Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02467715 2004-05-19
TITLE OF THE INVENTION
COMPOSITIONS AND METHODS FOR INHIBITING BONE RESORPTION
BACKGROUND OF THE INVENTION
FIELD OF THE INVENTION
The present invention relates to compositions comprising a bisphosphonate
compound
and a vitamin D compound. The present invention also relates to methods of
using such compositions for
example to treat, reduce, inhibit or prevent abnormal bone resorption in
mammals. The present invention
further relates to methods of making bisphosphonate and vitamin D
compositions.
RELATED ART
A variety of disorders in humans and other mammals involve or are associated
with
abnormal bone resorption. Among the most common of these disorders is
osteoporosis, which is a
systemic skeletal disease characterized by a low bone mass and
microarchitectural deterioration of bone
tissue, with a consequent increase in bone fragility and susceptibility to
fracture. Osteoporosis is
becoming a worldwide pandemic, with marked increases in its occurrence
coinciding with the worldwide
increase of longevity.
A principal cell type responsible for bone resorption is the multinucleated
cell called the
osteoclast. Bisphosphonates are well known as selective inhibitors of
osteoclastic bone resorption.
Bisphosphonates are believed to bind to hydroxyapatite in bone and to inhibit
the bone resorptive activity
of osteoclasts through their intracellular action. See, e.g., H. Fleisch,
Bisphosphonates In Bone Disease,
From The Laboratory To The Patient, 4th Edition, Academic Press (2000). It has
also been reported that
bisphosphonates bind to bone and then are released into the resorption lacuna
during resorption. After
this they are taken up by the osteoclast, and subsequently inhibit the enzyme
farnesyl diphosphate
synthase. This intracellular action in turn prevents the isoprenylation
(farnesylation and
geranylgeranylation) of GTPases, signaling proteins that attach to membrane
vesicles. The family of
geranylgeranylated small GTPases include those that direct the formation of
the ruffled border - the
organelle of active bone resorption. See A.A. Reszka and G.A. Rodan,
"Bisphosphonate Mechanism of
Action," Curr Rheumatol Rep. 5(1):65-74 (February, 2003).
Bisphosphonates are understood to be useful in preventing bone loss associated
with a
number of conditions. For example, bisphosphonates are known to be useful in
the prevention of bone
loss and in the treatment of diseases such as, but not limited to,
osteoporosis, osteopenia, metastatic bone
disease, multiple myeloma, periodontal disease, tooth loss,
hyperparathyroidism, rheumatoid arthritis,
CA 02467715 2004-05-19
Paget's disease, osteonecrosis, osteoarthritis, periprosthetic bone loss or
osteolysis, and hypercalcemia of
malignancy. All of these conditions are characterized by bone loss, resulting
from an imbalance between
bone resorption - i.e., breakdown - and bone formation.
Alendronate sodium is one of the most potent bisphosphonates currently
available, and
does not impair bone mineralization at doses which maximally inhibit bone
resorption. It has also been
found that the increase in bone mineral density observed with the
administration of alendronate is
positively associated with a decrease in vertebral and non-vertebral
(including the hip) fractures, a
decrease in spinal deformity and a retention of height. This indicates that
when administered for a
substantial period of time, alendronate decreases bone turnover acting
positively to produce a
strengthened bone. Alendronate sodium is approved in more than 90 countries
for the treatment of
osteoporosis in postmenopausal women. Alendronate sodium is also approved for
the treatment of
osteoporosis in men, glucocorticoid-induced osteoporosis, and Paget's disease
of bone. Evidence
suggests that other bisphosphonates such as ibandronate, minodronate,
pamidronate, risedronate,
tiludronate and zoledronate, have many properties in common with alendronate,
including high potency
as inhibitors of osteoclastic bone resorption.
Despite their therapeutic benefits, bisphosphonates are poorly absorbed (on
the order of
about 1%) from the gastrointestinal tract. See" e.g., B.J. Gertz et al.,
Clinical Pharmacology of
Alendronate Sodium, Osteoporosis Int., Suppl. 3: S13-16 (1993) and B.J. Gertz
et al., Studies ofthe oral
bioavailability of alendronate, Clinical Pharmacology & Therapeutics, vol. 58,
number 3, pp. 288-298
(September 1995). It is understood that food, as well as many other substances
that may be ingested
concomitantly (including beverages such as mineral water, and even some
excipients used to formulate
dosing vehicles) can adversely affect bisphosphonate absorption. Intravenous
administration has been
used to ensure that the entire dose reaches the circulation. However,
intravenous administration is costly
and inconvenient, especially when the subject must be given an intravenous
infusion lasting several hours
on repeated occasions. Unlike oral administration, intravenous administration
of bisphosphonates is
associated with acute renal injury if administered too rapidly.
