Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
1
INHIBITORS OF THE ICE/ced-3 FAMILY OF CYSTEINE PROTEASES
TECI~1ICAL FIELD
The present invention relates to novel classes of compounds which are
inhibitors of interleukin-1 (3 converting enzyme and related proteases
("ICE/ced-3
family of cysteine proteases"), as well as to pharmaceutical compositions
comprising
these compounds and to methods of using such pharmaceutical compositions.
BACKGROUND OF THE INVENTION
Interleukin 1 ("IL-1") is a major pro-inflammatory and
immunoregulatory protein that stimulates fibroblast differentiation and
proliferation,
the production of prostaglandins, collagenase and phospholipase by synovial
cells and
chondrocytes, basophil and eosinophil degranulation and neutrophil activation.
Oppenheim, J.H. et al., Immunology Today 7:45-56 (1986). As such, it is
involved in
the pathogenesis of chronic and acute inflammatory and autoimmune diseases. IL-
1 is
predominantly produced by peripheral blood monocytes as part of the
inflammatory
response. Mosely, B.S. et al., Proc. Nat. Acad. Sci. X4:4572-4576 (1987);
Lonnemann, G. et al., Eur. J. Immuhol. 19:1531-1536 (1989).
IL-1 (3 is synthesized as a biologically inactive precursor, proIL-1 (3.
ProIL-1 (3 is cleaved by a cysteine protease called interleukin-1 (3
converting enzyme
("ICE") between Asp-116 and Ala-117 to produce the biologically active C-
terminal
fragment found in human serum and synovial fluid. Sleath, P.R. et al., J.
Biol. Chem.
X65:14526-14528 (1992); A.D. Howard et al., J. Immunol. 147:2964-2969 (1991).
ICE is a cysteine protease localized primarily in monocytes. In
addition to promoting the pro-inflammatory and immunoregulatory properties of
IL-1 (3, ICE, and particularly its homologues, also appear to be involved in
the
regulation of cell death or apoptosis. Yuan, J. et al., Cell 75:641-652
(1993); Miura,
M. et al., Cell 75:653-660 (1993); Nett-Giordalisi, M.A. et al., J. Cell
Biochem.
17B:117 (1993). In particular, ICE or ICE/ced-3 homologues are thought to be
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
2
associated with the regulation of apoptosis in neurogenerative diseases, such
as
Alzheimer's and Parkinson's disease. Marx, J. and M. Baringa, Science 259:760-
762
(1993); Gagliardini, V. et al., Science 263:826-828 (1994).
Thus, disease states in which inhibitors of the ICE/ced-3 family of
cysteine proteases may be useful as therapeutic agents include: infectious
diseases,
such as meningitis and salpingitis; septic shock, respiratory diseases;
inflammatory
conditions, such as arthritis, cholangitis, colitis, encephalitis,
endocerolitis, hepatitis,
pancreatitis and reperfusion injury, ischemic diseases such as the myocardial
infarction, stroke and ischemic kidney disease; immune-based diseases, such as
hypersensitivity; auto-immune diseases, such as multiple sclerosis; bone
diseases; and
certain neurodegenerative diseases, such as Alzheimer's and Parkinson's
disease.
Such inhibitors are also useful for the repopulation of hematopoietic cells
following
chemo- and radiation therapy and for prolonging organ viability for use in
transplantation.
ICE/ced-3 inhibitors represent a class of compounds useful for the
control of the above-listed disease states. Peptide and peptidyl inhibitors of
ICE have
been described. However, such inhibitors have been typically characterized by
undesirable pharmacologic properties, such as poor oral absorption, poor
stability and
rapid metabolism. Planner, J.J. and D.W. Norbeck, in Drug Discovery
Technologies,
C.R. Clark and W.H. Moos, Eds. (Ellis Horwood, Chichester, England, 1990), pp.
92-126. These undesirable properties have hampered their development into
effective
drugs.
Accordingly, the need exists for compounds that can effectively inhibit
the action of the ICE/ced-3 family of proteases, for use as agents for
preventing
~ unwanted apoptosis and for treating chronic and acute forms of IL-1 mediated
diseases, such as inflammatory, autoimmune or neurodegenerative diseases. The
present invention satisfies this need and provides further related advantages.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
3
SUMMARY OF THE INVENTION
In general, the compounds of this invention incorporate an aryl or
heteroaryl substituted acyl group as a dipeptide mimetic. The resulting
compounds
exhibit improved properties relative to their peptidic counterparts, for
example, such
as improved cell penetration or improved absorption and metabolic stability
resulting
in enhanced bioavailability.
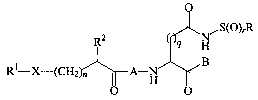
One aspect of the instant invention is the compounds of the Formula I:
O
R2 q _N~S(O)rR
( H
B
R1-X-(CH2)n A_H
O O
Formula I
wherein A, B, X, n, q, ~, R, Rl and R2 are as defined below, as well as
pharmaceutically acceptable salts thereof.
A further aspect of the instant invention is a pharmaceutical
composition comprising a compound of the above Formula I and a
pharmaceutically-acceptable carrier therefor.
Another aspect of this invention involves a method for treating an
autoimmune disease comprising administering an effective amount of a
pharmaceutical composition discussed above to a patient in need of such
treatment.
Yet another aspect of the instant invention is a method for treating an
inflammatory disease comprising administering an effective amount of a
pharmaceutical composition discussed above to a patient in need of such
treatment.
A further aspect of the instant invention is a method for treating a
neurodegenerative disease comprising administering an effective amount of a
pharmaceutical composition discussed above to a patient in need of such
treatment.
Another aspect of the instant invention is a method of preventing
ischemic injury to a patient suffering from a disease associated with ischemic
injury
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
4
comprising administering an effective amount of the pharmaceutical composition
discussed above to a patient in need of such treatment.
A further aspect of the instant invention is a method for expanding of
hematopoietic cell populations and/or enhancing their survival by contacting
the cells
with an effective amount of the pharmaceutical composition discussed above.
Cell
populations included in the method of the invention include (but are not
limited to)
granulocytes, monocytes, erthrocytes, lymphocytes and platelets for use in
cell
transfusions.
An alternate aspect of the instant invention is a method of prolonging
the viability of an organ that has been removed from the donor for the purpose
of a
future transplantation procedure, which comprises applying an effective amount
of the
pharmaceutical composition discussed above to the organ, thereby prolonging
the
viability of the organ as compared to an untreated organ. The organ may be an
intact
organ, or isolated cells derived from an organ (e.g., isolated pancreatic
islet cells,
isolated dopaminergic neurons, blood or hematopoietic cells).
These and other aspects of this invention will be evident upon reference
to the following detailed description.
DESCRIPTION OF DRAWING
Figure 1. Structure of sulfonamide (substituted)acyl dipeptidyl
ICE/ced-3 family of inhibitor compounds.
DETAILED DESCRIPTION OF THE INVENTION
As mentioned above, one aspect of the instant invention is the
compounds of the Formula I:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
O
R2 q _NiS(~)rR
( H
B
R1-X-(CHZ)n A-H
O O
Formula I
wherein:
n is 0, 1 or 2;
q is 1 or 2;
5 r is 1 or 2;
R is lower alkyl, alkyl, cycloalkyl, (cycloalkyl)alkyl, phenyl,
substituted phenyl, phenylalkyl, substituted phenylalkyl, naphthyl,
substituted naphthyl, (1 or 2 naphthyl)alkyl, substituted (1 or 2
naphthyl)alkyl, heteroaryl, substituted heteroaryl, (heteroaryl)alkyl,
substituted (heteroaryl)alkyl, NRa(Rb) or ORS;
Rl is phenyl, substituted phenyl, naphthyl, substituted naphthyl,
heteroaryl, or substituted heteroaryl;
R2 is hydrogen, alkyl, cycloalkyl, phenyl, substituted phenyl,
(CH2)PC02R3, (CH2)n,NH2, (CH2)mNHCORI~, (CH2)n,N(C=NH)NH2,
(CHz)pORll, (CH2)pSRla, (CH2)mcycloalkyl, (CH2)mphenyl,
(CH2)",(substituted phenyl), (CHz)m(1 or 2-naphthyl), or
(CH2)mheteroaryl, wherein heteroaryl includes (but is not limited to)
substituted or unsubstituted pyridyl, thienyl, furyl, thiazolyl,
imidazolyl, pyrazolyl, isoxazolyl, pyrazinyl, pyrimidyl, triazinyl,
tetrazolyl, and indolyl;
X is CH2, C=O, O, S, NH, C=ONH or CHZOC=ONH;
A is a natural or unnatural amino acid of Formula IIa-i:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
6
s
R4 R4a i 5 O R
\N ~ /N~ N
H
O
IIa IIb IIc O
R~ Rs
Rs. ~ Ni
~/N
/N
O
O
IId IIe IIf
H (CHz)c
R \N ~CE"~z)a \ ~CHz)a H
'/N C/N ~CHz)b ~/N tCHz)b
O IIg ~ O IIh ~ ~ O IIi
B is a hydrogen atom, a deuterium atom, C1_io straight chain or
branched alkyl, cycloalkyl, phenyl, substituted phenyl, naphthyl,
substituted naphthyl, 2-benzoxazolyl, substituted 2-oxazolyl,
(CHz)mcycloalkyl, (CHz)mphenyl, (CHz)",(substituted phenyl),
(CH2)m(1 or 2-naphthyl), (CH2)mheteroaryl, halomethyl, CO2R13,
CONRIaRIS, CHzZRI6, CH20C0(aryl), CH20C0(substituted aryl),
CH20C0(heteroaryl), CH20C0(substituted heteroaryl), or
CH20P0(Ri~)R18, where Z is an oxygen or a sulfur atom, or B is a
group of the Formula IIIa-c:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
7
H2 H2 H2
~C~O ~ CEO ~ CEO
R22
R19 ' R21 0 \ R21 R23 ~ R21
~ Y2 Y3 \\
20 2~ 19
R Y O R ~ O
R2o
IIIa IIIb IIIc
and wherein
Ra and Rb are the same or different and independently
hydrogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, phenyl, substituted
phenyl, phenylalkyl, substituted phenylalkyl, naphthyl, substituted
naphthyl, (1 or 2 naphthyl)alkyl, substituted (1 or 2 naphthyl)alkyl,
heteroaryl, substituted heteroaryl, (heteroaryl)alkyl, or substituted
(heteroaryl)alkyl, with the proviso that Ra and Rb cannot both be
hydrogen;
R~ is alkyl, cycloalkyl, (cycloalkyl)alkyl, phenyl, substituted
phenyl, phenylalkyl, substituted phenylalkyl, naphthyl, substituted
naphthyl, (1 or 2 naphthyl)alkyl, substituted (1 or 2 naphthyl)alkyl,
heteroaryl, substituted heteroaryl, (heteroaryl)alkyl, or substituted
(heteroaryl)alkyl;
R3 is hydrogen, alkyl, cycloalkyl, (cycloalkyl)alkyl,
phenylalkyl, or substituted phenylalkyl;
R4 is alkyl, cycloalkyl, phenyl, substituted phenyl, (CH2)mNH2,
(CHz)mNHCORIO, (CH2)mN(~ ~)~2~ (CH2)pCO2R3~ (CH2)pORt l,
(CH2)pSRl2, (CH2)mcycloalkyl, (CH2)mphenyl, (CH2)m(substituted
phenyl), (CHz)m(1 or 2-naphthyl), or (CH2)mheteroaryl, wherein
heteroaryl includes (but is not limited to) pyridyl, thienyl, furyl,
thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, pyrazinyl, pyrimidyl,
triazinyl, tetxazolyl, and indolyl,
R4a is hydrogen or methyl, or
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
8
R4 and R4a taken together are -(CH2)d- where d is an integer
from 2 to 6;
RS is phenyl, substituted phenyl, (CHZ)pphenyl,
(CHZ)p(substituted phenyl), cycloalkyl, or benzofused
cycloalkyl;
R6 is hydrogen, alkyl, cycloalkyl, phenyl, substituted
phenyl,
(CH2)mcycloalkyl, (CHZ)mphenyl, (CH2)m(substituted
phenyl), or
(CH2)m(1 or 2-naphthyl);
R' is hydrogen, fluorine, oxo (i. e., =O), alkyl,
cycloalkyl,
phenyl, substituted phenyl, naphthyl, (CH2)mcycloalkyl,
(CHz)mphenyl,
(CH2)m(substituted phenyl), (CH2)m(1 or 2-naphthyl),
ORII, SR12, or
NHCORIO;
R8 is hydrogen, oxo, alkyl, cycloalkyl, phenyl,
substituted
phenyl, naphthyl, (CH2)mcycloalkyl, (CH2)mphenyl,
(CH2)",(substituted
phenyl), or (CH2)m(1 or 2-naphthyl);
R9 is alkyl, cycloalkyl, (CHZ)mcycloalkyl, (CH2)mphenyl,
(CHZ)m(substituted phenyl), (CH2)m(1 or 2-naphthyl),
or CORIO;
Rl is hydrogen, alkyl, cycloalkyl, phenyl, substituted
phenyl,
naphthyl, (CH2)mcycloalkyl, (CHZ)mphenyl, (CH2)m(substituted
phenyl), (CH2)m(1 or 2-naphthyl), OR13, or NRI~RIS;
Rl r is hydrogen, alkyl, cycloalkyl, phenyl, substituted
phenyl,
naphthyl, (CHZ)mcycloalkyl, (CH2)mphenyl, (CH2)m(substituted
phenyl), or (CH2)m(1 or 2-naphthyl);
R12 is alkyl, cycloalkyl, phenyl, substituted phenyl,
naphthyl,
(CH2)mcycloalkyl, (CH2)mphenyl, (CH2)",(substituted
phenyl), or
(CHZ)m( 1 or 2-naphthyl);
R13 is alkyl, cycloalkyl, (CHZ)mcycloalkyl, (CHZ)mphenyl,
(CH2)",(substituted phenyl), or (CH2)m(1 or 2-naphthyl);
R14 is hydrogen, alkyl, cycloalkyl, phenyl, substituted
phenyl,
naphthyl, substituted naphthyl, (CHZ)mcycloalkyl,
(CHZ)mphenyl,
(CHZ)m(substituted phenyl), or (CH2)m(1 or 2-naphthyl);
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
9
Rls is hydrogen or alkyl; or
R14 and Rls taken together form a five, six or seven membered
carbocyclic or heterocyclic ring, such as morpholine or N-substituted
piperazine;
R16 is phenyl, substituted phenyl, naphthyl, substituted
naphthyl, heteroaryl, (CH2)mphenyl, (CH2)m(substituted phenyl),
(CHz)m(1 or 2-naphthyl), or (CHZ)mheteroaryl;
Rl' and Rl8 are independently alkyl, cycloalkyl, phenyl,
substituted phenyl, naphthyl, or phenylalkyl, substituted phenylalkyl,
or (cycloalkyl)alkyl;
R19 and RZ° are independently hydrogen, alkyl, phenyl,
substituted phenyl, (CH2)mphenyl, or (CH2)m(substituted phenyl), or
Rl9 and R2° taken together are -(CH=CH)2-;
Ral is hydrogen, alkyl, phenyl, substituted phenyl,
(CHz)mphenyl, (CH2)",(substituted phenyl);
R22' Ra3 and R24 are independently hydrogen or alkyl;
Yl 1S CH2~ (CH2)2~ (CH2)3~ Or S;
Yz 1S ~ Or NR24;
Y3 1S CH2, C), Or NR24;
a is 0 or 1 and b is 1 or 2, provided that when a is 1 then b is 1;
c is 1 or 2, provided that when c is 1 then a is 0 and b is l;
m is l, 2, 3 or 4; and
p is 1 or 2;
or a pharmaceutically acceptable salt thereof.
