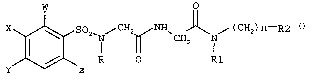Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
1
Nouveaux dérivés de la N(phén~rlsulfonvl)alyrcine et leur utilisation en
théraaeutiaue.
La présente invention concerne de nouveaux composés de la N
(phénylsulfonyl)glycine, leur procédé de préparation et leur utilisation pour
obtenir
s des compositions pharmaceutiques.
Ces nouveaux composés sont utiles en thérapeutique, particulièrement pour
le traitement de la douleur.
Art antérieur
On connaît déjà des composés comportant dans leur structure un
~o groupement du type arylsulfonamide et la glycine. Par exemple on peut citer
selon
EP 236 163 et EP 236 164 des dérivés N-a-arylsulfonylaminoacyl-p-amidino-
phényl
alaninamides qui sont des inhibiteurs sélectifs de la thrombine et sont utiles
comme
anti-thrombotiques. On connaît aussi, selon EP 614 911, des composés de
structure
assez proche des précédentes, comportant simultanément un groupe
arylsulfamoyle
1s et un groupe phénylamidine substitué, qui ont la propriété de se fixer sur
les
récepteurs du neuropeptide Y et qui peuvent présenter une utilité pour soigner
l'hypertension, l'angine de poitrine, !'athérosclérose, la dépression,
l'anxiété,
l'inflammation, l'allergie ou les surcharges graisseuses.
EP 558 961 suggère également l'utilisation de composés du type
2o arylsulfonamide d'acides aminés substitués pour le traitement de la
thrombose en
raison de propriétés anticoagulantes.
Des études relatives aux propriétés antithrombotiques de composés
présentant dans leur structure un groupe arylsulfonamide et un groupe
phénylamidine, ont également été publiées dans Pharmazie 1984 vol. 39 (5)
pages
2s 315-317 et Pharmazie 1986 vol 41 (4) p 233-235.
Dans un même domaine d'activité pharmacologique, WO 92/16549 A1 décrit
des dérivés de la phénylalanine comportant un groupe arylsulfonamide, qui sont
des inhibiteurs de protéinase, notamment des inhibiteurs de la thrombine.
On connaît aussi, selon WO 97/25315, des composés de structure N-
30 (arylsulfonyl)amino-acides, utiles pour traiter les maladies
inflammatoires.
Dans un domaine thérapeutique différent, WO 00/34313 décrit des peptides
qui peuvent comporter en extrémité de chaîne un groupe arylsulfonyl et qui
sont
revendiqués pour leur aptitude à inhiber la procollagène-C-protéinase. On
connaît
également par la publication ). Chem. Soc., Perkin Trans. 1(1986), (9) p 1655-
64,
3s des composés de structure proche qui sont présentés comme des inhibiteurs
de
l'élastase pancréatique porcine.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
2
Objet de l'invention
L'invention concerne de nouveaux composés comportant l'enchaînement N
(arylsulfonyl)glycyl-glycine substitué, lesdits composés étant notamment
utiles en
tant que principes actifs de médicaments destinés au traitement de la douleur,
s particulièrement les hyperalgésies et les algésies majeures.
Description
Selon la présente invention, on propose, en tant que produit industriel
nouveau, un composé de N-(phénylsulfonyl) glycine caractérisé en ce qu'il est
choisi
parmi l'ensemble constitué par
io i) les composés de formule
w o
SO / CHz ~~ ~ / C CHz ) n-Ra
CH2
R O R1
I
dans laquelle
W représente un atome de chlore,
Is X représente un atome d'hydrogène, un groupe méthyle ou un atome de chlore,
Y et Z représentent chacun indépendamment un atome d'hydrogène ou un atome
de chlore, ou
X et W ou X et Y forment ensemble, avec les atomes de carbone auxquels ils se
rattachent un noyau phényle,
2o R représente un atome d'hydrogène, un groupe allyle ou un groupe alkyle en
Cl-C4
non substitué ou substitué par un groupe phényle, un groupe méthoxy, un groupe
pyridinyle, un groupe carboxamide ou un groupe N-méthylcarboxamide,
Ri représente un atome d'hydrogène, un groupe alkyle en Cl-C4 ou
un groupe (CHz)m-R'z
25 n et m représentent chacun indépendamment 1, 2, 3 ou 4,
Ra et R'a représentent chacun indépendamment
un groupe
N/
I
R
3
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
3
un groupe
un groupe
un groupe
un groupe
CH -N N-R
2 ~ 3
R
/ 4
CH -N
2
R
s
CHZ-~ z
CHZ
N
un groupe
CH -O-R
2 s
un groupe
un groupe
R
8
N , ~u
R
et
1s R3 représente un atome d'hydrogène ou un groupe alkyle en Cl-C4,
R4 représente un atome d'hydrogène, un groupe COCH3, un groupe COOCH3, ou un
groupe alkyle en Cl-C4 ,
R5 représente un atome d'hydrogène ou un groupe alkyle en Cl-C4, non substitué
ou
substitué par un groupe phényle,
2o R6 représente un atome d'hydrogène ou un groupe CONHCZHS,
R, représente un atome d'hydrogène,
un groupe
// H
-C
NH
2
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
4
un groupe
~~ oH
-c
NH
z
un groupe
// -O-COCH3
-C
\
NH
z
un groupe
lN
-C
\
OU
r
un groupe CONHCH3
R8 représente un atome d'hydrogène, un groupe NHz, ou un groupe alkyle en Cl-
C4
1~ ,
p=4, 5ou6
ü) les sels d'addition des composés de formule I ci-dessus avec un acide.
Selon l'invention, on préconise aussi un procédé pour la préparation des
composés de formule I ainsi que de leurs sels d'addition.
1s On préconise également l'utilisation d'une substance choisie parmi les
composés de formule I et leurs sels d'addition non toxiques pour la
préparation d'un
médicament, utile en thérapeutique humaine ou animale, destiné à la prévention
ou
au traitement de pathologies liées à la douleur, notamment les hyperalgésies
consécutives à un état inflammatoire ou les algésies majeures liées à d'autres
états
2o pathologiques tels que, par exemple, le cancer.
Description détaillée
Dans la formule I, on entend par groupe alkyle en Cl-C4' une chaîne
hydrocarbonée ayant de 1 à 4 atomes de carbone, linéaire ou ramifiée, ou bien
encore cyclique. Un groupe alkyle en Ci-C4 est par exemple un groupe méthyle,
2s éthyle, propyle, butyle, 1-méthyl-éthyle, 1-méthylpropyle, 2-méthylpropyle,
1,1-
diméthyléthyle ou cyclopropylméthyle.
On entend par groupe alkyle en C~-C4 substitué par un groupe phényle, un
groupe alkyle en Cl-C4 dont l'un des atomes d'hydrogène est substitué par un
groupe phényle. Un tel groupe est par exemple un groupe phénylméthyle, un
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
groupe 2-(phényl)-éthyle, un groupe 1-(phényl)éthyle, un groupe phénylpropyle
ou
un groupe phénylbutyle.
Lorsque Rz ou R'2 représentent un cycle pipéridine, éventuellement substitué
par un groupe R3, les positions de liaison sur ce cycle peuvent se faire par
l'un
s quelconque des sommets substituables.
Lorsque Rz ou R'2 représentent un cycle pyridine, éventuellement substitué
par un groupe R8, les positions de liaison et de substitution peuvent se faire
sur l'un
quelconque des carbones du cycle.
Lorsque Rz ou R'a représentent un cycle phényle substitué par un groupe R~
o difFérent de H, !a position relative des substituants peut étre ortho, méta
ou para,
avec une préférence pour la position para.
Par sels d'addition, on entend les sels d'addition obtenus par réaction d'un
composé de formule I contenant au moins une fonction basique sous sa forme non
salifiée, avec un acide minéral ou organique. De préférence, ü s'agira de sels
d'addition pharmaceutiquement acceptables.
Parmi les acides minéraux convenant pour salifier un composé basique de
formule I, on préfère les acides chlorhydrique, bromhydrique, phosphorique et
sulfurique. Parmi les acides organiques convenant pour salifier un composé
basique
de formule I, on préfère les acides méthanesulfonique, benzènesulfonique,
2o toluènesulfonique, malëique, fumarique, oxalique, citrique, tartrique,
lactique et
trifluoroacétique.
Parmi les 1 composés selon la présente invention, on préfère ceux dans
lesquels R représente un groupe phényléthyle ou un groupe méthyle ou un groupe
acétamide et ceux dans lesquels l'un des substituants Rl ou R2 comprend dans
sa
2s structure un groupe 5-imidazolyle ou un groupe « amidinyle », étant entendu
que
l'on comprend par groupe « amidinyle » un groupe qui contient dans sa
structure
les atomes liés
N-
-C
NH
Ainsi le groupe « amidinyle » inclut par exemple les groupes amidine, 2-
3o imidazolyle ou 4,5-dihydro-2-imidazolyle.
Parmi les composés de l'invention, on préfère aussi ceux dans lesquels W
représente un atome de chlore, X représente un groupe méthyle ou un atome de
chlore, Y représente un atome d'hydrogène ou un atome de chlore et Z
représente
un atome d'hydrogène.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
6
Selon l'invention, on préconise un procédé général de préparation des
composés de l'invention comprenant les étapes consistant à
1) faire réagir le chlorure de benzènesulfonyle de formule
x w
Y ~ ~ SOzCl
z II
dans laquelle W, X, Y, Z sont tels que définis précédemment
Io
avec une amine de formule RNHZ dans laquelle R représente un groupe te! que
décrit précédemment, dans un solvant tel que le dichlorométhane et en présence
d'une base aprotique telle que la triéthylamine, pour obtenir un dérivé de
formule
x
Y ~ W
~NH~
Soz R
III
2) faire réagir le composé de formule III obtenu ci-dessus avec l'ester
éthylique de
la N-(2-chloroacétyl)-glycine
ClCHz-CO-NH-CHz-COz-CzHs
IV
dans un solvant tel que le diméthylformamide et en présence d'une base telle
que
par exemple le carbonate de potassium, pour obtenir un dérivé de formule
x w i o
o_c2H5
~ \ /~
Y ~ ~ SOz CHz N
H
O
z V
3) hydrolyser la liaison ester du composé de formule V, par action d'une base
forte
telle que la potasse en présence d'eau et éventuellement d'un solvant
organique
miscible, pour obtenir une glycine N-substïtuée de formule VI
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
7
x w ! e
~N ~ ~ CHz OH
Y 5Oz CHz N
H
O
VI
4) faire réagir la glycine N-substituée VI obtenue ci-dessus, avec une amine
primaire ou secondaire de formule
/(CHz ) n-Rb
H-N
Ra VII
s dans laquelle
n représente 1, 2, 3 ou 4,
Ra représente un atome d'hydrogène ou un groupe (CHZ)mR'b dans lequel m
représente 1, 2, 3 ou 4
Rb et R'b représentent chacun indépendamment un atome d'hydrogène,
Io un groupe
un groupe
CH2 ~ 2 ) P
dans lequel p représente 4, 5 ou 6 ;
R
/ 4
CH -N
z
R .
s
dans lequel R4 représente un groupe aminoprotecteur, COCH3, COOCH3, ou un
1s groupe alkyle en Cl-C4 et RS représente un groupe alkyle en Cl-C4
éventuellement
substitué par un groupe phényle ;
un groupe
J
N
R
3
dans lequel R3 représente un groupe aminoprotecteur ou un groupe alkyle en Cl-
C4 ;
2o un groupe
R
a
N
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
8
dans lequel R$ représente H, Ci-C4 alkyle ou NHRc dans lequel Rc représente un
groupe aminoprotecteur ;
un groupe
CH -N N-R
2 ~ 3
s dans lequel R3 représente un groupe alkyle en C~-C4 ou un groupe
aminoprotecteur ;
un groupe
CH -O-R
2 6
dans lequel R6 représente H ou CONHC2H5 ;
1o ou un groupe
R
dans lequel R~ représente H, CN ou CONHCH3 dans un solvant tel que par exemple
le dichlorométhane, en présence d'au moins un agent de couplage comme le 1-(3-
diméthylaminopropyl)-3-éthyl-carbodümide (EDCI) ou le 1-hydroxy-7-
Is azabenzotriazole (HOAT), pour obtenir la glycinamide de formule VIII
x w
~r
c
Y SOz ~ ~NH~ /NC ~CHz) n-Rb
R C Ra
° VIII
dans laquelle Ra, Rb et n conservent la même signification que ci-dessus,
5) si nécessaire, faire réagir le composé de formule VIII obtenu ci-dessus de
façon
à remplacer le ou les groupes) aminoprotecteurs R3 ou R~ par un atome
2o d'hydrogène, par exemple, si le groupe R3 est un groupe t-butyloxycarbonyl
(Boc), par action de l'acide trifluoroacétique en présence d'anisole et dans
un
solvant, pour obtenir le composé de formule VIII dans laquelle les
substituants
conservent la même signification que précédemment à l'exception de R3 et R~
qui
représentent un atome d'hydrogène
2s 6) et, ou, si nécessaire, si le groupe R, est présent et représente un
groupe cyano,
faire réagir le composé de formule VITI
ao) avec l'éthylènediamine, en présence de soufre, pour transformer le
groupe R~ en groupe 4,5-dihydro-2-imidazolyle ; ou successivement
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
9
a) avec de l'hydroxyiamine, dans un solvant tel que le DMSO pour
transformer le groupe R7 en groupe (amino)(hydroxyimino)méthyl,
b) puis avec de l'anhydride acétique, dans un solvant pour convertir le
groupe R7 en groupe (acétoxyimino)(amino)méthyl,
s c) puis avec de l'hydrogène en présence d'un catalyseur d'hydrogénation tel
que du charbon palladié, dans un solvant tel que le méthanol, pour
transformer le groupe R~ en groupe aminoiminométhyl,
d) si nécessaire faire réagir le composé ainsi obtenu de façon à remplacer le
(ou les) groupes) aminoprotecteurs R3 ou R~ par un atome d'hydrogène
to et ainsi obtenir le composé de formule I
w o
X \ Soz ~N CHz NH C ~N/ ( CHz ) n -Rz
z
/ R R1
Y z
7) si nécessaire, faire réagir Je composé de formule I obtenu précédemment,
lorsque celui-ci comporte une fonction basique, avec un acide minéral ou
organique pour obtenir le sel d'acide.
1s Selon l'invention, on préconise également un procédé de préparation des
composés de formule I dans lesquels au moins l'un des substituants Rl et RZ
comporte une fonction amine primaire ou secondaire (notamment les composés de
formule I comportant un groupe dans lequel R3 ou R4 ou RS est un atome
d'hydrogène), ledit procédé dit en phase solide consistant à
a) fixer une diamine de formule générale
~(CHz ) X-CH- (CHz ) Y-NH-R13
H-
R Riz
11 IX
2s dans laquelle Ril et Rua représentent chacun indépendamment un atome
d'hydrogène ou un groupe alkyle en Ci-C4, ou forment ensemble une chaîne
alkylène en Cl-C3,
x représente 2 ou 3,
y représente 0 ou 1
3o R13 représente un groupe aminoprotecteur, tel que par exemple un groupe
Fmoc,
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
sur une résine polystyrènique fonctionnalisée à l'aide d'un groupement chloro-
trityl
représenté par la formule
Cl-
X
s dans laquelle "Polymer" représente le polymère styrénique, ladite résine
étant
dénommée en abrégé par la suite Res-CI, en présence d'une amine tertiaire et
d'un
solvant de la diamine, pour obtenir la résine greffée de structure
/(CHz) x\
Res- ~ -CH (CHz ) Y NH Rl s
R Ri /z
11 XI
dans laquelle R11, Ri2, x, y et R13 conservent la même signification que ci-
dessus,
b) déprotéger la fonction amine protégée par le groupe aminoprotecteur R13,
par
exemple en faisant réagir la résine de formule XI avec la pipéridine en
présence
d'un solvant si le groupe R13 est un groupe Fmoc, de façon à obtenir la résine
Is greffée de formule
Res- ~ (CHz ) X-CI - (CHz ) Y-~z
R R
XII
dans laquelle Rll, RiZ, x et y conservent la même signification que
précédemment ;
2o c) faire réagir la résine de formule XII avec le chlorure de 2-
nitrobenzènesulfonyle
en présence d'un solvant et d'une base aprotique, telle que par exemple la
düsopropyléthylamine (DIPEA), pour obtenir la résine de formule
Res-N (CHz ) X-CH- (CHz ) -NH-SO
Y 2
R Riz O N
XIII
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
11
dans laquelle Ril, Rl2r x et y conservent la même signification que
précédemment ;
d) faire réagir la résine de formule XIII avec un alcool de formule générale
XIV
HO ( CH ) n-R
s 2 b XIV
dans laquelle
n représente 1, 2, 3 ou 4 et Rb représente
un groupe
CHz-~ z)p
dans lequel p représente 4, 5 ou 6
un groupe
R
4
CH -N
z
R
s
dans lequel R4 représente un groupe aminoprotecteur, C0CH3, C00CH3,
1s ou un groupe alkyle en Cl-C4 et RS représente un groupe alkyie en Cl-C4
éventuellement substitué par un groupe phényle ;
un groupe
N/
R
3
dans lequel R3 représente un groupe aminoprotecteur ou un groupe alkyle en Ci-
C4
2o un groupe
R
8
N
dans lequel R$ représente H, Cl-C4 alkyle ou NHRc dans lequel Rc représente un
groupe aminoprotecteur,
un groupe
CHz- ~ -R3
2s
dans lequel R3 représente un groupe alkyle en C1-C4 ou un groupe
aminoprotecteur,
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
12
en présence d'un solvant, de triphénylphosphine et d'un agent de couplage tel
que
le düsopropylazodicarboxylate (DIAD) pour obtenir la résine grefFée de formule
XV
/soz
Res- ~ ( CHz ) X -CI - ( CHz ) Y -N
Caf
Rli Riz ( CHz ) n -R
b
s dans laquelle Ril, Rlz, x, y, n et Rb conservent la même signification que
précédemment ;
e) faire réagir la résine de formule XV avec le thiophénol en présence d'un
solvant
et de triéthylamine de façon à éliminer le groupe 2-nitrobenzènesulfonyle et
1o obtenir la résine grefFée de formule
Res- ~ (CHz ) X CI - (CHz ) y-NH- (CHz ) n'-'-Rb
R R
11 1 z XVI
dans laquelle Ril, Rlz, x, y, n et Rb conservent la même signification que
précédemment ;
fj faire réagir la résine de formule XVI avec l'acide de formule VI obtenu
selon les
étapes 1 à 3 du procédé général décrit précédemment, en présence d'un solvant
et d'agents de couplage tels que le düsopropylcarbodümide (DIC) et
l'hydroxybenzotriazole (NOB'~, pour former la liaison amide et obtenir la
résine
2o de formule XVII
Res ! CHz ) X ! CHz ) O H
~CH~ ~N/C\C/N\C/CHz~ / SOz
Hz II N
O
Rll Riz ! CHz ) n-Rb R
Y
XVII
dans laquelle W, X, Y, Z, Rll, Riz, Rb, x, y et n conservent la même
signification que
2s précédemment ;
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
13
g) faire réagir la résine de formule XVII avec l'acide trifluoroacétique en
présence
d'un solvant, de façon à rompre la liaison de greffage sur la résine et,
simultanément, éliminer s'il existe, un groupement aminoprotecteur compris
dans
le groupe Rb et ainsi obtenir le composé de formule XVTII selon l'invention,
sous
s forme de sel avec l'acide trifluoroacétique
w o
X \ SOZ\N/CHz ~ /NH~ ~C\ / (CHz) n-Rb
CH2
/ R p ( CHZ ) y - j H- ( CHz ) X -NH-Rli
Y
Ria
XVIII
dans laquelle R, Rll, Ri2, x, y et n conservent la même signification que
1o précédemment et Rb représente
un groupe
CH2-~ a)P
dans lequel p représente 4, 5 ou 6
15 un groupe
R
n
CH -N
z
R
s
dans lequel R4 représente un atome d'hydrogène, COCH3, COOCH3,
ou un groupe alkyle en Cl-C4 et RS représente un groupe alkyle en Cl-C4
éventuellement substitué par un groupe phényle ;
2o un groupe
N/
R
3
dans lequel R3 représente un atome d'hydrogène ou un groupe alkyle en Cl-C4
un groupe
R
/ a
N
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
14
dans lequel R$ représente un atome d'hydrogène,
un groupe alkyle en Ci-C4 ou NHz
un groupe
CH -N N-R
2 ~ 3
s dans lequel R3 représente un atome d'hydrogène, un groupe alkyle en Cl-C4 .
