Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
ANTIVIRAL PROTEASE INHIBITORS
Technical field
This invention relates to novel protease inhibitors and in particular to
inhibitors of the
aspartate protease possessed by certain retroviruses, notably HN. The
invention further
relates to the use of such protease inhibitors in the treatment of conditions
caused by
retroviruses and in the preparation of medicaments for this purpose. The
invention also
relates to novel synthesis methodology for the facile preparation of protease
inhibitors and
similar chemical structures.
Background of the Invention
Many biological processes are dependent upon the accurate enzymatic abscission
of
polypeptides at particular amino acid sequences. An example of such an
operation is the
post-translational processing of the gag and gag-pol gene products of the
human
immunodeficiency virus HIV to allow for the organisation of core structural
proteins and
release of viral enzymes. The enzyme responsible for this task, HIV protease,
is a virally
encoded homodimeric protease belonging to the aspartic protease family of
enzymes. The
human renin and pepsin enzymes also belong to this family. Inhibition of the
HIV protease
in cell culture prevents viral maturation and replication and thus this enzyme
represents an
attractive target for antiviral therapy against HIV in humans.
WO 94/13629 describes HIV protease inhibitors based on mannitol esters. Our
copending
application no. W098/45330 describes sugar based protease inhibitors based on
a mannitol
skeleton, but wherein the P1 groups are ethers. While the latter compounds are
fairly potent
and very easy to synthesise, the nature of the HUIV replicative process with
its extremely
error prone transcription and rapid generation of drug escape mutants means
that further
more potent compounds with DMPK properties amenable to good patient compliance
are
required in the HIV armamentarium.
Brief description of the invention
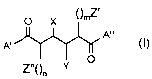
A first aspect of the invention provides novel compounds of the formula I:
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
2
O X ()mZ,
A"
A'
O
Y
wherein:
A' and A" are independently the same or different group of the formula II:
R"'
i ,
~N~Q~ / R
M
R"
II
wherein:
R' is H, CH3, C(CH3)z, -ORa, -N(Ra)z, -N(Ra)ORa or -DP
R"' is H or CH3; Ra is H, C,-C3 alkyl;
D is a bond, C,_3 alkylene, -C(=O)-, -S(O)- or -S(O)z- ;
P is an optionally substituted, mono or bicyclic carbo- or heterocycle;
R" is H, any of the sidechains found in the natural amino acids, C,-aalkyl,
carboxacetamide,
or a group (CHz)"DP;
M is a bond or -C(=O)N(R"')-;
Q is absent, a bond, -CH(OH)- or -CHz-;
or R" together with Q , M and R' define an optionally substituted S or 6
membered carbo- or
heterocyclic ring which is optionally fused with a further 5 or 6 membered
carbo- or
heterocyclic ring;
X is H, OH, OCH3;
Y is H, OH, OCH3,
Z' and Z" are independently an optionally substituted mono-or bicyclic carbo-
or
heterocycle;
() is a methylene group;
n and m are independently 0, 1 or 2;
and pharmaceutically acceptable salts and prodrugs thereof.
Compounds of the formula I are active inhibitors of aspartyl proteases, such
as those from
HIV. Further aspects of the invention thus provide:
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
- a pharmaceutical formulation comprising a compound of the formula I in
admixture with a pharmaceutical acceptable carrier or diluent;
- the use of a compound of the formula I in the manufacture of a medicament
for the
prophylaxis or treatment of conditions, such as AIDS caused by retroviruses,
such
as HIV; and
- a method for treating conditions caused by retroviruses, especially AIDS in
humans, comprising administering a compound of formula I to a subject
afflicted
with said condition.
The compounds have a relatively low molecular weight and should therefore
provide good
oral absorption properties in mammals.
For ease of synthesis it is generally preferred that the terminal amines A'
and A" are
identical. However, although the target enzyme is a symmetric dimer, thus
implying a tight
interaction with symmetric compounds, it can in some circumstances be
advantageous for
resistance or pharmacokinetic reasons etc to have asymmetric terminal amines.
Where is it is
desired to have an asymmetric compound, that is where the A' and A" groups
differ, it will
generally be most convenient to add the respective A' and A" groups
sequentially.
This may be achieved by the use of different protecting/leaving groups on the
diepoxydicarboxylic acid in conjunction with manipulation of the reagent
concentrations,
reaction conditions, speed of addition etc to provide a monoprotected,
monoamidated
diepoxycarboxylic acid followed by displacement of the remaining
protecting/leaving group
with the second amine.
Alternatively the differential terminal amination can be achieved with a solid
phase
synthesis where the unprotected or partially protected diepoxydicarboxy acid
is secured to a
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
4
solid phase substrate, such as polymer beads of which many are known in solid
phase
chemistry, for instance Merrifield resin, in conjunction with an appropriate
linker structure.
Immobilization of the diepoxydicarboxylic acid in this fashion will only allow
amination of
one carboxy group. Cleavage from the resin/linker releases the other carboxy
terminal
which is subsequently amidated with the second terminal amine using
conventional peptide
chemistry.
Preparation of compounds of Formula I in which X is hydrogen can be carried
out by
deoxygenation analogously to Examples 2 and 26 of WO 9845330. Preferably X and
Y are
not both H.
A preferred stereochemistry is the 2R, 3R, 4R, SR form, the synthesis of which
is shown in
the examples. A further preferred stereochemistry is
O x ()mZ'
A"
A'
O
Z~~~)m Y
which is readily accessible:
Z~ )n
O
metathesis reaction
/ 'A A ~ A
Z~ )n Z~ ) O
n
cis hydroxylation O OH Z~ ~n
A
A
H O
Z~ )n
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
Carbocyclic groups for R' as -DP and/or Z'/Z" and/or the optional substituents
thereto may
be saturated, unsaturated or aromatic and include monocyclic rings such as
phenyl,
cyclohexenyl, cyclopentenyl, cyclohexanyl, cyclopentanyl, or bicyclic rings
such as indanyl,
napthyl and the like.
Heterocyclic groups for R' as -DP and/or Z'/Z" and/or the optional
substituents thereto may
be saturated, unsaturated or aromatic and have 1 to 4 hetero atoms including
monocyclic
rings such as furyl, thienyl, pyranyl, pyrrolyl, pyrrolinyl, pyrrolidinyl,
pyrazolyl,
pyrazolinyl, pyrazolidinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyridyl,
piperidinyl,
pyrazinyl, piperazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolidinyl,
isoxazolyl,
isoxazolidinyl, morpholinyl, thiazolyl, thiazolidinyl, isothiazolyl,
isothiazolidinyl, and the
like or bicyclic rings especially of the above fused to a phenyl ring such as
indolyl,
quinolinyl, isoquinolinyl, benzimidazolyl, benzothiazolyl, benzoxazolyl,
benzothienyl etc.
The carbo or heterocyclic ring may be bonded via a carbon or via a hetero
atom, typically a
nitrogen atom, such as N-piperidyl, N-morpholinyl etc.
Preferred embodiments of Formula II for the A'/A" groups of the compounds of
the
invention include those of the formula IIa or IIe:
0
b.J~ p ~(-~ R,
R"
IIa
where n is 1 or 2 and R' is alkyloxy, preferably methyloxy, or those where n
is 0 and R' is
methyl.
Other preferred groups of formula II include IIb below
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
6
OH
/N N'CH
3
- O
R"
Ilb
An alternative preferred configuration for the A'/A" groups of the compounds
of the
invention includes groups of the formula IIc:
/N~/Q\ .
R
R~~ IIc
where Q is a bond, methylene or-C(OH)- and R' is -ORa, -N(Ra) Z, -NRa ORa,
where Ra is H
or C~-C3 alkyl, or a carbo- or heterocyclic group including N-piperidine, N-
morpholine, N-
piperazine, pyrrolyl, imidazolyl, pyrazolyl, pyridyl, pyrimidyl, pyrazinyl etc
.