As used herein, the term "alkyl" means a straight or branched Cl to C$
carbon chain such as methyl, ethyl, tert-butyl, iso-propyl, n-octyl, and the
like. The
term "lower alkyl" means a straight or branched Cl to C6 carbon chain, such as
methyl, ethyl, iso-propyl, and the like.
The term "cycloalkyl" means a mono-, bi-, or tricyclic ring that is
either fully saturated or partially unsaturated. Examples of such a ring
include
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl,
cyclooctyl,
cis- or trans decalin, bicyclo[2.2.1]hept-2-ene, cyclohex-1-enyl, cyclopent-1-
enyl, 1,4-
cyclooctadienyl, and the like.
The term "(cycloalkyl)alkyl" means the above-defined alkyl group
5 substituted with one of the above cycloalkyl rings. Examples of such a group
include
(cyclohexyl)methyl, 3-(cyclopropyl)-n-propyl, 5-(cyclopentyl)hexyl, 6
(adamantyl)hexyl, and the like.
The term "substituted phenyl" specifies a phenyl group substituted with
one or more substituents chosen from halogen, hydroxy, protected hydroxy,
cyano,
10 nitro, trifluoromethyl, alkyl, alkoxy, acyl, acyloxy, carboxy, protected
carboxy,
carboxymethyl, protected carboxymethyl, hydroxymethyl, protected
hydroxymethyl,
amino, protected amino, (monosubstituted)amino, protected
(monosubstituted)amino,
(disubstituted)amino, carboxamide, protected carboxamide, N-(lower
alkyl)carboxamide, protected N-(lower alkyl)carboxamide, N,N-di(lower
alkyl)carboxamide, N-((lower alkyl)sulfonyl)amino, N-(phenylsulfonyl)amino or
by a
substituted or unsubstituted phenyl group, such that in the latter case a
biphenyl or
naphthyl group results, or wherein two adjacent alkyl substituents on the
phenyl ring
taken together from a cycloalkyl to yield, for example, teterahydronaphthyl or
indanyl.
Examples of the term "substituted phenyl" includes a mono-, di-, tri-,
tetra- or penta(halo)phenyl group such as 2-, 3- or 4-chlorophenyl, 2,6-
dichlorophenyl,
2,5-dichlorophenyl, 3,4-dichlorophenyl, 2-,3- or 4-bromophenyl, 3,4-
dibromophenyl,
3-chloro-4-fluorophenyl, 2-, 3- or 4-fluorophenyl, 2,4,6-trifluorphenyl,
2,3,5,6
tetrafluorphenyl, 2,3,4,5-tetrafluorophenyl, 2,3,4,5,6-pentafluoropheny, and
the like; a
mono or di(hydroxy)phenyl group such as 2-, 3-, or 4-hydroxyphenyl,
2,4-dihydroxyphenyl, the protected-hydroxy derivatives thereof and the like; a
nitrophenyl group such as 2-, 3-, or 4-nitrophenyl; a cyanophenyl group, for
example,
2-,3- or 4-cyanophenyl; a mono- or di(alkyl)phenyl group such as 2-, 3-, or
4-methylphenyl, 2,4-dimethylphenyl, 2-, 3- or 4-(iso-propyl)phenyl, 2-, 3-, or
4-ethylphenyl, 2-, 3- or 4-(n-propyl)phenyl and the like; a mono or
di(alkoxy)phenyl
group, for example, 2,6-dimethoxyphenyl, 2-, 3- or 4-(iso-propoxy)phenyl, 2-,
3- or
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
11
4-(t-butoxy)phenyl, 3-ethoxy-4-methoxyphenyl and the like; 2-, 3- or
4-trifluoromethylphenyl; a mono- ox dicarboxyphenyl or (protected
carboxy)phenyl
group such as 2-, 3- or 4-carboxyphenyl or 2,4-di(protected carboxy)phenyl; a
mono-
or di(hydroxymethyl)phenyl or (protected hydroxymethyl)phenyl such as 2-, 3-
or
4-(protected hydroxymethyl)phenyl or 3,4-di(hydroxymethyl)phenyl; a mono- or
di(aminomethyl)phenyl or (protected aminomethyl)phenyl such as 2-, 3- or
4-(aminomethyl)phenyl or 2,4-(protected aminomethyl)phenyl; or a mono- or
di(N-(methylsulfonylamino))phenyl such as 2, 3 or
4-(N-(methylsulfonylamino))phenyl. Also, the term "substituted phenyl"
represents
disubstituted phenyl groups wherein the substituents are different, for
example,
3-methyl-4-hydroxyphenyl, 3-chloro-4-hydroxyphenyl, 2-methoxy-4-bromophenyl,
4-ethyl-2-hydroxyphenyl, 3-hydroxy-4-nitrophenyl, 2-hydroxy-4-chlorophenyl,
and
the like.
The term "phenylalkyl" means one of the above phenyl groups attached
to one of the above-described alkyl groups, and the term "substituted
phenylalkyl
means that either the phenyl or the alkyl, or both, are substituted with one
or more of
the above-identified substituents. Examples of such groups include 2-phenyl-1-
chloroethyl, 2-(4'-methoxyphenyl)ethyl, 4-(2',6'-dihydroxy phenyl)n-hexyI, 2-
(S'-
cyano-3'-methoxyphenyl)n-pentyl, 3-(2',6'-dimethylphenyl)n-propyl, 4-chloro-3-
aminobenzyl, 6-(4'-methoxyphenyl)-3-carboxy(n-hexyl), 5-(4'-
aminomethylphenyl)~3-
(aminomethyl)n-pentyl, 5-phenyl-3-oxo-n-pent-1-yl, (4-hydroxynapth-2-
yl)methyl,
and the like.
The term "substituted naphthyl" means a naphthyl group substituted
with one or more of the above-identifed substituents, ,and the term "(1 or 2
naphyl)alkyl" means a naphthyl attached to one of the above-described alkyl
groups at
the 1 or 2 position.
The terms "halo" and "halogen" refer to the fluoro, chloro, bromo or
iodo groups. These terms rnay also be used to describe one or more halogens,
which
are the same or different. Preferred halogens in the context of this invention
are
chloro and fluoro.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
12
The term "aryl" refers to aromatic five and six rnembered carbocyclic
rings. Six membered rings are preferred.
The term "heteroaryl" denotes optionally substituted aromatic
five-membered or six-membered heterocyclic rings that have I to 4 heteroatoms,
such
as oxygen, sulfur and/or nitrogen atoms, in particular nitrogen, either alone
or in
conjunction with sulfur or oxygen ring atoms.
The following ring systems are representative examples of the
heterocyclic radicals denoted by the term "heteroaryl" (whether substituted or
unsubstituted): thienyl, furyl, pyrrolyl, pyrrolidinyl, imidazolyl,
isoxazolyl, triazolyl,
thiadiazolyl, oxadiazolyl, tetrazolyl, thiatriazolyl, oxatriazolyl, pyridyl,
pyrimidyl,
pyrazinyl, pyridazinyl, oxazinyl, triazinyl, thiadiazinyl~ tetrazolo, 1,5-
[b]pyridazinyl
and purinyl, as well as benzo-fused derivatives, for example, benzoxazolyl,
benzothiazolyl, benzimidazolyl and indolyl.
Substituents for the above optionally substituted heteroaryl rings are
from one to three halo, trihalomethyl, amino, protected amino, amino salts,
mono-substituted amino, di-substituted amino, carboxy, protected carboxy,
carboxylate salts, hydroxy, protected hydroxy, salts of a hydroxy group, lower
alkoxy,
lower alkylthio, lower alkyl, substituted alkyl, cycloalkyl, substituted
cycloalkyl,
(cycloalkyl)alkyl, substituted (cycloalkyl)alkyl, phenyl, substituted phenyl,
phenylalkyl, and substituted phenylalkyl groups.
Substituents for the heteroaryl group are as defined above, or as set
forth below. As used in conjunction with the above substituents for heteroaryl
rings,
"trihalomethyl" can be trifluoromethyl, trichloromethyl, tribromomethyl or
triiodomethyl, "lower alkoxy" means a Cl to C4 alkoxy group, similarly, "lower
alkylthio" means a Cl to C4 alkylthio group. The term "substituted lower
alkyl"
means the above-defined lower alkyl group substituted from one to three times
by a
hydroxy, protected hydroxy, amino, protected amino, cyano, halo,
trifluoromethyl,
mono-substituted amino, di-substituted amino, lower alkoxy, lower alkylthio,
carboxy,
protected carboxy, or a carboxy, amino, and/or hydroxy salt:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
13
As used in conjunction with the substituents for the heteroaryl rings,
the terms "substituted (cycloalkyl)alkyl" and "substituted cycloalkyl" are as
defined
above substituted with the same groups as listed for a "substituted alkyl"
group. The
term "(monosubstituted)amino" refers to an amino group with one substituent
chosen
from the group consisting of phenyl, substituted phenyl, alkyl, substituted
alkyl, C1 to
C7 acyl, C2 to C~ alkenyl, Ca to C~ substituted alkenyl, Ca to C7 alkynyl, C~
to CI6
alkylaryl, C~ to C16 substituted alkylaryl and heteroaryl group. The
(monosubstituted)amino can additionally have an amino-protecting group as
encompassed by the term "protected '(monosubstituted)amino." The term
"(disubstituted)amino" refers to amino groups with two substituents chosen
from the
group consisting of phenyl, substituted phenyl, alkyl, substituted alkyl, C1
to C~ acyl,
C2 to C~ alkenyl, C2 to C7 alkynyl, C~ to CI6 alkylaryl, C~ to C16 substituted
alkylaryl
and heteroaryl. The two substituents can be the same or different. The term
"heteroaryl(alkyl)" denotes an alkyl group as defined above, substituted at
any
position by a heteroaryl group, as above defined.
Furthermore, the above optionally substituted five-membered or
six-membered heterocyclic rings can optionally be fused to a aromatic 5-
membered or
6-membered aryl or heteroaryl ring system. For example, the rings can be
optionally
fused to an aromatic 5-membered or 6-membered ring system such as a pyridine
or a
triazole system, and preferably to a benzene ring.