En variante du procédé sur phase solide décrit ci-dessus, certains composés
selon l'invention peuvent être préparés en effectuant les étapes consistant à
a) faire réagir une résine greffée de formule XII obtenue selon l'étape (b) du
o procédé décrit ci-dessus, avec un acide de formule
HOOC-R
XIX
dans laquelle Rb représente un groupe N-Boc-pipéridine, dans un solvant et en
présence d'agents de couplage tels que le düsopropylcarbodümide et le 1-
hydroxybenzotriazole, pour obtenir la résine de formule XX
Res- ~ (CHZ ) X CI - (CH2 ) Y-NH- -R
R R O
1 1 1 2
dans laquelle R11, Rlz, R6, x et y conservent la même signification que dans
les
composés de départ ;
2o b) réduire la fonction amide de la résine greffée de formule XX par action
du
complexe borane-diméthylsulfure, en présence d'un solvant, pour obtenir la
résine de formule XXI
Res- ~ (CHz ) X-CI - (CH2 ) Y-NH-CHZ Rb
R R
11 12 XXI
2s dans laquelle R11, Riz, x, y et Rb conservent la même signification que
précédemment ;
c) faire réagir l'amine supportée de formule XXI, avec l'acide de formule VI
obtenu
selon les étapes 1 à 3 du procédé général, dans des conditions opératoires
3o analogues à celles décrites pour réaliser l'étape f du procédé en phase
solide
précédent et obtenir ainsi la résine de formule
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
Res (CH ) ~ H W
\" / (CHz\ ~C~ ~N~ ~CHz SOz X
N I H N C C ~N~
ii
Rii Riz ( CHz ) n-Rb z 0 R
z Y
XXII
dans laquelle Ril, R12, x, y, Rb conservent la même signification que
précédemment,
5 et R représente un groupe alkyle en Cl-C4, éventuellement substitué par un
groupe
phényle,
d) faire réagir la résine ainsi obtenue avec l'acide trifluoroacétique, de
façon à
rompre la liaison de greffage sur la résine et éliminer le groupement
aminoprotecteur pour obtenir le composé de formule XXIII selon l'invention,
sous
io forme de son sel avec l'acide trifluoroacétique
X \ 50z~N~CHz ~ /NH~ C~ / (CHz) ~N
l ~HZ ~
R 0 ( CH2 ) Y - i H- ( CHz ) x -NH-Rli
Y z
Riz
XXIII
15 Dans la partie expérimentale relatant les synthèses effectuées en phase
solide, les groupements aminoprotecteurs ainsi que certains solvants et
certains
réactifs seront désignés en abrégé de façon conventionnelle
Fmoc = 9-fluorenylméthyloxycarbonyl
Boc = 1,1-diméthyléthoxycarbonyl
2o DIPEA = N,N-düsopropyléthylamine
DIC = düsopropylcarbodümide
DIAD = düsopropylazodicarboxylate
HOBT = 1-hydroxybenzotriazole hydrate
DCM = dichlorométhane
THF = tétrahydrofurane
DMF = diméthylformamide
Le support solide (résine) est, sauf indication contraire, un polymère
styrénique (PS) réticulé à l'aide de 1 % de divinylbenzène et fonctionnalisé
par un
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
16
groupe chlorotrityl. Ce support solide permet de fixer une amine substituée
RNHZ en
formant la résine substituée
~l + NH2-R ~ L~H-R
s Pour simplifier le texte, le support solide sera par la suite noté Res-,
précédé
de la position de substitution sur R.
A titre d'exemple, le composé de formule
S ~ ~ N CH -NH
2 2
1o dans laquelle PS représente le support polystyrène sera dénommé : 4-
(aminométhyl)-1-Res-pipéridine.
Dans les descriptions de modes opératoires relatifs à des synthèses en phase
solide, les dispositifs d'agitation sont toujours des agitateurs à mouvement
orbital,
sans agitateur à l'intérieur du vase de réaction.
15 L'identification et la pureté des nouveaux composés préparés en phase
solide sont déterminés au moyen d'une analyse par couplage LC/MS
(chromatographie en phase liquide couplée à un spectromètre de masse). Sauf
indication contraire, la chromatographie est réalisée sur une chaîne Hewlett
Packard
HP1100 équipée d'une colonne 50x4,6 mm remplie de phase stationnaire de type
2o silice greffée C1B, 3,5 ou 5 pm (par exemple référencée SYMMETRY chez
WATERS).
La colonne est thermostatée à 30 °C. La phase mobile, réglée sur un
débit de 0,4
ou 1 ml/mn, est un gradient des solvants A et B suivants
A : eau distillée contenant 0,05 % d'acide trifluoroacétique
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
17
B : acétonitrile contenant 0,05 % d'acide trifluoroacétique
Les différentes conditions de gradient mises en oeuvre pour les analyses sont
les suivantes (les valeurs indiquées dans le tableau sont la proportion en %
de
solvant B dans le mélange A + B).
t mn 0 5 6 7 8 9 10 12 colonne dbit ml/mn
Grad. 25 90 90 25 25 I 1
A
Grad. 30 90 90 30 30 I 1
B
Grad. 30 90 90 30 30 II 1
C
Grad. 30 30 90 30 30 III 0 4
D
Colonne I: colonne 50x4,6 mm, silice greffée Ci8 3,5p.m
(SYM M ETRY/WATERS)
Colonne II: colonne 50x4,6 mm, silice greffée Cl8 5p.m
(SYMMETRY/WATERS)
Colonne III : colonne 50x3 mm, silice greffée Cl$ ODB 3~.m (UPTISPHERE)
Le spectrographe de masse est un appareil PERICIN.ELMER SCIEX API 150
MCA avec détection par ionisation positive APCI+.
Le résultat analytique, indiqué LC/MS après chaque préparation ou exemple
1s mentionne les conditions de l'analyse (Grad. X) et le temps de rétention du
composé exprimé en mn et fraction de mn.
L'invention sera mieux comprise à la lecture des exemples de préparation
ainsi que des résultats d'essais pharmacologiques réalisés avec des composés
selon
l'invention. Ces exemples non limitatifs n'ont pour but que l'illustration de
l'invention
2o et ne sauraient en aucun cas en limiter la portée.
Parmi les abréviations utilisées dans les descriptions suivantes, mM signifie
millimole (10-3 mole).
PREPARATION I
2s 2,4-dichloro-3-méthyl-N-(2-phényléthyl)-benzènesulfonamide
On prépare une solution de 59,6 g (0,23 mole) de chlorure de 2,4-dichloro-
3-méthylbenzènesulfonyle dans 500 ml de dichlorométhane et on ajoute 25,2 g
(0,25 mole) de triéthylamine, puis, goutte à goutte, 31,4 ml (0,25 mole) de 2-
phényléthylamine. Le mélange réactionnel est maintenu sous agitation pendant
15
3o heures à température ambiante, puis lavé successivement avec une solution
normale d'acide chlorhydrique, une solution saturée de bicarbonate de sodium
puis
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
18
à l'eau. La phase organique est séchée sur sulfate de magnésium, puis
concentrée
sous pression réduite. Le résïdu obtenu est cristallisé dans l'éther de
pétrole. On
obtient ainsi 63,9 g du produit attendu sous forme d'un solide blanc
(rendement =
81 %).
s F=75°C
PREPARATION II
N-[2-[[(2,4-dïchloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino]acétyl]glycine, éthyl ester
1o On ajoute 24,05 g (0,174 M) de carbonate de potassium, puis 31,23 g
(0,174 M) de l'ester éthylique de la N-chloroacétylglycine à une solution de
47 g
(0,139 M) du composé obtenu selon la préparation I dans 300 ml de DMF. Le
mélange réactionnel est agité à température ambiante pendant 28 heures puis
versé sur de l'eau.
15 Le précipité formé est séparé par filtration puis repris en solution dans
l'acétate
d'éthyle. Cette phase organique est lavée par une solution normale d'acide
chlorhydrique, puis par une solution saturée de bicarbonate de sodium puis à
l'eau.
Aprés séchage sur sulfate de sodium puis concentration sous pression réduite,
on
obtient un solide que l'on recristallise dans l'alcool isopropylique. On
obtient ainsi
20 45,4 g du produit attendu sous forme d'un soude blanc (rendement = b7 %).
F = 100 °C
PREPARATION III
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
25 phényléthyl)amino]acétyl]glycine
On prépare une solution de 48,7 g (99,9 mM) de l'ester obtenu selon la
préparation II dans 150 ml de dioxanne, on ajoute 150 ml d'une solution
normale
de soude puis on agite le mélange à 50 °C pendant 3 heures. Le milieu
réactionnel
est ensuite concentré sous pression réduite et le résidu est repris à l'eau et
acidifié
3o à l'aide d'une solution normale d'acide chlorhydrique. Le mélange est
extrait à
l'acétate d'éthyle, la phase organique obtenue est lavée à l'eau, séchée puis
concentrée sous pression réduite. Le résidu est cristallisé dans l'éther de
pétrole. On
obtient ainsi 37 g du produit attendu sous forme d'un solide fin blanc
(rendement
81 °lo).
35 F = 110 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
19
PREPARATION IV
Acide [3-[[(4-cyanophényl)méthyl]amino]propyl]carbamique, 1,1-
diméthyléthyl ester
On prépare une solution de 2,61 g (15 mM) de l'ester 1,1-diméthyléthylique
s de l'acide (3-aminopropyl)carbamique dans 10 ml d'éthanol et on ajoute 1 g
(5,1
mM) de 4-(bromométhyl)benzonitrile en suspension dans 10 ml d'éthanol. Le
mélange réactionnel est agité à reflux dans du solvant pendant 18 heures puis
concentré sous pression réduite. Le résidu est purifié par chromatographie sur
gel
de silice en éluant à l'aide d'un mélange dichlorométhane/méthanol/ammoniaque
Io (98/2/0,2 ; v/v/v). On obtient ainsi 1,3 g du produit attendu sous forme
d'un solide
blanc (rendement = 88%).
F=64°C
PREPARATION V
is Acide [4-[[(4-cyanophényl)méthyl]amino]butyl]carbamique, 1,1-
diméthyléthyl ester
En opérant de façon analogue à la préparation IV, au départ de l'ester 1,1-
diméthyléthylique de l'acide (4-aminobutyl)carbamique, on obtient le produit
attendu sous forme d'un solide blanc (rendement = 87 %).
20 F = 48-50 °C
PREPARATION VI
Acide [4-[[(4-cyanophényl)méthyl][2-[[2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétyl]amino]acétyl]amino]-
2s butyl]carbamique, 1,1-diméthyléthyl ester
On prépare une solution de 0,4 g (0,871 mM) de l'acide obtenu selon la
préparation IIT, dans 20 ml de dichlorométhane, on ajoute 0,18 g (0,958 mM) de
chlorhydrate de 1-(3-diméthylaminopropyl)-3-éthyl-carbodümide (EDCI), puis
0,13 g
(0,958 mM) de 1-hydroxy-7-azabenzotriazole (HOAT). Le mélange réactionnel est
3o agité à température ambiante pendant 20 minutes puis on ajoute 0,1 g (1 mM)
de
triéthylamine et 0,29 g (0,958 mM) de l'amine obtenue selon la préparation V.
On
agite le mélange pendant 48 heures à température ambiante puis on le verse sur
de
l'eau. Après décantation et élimination de la phase aqueuse, la phase
organique est
séchée sur sulfate de magnésium puis concentrée sous pression réduite. Le
résidu
3s est purifié par chromatographie sur gel de silice en éluant à l'aide d'un
mélange
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
dichlorométhane/méthanol (8/2 ; v/v). On obtient ainsi 0,48 g du produit
attendu
sous forme d'un solide blanc amorphe (rendement = 74 %).
F=87%
5 PREPARATION VII
Acide [4-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][2-[[2-[[(2,4-
dichioro-3-méthylphényl)sulfonyl] (2-phényléthyt) amino]acétyl]amino]-
acétyl]amino] butyl] carbamique, 1,1- diméthyléthy) ester
On prépare une solution de 0,45 g (0,604 mM) du composé obtenu selon la
1o préparation VI, dans 10 ml de DMSO et on ajoute 0,15 g (2,1 mM) de
chlorhydrate
d'hydroxylamine et 0,427 g (4,2 mM) de triéthylamine. Le mélange réactionnel
est
agité 24 heures à température ambiante. On ajoute à nouveau 0,15 g de
chlorhydrate d'hydroxylamine et 0,427 g de triéthylamine et on agite encore
pendant 24 heures. Le mélange est ensuite versé sur de l'eau et il se forme un
15 précipité que l'on filtre, rince à l'eau et sèche sous pression réduite. On
obtient ainsi
0,43 g du composé attendu sous forme d'un solide blanc (rendement = 91 %).
F = 102 °C
PREPARATION VIII
2o Acide [4-[[[4-[[(acétyloxy)imino](amino)méthyl]phényl]méthyl][2-[[2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétyl]-
amino]acëtyl] amino]butyl] carbamique, 1,1-diméthyléthyl ester
On ajoute 175 mg (1,6 mM) d'anhydride acétique à une solution de 0,42 g
(0,54 mM) du composé obtenu selon la préparation VII dans 20 ml de
dichlorométhane. Après 20 heures sous agitation à température ambiante (20 à
25
°C) on lave le mélange réactionnel à l'aide d'une solution saturée de
bicarbonate de
sodium puis à l'eau. La phase organique est séchée sur sulfate de magnésium et
concentrée sous pression réduite. On obtient ainsi 0,43 g du produit attendu
sous
forme d'un solide blanc amorphe (rendement : 97 %).
3o F = 100 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
21
PREPARATION IX
Acide [4-[[[4-(aminoiminométhyl)phényl]méthyl][2-[[2-[[(2,4-dichloro-
3-méthyiphényl)sulfonyl](2-phényléthyi)amino]acétyl]amino]acétyl]-
amino]butyl]carbamique, 1,1-diméthyléthyl ester
On prépare une solution de 0,39 g (0,476 mM) du composé obtenu selon la
préparation VIII dans 30 ml de méthanol et on ajoute 40 mg de charbon platiné
(à
5 % de Pt). Le mélange est agité sous atmosphère d'hydrogène, à pression
atmosphérique et à température ambiante pendant 4 heures. Le catalyseur est
éliminé par filtration et le filtrat est concentré sous pression réduite. Le
résidu est
1o purifié par chromatographie sur gel de silice grefFé NH2 (Lichroprep NHZ-40-
63 p.m),
en éluant à l'âide d'un mélange dichlorométhane/méthanol (96/4 ; v/v). On
obtient
ainsi 0,37 g du produit attendu sous forme d'un solide blanc (rendement = 100
%).
F=122°C
Exemple 1
N-[2-[(4-aminobutyl)[[4-(aminoiminométhyl)phényl]méthyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétamide, bis trifluoroacétate
On prépare un mélange de 0,36 g (0,473 mM) du composé obtenu selon la
2o préparation IX et de 0,051 g (0,473 mM) d'anisole et on ajoute 1,5 ml
d'acide
triffuoroacétique. La solution obtenue est agitée pendant 4 heures à
température
ambiante, puis concentrée sous pression réduite. On ajoute ensuite 5 ml de
toluène
au résidu et l'on concentre à nouveau sous pression réduite afin de chasser
l'excédent d'acide trifluoroacétique. Le résidu solide est trituré avec de
l'éther
2s diéthylique et la phase liquide est éliminée. Le produit solide résiduel
est repris en
solution dans 10 ml d'eau distillée et fa solution est filtrée et lyophilisée.
On obtient
ainsi 0,28 g du produit attendu sous forme d'un solide fin et léger blanc
(rendement
= 67 %).
F=123°C
PREPARATION X
Acide [3-[[(4-cyanophényl)méthyi][2-[[Z-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](phényléthyl)amino]acétyl]amino]acétyl]amino]propyl]-
carbamique, 1,1-diméthyléthyl ester
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
22
En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation IV, on obtient le produit attendu sous forme d'un
solide
blanc amorphe (rendement = 45 %).
F=80°C
PREPARATION XI
Acide [3-[[[4-amino(hydroxyimino)méthyl]phényl]méthyl][2-[[2-[[(2,4-
dichloro-3-méthylphényl)sulfonyl](phényléthyl)amino]acétyl]amino]-
acétyl]amino]propyl]carbamique, 1,1-diméthyléthyl ester
Io En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation X, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 96 %).
F = 112 °C
PREPARATION XII
Acide [3-[[[4-[[(acétyloxy)imino)méthyl]phényl]méthyl][2-[[2-[[(2,4-
dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétyl]amino]-
acétyl]amino]propyl]carbamique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon la préparation XI, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 93 %).
F=92°C
PREPARATION XIII
2s Acide [3-[[[4-(aminoiminométhyl)phényl]méthyl][2-[[2-[[(2,4-dichloro-
3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétyl]amino]acétyl]-
amino]propyl]carbamique, i,i-diméthyléthyl ester
En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon la préparation XII, on obtient le produit attendu sous forme d'un
3o solide blanc (rendement = 69 %).
F = 106 °C
Exemple 2
N-[2-[(3-aminopropyl)[[4-(aminoiminométhyl)phényl]méthyl]amino]-2-
35 oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthy1)-
amino]acétamide, bis trifluoroacétate
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
23
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la préparation XIII, on obtient le produit attendu sous forme d'un
solide blanc
fin et léger (rendement = 93 %).
F=128°C
PREPAitATION XIV
4-[[[3-(1-pyrrolidinyl)propyl]amino]méthyl]benxonitrile
On prépare une solution de 1,96 g (15,3 mM) de 1-(3-
aminopropyl)pyrrolidine dans 25 ml de toluène et on ajoute 2 g (15,3 mM) de 4-
lo cyanobenzaldéhyde. La solution est chauffée à reflux sous agitation et
l'eau formée
par la réaction est éliminée au moyen d'un appareil de Dean-Stark. La réaction
dure
environ 6 heures. Le solvant est ensuite chassé sous pression réduite et le
résidu
est repris en solution dans 25 ml de méthanol. On ajoute 0,58 g (15,3 mM) de
borohydrure de sodium et on maintient le milieu réactionnel sous agitation
pendant
Is 20 heures à température ambiante. Le mélange est ensuite concentré sous
pression
réduite, le résidu est repris dans du dichlorométhane et la phase organique
obtenue
est lavée à l'eau 2 fois, puis séchée sur sulfate de magnésium et concentrée
sous
pression réduite. On obtient ainsi le produit attendu sous forme d'une huile
orange
(rendement = 95 %).
2o RMN (1H, 300 MHz, CDCI3 ) : 7,78 (d, 2H) ; 7,52 (d, 2H) ; 3,74 (s, 2H) ;
2,49
(m, 2H) ; 2,35 (m, 6H) ; 1,62 (m, 6H).
PREPARATION XV
N-[2-[[(4-cyanophényl)méthyl][3-(1-pyrrolidinyl)propyl]amino]-2-
2s oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino] acétamide
En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation XIV, on obtient le produit attendu sous forme
d'une
poudre amorphe blanche (rendement = 69 %).
30 F=88°C
Exemple 3
N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][3-(1-
pyrrolidinyl)propyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
35 phényl)sulfonyl](2-phényléthyl)amino]acétamide
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
24
En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XV, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 90 %).
F=92°C
Exemple 4
N-[2-[[[4-[[(acétyloxy)imino](amino)méthyl]phényl]méthyl][3-(1-
pyrrolidinyl)propy!]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide
o En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon l'exemple 3, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 79 %).
F=90°C
Exemule 5
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][3-(1-pyrrolidinyl)propyl]
amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon l'exemple 4, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 39 %).
F = 105 °C
Exemple 6
2s N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][3-(1-pyrrolidinyl)propyl]
amino]-2-oxoéthy!]-2-[[(2,4-dichtoro-3-méthylphényl)sutfonyl](2-
phényléthyl)amïno]acétamide, dichlorhydrate
On prépare une solution de 80 mg (0,11 mM) du composé obtenu selon
l'exemple 5 dans 5 ml de méthanol et on ajoute 1 ml d'une solution saturée de
3o chlorure d'hydrogène dans l'éther éthylique. Le mélange est agité pendant 1
heure
puis concentré sous pression réduite. Le résidu solide est repris en solution
dans 5
cm3 d'eau distillée, la solution est filtrée puis lyophilisée. On obtient
ainsi 88 mg du
produit attendu sous forme d'un fin solide blanc (rendement = 100 %).
F=145°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
PREPARATION XVI
4-[[[2-(1-pyrrolidinyl)éthyl]amino]méthyl]benzonitrile
En opérant de façon analogue à la préparation XIV, au départ de 1-(2-
aminoéthyl)pyrrolidine, on obtient le produit attendu sous forme d'une huile
jaune
s (rendement = 77 %).