A favoured subset of compounds within formula IIc has the formula IId:
/ N ~\ Ra
Rd Ild
where Rd is hydrogen or methyl (that is a valyl or isoleucyl side chain) and
Re is
N
or
N
where X is methylene, O, S, S=O, S(=O)z or NH or Re is -N(CH3)Z, -NHOH, -
NHOMe, -
NHOEt, -NMeOH, -NMeOMe etc.
In each of formulae IIa, IIb and IIc, R" is hydrogen, methyl, ethyl,
isopropyl, cycloalkyl such
as cyclopropyl, cyclobutyl or cyclohexyl, cycloalkenyl, benzyl,
carboxacetamide or 4-
imidazolylmethy, any of which may be substituted as defined above. Preferred
R" groups
include the side chains found in the natural amino acids, especially those of
leucine,
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
7
asparagine, histidine or proline. The most preferred R" groups for formula
IIa, IIb, IIc and
IId are the tertiary leucyl, isoleucyl and especially the valyl side chain.
R' will vary depending on the nature of Q and/or M, if present, and may for
instance be
selected from hydrogen, methyl, ethyl, isopropyl, Re as defined above,
valinol, a heterocycle
such as pyridyl, thiazole, oxazole, imidazole, N-piperidine, N-morpholine, N-
piperazine,
pyrrolyl, imidazolyl, pyrazolyl, pyrimidyl, pyrazinyl, any of which R' groups
may be
substituted as defined for Z'/Z" below.
Further favoured A'/A" groups include those of formula II where R", Q, M and
R' together
define an optionally substituted 5 or 6 membered carbo- or heterocylic ring. A
preferred
group within this definition include groups within formula III:
R... R2 R1
R6
N ~~,.
III
where
R"' is as defined above,
R~ is H, NR4R4, C(=O)R3, CR3R4 or a monocyclic, optionally substituted carbo-
or
heterocycle;
RZ is OH, or together with R' is =O, or if R' is NR4R4, then Rz may be H;
R3 is H, halo, C,-C3 alkyl, ORS, NR4R4;
R4 is H, C1-C3 alkyl;
RS is H or a pharmaceutically acceptable ester;
R6 is OH, NHZ, carbamoyl or carboxy;
R' is hydrogen, C,-C4 straight or branched alkyl or together with the adjacent
carbon atoms
forms a fused phenyl or heteroaromatic ring;
Preferred groups of formula III include aminoindanol and 1-amino-azaindan-2-
ol, that is
moieties of the formulae:
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
OH OH
~/ H ~/ H
or
\ / \ /N
Optional substitutents for the carbo- or heterocyclic moiety of Z'/Z" or A'/A"
include one to
three substituents such as halo, amino, mercapto, oxo, vitro, NHC,-C6 alkyl,
N(C~-C6 alkyl)z,
C1-C6 alkyl, C,-C6 alkenyl, C~-C6 alkynyl, C,-C6 alkanoyl, C,-C6 alkoxy,
thioC,-C6 alkyl,
thioC,-C6 alkoxy, hydroxy, hydroxyC,-C6 alkyl, haloC,-C6 alkyl, aminoC,-C6
alkyl, C,-C6
alkyl, cyano, carboxyl, carbalkoxy, carboxamide, carbamoyl, sulfonylamide,
benzyloxy,
morpholyl-C,-C6 alkyloxy, a monocyclic carbo- or heterocycle, as defined
above, a carbo- or
heterocyclic group spaced by alkyl, such as C,_3 alkylaryl, etc.
The preferred definitions for -()~,Z' and -()~, Z" include benzyl and
especially phenethyl
each unsubstituted or substituted with 1, 2 or 3 substituents, especially 1
selected from
fluoro, chloro, hydroxy, amino, -NH(C~_6 alkyl), -N(C~_6 alkyl)z, -NPh(C~_6
alkyl), -NHPh,
methoxy, cyano, hydroxymethyl, aminomethyl, alkylsulfonyl, carbamoyl,
morpholinethoxy,
benzyloxy, benzylamide etc. Other possibilities exhibiting the great freedom
in this area are
shown in WO 98/45330. It will be apparent that the substituent to Z' and/or Z"
may
comprise a ring structure (which substituent ring structure is itself
substituted as defined
herein) such as phenyl or a S or 6 membered heterocycle containing one or two
hetero atoms
such as thiophene, pyridine etc. The preparation of useful heterocyclic
substituents for Z'
and Z" phenyl are described in Tetrahedron Letters 1997 6359-6359-6367 and J
Org Chem
62 (1997) 1264 and 6066, including N-morpholine, N-piperidine, N-piperazine,
N'-methyl-
N-piperazine, N-pyrrolidone, N-pyrrolidine and the like.
Such substituents may be in the meta but especially the ortho or para
positions of Z'/Z",
with small groups such as fluoro being favoured for the ortho and meta and
with extensive
freedom for larger groups in the para such as (optionally substituted) cyclic
substituents,
including the N-bonded rings in the immediately preceding paragraph. The whole
Z' and Z"
group or their respective carbo-or heterocyclic moiety may be different but
for ease of
synthesis it is convenient if they are the same.
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
9
Appropriate pharmaceutically acceptable salts, both for A'/A" as a free acid
or for other
charged groups along the compound of formula I include salts of organic
carboxylic acids
such as acetic, lactic, gluconic, citric, tartaric, malefic, malic,
pantothenic, isethionic, oxalic,
lactobionic, and succinic acids, organic sulfonic acids such as
methanesulfonic acid,
ethanesulfonic acid, benzenesulfonic acid, p-chlorobenzenesulfonic acid and p-
toluenesulfonic acid; and inorganic acids such as hydrochloric, hydroiodic,
sulfuric,
phosphoric and sulfamic acids.
Prodrugs of the invention are derivatives that release a compound of formula I
in vivo,
generally by hydrolysis or other metabolic interaction in the intestine, liver
or plasma.
Typical prodrugs are esters formed on free hydroxy groups in the compounds.
Appropriate pharmaceutically acceptable esters include C,-Czz fatty acid
esters, where the
fatty acid is unsaturated, monounsaturated or multiply unsaturated. Saturated
fatty acid
esters include short chains such as acetyl or butyryl or long chain such as
stearoyl.
Unsaturated fatty acid esters are preferably in the c~-9 series, such as
palmitoleic or linolenic
esters. Other esters include C~-C6 alkylaryl esters such as benzyl or
methylpyridyl or esters
of phosphoric acid, such as monophosphate. Alternative esters include the
corresponding
fatty acid or alkylaryl carbonate, carbamate or sulphonic esters.
Particularly favoured prodrugs include those described in W099/41275,
especially when A'
and A" comprise an hydroxy function such as aminoindanol.
Intermediate compounds of formula III can be prepared by the following
reaction scheme:
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
OH Y OH Y O
HZN ."" 1 1 -CN
N-protection , N ~.." N ~,." H~
X Oxidation X
HO O . N
s CN HO N ~ ~NH
,N."" R Ho
H R=NH2 ~ ri
X ydrolysis N"" R=NHA BuaSnN3 N ""
R=OH
O
N "" Allylmagnesium- y HO I Y HO OH
X bromide 1 1
N .,", /' 1. 03 N .,",
X --~ X' -.
2. reduction
HO F HO NHz HO H
N."" N."" N..",
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
11
OOR
CN
/ COOR / \ Aminohydroxylation
OX. _
N
HO COOR Hp CN
Fi N ~~ Fi N '~,
or
R
N" CISi(CH3)3 X ~ "" ~ E N"" E
X
1-amino-azaindan-2-of P-2 filling groups can be prepared analogously to J Med
Chem 1991
1228-1230:
Cr03 O
H2S04/AcOH / POC13
w ~ w ~ w
N N N
NH2
~~~~~ OH
N
In treating conditions caused by retroviruses, the compounds of formula I are
preferably
administered in an amount to achieve a plasma level of around 10 to 1000 nM
and more
preferably 100 to 500 nM. This corresponds to a dosage rate, depending on the
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
12
bioavailability of the formulation of the order 0.001 to 100 mg/kg/day,
preferably 10 to 50
mg/kg/day.