If, instead of intravenous administration, oral administration of the
bisphosphonate is
desired, higher doses may be administered to compensate for the low
bioavailability from the
gastrointestinal tract. To offset this low bioavailability, it is generally
recommended that the subject take
the bisphosphonate on an empty stomach and fast for at least 30 minutes
afterwards. However, many
subjects find such fasting on a daily basis to be inconvenient.
Bisphosphonate therapy has been associated with hypocalcaemia. During
treatment with
bisphosphonates, the early inhibition of bone resorption can induce a decrease
in serum calcium, which
occurs within hours, days or weeks of the start of treatment. The serum
calcium decrease can persist for
many weeks to months following the initiation of treatment and can be
prominent in subjects having a
-2-
CA 02467715 2004-05-19
deficiency in vitamin D. The hypocalcaemia response to bisphosphonate therapy
can occasionally be
severe enough to be symptomatic and warrant clinical intervention,
particularly in patients with
hypoparathyroidism (See, e.g., Vasikaran, S.D., Ed., Bisphosphonates: An
Overview with Special
Reference to Alendronate, Ann. Clin. Biochem. (2001) 38: 608-623). As there
are no substantial body
stores of calcium outside of bone, the calcium required for new bone formed
after treatment with
alendronate is initiated must be absorbed from the diet - either from food or
calcium supplements.
Vitamin D is required for normal calcium absorption. Thus, adequate vitamin D
and calcium intake is
desirable for subjects using bisphosphonates. Adequate vitamin D levels become
even more important
when calcium needs are elevated due to the net influx of calcium into bone
that occurs as a result of
bisphosphonate therapy during effective osteoporosis treatment. As a result,
adequate vitamin D and
calcium intake is desirable for subjects using bisphosphonates.
Vitamin D compounds comprise a group of fat soluble secosteriods that are
found in very
few foods naturally, and they are photosynthesized in the skin of vertebrates
by the action of solar UV
radiation. While vitamin D may come in several forms, the most physiologically
relevant forms are
vitamin D3 (cholecalciferol) and vitamin DZ (ergocalciferol). The latter is
formed when the yeast and
plant sterol, ergosterol, is exposed to UV radiation, while the former
originates from 7-dehydrocholesterol
and is synthesized in the skin. The metabolic pathway for vitamin D3 and
vitamin DZ is similar, and their
biological efficacy in humans is similar, their main function being the
maintenance of serum calcium and
phosphorous concentrations within normal ranges. Vitamin D3 is the obligate
precursor of the hormone
calcitriol (also called 1,25-dihydroxycholecalciferol or 1,25-dihydroxyvitamin
D3), whose principal
action is to enhance the ability of the small intestine to absorb calcium, and
retain phosphate from the
diet. The hormone-like metabolite of ergocalciferol is 1,25-
dihydroxyergocalciferol (1,25-
dihydroxyvitamin Dz). When dietary calcium intake is insufficient to satisfy
the body's needs,
parathyroid hormone (PTH), along with the hormonal metabolite of vitamin D3,
calcitriol, mobilizes
monocytic stem cells in the bone marrow to become mature osteoclasts. These
osteoclasts are themselves
stimulated by a variety of cytokines and other factors to increase the
mobilization of calcium stores from
the bone.
The naturally-occurring forms of cholecalciferol and ergocalciferol are
biologically
inactive precursors of the hydroxylated biologically active metabolites of
vitamin D. Because vitamin D
is lipid soluble, it may be stored in fat tissues in the body or metabolized
to the principal storage
metabolite 25-hydroxyvitamin D and stored in other organs. The 25-
hydroxyvitamin D is transported in
blood plasma and metabolized by the body when needed. Specifically, as shown
in the example of
cholecalciferol in FIG. 1, 7-dehydrocholesterol in the skin converts to pre-
vitamin D3 (an isomer of
vitamin D3, not shown in FIG. 1) upon exposure to sunlight, and then to
vitamin D3 (cholecalciferol).
Cholecalciferol is then metabolized in the liver to form 25-
hydroxycholecalciferol, also known as 25-
-3-
CA 02467715 2004-05-19
hydroxy vitamin D3, and this is further metabolized in the kidney to the
hormonal form 1,25-
dihydroxycholecalciferol, also known as calcitriol. Although not depicted in
FIG. 1, other metabolites of
vitamin D (Dz or D3) include la -hydroxy vitamin D, 24,25 dihydroxy vitamin D,
and la, 24,25-
trihydroxy vitamin D. Only calcitriol is fully biologically active;
cholecalciferol and the metabolites
identified above show little or no biological activity.
A primary biological function of vitamin D (both Dz and D3) is to help
maintain calcium
homeostasis by increasing the intestine's efficiency in absorbing dietary
calcium. 1t helps to ensure that
the amount of calcium absorbed is adequate to maintain blood calcium in the
normal range and adequate
to maintain skeletal mineralization. Adequate vitamin D intake facilitates
intestinal absorption of
calcium, and plays an important role in regulating calcium metabolism and in
the mineralization of the
skeleton.