RMN (1H, 300 MHz) : 7,7 (d, 2H) ; 7,5 (d, 2H) ; 3,77 (s, 2H) ; 2,50 (m, 4H) ;
1,66 (m, 4H)
1o PREPARATION XVII
N-[2-[[(4-cyanophényl)méthyl][2-(1-pyrrolidinyl)éthyl]amino]-2-oxo-
éthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
acétamide
En opérant de façon analogue à la préparation VI, au départ du composé
1s obtenu selon la préparation XVI, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 77 %).
F=65°C
Exemple 7
20 N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][2-(1-
pyrrolidinyl)éthyl]amino]-2-oxoéthyl]-2-[[(2,4-dïchloro-3-métlZyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XVII, on obtient le produit attendu sous forme
d'un
2s solide blanc (rendement = 97 %).
F=85°C
Exemple 8
N-[2-[[[4-[[(acétyloxy)imino](amino)méthyl]phényl]méthyl][2-(1-
3o pyrrolidinyl)éthyl]amino]-2-oxoéthyl]-2-[[(2,4-dïchloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon l'exemple 7, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 91 %).
3s F = 82 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
26
Exemple 9
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][2-(1-pyrrolidinyl)éthyl]-
amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino] acétamide
s En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon l'exemple 8, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 52 %).
F=106°C
1o Exemple 10
N-j2-[[[4-(aminoiminométhyl)phényl]méthyl][2-(1-pyrrolidinyl)éthyl]-
amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
1s selon l'exemple 9, on obtient le produit attendu sous forme d'une poudre
fine
blanche (rendement = 94 %).
F = 140 °C
PREPARATION XVIII
20 4-[[[4-(1-pyrrolidinyl)butyl]amino]méthyl]benzonitrile
En opérant de façon analogue à la préparation XIV, au départ de 1-(4-
aminobutyl)pyrrolidine, on obtient le produit attendu sous forme d'une huile
orange
(rendement = 81 %).
RMN (1H, 300 MHz, CDCI3) : 7,77 (d, 2H) ; 7,51 (d, 2H) ; 3,75 (s, 2H) ; 2,38
25 (m, 8H) ; 1,67 (m, 4H) ; 1,44 (m, 4H~
PREPARATION XIX
N-[2-[[(4-cyanophényl)méthyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxo-
éthyl]-~-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
3o amino]acétamide
En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation XVIII, on obtient le produit attendu sous forme
d'un
solide amorphe blanc cassé (rendement = 62 %).
F=70°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
27
Exemple 11
N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][4-(1-
pyrrolidinyl)butyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide
s En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XIX, on obtient le produit attendu sous forme d'un
solide blanc (rendement = 96 %).
F=92°C
1o Exemple 12
N-[2-[[[4-[[(acétyloxy)imino](amino)méthyl]phényl]méthyl][4-(1-
pyrrolidinyl)butyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyi](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VIII, au départ du composé
Is obtenu selon l'exemple 11, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 90 %).
F=88°C
Exemple 13
2o N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][4-(1-pyrrolidinyl)butyl]-
amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino] acétamide
En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon l'exemple 12, on obtient le produit attendu sous forme d'un
solide
2s blanc amorphe (rendement = 40 %).
F = 155 °C
Exemple 14
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][4-(1-pyrrolidinyl)butyl]-
3o amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 13, on obtient le produit attendu sous forme d'un solide fin
et léger
blanc (rendement = 100 %).
35 F = 155 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
28
PREPARATION XX
4-[[[4-(diméthylamïno)butyl]amino]méthyl]benzonitrile
En opérant de façon analogue à la préparation XIV, au départ de N-N-
diméthyl-1,4-butanediamine, on obtient le produit attendu sous forme d'une
huile
s jaune (rendement = 77 %).
RMN (1H, 300 MHz, CDCI3) : 7,76 (d, 2H) ; 7,52 (d, 2H) ; 3,74 (s, 2H) ; 2,49
(m, 2H) ; 2,14 (m, 2H) ; 2,08 (s, 6H) ;
1,39 (m, 4H).
1o PREPARATION XXI
N-[2-[[(4-cyanophényl)méthyl][4-(diméthylamino)butyl]amino]-2-oxo-
éthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyi)amino]-
acétamide
En opérant de façon analogue à la préparatïon VI, au départ du composé
1s obtenu selon la préparation XX, on obtient le produit attendu sous forme
d'un solide
amorphe blanc (rendement = 68 %).
F=60°C
Exemgle 15
2o N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][4-(diméthyl-
amino)butyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyiphënyl)-
sulfonyl](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XXI, on obtient le produit attendu sous forme d'un
2s solide blanc (rendement = 93 %).
F=92°C
Exemple 16
N-[2-[[[4-j[(acétyloxy)imino](amino)méthyl]phényl]méthyl][4-
30 (diméthylamino)butyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon l'exemple 15, on obtient le produit attendu sous forme d'un
solide
blanc amorphe (rendement = 100 %).
3s F = 55 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
29
Exemple 17
N-[2-[((4-(aminoiminométhyl)phényl]méthyt](4-(diméthylamino)butyl]-
amino]-2-oxoéthyl]-2-([(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phénytéthyl)amino] acétamide
s En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon l'exemple 16, on obtient le produit attendu sous forme d'un
solide
blanc amorphe (rendement = 83 %).
F=78°C
1o Exemple i8
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][4-(diméthylamino)butyl]-
amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
~s selon l'exemple 17, on obtient le produit attendu sous forme d'un solide
fin et léger
blanc (rendement = 91 %).
F=148°C
PREPARATION XXII
20 4-[[[3-(diméthylamino)propyl]amino]méthyl]benzonitrile
En opérant de façon analogue à la préparation XIV, au départ de N-N-
diméthyl-1,3-propanediamine, on obtient le produit attendu sous forme d'une
huile
orange (rendement = 40 %).
RMN (1H, 300 MHz, DMSO) : 7,91 (d, 2H) ; 7,53 (d, 2H) ; 3,74 (s, 2H) ; 3,3
25 (m, 1H) ; 2,48 (t, 2H) ; 2,21 (t, 2H) ;
2,08 (s, 6H) ; 1,53 (m, 2H).
PREPARATION XXIII
N-(2-[((4-cyanophényl)méthyl][3-(diméthylamino)propyl]amino]-2-oxo-
3o éthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyt](2-phénytéthyl)amino]-
acétamide
En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation XXII, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 51 %).
35 F = 70 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
Exemple 19
N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][3-(diméthyl-
amino)propyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)-
sulfonyl](2-phényléthyl)amino]acétamide
s En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XXIII, on obtient ie produit attendu sous forme
d'un
solide blanc (rendement = 98 %).
F = 56-58 °C
Io Exemple 20
N-[2-[[[4-[amino(hydroxyimino)méthyl]phënyl]mëthyl][3-(dimëthyl-
amino)propyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)-
sulfonyl](2-phénytéthyl)amino]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
z5 selon l'exemple 19, on obtient le produit attendu sous forme d'un solide
fin blanc
(rendement = 98 %).
F = 142 °C
Exemule 21
ao N-[2-[[[4-[[(acétyloxy)imino](amino)méthyl]phényl]méthyl][3-
(diméthylamino)propyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon l'exemple 19, on obtient le produit attendu sous forme d'un
solide
2s blanc (rendement = 71 %).
F=90°C
Exemple 22
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][3-(diméthylamino)-
so propyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]-
(2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon l'exemple 21, on obtient le produit attendu sous forme d'une
poudre
amorphe blanche (rendement = 73 %).
F=114°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
31
Exemple 23
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl][3-
(dïméthylamino)propyi]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 22, on obtient le produit attendu sous forme d'un solide fin
blanc
(rendement = 98 %).
F = 157 °C
1o PREPARATION XXIV
Acide 4-[[[(4-cyanophényl)méthyl]amino]méthyl]-1-pipéridine-
carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XIV, au départ de l'ester t-
butylique de l'acide 4-(aminométhyl)-1-pipéridinecarboxylique, on obtient le
produit
1s attendu sous forme d'une huile jaune (rendement = 88 %).
RMN (1H, 300 MHz, DMSO) : 7,76 (d, 2H) ; 7,52 (d, 2H) ; 3,90 (d, 2H) ; 3,74
(s, 2H) ; 2,66 (m, 1H) ; 2,30 (d, 2H) ;
1,68 (d, 2H) ; 1,54 (m, 2H) ; 1,37 (s, 9H) ;
0,95 (m, 2H).
PREPARATION XXV
Acide 4-[[[(4-cyanophényl)méthyl][2-[[2-[[(2,4-dichloro-3-méthyl-
phényl)sulfonyl](2-phényléthyl)amino]acétyl]amino]acétyl]amino]-
méthyl]-1-pipéridine carboxylique, l,1-diméthyléthyl ester
2s En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation XXIV, on obtient (e produit attendu sous forme
d'une
huile visqueuse (rendement = 84 %).
RMN (1H, 300 MHz, DMSO) : 8,88 (m, 1H) ; 8,54 (m, 2H) ; 8,43 (d, 1H) ; 8,2Z
(d,
1H) ; 8,08 (d, 2H) ; 7,83 (m, 5H) ; 5,52 (s, 1H) ; 5,37 (s, 1H) ; 4,82 (d, 2H)
; 4,75
(d, 1H) ; 4,58 (m, 3H) ; 4,14 (m, 2H) ; 3,98 (m, 2H) ; 3,86 (d, 2H) ; 3,40 (m,
3H) ;
3,05 et 2,96 (2s, 3H) ; 2,22 (m, 2H) ; 2,045 (d, 9H) ; 1,68 (m, 2H).
PREPARATION XXVI
Acide 4-[[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl][2-[[2-[[(2,4-
3s dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétyl]amino]-
acétyl]amino]méthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl ester
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
32
En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XXV, on obtient le produit attendu sous forme d'un
solide beige amorphe (rendement = 84 %).
F = 100 °C
PREPARATION XXVII
Acide 4-[[[[4-[[(acétyloxy)imino](amino)méthyl]phény!]méthyl][Z-[[2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétylJ-
amino]acétyl]amino]méthyl]-1-pipéridinecarboxylique, 1,1-diméthyl-
1 o éthyl ester
En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon la préparation XXVI, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 89 %).
F = 110 °C
PREPARATION XXVIII
Acide 4-[[[[4-(aminoiminométhyl)phényl]méthyl][2-[[2-[[(2,4-dichloro-
3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétyl]amino]acétyl]-
amino]méthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl ester
2o En opérant de façon analogue à la préparation IX, au départ du composé
obtenu selon la préparation XXVII, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 98 %).
F=140°C
Exemple 24
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl](4-pipéridinyléthyl)amino]-
2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphény!)sulfonyl](2-phényléthyl)-
amino] acétamide, bis trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
3o selon la préparation XXVIII, on obtient le produit attendu sous forme d'un
solide
blanc
(rendement = 88 %).
F = 130 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
33
PREPARATION XXIX
Acide 4-([[(2,4-dinitrophényl)sulfonyl]amino]méthyl]-1-pipéridine-
carboxylique, 1,1-diméthyléthyl ester
On prépare une solution de 5,36 g (25 mM) de l'ester 1,1-diméthyléthylique
de l'acide 4-(aminométhyl)-1-pipéridinecarboxylique dans 60 ml de
dichlorométhane
et on ajoute une solution de 6,66 g (25 mM) de chlorure de 2,4
dinitrobenzènesulfonyle dans 40 ml de dichlorométhane, puis 2,52 g (25 mM) de
pyridine. Le mélange réactionnel est agité pendant 18 heures à température
ambiante puis lavé successivement avec une solution 0,1 N d'acide
chlorhydrique,
1o une solution saturée de bicarbonate de sodium et à l'eau pure. Après
séchage sur
sulfate de sodium, la phase organique est concentrée sous pression réduite et
le
résidu est purifié par chromatographie sur gel de silice ~en éluant à l'aide
d'un
mélange cyclohexane/acétate d'éthyle (6/4 ; v/v). On obtient ainsi 6,9 g du
produit
attendu sous forme d'un solide jaune (rendement = 62 %).
F=148°C
PREPARATION XXX
Acide 4-[[[4-[[(1,1-diméthyléthoxy)carbonyl]amino]butyl]amino]-
méthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl ester, chlorhydrate
2o a) on prépare une solution de 1,33 g (3 mM) du composé obtenu selon la
préparation XXIX dans 20 ml de tétrahydrofurane. On ajoute 1,57 g
(6 mM) de triphénylphosphine, une solution de 1,13 g (6 mM) de l'ester
1,1-diméthyléthylique de l'acide (4-hydroxybutyl)carbamique dans 20 ml
de toluène puis 1g (6 mM) de diéthylazodicarboxylate. Le mélange est
agité pendant 4 heures à température ambiante. On ajoute alors 10 g de
gel de silice pour chromatographie et on concentre sous pression réduite.
Le résidu poudreux est ensuite soumis à une chromatographie
préparative sur gel de silice en éluant à l'aide d'un mélange acétate
d'éthyle/hexane (4/6 ; v/v). On obtient ainsi 1,86 g de l'ester 1,1-
3o diméthyléthylique de l'acide 4-[[[4-[[(1,1-diméthyléthoxy)carbonyl]-
amino]butyl][(2,4-dinitrophényl)sulfonyl]amino]méthyl]-1-pipéridine-
carboxylique mis en réaction ensuite sans purification complémentaire
b) le composé obtenu ci-dessus est mis en solution dans 20 ml de
dichlorométhane puis on ajoute 0,6 g (6 mM) de triéthyiamine et 0,36 g
(3, 9 mM) d'acide thioglycolique. Le mélange est agité pendant 2 heures
à température ambiante puis lavé avec une solution de soude diluée. La
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
34
phase organique est séchée sur sulfate de sodium puis concentrée sous
pression réduite. Le résidu mélangé est agité avec 25 ml d'éther éthylique
et le mélange est filtré. Le solide est éliminé et on ajoute au filtrat 1 ml
d'une solution de chlorure d'hydrogène dans l'éther éthylique. Le précipité
formé est filtré et séché. On obtient ainsi 0,85 g du produit attendu sous
forme d'une poudre blanche (rendement = 67 %).
F = 156 °C
PREPARATION XXXI
1o Acide 4-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényl-
éthyl)amino]acétyl]amino]acétyl][4-[[(1,1-diméthyléthoxy)carbonyl]-
amino]butyl]amino]méthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl
ester
En opérant de façon analogue à la préparation VI, au départ du composé
Is obtenu selon la préparation )00C, on obtient le produit attendu sous forme
d'une
huile jaune (rendement = 63 %).
Exemple 25
N-[2-[(4-aminobutyl)(4-pipéridinylméthyl)amino]-2-oxoéthyl]-2-[[(2,4-
2o dichtoro-3-méthylphényt)sutfonyt](2-phénytéthyl)amino]acétamide, bis
trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la préparation XXXI, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 58 %).
25 F = 90 °C
PREPARATION X~CII
Acide 4-[[[4-(acétyloxy)butyl]amino]méthyl]-1-pipéridinecarboxylique,
1,1-diméthyléthyl ester
3o a) on prépare une solution de 0,444 g (1 mM) du composé obtenu selon la
préparation XXIX dans 5 ml de diméthylformamide. On ajoute 0,48 g (2 mM)
d'acétate de 4-iodobutyle et 0,69 g (5 mM) de carbonate de potassium. Le
mélange réactionnel est agité 24 heures à température ambiante puis dilué par
50 ml d'acétate d'éthyle et lavé successivement par une solution 0,1 N d'acide
3s chlorhydrique, une solution saturée de bicarbonate de soude puis à l'eau.
La
phase organique est séchée sur sulfate de sodium puis concentrée sous pression
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
réduite. Le résidu est purifié par chromatographie sur gel se silice en éluant
à
l'aide d'un mélange cyclohexane/acétate d'éthyle (6/4 ; v/v). On obtient ainsi
0,23 g de l'ester 1,1-diméthyléthylique de l'acide 4-[[[4-
(acétyloxy)butyl][(2,4
dinitrophényl)sulfonyl]amino]méthyl]-1-pipéridinecarboxyüque sous forme d'une
s huile jaune orangée (rendement = 41 %)
b) le composé obtenu ci-dessus est ensuite traité par l'acide thioglycolique
selon un
procédé analogue à la préparation X30C (b), ia purification étant faite sur le
composé non salifié, au moyen d'une chromatographie sur gel de silice en
éluant
à t'aide d'un mélange dichlorométhane/méthanol/ammoniaque (8/2/0,5 ; v/v/v).
1o Le produit est obtenu sous forme d'une huile rouge (rendement = 91 %).
RMN (1H, 300 MHz, DMSO) : 3,98 (t, 2H) ; 3,91 (m, 2H) ; 2,66 (m, 2H) ; 2,51
(m,
2H) ; 2,39 (d, 2H) ; 1,99 (s, 3H) ; 1,67-1,53 (m, 5H) ; 1,45 (m, 2H) ; 1,38
(s, 9H) ;
0,95 (m, 2H).
15 PREPARATION X~CIII
Acide 4-[[[4-(acétyloxy)butyl][2-[[2-[[(2,4-dichloro-3-méthylphényl)-
sulfonyl](2-phényléthyl)amino]acétyl]amino]acétyl]amino]méthyl]-1-
pipéridinecarboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation VI, au départ de la
2o préparation ?OCXII, on obtient le produit attendu sous forme d'un solide
mal
cristallisé (r endement = 42 %).
RMN (1H, 300 MHz, DMSO) : 8,18 (m, 1H) ; 7,86 (d, 1H) ; 7,57 (d, 1H) ; 7,12
(m,
3H) ; 7,04 (m, 2H) ; 4,17 (s, 2H) ; 3,99 (m, 2H) ; 3,93 (m, 4H) ; 3,45 (m, 2H)
;
3,24 (m, 2H) ; 3,13 (m, 2H) ; 2,74 (t, 2H) ; 2,60 (m, 2H) ; 2,35 (s, 3H) ;
1,96 (s,
25 3H) ; 1,76 (m, 1H) ; 1,55 (m, 4H) ; 1,48 (m, 2H) ; 1,35 (s, 9H) ; 0,98 (m,
2H).
Exemple 26
N-[2-[(4-hydroxybutyl)(4-pipéridinylméthyl)amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
3o acétamide, trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ de la préparation
X~CIII, on obtient le produit attendu sous forme d'un solide blanc (rendement
= 41
%).
F=80°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
36
PREPARATION ~CXIV
Acide 4-[[[4-(acétylamino)butyl]amino]méthyl]-1-pipéridine-
carboxylique, 1,1-diméthyléthyl ester
a) on prépare une solution de 0,64 g (3 mM) de l'ester 1,1-diméthyléthylique
de
s l'acide 4-(aminométhyl)-1-pipéridinecarboxylique dans 10 ml d'acétonitrile
et on
ajoute 0,55 g (4 mM) de carbonate de potassium et 0,7 g (2,5 mM) de N-(4-
bromobutyl)phtalimide. Le mélange réactionnel est chauffé à reflux pendant
16 heures puis concentré sous pression réduite. Le résidu est repris en
solution
dans l'acétate d'éthyle et la phase organique est lavée par une solution
saturée
1o de bicarbonate de sodium, puis séchée sur sulfate de sodium et concentrée
sous
pression réduite. Après purification par chromatographie sur gel de silice en
éluant à l'aide d'un mélange dichlorométhane/méthanol/düsopropylamine
(90/10/2 ; v/v/v), on obtient le N-[4-[[[1-[(1,1-diméthyléthoxy)carbonyl]-4
pipéridinyl]méthyl]amino]butyl]phtalimide sous forme d'un solide mal
cristallisé
1s (rendement = 49 %).
b) on prépare une solution de 0,49 g (1,18 mM) du composé obtenu ci-dessus
dans
ml de tétrahydrofuranne et on ajoute 0,15 g (1,5 mM) de triéthylamine et
0,24 g (1,4 mM) de chloroformiate de benzyle. Le mélange réactionnel est
maintenu sous agitation pendant 24 heures à température ambiante, puis dilué
2o par 60 ml d'acétate d'éthyle. La phase organique obtenue est lavée par une
solution diluée d'acide chlorhydrique, puis par une solution saturée de
bicarbonate de sodium, séchée sur sulfate de sodium et concentrée sous
pression réduite. Le résidu est purifié par chromatographie sur gel de silice
en
éluant à l'aide d'un mélange cyclohexane/acétate d'éthyle (6/4 ; v/v). On
obtient
25 ainsi 0,51 g de N-[4-[[[1-(1,1-diméthyléthoxy)carbonyl]-4-
pipéridinyl]méthyl]
[(phénylméthoxy)carbonyl]amino]butyl]phtalimide (rendement = 78 %).
c) 0,11 g (0,2 mM) du composé obtenu ci-dessus sont dissous dans 1 ml
d'éthanol
et on ajoute 0,02 g (0,4 mM) d'hydrate d'hydrazine. Le mélange est chauffé
pendant 3 heures à reflux puis concentré sous pression réduite. Le résidu est
3o purifié par chromatographie en éluant à l'aide d'un mélange
dichlorométhane/méthanol/düsopropylamine (9/1/0 ; v/v/v). On obtient ainsi 66
mg d'ester 1,1-diméthyléthylique de l'acide 4-[[(4-aminobutyl)[(phénylméthoxy)
carbonyl]amino]méthyl]-1-pipéridinecarboxylique (rendement = 78 %).
d) on prépare une solution de 0,17 g (0,4 mM) du composé obtenu selon l'étape
3s ~ précédente dans 1 ml de pyridine et on ajoute 51 mg (0,5 mM) d'anhydride
acétique. Le mélange est agité pendant 48 heures à température ambiante puis
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
37
dilué avec 10 ml d'acétate d'éthyle. La phase organique est lavée en milieu
acide,
puis avec une solution de bicarbonate de sodium et séchée sur sulfate de
sodium. Après concentration sous pression réduite, on obtient 0,18 g de
l'ester
1,1-diméthyléthylique de l'acide 4-[[[4-(acétylamino)butyl][(phénylméthoxy)
s carbonyl]amino]méthyl]-1-pipéridinecarboxylique (rendement = 96 %)
e) on prépare une solution de 1,04 g du composé obtenu selon l'étape
précédente
dans 10 ml d'éthanol et on ajoute 100 mg de charbon palladié à 10 % Pd. Le
mélange est agité sous atmosphère d'hydrogène à pression atmosphérique
pendant 3 heures puis le catalyseur est éliminé par filtration. Le filtrat est
1o concentré sous pression réduite et on obtient ainsi 0,58 g du composé
attendu
sous forme d'une huile jaune pâle (rendement = 79 %).