The term "pharmaceutically-acceptable salt" encompasses those salts
that form with the carboxylate anions and includes salts formed with the
organic and
inorganic cations such as those chosen from the alkali and alkaline earth
metals, (for
example, lithium, sodium, potassium, magnesium, barium and calcium); and
ammonium ion; and the organic cations (for example, dibenzylammonium,
benzylammonium, 2-hydroxyethylammonium, bis(2-hydroxyethyl)ammonium,
phenylethylbenzylammonium, dibenzylethylenediammonium, and like cations.)
Other
cations encompassed by the above term include the protonated form of procaine,
quinine and N-methylglucosamine, the protonated forms of basic amino acids
such as
glycine, ornithine, histidine, phenylglycine, lysine, and arginine.
Furthermore, any
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
14
zwitterionic form of the instant compounds formed by a carboxylic acid and an
amino
group is referred to by this term. A preferred cation for the carboxylate
anion is the
sodium cation. Furthermore, the term includes salts that form by standard acid-
base
reactions with basic groups (such as amino groups) and includes organic or
inorganic
acids. Such acids include hydrochloric, sulfuric, phosphoric, acetic,
succinic, citric,
lactic, malefic, fumaric, palmitic, cholic, pamoic, mucic, D-glutamic, D-
camphoric,
glutaric, phthalic, tartaric, lauric, stearic, salicyclic, methanesulfonic,
benzenesulfonic,
sorbic, picric, benzoic, cinnamic, and the like acids.
The compounds of Formula I may also exist as solvates and hydrates.
Thus, these compounds may crystallize with, for example, waters of hydration,
or one,
a number of, or any fraction thereof of molecules of the mother liquor
solvent. The
solvates and hydrates of such compounds are included within the scope of this
invention.
The term "carboxy-protecting group" as used herein refers to one of the
ester derivatives of the carboxylic acid group commonly employed to block or
protect
the carboxylic acid group while reactions are carried out on other functional
groups on
the compound. Examples of such carboxylic acid protecting groups include t-
butyl,
4-nitrobenzyl, 4-methoxybenzyl, 3,4-dimethoxybenzyl, 2,4-dimethoxybenzyl,
2,4,6-trimethoxybenzyl, 2,4,6-trimethylbenzyl, pentamethylbenzyl,
3,4-methylenedioxybenzyl, benzhydryl, 4,4'-dimethoxytrityl, 4,4',4"-
trimethoxytrityl,
2-phenylpropyl, trimethylsilyl, t-butyldimethylsilyl, pheriacyl, 2,2,2-
trichloroethyl,
(3-(trimethylsilyl)ethyl, (3-(di(n-butyl)methylsilyl)ethyl, p-
toluenesulfonylethyl,
4-nitrobenzylsulfonylethyl, allyl, cinnamyl, 1-(trimethylsilylmethyl)-propenyl
and like
moieties. The species of carboxy-protecting group employed is not critical so
long as
the derivatized carboxylic acid is stable to the conditions of subsequent
reactions) and
can be removed at the appropriate point without disrupting the remainder of
the
molecule. Further examples of these groups are found in C.B. Reese and E.
Haslam,
"Protective Groups in Organic Chemistry," J.G.W. McOmie, Ed., Plenum Press,
New
York, NY, 1973, Chapter 5, respectively, and T.W. Greene and P.G.M. Wuts,
"Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New
York,
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
NY, 1991, Chapter 5, each of which is incorporated herein by reference. A
related
term is "protected carboxy," which refers to a carboxy group substituted with
one of
the above carboxy-protecting groups.
The term "hydroxy-protecting group" refers to readily cleavable groups
5 bonded to hydroxyl groups, such as the tetrahydropyranyl, 2-methoxyprop-2-
yl,
1-ethoxyeth-1-yl, methoxymethyl, (3-methoxyethoxymethyl, methylthiomethyl,
t-butyl, t-amyl, trityl, 4-methoxytrityl, 4,4'-dimethoxytrityl, 4,4',4"-
trimethoxytrityl,
benzyl, allyl, trimethylsilyl, (t-butyl)dimethylsilyl, 2,2,2-
trichloroethoxycarbonyl, and
the like.
10 Further examples of hydroxy-protecting groups are described by C.B.
Reese and E. Haslam, "Protective Groups in Organic Chemistry," J.G.W. McOmie,
Ed., Plenum Press, New York, NY, 1973, Chapters 3 and 4, respectively, and
T.W.
Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis," Second
Edition,
John Wiley and Sons, New York, NY, 1991, Chapters 2 and 3. A preferred
15 hydroxy-protecting group is the tert-butyl group. The related term
"protected
hydroxy" denotes a hydroxy group bonded to one of the above hydroxy-protecting
groups.
The term "amino-protecting group" as used herein refers to substituents
of the amino group commonly employed to block or protect the amino
functionality
while reacting other functional groups of the molecule. The term "protected
(monosubstituted)amino" means there is an amino-protecting group on the
monosubstituted amino nitrogen atom.
Examples of such amino-protecting groups include the formyl ("For")
group, the trityl group, the phthalimido group, the trichloroacetyl group, the
trifluoroacetyl group, the chloroacetyl, bromoacetyl, and iodoacetyl groups,
urethane-
type protecting groups, such as t-butoxycarbonyl ("Boc"), 2-(4-
biphenylyl)propyl-2-
oxycarbonyl ("Bpoc"), 2-phenylpropyl-2-oxycarbonyl ("Poc"), 2-(4-
xenyl)isopropoxycarbonyl, 1,1-diphenylethyl-1-oxycarbonyl, 1,1-diphenylpropyl-
1-
oxycarbonyl, 2-(3,5-dimethoxyphenyl)propyl-2-oxycarbonyl ("Ddz"), 2-(p-
toluyl)propyl-2-oxycarbonyl, cyclopentanyloxycarbonyl, 1-methylcyclopentanyl-
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
16
oxycarbonyl, cyclohexanyloxy-carbonyl, 1-methyl-cyclohexanyloxycarbonyl, 2-
methylcyclohexanyl-oxycarbonyl, 2-(4-toluylsulfonyl)ethoxycarbonyl, 2-
(methylsulfonyl)ethoxycarbonyl, 2-(triphenylphosphino)-ethoxycarbonyl, 9-
fluorenylmethoxycarbonyl ("Fmoc"), 2-(trimethylsilyl)ethoxycarbonyl,
S allyloxycarbonyl, 1-(trimethylsilylmethyl)prop-1-enyloxycarbonyl, S-
benzisoxalylmethoxycarbonyl, 4-acetoxybenzyl-oxycarbonyl, 2,2,2-
trichloroethoxycarbonyl, 2-ethynyl-2-propoxycarbonyl,
cyclopropylrnethoxycarbonyl,
isobornyloxycarbonyl, 1-piperidyloxycarbonyl, benzyloxycarbonyl ("Cbz"), 4-
phenylbenzyloxycarbonyl, 2-methylbenzyloxycarbonyl, a-2,4,5,-tetramethylbenzyl-
oxycarbonyl ("Tmz"), 4-methoxybenzyloxycarbonyl, 4-fluorobenzyloxycarbonyl, 4-
chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2-chlorobenzyloxycarbonyl,
2,4-dichlorobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 3-
bromobenzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, 4-cyanobenzyloxycarbonyl, 4-
(decyloxy)benzyloxycarbonyl and the like; the benzoylmethylsulfonyl group, the
1S 2,2,5,7,8-pentamethylchroman-6-sulfonyl group ("PMC"), the dithiasuccinoyl
("Dts")
group, the 2-(nitro)phenyl-sulfenyl group ("Nps"), the diphenylphosphine oxide
group, and like amino-protecting groups. The species of amino-protecting group
employed is not critical so long as the derivatized amino group is stable to
the
conditions of the subsequent reactions) and can be removed at the appropriate
point
without disrupting the remainder of the molecule. Preferred amino-protecting
groups
are Boc, Cbz and Fmoc. Further examples of amino-protecting groups embraced by
the above term are well known in organic synthesis and the peptide art and are
described by, for example, T.W. Greene and P.G.M. Wuts, "Protective Groups in
Organic Synthesis," 2nd ed., John Wiley and Sons, New York, NY, 1991, Chapter
7,
2S M. Bodanzsky, "Principles of Peptide Synthesis," 1st and 2nd revised Ed.,
Springer-
Verlag, New York, NY, 1984 and 1993, and J.M. Stewart and J.D. Young, "Solid
Phase Peptide Synthesis," 2nd Ed., Pierce Chemical Co., Rockford, IL, 1984, E.
Atherton and R.C. Shephard, "Solid Phase Peptide Synthesis - A Practical
Approach"
IRL Press, Oxford, England (1989), each of which is incorporated herein by
reference.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
17
The related term "protected amino" defines an amino group substituted with an
amino-protecting group discussed above.
The terms "natural and unnatural amino acid" refers to both the
naturally occurring amino acids and other non-proteinogenic a-amino acids
commonly utilized by those in the peptide chemistry arts when preparing
synthetic
analogues of naturally occurring peptides, including D and L forms. The
naturally
occurring amino acids are glycine, alanine, valine, leucine, isoleucine,
serine,
methionine, threonine, phenylalanine, tyrosine, tryptophan, cysteine, proline,
histidine, aspartic acid, asparagine, glutamic acid, glutamine, y-
carboxyglutamic acid,
arginine, ornithine and lysine. Examples of unnatural alpha-amino acids
include
hydroxylysine, citrulline, kynurenine, (4-aminophenyl)alanine, 3-(2'-
naphthyl)alanine,
3-(1'-naphthyl)alanine, methionine sulfone, (t-butyl)alanine, (t-
butyl)glycine, 4-
hydroxyphenyl-glycine, aminoalanine, phenylglycine, vinylalanine, propargyl-
gylcine,
1,2,4-triazolo-3-alanine, thyronine, 6-hydroxytryptophan, 5-hydroxytryptophan,
3-
hydroxy-kynurenine, 3-aminotyrosine, trifluoromethylalanine, 2-thienylalanine,
(2-(4-
pyridyl)ethyl)cysteine, 3,4-dimethoxy-phenylalanine, 3-(2'-thiazolyl)alanine,
ibotenic
acid, 1-amino-1-cyclopentane-carboxylic acid, 1-amino-1-cyclohexanecarboxylic
acid, quisqualic acid, 3-(trifluoromethylphenyl)alanine, (cyclohexyl)glycine,
thiohistidine, 3-methoxytyrosine, norleucine, norvaline, alloisoleucine,
homoarginine,
thioproline, dehydro-proline, hydroxyproline, homoproline, indoline-2-
carboxylic
acid, 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid, 1,2,3,4-
tetrahydroquinoline-2-
carboxylic acid, a-amino-n-butyric acid, cyclohexylalanine, 2-amino-3-
phenylbutyric
acid, phenylalanine substituted at the ortho, meta, or para position of the
phenyl
moiety with one or two of the following groups: a (C1 to C4)alkyl, a (C1 to
C4)alkoxy,
a halogen or a nitro group, or substituted once with a methylenedioxy group;
[3-2- and
3-thienylalanine; [3-2- and 3-furanylalanine; (3-2-, 3- and 4-pyridylalanine;
(3-
(benzothienyl-2- and 3-yl)alanine; (3-(1- and 2-naphthyl)alanine; O-alkylated
derivatives of serine, threonine or tyrosine; S-alkylated cysteine, S-
alkylated
homocysteine, the O-sulfate, O-phosphate and O-carboxylate esters of tyrosine;
3-
(sulfo)tyrosine, 3-(carboxy)tyrosine, 3-(phospho)tyrosine, the 4-methane-
sulfonic acid
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
18
ester of tyrosine, 4-methanephosphonic acid ester of tyrosine, 3,5-
diiodotyrosine, 3-
nitrotyrosine, s-alkyllysine, and delta-alkyl ornithine. Any of these a-amino
acids
may be substituted with a methyl group at the alpha position, a halogen at any
position
of the aromatic residue on the a-amino side chain, or an appropriate
protective group
at the O, N, or S atoms of the side chain residues. Appropriate protective
groups are
discussed above.
The compounds of this invention may be modified by appropriate
functionalities to enhance selective biological properties. Such modifications
are
known in the art and include those which increase biological penetration into
a given
biological system (e.g., blood, lymphatic system, central nervous system),
increase
oral availability, increase solubility to allow administration by injection,
alter
metabolism and alter rate of exertion. ,In addition, the compounds may be
altered to
pro-drug form such that the desired compound is created in the body of the
patient as
the result of the action of metabolic or other biochemical processes on the
pro-drug.
Some examples of pro-drug forms include ketal, acetal, oxime, and hydrazone
forms
of compounds which contain ketone or aldehyde groups, especially where they
occur
in the group donated as "A" in Formula I or the modified aspartic acid residue
attached to the group denoted as "A".
With regard to the q and r groups of Formula I, typical embodiments
include compounds wherein q is 1 and ~ is 2.