Vitamin D insufficiency and deficiency are recognized as causes of metabolic
bone
disease in adults. Vitamin D insufficiency is characterized by the impairment
of calcium and phosphate
absorption but no impairment of normal bone mineralization and is typically
associated with a serum 25-
I 5 hydroxy vitamin D level between about <9 to about 30 nglmL. Vitamin D
deficiency is characterized by
severely impaired calcium absorption, secondary hyperparathyroidism,
hypophosphatemia, low or low
normal blood calcium, and impaired bone mineralization. Serum 25-hydroxy
vitamin D levels are usually
about < 9 ng/mL. Vitamin D insufficiency and deficiency result in increased
parathyroid hormone
(PTH), which in turn causes increased osteoclastic activity, urinary phosphate
loss and calcium
mobilization from bone. This in turn can aggravate osteoporosis, especially in
older adults, as impaired
bone mineralization results in independent and additional reductions in bone
strength. Sustained vitamin
D insufficiency is thought to be an important cause of gradual bone loss.
Depending on the degree of the
vitamin D and calcium deficiency, the histological picture may either be one
of osteomalacia,
osteoporosis or a combination of the two.
The prevalence of vitamin D insufficiency and deficiency creates a need for
additional
vitamin D intake in the patient populations prone to, or suffering from,
conditions such as osteoporosis or
osteopenia and in the subjects undergoing bisphosphonate therapy for these
conditions. In subjects
undergoing bisphosphonate therapy, and in particular those subjects with
inadequate dietary calcium
intake or inadequate calcium absorption, there is a need for adequate vitamin
D to facilitate bone
formation and mineralization, while minimizing the potential for or occurrence
of vitamin D
insufficiency. Some form of increasing vitamin D intake is often used in
clinical trials of bone resorption
compounds and recommended on product labels and in product package circulars.
However,
approximately 30% of the osteoporotic patients in, for example, the United
States have some degree of
vitamin D insufficiency and prevalence increases with age.
-4-
CA 02467715 2004-05-19
Currently, subjects taking oral bisphosphonates and requiring vitamin D are
advised to
take two separate products at two different times. Vitamin D formulations are
most commonly taken
daily, while bisphosphonates may be administered daily, weekly, monthly or at
longer intervals. As a
result, many patients in treatment for osteoporosis or osteopenia fail to take
vitamin D despite being
advised to do so. Typically, vitamin D cannot be taken simultaneously with
bisphosphonates, simply due
to the fact that bisphosphonate absorption is so poor, and that most
bisphosphonate oral dosage regimens
require a 30 minute time interval between ingestion of the bisphosphonate and
other substances
(including but not limited to vitamin D). As a result, patient compliance with
dosing regimens that
require a separate administration of a vitamin D compound at some time
interval either before or after
bisphosphonate administration is not high. (Some bisphosphonate compounds
require administration
prior to ingestion of foods, and therefore any vitamin D administration would
have to occur at some time
interval after bisphosphonate administration). While it is possible for
patients to take vitamin D before or
after taking their bisphosphonate dosages, there is evidence that many
patients do not do so. A 1998
marketing study showed that while 75-85% of physicians prescribing alendronate
also recommended
vitamin D supplementation, only 57% of osteoporotic patients actually
complied.
Although vitamin D can also be administered in the form of a multi-vitamin, in
the
United States, for example, many over-the-counter oral vitamin D formulations
are not sold in the dosage
units required for dosing less frequently than daily. And, if patients self
administer vitamin D
simultaneously with their bisphosphonate dosage, it is possible that the type
of vitamin D administered
could interfere with and further reduce bisphosphonate absorption since many
vitamin D compounds
formulated for osteoporotic patients contain calcium which reduces the
absorption of a bisphosphonate.
The patent literature includes patents and published patent applications that
disclose
vitamin D3 or vitamin DZ or their metabolites or analogs in combination with a
bisphosphonate. See, e.g.,
U.S. Patent Nos. 4,230,700, 4,330,537 and 4,812,304; European Patent Nos. EP 0
381 296 and EP 0 162
S l0; International Patent Publication Nos. WO 90/01321, WO 92/21355, WO
01/28564, WO 01/97788
and WO 03/086415; European Patent Publication No, EP 1 051 976; Japanese
Patent Publication Nos. 7-
330613 and 1 1-60489; U.S. Patent Application Nos. US 2003/0139378 Al, and US
2003/0225039 A 1.
These patents and publications, however, do not disclose or enable a
composition, product or formulation
(and, most particularly, a tablet) comprising a bisphosphonate compound and a
vitamin D compound that
is useful for continuous oral administration at intervals, such as once
weekly, that are less frequent than
daily and more frequent than six months or longer. These patents and
publications also do not disclose or
enable treating, inhibiting, reducing or preventing osteoporosis and other
conditions associated with
abnormal bone resorption by administering such bisphosphonate/vitamin D
compositions at intervals less
frequent than daily and more frequent than six months.