In keeping with the usual practice with HIV inhibitors it is advantageous to
co-administer
one to three additional antivirals, such as AZT, ddI, ddC, D4T, ritonavir,
saquinavir,
indinavir, nelfinavir, efavirenz, delavirdine, nevirapine, trovirdine, PFA,
H2G etc. The
molar ratio for such co-administered antivirals will generally be chosen to
reflect the
respective ECso performances of the antiviral. Molar ratios of 25:1 to 1:25,
relative to the
compound of formula I will often be convenient.
While it is possible for the active agent to be administered alone, it is
preferable to present it
as part of a pharmaceutical formulation. Such a formulation will comprise the
above defined
active agent together with one or more acceptable carriers and optionally
other therapeutic
ingredients. The carriers) must be acceptable in the sense of being compatible
with the
other ingredients of the formulation and not deleterious to the recipient.
The formulations include those suitable for oral, rectal, nasal, topical
(including buccal and
sublingual), vaginal or parenteral (including subcutaneous, intramuscular,
intravenous and
intradermal) administration. The formulations may conveniently be presented in
unit dosage
form, e.g. tablets and sustained release capsules, and may be prepared by any
methods well
known in the art of pharmacy.
Such methods include the step of bringing into association the above defined
active agent
with the carrier. In general, the formulations are prepared by uniformly and
intimately
bringing into association the active agent with liquid carriers or finely
divided solid carriers
or both, and then if necessary shaping the product.
Formulations for oral administration in the present invention may be presented
as discrete
units such as capsules, cachets or tablets each containing a predetermined
amount of the
active agent; as a powder or granules; as a solution or a suspension of the
active agent in an
aqueous liquid or a non-aqueous liquid; or as an oil-in-water liquid emulsion
or a water in
oil liquid emulsion and as a bolus etc.
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
13
With regard to compositions for oral administration (e.g. tablets and
capsules), the term
suitable carrier includes vehicles such as common excipients e.g. binding
agents, for
example syrup, acacia, gelatin, sorbitol, tragacanth, polyvinylpyrrolidone
(Povidone),
methylcellulose, ethylcellulose, sodium carboxymethylcellulose,
hydroxypropylmethylcellulose, sucrose and starch; fillers and carriers, for
example corn
starch, gelatin, lactose, sucrose, microcrystalline cellulose, kaolin,
mannitol, dicalcium
phosphate, sodium chloride and alginic acid; and lubricants such as magnesium
stearate and
other metallic stearates, stearic acid, silicone fluid, talc waxes, oils and
colloidal silica.
Flavouring agents such as peppermint, oil of wintergreen, cherry flavouring or
the like can
also be used. It may be desirable to add a colouring agent to make the dosage
form readily
identifiable. Tablets may also be coated by methods well known in the art.
A tablet may be made by compression or moulding, optionally with one or more
accessory
ingredients. Compressed tablets may be prepared by compressing in a suitable
machine the
active agent in a free flowing form such as a powder or granules, optionally
mixed with a
binder, lubricant, inert diluent, preservative, surface-active or dispersing
agent. Moulded
tablets may be made by moulding in a suitable machine a mixture of the
powdered
compound moistened with an inert liquid diluent. The tablets may be optionally
be coated or
scored and may be formulated so as to provide slow or controlled release of
the active agent.
Formulations suitable for topical administration include lozenges comprising
the active
agent in a flavoured base, usually sucrose and acacia or tragacanth; pastilles
comprising the
active agent in an inert base such as gelatin and glycerin, or sucrose and
acacia; and
mouthwashes comprising the active agent in a suitable liquid carrier.
Formulations suitable for topical administration to the skin may be presented
as ointments,
creams, gels, and pastes comprising the active agent and a pharmaceutically
active carrier.
An exemplary topical delivery system is a transdermal patch containing the
active agent.
Formulations for rectal or vaginal administration may be presented as a
suppository or
pessary with a suitable base comprising, for example, cocoa butter or a
salicylate. Other
vaginal preparations can be presented as tampons, creams, gels, pastes, foams
or spray
formulations containing, in addition to the active agent, such carriers as are
known in the art
to be appropriate.
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
14
Formulations suitable for nasal administration wherein the carrier is a solid
include a coarse
powder having a particle size, for example, in the range 20 to S00 microns
which is
administered in the manner in which snuff is taken, i.e. by rapid inhalation
from a container
of the powder held up close to the nose. Suitable formulations wherein the
carrier is a liquid
for administration, for example, as a nasal spray or as nasal drops, include
aqueous or oily
solutions of the active agent.
Formulations suitable for parenteral administration include aqueous and non-
aqueous sterile
injection solutions which may contain antioxidants, buffers, bacteriostats and
solutes which
render the formulation isotonic with the blood of the intended recipient; and
aqueous and
non-aqueous sterile suspensions which may include suspending agents and
thickening
agents. The formulations may be presented in unit-dose or multi-dose
containers, for
example sealed ampoules and vials, and may be stored in freeze-dried
(lyophilized)
condition requiring only the addition of the sterile liquid carrier, for
example water for
injection, immediately prior to use. Extemporaneous injection solutions and
suspensions
may be prepared from sterile powders, granules, and tablets of the kind
previously
described.
Detailed Description of Embodiments of the Invention.
The invention will now be further illustrated by reference to the following
non-limiting
Examples, Figures 1 and 2 which depict synthesis schemes for preparing
intermediates, and
Figure 3 which is a synthesis scheme for compounds of the invention.
General
Reference numerals refer to the compounds in Figures 1-3.
All glassware was dried over an open flame before use in connection with an
inert
atmosphere. Concentrations were performed under reduced pressure at <40
°C (bath
temperature). Thin layer chromatography was performed using silica gel 60 F-
254 plates
with detection by UV and charring with 8% sulphuric acid and/or ninhydrin in
ethanol.
Silica gel (0.040-0.063 mm) was used for column chromatography. Me4Si (0.0
ppm) was
used as an internal standard in IH NMR and Me4Si or CDC13 (77.0 ppm) were used
in'3C
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
NMR. Melting points are uncorrected. Unless stated otherwise, all materials
were obtained
from commercial suppliers and used without further purification. Yields are
not optimised.
5 Intermediates
(4R,SS~-Indano[1,2-d]-oxazolidin-2-one (1~
(1R,2,S~-(+)-cis-1-Amino-2-indanol (18.4 g, 0.125 mol) was dissolved in CHZCIZ
(1350 mL)
under a nitrogen atmosphere. The temperature was lowered to 0 °C before
addition of
10 triphosgene (14.8 g, 49.9 mmol, 0.4 equiv.) and NEt3 (35.0 mL, 0.251 mol,
2.0 equiv.).
Stirring was continued at 0 °C for 1.5 h, when TLC (Rf0.39, CHCl3-MeOH
15:1 ) showed
completion of the reaction. The mixture was concentrated to 675 mL and washed
with
NH4C1 (lx) and water (2x) and the combined aqueous layers were extracted with
EtOAc
(2x). The organic layers were combined with the mixture from above, dried
(MgS04) and
15 concentrated to give 1 (20.9 g, 0.119 mol, 95%) as white crystals: [D]D +71
(c 0.65, EtOAc);
mp 200-201 °C;'H NMR (300 MHz, CDC13 and CD30D) 0 3.32 (dd, 1 H, J=
1.76, 18.0
Hz), 3.43 (dd, 1 H, J= 6.15, 18.0), 3.74 (s, 1 H), 5.18 (d, 1 H, J= 7.47 Hz)
and 5.42 (ddd, 1
H, J= 1.76, 6.15, 7.47);'3C NMR (67 MHz, CDC13 and CD30D) ~ 38.5, 61.0, 80.5,
124.6,
125.3, 127.6, 129.1, 139.5, 140.1 and 159.9. Anal Calcd for C,oH9N02: C, 68.6;
H, 5.2; N,
8Ø Found: C, 68.4; H, 5.2; N, 8Ø
N(1-Oxo-4-phen l~tXl)-(4R,5S~-indano[l,2-d]-oxazolidin-2-one (2).