Compounds of this invention with respect to the n, R, RI, RZ and X
groups in Formula I include those wherein:
R is lower alkyl (such as methyl);
Rl is substituted phenyl (such as 2-substituted phenyl),
naphthyl, or substituted naphthyl;
Ra is hydrogen, lower alkyl, (CH2)pCOzR3, (CHZ)mphenyl,
(CH2)m(substituted phenyl), (CH2)m(1 or 2-naphthyl), or
(CH2)mtetrazolyl, where p is 1 or 2, m is 1 or 2;
X is O or NH; and
nis0orl.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
19
Other compounds of this invention with respect to the RI, RZ and X
groups in Formula I include those wherein:
Rl is substituted phenyl, naphthyl, or substituted naphthyl;
R2 is (CH~)mtetrazolyl, where m is 1 or 2; and
X is C=ONH.
Compounds of this invention with respect to the A group in Formula I
include those of Formula IIa wherein:
Rø is lower alkyl, cycloalkyl, phenyl, substituted phenyl,
(CH2)m~2~ (CHa)rORll, (CHa)PSR12, (CHa)mcYcloalkyl,
(CH2)mphenyl, (CH2)m(substituted phenyl), or (CH2)m(1 or 2-naphthyl);
Rl1 is hydrogen, lower alkyl, cycloalkyl, phenyl, substituted
phenyl, naphthyl, (CH2)mcycloalkyl, (CH2)mphenyl, (CH2)m(substituted
phenyl), or (CH2)m(1 or 2-naphthyl);
R12 is lower alkyl, cycloalkyl, phenyl, substituted phenyl,
1 S naphthyl, (CH2)mcycloalkyl, (CHZ)mphenyl, (CH2)m(substituted
phenyl), or (CHZ)m(1 or 2-naphthyl); and
m is l, 2, 3, 4 and p is 1 or 2.
Compounds of this invention with respect to the A group in Formula I
also include those of Formula IIb wherein:
RS is phenyl, substituted phenyl, (CH2)pphenyl,
(CHa)p(substituted phenyl), cycloalkyl, or 2-indanyl; and
pislor2.
Another group of compounds with respect to the A group in Formula I,
include those of Fornmla IId wherein:
R' is hydrogen, fluorine, cycloalkyl, phenyl, substituted phenyl,
naphthyl, (CH2)mcycloalkyl, (CH2)mphenyl, (CH~)m(substituted
phenyl), (CH2)m(1 or 2-naphthyl), ORII~ or SR12;
Rll and R12 are independently cycloalkyl, phenyl, substituted
phenyl, naphthyl, (CHZ)mcycloalkyl, (CH2)mphenyl, (CH2)~"(substituted
phenyl), or (CH2)m(1 or 2-naphthyl); and
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
m is 1, 2, 3 or 4.
A forth group of compounds with respect to the A group in Formula I
include those of Formula IIe wherein:
R$ is hydrogen, oxo, cycloalkyl, phenyl, substituted phenyl, or
5 naphthyl; and
Yl is CHz, (CHZ)2, (CH2)3, or S.
Another group of compounds with respect to the A group in Formula I
include those of Formula IIh wherein:
a is 0 and b is 1 or 2.
10 Compounds of this invention with respect to the B group in Formula I
include those wherein:
B is hydrogen, 2-benzoxazolyl, substituted 2-oxazolyl,
CH2Z1R6, CHZOCO(aryl), or CHZOPO(Rl~)Rl$, where Z is O or S;
R16 is phenyl, substituted phenyl, naphthyl, substituted
15 naphthyl, heteroaryl, (CH2)mphenyl, (CH2)",(substituted phenyl),
(CH2)m(1 or 2-naphthyl), or (CH2)mheteroaryl; and
Rl' and Rl$ are independently alkyl, cycloalkyl, phenyl,
substituted phenyl, naphthyl, phenylalkyl, substituted phenylalkyl and
(cycloalkyl)alkyl.
20 Another group of compounds with respect to the B group in Formula I
include those of Formula IIIa-c wherein:
Y2 is O or NRZa;
Y3 is CH2, O, or NR2a;
R19 and R2° are independently hydrogen, alkyl, phenyl, or RIs
and RZ° taken together are -(CH=CH)2-;
R2I is hydrogen, alkyl, phenyl, substituted phenyl,
(CHz)mphenyl, or (CH2)",(substituted phenyl); and
R2z, Ras and R24 are independently hydrogen or alkyl.
The compounds of Formula I may be synthesized using conventional
techniques as discussed below. Advantageously, these compounds are
conveniently
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
21
synthesized from readily available starting materials to form a carboxylic
acid
intermediate as represented in the following Reaction Schemes 1 and 2. To this
end,
in the following synthetic schemes, q is 1, and corresponding compounds
wherein q is
2 may be made in the same manner by employing the corresponding ethylene (-
CH2CHz-) starting material in place of the methylene (-CH2-) moiety.
One synthetic route for synthesizing the carboxylic acid intermediate is
set forth in the following Scheme 1:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
22
SCHEME 1 Rz
PG-A-OH + H N-C H-A-OR + R~-X-~CHz)~CO H
2 2
(Formula IV) (Formula V) (Formula IX) (Formula VII)
STEP A STEP D
Rz
H
PG-A-N-C
( Formula VI ) R~ X-~CHz)n ~A-ON
2 O
(Formula X)
STEP B R~-X-tCHz)n COgH
STEP E HZN-C
(Formula VII) (Formula V)
Rz
R~-X-~CHz)~A-N-C
O
(Formula VIII)
STEP C
C02R
R2
B
R~-X-(CHz)n A-N
H
O O
(Formula Ia)
In the above Scheme 1, R' represents hydrogen or a carboxy-protecting
group, wherein the carboxy-protecting group is as defined above. "PG" stands
for an
amino-protecting group, and "A" stands for a natural or unnatural amino acid
of
Formula IIa through IIi, as discussed above. In addition, Formula (V) above
(i.e.,
H2N-C) represents a modified aspartic acid residue of Formulas Va through Vd:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
23
O
C02R1 O
O
H2N H2N ~ N~ N ~ ~a
H
OR'
Formula Va; Formula Vb;
C02R' CO~,R'
B B
H2N HaN
OH O
Formula Vc; or Formula Vd.
The modified aspartic acids of Formula Va-d can be prepared by
methods well known in the art. See, for example, European Patent Application
519,748; PCT Patent Application No. PCTBP92/02472; PCT Patent Application No.
PCT/LTS91/06595; PCT Patent Application No. PCT/US91/02339; European Patent
Application No. 623,592; World Patent Application No. WO 93/09135; PCT Patent
Application No. PCT/LTS94/08868; European Patent Application No. 623,606;
European Patent Application No. 618,223; European Patent Application No.
533,226;
European Patent Application No. 528,487; European Patent Application No.
618,233;
IO PCT Patent Application No. PCT/EP92/02472; World Patent Application No. WO
93/09135; PCT Patent Application No. PCT/LJS93/03589; and PCT Patent
Application No. PCT/US93/00481, all of which are herein incorporated by
reference.
The coupling reactions carried out under Step A are performed in the
presence of a standard peptide coupling agent such as the combination of the
combination of dicyclohexylcarbodiimide(DCC) and 1-hydroxy-
benzotriazole(HOBt),
as well as the BOP (benzotriazolyloxy-tris-(dimethylamino)phosphonium
hexafluorophosphate) reagent, pyBOP (benzotriazolyloxy-tris(N-
pyrolidinyl)phosphoniumhexafluorophosphate), HBTU (O-benzotriazolyly-
tetramethylisouronium-hexafluorophosphate), and EEDQ (1-ethyloxycarbonyl-2-
ethyloxy-1,2-dihydroquinoline) reagents, the combination of
1-ethyl(3,3'-dimethyl-1'-aminopropyl)carbodiimide (EDAC) and HOBt, and the
like,
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
24
as discussed in J. Jones, "Amino Acid and Peptide Synthesis," Steven G. Davis
ed.,
Oxford University Press, Oxford, pp. 25-41 (1992); M. Bodanzky, "Principles of
Peptide Synthesis," Hafner et al. ed., Springer-Verlag, Berlin Heidelberg, pp.
9-52 and
pp. 202-251 (1984); M. Bodanzky, "Peptide Chemistry, A Practical Textbook,"
Springer-Verlag, Berlin Heidelberg, pp. 55-73 and pp. 129-180; and Stewart and
Young, "Solid Phase Peptide Synthesis," Pierce Chemical Company, (1984), all
of
which are herein incorporated by reference. The amino protecting group is then
removed and the resulting amine is coupled to the (substituted) carboxylic
acid of
Formula VII (Step B). Again, this coupling reaction uses the standard peptide
coupling reactions mentioned above.
Alternatively, the (substituted)carboxylic acid of Formula VII can be
coupled to an amino ester of Formula IX (Step D). Again, this coupling
reaction uses
the standard peptide coupling reactions mentioned above. In Formula IX, the
group R
is a carboxyl protecting group such as methyl, allyl, benzyl or tert-butyl.
After
removal of the carboxyl protecting group under standard conditions well known
in the
art, the resulting carboxylic acid is coupled to amine V using the standard
peptide
coupling methods described above (Step E).
In the case where the coupling reaction depicted by either Step A or
Step E was carried out with the amino alcohol of Formula Vc, the alcohol
moiety must
be , oxidized to the corresponding carbonyl compound prior to removal of the
protecting groups. Preferred methods for the oxidation reaction include Swern
oxidation (oxalyl chloride-dimethyl sulfoxide, methylene chloride at -
78°C followed
by triethylamine); and Dess-Martin oxidation (Dess-Martin periodinane, t-
butanol,
and methylene chloride.) The protecting groups contained in substructures of
the
Formula Va-d, VII and A are removed by methods well known in the art. These
reactions and removal of some or all of the protecting groups are involved in
Step C in
the above Scheme 1.
An alternative synthetic route for synthesizing the carboxylig acid
intermediate having a protected carboxy group is set forth in the following
Scheme 2:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
SCHEME 2
C02R3 CO2R3
STEP F
PG-A-OH '~' H2N C02R -> PG-A-H CO2R
(Formula IV) (Formula XI) (Formula XII)
R2
C02R
~--X- C H )~ CO H
R ( 2n 2 R
(Formula VII) STEP H
R~-X-(CHa)n ~A-H C02R >
STEP IIG
O
(Formula XIII)
C02R
R2
STEP I
R~-X-(CH2)n ~A-H ~ ~ Br >
~O~ O
(Formula XIV)
C02R
R2
B
R'~-X-(CH2)n A
O O
(Formula Ia)
In the above Scheme 2, "PG" stands for an amino protecting group and
"A" stands for a natural or unnatural amino acid of formula IIa through IIi,
as
5 discussed above. The R' is a carboxyl protecting group such as
trimethylsilyl, methyl,
allyl, benzyl or tert-butyl.
The coupling reactions carried out under Step F and Step G are
performed in the presence of a standard peptide coupling agent as discussed
above. In
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
26
Step G, the amino protecting group must be removed prior to the coupling step.
In
Step H the alpha-carboxy protecting group R of the compound of Formula XIII is
selectively removed and the resulting mono-carboxylic acid treated
sequentially with
diazomethane and hydrobromic acid to give the alpha-bromoketone of Formula
XIV.
In Step I, the bromoketone of Formula XIV is treated with either
R16Z-H, (aryl)-COZH, (heteroaryl)-C02H, or R17(Rl$)P02H in the presence of an
inorganic base such as potassium carbonate or potassium fluoride in an inert
solvent
such as dimethyl formamide to give the corresponding compound of Formula Ia in
which B is CHZZR'6, CHZOCO(aryl), CH20C0(heteroaryl), or CH20P0(Rl~)R18,
respectively. Compounds of Formula Ia in which B is a fragment of Formula III
may
also be prepared in a similar fashion. The protecting groups contained in
substructures of the Formula VII, XI and A are removed by methods well known
in
the art. These reactions and removal of some or all of the protecting groups
are
involved in Step I in the above Scheme 2.
An alternative method for the preparation of the carboxylic acid
intermediate of Formula Ia in which R' and B are both hydrogen (i.e., Formula
Ib
below) is set forth in Scheme 3:
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
27
SCHEME 3
co2tBu
H / O~CO~H
Fmoc-H
STEP J
N~ \
NH
(Formula XV) p'" N
H
O
C02tBu
N
H
H / O
Fmoc-H
STEP K
~~NH \
Fmoc-A-OH
(Formula XVI) O' N (Formula IVa)
H
O
C02tBu
H
H O
Fmoc-A-~
STEP I,
N~ NH
R~ X-(CHZ)nCH(RZ)COZH
(Formula XVII) O~ (Formula VII)
O
CO~tBu
RZ H
H O
Ft~-X-(CHz)n A'
O N~ \ STEP M
NH
(Formula XVIII) 0~ N
H
COzH
R2
H
R~-X-(CH2)n
O O
(Formula Ib)
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
28
In Scheme 3, Fmoc is the amino protecting group 9-
fluorenylmethoxycarbonyl and the shaded circle labeled "PS" represents
polystyrene
resin.