-5-
CA 02467715 2004-05-19
As a result, there is a need for a combination product comprising a
bisphosphonate
compound and a vitamin D compound, including to enhance the overall efficacy
of bisphosphonate
treatment by helping to assure adequate vitamin D intake to facilitate calcium
absorption. There is also a
need for a vitamin D and bisphosphonate product to facilitate normal bone
formation and mineralization
white reducing or minimizing the potential for or occurrence of complications
associated with vitamin D
insufficiency, such as hypocalcemia and osteomalacia. There is a need for a
bisphosphonate and vitamin
D product to provide an amount of vitamin D nutrition to facilitate normal
bone formation and
mineralization in subjects undergoing bisphosphonate therapy. There is also a
need for a bisphosphonate
and a vitamin D combination for oral administration according to a continuous
dosing schedule at dosing
intervals less frequent than daily dosing and more frequent than dosing at 6
months or longer intervals.
There is a need for a single product comprising vitamin D and a
bisphosphonate, suitable for once-weekly
dosing, to increase the convenience of vitamin D intake and to increase
patient compliance with
recommended vitamin D nutrition during bisphosphonate therapy. Furthermore,
there is a need for
methods of preparing and administering such vitamin D/bisphosphonate
compositions.
IS
SUMMARY OF THE INVENTION
The present invention provides pharmaceutical compositions comprising a
bisphosphonate compound and a vitamin D compound. Embodiments of the present
invention, for
example, include pharmaceutical compositions comprising a bisphosphonate
compound, or
pharmaceutically acceptable salts, derivatives or hydrates of the
bisphosphonate, or mixtures thereof, and
a vitamin D compound, such as a pharmaceutical grade vitamin D compound. In
embodiments, the
vitamin D compound comprises cholecalciferol. In embodiments, the
bisphosphonate comprises, for
example, alendronate, a pharmaceutically acceptable salt of alendronate (for
example, sodium, potassium,
calcium, magnesium, or ammonium, or a hydrate of any of those salts), such as
alendronate monosodium,
alendronate monosodium monohydrate, or alendronate monosodium trihydrate. In
an embodiment of the
present invention, the pharmaceutical composition comprises cholecalciferol
and alendronate
monosodium trihydrate. (See FIG. 2) Compositions of the present invention may
be in the form of
compressed, coated or uncoated tablets, capsules, elixirs, emulsions, or other
acceptable dosage forms.
In embodiments of compositions of the present invention, the bisphosphonate
(or
pharmaceutically effective salts, derivatives or hydrates thereof, or mixtures
thereof) is present in
pharmaceutically effective amounts, for example from about 0.05 mg to about
1120 mg. 1n the same or
other embodiments of the compositions of the present invention, the vitamin D
compound is present in
pharmaceutically effective amounts, for example from about 100 IU to about
60,000 IU of a vitamin D
compound (40 IU of vitamin D has a mass of approximately 1 microgram). In
other embodiments, the
present invention relates to a pharmaceutical composition comprising from
about 100 IU to 36,0001U of
-6-
CA 02467715 2004-05-19
a vitamin D compound, and from about 5 mg to about 560 mg, on a bisphosphonic
acid active basis, of a
bisphosphonate, pharmaceutically acceptable salts, derivatives or hydrates
thereof, or mixtures thereof.
1n other embodiments, the present invention relates to a pharmaceutical
composition comprising from
about 100 IU to 28,0001U of a vitamin D compound, and from about 5 mg to about
280 mg, on a
bisphosphonic acid active basis, of the bisphosphonate, pharmaceutically
acceptable salts, derivatives or
hydrates thereof, or mixtures thereof. In other embodiments, the present
invention relates to a
pharmaceutical composition comprising from about 100 IU to 8,400 IU of a
vitamin D compound, and
from about 5 mg to about 280 mg, on a bisphosphonic acid active basis, of the
bisphosphonate,
pharmaceutically acceptable salts, derivatives or hydrates thereof or mixtures
thereof. In other
embodiments, the present invention relates to a pharmaceutical composition
comprising from about 100
IU to 5,600 IU of a vitamin D compound, and from about 5 mg to about 280 mg,
on a bisphosphonic acid
active basis, of the bisphosphonate, pharmaceutically acceptable salts,
derivatives or hydrates thereof, or
mixtures thereof. In other embodiments, the present invention relates to a
pharmaceutical composition
comprising from about 100 IU to 4,200 IU of a vitamin D compound, and from
about 5 mg to about 280
l 5 mg, on a bisphosphonic acid active basis, of the bisphosphonate,
pharmaceutically acceptable salts,
derivatives or hydrates thereof, or mixtures thereof. An embodiment of the
present invention is a
pharmaceutical composition comprising about 2,800 IU of a vitamin D and about
70 mg, on an
bisphosphonic acid active basis, of a bisphosphonate or pharmaceutically
acceptable salts, derivatives or
hydrates of a bisphosphonate, or mixtures thereof. An embodiment of the
present invention is a
pharmaceutical composition comprising about 2,800 IU of cholecalciferol and
about 70 mg, on an
alendronic acid active basis, of alendronate or pharmaceutically acceptable
salts, derivatives or hydrates
of alendronate, or mixtures thereof. In a further embodiment, the
cholecalcifeo) is pharmaceutical grade.