To a stirred solution of 4-phenylbutyric acid (35.0 g, 0.214 mol) and SOCIz
(98 mL, 1.34
mol, 6.3 equiv.) was added pyridine (0.64 mL, 7.9 mmol, 0.04 equiv.). The
solution was
stirred at room temperature for 30 min and at 40 °C for an additional 1
h before it was
concentrated to give 4-phenylbutanoyl chloride (38.3 g, 0.210 mol, 98%) as a
yellow oil,
which was used in the next step without further purification: 'H NMR (300 MHz,
CDC13) ~
2.03(dt,2H,J=7.6,7.3Hz),2.67(t,2H,J=7.6Hz),2.88(t,2H,J=7.3Hz)and7.15-
7.32 (m, 5 H);'3C NMR (67 MHz, CDC13) ~ 26.4, 34.0, 46.1, 126.2, 128.3, 128.4,
140.2
and 173.4.
NaH (1.61 g, 67.1 mmol, 1.1 equiv.) was suspended in DMF (150 mL) under an
argon
atmosphere and the temperature was lowered to 0 °C. A dropping funnel
was charged with
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
16
the chiral auxiliary 1 (10.7 g, 61.2 mmol) in DMF (90 mL), a second dropping
funnel was
charged with 4-phenylbutanoyl chloride (13.4 g, 73.4 mmol, 1.2 equiv.) in DMF
(90 mL)
and the two solutions were then added simultaneously to the stirred NaH
suspension during
30 min. Stirring was continued at 0 °C for an additional 3 h (Rf0.56,
toluene-EtOAc 10:1),
before slow addition of MeOH and H20. The layers were separated after dilution
with
toluene and HzO. The organic layer was washed with Hz0 (2x), followed by
extraction of
the combined aqueous layers with toluene (lx). Finally, the combined organic
layers were
dried (NaZS04) and concentrated. Purification by column chromatography
(toluene; toluene-
EtOAc 40:1 ) gave 2 (14.3 g, 44.5 mmol, 73%) as a colourless syrup:
[a]2°D -209 (c 0.948,
CHC13);'H NMR (300 MHz, CDCl3) 0 1.98-2.08 (m, 2 H), 2.70 (t, 2 H, J= 7.69),
3.00
(ddd, 2 H, J= 1.76, 7.47, 9.23 Hz), 3.37 (d, 2 H, J= 3.52 Hz), 5.26 (ddd, 1 H,
J= 3.08, 4.40,
7.47 Hz), 5.91 (d, 1 H, J= 7.03 Hz), 7.16-7.37 (m, 8 H) and 7.62 (d, 1 H, J=
7.03 Hz);'3C
NMR (67 MHz, CDC13) 0 26.0, 34.8, 35.2, 38.0, 63.0, 78.0, 125.2, 126.0, 127.2,
128.2,
128.4, 128.5, 129.8, 139.2, 139.5, 141.6, 153.0 and 173.4. Anal Calcd for
CZ°H19NO3: C,
74.8; H, 6.0; N, 4.4. Found: C, 74.9; H, 6.0; N, 4.3.
N [(2,51-2-(H droxymeth~)-1-oxo-4-phenylbut~]~4R.5 -indano[1.2-dJ-oxazolidin-2-
one
TiCl4 (3.6 mL, 32.8 mmol, 1.05 equiv.) was added to a stirred solution of
compound 2 (10.0
g, 31.1 mmol) in CHzCIz (525 mL), under an argon atmosphere, with the
temperature kept at
0 °C. A yellow precipitate was formed within 30 min, DIEA (5.5 mL, 31.9
mmol, 1.0 equiv.)
was added (the colour changed from yellow to red), and stirring was continued
at 0 °C for 1
h. Trioxane (2.08 g, 23.1 mmol, 0.74 equiv.) and TiCl4 (4.5 mL, 40.9 mmol, 1.3
equiv.) were
added, and stirring was continued until TLC (Rf0.15, toluene-EtOAc 10:1)
showed complete
reaction (3 h). During this time the colour of the reaction changed from dark
red to light
brown. Neutralisation (pH=7) of the cold reaction was performed by adding sat.
aqueous
NaHC03 (500 mL). The layers were separated and the aqueous layer was extracted
with
CH2ClZ (4x). The combined organic layers were dried (Na2S04) and concentrated.
The crude
product was immediately purified using column chromatography (toluene; toluene-
EtOAc
10:1) to give alcohol 3 (8.1 g, 23.0 mmol, 74%) as a colourless syrup, which
solidified upon
standing: [0]z°p -174 (c 1.19, CHC13);'H NMR (300 MHz, CDC13) ~ 1.58
(s, 1 H), 1.90-
2.02 (m, 1 H), 2.15-2.24 (m, 1 H), 2.63-2.84 (m, 2 H), 3.36 (d, 2 H, J= 3.52
Hz), 3.84 (m, 2
H), 3.94-4.02 (m, 1 H), 5.14-5.19 (m, I H), 5.72 (d, I H, J= 7.03 Hz), 7.18-
7.36 (m, 8 H)
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
17
and 7.58 (d, 1 H, J= 7.47 Hz);'3C NMR (67 MHz, CDC13) X29.9, 33.8, 37.7, 45.2,
63.2,
63.8, 78.2, 125.1, 126.0, 127.1, 128.2, 128.3, 128.6, 129.8, 138.9, 139.3,
141.2, 152.9 and
175.9. Anal Calcd for Cz,H2,N04: C, 71.8; H, 6.0; N, 4Ø Found: C, 71.6; H,
6.1; N, 4Ø
N [f2SL(Benzyloxymeth~)-1-oxo-4-phen r~ lbut~44R.5S~-indanof 1,2-dJ-oxazolidin-
2-one
To a stirred solution of alcohol 3 (18.2 g, 51.7 mmol) and benzyl 2,2,2-
trichloroacetimidate
(11.6 mL, 62.4 mmol, 1.2 equiv.) in 1,4-dioxane (900 mL) was added TMS-OTf
(0.9 mL,
4.96 mmol, 0.1 equiv.) dropwise under argon. Within 1 h the colour changed to
brown-
yellow and TLC (Rf0.47, toluene-EtOAc 10:1) showed completion of the reaction.
The
reaction mixture was filtered through a pad of SiOz/NaHC03/Si02 in a glass
filter-funnel
and concentrated. Purification by column chromatography (toluene-EtOAc 40:1 )
gave the
benzylated compound 4 (20.2 g, 45.8 mmol, 89%) as a colourless syrup:
[~]2°D -117 (c 1.16,
CHCI3);'H NMR (300 MHz, CDC13) ~ 1.87-1.98 (m, 1H), 2.15-2.27 (m, 1 H), 2.61-
2.78
(m, 2 H), 3.35 (d, 2 H, J= 3.08 Hz), 3.64 (dd, 1 H, J= 5.27, 9.22 Hz), 3.78
(dd, 1 H, J=
7.03, 9.22 Hz) 4.23 (m, 1 H), 4.41 (s, 2 H), 5.13-5.17 (m, 1 H), 5.78 (d, 1 H,
J= 7.03 Hz),
7.11-7.47 (m, 14 H) and 7.55 (d, 1 H, J= 7.91 Hz);'3C NMR (67 MHz, CDCl3) ~
30.3,
33.7, 37.8, 43.2, 63.0, 71.2, 72.9, 77.8, 125.0, 126.0, 127.2, 127.3, 128.0,
128.2, 128.3,
128.6, 129.7, 138.1, 139.1, 139.3, 141.4, 152.6 and 174.8. Anal Calcd for
Cz8H2~N04: C,
76.2; H, 6.2; N, 3.2. Found: C, 76.2; H, 6.2; N, 3.1.