The coupling of the acid of Formula XV to a primary amine on solid
support, preferably aminomethyl polystyrene, is carried out using standard
peptide
coupling agents, preferably using benzotriazolyloxy-tris(N-
pyrolidinyl)phosphoniumhexafluorophosphate (pyBOP) in a inert solvent such as
dimethylformamide or N-methyl pyrrolidone (Step ~. After removal of the Fmoc
protecting group of XVI by treatment with pyrrolidine-dimethylfonmamide, the
resulting amine is coupled to Fmoc-amino acid of Formula IVa using standard
peptide
coupling conditions as discussed above (Step K).
In Step L the Fmoc protecting group of the compound of Formula XVII
is removed again by treatment with pyrrolidine-dimethylformamide and the
resulting
amine coupled to the (substituted)carboxylic acid of Formula VII again using
standard
peptide coupling conditions as discussed above. The tert-butyl ester of the
compound
of Formula XVIII is removed by treatment with trfluoroacetic acid-methylene
chloride
in the presence of a trapping agent such as anisole and the resulting acid
cleaved from
the solid support by treatment with 37% aqueous formaldehyde/acetic
acid/tetrahydrofuran/ trifluoroacetic acid, preferably in a ratio of
1/1/5/0.025, to give
the aspartyl aldehyde of Formula Ib (Step M).
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
29
Once synthesized by, for example, the above techniques, the carboxylic
acid intermediate of Formula Ia (R'=H) may then be converted to compounds of
Formula I according to the following Reaction Scheme 4:
SCHEME 4
R2 ( 9 OH Rz ( 9 N~S(0)rR
H
Rl-X-(CHz)~A-H $ ~ R'-X-(CH2)~A-H
I0I O IO' O
Formula Ia Formula I
In Reaction Scheme 4, conversion of the carboxylic acid intermediate
to the corresponding sulfonimide is typically accomplished by utilizing an
intermediate having a protected carboxyl group through which the B moiety is
attached. For example, in place of the -C(=O)B moiety, a hydroxy-protected
group
may be employed, such as -C(OTHP)B. As represented by Step N in Reaction
Scheme 5, this hydroxy-protected intermediate, Formula Ic, may be converted to
the
corresponding sulfonimide intermediate of Formula Id by treatment with CDI (2
eq.)
in THF at room temperature for 3 hours, followed by H2NS(O)rR (2 eq.) in DBU
(2
eq.) at room temperature for 4 hours.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
SCHEME 5
0
~S(O)rR
RZ C q ~OH STEP N RZ ( q 'H
B ---~ A-N B
Rl X-(CHz)~A-H Rl X-(CHz)n H
~O~ THP O THP
Formula Ic Formula Id
STEP O
~S(~)rR z ( N~s(~)rR
RZ ( q 'H STEP P R q H
B
Rl X-'(CH2)n A-H B Rl X-(CHZ)~A-H
O O O H
Formula I Formula Ie
The sulfonimide intermediate of Formula Id is reacted in Step O with
5 TsOH (0.4 eq.) in methanol at room temperature for 30 minutes to de-protect
the
alcohol of Formula Ie, which in Step P may be converted to the corresponding
carbonyl of Formula I by employing the Dess-Martin periodinane reagent and DCM
at
room temperature for 30 minutes.
Alternatively, a stabilize sulfonamide ring may first be formed and then
10 added to the remainder of the molecule via amide bond formation with the
carboxy
terminus of the natural or unnatural amino acid A, as illustrated in Reaction
Scheme 6
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
31
SCHEME 6
O
Rz
~9 \
NS O
Rl-X-(CHz)n A-C02H H2N ( )rR
O B OR'
O
~~ S(O)rR
R2
B
R1- X-(CHz)n
O O
Formula I
This reaction is further illustrated in Example 3 below.
Depending on the choice of solvent and other conditions known to the
practitioner skilled in the art, compounds of this invention may also take a
cyclized
form, which forms are included in the instant invention. In particular, when B
is
hydrogen compounds of Formula I may exist in the cyclic Formula I' shown
below:
0 0
Rz n'iS(O)rR Rz
i
( q ~N ( q ~NS(O),R
R~-X-(CHz)n A- 8 ~-- R~'X-(CHz)n~A-
OH
O O O
(Formula I) (Formula I')
When B is a moiety other than hydrogen, and depending upon the choice of
solvents
(e.g., R'OH), the compounds of the cyclic form also include compounds having
Formula I" as shown below.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
32
0 0
R2 ( N..S(O)rR RZ
q ~I-i ( q ~NS(O),R
A g
g ~ R~-X-(CHZ)n~
R~-X-(CHZ)n'~--A-~ OR
O
O O
(Formula I) (Formula I')
In addition, it should be understood that the equilibrium forms of the
compounds of this invention may include tautomeric forms. All such forms of
these
compounds are expressly included in the present invention.
Pharmaceutical compositions of this invention comprise any of the
compounds of the present invention, and pharmaceutically acceptable salts
thereof,
with any pharmaceutically acceptable carrier, adjuvant or vehicle (hereinafter
collectively referred to as "pharmaceutically-acceptable carriers").
Pharmaceutically
acceptable carriers, adjuvants and vehicles that may be used in the
pharmaceutical
compositions of this invention include, but are not limited to, ion exchange,
alumina,
aluminum stearate, lecithin, serum proteins, such as human serum albumin;
buffer
substances such as the various phosphates, glycine, sorbic acid, potassium
sorbate,
partial glyceride mixtures of saturated vegetable fatty acids; water, salts or
electrolytes, such as protamine sulfate, disodium hydrogen phosphate,
potassium
hydrogen phosphate, sodium chloride, and zinc salts; colloidal silica,
magnesium
trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene
glycol,
sodium carboxymethylcellulose, polyarylates, waxes,
polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool
fat,
and the like.
The pharmaceutical compositions of this invention may be
administered orally, parenterally, by inhalation spray, topically, rectally,
nasally,
buccally, vaginally or by an implanted reservoir. Oral and parenteral
administration
are preferred. The term "parenteral" as used herein includes subcutaneous,
intracutaneous, intravenous, intramuscular, intra-articular, intrasynovial,
intrasternal,
intrathecal, intralesionai and intracranial injection or infusion techniques.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
33
The pharmaceutical compositions may be in the form of a sterile
injectable preparation, for example, as a sterile injectable aqueous or
oleaginous
suspension. This suspension may be formulated according to techniques known in
the
art using suitable dispersing or wetting agents (such as, for example, Tween
80) and
suspending. agents. The sterile injectable preparation may also be a sterile
injectable
solution or suspension in a non-toxic parenterally-acceptable diluent or
solvent, for
example, as a solution in 1,3-butanediol. Among the acceptable vehicles and
solvents
that may be employed are mannitol, water, Ringer's solution and isotonic
sodium
chloride solution. In addition, sterile, fixed oils are conventionally
employed as a
solvent or suspending medium. For this purpose, any bland fixed oil may be
employed including synthetic mono- or diglycerides. Fatty acids, such as oleic
acid
and its glyceride derivatives are useful in the preparation of injectables, as
are natural
pharmaceutically-acceptable oils, such as olive oil or castor oil, especially
in their
polyoxyethylated versions. These oil solutions or suspensions may also contain
a
long-chain alcohol diluent or dispersant.
The pharmaceutical compositions of this invention may be orally
administered in any orally acceptable dosage form including, but not limited
to,
capsules, tablets, and aqueous suspensions and solutions. In the case of
tablets for
oral use, carrier which are commonly used include lactose and corn starch.
Lubricating agents, such as magnesium stearate, are also typically added. For
oral
administration in capsule form useful diluents include lactose and dried corn
starch.
When aqueous suspensions are administered orally, the active ingredient is
combined
with emulsifying and suspending agents. If desired, certain sweetening and/or
flavoring and/or coloring agents may be added.
The pharmaceutical compositions of this invention may also be
administered in the form of suppositories for rectal administration. These
compositions can be prepared by mixing a compound of this invention with a
suitable
non-irritating excipient which is solid at room temperature but liquid at the
rectal
temperature. Such materials include, but are not limited to, cocoa butter,
beeswax and
polyethylene glycols.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
34
Topical administration of the pharmaceutical compositions of this
invention is especially useful when the desired treatment involves areas or
organs
readily accessible to topical application. For application topically to the
skin, the
pharmaceutical composition should be formulated with a suitable ointment
containing
the active components suspended or dissolved in a carrier. Carriers for
topical
administration of the compounds of this invention include, but are not limited
to,
mineral oil, liquid petroleum, white petroleum, =propylene glycol,
polyoxyethylene,
polyoxypropylene compound, emulsifying wax and water. Alternatively, the
pharmaceutical composition can be formulated with a suitable lotion or cream
containing the active compound suspended or dissolved in a carrier. Suitable
carriers
include, but are not limited to, mineral oil, sorbitan monostearate,
polysorbate 60,
cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and
water. The
pharmaceutical compositions of this invention may also be topically applied to
the
lower intestinal tract by rectal suppository formulation or in a suitable
enema
formulation. Topically-applied transdermal patches are also included in this
invention.
The pharmaceutical compositions of this invention may be
administered by nasal aerosol or inhalation. Such compositions are prepared
according to techniques well-known in the art of pharmaceutical formulation
and may
be prepared as solutions in saline, employing benzyl alcohol or other suitable
preservatives, absorption promoters to enhance bioavailability, fluorocarbons,
and/or
other solubilizing or dispersing agents known in the art.
The compounds of this invention may be used in combination with
either conventional anti-inflammatory agents or with matrix metalloprotease
inhibitors, lipoxygenase inhibitors and antagonists of cytokines other than IL-
1 [3.
The compounds of this invention can also be administered in
combination with immunomodulators (e.g., bropirimine, anti-human alpha
interferon
antibody, IL-2, GM-CSF, methionine enkephalin, interferon alpha,
diethyldithiocarbamate, tumor necrosis factor, naltrexons and rEPO) or with
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
prostaglandins, to prevent or combat IL-1-mediated disease symptoms such as
inflammation.
When the compounds of this invention are administered in combination
therapies with other agents, they may be administered sequentially or
concurrently to
5 the patient. Alternatively, pharmaceutical compositions according to this
invention
may be comprised of a combination of a compound of Formula I and another
therapeutic or prophylactic agent mentioned above.
The disease states which may be treated or prevented by the instant
pharmaceutical compositions include, but are not limited to, inflammatory
diseases,
10 autoimmune diseases and neurodegenerative diseases, and for inhibiting
unwanted
apoptosis involved in ischemic injury, such as ischemic injury to the heart
(e.g.,
myocardial infarction), brain (e.g., stroke), and kidney (e.g., ischemic
kidney disease).
As a consequence of their ability to inhibit apoptosis, the present
pharmaceutical
compositions are also useful for the repopulation of hematopoietic cells of a
patient
15 following chemotherapy. Methods of administering an effective amount of the
above-described pharmaceutical compositions to mammals, also referred to
herein as
patients, in need of such treatment (that is, those suffering from
inflammatory
diseases, autoimmune diseases, neurodegenerative diseases and for the
repopulation of
hematopoietic cells in cancer patients who have undergone chemotherapy) are
another
20 aspect of the instant invention. Finally, as a further consequence of their
ability to
inhibit apoptosis, the instant pharmaceutical compositions may be used in a
method to
prolong the viability of organs to be used in transplantations.
Inflammatory disease which may be treated or prevented include, for
example, septic shock, septicemia, and adult respiratory distress syndrome.
Target
25 autoimmune diseases include, for example, rheumatoid, arthritis, systemic
lupus
erythematosus, scleroderma, chronic thyroiditis, Graves' disease, autoimmune
gastritis, insulin-dependent diabetes mellitus, autoimmune hemolytic anemia,
autoimmune neutropenia, thrombocytopenia, chronic active hepatitis, myasthenia
gravis and multiple sclerosis. Target neurodegenerative diseases include, for
example,
30 amyotrophic lateral sclerosis, Alzheimer's disease, Parkinson's disease,
and primary
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
36
lateral sclerosis. The pharmaceutical compositions of this invention may also
be used
to promote wound healing. Target diseases associated with harmful, apoptosis,
in
other words, those associated with ischemic injury, includes myocardial
infarction,
stroke, and ischemic kidney disease. The pharmaceutical compositions of this
invention may also be used to treat infectious diseases, especially those
involved with
viral infections.
The term "effective amount" refers to dosage levels of the order of
from about 0.05 milligrams to about 140 milligrams per kilogram of body weight
per
day for use in the treatment of the above-indicated conditions (typically
about 2.5
milligrams to about 7 grams per patient per day). For example, inflammation
may be
effectively treated by the administration of from about 0.01 to 50 milligrams
of the
compound per kilogram of body weight per day (about 0.5 milligrams to about
3.5
grams per patient per day).
The amount of the compounds of Formula I that may be combined with
the carrier materials to produce a single dosage form will vary depending upon
the
host treated and the particular mode of administration. For example, a
formulation
intended for the oral administration of humans may contain from 0.5 milligrams
to 5
grams of a compound of Formula I combined with an appropriate and convenient
amount of a pharmaceutically-acceptable carrier which may vary from about 5 to
about 95 percent of the total composition. Dosage unit forms will generally
contain
between from about 1 milligram to about 500 milligrams of an active compound
of
Formula I.