An example of a composition of the present invention is a tablet comprising
alendronate
sodium, cholecalciferol or cholecalciferol granules containing an appropriate
equivalent amount of
cholecalciferol and additional excipients, such as suitable fillers, diluents,
binders, lubricants, glidants,
disintegrants and the like. A further example of a composition of the present
invention is a tablet
comprising alendronate sodium, cholecalciferol or cholecalciferol granules
containing an appropriate
equivalent amount of cholecalciferol, lactose, lactose anhydrous,
microcrystalline cellulose, colloidal
silicon dioxide, croscarmellose sodium, and magnesium stearate. Compositions
of the present invention
may be formulated to meet various purity and stability criteria, for example,
to comprise less than 1% by
weight of each isomer of cholecalciferol (relative to total cholecalciferol),
after storage for 24 months at
about < 30 °C and about < 30 % relative humidity. Compositions of the
present invention may also be
formulated to comprise less than about 5% total degradants of cholecalciferol
after storage for 24 months
at about < 30 °C and about < 30 % relative humidity, For storage
purposes, the effect that environmental
_7_
CA 02467715 2004-05-19
humidity may have on the purity and stability of the composition can be
eliminated by using appropriate
packaging, such as aluminum foil blister packs or HDPE bottles with dessicant.
In addition, the present invention encompasses methods of manufacturing
compositions
and dosage forms disclosed in this specification, as well as products made
according to those methods.
An embodiment of such a method comprises preparing a powder blend comprising a
bisphosphonate,
such as alendronate, compacting the powder blend to form a mixture, milling
and blending the mixture
with a vitamin D compound to form a final granule blend, and compressing the
final granule blend, for
example, to form tablets. In a further embodiment, the final granule blend may
be lubricated before it is
compressed. In an embodiment, the powder blend comprises alendronate,
colloidal silicon dioxide,
lactose anhydrose, microcrystalline cellulose, croscarmellose sodium and
magnesium stearate. In another
embodiment, the roller compacting of the powder blend forms compacted ribbons,
which may be milled,
blended with cholecalciferol granules, lubricated, and compressed into a solid
dosage form. The
advantages of the methods of manufacturing of the present invention include
increased stability of the
vitamin D compound in a bisphosphonate/vitamin D composition, as well as
increased uniformity of the
particle size of the individual compounds used in the compositions of the
present invention.
Additionally, the alendronate powder blend may be pre-blended with one or more
of the
excipients first, blended with the rest of the excipients, and then roller
compacted.
Other methods of manufacturing bisphosphonate granules, such as, for example
alendronate granules, include but are not limited to, slugging, as well as wet
granulation methods. If the
bisphosphonate granules are manufactured using slugging, then the
bisphosphonate (such as alendronate)
powder blend may be compressed into a non-ribbon compact and then milled into
granules, which may
then be blended with a vitamin D compound to form a granule blend, which may
then be compressed into
a solid dosage form (e.g., a tablet). Alternatively, the bisphosphonate and
all of the excipients may be wet
granulated with a granulating liquid (e.g., water), then dried, milled,
blended with a vitamin D compound,
and processed to a final dosage form (e.g., a tablet).
In addition to the above mentioned methods, a direct blend method can also be
employed
by blending the bisphosphonate, all of the excipients, and the vitamin D
compound together and then
compressing into a tablet or encapsulting into a capsule or other solid dosage
forms. Further description
of possible methods for manufacturing the bisphosphonate granule are described
in U.S. Patent
5,358,941; U.S. Patent 5,882,656 and PCT Publication WO 95/29679.
It is also possible to manufacture the bisphosphonate/vitamin D composition as
described
above and then perform a drying step in order to reduce the moisture level of
the composition.
Additionally, it is possible to package the composition with a dessicant in
order to reduce the moisture
level.
_g_
CA 02467715 2004-05-19
The present invention also encompasses methods for preventing, reducing,
inhibiting or
treating metabolic bone diseases. Metabolic bone diseases include, but are not
limited to, osteoporosis,
post-menopausal osteoporosis, steroid-induced osteoporosis, male osteoporosis,
other disease-induced
osteoporosis, idiopathic osteoporosis, and glucocorticoid-induced
osteoporosis. The present invention
also encompasses methods for preventing, reducing, inhibiting or treating
osteoporosis, conditions
associated with osteoporosis, and other diseases and conditions associated
with abnormal bone resorption.