(2R -L2-(Benzyloxymethyl)-4-phenyl-1-butanol (5).
The benzylated compound 4 (3.85 g, 8.72 mmol) was dissolved in THF (90 mL)
under an
argon atmosphere, the temperature was lowered to ~0 °C and LAH (3.38 g,
89.0 mmol, 10
equiv.) was added. Stirring was continued at-60 °C for 30 min and at 0
°C for 1 h (Rf0.2,
toluene-EtOAc 9:1). The reaction mixture was acidified by the addition of cold
0.6 M HCI.
The suspension was extracted with EtzO (5x) and the combined organic layers
were dried
(MgS04) and concentrated. Purification by column chromatography (toluene-EtOAc
3:1)
gave alcohol 5 (1.96 g, 7.25 mmol, 83%) as a colourless oil: [~]Z°D +14
(c 0.90, CHCl3);'H
NMR (400 MHz, CDC13) ~ 1.57-1.71 (m, 2 H), 1.86-1.93 (m, 1 H), 2.49 (s, 1 H),
2.57-2.67
(m, 2 H), 3.50-3.53 (m, 1 H), 3.62-3.69 (m, 2 H), 3.74-3.78 (m, 1 H), 4.51 (q,
2 H, J= 12.1
and 17.6 Hz) and 7.15-7.36 (m, 10 H);'3C NMR (67 MHz, CDC13) 0 29.8, 33.3,
40.1, 65.5,
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
18
73.4, 125.8, 127.6, 127.7, 128.3, 128.4, 138.0 and 142.1. Anal Calcd for
C,gHzz02: C, 80.0;
H, 8.2. Found: C, 80.1; H, 8.2.
(2.5~-2-[(Benzyloxymeth~l-4-phen l~but~] (2R)-3,3,3-Trifluoro-2-methox ~-~2-
phen ~~1-
propanoate.
Moshers ester of alcohol 5 was prepared according to the method described by
Wipf and
Fritch, using 26 mg (0.096 mmol) of 5 in 1.4 ml of CHZCIz together with 580 ~L
of pyridine
and 210 ~ L of (S~-(+)-0-methoxy-0-(trifluoromethyl)phenylacetic acid chloride
with
stirring during 40 min. Purification by column chromatography (toluene-EtOAc
40:1) gave
the ester (38 mg, 0.078 mmol, 81%) as an oil:'3C NMR (75 MHz, CDCl3) ~ 29.9,
33.1,
37.8, 55.4, 66.4, 69.6, 73.2, 121.3, 125.2, 125.8, 127.2, 127.4, 127.5, 128.2,
128.3, 129.5,
132.2, 138.1, 141.5 and 166.4.
2-[(Benzylox~yl)-4-phenylbut~]~2R)-3,3,3-Trifluoro-2-methox ~-~2-phen ~Ll-
propanoate.
Moshers ester of racemic alcohol 5 (ref) was prepared as described above,
using 20 mg
(0.074 mmol) of racemic 5, which gave the racemic ester (28 mg, 0.058 mmol,
78%) as an
oil: '3C NMR (75 MHz, CDCl3) 0 29.8, 29.9, 33.0, 37.8 (2 C), 55.4, 66.2, 66.4,
69.6, 69.7,
73.2, 121.4, 125.2, 125.8, 127.2, 127.4 (2 C), 127.5, 128.2, 128.3, 129.5,
138.1 and 141.5.
(2S1-2-(BenzXlox~yl)-4-phenylbutanal (6).
DMSO (4.03 mL, 56.7 mmol, 2.2 equiv.) in CHZCIz (11.3 mL) was added to a
solution of
oxalyl chloride (2.43 mL, 28.3 mmol, 1.1 equiv.) in CHzCIz (64 mL) at-70
°C under argon.
After stirring for 5 min, alcohol 5 (6.97 g, 25.8 mmol) dissolved in CHZCIz
(26 mL) was
added dropwise (20 min). Stirring was continued for an additional 20 min. NEt3
(7.18 mL,
54.0 mmol, 2.1 equiv.) was added, and the reaction mixture was stirred during
5 min at-70
°C, and subsequently during 1 h while the reaction mixture slowly was
allowed to attain
room temperature (Rf0.35, pentane-EtOAc 15:1 and Rf0.57, toluene-EtOAc 9:1).
Dilution
with HzO, extraction of the aqueous layer with CHZCIz (3x), drying (MgS04) and
concentration gave the crude aldehyde 6. Purification by column chromatography
(pentane-
EtOAc 25:1; 15:1 and 10:1) gave 6 (5.64 g, 21.0 mmol, 82%) as a transparent
oil, which was
used immediately in the next step: [0]2°D +17 (c 1.1, CHC13);'H NMR
(300 MHz, CDC13)
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
19
D 1.72-1.86 (m, 1 H), 1.97-2.12 (m, 1 H), 2.48-2.58 (m, 1 H), 2.62 (t, 2 H, J=
7.97 Hz),
3.67 (d, 2 H, J= 5.50 Hz), 4.47 (d, 2 H, J= 1.65 Hz), 7.12-7.40 (m, 10 H) and
9.68 (s, 1 H);
isC NMR (75 MHz, CDC13) 0 27.3, 32.9, 51.3, 68.3, 73.1, 125.8, 127.4, 127.5,
128.1, 128.2
(2 C), 137.6, 140.9 and 203.1. Anal Calcd for Cl8HZO0z ~'/4 HzO: C, 79.2; H,
7.6. Found: C,
79.4; H, 7.6.
Reduction of aldehyde 6 with borane-dimethylsulflde complex BMS~
Aldehyde 6 was reduced with BMS according to the method described by Brown et
al.,
using 64 mg (0.238 mmol) of 6 in 2.0 ml of diethyl ether together with 60 OL
of BMS with
stirring at 0 °C during 30 min, followed by stirring at room
temperature during 2.5 h.
Purification by column chromatography (toluene-EtOAc 3:1) gave alcohol 5 (50
mg, 0.185
mmol, 78%) as an oil. Alcohol 5 (20 mg, 0.074 mmol) was converted to Moshers
ester (28
mg, 0.058 mmol, 78%) according to the method described above.'3C NMR spectral
data
were in agreement with those reported above for (2,5~-2-[(benzyloxymethyl)-4-
phenylbutyl]
(2R)-3,3,3-trifluoro-2-methoxy-2-phenyl-propanoate.
(2,5~-2- { (4R, S,S~-5-[( 1,5~-1-(Hydroxymethyl)-3 -phenylpropyl ]-2,2-d i
methyl-1,3-dioxolan-4-
yl}-4'-phenyl-1-butanol (9 a~,
(2S'L~(4R.SR)-5~(1,5~-1-(hydroxymethyll-3-phenylprop~]-2,2-dimethyl-1.3-
dioxolan-4-
~1-4-phenyl-1-butanol (9 b) and
(2,SZ~,~4R.SR)-5-(( l,S~l-1-(h~~~)-3-phen~prop~]-2.2-dimethyl-1,3-dioxolan-4-
yl~phen,Z-1-butanol (9 c~
Zinc dust (pre-washed with 1M HC1, EtOH, acetone and CHzCl2) (2.72 g, 41.6 mg-
atom, 2.0
equiv.) was added to a solution of VC13 (THF)3 in CHzCIz (50 mL, 0.5 M, 25
mmol, 1.2
equiv.), under an argon atmosphere, changing the colour of the solution from
deep-red to
violet. The mixture was stirred at room temperature during 80 min, while the
colour slowly
changed from violet to black-green. 1,3-Dimethyl-2-imidazolidinone (16.0 mL,
146 mmol,
7.0 equiv.) was added, and stirring was continued during 1 S min, before
dropwise addition
(40 min) of aldehyde 6 (5.59 g, 20.8 mmol) dissolved in CHZCIZ (56 mL) which
gave a
brown reaction mixture. The reaction was quenched after 20 h (Rf7 a 0.32 and
Rf7 b, 7 c
0.21, toluene-EtOAc 9:1), by the addition of 1 M HCI (127 mL). Stirring of the
two-phase
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
mixture during 30 min gave a turquoise aqueous layer and a brown organic
layer, which
were separated. Additional extraction of the aqueous layer with CHZC12 (3x),
drying
(MgS04), concentration and subsequent separation by column chromatography
(toluene;
5 toluene-EtOAc 25:1 and 15:1) gave 7 a (2.29 g) and a mixture of 7 b and 7 c
(2.16 g), all of
which were used in the next step without further purification.