It will be understood, however, that the specific "effective amount" for
any particular patient will depend upon a variety of factors including the
activity of
the specific compound employed, the age, body weight, general health, sex,
diet, time
of administration, route of administration, rate of excretion, drug
combination and the
severity of the particular disease undergoing prevention or therapy.
Although this invention focuses on the use of the compounds disclosed
herein for preventing and treating IL-1-mediated diseases, the compounds of
this
invention can also be used as inhibitory agents for other cysteine proteases.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
37
The compounds of this invention are also useful as commercial
reagents which effectively bind to the ICE/ced-3 family of cysteine protease
or other
cysteine proteases. As commercial reagents, the compounds of this invention,
and
their derivatives, may be used to block proteolysis of a target peptide or may
be
derivatized to bind to a stable resin as a tethered substrate for affinity
chromatography
applications. These and other uses which characterize commercial cystine
protease
inhibitors will be evident to those of ordinary skill in the art.
In order that this invention be more fully understood, the following
examples are set forth. These examples are for the purpose of illustration
only and are
not to be construed as limiting the scope of the invention in any way.
In the following Examples, proton NMR spectra were obtained at 300
MHz; chemical shifts are quoted downfield from internal tetramethylsilane.
EXAMPLE 1
Assay for Inhibition of ICE/ced-3 Protease Family Activity
A. Determination of ICso Values
Fluorescence enzyme assays detecting the activity of the compounds of
Formula I utilizing the recombinant ICE and CPP32 enzymes are perfornied
essentially according to Thornberry et al. (Nature 356:768:774 (1992)) and
Nicholson
et al. (Nature 376:37-43 (1995)) respectively, (herein incorporated by
reference) in 96
well microtiter plates. The substrate is Acetyl-Tyr-Val-Ala-Asp-amino-4-
methylcoumarin (AMC) for the ICE assay and Acetyl-Asp-Glu-Val-Asp-amino-4-
methylcoumarin for the CPP32, Mch2, Mch3 and MchS assays. Enzyme reactions are
run in ICE buffer (25 mM HEPES, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, pH
7.5) containing 2 mM DTT at room temperature in duplicate. The assays are
performed by mixing the following components:
50 ~L ICE, Mch2, MchS, CPP32 (18.8, 38, 8.1 and 0.153 nM
concentrations, respectively) or Mch3 (1 unit) enzyme in ICE buffer
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
38
containing either 8.0 (ICE, Mch2, Mch3, CPP32) or 20 (MchS) mM
DTT;
50 wL compound of Formula 1 or ICE buffer (control); and
100 ~L of 20 ~,M substrate.
The enzyme and the compound of Formula I to be assayed are allowed
to preincubate in the microtitre plate wells for 30 minutes at room
temperature prior to
the addition of substrate to initiate the reaction. Fluorescent AMC product
formation
is monitored for one hour at room temperature by measuring the fluorescence
emission at 460 nm using an excitation wavelength of 360 nm. The fluorescence
change in duplicate (control) wells are averaged and the mean values are
plotted as a
function of inhibitor concentration to determine the inhibitor concentration
producing
50% inhibition (ICso).
B. Determination of the dissociation constant Ki and irreversible rate
constant k~ for irreversible inhibitors
For the irreversible inhibition of a ICE/ced-3 Family Protease enzyme
with a competitive irreversible inhibitor; using the model represented by the
following
formulas:
K; k 3
E + I ~ EI --------> E-I
KS ks
E+S -.~ ES -_.__._.> E+S
The product formation at time t may be expressed as:
fPl t ° fEl T ~S~K ' ~- -k 3 t/(1 + ~ (i + ~-~)
[I]K S k3 I-a LIl KS
Equation 1
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
39
where E, I, EI and E-I denote the active enzyme, inhibitor, non-covalent
enzyme-
inhibitor complex and covalent enzyme-inhibitor adduct, respectively. The K;
value is
the overall dissociation constant of the reversible binding steps, and k3 is
the
irreversible rate constant. The [S] and KS values are the substrate
concentration and
dissociation constant of the substrate bound to the enzyme, respectively. [E]T
is the
total enzyme concentration.
EXAMPLE 2
Synthesis of Representative Compound
SO2CH3 F
F
I
/ O T O ~ / F
~ O F
"Compound No. I"
Bromomethylketone (21:
4-Methylmorpholine (0.75 mL, 6.8 mmol) was added to a solution of
Fmoc-Asp(OBn)-OH (1) (2.03 g, 4.55 mmol) in 50 mL of dry THF at -10°C
under an
atmosphere of nitrogen, followed by the addition of isobutyl chloroformate
(0.78 mL,
6.0 mmol), and the solution was stirred for 20 minutes. The resulting white
precipitate was removed by filtration and the filtrate was cooled to
0°C. In a separate
flask, 1-methyl-3-nitro-1-nitrosoguanidine (1.08 g, 7.36 mmol) was added to a
vigorously stirred mixture of diethyl ether (14 mL) and 40% KOH (8 mL) at
0°C. The
resulting mixture was stirred for 10 minutes and the layers were allowed to
separate.
The ether layer was transferred via plastic pipette to the original filtrate
in THF and
the reaction mixture was stirred for 30 minutes. Then, 48% HBr in water (2.10
mL)
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
was added and the reaction mixture was warmed to room temperature over 15
minutes. The solution was diluted with ethyl acetate, washed twice with
saturated
aqueous sodium bicarbonate, once with brine, dried (MgS04), and concentrated.
The
resulting crude product was purified by flash chromatography on silica gel,
eluting
5 with 35% ethyl acetate-hexanes, to afford 1.73 g (73%) of (2) as a white
solid. 1H-
NMR (300 MHz, CDC13): 8 7.77 (d, J=8 Hz, 2H), 7.58 (d, J=8 Hz, 2H), 7.45-7.29
(m,
9H), 5.77 (d, J=9 Hz, 1H), S.I2 (s, 2H), 4.79-4.71 (m, IH), 4.63-4.42 (m, 2H),
4.21 (t,
J=6 Hz, 1H), 4.04 (s, 2H), 2.97 (ABXq, J=17, 5 Hz, 2H).
Ketone 3
10 Sodium iodide (108 mg, 0.720 mmol) was added to a solution of 2
(I.72 g, 3.28 mmol) in 10 mL of acetone at room temperature, followed by the
addition of the potassium salt of 2,3,5,6-tetrafluorophenol (704 mg, 3.45
mmol) and
the resulting mixture was stirred for one hour. The reaction mixture was
diluted with
ethyl acetate, washed twice with brine, dried (MgSO~), and concentrated. The
crude
15 product was purified by flash chromatography on silica gel, eluting with
1:1:3
dichloromethane/diethyl ether/hexanes, to provide 1.60 g (80%) of (3) as a
white
solid. IH-NMR (300 MHz, CDC13): 8 7.76 (d, J=8 Hz, 2H), 7.58 (d, J=8Hz, 2H),
7.44-7.27 (m, 9H), 6.85-6.73 (m, 1H), 5.73 (d, J=9 Hz, 1H), 5.15-4.92 (m, 4H),
4.75-
4.67 (m, IH), 4.61-4.42 (m, 2H), 4.2I (t, J=6 Hz, 1H), 3.00 (ABXq, J=I8, 4 Hz,
2H).
20 Alcohol (4):
Sodium borohydride (121 mg, 3.20 mmol) was added to a solution Qf 3
(1.60 g, 2.63 mmol) in 7 mL of dry methanol and 7 mL of dry THF at 0°C
and the
resulting mixture was stirred for 30 minutes. The reaction mixture was
quenched with
saturated aqueous ammonium chloride solution, extracted three times with
25 dichloromethane, and the combined dichloromethane layers were washed once
with
brine, dried (MgS04), and concentrated. The crude product was purified by
flash
chromatography on silica gel, eluting with SO% ethyl acetate-hexanes, to give
1.39 g
(87%) of (4) as a white solid. 1H-NMR (300 MHz, CDC13): b 7.78-7.74 (m, 2H),
7.57
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
41
(d, J=7 Hz, 2H), 7.44-7.27 (m, 9H), 6.87-6.75 (m, 1H), 5.62 (d, J=9 Hz, 0.3H),
5.44
(d, J=9 Hz, 0.2H), 5.29-5.23 (m, O.SH), 5.16-5.11 (m, 1H), 4.69 (d, J=6 Hz,
1H), 4.59-
4.37 (m, 4H), 4.30-4.04 (m, 3H), 3.35-3.09 (m, 1H), 2.94-2.41 (m, 2H).
THP Ether (5l:
3,4-Dihydro-2H-pyran (0.31 mL, 3.4 mmol) and pyridinium p-
toluenesulfonate (111 mg, 0.441 mmol) were added to a solution of 4 (1.39 g,
2.28
mmol) in 12 mL of dry dichloromethane and the resulting solution was stirred
at room
temperature for 16 hours. The reaction mixture was diluted with ethyl acetate,
washed
twice with saturated aqueous sodium bicarbonate solution, once with brine,
dried
(MgSO4), and concentrated. The crude product was purified by flash
chromatography
on silica gel, first eluting with 15% ethyl acetate-hexanes and then with SO%
ethyl
acetate-hexanes, to afford 1.09 g (69%) of (5) as a colorless oil. 1H-NMR
(300, MHz,
CDCl3): 8 7.76 (d, J=7 Hz, 2H), 7.62-7.55 (m, 2H), 7.42-7.27 (m, 9H), 6.84-
6.71 (m,
1H), 6.21 (d, J=9 Hz, 0.3H), 5.65 (d, J=9 Hz, 0.2H), 5.33-5.27 (m, O.SH), 5.13
(t, J=3
Hz, 2H), 4.72-4.04 (m, 8H), 3.91-3.73 (m, 1H), 3.51-3.36 (m, 1H), 2.98-2.57
(m, 2H),
1.86-1.61 (m, 2H), 1.57-1.43 (m, 4H).
Amine 6
Piperidine (0.50 mL, 5.1 mmol) was added to a solution of 5 (1.09 g,
1.57 mmol) in 10 mL of dry DMF at room temperature and the resulting solution
was
stirred for 5 minutes. The reaction mixture Was diluted with ethyl acetate,
washed
once with saturated aqueous ammonium chloride solution, twice with water, once
with
brine, dried (MgS04), and concentrated. The crude product was purified by
flash
chromatography on silica gel, first eluting with 50% ethyl acetate-hexanes and
then
with 80% ethyl acetate-hexanes, to provide 544 mg (74%) of (6) as a colorless
oil.
1H-NMR (300 MHz, CDC13): S 7.39-7.29 (m, SH), 6.82-6.70 (m, 1H), 5.15 (s, 2H),
4.78-4.63 (m, 1H), 4.53-4.26 (m, 2H), 4.03-3.79 (m, 2H), 3.71-3.43 (m, 2H),
2.80-
2.43 (m, 2H), 1.85-1.66 (m, 2H), 1.57-1.45 (m, 4H).
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
42
Dipeptide (7):
Amine 6 (S44 mg, 1.15 mmol) and Fmoc-Val-OH (433 mg, 1.28
mmol) were dissolved in 30 mL of dry dichloromethane. 1-Hydroxybenzotriazole
hydrate (237 mg, 1.76 mmol) was added to this solution, followed by the
addition of
S 4-methylmorpholine (0.19 mL, 1.7 mmol) and 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride (271 mg, 1.41 mmol), and the resulting mixture
was
stirred at room temperature for 16 hours. The reaction mixture was diluted
with ethyl
acetate, washed once with saturated aqueous ammonium chloride solution, once
with
saturated aqueous sodium bicarbonate solution, once with brine, dried (MgS04),
and
concentrated. The crude product was purified by flash chromatography on silica
gel,
eluting with 3S% ethyl acetate-hexanes, to give 837 mg (91%) of (7) as a white
solid.
1H-NMR (300 MHz, CDC13): 8 7.76 (d, J=BHz, 2H), 7.62-7.56 (m, 2H), 7.42=7.27
(m,
9H), 6.91-6.71 (m, 2H), 5.39-5.30 (m, 1H), 5.12-S.OS (m, 2H), 4.74-3.78 (m,
10H),
3.50-3.36 (m, 1H), 2.97-2.61 (m, 2H), 2.19-2.06 (m, 1H), 1.82-1.68 (m, 2H),
1.52-
1S 1.40 (m, 4H), 0.98-0.87 (m, 6H).
Amine 8
Piperidine (0.35 mL, 3.6 mmol) was added to a solution of 7 (830 mg,
1.0S mmol) in 7 mL of dry DMF at room temperature and the resulting solution
was
stirred for S minutes. The reaction mixture was diluted with ethyl acetate,
washed
once with saturated aqueous ammonium chloride solution, twice with water, once
with
brine, dried (MgS04), and concentrated. The crude product was purified by
flash
chromatography on silica gel, first eluting with SO% ethyl acetate-hexanes and
then
with 20% methanol-dichloromethane, to provide S97 mg (100%) of (8) as a yellow
oil. 1H-NMR (300 MHz, CDC13): 8 8.07-7.63 (m, 1H), 7.40-7.27 (m, SH), 6.83-
6.71
(m, 1H), 5.17-5.07 (m, 2H), 4.76-4.62 (m, 1H), 4.60-4.26 (m, 2H), 4.24-4.09
(m, 2H),
3.92-3.80 (m, 1H), 3.52-3.39 (m, 1H), 3.22-3.16 (m, 1H), 2.97-2.60 (m, 2H),
2.31-
2.16 (m, 1H), 1.84-1.64 (m, 2H), 1.59-1.44 (m, 4H), 0.98-0.94 (m, 3H), 0.81-
0.76 (m,
3H).