Such other diseases and conditions may include, as further examples,
metastatic bone disease,
hypercalcemia of malignancy, periprosthetic osteolysis, inflammatory
arthritis, and other diseases and
conditions identified herein in a human or other mammal. Additionally, the
present invention relates to a
method for eliciting a disease modifying effect on an arthritic condition in a
mammal which comprises
administering to the mammal a therapeutically effective amount of a vitamin
D/bisphosphonate
composition. The present invention also relates to methods for eliciting a
disease modifying effect on
subchondral bone sclerosis, preventing osteophyte formation or progression and
preventing joint
destruction in a mammal, which comprise administering to the mammal a
therapeutically effective
amount of a vitamin D/bisphosphonate composition. The present invention also
encompasses a method
for reducing the risk of bone fractures in a mammal which comprises
administering a unit dosage of the
vitamin D/bisphosphonate composition.
Embodiments of such methods encompass administering the compositions of the
present
invention to mammals, including humans. Such compositions may be administered
at intervals of once-
weekly, bi-weekly, monthly, twice-monthly, and bi-monthly. In such methods,
vitamin D is provided by
compositions of the present invention during bisphosphonate therapy while
minimizing the occurrence of
or potential for the complications associated with vitamin D insufficiency.
Accordingly, compositions
and methods of the present invention may be useful in mammals identified as
having or being susceptible
to vitamin D insufficiency or deficiency, or desiring adequate amounts of
vitamin D. In an embodiment,
once-weekly dosing to treat osteoporosis or another disease or condition
associated with abnormal bone
resorption and to minimize the risk or complications from vitamin D
insufficiency, is maintained on a
continuous schedule until the desired therapeutic effect is achieved. An
embodiment of the methods of
the present invention includes administering once weekly, to a mammal
suffering from osteoporosis, a
tablet comprising about 2,800 IU cholecalciferol and about 70 mg alendronate
or pharmaceutically
acceptable salts, derivatives or hydrates of alendronate, or mixtures thereof.
1n embodiments, the
therapeutic effect of once-weekly administration of the vitamin D compound of
a composition of the
present invention is substantially similar to the therapeutic effect of a
recommended daily dosage of
vitamin D, for example, 400 IU, 600 IU or 800 IU vitamin D daily.
The present invention additionally encompasses methods for measuring
cholecalciferol in
the pharmaceutical compositions (e.g., stability) comprising cholecalciferol
and a bisphosphonate. An
-9-
CA 02467715 2004-05-19
embodiment of such a method comprises extracting cholecalciferol from such a
composition into a first
solution to form a second solution, separating a sample containing
cholecalciferol from the second
solution, and detecting an amount of the cholecalciferol in the sample, for
example, using reverse-phase
high performance liquid chromatography. Embodiments of such methods provide
increased measurement
sensitivity and may advantageously be used with compositions of the present
invention to distinguish
between cholecalciferol and pre-cholecalciferol, or between isomers of
cholecalciferol and pre-
cholecalciferol, or to detect cholecalciferol or pre-cholecalciferol ester
adducts.
The present invention further encompasses methods for measuring
cholecalciferol in
plasma after administration of the bisphosphonate/cholecalciferol compositions
of the present invention.
An embodiment of such a method comprises administering to a mammal a
composition comprising
alendronate and cholecalciferol, obtaining from the mammal a plasma sample,
extracting the
cholecalciferol from the plasma sample to form a first solution, reacting the
cholecalciferol in the first
solution with a dienophile to form one or more diets-alder addition products
of cholecalciferol, separating
the diets-alder addition products of cholecalciferol using high performance
liquid chromatography
(HPLC) separation, and detecting an amount of cholecalciferol in the sample
using mass spectroscopy.
Embodiments of such methods provide an increased measurement sensitivity and
may advantageously be
used with the compositions of the present invention, for example, to measure
the pharmacokinetic effects
of administration of the compositions of the present invention.
The present invention also provides methods of measuring the pharmacokinetic
effect
over time of administering the compositions of the present invention,
including, for example, as reflected
by the total urinary excretion, area under the serum-concentration-versus-time
curve (AUC), steady state
maximum plasma concentration (C,"~), time of C",~ (Tm~), and plasma
concentration median apparent
half life (t,n) of a tablet comprising about 70 mg alendronate and about 2,800
IU cholecalciferol.
The present invention can comprise, consist of, or consist essentially of the
essential as
well as optional ingredients, components, steps and methods described or
claimed herein. Further
features, advantages and embodiments of the invention, its nature and various
advantages, will become
more apparent from the following detailed description, and from practice of
the invention.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 depicts the metabolism of vitamin D3.
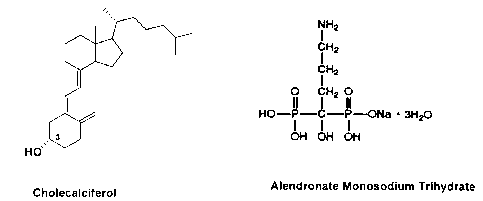
FIG. 2 depicts the chemical structures of cholecalciferol and alendronate
monosodium
trihydrate.