(7 a): ~3C NMR (75 MHz, CDC13) 026.6, 31.3, 33.8, 34.0, 38.4, 39.8, 70.2,
72.9, 73.5, 73.6
(2 C), 75.9, 125.6, 127.5, 127.7, 127.8, 128.1, 128.2 (2 C), 128.3 (2 C),
137.4, 137.7, 142.2
and 142.4.
10 (7 b and 7 c):'3C NMR (75 MHz, CDC13) Q28.5, 31.1, 33.5, 33.8, 40.7 (2 C),
70.3, 70.4,
72.6, 73.4, 73.7, 73.9, 125.7 (2 C), 127.5, 127.6 (2 C), 127.7, 128.2 (2 C),
128.3 (2 C),
137.7, 142.2 and 142.1.
Std II. lsopro~ liy dene~rotection.
2,2-Dimethoxypropane (2.5 equiv.) and (~)-10-camphorsulfonic acid (0.8 equiv.)
were
15 added to a solution of 7 a-c (1.0 equiv.) in acetone (5.3 mL/mmol) under
nitrogen. After
stirring for 30 min, when TLC (Rf 8 a 0.63 and Rf 8 b, 8 c 0.72, toluene-EtOAc
9:1 ) showed
completion of the reaction, the reaction mixture was poured into sat. aqueous
NaHC03 and
extracted with EtOAc (3x). Activated charcoal and NaS04 were added to the
organic layer,
which was stirred for 10 min before filtration through a pad of Celite and
NaS04, and
20 concentration. Column chromatography (toluene; toluene-EtOAc 20:1 and 9:1)
gave the
crude product, which was used in the next step without further purification.
(8 a). The isopropylidene protected compound was prepared according to step
II, using 2.29
g of 7 a, and isolated as a white solid (2.29 g):'3C NMR (75 MHz, CDC13) 0
25.1, 26.2,
29.6, 31.0, 33.0, 33.3, 38.0, 38.2, 69.6, 71.1, 73.0, 73.1, 77.1, 77.5, 106.9,
125.6 (2 C), 127.3
(2 C), 127.4, 128.1, 128.2 (3 C), 128.3, 128.4, 138.5, 138.8, 142.3 and 142.4.
(8 b and 8 c). The isopropylidene protected compounds was prepared according
to step II,
using 2.16 g of the mixture 7 b and 7 c, and isolated a transparent syrup
(1.95 g). After
column chromatography a small amount of alcohol 5 (265 mg, 0.980 mmol) was
isolated as
well.'3C NMR (75 MHz, CDC13) ~ 27.4, 28.6, 31.5, 33.4, 33.8, 40.2, 40.7, 69.4,
70.7, 72.9,
73.0, 79.1, 79.3, 107.5, 107.8, 125.6, 125.7, 127.3 (2 C), 127.5, 128.1, 128.2
(2 C), 128.3,
138.4 (2 C), 142.2 and 142.3.
Step III. Hydrogenolysis.
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
21
Compounds 8 a-c (1.0 equiv.) was dissolved in EtOAc (23 mL/mmol), and NaHC03
(3.0
equiv.), a minor amount of Hz0 and Pd/C (0.8 g) was added. The suspension was
stirred at
room temperature under a hydrogen atmosphere. After 19 h, 39 h and 47 h, the
suspension
was filtered through a pad of Celite and NazS04, fresh reagents were added and
stirring
under hydrogen was continued. When TLC showed completion of the reaction (Rf9
a 0.33,
Rf 9 b, 9 c 0.31, toluene-EtOAc 1:1) after 63 h, filtration and concentration
followed by
column chromatography (toluene-EtOAc 2:1 ) gave compounds 9 a-c.
(9 a). The title compound was prepared according to step III, using 2.47 g of
8 a, and
isolated as a transparent syrup (1.27 g, 3.19 mmol): [D]z°D +91 (c 1.4,
CHCI3);'H NMR
(300 MHz, CDC13) ~ 1.26 (s, 3 H), 1.37 (s, 3 H), 1.59-1.71 (m, 4 H), 1.90 (s,
1 H), 1.97 (s,
1H), 2.12-2.28 (m, 3 H), 2.49-2.67 (m, 3 H), 3.57-3.77 (m, 4 H), 4.03-4.08 (m,
1 H), 4.29-
4.32 (dd, 1 H, J= 2.64, 6.59 Hz) and 7.03-7.25 (m, 10 H);'3C NMR (75 MHz,
CDC13) 0
25.0, 26.1, 27.3, 30.4, 32.8, 33.1, 38.6, 39.5, 64.0, 64.2, 79.2, 81.0, 108.1,
125.8, 126.0,
128.3 (2 C), 128.4, 128.5, 141.4 and 141.9. Anal Calcd for Cz5H3a0a ~ '/4 HzO:
C, 74.5; H,
8.6. Found: C, 74.5; H, 8.6.
(9 b and 9 c). The mixture of the title compounds was prepared according to
step III, using
1.77 g of the mixture of 8 b and 8 c, and isolated as a transparent syrup
(1.03 g, 2.58 mmol):
'3C NMR (75 MHz, CDC13) 0 26.9, 27.1, 27.3, 30.6, 33.3, 33.5, 40.6, 41.1,
62.2, 63.4, 79.9,
81.9, 108.8, 108.3, 125.8 (2 C), 128.2 (2 C), 128.3, 141.5 and 141.8. Anal
Calcd for
CzsH3a0a ~ 1/3 HzO: C, 74.2; H, 8.6. Found: C, 74.3; H, 8.7.
Diol 9 a and the mixture of diols 9 b and 9 c were isolated with a total yield
of 64% over
three steps (2.30 g 5.77 mmol). A small amount of alcohol 5 (265 mg, 0.980
mmol) was also
isolated.
General method for the preparation of compounds 10 a-c.
Method I. The diol 9 (1.0 equiv.) was dissolved in a stirred mixture of CH3CN
(4
mL/mmol), CHZCIz (4 mL/mmol) and Hz0 (6 mL/mmol). RuCl3 ~ xHzO (0.05 equiv.)
and
NaI04 (9.0 equiv.) were added. TLC (toluene-EtOAc 1:3) showed completion of
the
reaction after vigorous stirring for 105 min. The reaction mixture was diluted
with CHZCIz
(lx volume) and stirred for an additional 10 min. Water was added (lx volume)
and the
aqueous layer was extracted with EtOAc (4x). The combined organic layers were
dried
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
22
(MgS04), and concentrated to provide the corresponding carboxylic acid, which
was
immediately activated without further purification.
A mixture of the carboxylic acid, N,N disuccinimidyl carbonate (S.0 equiv.)
and pyridine
(6.9 equiv.) in CH3CN (16 mL/mmol) was stirred under an argon atmosphere. When
TLC
showed completion of the reaction after 23 h (RflO a 0.63, Rf 10 b 0.47 and Rf
10 c 0.58,
toluene-EtOAc 1:1), toluene ('/z x volume) was added and the reaction mixture
was
concentrated. The residue was dissolved in EtOAc, extracted with water (5x)
and the
combined aqueous layers were washed with EtOAc (3x). The combined organic
layers were
dried (Na2S04), concentrated and purificated by column chromatography (toluene-
EtOAc
4:1) to provide the disuccinimidyl esters 10 a-c.
Succinimid~2R)-2-[(4R.S,S~-S-(3-phen~prop~ 1Rl-1-succinimid~ycarbon~)-2.2-
dimethyl-1.3-dioxolan-4-yl]-4-phen~butanoat (10 a).