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
43
Dipeptide (91:
Amine 8 (578 mg, I.O1 mmol) and (1-naphthoxy)acetic acid (226 mg,
1.12 mmol) were 'dissolved in 25 mL of dry dichloromethane. 1-
Hydroxybenzotriazole hydrate (211 mg, 1.56 mmol) was added to this solution,
followed by the addition of N-methylmorpholine (0.17 mL, 1.5 mmol) and 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (239 mg, 1.25 mmol),
and
the resulting mixture was stirred at room temperature for 16 hours. The
reaction
mixture was diluted with ethyl acetate, washed once with saturated aqueous
ammonium chloride solution, once with saturated aqueous sodium bicarbonate
solution, once with brine, dried (MgSOd), and concentrated. The crude product
was
purified by flash chromatography on silica gel, eluting with 35% ethyl acetate-
hexanes, to give 682 mg (89%) of (9) as a white foam. 1H-NMR (300 MHz, CDC13):
8 8.28-8.2I (m, 1H), 7.85-7.80 (m, 1H), 7.57-7.49 (m, 3H), 7.40-7.22 (m, 8H),
6.96-
6.42 (m, 2H), S.I2-5.05 (m, 2H), 4.80-4.59 (m, 3H), 4.58-4.24 (m, 3H), 4.23-
4.06 (m,
2H), 3.96-3.77 (m, 1H), 3.56-3.38 (m, 1H), 2.97-2.61 (m, 2H), 2.24-2.10 (m,
1H),
1.84-1.66 (m, 2H), 1.55-1.40 (m, 4H), 0.98-0.87 (m, 6H).
Acid 10
10% Palladium on carbon (170 mg) was added to a solution of 9 (650
mg, 0.861 mmol) in anhydrous methanol (15 mL) under an atmosphere of nitrogen
and the flask was then evacuated with the house vacuum. The mixture was
stirred
under a balloon of hydrogen gas for 75 minutes, then filtered through Celite,
and
eluted with methanol. The solution was concentrated to afford 345 mg (94%) of
(10)
as a white solid.
Methyl sulfonimide (I11:
1,1'-Carbonyldiimidazole (146 mg, 0.900 mmol) was added to a
solution of 10 (300 mg, 0.451 mmol) in dry THF (7 mL) under an atmosphere of
nitrogen, and the reaction mixture was stirred for 3 hours. The mixture was
cooled to
0°C, and the methanesulfonamide (86 mg, 0.90 mmol) was added, followed
by the
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
44
addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (0.135 mL, 0.903 mmol). The
resulting mixture was stirred at room temperature for 4 hours. The reaction
mixture
was diluted with ethyl acetate, washed once with 1 N HCI solution, twice with
water,
once with brine, dried (MgS04), and concentrated. The residue was
reconcentrated
S from dichloromethane to provide 314 mg (98%) of (11) as a white solid. 1H-
NMR
(300 MHz, CDCl3): 8 10.22-9.99 (m, 1H), 8.24-8.17 (m, 1H), 7.85-7.69 (m, 1H),
7.59-
7.47 (m, 3H), 7.40-7.27 (m, 2H), 6.94-6.66 (m, 2H), 4.85-4.62 (m, 4H), 4.SS-
4.02 (m,
6H), 3.59-3.45 (m, 1H), 3.22-3.19 (m, 3H), 2.89-2.SS (m, 2H), 2.21-2.09 (m,
1H),
1.88-1.67 (m, 2H), 1.63-1.43 (m, 4H), 1.00-0.86 (m, 6H).
Alcohol X12):
p-Toluenesulfonic acid (34 mg, 0.18 mmol) was added to a solution of
11 (302 mg, 0.426 mmol) in anhydrous methanol (S mL) and the reaction mixture
was
stirred at room temperature for 30 minutes. The mixture was diluted with ethyl
acetate, washed twice with water, once with brine, dried (MgS04), and
concentrated to
1S afford 249 mg (93%) of (12) as a white solid. IH-NMR (300 MHz, DMSO): 8
8.23-
8.18 (m, 2H), 8.04-7.88 (m, 2H), 7.61-7.50 (m, 4H), 7.43-7.37 (m, 1H), 6.90
(d, J=8
Hz, 1H), 5.61 (d, J=5 Hz, 0.3H), 5.48 (d, J=6 Hz, 0.7H), 4.83-4.72 (m, 2H),
4.58-3.75
(m, SH), 3.17 (s, 1H), 3.13 (s, 2H), 2.74-2.37 (m, 2H), 2.03-1.91 (m, 1H),
0.84-0.76
(m, 6H).
Methylsulfonimide ("Compound No. 1"l: ,
Dess-Martin periodinane (203 mg, 0.479 mmol) was added to a
solution of 12 (233 mg, 0.373 mmol) in 7 mL of dry dichloromethane, and the
reaction mixture was stirred at room temperature for 30 minutes. The mixture
was
diluted with ethyl acetate, washed twice with water, once with brine, dried
(MgS04),
2S and concentrated. The crude product was purified by flash chromatography,
eluting
first with 60% ethyl acetate-hexanes and then with 80% ethyl acetate-hexanes,
to
provide 134 mg (S8%) of Compound No. 1 as a mixture of diastereomers and as a
white solid. 1H-NMR (300 MHz, DMSO): 8 8.84 (d, J=7 Hz, 1H), 8.28-8.06 (m,
2H),
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
7.91-7.87 (m, 1H), 7.58-7.49 (m, SH), 7.39 (t, J=8 Hz, 1H), 6.89 (d, J=8 Hz,
1H), 5.23
(s, 1H), 4.93-4.42 (m, 4H), 4.28-4.19 (m, 1H), 3.28 (s, 1.3H), 3.19 (s, 1.7H),
2.96-2.59
(m, 2H), 2.06-1.96 (m, 1H), 0.89-0.80 (m, 6H); MS (ESI) m/e 654 [(M+) -1].
EXAMPLE 3
5 Alternative Synthesis of Compounds of Formula I
This example illustrates synthesis of compounds of Formula I by
formation of a stabilized sulfonamide ring, followed by addition via amide
bond
formation to the remainder of the compound. In the following representative
10 examples, q is I, r is 2 and R is methyl.
Example Scheme 3a
0 0 o
~OtBu ~ ~NHSOZCH3 d ~ ~NHSOZCH3
CbzNH OH CbzNH N(OMe)Me CbzNH H
O O O
O '
CONHSOZCH3
e,f NSOZCH3 Rz O
9,h II H
--~ HzN ' R1-X-(CHz)n NH'A~NH~('
O O
O
Starting with the commercially available Z-Asp(OtBu)-OH, the
Weinreb amide is formed, followed by hydrolysis of the t-butyl ester. The beta
carboxylic acid is then coupled with methyl sulfonamide (or other substituted
sulfonamide), followed by reduction of the Weinreb amide to the aldehyde. Acid-
catalyzed acetal formation using ethanol is assisted by cyclization of the
sulfonamide
to form a stable 5-membered ring. The carbobenzyloxy urethane is then removed,
the
aspartyl intermediate coupled to the substituted acyl peptide of choice, then
the acetal
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
46
deprotected. (a) EDC1, HOBt, N-Me Morpholine, HC1H2N(OCH3)CH3, CH2C12,
0°C-
RT; (b) TFA, anisole, CH2C12, RT; (c) EDCI, DMAP (cat.), CH3S02NH2, CHZC12,
RT; or i. I CDI; ii. GH3S02NHz, DBU, 0°C, (d) LAH, THF,
0°C; (e) Ethanol,
CH(OEt)3, p-TsOH, toluene reflux; (f) Hz; Pd/C, RT; (g) Rl-X-(CH2)n
CH(RZ)CO(amino acid)C02H, EDCI, HOBt, N-Me Morpholine, CH2C12, 0°C-
RT;
(h) TFA, anisole, CH2C12, H20 RT.
Example Scheme 3b
0 0 0
'OtBu a-c ~NHSO~CH3 d ~NHSOzCH3
FmocNH ~H FmocNH N(OMe)Me FmocNH H
O 0 O
O
' CONHSO~CH3
elf NSOzCH3 9~ R~ O
H2N O R~-X-(CH~)n NKA~NH~H
O
O
Starting with the commercially available Fmoc-Asp(OtBu)-OH, the
Weinreb amide is formed, followed by hydrolysis of the t-butyl ester. The beta
carboxylic acid is then coupled with methyl sulfonamide (or other substituted
sulfonamide), followed by reduction of the Weinreb amide to the aldehyde. Acid-
catalyzed acetal formation using benzyl alcohol is assisted by cyclization of
the
sulfonamide to form a stable 5-membered ring. The fluorenylmethyloxy urethane
is
then removed, the aspartyl intermediate coupled to the substituted acyl
peptide of
choice, then the acetal deprotected. (a) EDCI, HOBt, N-Me Morpholine,
HC1H2N(OCH3)CH3, CH2C12, 0°C-RT; (b) TFA, anisole, CHZCl2, RT; (c)
EDCI,
DMAP (cat.), CH3S02NHa, CHzCl2, RT; (d) LAH, THF, 0°C; (e) Benzyl
alcohol, p-
TsOH, toluene reflux; (f) Et2NH, DMF, RT; (g) Rl-X-(CH2)"CH(R2)CO(amino
acid)C02H, EDCI, HOBt, N-Me Morpholine, CHzCl2, 0°C-RT; (h) H2; Pd/C,
RT.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
47
Example Scheme 3c
0
OtBu F F O 0
'NHSOzCH3
CbzNH 0 ~ ~ ~ c ; NSOZCH3
p CbzNH OTFP CbzNH pTFP
F' F
TFP, tetrafluorouphenyl
O
CONHSOZCH3
NSO2CH3 e,f H
HZN O OTFP ~ R1-XyCHy)n~N 'A~H~OTFP
O O
Starting from the readily available intermediate, modified by addition
of tetrafluorophenyl, the t-butyl ester is hydrolyzed, followed by coupling
with methyl
sulfonamide (or other substituted sulfonamide). Acid-catalyzed ketal formation
using
ethanol is assisted by cyclization of the sulfonamide to form a stable 5-
membered
ring. The carbobenzyloxy urethane is then removed, the aspartyl intermediate
coupled
to the substituted aryl peptide of choice, then the ketal deprotected. (a)
TFA, anisole,
CHZC12, RT; (b) EDCI, DMAP (cat.), CH3SO2NHa, CHZCl2, RT; or i. CDI; ii.
CH3SO2NH2, DBU, 0°C; (c) Ethanol, CH(OEt)3, p-TsOH, toluene reflux;
(d) H2;
Pd/C, RT; (e) Rl-X(CH2)"CH(R2)CO(amino acid)COZH, EDCI, HOBt, N-Me
Morpholine, CH2C12, 0°C-RT; (~ TFA, anisole, CH2C12, H20, RT.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
48
Example Scheme 3d
0
OfBu F F O
'NHSO CH
FmocNH 0 ~ ~ ~ z s
O FmocNH OTFP
F F O
TFP, tetrafluorouphenyl
O
d NSOzCH3 ef Rz H p CONHSOZCH3
HzN OTFP ~ Rt XWCHz)~N A~H~OTFP
O
O O
. Starting from the readily available intermediate, modified by addition
of tetrafluorophenyl, the t-butyl ester is hydrolyzed, followed by coupling
with methyl
sulfonamide (or other substituted sulfonamide). Acid-catalyzed ketal formation
using
ethanol is assisted by cyclization of the sulfonamide to form a stable 5-
membered
ring. The fluorenylmethyloxy urethane is then removed, the aspartyl
intermediate
coupled to the substituted acyl peptide of choice, then the ketal deprotected.
(a) TFA,
anisole, CH2C12, RT; (b) EDCI, DMAP (cat.), CH3S02NH2, CH2C12, RT; (c)
Ethanol,
CH(OEt)3, p-TsOH, toluene reflux; (d) Et2NH, DMF, RT; (e) Rl-X-(CH2)"
CH(RZ)CO(amino acid)C02H, EDCI, HOBt, N-Me Morpholine, CHZCIz, 0°C-
RT;
(fj H2; Pd/C, RT.
EXAMPLE 4
Representative Compounds
The. representative compounds listed in the following Table 1 may be
made according to the procedures set forth in Examples 2 and 3.
