methylpiperazin-1-yl)phenyl]-(2,7-dimethyl-1,2,3,4-tetrahydro-
acridin-9-yl)amine
Following the procedure of Example 26, but substituting 2-amino-5-
s methylbenzoic acid for 2-aminobenzoic acid, afforded the title compound (4
%).
'H NMR (CDC13, 500 MHz): 8.10 (1H, m), 7.43 (2H, m), 6.85 (2H, m),
6.83 (2H, m), 6.27 (1 H, br s), 3.37 (1 H, m), 3.18 (4H, m), 2.78 (1 H, m),
2.61
(4H, m), 2.37 (3H, s), 2.33 (3H, s), 2.18 (1 H, m), 2.02 (1 H, m), 1.91 (1 H,
m),
1.52 (1 H, m), 1.09 (1 H, m), 1.10 (3H, d); MS (ESI+ TOF): 401 (M+)
EXAMPLE 29
(8-Fluoro-1,2,3,4-tetrahydroacridin-9-yl)-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure of Example 26, but substituting 2-amino-6-
fluorobenzoic acid for 2-aminobenzoic acid, and cyclohexanone for 4-
methylcyclohexanone, afforded the title compound (3 %).
'H NMR (CDC13, 500 MHz): 7.89 (1 H, m), 7.51 (1 H, m), 7.05 (1 H, m),
6.87 (2H, m), 6.82 (2H, m), 3.22 (4H, m), 3.14 (2H, m), 2.66 (4H, m), 2.41
(3H,
s), 2.34 (2H, m), 1.88 (2H, m), 1.67 (2H, m); MS (ESI+ TOF): 391 (M+)
EXAMPLE 30
[4-(4-Methylpiperazin-1-yl)phenyl]-(1,1,3,3-tetramethyl-1,2,3,4-
tetrahydroacridin-9-yl)amine
1.49 ml (10 mmol) of ethyl 2-aminobenzoate and 1.73 ml (10 mmol) of
3,3,5,5-tetramethyl-cyclohexanone were mixed with 20 ml of toluene. 20 mg
(0.1 mmol) of para-toluenesulfonic acid was added, and the reaction mixture
was refluxed with continuous removal of the water produced in the reaction.
After 9 h, toluene was removed under vacuum, and the residue was refluxed in
10 ml of phenyl ether. After cooling, the precipitate was filtered and washed
3o with diethyl ether to obtain 1,1,3,3-tetramethyl-1,2,3,4-tetrahydro-9(10H)-
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
34
acridone (18 %). Following the procedure of Step 3 in Example 1, but
substituting 1,1,3,3-tetramethyl-1,2,3,4-tetrahydro-9(10H)acridone for 9-
(10H)acridone, gave 9-chloro-1,1,3,3-tetramethyl-1,2,3,4-tetrahydroacridine
(21
%), which was reacted with 4-(4-methylpiperazin-1-yl)aniline (according to the
procedure of Step 4 in Example 1 ), to afford the title compound (3 %, overall
yield 0,1 %).
MS (ESI+ TOF): 429 (M+)
EXAMPLE 31
(1,4-Methano-1,2,3,4-tetrahydroacridin-9-yl)-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure of Example 30, but substituting norcamphore for
3,3,5,5-tetramethyl-cyclohexanone, afforded the title compound (overall yield
1
%).
~5 'H NMR (CDC13, 500 MHz): 7.99 (1 H, m), 7.84 (1 H, m), 7.58 (1 H, m),
7.38 (1 H, m), 7.02 (2H, m), 6.91 (2H, m), 6.13 (1 H, s), 3.44 (1 H, m), 3.21
(4H,
m), 3.00 (1 H, m), 2.61 (4H, m), 2.37 (3H, s), 1.97 (1 H, m), 1.77 (2H, m),
1.48
(2H, m), 1.26 (1 H, m); MS (ESI+ TOF): 385 (M+)
2o EXAMPLE 32
[4-(4-Methylpiperazin-1-yl)phenyl]-(1,2,3,4,5,6,7,8-octahydroacridin-
9-yl)amine
Following the procedure of Example 30, but substituting ethyl 2-amino-1-
cyclohexene-1-carboxylate for ethyl 2-aminobenzoate, and cyclohexanone for
25 3,3,5,5-tetramethylcyclo-hexanone, afforded the title compound (overall
yield
0,2 %).