FIG. 3 depicts the chemical structures of vitamin DZ and of vitamin D3.
FIG. 4 shows a schematic diagram summarizing an embodiment of a method of
preparing
compositions of the present invention.
- 10-
CA 02467715 2004-05-19
vitamin D3.
FIG. 5 depicts thermal and photochemical isomerizations and
transesterifications of
FIG. 6 shows the results of a radiolabel study of vitamin D3 degradation,
DETAILED DESCRIPTION OF THE INVENTION
The term "abnormal bone resorption," as used herein, means a degree of bone
resorption
that exceeds the degree of bone formation, either locally, or in the skeleton
as a whole, or alternatively,
can be associated with the formation of bone having an abnormal structure.
"Arthritic condition" or "arthritic conditions" refers to a disease wherein
lesions, some of
which are inflammatory, are confined to the joints or any inflammatory
conditions of the joints, most
notably rheumatoid arthritis. (Academic Press Dictionary of Science
Technology; Academic Press; 1st
edition, January 15, 1992). An arthritic condition can be caused by
inflammation, trauma or infection.
The compositions of the present invention are also useful, alone or in
combination, to treat or prevent
arthritic conditions or symptoms/diseases involving arthritis, such as
amyloidosis; ankylosing spodylitis;
bacterial arthritis; basic calcium phosphate crystal deposition disease;
Behcet's disease; bursitis and
tendinitis; CPPD deposition disease; calcific tendonitis; carpal tunnel
syndrome; Ehlers-Danlos
syndrome; enteropathic arthritis; Felty's syndrome; fibromyalgia; gout; fungal
arthritis;
hemoglobinopathy; hemophilic arthropathy; hypertrophic osteoarthropathy;
infectious arthritis;
inflammatory bowel disease; juvenile arthritis; juvenile rheumatoid arthritis;
lupus erythematosus; lyme
disease; marfan syndrome; mixed connective tissue disease; multicentric
reticulohistocytosis, myopathies;
myositis; osteoarthritis; osteonecrosis; osteonecrosischondrodystrophy;
polyarteritis; polymyalgia
rheumatica; psoriatic arthritis; Raynaud's phenomenon; reflex sympathetic
dystrophy syndrome; Reiter's
syndrome; relapsing polychondritis; rheumatoid arthritis; rheumatic fever;
sarcoidosis; septic arthritis;
scleroderma; Sjogren's syndrome; spondyloepiphyseal dysplasia; systemic lupus
erythematosus; and viral
arthritis. Unlike rheumatoid arthritis, osteoarthritis is a connective tissue
disease, with pathology arising
from mechanical insult-induced articular cartilage degeneration, subchondral
bone remodeling and
limited synoviticc inflammation response. The net outcome of these activities
is joint deformity
secondary to erosion of articular cartilage, peri-articular endochondral
ossification/osteophytosis,
subchondral bone sclerosis and cyst formation. See, Oettmeier, R., and K.
Abendroth, 1989,
"Osteoarthritis and bone: osteologic types of osteoarthritis of the hip,"
Skeletal Radiol. 18:165-74; Cutolo
M, Seriolo B, Villaggio B, Pizzorni C, Craviotto C, Sulli A. Ann. N.Y. Acad.
Sci. 2002 Jun;966:131-42;
Cutolo, M. Rheum Dis Clin North Am2000 Nov;26(4):881-95; Bijlsma JW, Van den
Brink HR. Am J
Reprod Tmmunol 1992 Oct-Dec;28(3-4):231-4; Jansson L, Holmdahl R.; Arthritis
Rheum 2001
Sep;44(9):2168-75; and Purdie DW. Br Med Bull 2000;56(3):809-23; See also
Merck Manual, 17th
edition, pp. 449-451. An embodiment of the present invention encompasses the
treatment, reduction,
-11-
CA 02467715 2004-05-19
inhibition or prevention of an arthritic condition which comprises
administering a therapeutically
effective amount of a composition of the present invention. Another embodiment
is the treatment,
reduction, inhibition or prevention of osteoarthritis which comprises
administering a therapeutically
effective amount of a composition of the present invention.
The term "bisphosphonate," as used herein, corresponds to the chemical
formula:
HO R~ OH
O P C P O
HO R2 OH
where R, is independently selected from the group consisting of H, OH, and CI,
Rz is independently
selected from CH3, Cl, CHZCHZNHz, (CHz)3NHz, CHz-3-pyridyl, CHz-S-phenyl-Cl,
CHZCHzN(CH3)(pentyl), CHz-imidazole, CHz-2-imidazo-pyridinyl, N-(cycloheptyl),
CHzCH(CH3)z,
(CHz)SNHz, and CHz-1-pyrrolidinyl, and combinations thereof. In embodiments of
the present invention,
R, is OH and Rz is a 3-aminopropyl moiety, so that the resulting compound is a
4-amino-I-
hydroxybutylidene-l,l-bisphosphonate, i.e., alendronate.