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
49
Table 1
Representative Compounds
i,aCH3
R1
~O~A-
~ ~O
No.A B R'
2 NHCH(CHZCH(CH3)z)COCHzF 1-naphthyl
3 NHCH(CH(CH3)z)CO CHzF 1-naphthyl
4 NHCH(CH(CH3)2)CO CHZOCO(2,4-diCl-Ph)1-naphthyl
NHCH(CH(CH3)2)CO CH20(2,6-diF-Ph) 1-naphthyl
6 NHCH(CH(CH3)Z)CO CHzO(2,4,6-triF-Ph)1-naphthyl
7 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)1-naphthyl
8 NHCH(CH(CH3)2)CO CH20(6-Me-2-pyron-4-yl)1-naphthyl
9 NHCH(CH(CH3)z)CO CHZO(2-Ph-5,6-benzopyran-1-naphthyl
4-on-3-yl)
NHCH(CH(CH3)2)CO CHZOPO(Me)Ph 1-naphthyl
11 NHCH(CH(CH3)Z)CO CHZOPOPh2 1-naphthyl
12 NHCH(CH(CH3)2)CO CH20(2-CF3-pyrimidin-4-yl)1-naphthyl
13 NHCH(CH(CH3)z)CO CHZO(5-COzMe-isoxazol-3-1-naphthyl
yl)
14 NHCH(CH(CH3)z)CO CHZOPO(Me)(1-naphthyl)1-naphthyl
NHCH(CH2CH(CH3)2)COCHZOPOPh2 1-naphthyl
16 NHCH(CHZCH(CH3)2)COCHZOCO(2,6-diCl-Ph)1-naphthyl
17 NHCH(CH2CH(CH3)2)COCHzO(2,4,6-triF-Ph)1-naphthyl
18 NHCH(CHZCH(CH3)Z)COCHZO(2,3,5,6-tetraF-Ph)1-naphthyl
19 NHCH(CHZCH(CH3)2)COCHZOPO(Me)Ph 1-naphthyl
NHCH(CH3)CO CH20(2-F-Ph) (2-Ph)Ph
21 NHCH(CH3)CO CHZOCO(2,6-di-CI-Ph)(2-Ph)Ph
22 NHCH(CH3)CO CHZOPOPhz (2-Ph)Ph
23 NHCH(CH3)CO CHzO(2-F-Ph) (2-t-Bu)Ph
24 NHCH(CH3)CO CHZOPOPhz (2-t-Bu)Ph
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
No.A B R'
25 NHCH(CH3)CO CHZOCO(2,3,5,6-tetra-Cl-1-naphthyl-CHZ
Ph)
26 NHCH(CH3)CO CHzOCO(2,6-di-Cl-Ph)1-naphthyl-CHZ
27 NHCH(CH3)CO CHZOPOPhz 1-naphthyl-CHZ
28 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)1-naphthyl-CHZ
29 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)PhCH2
30 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)Ph(CHZ)z
31 NHCH(CH(CH3)Z)CO CH20(2,3,5,6-tetraF-Ph)Ph2CH
32 NHCH(CH(CH3)Z)CO CHZO(2,3,5,6-tetraF-Ph)Ph
33 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2-Ph)Ph
34 NHCH(CH(CH3)2)CO CHZO(2,3,5,6-tetraF-Ph)(2-PhCH2)Ph
35 NHCH(CH(CH3)Z)CO CH20(2,3,5,6-tetraF-Ph)(3-Ph0)Ph
36 NHCH(CH(CH3)2)CO CHZO(2,3,5,6-tetraF-Ph)4-Cl-1-naphthyl
37 NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)2-anthryl
38 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)2-benzimidazolyl
39 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)1-adamantanyl
40 NHCH(CH(CH3)Z)CO CHZO(2,3,5,6-tetraF-Ph)(2-F)Ph
41 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(4-F)Ph
42 NHCH(CH(CH3)2)CO CHzO(2,3,5,6-tetraF-Ph)(2-CF3)Ph
43 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2-t-Bu)Ph
44 NHCH(CH(CH3)Z)CO CH20(2,3,5,6-tetraF-Ph)(4-n-heptyl)Ph
45 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2-CH30)Ph
46 NHCH(CH(CH3)2)CO CHzO(2,3,5,6-tetraF-Ph)(2-Ph0)Ph
47 NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)2-naphthyl
48 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)5,6,7,8-tetrahydro-i-
naphthyl
49 NHCH(CH(CH3)Z)CO CH20(2,3,5,6-tetraF-Ph)1-anthryl
50 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)2-pyridinyl
51 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)4-pyridinyl
52 NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)2,3,5,6-tetrafluoro-4-
pyridinyl
53 IVHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)2-pyrazinyl
54 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)1,2,3,4-tetrahydro-1-
naphthyl
NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)(2-Cl)Ph
56 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2-Br)Ph
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
51
No.A B R1
57 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2-I)Ph
58 NHCH(CH(CH3)2)CO CHzO(2,3,5,6-tetraF-Ph)(2,6-di-F)Ph
59 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2,5-di-t-Bu)Ph
60 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)5-indanyl
61 NHCH(CH(CH3)2)CO CHZO(2,3,5,6-tetraF-Ph)(3,4,5-tri-Me0)PhCH2
62 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)methyl
63 NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)n-heptyl
64 NHCH(CH(CH3)2)CO CHZO(2,3,5,6-tetraF-Ph)t-octyl
65 NHCH(CH(CH3)2)CO CHZO(2,3,5,6-tetraF-Ph)cyclo-hexyl
66 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)5-Ph-3-pyrazolyl
67 NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)(2-F-4-I)Ph
68 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)(2,3,4,5-tetra-F)Ph
69 NHCH(CH(CH3)z)CO CHzO(2,3,5,6-tetraF-Ph)(2,3,4,6-tetra-F)Ph
70 NHCH(CH(CH3)2)CO CHzO(2,3,5,6-tetraF-Ph)(2,3,5,6-tetra-Cl)Ph
71 NHCH(CH(CH3)2)CO CHZO(2,3,5,6-tetraF-Ph)(2,3,4,5,6-penta-F)Ph
72 NHCH(CH(CH3)Z)CO CHzO(2,3,5,6-tetraF-Ph)PhZN
73 NHCH(CH(CH3)z)CO CH20(2,3,5,6-tetraF-Ph)PHCHz(Ph)N
74 NHCH(CH(CH3)2)CO CH20(2,3,5,6-tetraF-Ph)PhCH20
75 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(2-t-Bu)Ph
76 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(2-CF3)Ph
77 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(2-Ph)Ph
78 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(2-PhCH2)Ph
79 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(2-PhO)Ph
80 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(3-Ph0)Ph
81 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)5,6,7,8-tetrahydro-1-
naphthyl
82 NHCH(CH3)CO CHZO(2,3,5,6-tetraF-Ph)1-naphthyl
83 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)Ph
84 NHCH(CH3)CO CHzO(2,3,5,6-tetraF-Ph)(2,6-di-F)Ph
85 NHCH(CH3)CO CHZO(2,3,5,6-tetraF-Ph)(4-Ph)Ph
86 NHCH(CH3)CO CH20(2,3,5,6-tetraF-Ph)(4-MeO)Ph
87 NHCH(CH3)CO CHzO(2,3,5,6-tetraF-Ph)PhZCH
88 NHCH(CHzcyclohexyl)COCH20(2,3,5,6-tetraF-Ph)(2-Ph0)Ph
89 NHCH(CHzcyclohexyl)COCH20(2,3,5,6-tetraF-Ph)(2-Ph)Ph
I
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
52
No. A B RI
90 NHCH(CHzcyclohexyl)CO CH20(2,3,5,6-tetraF-Ph) (2-PhCHz)Ph
91 NHCH(CHzcyclohexyl)CO CHZO(2,3,5,6-tetraF-Ph) 1-naphthyl
92 NHCH(CHzcyclohexyl)CO CH20C0(2,6-diCl-Ph) 5,6,7,8-tetrahydro-1-
naphthyl
93 NHCH(CHzcyclohexyl)CO CH20(2,3,5,6-tetra-F-Ph) 5,6,7,8-tetrahydro-1-
naphthyl
94 NHCH(CHzcyclohexyl)CO CHzOPO(Me)Ph 5,6,7,8-tetrahydro-1-
naphthyl
95 NHCH(CHzcyclohexyl)CO CHZOPOPhz 5,6,7,8-tetrahydro-1-
naphthyl
96 NHCH(CHzcyclohexyl)CO CHZOPO(Me)Ph (2-PhCHz)Ph
97 NHCH(CHzcyclohexyl)CO CHZOPOPhz (2-PhCHz)Ph
98 NHCH(CHzcyclohexyl)CO CHZOPO(Me)Ph (2-Ph)Ph
99 NHCH(CHzcyclohexyl)CO CHzOPOPhz (2-Ph)Ph
100 CHzO(2,3,5,6-tetra-F-Ph) 1-naphthyl
" ,N
O
101 CH20(2,3,5,6-tetra-F-Ph) 1-naphthyl
2 'N
O
102 NHCH(cyclohexyl)CO CHzO(2,3,5,6-tetra-F-Ph) 1-naphthyl
103 Norleucine CH20(2,3,5,6-tetra-F-Ph) 1-naphthyl
104 (t-butyl)glycine CHzO(2,3,5,6-tetra-F-Ph) 1-naphthyl
105 (t-butyl)alanine CH20(2,3,5,6-tetra-F-Ph) 1-naphthyl
106 Phenylglycine CH20(2,3,5,6-tetra-F-Ph) 1-naphthyl
107 Phenylalanine CH20(2,3,5,6-tetra-F-Ph) 1-naphthyl
108 Homophenylalanine CHZO(2,3,5,6-tetra-F-Ph) 1-naphthyl
109 1-aminocyclopentane CHZO(2,3,5,6-tetra-F-Ph) 1-naphthyl
carboxylic acid
110 NHCH(CHzCH2SOCH3)CO CH20(2,3,5,6-tetra-F-Ph) 1-naphthyl
111 H 1-naphthyl
2 'N
O
1I2 NHCH(CH(CH3)z)CO H 2-(1H-tetrazol-5-yl)Ph
113 NHCH(CH(CH3)z)CO H 1-adamantanyl
114 NHCH(CH(CH3)z)CO ~ H Ph
115 NHCH(CH(CH3)z)CO H PhCHz
116 NHCH(CH(CH3)z)CO H Ph(CHz)z
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
53
No.A B R1
117NHCH(CH(CH3)z)CO H (2-CF3)Ph
118NHCH(CH(CH3)Z)CO H (2-t-Bu)Ph
119NHCH(CH(CH3)2)CO H (2-Ph)Ph
120NHCH(CH(CH3)2)CO H (2-PhCH2)Ph
121NHCH(CH(CH3)z)CO H (2-Ph0)Ph
122NHCH(CH(CH3)Z)CO H 2-naphthyl
123NHCH(CH(CH3)2)CO H 1-naphthyl
124NHCH(CH(CH3)z)CO H 4-Cl-1-naphthyl
125NHCH(CH(CH3)2)CO H 5,6,7,8-tetrahydro-1-
naphthyl
126NHCH(CH(CH3)2)CO H 1,2,3,4-tetrahydro-1-
naphthyl
127NHCH(CH(CH?)2)CO H (1-naphthyl)CHz
128NHCH(CHZCH(CH3)2)COH 1-naphthyl
I
EXAMPLE 5
Activity of Representative Compound
The activity of a representative compound of this invention (i.e.,
Compound No. 1) was evaluated according to the procedures disclosed in Example
1.
More specifically, the ICSO and K; for Compound No. 1 were determined as set
forth
above. The ICSO results are provided in Table 2, as run against Cbz-ValAlaAsp-
H as a
reference control.
Table 2
Cpd. Csp-1 Csp-3 Csp-6 Csp-7 Csp-8 lCsp-9
No.
ICSOOM) ICsoOM) ICso(!~M)ICsoOM)ICsoOM) ICso(w~
1 0.004 0.002 0.002 0.004 0.006 0.005
Reference0.064 47.0 >10 >10 2.96 0.87
CA 02406247 2002-10-16
WO 01/79162 PCT/USO1/12563
54
The equations set forth in Example 1 were also used determine the K;
values of inhibitor (i.e., Compound No. 1) bound to a ICE/ced-3 family
protease.
Thus, a continuous assay was run for sixty minutes at various concentrations
of the
inhibitor and the substrate. The assay was formulated essentially the same as
described above for generating the data in Table 2, except that the reaction
was
initiated by adding the enzyme to the substrate-inhibitor mixture. The K;
values were
obtained by simulating the product AMC formation as a function of time
according to
Equation 1. The results of this second assay are set forth below in Table 3,
wherein
the reference compound was Cbz-ValAlaAsp-CHZF.
I0 Table 3
Cpd. No. Csp-1 Csp-3 Csp-6 Csp-8
K;(p,M) K;(pM) K;(pM) K;(pM)
1 0.20 0.08 0.40 0.60
referenceO.O1S 0.820 O.S94 0.018
Although the invention has been described with reference to the
examples provided above, it should be understood that various modifications
can be
made without departing from the spirit of the invention. Accordingly, the
invention is
limited only by the claims.