RMN 1H (300 MHz, DMSO) : 7,80 (m, 1H) ; 4,36 (m, 1H) ; 3,88 (m, 2H) ; 2,99 (m,
2H) ; 2,65 (m, 2H) ; 2,46 (m, 2H) ; 2,36 (d, 2H) ; 1,87 (s, 3H) ; 1,64 (m, 3H)
; 1,50
(m, 4H) ; 1,38 (s, 9H) ; 0,96 (m, 2H).
1s
PREPARATION X)OCV
Acide 4-[[[4-(acétylamino)butyl][2-[[2-[[(2,4-dichloro-3-méthylphényl)-
sulfonyl](2-phényléthyl)amino]acétyl]amino]acétyl]amino]méthyl]-1-
pipéridinecarboxylique, 1,1-diméthyléthyl ester
2o En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation ~CXIV (e), on obtient le produit attendu sous
forme
d'un produit pâteux (rendement = 87 %).
RMN 1H (250 MHz, DMSO) : 8,16 (m, 1H) ; 7,86 (d, 2H) ; 7,82 (m, 1H) ; 7,57 (d,
2H) ; 7,12 (m, 3H) ; 7,04 (m, 2H) ; 4,17 (s, 2H) ; 3,95 (m, 4H) ; 3,46 (m, 4H)
;
2s 3,23 (m, 2H) ; 3,15 (m, 2H) ; 3,04 (m, 2H) ; 2,73 (m, 2H) ; 2,38 (s, 3H) ;
1,78 (s,
3H) ; 1,52 (m, 5H) ; 1,40 (m, 2H) ; 1,37 (s, 9H) ; 1,02 (m, 2H).
Exemple 27
N-[2-[[4-(acétylamino)butyl](4-pipéridinylméthyl)amino]-2-oxoéthy1]-2-
30 [[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
acétamide, trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la préparation )OOCV, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 93 %).
3s F = 100 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
38
PREPARATION 70CXVI
N-méthyl-4-[([3-(1-pyrrolidinyl)propyl]amino]méthyl]benzamide
En opérant de façon analogue à la préparation IV, au départ de N-(3-
aminopropyl)pyrrolidine et de 4-formyl-N-méthyl-benzamide, on obtient le
produit
attendu sous forme d'une huile jaune (rendement = 35 %).
RMN iH (300 MHz, DMSO) : 8,36 (m, 1H) ; 7,80 (d, 2H) ; 7,37 (d, 2H) ; 3,7 (s,
2H) ; 2,76 (d, 3H) ; 2,46 (m, 8H) ; 2,26 (m, iH) ; 1,63 (m, 4H) ; 1,55 (m,
2H).
Exemple 28
io 4-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétyl]amino]acétyl][3-(1-pyrrolidinyl)propyl]amino]méthyl]-N-
méthyl-benzamide
En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation )OCXVI, on obtient le produit attendu sous forme
d'un
i5 solide blanc cassé (rendement = 31 %).
F=80°C
Exemple 29
4-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthy1)-
2o amino]acétyl] amino]acétyl][3-(1-pyrrolidinyl)propyl]amino]méthyl]-N-
rncthyl-benzamide, chlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 28, on obtient le produit attendu sous forme d'une poudre fine
blanche (rendement = 86 %).
25 F = 110 °C
PREPARATION X)OCVII
6-amino-N-[3-(1-pyrrolidinyl)propyl]nicotinamide
On prépare une solution de 0,8 g (6,24 mM) de 1-(3-aminopropyl)pyrrolidine
3o dans 40 ml de dichlorométhane et on ajoute 1,89 g (18,7 mM) de
triéthylamine et
1,2 g (6,24 mM) de chlorure de 6-aminonicotinoyle (sous forme de
chlorhydrate).
Le mélange réactionnel est agité pendant 48 heures à température ambiante. Le
précipité formé est isolé par filtration, rincé avec du dichlorométhane puis
séché. On
obtient 0,75 g du produit attendu sous forme d'un solide beige (rendement 50
%).
3s F=90°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
39
PREPARATION )O(XVIII
6-amino-N-[3-(1-pyrrolidinyl)propyl]-3-pyridineméthanamine
On prépare une suspension de 0,73 g (2,94 mM) du composé obtenu selon
la préparation )00C1/II dans 50 ml de dichlorométhane. On ajoute goutte à
goutte
10,3 ml (20,6 mM) d'une solution 2M de complexe borane/sulfure de diméthyle
dans le tétrahydrofurane. Le mélange réactionnel est agité pendant 24 heures à
température ambiante. On ajoute 15 ml d'une solution d'acide chlorhydrique 5N,
puis 15 ml d'eau, puis 100 ml de méthanol. Le mélange est agité pendant 20
heures
à température ambiante puis concentré sous pression réduite. Le résidu est
purifié
io par chromatographie sur silice greffée NHa (Lichroprep NHZ) en éluant à
l'aide d'un
mélange dichlorométhane/méthanol (98/2 ; v/v). On obtient ainsi 0,15 g du
produit
attendu sous forme d'un solide blanc (rendement = 22 %).
F = 45-47 °C
Exemple 30
N-[2-[[(6-amino-3-pyridinyl)méthyl][3-(1-pyrrolidinyl)propyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétamide
En opérant de façon analogue à la préparation VI, au départ du composé
obtenu selon la préparation )OOCVIII, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 42 %).
F=80°C
Exemple 31
2s N-[2-[[(6-amino-3-pyridinyl)méthyl][3-(1-pyrrolidinyl)propyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 30, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 98 %).
F=142°C
Exemple 32
N-[2-[bis[3-(diméthylamino)propyl]amino]-2-oxoéthyl]-2-[[(2,4-
3s dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétamide
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
En opérant de façon analogue à la préparation VI, au départ de bis[3-
(diméthylamino)propyl~amine, on obtient le produit attendu sous forme d'une
huile
jaune (rendement = 47 %).
RMN 1H (300 MHz, DMSO) : 8,15 (m, 1H) ; 7,86 (d, 1H) ; 7,56 (d, 1H) ; 7,12 (m,
5 3H) ; 7,0 (m, 2H) ; 4,17 (s, 2H) ; 4,0 (d, 2H) ; 3,49 (t, 2H) ; 3,25 (m, 4H)
; 2,71 (t,
2H) ; 2,38 (s, 3H) ; 2,19 (m, 4H) ; 2,11 (s, 6H) ; 2,09 (s, 6H) ; 1,15 (m,
4H).
Exemule 33
N-[2-[bis[3-(diméthylamino)propyl]amino]-2-oxoéthyl]-2-[[(2,4-
lo dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétamide,
dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 32, on obtient le produit attendu sous forme d'un solide fin
léger
blanc (rendement = 90 %).
15 F = 100 °C
PREPARATION XXXIX
Acide 4-[[[2-j[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino]acétyl]amino]acétyl][3-(diméthylamino)propyl]-
2o amino]méthyl]benzènecarboximidique, éthyl ester, chlorhydrate.
On pr épare une solution de 0,8 g (1,215 mM) du composé obtenu selon la
préparation XXIII dans 50 ml d'éthanol. Cette solution est refroidie à 0
°C dans un
bain de glace puis saturée par un courant de chlorure d'hydrogène gazeux. Le
mélange réactionnel est ensuite agité pendant 48 heures à température ambiante
2s puis concentré sous pression réduite. Le précipité formé est séparé par
filtration,
lavé à l'éther et séché. On obtient 0,65 g du produit attendu sous forme de
cristaux
blancs (rendement = 68 %).
F = 45-46 °C
3o Exemple 34
2-[[(2,4-dichloro-3-méthylphény!)sulfonyl](2-phényléthyl)amino]-N-[2-
[[[4-(4,5-dihydro-iH-imidazol-2-yl)phényl]méthyl][3-(diméthylamino)-
propyl]amino]-2-oxoéthyl]acétamide
On prépare une solution de 0,057 g (0,95 mM) d'éthylènediamine dans 30
35 ml d'éthanol. On ajoute, goutte à goutte, à la température de reflux de
l'éthanol,
0,64 g (0,91 mM) du composé obtenu selon la préparation XXXIX en solution dans
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
41
50 ml d'éthanol, on maintient le mélange réactionnel à reflux pendant 48
heures
puis on concentre sous pression réduite. Le résidu est repris dans du
dichlorométhane et la phase organique obtenue est lavée à l'eau puis séchée
sur
sulfate de sodium et concentrée sous pression réduite. Le produit brut obtenu
est
s purifié par chromatographie sur silice greffée NH~ en éluant à l'aide d'un
mélange
toluène/2-propanol (95/5 ; v/v). On obtient ainsi 0,18 g du produit attendu
sous
forme d'un solide amorphe blanc crème (rendement = 31 %).
F=75°C
1o Exemule 35
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[[4-(4,5-dihydro-1H-imidazol-2-yl)phényl]méthyl][3-(diméthylamino)-
propyl]amino]-2-oxoéthyl]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
Is selon l'exemple 34, on obtient le produit attendu sous forme d'un solide
fin blanc
(rendement = 93 %).
F = 14~ °C
PREPARATION XL
2o N-[2-[[(4-cyanophényl)méthyl]méthylamino]-2-oxoéthyl]-2-[[(2,4
düchloro-3-mét:ylphényl)sulfonyl](2-phényléthy!)a:r~ino]acétamide
En opérant de façon analogue à la préparation VI, au départ de 4
(méthylaminométhyl)benzonitrile, on obtient le produit attendu sous forme d'un
solide blanc (rendement = 75 %).
2s F = 72 °C
Exemple 36
N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl]méthylamino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
3o amino]acétamide
En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XL, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 98 %).
F=97°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
42
Exemple 37
N-[2-[[[4-[[(acétoxy)imino](amino)méthyl]phényl]méthyl]méthyl-
amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino] acétamide
s En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon l'exemple 36, on obtient le produit attendu sous forme d'un
solide
blanc amorphe (rendement = 82 %).
F=50°C
1o Exemple 38
N-[2-[[[4-(aminoiminométhyl)phényl]méthyl]méthylamino]-2-oxoéthyl]-
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
acétamide
En opérant de façon analogue à la préparation IX, au départ du composé
1s obtenu selon l'exemple 37, on obtient le produit attendu sous forme d'un
solide
blanc amorphe (rendement = 95 %).
F = 102 °C
Exemple 39
2o N-[2-[[[4-(aminoirninométhyl)phényl]méthyl]méthylamino]-2-oxoéthyl]-
2-[[(~,4-dichloro-3-mëthylphényl)sulfonyl](2-phényléthyl)amino]-
acétamide, chlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 38, on obtient le produit attendu sous forme d'un solide fin
blanc
25 amorphe (rendement = 88 %).
F = 130 °C
PREPARATION XLI
Acide 4-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
3o phényléthyl)amino]acétyl]amino]acétyl]méthylamino]méthyl]benzene-
carboximidique, éthyl ester, chlorhydrate.
En opérant de façon analogue à la préparation XXXIX, au départ du composé
obtenu selon la préparation XL, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 60 %).
35 F = 47-48 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
43
Exemple 40
2-[[2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[[4-(4,5-dihydro-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxo-
éthyl]acétamide
s En opérant de façon analogue à l'exemple 34, au départ du composé obtenu
selon la préparation XLI, on obtient le produit attendu sous forme d'un solide
écru
(rendement = 17 %).
F=80°C
1o Exemple 41
2-[[2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[[4-(4,5-dihydro-iH-imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]acétamide, chlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
1s selon l'exemple 40, on obtient le produit attendu sous forme d'un solide
fin blanc
(rendement = 90 %).
F=100°C
PREPARATION XLII
2o N-[2-[[(4-cyanophényl)méthyl]amino]-2-oxoéthyl]-2-[[(2,4-dichloro-3-
iréthylphényl)sulfonyl](2-phényléthyl)amino]acétamide
En opérant de façon analogue à la préparation VI, au départ de 4-
(aminométhyl)-benzonitrile, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 52 %).
2s F = 82 °C
Exemple 42
N-[2-[[[4-[amino(hydroxyimino)méthyl]phényl]méthyl]amino]-2-oxo-
éthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
3o acétamide
En opérant de façon analogue à la préparation VII, au départ du composé
obtenu selon la préparation XLII, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 98 %).
F=100°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
44
Exemple 43
N-[2-[[[4-[[(acétyloxy)imino](amino)méthyl]phényl]méthyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétamide
s En opérant de façon analogue à la préparation VIII, au départ du composé
obtenu selon l'exemple 42, on obtient le produit attendu sous forme d'un
solide
amorphe blanc (rendement = 50 %).
F=104°C
1o Exemple 44
N-2-[[[4-(aminoiminométhyl)phényl]méthyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
acétamide
En opérant de façon analogue à la préparation IX, au départ du composé
~s obtenu selon l'exemple 43, on obtient le produit attendu sous forme d'un
solide
blanc cassé (rendement = 91 %).
F = 130 °C
Exemple 45
2o N-2-[[[4-(aminoiminométhyl)phényl]méthyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-
acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon l'exemple 44, on obtient le produit attendu sous forme d'un solide blanc
25 (rendement = 83 %).
F = 140 °C
PREPARATION XLIII
Acide 4-[[[(9H-fluoren-9-yl-méthoxy)carbonyl]amino]méthyl]-1-
3o pipéridine carboxylique, 1,1-diméthyléthylester ou [4-(Fmoc-
aminométhyl)-1-Boc-pipéridine]
On prépare une solution de 8,66 g (40,5 mM) de 4-(aminométhyl)-1-Boc-
pipéridine dans 100 ml de DCM. On ajoute 5,26 g (40,5 mM) de DIPEA et une
solution de 10,47 g (40,5 mM) de chloroformate de 9H fluoren-9-yle (ou Fmoc-
CI)
3s dans 50 ml de DCM. Le mélange réactionnel est agité à température ambiante
pendant 1 heure, lavé par une solution saturée d'hydrogénosulfate de potassium
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
puis à l'eau jusqu'à neutralité. La phase organique est séchée puis concentrée
sous
pression réduite. On obtient ainsi 16,9 g du composé attendu qui est utilisé
sans
autre purification pour l'étape suivante.
5 PREPARATION XLIV
Acide (4-pipéridinylméthyl)carbamique, 9H-fluoren-9-yl-méthyl ester
(ou : 4-(Fmoc-aminométhyl)pipéridine), trifluoroacétate
On prépare une solution de 0,5 g (1,15 mM) du composé obtenu selon la
préparation XLIII dans 10 ml de DCM et on ajoute 3 ml d'acide
trifluoroacétique. Le
1o mélange réactionnel est agité à température ambiante pendant 2 heures puis
concentrée sous pression réduite. Le résidu est repris dans.du toluène et à
nouveau
concentré sous pression réduite. Le résidu d'évaporation est trituré dans 10
ml
d'éther éthylique et le solide cristallisé formé est séparée par filtration,
lavé avec 5
ml d'éther éthylique puis séché sous vide. On obtient ainsi 0,45 g du produit
ts attendu sous forme d'un solide blanc (rendement = 87 %).
F = 167 °C
PREPARATION XLV
Acide [(1-Res-4-pipéridinyt)méthyl]carbamique, 9H fluoren-9-yl-méthyl
2o ester (ou : 4-(Fmoc-amino-méthyl)1-Res-pipéridine)
On prépare une suspension de 5,36g de résine fonctionnalisée (copolymère
styrène à 1 % de~divinylbenzène fonctionnalisé par un groupe chlorotrityl,
chargé à
2,05 mM/g en chlore actif obtenu auprès de la société Novabiochem) soit 11 mM,
dans 40 ml de DCM. On ajoute 5,69 g (44 mM) de DIPEA, puis une solution de
7,43
25 g (16,5 mM) du composé obtenu selon la préparation XLIV. Le mélange
réactionnel
est agité à l'aide d'un agitateur orbital pendant 18 heures à température
ambiante.
La résine est séparée par filtration et rincée successivement par 10 ml de
DMF, 10
ml de méthanol, 10 ml de DCM, 10 ml de méthanol, 10 ml de DCM et 10 ml d'éther
éthylique. Après séchage, la résine est utilisée directement pour réaliser
l'étape
30 suivante.
PREPARATION XLVI
Acide 2-[[[(1-Res-4-pipéridinyl)méthyl]amino]carbonyl]-1-pipéridine
carboxylique, 1,1-diméthyléthyl ester ou [N-[(1-Res-4-pipéridinyl)-
35 méthyl]-1-Boc-2-pipéridinecarboxamide]
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
46
On prépare une suspension de 0,158 g (0,2 mM) de la résine obtenue selon
la préparation XLV (taux de greffage : 1,27 mM/g) dans 5 ml d'une solution à
20
de pipéridine dans la DMF. Le mélange réactionnel est agité pendant 5 heures à
température ambiante puis filtré. La résine est lavée successivement avec 3 m!
de
DMF, 3 ml de DCM, puis 3 ml de DMF, et remise en suspension dans 5 ml de DMF.
On ajoute alors 0,155 g (1,2 mM) de DIPEA, 0,138 g (0,6 mM) d'acide N-Boc-2-
pipéridinecarboxylique, 0,076 g (0,6 mM) de HOBT et 0,075 g (0,6 mM) de DIC.
Le
mélange est agité pendant 22 heures à température ambiante puis filtré. La
résine
est Lavée successivement par 3 ml de DMF, 3 ml de méthanol, 3 ml de THF, 3 ml
de
1o méthanol, 3 ml de THF et 5 ml de DCM, puis séchée. La résine sèche est
utilisée
directement pour l'étape suivante.
PREPARATION XLVII
Acide 2-[[[(1-Res-4-pipéridinyl)méthyl]amino]méthyl]-1-pipéridine
Is carboxylique, 1,1-diméthyléthyl ester ou 2-[[[(1-Res-4-pipéridinyl)
méthyl]amino]méthyl]-1-Boc-pipéridine
On prépare une suspension de 0,2 mM de la résine obtenue selon la
préparation XLVI dans 2 ml de THF et on ajoute 0,083 g (0,8 mM) de triméthyle
borate puis 2 ml d'une solution 2M du complexe borane/sulfure de diméthyle
dans
20 l'éther éthylique. Le mélange est agité à température ambiante pendant 23
heures.
La résine est séparée par filtration, lavée par 3 ml de DCM, puis 3 ml de THF
et
remise en réaction en présence de 2 ml de THF, 0,083 g (0,8 mM) de
triméthylborate et 2 ml de la solution 2M du complexe borane/sulfure de
diméthyle
dans l'éther, pendant 72 heures à température ambiante. La résine est séparée
par
25 filtration, lavée par 3 ml de DCM, puis par 3 ml de THF et agitée en
présence de
2 ml de THF et 0,47 g (8 mM) de propylamine pendant 24 heures. La résine est
filtrée, lavée par 3 ml de DMF, 3 ml de méthanol, 3 ml de THF, 3 ml de
méthanol,
3 mi de THF et 4 ml de DCM. Après séchage, la résine est utilisée directement
à
l'étape suivante.