MS (ESI+ TOF): 377 (M+)
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
EXAMPLE 33
(3-Ethyl-2-methylquinolin-4-yl)-methyl-[4-(4-methylpiperazin-1-
yl)phenyl]amine
Following the procedure of Step 1 in Example 1, but substituting (3-ethyl-
5 2-methylquinolin-4-yl)-[4-(4-methylpiperazin-1-yl)phenyl]amine (Example 9)
for
N-(4-nitrophenyl)piperazine, afforded the title compound (overall yield 12 %).
~H NMR (CDC13, 500 MHz): 8.03 (1 H, m), 7.58 (2H, m), 7.34 (1 H, m),
6.81 (2H, m), 6.42 (2H, m), 3.33 (3H, s), 3.07 (4H, m), 2.80 (3H, s), 2.71
(2H, q,
J = 7.50Hz), 2.57 (4H, m), 2.34 (3H, s), 1.12 (3H, t, J = 7.50 Hz); MS (ESI+
~ o TOF): 375 (M+)
EXAMPLE 34
Acridin-9-yl-methyl-[4-(4-(methylpiperazin-1-yl)phenyl]amine
Following the procedure of Step 1 in Example 1, but substituting acridin-
15 9-yl-[4-(4-methyl-piperazin-1-yl)-phenyl]-amine (Example 1 ) for N-(4-
nitrophenyl)piperazine, afforded the title compound (overall yield 9 %).
'H NMR (CD30D, 500 MHz): 7.8 (2H, br s), 7.54 (2H, m), 7.48 (2H, m),
6.98 (4H, m), 6.79 (2H, m), 3.77 (3H, s), 3.18 (4H, m), 2.65 (4H, m), 2.36
(3H,
s); MS (E1+): 383 (M+)
The compounds of the invention show interesting pharmacological
properties, namely, they exhibit antagonistic affinity for alpha-2
adrenoceptors.
This activity is demonstrated in the pharmacological tests presented below.
EXPERIMENT I: Binding affinity
The affinity of test compounds for the three human alpha-2-adrenoceptor
subtypes (alpha-2A, alpha-2B and alpha-2C) was determined in binding
competition assays with 3H-rauwolscine. The biological material consisted of
membranes from Shionogi S115 cells stably transfected with one of the three
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
36
human alpha-2 subtypes (A. Marjamaki et al., Biochem. Biophys. Acta,
vo1.1134, 1992, p.169). The membrane suspension (about 10 ~g total protein
per sample) and 1 nM of 3H-rauwolscine (specific activity 75-85 Ci/mmol) were
incubated with a minimum of six concentrations of the test compound in a total
volume of 90 ~I (50 mM KH2P04, pH 7.5, at room temperature). Non-specific
binding was defined by 100 ~M oxymetazoline and corresponded to 4-10 % of
the total binding. After 30 min at room temperature, the incubation was
terminated by rapid filtration (TomTec 96 harvester) through presoaked GF/B
glass-fiber mats (Wallac Oy) and three washes with ice-cold 50 mM KH2P04
(pH 7.5, at room temperature). After drying, a solid scintillate (Meltilex;
Wallac
Oy) was melted on filter mats, and the radioactivity was measured (BetaPlate;
Wallac Oy). The analysis of the experiments was carried out by non-linear
least
square curve fitting. 1C50 's were converted to Ki's by using the equation of
Cheng-Prussoff
~5 (K; = ICSO / (1 + [3H-ligand] / Kd, 3H-ligand))~ The K; values for Compound
1
obtained in a minimum of three independent experiments were:
alpha-2A Adrenoceptor: 3150 ~ 50 nM
alpha-2B Adrenoceptor: 1470 ~ 130 nM
alpha-2C Adrenoceptor: 28 ~ 2 nM
EXPERIMENT II: Antagonist activity