The title compound was prepared according to method I, using 1.14 g (2.87
mmol) of diol 9
a, and isolated as a white foam in 22% yield (393 mg, 0.633 mmol):
[~]z°D +19 (c 0.35,
CDC13);'3C NMR (75 MHz, CDCl3) ~ 25.1, 25.7, 26.8, 31.8, 32.0, 32.5, 32.6,
42.2, 43.5,
76.0, 77.8, 108.9, 125.2, 125.9 (2 C), 128.1, 128.2, 128.3, 128.5, 128.7,
128.9, 140.9, 141.1,
168.2, 168.5, 168.6 and 168.8. Anal Calcd for C33H36N2010~ C, 63.9; H, 5.9; N,
4.5. Found:
C, 63.8; H, 5.9; N, 4.4.
Succin i midi(2R)-2-[(4R,SR)-5-(3-phen~propyl-( 1 R)-1-succinimidyloxycarbon~)-
2,2-
dimethyl-1,3-dioxolan-4-~]-4-nhenylbutanoat (10 b) and
succinimidyl~2R)-2-I(4S,S,S~-5~3-phen~prop~rl-(1R)-1-succinimid~ ca~~r~-2,2-
dimethyl-1,3-dioxolan-4-yl]-4 phenylbutanoat (10 c1.
The title compounds, separated by column chromatography, were prepared
according to
method I, using 965 mg (2.42 mmol) of the mixture of diols 9 b and 9 c, and
isolated as
white foams in 43% total yield (10 b: 126 mg, 0.203 mmol; 10 c: 527 mg, 0.849
mmol).
(10 b): [D]2°p +86 (c 0.40, CDC13);'3C NMR (75 MHz, CDC13) 0 25.7,
27.4, 29.8, 32.7,
45.6, 78.8, 110.4, 126.0, 128.1, 128.3, 128.6, 128.9, 140.8, 168.3 and 168.5.
Anal Calcd for
C33H36N2~10 ' '/4 H2O: C, 63.4; H, 5.9; N, 4.5. Found: C, 63.4; H, 5.7; N,
4.3.
(10 c): [~]z°D-5.3 (c 0.75, CDC13);'3C NMR (75 MHz, CDC13) ~ 25.7,
27.6, 27.9, 31.4,
31.7, 32.6, 33.0, 45.0, 78.5, 80.2, 110.3, 125.2, 125.9, 126.1, 128.1, 128.3,
128.4, 128.6,
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
23
128.9, 140.4, 167.5 and 168.6. Anal Calcd for C33H36NZOio~ C, 63.9; H, 5.9; N,
4.5. Found:
C, 63.8; H, 5.9; N, 4.5.
General method for the preparation of compounds 12 a and 12 b.
Method II. The disuccinimidyl esters 10 a-c (1.0 equiv.) and (1S,2R)-(-)-cis-1-
amino-2-
indanol (6.3 equiv.) were dissolved in 1,2-dichloroethane (30 mL/mmol) under a
nitrogen
atmosphere. The reaction was stirred at 60 °C until TLC (Rf 11 a 0.31,
Rf 11 b 0.43 and Rf
11 c 0.19, toluene-EtOAc 1:1) showed completion of the reaction (16-20 h).
Concentration
followed by column chromatography (toluene-EtOAc 3:1 and 1:1) gave the amides
11 a-c.
A mixture of FeCl3 ~ 6H20 (3.5 equiv.) and the amides 11 a and 11 b (1.0
equiv.) were
dissolved in warm CHZCIz (15 mL/mmol) and stirred at room temperature for 1 h
and 8 h (Rf
12 a 0.18, Rfl2 b 0.21, CHC13-MeOH 20:1), respectively. Sat. aqueous NaHC03
(lx
volume) was added and the aqueous layer was extracted with CHZCIz (3x) and
EtOAc (3x).
Drying (MgS04), concentration, and purification by column chromatography
(0H013;
CHCl3-MeOH 40:1 and 30:1), followed by trituration from EtOAc, gave the target
compounds 12 a and 12 b.
(11 a): '3C NMR (75 MHz, CDC13) 0723.3, 25.4, 32.7, 32.9, 33.4, 34.0, 39.2,
39.7, 46.1,
46.7, 58.0, 58.2, 73.4, 73.6, 77.4, 77.7, 107.5, 123.6, 125.0, 125.3, 125.4,
125.9, 126.0,
126.8, 127.1, 128.1, 128.3 (2 C), 128.4, 128.6, 139.8, 140.1, 140.4, 141.1,
141.5, 173.2 and
174.7.
(11 b): '3C NMR (75 MHz, CDC13) 027.3, 29.6, 33.4, 39.5, 49.8, 57.7, 73.5,
79.6, 108.9,
124.7, 125.2, 126.0, 127.2, 128.3, 128.5, 140.0, 140.2, 141.0 and 172.9.
(11 c): 29% yield (73 mg, 0.106 mmol); [D]ZED +15 (c 0.48, CHC13-MeOH 1:1);'H
NMR
(400 MHz, CDC13 and CD30D) ~ 1.40 (s, 6H), 1.77-1.84 (m, 2 H), 2.00-2.10 (m, 2
H),
2.60-2.68 (m, 4 H), 2.74-2.82 (m, 2 H), 2.96 (d, 2 H, J= 16.41 Hz), 3.17 (dd,
2 H, J= 4.69,
16.02 Hz), 3.67 (d, 2 H, J= 3.12 Hz), 4.17 (d, 2 H, J= 5.86 Hz), 4.61-4.64 (m,
2 H), 5.42 (d,
2 H, J= 5.08 Hz) and 7.15-7.35 (m, 18 H);'3C NMR (75 MHz, CDC13 and CD30D)
Q26.9,
31.5, 33.1, 39.6, 50.6, 57.2, 72.5, 79.7, 108.9, 124.0, 124.9, 125.7, 126.5,
127.7, 128.0,
128.1, 139.9, 140.4, 140.7 and 173.2. Anal Calcd for C43H48NzO6 ~ 1 HzO: C,
73.1; H, 7.1;
N, 4Ø Found: C, 72.9; H, 6.8; N, 4Ø
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
24
N1.N6-D~(2R)-hydroxy~lSj-indanyl]-(2R,3R,4S.5R -2,5-di(2-phen~~ -L
dihydroxyhexanediamide (12 a)
The title compound was prepared according to method II, using 195 mg (0.314
mmol) of the
disuccinimidyl ester 10 a, and isolated as a white solid in 47% yield (96 mg,
0.148 mmol):
[~]z°D +20 (c 0.94, CHC13-MeOH 1:1);'H NMR (400 MHz, CDC13 and CD30D) D
1.97-
2.23 (m, 4 H), 2.61-2.85 (m, 6 H), 2.92-2.99 (m, 2 H), 3.12-3.19 (m, 2 H),
3.69 (bs, 1 H),
3.91-3.94 (m, 1 H), 4.48 (s, 6 H), 4.54-4.61 (m, 2 H), 5.38-5.41 (m, 2 H) and
7.13-7.30 (m,
18 H);'3C NMR (75 MHz, CDC13 and CD30D) ~ 33.3 (2 C), 39.3, 47.0, 56.8, 56.9,
72.3,
72.4, 72.7, 73.4, 123.6, 123.8, 124.7, 125.3, 125.4, 126.5, 127.5, 127.8 (3
C), 139.7, 139.8,
140.2, 141.1, 141.5 and 175.9. Anal Calcd for C4oH4aNz06 ~ 1 '/~ HZO: C, 71.1;
H, 7.0; N,
4.2. Found: C, 71.3; H, 6.7; N, 4.1.
N1 ,N6-Di [(2R)-h~y~ 1 S)-indan~]-(2R.3R,4R,5R1-2,5-di(2-phem~leth~)-3,4-
dihydroxyhexanediamide (12 b).