PREPARATION XLVIII
Acide 2-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényl-
éthyl)amino]acétyl]amino]acétyl][(1-Res-4-pipéridinyl)méthyl]amino]-
méthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl ester
On prépare une suspension de 0,2 mM de la résine obtenue selon la
préparation XLVII dans 4 ml de DMF. On ajoute une solution de 0,081 g (0,6 mM)
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
47
de HOBT dans 1 ml de DMF, 0,076 g (0,6 mM) de DIC, 0,159 g (1,2 mM) de DIPEA
puis une solution de 0,276 g de l'acide obtenu selon la préparation III dans 1
ml de
DMF. Le mélange réactionnel est agité pendant 12 heures à 50 °C puis 10
heures à
température ambiante. La résine est séparée par filtration et lavée
successivement
avec 4 ml de DMF, 4 ml de DCM puis 4 ml de DMF. LA résine est ensuite soumise
à
un nouveau cycle de couplage avec l'acide, dans les mêmes conditions, puis
lavée
par 4 ml de DMF, 4 ml de méthanol, 4 mi de THF, 4 ml de méthanol, 4 mi de THF
et 4 ml de DCM et séchée.
Exemple 46
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
oxo-2-[(2-pipéridinylméthyl)(4-pipéridinylméthyl)amino]éthyl]-
acétamide, bis trifluoroacétate
On prépare une suspension de 0,2 mM de la résine obtenue selon la
préparation XLVIII dans 4 ml de DCM et on ajoute 0,4 ml d'acide
trifluoroacétique.
Le mélange est agité pendant 1,S heures à température ambiante puis la résine
est
filtrée et rincée à l'aide de 5 ml de DCM puis 5 ml de méthanol. Les filtrats
réunis
sont concentrés sous flux d'azote et le résidu d'évaporation est purifié par
chromatographie préparative HPLC, à l'aide d'une colonne 250x20 mm chargée de
2o phase stationnaire INERTSIL PREP. ODS obtenue auprès de la société G.L.
Sciences
Ine., et en éluant à l'aide d'un mélange eau/acétonitrile en gradient et en
présence
de 0,05 % d'acide trifluoroacétique. On obtient ainsi 117 mg du produit
attendu.
LC/MS (Grad. C) : 2,32 mn
En suivant le cycle des étapes de la préparation XLVI à l'exemple 46 et en
2s modifiant la nature de !'acide mis en oeuvre lors de la préparation XLVI,
on obtient
les composés des exemples suivants
Exemple 47
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
30 oxo-2-[bis(4-pipéridinylméthyl)amino]éthyl]acétamide, bis trifluoro-
acétate
LC/MS (Grad C) : 2,17 mn
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
48
Exemule 48
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[(1-méthyl-4-pipéridinyl)méthyl](4-pipéridinylméthyl)amino]-2-oxo-
éthyl]acétamide, bis trifluoroacétate
LC/MS (Grad C) : 2,20 mn
Exemple 49
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[(1-éthyl-4-pipéridine)méthyl](4-pipéridinylméthyl)amino]-2-oxoéthyl]-
lo acétamide, bis trifluoroacétate
LC/MS (Grad C) : 2,30 mn
PREPARATION IL
N-[(1-Res-4-pipéridinyl)méthyl]-2-nitro-benzènesulfonamide
is On prépare une suspension de 0,158 g (0,2 mM) de la résine obtenue selon
la préparation XLV (le taux de greffage est de 1,27 mM/g) dans 5 ml d'une
solution
à 20 % de pipéridine dans la DMF. Le mélange réactionnel est agité à
température
ambiante pendant 5 heures puis filtré. La résine est lavée sur filtre par 2 ml
de DMF
puis 2 ml de DCM, puis mise en suspension dans 5 ml de DCM. On ajoute 0,077 g
20 (0,6 mM) de DIPEA puis une solution de 0,133 g (0,6 mM) de chlorure de 2-
nitro-
b~nzènesulfonyle dans 2 ml de DCM. Le mélange réactionnel est agité pendant 30
heures à température ambiante puis filtré. La résine est lavée successivement
par
chaque fois 2 ml de DMF, méthanol, THF, méthanol, THF et DCM, puis utilisée
pour
l'étape suivante.
PREPARATION L
Acide 4-[2-[[(2-nitrophényl)sulfonyl][1-Res-4-pipéridinyl)méthyl]-
amino]éthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl ester
On prépare une suspension de 0,2 mM de la résine obtenue selon la
3o préparation IL dans 1 ml de THF et on ajoute 0,52 g (2 mM) de
triphénylphosphine
en solution dans 2 ml de THF, puis une solution de 0,46 g (2 mM) de l'ester
1,1
diméthyléthylique de l'acide 4-(2-hydroxyéthyl)-1-pipéridinecarboxylique dans
1 ml
de THF, puis 0,20 g (1 mM) de DIAD. Le mélange est agité pendant 30 mn à
température ambiante et on ajoute à nouveau 0,20 g (1 mM) de DIAD. Le mélange
3s est agité à température ambiante pendant 20 heures. La résine est filtrée,
lavée par
2 ml de DCM puis 2 ml de THF et soumise à un nouveau cycle d'alkylation dans
les
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
49
mêmes conditions. La résine est ensuite séparée et lavée successivement par
chaque fois 2 ml de DMF, méthanol, THF, méthanol, THF et DCM. La résine
greffée
ainsi obtenue est utilisée à l'étape suivante.
PREPARATION LI
Acide 4-[2-[[(1-Res-4-pipéridinyl)méthyl]amino]éthyt]-i-pipéridine-
carboxylique, 1,1-diméthyléthyl ester
On prépare une suspension de 0,2 mM de ia résine obtenue selon ia
préparation L dans 5 ml de DMF. On ajoute 0,22 g (2 mM) de thiophénol puis
0,12 g (1,2 mM) de triéthylamine. Le mélange réactionnel est agité pendant
22 heures à température ambiante, puis la résine est séparée par filtration et
rincée
successivement avec chaque fois 2 ml de DMF, méthanol et THF. On la soumet une
seconde fois au cycle réactionnel décrit ci-dessus et on fa lave sur fritre
successivement par chaque fois 2 ml de DMF, méthanol, THF, méthanol, THF et
DCM. La résine ainsi obtenue est utilisée pour l'étape suivante.
PREPARATION LII
Acide 4-[2-[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényl-
éthyi)amino]acétyl]amino]acétyl][(1-Res-4-pipéridiny!)méthyl]amino]-
2o éthyl]-1-pipéridinecarboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XLVIII, au départ de la résine
obtenue selon la préparation LI, on obtient la résine attendue.
Exemule 50
2s 2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
oxo-2-[[2-(4-pipéridinyl)éthyl](4-pipéridinylméthyl)amino]éthyl]-
acétamide, bis trifluoroacétate
En opérant de façon analogue à l'exemple 46, au départ du composé obtenu
selon la préparation LII, on obtient le produit attendu.
3o LC/MS (Grad B) : 2,22 mn
En opérant de façon analogue à la série des étapes allant de la préparation L
à l'exemple 50 et en modifiant la nature de l'amino-alcool introduit à la
préparation
L, on obtient les composés selon l'invention suivants :
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
Exemple 51
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N- j2-
oxo-2-[[2-(1-pipéridinyl)éthyl](4-pipéridinylméthyl)amino]éthy1]-
acétamide, bis trifluoroacétate
5 LC/MS (Grad B) : 2,45 mn
Exemple 52
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
oxo-2-[ j2-(1-pyrrolinyl)éthyl](4-pipéridinylméthyl)amino]éthyl]-
1o acétamide, bis trifluoroacétate
LC/MS (Grad B) : 2,42 mn
Exemele 53
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
i5 [[2-(hexahydro-iH azepin-1-yl)éthyl](4-pipéridinylméthyl)amino]-2-
oxo-éthyl] acétamide, bis trifluoroacétate
LC/MS (Grad B) : 2,57 mn
Exemple 54
20 2-j[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-j2-
j j(1-méthyl-3-pipéridinyl)méthyl](4-pipéridinylméthyl)amino;-2-oxo-
éthyl] acétamide, bis trifluoroacétate
LC/MS (Grad B) : 2,35 mn
25 Exemple 55
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[ j(1-méthyl-2-pipéridinyl)méthyl](4-pipéridinylméthyl)amino]-2-
oxoéthyl] acétamide, bis trifluoroacétate
LC/MS (Grad B) : 2,43 mn
Exemple 56
2-[ j(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[(3-aminopropyl)(4-pipéridinylméthyl)amino]-2-oxoéthyl] acétamide, bis
trifluoroacétate
LC/MS (Grad B) : 2,20 mn
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
51
Exemple 57
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[3-(diméthylamino)propyl](4-pipéridinylméthyl)amino]-2-oxoéthyl]
acétamide, bis trifluoroacétate
s LC/MS (Grad A) : 2,78 mn
Exemple 58
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2
[[2-[méthyl(phénylméthyl)amino)éthyl](4-pïpéridinylméthyl)amino]-2
io oxoéthyl] acétamide, bis trifluoroacétate
LC/MS (Grad B) : 2,78 mn
Exemple 59
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
is [[2-(4-méthyl-1-piperazinyl)éthyl](4-pipéridinylméthyl)amino]-2-oxo-
éthyl] acétamide, bis trifluoroacétate
LC/MS (Grad B) : 1,93 mn
Exemple 60
20 2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
oxo-2-[(4-pipéridinylméthyl)(4-pyridinylméthyl)amino]éthyl]-acétamide,
bis trifluoroacétate
LC/MS (Grad A) : 2,75 mn
25 PREPARATION LIII
N-Res-1,4-butanediamine
On prépare une suspension de 7,32 g (15 mM) de résine fonctionnalisée
(analogue à celle mise en oeuvre pour la préparation XLV) dans 60 ml de DCM.
On
ajoute 3,88 g (30 mM) de DIPEA et 2,65 g (30 mM) de 1,4-butanediamine. Le
3o mélange réactionnel est agité pendant 18 heures à température ambiante puis
la
résine est filtrée et lavée successivement par chaque fois 15 ml de DCM,
méthanol,
DCM et éther éthylique. Elle est ensuite utilisée pour l'étape suivante.
PREPARATION LIV
3s N-Res-N'-[(2-nitrophényl)sulfonyl]-1,4-butanediamine
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
52
On prépare une suspension de 0,2 mM de la résine obtenue selon la
préparation LIII, dans 5 ml de DCM et on ajoute 0,077 g (0,6 mM) de DIPEA,
puis
0,133 g (0,6 mM) de chlorure de 2-nitrobenzènesulfonyle dans 2 ml de DCM. Le
mélange réactionnel est agité 30 heures à température ambiante puis filtré. La
s résine est lavée successivement par chaque fois 2 ml de DMF, méthanol, THF,
méthanol, THF et DCM, puis utilisée pour l'étape suivante.
En opérant ensuite de façon analogue aux étapes décrites pour les
préparations L, LI, LII et l'exemple 50, au départ de la résine obtenue selon
la
préparation LIV et en modifiant de façon appropriée la nature de l'alcool lors
de la
1o première étape, on obtient les composés selon l'invention suivants
Exemple 61
N-[2-[(4-aminobutyl)[2-(diméthylamino)éthyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl) amino]
is acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,72 mn
Exemple 62
N-[2-[(4-aminobutyl)[3-(diméthylamino)propyl]amino]-2-oxoéthyl]-2-
2o [[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl) amino]
acÉ~amide, bis trifluoroacétate
LC/MS (Grad A) : 2,70 mn
Exemple 63
2s N-[2-[(4-aminobutyl)[2-(1-pyrrolidinyl)éthyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl) amino]
acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,80 mn
3o Exemple 64
N-[2-[(4-aminobutyl)[2-(1-pipéridinyl)éthyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]
acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,88 mn
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
53
Exemple 65
N-[2-[(4-aminobutyl)[2-(4-pipéridinyl)éthyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichlpro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]
acétamide, bis trifluoroacétate
s LC/MS (Grad A) : 2,72 mn
Exemple 66
N-[2-[(4-aminobutyl)(3-pipéridinylméthyl]amino]-2-oxoéthyl]-2-[[(2,4
dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amïno] acétamide, bis
io trifluoroacétate
LC/MS (Grad A) : Z,72 mn
Exemple 67
N-[2-[(4-aminobutyl)[(1-méthyl-3-pipéridinyl)méthyl]amino]-2-oxo
1s éthyl]-2-[[(2,4-dichloro-3-méthylphény!)sulfonyl](2-phényléthyl)amino]
acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,75mn
Exemple 68
2o N-[2-[(4-aminobutyl)[(1-méthyl-2-pipéridinyl)méthyl]amino]-2-oxo-
é~~; y!]-~-[[(2,4-dichlpro-3-méthylphényl)sulfonyl](2-phényléthy!)amino]
acétamide, bïs trifluoroacétate
LC/MS (Grad A) : 2,8Zmn
2s Exemple 69
N-[2-[(4-aminobutyl)[2-(hexahydro-iH-azepin-1-yl)éthyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-
phényléthyl)amino] acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,98mn
Exemple 70
N-[2-[(4-aminobutyl)[2-(4-méthyl-1-piperazinyl)éthyl]amino]-2-oxo-
éthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]
acétamide, bis trifluoroacétate
3s LC/MS (Grad A) : 2,48mn
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
54
Exemple 71
N-[2-[(4-aminobutyl)[2-(4-pyridinyl)éthyl]amino]-2-oxoéthyl]-2-[[(2,4-
dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino) acétamide, bis
trifluoroacétate
s LC/MS (Grad C) : 3,02mn
Exemple 72
N-[2-[(4-aminobutyl)[3-(4-pyridinyl)propyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]
io acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,73mn
Exemple 73
N-[2-[(4-aminobutyl)[3-(3-pyridinyl)propyl]amino]-2-oxoéthyl]-2-
1s [[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]
acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,75mn
Exemple 74
2o N-[2-[(4-aminobutyl)[3-(2-pyridinyl)propyl]amino]-2-oxoéthyl]-2-
[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]
acétamide, bis trifluoroacétate
LC/MS (Grad A) : 2,77mn
25 Exemule 75
N-[2-[(4-aminobutyl)[2-[[(éthylamino)carbonyl]oxy]éthyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényl-
éthyl)amino]acétamide, trifluoroacétate
LC/MS (Grad D) : 5,93mn
Exemple 76
N-[2-[(4-aminobutyl)(3-[[(éthylamino)carbonyl]oxy]propyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétamide, trifluoroacétate
LC/MS (Grad D) : 5,97mn
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
Exemple 77
N-[2-[(4-aminobutyl)[4-[[(éthylamino)carbonyl]oxy]butyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
amino]acétamide, trifluoroacétate
5 LC/MS (Grad D) : 6,12mn
Exemple 78
N-[2-[(4-aminobutyl)[3-[[(méthoxy)carbonyl]amino]propyl]amino]-2-
oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)-
1o amino]acétamide, trifluoroacétate
LC/MS (Grad D) : 5,82mn
Exemple 79
N-[2-[(4-aminobutyl)[4-[[(méthoxy)carbonyl]amino]butyl]amino]-2-
15 oxoéthyl]-2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthy1)-
amino]acétamide, trifluoroacétate
LC/MS (Grad D) : 5,82mn
Exemple 80
2o N-[2-[(4-aminobutyl)(3-aminopropyl)amino]-2-oxoéthyl]-2-[[(2,4-
dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]acétamide, bis
trifluoroacétate
LC/MS (Grad A) : 2,65mn
25 PRÉPARATION LV
5-[3-[[4-(1-pyrrolidinyl)butyl]amino]propyl]-1-(triphénylméthyl)-1-H
imidazole
En opérant de façon analogue à la préparation XIV, au départ de 1-(4
aminobutyl)pyrrolidine et de 1(triphénylméthyl)-1-H-imidazole-5-propanal, on
30 obtient le produit attendu sous forme d'une huile jaune (rendement 35 %).
nD~2 = 1,596
PRÉPARATION LVI
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2
35 oxo-2-[[4-(1-pyrrolidinyl)butyl][3-[1-(triphénylméthyl)-1H-imidazol-5
yl]propyl]amino]éthyl]acétamide.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
56
On prépare une solution de 1,84 g (4mM) de l'acide obtenu selon la préparation
ITI
dans 20 ml d'acétonitrile et on ajoute une solution de 1,97 g (4 mM) de
l'amine
obtenue à la préparation LV dans 20 ml d'acétonitrile, puis 1,67 g (4,4 mM) de
HBTU (hexafluoro phosphate de O-benzotriazol-1-yl-N,N,N',N°-
tétraméthyluronium)
puis 0,59 g (4,4 mM) de HOBT et 0,57 g (4,4 mM) de düsopropyléthylamine. Le
mélange réactionnel est agité 20 heures à température ambiante, puis concentré
sous pression réduite. Le résidu est repris dans du dichlorométhane et la
phase
organique obtenue est lavée à l'aide d'une solution de soude diluée, puis à
l'eau,
puis séchée sur sulfate de sodium et concentrée sous pression réduite. Le
résidu est
1o purifié par chromatographie sur gel de silice en éluant à l'aide d'un
mélange
dichlorométhane/méthanol (95/5 ;v/v). On obtient ainsi 3,6 g du produit
attendu
sous forme d'un solide amorphe (rendement = 87 %).
F=72°C
i5 Exemple 81
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[3-(iH-irr~idazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxo-
éthyl]acétamide
On prépare une solution de 3,6 g (3,86 mM) du produit obtenu selon la
préparation
2o LV, dans 30 ml de dichlorométhane et on ajoute 0,42 g (3,86 mM) d'anisole
puis
ml d'acide tr ifluoroacétique. Le mélange réactionnel est agité pendant 20
heure
à température ambiante puis concentré sous pression réduite. On ajoute 100 ml
de
toluène au résidu et on concentre à nouveau sous pression réduite afin
d'entraîner
l'acide trifluoroacétique. Le résidu est purifié par chromatographie sur gel
de silice
en éluant à l'aide d'un mélange dichlorométhane/méthanol/ammoniaque (90/10/1 ;
v/v/v). On obtient ainsi 2,1 g du produit attendu sous forme d'un solide
amorphe
blanc (rendement = 78 %).
F = 114 °C
3o Exemple 82
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-phényléthyl)amino]-N-[2-
[[3-(1H-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxo-
éthyl]acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu selon
3s l'exemple 81, on obtient le produit attendu sous forme d'un solide blanc
(rendement
= 98 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
57
F = 122 °C
PREPARATION LVII
2,4-dichloro-N,3-diméthyl-benzènesulfonamide
s En opérant de façon analogue à la préparation I, au départ de chlorhydrate
de
méthylamine et d'un excès de triéthylamine, on obtient le produit attendu sous
forme d'un solide blanc (rendement = 83 %)
F = 112 °C
l0 PREPARATION LVIII
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]acétyl]-
glycine, éthyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon la préparation LVII, on obtient le produit attendu sous forme d'un
solide blanc
1s (rendement = 51 %)
F=120°C
PREPARATION LIX
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]acétyl]-
2o glycine
On prépûre une solution de 4,65 g (11 mM) de l'ester obtenu selon 1û
préparatio;;
LVIII dans 100 ml de THF et on ajoute une solution de 0,96 g (23 mM) de
lithine
dans 20 ml d'eau. Le mélange réactionnel est agité pendant 3 heures à 50
°C puis
concentré sous pression réduite. Le résidu est repris avec de l'eau et
acidifié à 5 °C
2s avec une solution 1N d'acide chlorhydrique. Le mélange est extrait par du
DCM et la
phase organique obtenue est lavée à l'eau, séchée et concentrée sous pression
réduite. On obtient ainsi 3,79 g du produit attendu sous forme d'un solide
blanc
(rendement = 93 %).
F=154°C
PREPARATION LX
Acide [(4-cyanophényl)méthyl]méthylcarbamique, phénylméthyl ester
On prépare un mélange de 7 g (47,9 mM) de [(4-cyanophényl)méthyl]méthanamine
dans 60 ml de DCM et on ajoute 5,8 g (57,5 mM) de triéthylamine. Le mélange
est
3s refroidi à 0 °C et on ajoute goutte à goutte une solution de 9,8 g
(57,5 mM) de
chloroformate de benzyle dans 20 ml de DCM. Le mélange est ensuite agité
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
58
pendant 20 heures à température ambiante puis lavé par une solution d'acide
chlorhydrique 0,1 N, puis par de l'eau, séché sur sulfate de sodium et
concentré
sous pression réduite. Le résidu est purifié par chromatographie sur gel de
silice en
éluant à l'aide d'un mélange toluène/acétate d'éthyle (95/5 ; v/v). On obtient
ainsi
s 11,4 g du produit attendu sous forme d'une huile (rendement = 87 %).
np2Z = 1,564
PREPARATION IJCI
Acide [[4-(4,5-dihydro-1H imidazol-2-yl)phényl]méthyl]méthyl-
1o carbamique, phénylméthyl ester
On prépare un mélange de 11,3 g (40 mM) du composé obtenu selon la préparation
LX dans 40 ml d'éthylènediamine et on ajoute 0,64 g (20 mM) de fleur de
soufre. Le
mélange réactionnel est agité pendant 2 heures à 100 °C puis refroidi.