Antagonist activity was determined as the ability of compounds to
competetively inhibit epinephrine-stimulated 35S-GTPyS binding to G proteins
(J.R. Jasper et al., Biochem. Pharmacol., vo1.55, 1998, p.1035) in membranes
of CHO cells stably transfected with one of the three human alpha-2 subtypes
(K. Pohjanoksa et al., Eur. J. Pharmacol., vo1.335, 1997, p.53). Membranes (5-
10 ~g of protein per sample) and 12 concentrations of test compound were
preincubated for 30 min at room temperature in 50 mM Tris, 5 mM MgCl2, 150
mM NaCI, 1 mM DTT, 1 mM EDTA, 10 ~M GDP, 30 ~M ascorbic acid, pH 7.4,
3o with a fixed concentration of epinephrine (5 ~M for alpha-2A, 15 ~M for
alpha-
2B, 5 ~M for alpha-2C). Then trace amounts of 35S-GTPyS (0.08 nM- 0.15 nM,
specific activity 1250 Ci/mmol) were added to the incubation mixture. After an
additional 30 min at room temperature, the incubation was terminated by rapid
vacuum filtration through glass fiber filter. Filters were washed three times
with
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
37
ml ice cold wash buffer (20 mM Tris, 5 mM MgCl2, 1 mM EDTA, pH 7.4, at
room temperature), dried and counted for radioactivity in a scintillation
counter.
Analysis of experiments was carried out by nonlinear least square fitting.
Experiments were repeated at least three times. The KB values of Compound 1
were found to correspond to:
alpha-2A Adrenoceptor: 1495 ~ 270 nM
alpha-2B Adrenoceptor: 2175 ~ 345 nM
alpha-2C Adrenoceptor: 16 ~ 6 nM
EXPERIMENT III: Antagonism of dexmedetomidine -induced
locomotor inhibition by atipamezole but not by Compound 1;
demonstration of in vivo alpha2C selectivity of Compound 1
Dexmedetomidine and atipamezole are very potent and specific alpha-2
adrenoceptor agonists and antagonists, respectively, which lack alpha-2
subtype selectivity (H. Scheinin et al., European Journal of Pharmacology,
Molecular Section, vol.151 (1 ), 1988, p.35-42). The alpha-2 agonist -induced
sedation is known to be an alpha-2A -mediated phenomenon that can be
antagonised by alpha-2 antagonists (J. Sallinen et al., Mol. Pharmacol.
Vo1.51,
1997, p.36-46, and A. Haapalinna et al., Naunyn-Schimiedeberg's Arch.
2o Pharmacol. Vo1.356, 1997, p.570-582). The sedative effect of alpha-2
agonists
in mice is measured by the inhibition on locomotor activity. Therefore, we
compared the ability of Compound 1 and atipamezole to antagonise the
dexmedetomidine -induced locomotor inhibition to evaluate the in vivo alpha-
2A adrenoceptor antagonism (and alpha-2C selectivity) of these compounds.
25 Spontaneous locomotor activity of a total of 76 male NMRI mice (B&K,
Sweden) was measured by placing individual animals into a polypropylene
animal cage (38 x 22 x 15 cm). The cages were surrounded with an infrared
photobeam frame system designed for activity measurements (Photobeam
Activity System PAS, Cage Rack, San Diego Instruments, San Diego, CA,
3o USA). The animals were injected with various doses of either Compound 1 or
atipamezole 20 min before injection of dexmedetomidine (50 nmol/kg s.c.).