The title compound was prepared according to method II, using 67 mg (0.108
mmol) of the
disuccinimidyl ester 10 c, and isolated as a white solid in 58% yield (41 mg,
0.063 mmol):
[O]z°p +30 (c 0.77, CHCl3-MeOH 1:1);'H NMR: (400 MHz, CDC13 and CD30D)
Q 1.99-
2.03 (m, 2 H), 2.05-2.15 (m, 2 H), 2.60-2.69 (m, 4 H), 2.76-2.84 (m, 2 H),
2.95 (dd, 2 H, J=
2.20, 16.48 Hz), 3.15 (dd, 2 H, J= 5.13, 16.48 Hz), 3.81 (d, 2 H, J= 8.79 Hz),
4.24 (s, 6 H),
4.60-4.63 (m, 2 H), 5.41 (d, 2 H, J= 5.13 Hz), and 7.16-7.33 (m, 18 H);'3C NMR
(100
MHz, CDC13 and CD30D) ~ 30.8, 33.4, 39.4, 50.8, 57.1, 71.6, 72.6, 124.0,
124.7, 125.5,
126.6, 127.6, 127.9, 139.8, 140.0, 141.2 and 174.6. Anal Calcd for C4oH4aNz06
~ 1 HzO: C,
72.1; H, 7.0; N, 4.2. Found: C, 72.4; H, 6.6; N, 4. I .
General method for the preparation of compounds 14 a and 14 b.
Method III. The disuccinimidyl esters 10 a-c (I.0 equiv.) and H-Val-NHMe (6.3
equiv.)
were dissolved in 1,2-dichloroethane (30 mL/mmol) under a nitrogen atmosphere.
The
reaction was stirred at 50 °C until TLC (Rfl3 a 0.34, Rfl3 b 0.41,
CHC13-MeOH 9:1)
showed completion of the reaction (22 h). Concentration followed by column
chromatography (CHC13-MeOH 20:1) gave the amides 13 a and 13 b.
A mixture of FeCl3 ~ 6H20 (3.5 equiv.) and the amides 13 a and 13 b ( 1.0
equiv.) were
dissolved in warm CHZCIz (17.5 mL/mmol) and stirred at room temperature. The
reaction
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
was followed with TLC (Rfl4 a 0.23, Rfl4 b 0.40, CHC13-MeOH 9:1), and every
other hour
the reaction was concentrated and new CHZCIz was added. When TLC showed
completion
of the reaction (14 a: 4 h and 14 b: 8 h) sat. aqueous NaHC03 (lx volume) was
added and
5 the aqueous layer was extracted with CHZC12 (3x) and EtOAc (3x). Drying
(MgS04),
concentration, and purification by column chromatography (0H013; CHC13-MeOH
30:1 and
20:1), followed by trituration from EtOAc, gave the target compounds 14 a and
14 b.
(13 a): '3C NMR (75 MHz, CDC13 and CD30D) ~ 19.0, 24.3, 25.6, 25.7, 25.8,
30.8, 31.0,
31.6, 32.0, 32.3, 33.0, 46.1, 46.5, 58.5, 59.0, 76.4, 77.9, 107.7, 125.8,
128.0, 128.2, 140.9,
10 141.6, 172.1, 172.2, 173.0 and 174Ø
(13 b): '3C NMR (75 MHz, CDC13) 018.6, 19.5, 26.2, 27.2, 29.7, 30.6, 33.6,
49.1, 58.8,
79.1, 109.1, 125.9, 128.3, 141.2, 171.6 and 172.6.
N1 ,N6-Di-[( 1 S)-2-meth~~methylcarbamo~)prop]'-(2R,3R,4S,5R)-2,5-di(2-
phenylethyl)-
15 3,4-dihydroxyhexanediamide (14 a~
The title compound was prepared according to method III, using 153 mg (0.247
mmol) of
the disuccinimidyl ester 10 b, and isolated as a white solid in 7% yield (10
mg, 0.017 mmol)
together with recovered 13 a (26 mg, 0.040 mmol). [~]Z°D -27 (c 0.32,
CHC13-MeOH 1:1);
'H NMR: (400 MHz, CDCl3 and CD30D) 070.92 (d, 3 H, J= 6.64 Hz), 0.97 (d, 9 H,
J= 6.64
20 Hz), 1.85-2.19 (m, 6 H), 2.54-2.72 (m, 6 H), 2.76 (s, 3 H), 2.77 (s, 3 H),
3.60-3.62 (m, 1 H),
3.70-3.73 (m, 1 H), 4.12-4.18 (m, 2 H), 4.56 (s, 6 H) and 7.16-7.55 (m, 10
H);'3C NMR
(100 MHz, CDC13 and CD30D) 0 17.8, 18.7, 18.8, 25.2, 28.4, 30.1, 30.3, 32.5,
33.2, 46.5,
58.3, 58.5, 72.5, 73.5, 125.2, 125.3, 127.6, 127.7, 127.8, 141.0, 141.3,
171.8, 171.9, 175.2
and 175.4. HRMS Calcd for C34H5°O6N4 [M+Na] 633.3628. Found [M+Na]
633.3604.
N1,N6-Di-[(1,S')-2-methyl-1-(methylcarbamo~)props]-(2R,3R,4R,SR -2,5-di(2-
phen~~Z
3.4-dih,~yhexanediamide (14 b).
The title compound was prepared according to method III, using 52 mg (0.084
mmol) of the
disuccinimidyl ester 10 b, and isolated as a white solid in 22% yield (11 mg,
0.018 mmol)
together with recovered 13 b (16 mg, 0.025 mmol). [~]2°D-23 (c 0.75,
CHCI3-MeOH 1:1);
'H NMR: (400 MHz, CDC13 and CD30D) 00.60-0.64 (m, 12 H), 1.61-1.74 (m, 6 H),
2.18-
2.26(m,6H),2.42(s,6H),3.34(d,2H,J=8.59Hz),3.76-3.78(m,2H),4.32(s,6H)and
6.81-7.27 (m, 10 H);'3C NMR (100 MHz, CDC13 and CD30D) 0 17.8, 18.5, 25.1,
29.9,
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
26
30.8, 33.0, 50.4, 58.7, 71.1, 125.1, 127.5, 141.0, 171.9 and 174.1. HRMS Calcd
for
C3aHso06Na [M+Na] 633.3628. Found [M+Na] 633.3638.
Biolo,~ical Example 1
Compounds arre tested for HIV protease activity in a spectrophotometric enzyme
assay
using the chromogenic substrate His-Lys-Ala-Arg-Val-Leu-p-nitro-Phe-Glu-Ala-
Nle-Ser-
amide and purified HIV proteinase. The rate of cleavage is followed by
continuously
registering the change in absorbance at 300 nm. The ICso representing the
compound
concentration which inhibits enzyme performance by 50% is calculated from the
dose
response curve. Extremely active compounds are best compared with affinity
constants (K;).
Compounds are also tested for HIV protease activity in cell culture. MT4 cells
grown in
RPMI 1640 cell culture medium including 10 % fetal calf serum are infected
with 10 TCID
HIV-1 per 2 x l Os cells and cultured for 6 days. XTT is added and the amount
of XTT
formazan produced in the following 6 hours represents the number of surviving
cells.
Results are expressed as the EDso, that is the concentration in pg/ml of the
compound of the
invention which suppresses viral replication by 50%. Additionally, activity
measurements
are run in the corresponding system further comprising 40% human serum to
mimic protein
binding effects in vivo.
Exam 1e K; nM EDSO M EDSO 40% HS
M
8b 1.1 0.093 0.39
Prior art 1 0.6 0.96 1.5
9b 0.40 1.9 1.4
Prior art 2 0.80 1.3 34
Example 1 of WO 98/45330:
2 Example 10 of W098/45330
CA 02458040 2004-02-19
WO 03/018537 PCT/SE02/01549
27
The compounds of the invention have a particularly good performance in the
presence of human serum relative to the closest compounds of the prior art .