On ajoute de
l'eau et on extrait par l'acétate d'éthyle. La phase organique est lavée à
l'eau,
1s séchée sur sulfate de sodium et concentrée sous pression réduite. Le résidu
est
purifié par chromatographie sur gel de silice en éluant à l'aide d'un mélange
dichlorométhane/méthanol/ammoniaque (95/5/0,05 ; v/v /v). On obtient ainsi 11
g
de produit attendu sous forme d'un solide blanc (rendement = 85 %).
F=84°C
2o PREPARATION I.XII
acide ~,5-dihydro-2-[4-[[méthyl[(phénylmétho~,r)carbonyl]amino]-
méthyl]phényl]-iH imidazole-1-carboxylique, 1,1-diméthyléthyl ester
On prépare une solution de 3,22 g (10 mM) du composé obtenu selon la
préparation LXI dans 45 ml de DCM, et on ajoute 1,34 g (11 mM) de N,N
2s diméthylaminopyridine, puis, goutte à goutte, une solution de 2,4 g (11 mM)
de dé
tert-butyl Bicarbonate dans 45 ml de DCM. Le mélange réactionnel est agité
pendant 2 heures à température ambiante puis lavé à I°aide d'une
solution d'acide
chlorhydrique 0,5 N, puis avec de l'eau. La phase organique est séchée sur
sulfate
de sodium puis concentrée sous pression réduite. Le résidu est cristallisé
dans
3o I°éther isopropylique puis filtré et séché. On obtient ainsi 4 g du
produit attendu
sous forme de fins cristaux blancs (rendement = 94 %).
F = 124 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
59
PREPARATION LXIII
Acide 4,5-dihydro-2-[4-[(méthylamino)méthyl]phényl]-1H-imidazole-1-
carboxylique, 1,1-diméthyléthyl ester
s On prépare un mélange de 4,23 g (10 mM) du composé obtenu selon la
préparation
LXII dans 80 ml de méthanol et on ajoute 0,4 g de charbon palladié (à 10 %
Pd).
Le mélange est agité sous atmosphère d'hydrogène à température ambiante et
pression atmosphérique pendant 2 heures. Le catalyseur est éliminé par
filtration
puis le filtrat est concentré sous pression réduite. Le résidu est purifié par
o chromatographie sur gel de silice en éluant à l'aide d'un mélange
dichlorométhane/méthanol/ammoniaque (90/10/0,1 ; v/v/v). On obtient ainsi 2,5
g
du produit attendu sous forme d'un solide blanc cassé (rendement = 90 %).
F=65°C
15 PREPARATION LXIV
Acide 2-[4-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl) sulfonyl] methyl-
amino] acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-
iH-imidazole-1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation VI, au départ des composés
obtenus
2o selon les préparations LIX et LXIII, on obtient le produit attendu sous
forme d'un
solide blanc (r endement = 90 %).
F=61 °C
Exemple 83
2s 2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]-N-[2-[[[4-(4,5-
dihydro-1H imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]-
acétamide
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation LXIV, on obtient après purification par chromatographie sur gel se
silice
30 (éluant : dichlorométhane/méthanol/ammoniaque ; 95/5/0,1 ; v/v/v), le
produit
attendu sous forme d'un solide blanc (rendement = 98 %).
F=72°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
Exemple 84
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]-N-[2-[[[4-(4,5-
dïhydro-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]-
acétamide, chlorhydrate
s En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon
l'exemple 83, on obtient le produit attendu sous forme d'une poudre blanche
(rendement = 88 %).
F = 162 °C
1o PREPARATION LXV
2,4-dichloro-3-méthyl-N-(1,1-diméthyléthyl)-benzènesulfonamide
En opérant de façon analogue à la préparation I, au départ de tert-butylamine,
on
obtient fe produit attendu sous forme d'un solide blanc (rendement = 84 %).
F = 150 °C
~5
PREPARATION I.XVI
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][1,1-diméthyléthyl]-
amino]acétyl]glycine, éthyl ester
On prépare une solution de 3 g (10 mM) du composé obtenu selon la préparation
2o LXV dans 80 ml de DMF anhydre et on ajoute 0,264 g (11 mM) d'hydrure de
sodium. Le mélange est agité pendant 1 heure à température ambiante puis on
ajoute goutte à goutte une solution de 2,98 g (11 mM) de l'ester éthylique de
la N-
(2-iodo-acétyl)glycine. Le mélange réactionnel est agité pendant 15 heures à
température ambiante puis versé sur de l'eau et extrait par l'acétate
d'éthyle, La
25 phase organique est lavée à l'eau, séchée et concentrée sous pression
réduite. Le
résidu est purifié par chromatographie sur gel de silice en éluant à l'aide
d'un
mélange méthylcyclohexane/acétate d'éthyle (6/4 ; v/v). On obtient ainsi 1,87
g du
produit attendu sous forme d'un solide blanc (rendement = 42 %).
F=180°C
PREPARATION LXVII
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][1,1-diméthyléthyl]amino]
acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ du composé obtenu
3s selon la préparation LXVI, on obtient le produit attendu sous forme d'une
poudre
blanche (rendement = 80 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
61
F = 178 °C
PREPARATION LXVIII
2-[[(2,4-dichloro-3-méthytphényf)sulfony(][i,i-diméthyféthyl]amino]-N-
s [2-oxo-2-[[4-(1-pyrrolidinyl)butyl][3-[1-(triphénylméthyl)-iH imidazol-
5-yl] propyl] amino] éthyt] acétamide
En opérant de façon analogue à la préparation LVI, au départ de l'acide obtenu
selon la préparation LXVII, on obtient ie produit attendu sous forme d'un
solide
beige (rendement = 95 %).
1o F = 85 °C
Exemple 85
2-[[(2,4-dichloro-3-méthylphény()sulfonyl]amino]-N-[2-[[3-(iH
imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxoéthyl]
1s acétamide
En opérant de façon analogue à l'exemple 81, au départ du composé obtenu selon
la préparation LXVIII, on obtient le produit attendu sous forme d'un solide
amorphe
blanc (rendement = 67 %).
F=88°C
Exerr:ple 86
2-[[(2,4-dichloro-3-méthylphényl)sulfonyt]amino]-N-[2-[[3-(iH
imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxoéthyl]
acétamide, dichlorhydrate
2s En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon
l'exemple 85, on obtient le produit attendu sous forme d'une poudre blanc
cassé
(rendement = 75 %).
F=55°C
PREPARATION LXIX
2,4-dichloro-N-(2-méthoxyméthyl)-3-méthyl-benzènesulfonamide
En opérant de façon analogue à la préparation I, au départ de 2-
méthoxyéthylamine, on obtient le produit attendu sous forme d'une huile jaune
(rendement = 78 %).
RMN 1H (300 MHz, DMSO) b : 8.00 (s, 1H, NH) ; 7.83 (d, 1H) ; 7.63 (d, 1H) ;
3.27
(t, 2H) ; 3.07 (s, 3H) ; 3.02 (t, 2H) ; 2.49 (s, 3H).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
62
PREPARATION LJOC
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyl)amino]
acétyl]glycine, ethyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
s selon la préparation LXIX, on obtient le produit attendu sous forme d'un
solide blanc
(rendement = 87 %).
F = 108 °C
PREPARATION ~OCI
io N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyl)amino]
acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ du composé obtenu
selon la préparation LXX, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 86 %).
15 F=140°C
PREPARATION L~OfII
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyl)amino]-N-
[2-oxo-2-[[4-(1-pyrrolidinyl)butyl][3-[1-(triphénylméthyl)-1H-imidazol-
20 5-yl]propyl]amino]éthyl]acétamide.
En opérant de façon analogue à la préparation LVI, au départ de l'acide obtenu
selon la préparation LXXI, on obtient le produit attendu sous forme d'un
solide fin
jaune (rendement = 66 %).
F=50°C
Exemple 57
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyl)amino]-N-
[2-[[3-(iH-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxo-
éthyl]acétamide
3o En opérant de façon analogue à l'exemple 81, au départ du composé obtenu
selon
la préparation LXXII, on obtient le produit attendu sous forme d'un solide
blanc écru
(rendement = 70 %).
F=50°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
63
Exemple 88
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyl)amino]-N-
[2-[[3-(iH-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxo-
éthyl]acétamide, dichlorhydrate
s En opérant de façon analogue à l'exemple 6, au départ du composé obtenu
selon
l'exemple 87, on obtient le produit attendu sous forme d'un solide blanc
(rendement
= 97 %).
F=92°C
1o PREPARATiON L~CIII
2,4-dichloro-3-méthyl-N-[2-(3-pyridinyl)éthyl)-benzènesulfonamide
En opérant de façon analogue à la préparation I, au départ de 2-(3-
pyridinyl)éthylamine, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 80 %).
I5 F = 106 °C
PREPARATION UOCIV
N-2-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-(3-pyridïnyl)éthyl]-
amino]acétyl]glycine, 1,1-diméthyléthyl ester
2o En opérant de façon analogue à la préparation II, au départ du composé
obtenu
selon la préparation LXXIII, on obtient le produit attendu sous forme d'un
solide
blanc écru (rendement = 36 %).
F=130°C
25 PREPARATION L)OCV
N-2-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-(3-pyridinyl)éthyl]-
amino]acétyl]glycine, chlorhydrate
On prépare une solution de 0,53 g (1,02 mM) de l'ester obtenu selon la
préparation
LXXIV dans 10 ml de dichlorométhane et on ajoute 10 ml d'une solution saturée
de
3o chlorure d'hydrogène dans le dioxane. Le mélange réactionnel est agité
pendant 10
jours à température ambiante puis le précipité formé est filtré et rincé avec
du
dichlorométhane et séché. On obtient ainsi 0,43 g du produit attendu, sous
forme
d'un solide blanc (rendement = 84 %).
F= 154°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
64
PREPARATION L)OCVI
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-(3-pyridinyl)éthyl]amino]-
N-[2-oxo-2-[[4-(1-pyrrolidinyl)butyl][3-[1-(triphénylméthyl)-iH-
imidazol-5-yl]propyl]amino]éthyl]acétamide.
s En opérant de façon analogue à la préparation LVI, au départ du composé
obtenu
selon la préparation LXXV, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 63 %).
F=82°C
1o Exemple 89
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-(3-pyrïdinyl)éthyl]amino]-
N-[2-[[3-(1H-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-
oxoéthyl]acétamide
En opérant de façon analogue à l'exemple 81, au départ du composé obtenu selon
~5 la préparation LXXVI, on obtient le produit attendu sous forme d'une pâte
beige
(rendement = 99 %).
RMN 1H (300 MHz, DMSO) b : 8.28 (m, 2H) ; 8.21 (m, 1H) ; 7.84 (d, 1H) ; 7.56
(d,
2H) ; 7.07 (t, 1H) ; 6.81 (d, 1H) ; 4.21 (d, 2H) ; 3.98 (dd, 2H) ; .49 (t, 2H)
; 3.27
(d, 4H) ; 3.15 (s, 4H) ; 3.03 (2 H) ; 2.74 (t, 2H) ; 2.49 (m 2H) ; 2.34 (s,
3H) ; 1.88
20 (m, 6H) ; I.53 (m, 4H).
Exemple 90
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-(3-pyridinyl)éthyl]amino]-
N-[2-[[3-(iH-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-
2s oxoéthyl]acétamide, tris trifluoroacétate
On prépare une solution de 0,14 g (0,202 mM) du composé obtenu selon l'exemple
89, dans 2 ml de méthanol et on ajoute 50 8.1 d'acide trifluoroacétique. Le
mélange
réactionnel est agité pendant 15 mn à température ambiante puis concentré sous
pression réduite. Le résidu est repris dans 20 ml d'eau et la solution obtenue
est
30 lyophilisée. On obtient ainsi 0,17 g du produit attendu sous forme d'un
solide blanc
(rendement = 81 %).
F=60°C
PREPARATION L.)OCVII
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]amino]acétamide
3s En opérant de façon analogue à la préparation I, au départ de 2-
aminoacétamide,
on obtient le produit attendu sous forme d'un solide blanc (rendement = 60 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
F=180°C
PREPARATION L.)OCVIII
N-[2-[(2-amino-2-oxoéthyl)[(2,4-dichloro-3-méthylphényl)sulfonyl]-
5 amino]acétyl]glycine, éthyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon la préparation LXXVII, on obtient le produit attendu sous forme d'une
poudre
blanche (rendement = 76 %).
F = 190 °C
PREPARATION UOCIX
N-[2-[(2-amino-2-oxoéthyl)[(2,4-dichloro-3-
méthylphényl)sulfonyl]amino]acétyl]
glycine
En opérant de façon analogue à la préparation LIX, au départ de la préparation
LXXVIII, on obtient le produit attendu sous forme d'un solide blanc (rendement
=
43 %).
F=94°C
PREPARATION LX)OC
Acidc 2-[4-[[[2-[[2-[(2-amino-2-uxoéthyl)[(2,4-dichloro-3-
méthylphényl)sulfonyl]amino]acétyl]amino]acétyl]méthylamino]méthyl]
phényl]-4,5-dihydro-1H-imidazole-1-carboxylique, 1,1-diméthyléthyl
ester
2s
En opérant de façon analogue à la préparation VI, au départ des composés
obtenus
selon les préparations LXXIX et LXIII, on obtient le produit attendu sous
forme d'un
solide blanc (rendement = 53 %).
F = 118 °C
Exemple 91
2-[(2-amino-2-oxoéthyl)[(2,4-dichloro-3-méthylphényl)sulfonyl]amino]-
N-[2-[[[4-(4,5-dihydro-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-
2-oxoéthyl]acétamide, trifluoroacétate
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
66
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation LXXX, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 83 %).
F = 130 °C
PREPARATION L)OOCI
N-méthyl-2,3,4-trichlorobenzènesulfonamide
En opérant de façon analogue à la préparation LVII, au départ de chlorure de
2,3,4-
trichlorobenzènesulfonyle, on obtient le produit attendu sous forme d'un
solide
1o beige (rendement = 53 %).
F = 134 °C
PREPARATION L)OOCII
N-[2-[méthyl-[(2,3,4-trichlorophényl)sulfonyl]amino]acétyl]glycine,
éthyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon la préparation LXXXI et de N-(iodoacétyl)glycinate d'éthyle, on obtient
le
produit attendu sous forme d'un solide blanc (rendement = 80 %).
F = 140 °C
~'REPARATION L)00(III
N-[2-[méthyl-[(2,3,4-trichlorophényl)sulfonyl]amino]acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ du composé obtenu
selon la préparation LXXXII, on obtient le produit attendu sous forme d'une
poudre
2s blanche (rendement = 93 %).
F = 13~ °C
PREPARATION L)OOCIV
2-[méthyl[(2,3,4-trichlorophényl)sulfonyl]amino]-N-[2-oxo-2-[[4-(1-
3o pyrrolidinyl)butyl][3-[1-(triphénylméthyl)-iH-imidazol-5-yl]propyl]-
amino]éthyl]acétamide
En opérant de façon analogue à la préparation LVI, au départ de l'acide obtenu
selon la préparation LXXXIII, on obtient le produit attendu sous forme d'un
solide
beige (rendement = 68 %).
35 F = 120 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
67
Exemple 92
N-[2-[[3-(1H-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2
oxoéthyl]-2-[méthyl[(2,3,4-trichlorophényl)sulfonyl]amino]acétamide,
bïs trifluoroacétate
s En opérant de façon analogue à l'exemple 81, mais en purifiant le composé
brut à
l'aide d'un mélange dichlorométhane/méthanol (90/10 ; v/v), on obtient le
produit
attendu sous forme d'un solide beige (rendement = 71 %).
F=76°C
1o PREPARATION L)00(V
N-(2-propényl)-2,4-dichloro-3-méthylbenzènesulfonamide
En opérant de façon analogue à la préparation I, au départ d'allylamine, on
obtient
le produit attendu sous forme d'un solide blanc (rendement = 77 %).
F=91 °C
PREPARATION IJOOCVI
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]acétyl]-
glycine, éthyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
2o selon la préparation LXXXV, on obtient le produit attendu sous forme d'un
solide
blanc (r endement = 87 %).
F=81 °C
PREPARATION L)OOCVII
2s N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]acétyl]-
glycine,
En opérant de façon analogue à la préparation LIX, au départ du composé obtenu
selon la préparation LXXXV, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 99 %).
3o F=138°C
PREPARATION L)00(VIII
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]-N-[2-oxo-
2-[[4-(1-pyrrolidinyl)butyl][3-[1-(triphénylméthyl)-1H imidazol-5-yl]-
3s propyl]amino]
éthyl]acétamide
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
68
En opérant de façon analogue à la préparation LVI, au départ du composé obtenu
selon la préparation LXXXVII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 40 %).
F=60°C
Exemple 93
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]-N-[2-[[3-
(iH-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amïno]-2-oxoéthyl]-
acétamide
o En opérant de façon analogue à l'exemple 81, au départ du composé obtenu
selon
la préparation LXXXVIII, on obtient le produit attendu sous forme d'un solide
amorphe (rendement = 75 %).
F=50°C
Exemple 94
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]-N-[2-[[3-
(1H-imidazol-5-yl)propyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxoéthyl]-
acétamide, dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu selon
l'exemple 93, on obtient le produit attendu sous forme d'un solide blanc
(rendement
= 80 %).
F=90°C
PREPARATION L.)OOCIX
N-méthyl-2,6-dichlorobenzènesulfonamide
En opérant de façon analogue à la préparation LVII, au départ du chlorure de
2,6-
benzènesulfonyle, on obtient le produit attendu sous forme d'un solide blanc
écru
(rendement = 99 %).
F = 115 °C
PREPARATION XC
N-[2-[[(2,6-dichlorophényl)sulfonyl]méthylamino]acétyl]glycine, éthyl
ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon la préparation LXXXIX, on obtient le produit attendu, utilisé sans
purifrcation
complémentaire pour l'opération suivante.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
69
PREPARATION XCI
N-[2-[[(2,6-dichlorophényl)sulfonyl]méthylamino]acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ de l'ester obtenu
selon la préparation XC, on obtient le produit attendu sous forme d'un solide
blanc
s écru (rendement = 76 %).
F = 157 °C
PREPARATION XCII
Acide 2-[4-[[[2-[[2-[[(2,6-dichlorophényl)sulfonyl]méthylamino] acétyl]
lo amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-1H-imidazole-1
carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation LXIV, au départ du composé
obtenu
selon la préparation XCI, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 57 %).
~s F=88°C
Exemple 95
2-[[(2,6-dichlorophényl)sulfonyl]méthylamino]-N-[2-[[[4-(4,5-dihydro-
iH-imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]acétamide,
2o trifluoroacétate
Ec. cpérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation XCII, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 81 %).
F=96°C
2s
PREPARATION XCIII
~y~-oxo-N-[3-(4-pyridinyl)propyl]-1-pyrrolidinebutanamide
En opérant de façon analogue à la préparation VI, au départ d'acide y oxo-1-
pyrrolidinebutanôique et de 4-pyridinepropanamine, on obtient le produit
attendu
3o sous forme d'un solide blanc (rendement = 56 %).
F = 110 °C
PREPARATION XCIV
N-[4-(1-pyrrolidinyl)butyl]-4-pyridinepropanamine
3s On prépare une solution de 640 mg (2 mmoles) du composé obtenu selon la
préparation XCIII dans 30 ml de tétrahydrofurane anhydre et on ajoute 505 mg
(13
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
mmoles) d'hydrure de lithium-aluminium. Le mélange est agité à reflux pendant
20
heures. On ajoute ensuite 20 ml de tétrahydrofurane puis 1 g de sulfate de
sodium
hydraté. Le mélange est agité pendant 30 mn à température ambiante puis
filtré. Le
filtrat est concentré sous pression réduite et le résidu huileux est purifié
par
s chromatographie sur gel de silice greffé Cl$ en éluant à l'aide d'un mélange
eau/acétonitrile/acide trifluoroacétique (90/10/5 ; v/v/v). On obtient ainsi
320 mg
du produit attendu sous forme d'une pâte jaune (rendement : 20 %).
RMN 1H (300 MHz, DMSO) 8 : 9.94 (s, 1H) ; 8.80 (d, 2H) ; 8.73 (s, 2H) ; 7.79
(d,
2H) ; 3.52 (m, 2H) ; 3.11 (m, 2H) ; 2.90 (m, 8H) ; 1.92 (m, 6H) ; 1.64 (m,
4H).