Spontaneous locomotor activity was measured 20 min after dexmedetomidine
injection.
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
38
The results presented in figure 1 show that atipamezole inhibited
dexmedetomidine -induced sedation with doses 0.3 and 1.0 ~mol/kg s.c. (p <
0.01 ) as was expected. In contrast, Compound 1 did not antagonise the alpha-
2-agonist -induced sedation at all, demonstrating the lack of alpha-2A
antagonism and the alpha-2C selectivity of Compound 1 in vivo.
EXPERIMENT IV: Stress-protective effect of Compound 1 in the
mouse forced swimming test
Exposure of a test animal to an intense stressful stimulus has been
~o observed to propagate a state of behavioural despair. This can be observed
for
example employing the forced swimming test, in which a rat or mouse is put
into a water-filled cylinder. After a vigorous period of attempts to escape
animals adopt an immobile floating posture; the extent of the immobile period
is
monitored and can be reduced by antidepressants and stress-protective
~5 agents. Transgenic mice that lack functional alpha-2C adrenoceptor
tolerated
swim-stress better than their similarly treated wild-type controls (J.
Sallinen et
al., Mol. Psychiatry, vol.4, 1999, p.443-452). Therefore, an increase in the
forced swimming activity can be used as a measure of in vivo alpha-2C
selective antagonism of a compound. The non-selective antagonist
2o atipamezole did not have a clear stress-protective effect and even
increased
vocalizations of test animals (T. Kauppila et al., Eur. J. Pharmacol.,
vo1.205,
1991, p.177-182). This may have resulted from the simultaneous alpha-2A
antagonistic activity of atipamezole, since conventionally non-selective alpha-
2
adrenoceptor antagonists, such as yohimbine, have been found to be
25 anxiogenic (S. Southwick et al., Arch. Gen. Psychiatry, vo1.54, 1997, p.749-
758).
The forced swim test was conducted as originally described employing
the stress sensitive (J. Crawley and L. Davis, Brain Research Bulletin, vol.8,
1982, p.609-612, and US-A-5 902 807) Balb/c mouse strain (B&K, Sweden).
3o The mice were administered either with vehicle (0.1 % DMSO, 5 ml/kg
subcutaneously), Compound 1 0.3 ~mol/kg, or atipamezole 0.3 ~mol/kg 40 min
before putting the mice into a vessel (10 cm diameter, 18.5 cm height, filled
with 25°C water up to 8 cm height). The cumulative activity of each
mouse was
measured between 2 and 6 min after introduction to the vessel. Only vigorous
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
39
attempts to escape (climbing) were registered. The mice (a total of 96) were
tested only once. The results are shown in Figure 2.
Mice administered with Compound 1 tolerated clearly better stress
induced propagation of behavioural despair when compared to vehicle injected
group of mice (1-way ANOVA, followed by LSD post hoc test; p = 0.013) as
was expected for an alpha-2C selective compound. Atipamezole did not have
any clear effect on the activity compared to control mice (p = 0.52). A
possible
marginal effect of atipamezole was expected, since this subtype-non-selective
alpha-2 antagonist blocks also the alpha-2C adrenoceptors. On the other hand
the employed 0.3 ~mol/kg dose of atipamezole was shown to have also
alpha2A antagonism in vivo (Figure 1 ). The employed doses of atipamezole
have also been shown to have clear neurochemical effects, i.e. to stimulate
noradrenaline release in brain (A. Haapalinna et al., Naunyn-Schimiedeberg's
Arch. Pharmacol. Vo1.356, 1997, p.570-582). Therefore, the results support
that
~s alpha-2A adrenoceptor antagonism of atipamezole can counteract the stress-
protective and antidepressant effects of alpha-2C antagonism in vivo (J.
Sallinen et al., Mol. Psychiatry, vol.4, 1999, p.443-452, and US-A-5 902 807).