Exemple 96
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]-N-[2-oxo-2-
[[3-(4-pyridinyl)propyl][4-(1-pyrrolidinyl)butyl]amino]éthyl]acétamide
En opérant de façon analogue à la préparation LVI, au départ de l'acide obtenu
Is selon la préparation LIX et de l'amine obtenue selon la préparation XCIV,
on obtient
le produit attendu sous forme d'une pâte incolore (rendement = 35 %).
RMN 1H (250 MHz, DMSO) 8 : 8.45 (m, 2H) ; 8.02 (t, 1H) ; 7.88 (d, 1H) ; 7.63
(d,
1H) ; 7.25 (m, 2H) ; 3.99 (s, 2H) ; 3.94 (t, 2H) ; 3.25 (m, 4H) ; 2.88 (s, 3H)
; 2.56
(m, 2H) ; 2.50 (s, 3H) ; 2.40 (m, 6H) ; 1.85 (m, 2H) ; 1.65 (s, 4H) ; 1.41 (m,
4H).
Exc.;,pl~ 97
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]-N-j2-oxo-2-
[[3-(4-pyridinyl)propyl][4-(1-pyrrolidinyl)butyl]amino]éthyl]acétamide,
dichlorhydrate
En opérant de façon analogue à l'exemple 6, au départ de l'acide obtenu selon
l'exemple 96, on obtient le produit attendu sous forme d'une pâte jaune
(rendement
= 68%).
RMN 1H (300 MHz, DMSO) 8 : 10.95 (m, 2H) ; 8.81 (t, 2H) ; 8.12 (t, 1H) ; 7.98
(dd, 2H) ; 7.89 (d, 1H) ; 7.65 (d, 1H) ; 4.00 (s, 4H) ; 3.96 (t, 2H) ; 3.49
(m, 2H) ;
3.33 (m, 4H) ; 3.07 (m, 2H) ; 2.91 (m, 2H) , 2.87 (s, 3H) ; 2.49 (s, 3H) ;
1.92 (m,
6H) ; 1.62 (m, 4H).
PREPARATION XCV
4-[[[4-(1-pyrrolidinyl)butyl]amino]méthyl]phénol
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
71
En opérant de façon analogue à la préparation XIV, au départ de 1-(4-
aminobutyl)pyrrolidine et de 4-(triméthylsilyloxy)benzaldéhyde, on obtient le
produit
attendu sous forme d'une huile orange (rendement = 99%)
RMN 1H (300 MHz, DMSO) 8 : 7.03 (d, 2H) ; 6.62 (d, 2H) ; 3.51 (s, 2H) ; 2.2-
2.6
s (m, 8H) ; 1.55-1.8 (m, 4H); 1.3-1.5 (m, 4H).
Exemple 98
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]méthylamino]-N-[2-[[(4
hydroxyphényl)methyl][4-(1-pyrrolidinyl)butyl]amino]-2-oxoéthyl]
1o acétamide
En opérant de façon analogue à l'exemple 96, au départ de l'amine obtenue
selon la
préparation XCV, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 33%).
F=104°C
PREPARATION XCVI
N-[2-[[(2-chlorophényl)sulfonyl]méthylamino]acétyl]glycine, éthyl ester
En opérant de façon analogue à la préparation LXXXIX, au départ de N-méthyl-2-
chlorobenzènesulfonamide, on obtient le produit attendu, utilisé sans
purification
2o complémentaire pour la synthèse suivante.
PREPARATION XCVII
N-[2-[[(2-chlorophényl)sulfonyl]méthylamino]acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ du composé obtenu
2s selon la préparation XCVI, on obtient le produit attendu sous forme d'un
solide
beige (rendement = 74 %).
F = 132 °C
PREPARATION XCVIII
3o Acide 2-[4-[[[2-[[2-[[(2-chlorophényl) sulfonyl]méthylamino] acétyl]-
amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-1H-imidazole-1-
carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation XCVII, on obtient le produit attendu sous forme d'un
solide
35 blanc (rendement = 45 %).
F=82°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
72
Exemple 99
2-[[(2-chlorophényl)sulfony!]méthylamino]-N-[2-[[[4-(4,5-dihydrô-iH-
imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]acétamide,
trifluoroacétate
s En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la
préparation XCVIII, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 80 %).
F=100°C
1o PREPARATION IC
Acide 2-[4-[[[2-[[2-[[(2,3,4-trichlorophényl) sulfonyl]méthylamino]
acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-1H
imidazole-1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
1s selon la préparation UOOCIII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 67 %).
F=94°C
Exemple 100
20 2-[[(2,3,4-trichlorophényl)sulfonyl]méthylamino]-N-[2-[[[4-(4,5-
dc:~ydra-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]acétamide, trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation IC, on obtient le produit attendu sous forme d'un solide blanc
2s (réndement = 60 %).
F=95°C
PREPARATION C
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]amino]-N-méthyl-acétamide
3o En opérant de façon analogue à la préparation I, au départ de 2-amino-N-
méthyl-
acétamide, on obtient le produit attendu sous forme d'un solide blanc
(rendement =
76 %).
F=148°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
73
PREPARATION CI
N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-(méthylamino)-2-
oxoéthyl]amino]acétyl]glycine, éthyl ester
En opérant de façon analogue à (a préparation II, au départ du composé obtenu
s selon la préparation C, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 63 %).
F = 140 °C
PREPARATION CII
1o N-[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-(méthylamino)-2-oxo-
éthyl]amino]acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ du composé obtenu
selon la préparation CI, on obtient le produit attendu sous forme d'un solide
blanc
écru (rendement = 81 %).
1 s F = 205 °C
PREPARATION CIII
Acide 2-[4-[8-[(2,4-dichloro-3-méthylphényl)sulfonyl]-2-méthyl-3,6,10-
trioxo-2,5,8,11-tétraazadodec-1-yl]phényl]-4,5-dihydro-1H-imidazole-1-
2o carboxylique, 1,1-diméthyléthyl ester
L~. or'rant de façon analogue à la préparation XCII, au départ du ccrnposé
obtenu
selon la préparation CII, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 53 %).
F = 105 °C
Exemple 101
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl][2-[[2-[[[4-(4,5-dihydro-1H
imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]amïno]-2-oxo-
éthyl]amino]-N-méthyl-acétamide, trifluoroacétate
3o En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la
préparation CIII, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 99 %).
F = 106 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
74
PREPARATION CIV
Acide 2-[4-[[[2-[[2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]-2-
propénylamino]acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-
dihydro-iH-imidazole-1-carboxylique, 1,1-diméthyléthyl ester
s En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation UOOCVII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 35 %).
F = 70 °C
1o Exemple 102
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]-N-[2-[[[4-
(4,5-dihydro-iH-imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxo-
éthyl]acétamide
En opérant de façon analogue à l'exemple 1 et en ajoutant de l'ammoniaque, au
15 départ du composé obtenu selon la préparation CIV, on obtient le produit
attendu
sous forme d'un solide blanc (rendement = 84 %).
F = 90 °C
Exemple 103
20 2-[[(2,4-dichloro-3-méthylphényl)sulfonyl]2-propénylamino]-N-[2-[[[4-
(4,5-ûit~ydro-iH-imidazol-2-yl)Phényl]méthyl]méthylamir~o]-'~_oxo-
éthyl]acétamide, chlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu selon
l'exemple 102, on obtient le produit attendu sous forme d'un solide blanc
2s (rendement = 54 %).
F=125°C
PREPARATION CV
Acide 2-[4-[8-[(2,4-dichloro-3-méthylphényl)sulfonyl]-2-méthyl-3,6-
3o dioxo-11-oxa-2,5,8-triazadodec-1-yl]phényl]-4,5-dihydro-iH imidazole-
1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XCII, au départ de l'acide
obtenu
selon la préparation UOCI, on obtient le produit attendu sous forme d'un
solide blanc
(rendement = 76 %).
35 F = 80 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
EXemple 104
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyl)amino]-N-
[2-[[[4-(4,5-dihydro-iH-imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]acétamide
s En opérant de façon analogue à l'exemple 102, au départ du composé obtenu
selon
la préparation CV, on obtient le produit attendu sous forme d'un solide blanc
écru
(rendement = 99 %).
F=76°C
1o Exemple 105
2-[[(2,4-dichloro-3-méthylphényl)sulfonyl](2-méthoxyéthyi)amino]-N-
[2-[[[4-(4,5-dihydro-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]acétamide, chlorhydrate
En opérant de façon analogue à l'exemple 6, au départ du composé obtenu selon
is l'exemple 104, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 85 %).
F=130°C
PREPARATION CVI
2o N-méthyl-2,3-dichlorobenzènesulfonamide
En opcïa~.t de façon analogue à la préparation LVII, au départ de chlorure de
2,3-
dichlorobenzènesulfonyle, on obtient le produit attendu qui est utilisé sans
purification complémentaire dans la synthèse suivante.
25 PREPARATION CVII
N-2-[[(2,3-dïchlorophényl)sulfonyl]méthylamino]acétyl]glycine, éthyl
ester
En opérant de façon analogue à la préparation LXXXII, au départ du composé
obtenu selon la préparation CVI, on obtient le produit attendu qui est utilisé
sans
3o purification complémentaire dans la synthèse suivante.
PREPARATION CVIII
N-2-[[(2,3-dichlorophényl)sulfonyl]méthylamino]acétyl]glycine
En opérant de façon analogue à la préparation LIX, au départ dû composé obtenu
3s selon la préparation CVII, on obtient (e produit attendu sous forme d'un
solide blanc
(rendement = 55 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
76
F=147°C
PREPARATION CIX
Acide 2-[4-[[[2-[[2-[[(2,3-dïchlorophényl)sulfonyl]méthylamino] acétyl]
s amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-iH-imidazole-1
carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation CVIII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 59 %).
1o F=88°C
Exemple 106
2-[[(2,3-dichlorophényl)sulfonyl]méthylamino]-N-[2-[[[4,5-dihydro-iH-
imidazole-2-yl)phényl]méthyl]méthylamino]2-oxoéthyl]acétamide,
i5 trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation CIX, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 80 %).
F = 100°C
PPE~'ARATION CX
N-[2-[[(2,4-dichlorophényl)sulfonyl]méthylamino]acétyl]glycine
En opérant de façon analogue aux préparations UOOCIX à XCI, au départ de
chlorure de 2,4-dichlorobenzènesulfonyle, on obtient le produit attendu sous
forme
2s d'un solide blanc (rendement = 30 %).
F = 179 °C
PREPARATION CXI
Acide 2-[4-[[[2-[[2-[[(2,4-dichlorophényl)sulfonyl]méthylamino]
3o acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-iH-
imidazole-1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation LXIV, au départ du composé
obtenu
selon la préparation CX, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 68 %).
35 F = 72 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
77
Exemple 107
2-[[(2,4-dichlorophényl)sulfonyl] méthylamino]-N-[2-[[[4-(4,5-dihydro-
iH imidazol-2-yl)phenyl]methyl]méthylamino]-2-oxoéthyl]acétamide,
trifluoroacétate
s En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la
préparation CXI, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 99 %).
F=90°C
1o Exemule 108
2-[[(2,4-dichlorophényl)sulfonyl]méthylamino]-N-[2-[méthyl[4-(1-
pyrrolidinyl)butyl]amino]-2-oxoéthyl]acétamide
En opérant de façon analogue à la préparation LVI, au départ de l'acide obtenu
selon la préparation LIX et de N-méthyl-1-pyrrolidinebutanamide, on obtient le
Is produit attendu sous forme d'une huile jaune (rendement = 91 %).
RMN 1H (300 MHz, DMSO) â : 8.01 (m, 1H) ; 7.89 (d, 1H) ; 7.64 (d, 1H) ; 3.99
(s,
2H) ; 3.94 (m, 2H) ; 3.26 (m, 6H) ; 2.91 (s) et 2.80 (s) (total 3H) ; 2.88 (s,
3H) ;
2.50 s, 3H) ; 2.38 (m, 2H) ; 1.65 (s, 4H) ; 1.44 (m, 4H).
2o Exemple 109
2-[[(2,4-dichlorophényl)sulfonyl]méthylamino]-N-[2-[methyl[4-(1-
pyrrolidinyl)butyl]amino]-2-oxoéthyl]acétamide, fumarate
On prépare une solution de 127 mg (0,25 mM) du composé obtenu selon l'exemple
108 dans 6 ml de méthanol et on ajoute 29 mg (0,25 mM) d'acide fumarique. Le
2s mélange réactionnel est agité 15 mn puis concentré sous pression réduite.
Le résidu
est dissout dans 10 ml d'eau puis lyophilisé. On obtient ainsi 145 mg du
produit
attendu sous forme d'un solide amorphe (rendement = 93 %).
RMN 1H (250 MHz, DMSO) 8 : 8.04 (m, 1H) ; 8.01 (d, 1H) ; 7.64 (d, 1H) ; 5.51
(s,
2H) ; 4.00 (s, 2H) ; 3.95 (t, 2H) ; 3.28 (m, 2H) ; 2..87 (m, 12H) ; 2.49 (s,
3H) ;
30 1.81 (s, 4H) ; 1.49 (m, 4H).
PREPARATION CXII
N-[2-[méthyl(1-naphtalénylsulfonyl)amino]acétyl]glycine
En opérant de façon analogue à la préparation CX, au départ du chlorure de 1-
3s naphtalènesulfonyle, on obtient le produit attendu sous forme d'un solide
jaune
(rendement = 40 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
78
F=120°C
PREPARATION CXIII
Acide 4,5-dihydro-2-[4-j[méthylj2-[[2-[méthyl(1-naphtalénylsulfonyl)
s amino]acétyl]amino]méthyl]phényl]-iH-imidazole-1-carboxylique, 1,1-
diméthyléthyl ester
En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation CXII, on obtient le produit attendu sous forme d'un
solide blanc
(rendement = 53 %).
o F=90°C
Exemple 110
N-[2-[[[4-(4,5-dihydro-iH imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]-2-[méthyl(1-naphtalénylsulfonyl)amino]acétamide,
is trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation CXIII, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 88 %).
F = 115 °C
PRÉPARATION CXIV
N-[2-[méthyl(2-naphtalénylsulfonyl)amino]acétyl]glycine
En opérant de façon analogue à la préparation CX, au départ du chlorure de 2-
naphtalènesulfonyle, on obtient le produit attendu sous forme d'un solide
blanc
2s (rendement = 54 %).
F=185°C
PRÉPARATION CXV
Acide 4,5-dihydro-2-[4-[[méthyl[2-[[2-[méthyl(2-naphtalénylsulfonyl)
3o amino]acétyl]amino]méthyl]phényl]-iH-imidazole-1-carboxylique, 1,1-
diméthyléthyl ester
En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation CXIV, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 62 %).
35 F=89°C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
79
Exemple 111
N-[2-[[[4-(4,5-dihydro-iH-imidazol-2-yl)phényl]méthyl]méthylamino]-
2-oxoéthyl]-2-[méthyl(2-naphtalénylsulfonyl)amino]acétamide, tri-
fluoroacétate
s En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la
préparation CXV, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 85 %).
F = 101 °C
1o PREPARATION CXVI
N-cyclopropyl-2,6-dichlorobenzènesulfonamide
En opérant de façon analogue à la préparation LXX)CIX, au départ de
cyclopropanamine, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 99 %).
1s F=76°C
PREPARATION CXVII
N-[2-[cyclopropyl[(2,6-dichlorophényl)sulfonyl]amino]acétyl]glycine,
éthyl ester
2o En opérant de façon analogue à la préparation XC, au départ du composé
obtenu
selon 1û préparation CXVI, on obtient le produit attendu sous forme d'un
sclide
jaune (rendement = 76 %).
F = 125 °C
2s PREPARATION CXVIII
N-[2-[cyclopropyl[(2,6-dichlorophényl)sulfonyl]amino]acétyl]glycine
En opérant de façon analogue à la préparation XCI, au départ du composé obtenu
selon la préparation CXVII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 62 %).
3o F = 164 °C
PREPARATION CXIX
Acide 2-[4-[[[2-[[2-[cyclopropyl[(2,6-dichlorophényl)sulfonyl]
amino]acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-
3s iH-imidazole-1-carboxylique, 1,1-diméthyléthyl ester
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation CXVIII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 55 %).
F=66°C
5
Exemple 112
2-[cyclopropyl[(2,6-dichlorophényl)sulfonyl]amino]-N-[2-[[[4-(4,5-
dihydro-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]-
acétamide, trïfluoroacétate
1o En opérant de façon analogue à l'exemple 1, au départ du composé obtenu
selon la
préparation CXIX, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 71 %).
F = 118 °C
15 PREPARATION CXX
N-cyclopropyl-2,3-dichlorobenzènesulfonamide
En opérant de façon analogue à la préparation CVI, au départ de
cyclopropanamine,
on obtient le produit attendu sous forme d'un solide blanc écru (rendement =
50
%).
2o F = 140 °C
PREPARATION CXXI
N-[2-[cyclopropyl[(2,3-dichlorophënyl)sulfonyl]amino]acétyl]glycine,
éthyl ester
2s En opérant de façon analogue à la préparation XC, au départ du composé
obtenu
selon la préparation CXX, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 89 %).
F = 155 °C
3o PREPARATION CXXII
N-[2-[cyclopropyl[(2,3-dichlorophényl)sulfonyl]amino]acétyl]glycine
En opérant de façon analogue à la préparation XCI, au départ du composé obtenu
selon la préparation CXXI, on obtient le produit attendu sous forme d'un
solide
blanc écru (rendement = 72 %).
3s F = 174 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
81
PREPARATION CXXIII
Acide 2-[4-[[[2-[[2-[cyclopropyl[(2,3-dichlorophényl)sulfonyl]
amino]acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-
iH-imidazole-1-carboxylique, 1,1-diméthyléthyl ester
s En opérant de façon analogue à la préparation XCII, au départ du composé
obtenu
selon la préparation CXXII, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 67 %).
F=98°C
1o Exemple 113
2-[cyclopropyl[(2,3-dichlorophényl)sulfonyl]amino]-N-[2-[[[4-(4,5-
dihydro-1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]acétamide, trifiuoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
i5 préparation CXXIII, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 84 %).
F=88°C
PREPARATION CXXIV
20 2-chloro-N-cyclopropyl-benzènesulfonamide
En opérant de façon analogue à la préparation CXVI, au départ de chlorur e de
2-
chlorobenzènesulfonyle, on obtient le produit attendu sous forme d'un solide
blanc
(rendement = 82 %).
F = 117 °C
PREPARATION CXXV
N-[2-[[(2-chlorophényl)sulfonyl]cyclopropylamino]acétyl]glycine, éthyl
ester
En opérant de façon analogue à la préparation CXXIV, au départ du composé
obtenu selon la préparation CXXIV, on obtient le produit attendu sous forme
d'un
solide blanc (rendement = 93 %).
F=98°C
PREPARATION CXXVI
N-[2-[[(2-chlorophényl)sulfonyl]cyclopropylamino]acétyl]glycine
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
82
En opérant de fiaçon analogue à la préparation XCI, au départ du composé
obtenu
selon la préparation CXXV, on obtient le produit attendu sous forme d'un
solide
jaune (rendement = 72 %).
F = 125 °C
PREPARATION CXXVII
Acide 2-[4-[[[2-[[2-[[(2-chlorophényl)sulfonyt]cyctopropylamino]acétyl]
amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-1H imidazole-1-
carboxylique, 1,1-diméthytéthyl ester
1o En opérant de façon analogue à la préparation CXII, au départ du composé
obtenu
selon la préparation CXXVI, on obtient le produit attendu sous forme d'un
solide
blanc (rendement = 75 %).
F=70°C
Exemple 114
2-[[(2-chlorophényl)sulfonyl]cyclopropylamino]-N-[2-[[[4-(4,5-dihydro-
1H-imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxoéthyl]acétamide,
trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
2o préparation CXXVII, on obtient le produit attendu sous forme d'un solide
blanc
(rerûement = 76 %).
F=106°C
PREPARATION CXXVIII
2s 2-[[(2,6-dichlorophényl)sulfonyl]amino]acétamide
En opérant de façon analogue à la préparation CXVI, au départ de 2-
aminoacétamide, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 54 %).
F=164°C
PREPARATION CXXIX
N-[2-[(2-amino-2-oxoéthyl)[(2,6-
dichlorophényl)sulfonyl]amino]acétyl]glycine, éthyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
3s selon la préparation CXXVIII, on obtient le produit attendu sous forme d'un
solide
beige (rendement = 31 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
83
F=178°C
PREPARATION CX~C
N-[2-[(2-amino-2-oxoéthyl)[(2,6-dichlorophényl)sulfonyl]amino]-
s acétyl]glycine,
En opérant de façon analogue à la préparation UOCV, au départ du composé
obtenu
selon la préparation CXXIX, on obtient le produit attendu sous forme d'une
pâte
beige (rendement = 99 %).