In general, the compounds of the invention exhibiting alpha-2 adrenoceptor
antagonistic activity may be useful for the treatment of diseases or
conditions
2o wherein alpha-2 antagonists are effective. For example, the compounds can
be
used to treat disorders of the central nervous system, male sexual impotence,
orthostatic hypotension, non-insulin dependent diabetes, and obesity, for
example, disorders of the central nervous system. The compounds can also be
used to reverse effects induced by alpha-2 agonists. Disorders of the central
25 nervous system treatable with the compounds of the invention include
depression, anxiety, post traumatic stress disorder, schizophrenia,
Parkinson's
disease, and other movement disorders.
The selective alpha-2C antagonists of the present invention may be
3o used for the treatment of various diseases or conditions of CNS-system
where
alpha2C-antagonists are indicated to be beneficial (see e.g. US-A-5 902 807,
J.
Sallinen et al., Neuroscience, vol. 86, 1998, p.959-965, J. Sallinen et al.,
J.
Neurosci., vo1.18, 1998, p.3035-3042, and M. Bjorklund et al., Molecular
Pharmacology, vo1.54, 1998, p.569-76, the contents of which are hereby
35 incorporated by reference), for example, in the treatment of schizophrenia
and
depression. Furthermore, the present alpha-2C antagonists can be used as
stress-protective agents, or as agents for the treatment of CNS-disorders
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
induced by stress, e.g. of post-traumatic stress disorder, as indicated, for
example, in US-A-5 902 807 cited above. Because alpha-2C antagonists
appear to stimulate central dopaminergic activity, they can be used as
antiparkinsonian agents in Parkinson's disease and other movement disorders.
Moreover, the present alpha-2C antagonists may also exhibit cognition
enhancing properties and thus may be used in the treatment of Alzheimer's
disease and other dementias.
Due to the selectivity of tissue distribution, the alpha-2C antagonists of
the invention have less or no undesirable side-effects, such as cardiovascular
10 effects.
The compounds of the invention may be administered enterally, topically
or parenterally.
The compounds of the invention may be formulated alone or together
with another active ingredient and/or together with a pharmaceutically
15 acceptable diluent, carrier and/or excipient in different pharmaceutical
unit
dosage forms, e.g. tablets, capsules, solutions, emulsions and powders etc.,
depending on the route of adminstration, using conventional techniques. The
pharmaceutically acceptable diluent, carrier and/or excipient can be selected
from those conventionally used in the field of pharmaceuticals noticing the
2o chosen route of administration.
The amount of the active ingredient in a dosage form may vary from, for
example, 0.01 to 75 weight-% depending on, for example, the type of the
dosageform.
The specific dose level of the compounds of the invention depends on
25 several factors such as the compound to be administered, the species, age
and
the sex of the subject to be treated, the condition to be treated and on the
route
and method of administration. Accordingly, the dosage for parenteral
administration is typically from 0.5 ~g/kg to 10 mg/kg per day and that for
oral
administration is from 5 ~g/kg to 100 mg/kg for an adult male.
3o The present invention further provides a compound of the invention for
use as alpha-2 antagonist. Furthermore, a method for the treatment of
diseases or conditions where alpha-2 antagonists, e.g. alpha-2C antagonists,
are indicated to be useful, e.g. a method for the treatment of diseases or
conditions of the central nervous system, is provided. In such a method a
CA 02400657 2002-08-16
WO 01/64645 PCT/FI01/00203
41
therapeutically effective amount of a compound of the invention is
administered
to a subject in need of such treatment. The use of the compounds of the
invention for the manufacture of a medicament to be used for the above
indications is also provided.
Those skilled in the art will appreciate that the embodiments described in
this application could be modified without departing from the broad inventive
concept. Those skilled in the art also understand that the invention is not
limited to the particular disclosed embodiments, but is intended to also cover
modifications to the embodiments that are within the spirit and scope of the
invention.