RMN 1H (250 MHz, DMSO) 8 : 8.61 (t, 1H) ; 7.68 (s, 1H) ; 7.61 (m, 2H) ; 7.52
(dd,
1H) ; 7.10 (s, 1H) ; 4.21 (s, 2H) ; 4.08 (s, 2H) ; 3.74 (d, 2H).
PREPARATION C700CI
Acide 2-[4-[[[2-[[2-[(2-amino-2-oxoéthyl)[(2,6-dichlorophényl)sulfonyl]
amino]acétyl]amino]acétyl]méthylamino]méthyl]phényl]-4,5-dihydro-
is 1H-imidazole-1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation VI, au départ des composés
obtenus
selon les préparations CXXX et LXIII, on obtient le produit attendu sous forme
d'une
pâte incolore (rendement = 15 %).
RMN 1H (300 MHz, CDCI3) 8 : 7.47 (m, 5H) ; 7.32 (m, 2H) ; 7.21 (m, 2H) ; 5.75
(s,
2o 1H) ; 4.61 (s, 2H) ; 4.26 (s, 2H) ; 4.1 (m, 4H) ; 3.96 (m, 4H) ; 2.87 (s,
3H) ; 1.27
(s, 9H).
Exemple 115
2-[(2-amino-2-oxoéthyl)[(2,6-dichlorophényl)sulfonyl]amino]-N-[2-[[[4-
2s (4,5-dihydro-iH-imidazol-2-yl)phényl]méthyl]méthylamino]-2-
oxoéthyl]acétamide, trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation CXXXI, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 90 %).
3o F=124°C
PREPARATION CX~OCII
2-[[(2,3-dichlorophényl)sulfonyl]amino]acétamide
En opérant de façon analogue à la préparation CVI, au départ de 2-
3s aminoacétamide, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 54 %).
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
84
F = 152 °C
PREPARATION C~OCITI
N-[2-[(2-amino-2-oxoéthyl)[(2,3-dichlorophényl)sulfonyl]amino]-
acétyl]glycine, éthyl ester
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon la préparation C?OCXII, on obtient le produit attendu sous forme d'un
solide
beige (rendement = 46 %).
F=208°C
PREPARATION CX~CIV
N-[2-[(2-amino-2-oxoéthyl)[(2,3-dichlorophényl)sulfonyl]amino]acétyl]-
glycine,
En opérant de façon analogue à la préparation L)OCV, au départ du composé
obtenu
selon la préparation CX?CXIII, on obtient le produit attendu sous forme d'un
solide
beige (rendement = 99 %).
F = 110 °C
PREPARATION CX)OCV
2o Acide 2-[4-[[[2-[[2-[(2-amino-2-oxoéthyl)[(2,3-dichlorophényl)
sulfo~:y~l]a:vina]acétyl]amino]acétyl]méthylamino]méthyl]phény!]-~,5-
dihydro-1H-imidazole-1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation VI, au départ des composés
obtenus
selon les préparations CX?OCIV et LXTII, on obtient le produit attendu sous
forme
2s d'un solide blanc (rendement = 64 %).
F = 118 °C
Exemule 116
2-[(2-amino-2-oxoéthyl)[(2,3-dichlorophényl)sulfonyl]amino]-N-[2-[[[4-
30 (4,5-dihydro-iH imidazol-2-yl)phényl]méthyl]méthylamino]-2-oxo-
éthyl]acétamide, trifluoroacétate
En opérant de façon analogue à l'exemple 1, au départ du composé obtenu selon
la
préparation C)OCXV, on obtient le produit attendu sous forme d'un solide blanc
(rendement = 71 %).
35 F = 122 °C
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
Les structures chimiques des composés selon l'invention décrits ci-dessus
sont résumées dans le tableau 1 dans le cas où R représente un groupe
phényléthyle, dans le tableau 2 dans le cas où R représente un atome
d'hydrogène
ou un groupe méthyle. et dans le tableau 3 dans le cas où le substituant X est
s difFérent d'un groupe méthyle.
TABLEAU I
cl o
H3C \ SO2~ /CHz NH ~"~ / tCH2 ) n-R
N ~ CHz
/ O R1
C1
Ex R,, R2 n Sel
-(CHZ)4-NHZ / \ ~NH 1 TFA
~
NHZ
2 -(CH~)3-NHz / \ ~NH 1 TFA
~
NHz
-(CHZ)3 N~ / \ N OH
~
NHZ
,N-O-COCH3
-(CHZ)3 N~ / \ C 1
~
NHZ
~ NH
-(CHZ)3 N, / / \ C 1
~
NH2
NH
g -(cH2)3 N\~ / \ c' 1 2HC1
NHz
7 =(CHZ)2 N, / / \ CN OH
N HZ
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
86
,N-O-COCH3
8 =(cH2)2 N~ / \ ~ 1
~
NHZ
,NH
-(cHZ)Z N~ / \ c 1
~
NHZ
1O -(cH2)2 N'~ / \ ~NH 1 2HCI
NHZ
N-OH
11 -(~Hz)4 N~ / \ ~ 1
NHZ
,N-O-COCH3
12 -(cHz)4 N~ / \ c 1
~
NHz
NH
13 -(cHZ>4 N~ / \ ~ 1
NHZ
NH
14 -(cH2)4 N~ / \ 1 2HC1
NHZ
,N-OH
15 -(CHa)4-N(CH3)z ~ \ c I 1
I ~
NHz
,N-O-COCH3
16 -(CHz)4-N(CH3)~ ~ \ 1
NHz
17 -(CHZ)a-N(CHs)z / \ NH
~
NHZ
18 -(CHZ)4-N(CH3)Z / \ NH 1 2HCI
NHz
19 -(CHZ)s-N(CH3)2 / \ CN-OH
NHz
20 -(CH~)3-N(CH3)Z / \ N-OH 1 2HCI
NHz
/ \ ,N-O-COCH3
C
~
NHZ
21 - CHI 3-N CH3 1
Z
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
87
,NH
22 -(CHz)s-N(CH3)z'/ \ ~NH
z
23 -(CHz)3-N(CH3)z/ ~ NH 1 HCI
2
~NHz
NH
24 -CHz~N-H / ~ c,' 1 2TFA
NHz
25 -(CHz)4-NHz --~N-H 1 2TFA
26 -(CHz)4-OH ~N-H 1 1TFA
27 -(CHz)4-NH-COCH3~N-H 1 1 TFA
2$ -(CHz)3 N~ / ~ CONH-CH3 1
29 -rcH2)3 N~ / ~ coNH-cH3 1 1HCI
N
3~ -~cHz)3 N~ ~ / NHz 1
31 -~cHz)3 N'J y / NH2 1 2HCI
32 - CHz 3-N CH3 -CHz-N CH3 z 2
z
33 -(CHz)3-N(CH3)z-CHz-N(CH3)z 2 2HCI
N
34 -(CHz)3-N(CHs)z~ / c,' ~ 1
N
N
35 -(CHz)3-N(CH3)z~ / c,' ~ 1 2HCI
N
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
88
N-OH
36 -CH3 ~ ~ c,'
NHZ
1
,N-O-COCH3
37 =CH3 ~ ~ 1
NHz
NH
38 -CH3 ~ ~ 1
NH2
39 -CH3 ~ ~ NH 1 1HCI
NHz
40 -CH3 \ ~ ~N~ 1
N
41 -CH3 ~ ~ N~ 1 1HCI
N
NH
42 H ~ ~ 1
NH2
N-O-COCH3
43 H ~ ~ c 1
'
NHZ
NH
44 H ~ ~ 1
NHz
45 H ~ ~ NH 1 1HCI
NHz
46 -CH2~ --~N-H 1 2TFA
N
H
47 -CHZ--( ,N-H --( ,N-H 1 2TFA
48 -cHz--CN-cH3 --CN-H 1 2TFA
4g -CHZ--( ,N-CZHS--( ,N-H 1 2TFA
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
89
50 -(~HZ)z~N-H --( ,N-H 1 2TFA
51 -(cHz)Z N~ N-H 1 2TFA
52 -(cHZ>2 N~ N-H 1 2TFA
53 -(cHz)Z N~ N-H 1 2TFA
54 -CH~-CH ~N-H 1 2TFA
3
55 -cHZ'~ --~N-H 1 2TFA
H3C
56 -(CHa)3 NHz N-H .
1 2TFA
57 ~~)3~(~~ ~N-H 1 2TFA
H3 /~
58 (cHz)2 N ~N-H 1 2TFA
59 -(cHZ>Z ~N-cH3 ~N-H 1 ~TFA
60 -cHz ~~~N --( N-H 1 2TFA
61 -(cHz)z N(CN3)z -CHZ-NHZ 3 2TFA
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
62 - t~~~ 3 N 3> -CHz-NHz 3 2TFA
z
63 -(cHz>z N~ -CHz-NHz 3 2TFA
64 -(cH2)z N~ -CHz-NHz 3 2TFA
65 -CHz-NHz 3 2TFA
-~~H )Z~N
2
H
66 N -CHz-NHz 3 2TFA
Ha
67 NcH3 _CHz_NHz 3 2TFA
112
68 -cH2--~~ -CHz-NHZ 3 2TFA
N
CH3
69 -(cHZ~2 N~ -CHz-NHz 3 2TFA
7p -(cHz~z ~N-CH3 -CHz-NHz 3 2TFA
71 -(cH2)2 \ ~N -CHz-NHz 3 2TFA
72 -(cNz)3 \ ~N -CHz-NHz 3 2TFA
73 -(cHz)3 \ ~ -CHz-NHz 3 2TFA
N
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
91
74 -(~Hz)3 N ~ -CHZ-NHa 3 2TFA
75 -(cH2)2 O--NH-C2H5-CHz-NHz 3 1TFA
76 -(cH2)3 O- NH-czHS-CHz_NHz 3 1TFA
77 -(cH2)4 O--NH-C2H5-CHZ-NHz 3 1TFA
-~cHZ~3 NHZ O-cH3-CHZ-NHZ 3 1TFA
%9 (CHz)4 NH--O-cH3
0 -CHZ-NHa 3 1TFA
gp -(cH2)3 NHz -CHI-NHz 3 2TFA
N
1. -(ONz) N~ ~ z \
2 '-(CHz) N~ ~ z \ N ~HC~
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
92
Tableau 2
Cl
H3C \ SOZ~N/CH2 NH\CH~N~ (CHZ ) n Rz
/ R O R1
Cl
Ex R Ri R2 n Sel
83 CH3 CH3 / \ ~ N, 1
, JN
84 CH3 CH3 / \ ~ N, 1 HCI
JN
85 H -(CH2)4 N~ .~ 2 ~ N 2
86 H -(CH2)4 N~ ~ ~ ~ , 2 2HCI
N
87 -CHI-CHI-0-CH3-(CHz)4 N~ ~ Z ~ , 2
N
88 -CH2-CHa-0-CH3-(CH2)4 N~ ~ 2 ~ ~ 2 2 HCI
N
89 -cxz-cx~ (CH2)4 N~ ~ Z ~ 1 2
\ ~ N
90 -N -(CH2)4 N~ ~~~ 2 3TFA
-cH2_~ \ 2 \ N
~
91 -CHI-CO-NH2 CH3 ~ ~ c'N~ 1 TFA
,,
N
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
93
Ex R R1 RZ n Sel
93 -CHZ-CH=CHz -(CH2)4 N~ ~ z ~
2
N
94 -CH2-CH=CHZ -(CH2)4 N
N 2 2HCI
2 N
96 CH3 -(CH2)4 N~ N
\ /
97 CH3 -(CH2)4 N~ 3 2HC1
/N
98 CH3 -(CH2)4 N~ o
\ /
101 -CHz-CO-NH-CH3CH3 / \ ~ N, 1 TFA
-~N
102 -CHZ-CH=CHI CH3 ~ ~ ~N~ 1
N
103 -CH2-CH=CHz CH3 ~ ~ ;N~ 1 HCI
N
104 CHI-CHz-O-CH3CH3 ~ ~ N~ 1
N V
105 CHI-CHZ-O-CH3CH3 ~ ~ c'N~ 1 HC!
~N
108 CH3 CH3 -~'N~ 3
z
109 CH3 CH3 -~-N~ 3 Fumarate
z
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
94
Tableau 3
Y
1
N/ II ~ ~ ~~ ~ TFA
H N
z O
Ex : X Y Z R
95 H H CI CH3
99 H H H CH3
100 CI CI CI CH3
106 CI H H CH3
107 H CI H CH3
112 H H CI
113 CI H H
114 ~ H H H
115 H H CI CH~CONHz
116 CI H H CHZCONHZ
Note : tous les composés cités dans le tableau 3 sont sous forme de sel avec
l'âcide
trifluoroacétique
Activité biologique
Les composés de la présente invention ont été évalués pour leur propriété
analgésique dans le test de douleur induite par le formaldéhyde chez la souris
(Shibata, M., Ohkubo, T., Takahashi, H. & R. Inoki. Modified formalin test:
characteristic biphasic pain response. Pain, 38, 347-352). En résumé, une
administration de formaldéhyde (0,92 % dans le sérum physiologique) est
effectuée
dans la patte arrière et la durée de léchage, qui reflète l'intensité de la
douleur, est
enregistrée de 0 à 5 min (lare phase) et de 15 à 30 min (2~de phase) après
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
l'injection. Le pourcentage d'inhibition de la seconde phase de léchage
induite par le
formaldéhyde est donné, pour quelques composés selon l'invention, dans le
tableau
suivant
Exemple Dose (mg/kg)Voie % d'inhibition
d'administrationde la
de phase de
lcha e
14 10 i.p. 93
82 10 i.p. 63
84 30 i.p; 57
25 30 s.c. 87
23 30 s.c. 98
24 10 s.c. 76
35 10 s.c. 92
s i.p. : intrapéritonéale
s.c. : sous-cutanée
Ces résultats témoignent d'une baisse très sensible de la douleur après
administration des composés.
io ~ui~e aux résultats de l'essai précédent, ies composés selon l'inve~ition
ont
été soumis à un test visant à démontrer leur mode d'action et mettant en jeu
le
récepteur B1 de la bradykinine.
Ce test utilise la veine ombilicale humaine et est réalisé selon le protocole
suivant
1s Des cordons ombilicaux humains de 15-25 cm de long sont récupérés juste
après la
délivrance et placés immédiatement dans un flacon contenant une solution de
Krebs
de composition (en mM): NaCI 119, KCI 4.7, KHaP04 1.18, MgS04 1.17, NaHC03 25,
CaCl2 2.5, Glucose 5.5, EDTA 0.026 puis stockés à 4°C.
Le cordon est disséqué sous solution de Krebs afin de dégager la veine
ombilicale.
2o La veine est nettoyée de tout tissu adhérent et coupée en petits anneaux de
3-4
mm de large. L'endothélium est enlevé précautionneusement par introduction
dans
la lumière du vaisseau d'un fin cathéter n°1, rendu légèrement abrasif.
Afin d'induire l'expression du récepteur Bl de la bradykinine , les segments
de veine
sont mis à incuber à 37°C dans une cuve de 25 ml pendant 16 heures dans
un
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
96
milieu de culture EMEM oxygénée par un mélange 95% OZ + 5% CO~ auquel on
ajoute des antibiotiques: pénicilline 10000 UI/ml et streptomycine 10000 UG /
ml.
Le lendemain, les anneaux de veine sont montés sur un support en acier
inoxydable, relié à un capteur isométrique et placés dans une cuve à organes
isolés
s de 8 ml thermostatée à 37 °C, contenant de la solution de Krebs
oxygénée par un
mélange 95 % O~ + 5 % COa .
Après une période de repos d'une heure pendant laquelle les anneaux sont
rincés 5
à 6 fois avec la solution de Krebs (maintenue à 37 °C pendant toute la
manipulation
et oxygénée par le mélange 95 % OZ + 5 % COa), la veine est soumise
1o progressivement à une tension de 1 g. Lorsque la tension est stable, après
45 min
environ, la solution de Krebs est remplacée par une solution hyperpotassique
(KPSS : à température de 37°C) de même composition, mais contenant du
KCI 125
mM et pas de NaCI.
Après une série de rinçages, repos et réajustement de la tension, la
contraction
Is maximale de chaque segment est déterminée par une nouvelle dépolarisation
avec
la solution de KPSS.
Après une nouvelle période de repos pendant laquelle la tension à 1g est
réajustée
constamment, les composés suivants sont ajoutés dans le bain d'organe isolé
Mépyramine (1 pM ), Atropine (1 pM ), Indométacine (3 ~rM), LNA (30 pM ),
2o Captopril (10 pM), DL-Thiorphan (1 pM ) et Nifédipine (0.1 pM ).
~0 ï~~inutes ûprès, la molécule à tester ou le solvant de la molécule est
ajouté dans
le bain d'organe isolé. Les molécules sont étudiées à 10 NM ; si une molécule
présente un degré d'activité suffisant, elle est étudiée à des concentrations
plus
faibles (ex :1 - 0.1 - 0.01 NM).
25 Après 15 minutes d'incubation, les segments de veine sont contractés par
l'ajout de
concentrations croissantes de des-Argl°-Kallidin (0.1 nM à 30000 nM)
dans la cuve.
Les EC5° (concentrations effectives d'agoniste requises pour produire
50% de la
réponse maximale obtenue avec le KPSS ) sont calculées par la méthode des
moindres carrés.
3o Le pKB = [-log KB] est obtenue à partir de l'équation
Kg= [A] / (concentration ratio-1)
où [A] est la concentration d'antagoniste et la (concentration ratio)
représente le
rapport entre l'ECS° en présence d'antagoniste, et l'ECS° en
l'absence d'antagoniste.
Selon ce test, les composés selon l'invention cités dans la description
présentent un
35 pKB compris entre 7 et 9.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
97
Les composés de la présente invention sont utiles pour le traitement de
diverses formes de douleur telles que l'hyperalgésie inflammatoire,
l'allodynie, la
douleur neuropathique associée, par exemple, au diabète, à des neuropathies
(constriction du nerf sciatique, lombalgies), à toute forme de traumatisme, à
une
s intervention chirurgicale (extraction dentaire, ablation des amygdales), à
une cystite
interstitielle, à une maladie inflammatoire du colon, à un cancer.
Par ailleurs, il a été vérifié que certains composés de la présente invention
réduisent significativement la migration de neutrophiles induite par injection
intra-
pleurale de carragénine chez la souris selon les modalités précédemment
décrites
(A.L.F. Sampaio, G.A. Rae, & M.G.M.O. Henriques. Participation of endogenous
endothelins in delayed eosinophil and neutrophil recruitment in mouse
pleurisy.
Intlamm. Res., 49, 170-176, 2000).
Ainsi, les composés de la présente invention peuvent-ils être utiles aussi
pour traiter toute pathologie associée à un recrutement de neutrophiles comme
par
~s exemple, le syndrome de détresse respiratoire aigu, le psoriasis, les
obstructions
pulmonaires chroniques, les maladies inflammatoires du colon, la polyarthrite
rhumatôide.
L'activité des composés selon l'invention, mise en évidence au cours des
tests biologiques, est significative de propriétés antalgiques et permet
d'envisager
leur utilisation en thérapeutique.
Selon !'invention, on préconise l'utilisation des composés définis par la
formule I, ainsi que de leurs sels avec des acides non toxiques, en tant que
principes actifs de médicaments destinés à un traitement chez les mammifères,
notamment chez l'homme, vis à vis de la douleur ou de certaines maladies
2s généralement caractérisées par une migration massive de neutrophiles.
Parmi les maladies qui peuvent être traitées au moyen d'une administration
d'une quantité thérapeutiquement efficace d'au moins l'un des composés de
formule
I, on peut citer les hyperalgésies inflammatoires, les douleurs
neuropathiques, les
douleurs associées à un traumatisme ou à un cancer, les maladies
inflammatoires
3o du côlon, la polyarthrite rhumatoïde, le psoriasis, les obstructions
pulmonaires
chroniques ou le syndrome de détresse respiratoire aiguë.
La dose de principe actif dépend du mode d'administration et du type de
pathologie ; elle est généralement comprise entre 0,05 et 10 mg/kg. En
fonction du
traitement envisagé, les composés de formule I ou leurs sels pourront être
associés
3s à d'autres principes actifs, et seront formulés avec des excipients
couramment
utilisés.
CA 02434124 2003-07-07
WO 02/053516 PCT/FR02/00033
98
Dans le but d'obtenir une action rapide, notamment lorsqu'il s'agit de traiter
une douleur, le mode d'administration du médicament se fera de préférence par
injection, par exemple par voie intramusculaire ou sous-cutanée.