Pharmaceutically acceptable salts, derivatives, and hydrates of the
bisphosphonates are
also encompassed by the compounds and methods of the present invention. Non-
limiting examples of
salts include those selected from the group consisting alkali metal, alkaline
metal, ammonium, and
mono-, di-, tri-, or tetra-C,-C3o-alkyl-substituted ammonium, including
sodium, potassium, calcium,
magnesium, and ammonium salts. Non-limiting examples of derivatives include
those selected from the
group consisting of esters and amides. Also encompassed within the scope of
the present invention are
the various hydrates and other solvates of bisphosphonates, and
pharmaceutically acceptable salts thereof.
Also encompassed within the scope of the present invention are hydrates of
alendronate, including but
not limited to, hydrates with water content between about one to twelve
percent, and their crystalline
forms. Non-limiting examples of hydrates of alendronate and other
bisphosphonates include the
monohydrate, dehydrate, trihydrate, hemihydrate, 1/4 hydrate, 1/3 hydrate, 2/3
hydrate, 3/4 hydrate, 5/4
hydrate, 4/3 hydrate, and 3/2 hydrate.
Non-limiting examples of bisphosphonates useful in the present invention
include the
following:
Alendronic acid, 4-amino-1-hydroxybutylidene-I,1-bisphosphonic acid.
Alendronate (also known as alendronate sodium or monosodium trihydrate, or by
the
trademark FOSAMAX~), 4-amino-1-hydroxybutylidene-1,1-bisphosphonic acid
monosodium trihydrate.
-12-
CA 02467715 2004-05-19
(Alendronic acid and alendronate are described, for example, in U.S. Patents
4,922,007, to Kieczykowski
et al., issued May 1, 1990, and 5,019,651, to Kieczykowski, issued May 28,
1991 ).
Cycloheptylaminomethylene-1,1-bisphosphonic acid, YM 175, Yamanouchi
(incadronate
or cimadronate), as described, for example, in U.S. Patent 4,970,335, to
Isomura et al., issued November
13, 1990.
1,1-dichloromethylene-1,1-diphosphonic acid (clodronic acid), and the disodium
salt
(clodronate, Procter and Gambie), are, for example, described in Belgium
Patent 672,205 (1966) and J.
Org. Chem 32, 411 1 (1967).
1-hydroxy-3-(1-pyrrolidinyl)-propylidene-I,1-bisphosphonic acid (EB-1053).
l0 I-hydroxyethane-l,l-diphosphonic acid (etidronic acid).
1-hydroxy-3-(N-methyl-N-pentylamino)propylidene-1,1-bisphosphonic acid, also
known
as BM-210955, Boehringer-Mannheim (ibandronate), is described, for example, in
U.S. Patent No.
4,927,814, issued May 22, 1990.
[1-hydroxy-2-imidazopyridin-(l,2-a)-3-ylethylidene]-bis-phosphonate
(minodronate).
15 6-amino-1-hydroxyhexylidene-l,l-bisphosphonic acid (neridronate).
3-(dimethylamino)-1-hydroxypropylidene-1,1-bisphosphonic acid (olpadronate).
3-amino-1-hydroxypropylidene-1,1-bisphosphonic acid (pamidronate).
[2-(2-pyridinyl)ethylidene]-l,l-bisphosphonic acid (piridronate) is described,
for
example, in U.S. Patent No. 4,761,406.
20 I-hydroxy-2-(3-pyridinyl)-ethylidene-l,l-bisphosphonic acid (risedronate).
(4-chlorophenyl)thiomethane-l,l-disphosphonic acid (tiludronate) as described,
for
example, in U.S. Patent 4,876,248, to Breliere et al., October 24, 1989.
i-hydroxy-2-(1H-imidazol-1-yl)ethylidene-1,1-bisphosphonic acid (zoledronate).
In embodiments of the present invention, the bisphosphonate is selected from
the group
25 consisting of alendronate, pharmaceutically acceptable salts, derivatives
and hydrates thereof, and
mixtures thereof. The pharmaceutically acceptable salt of alendronate may be
selected from the group
consisting of the sodium, potassium, calcium, magnesium, and ammonium salt of
alendronate, and may
be alendronate monosodium or a hydrate thereof, including for example
alendronate sodium monohydrate
or alendronate sodium trihydrate.
30 1n an embodiment, the compositions of the present invention comprise
alendronate
sodium (monosodium salt of 4-amino-1-hydroxybutylidene-1, 1-bisphosphate),
which is a member of the
nitrogen-containing bisphosphonate class of drugs.
It should be noted that the terms "bisphosphonate" and "bisphosphonates," as
used herein
in referring to the therapeutic agents of the present invention, are meant to
also encompass
35 diphosphonates, bisphosphonic acids, and diphosphonic acids, as well as
salts, derivatives and hydrates of
-13-
CA 02467715 2004-05-19
these materials. The use of a specific nomenclature in referring to the
bisphosphonate or bisphosphonates
is not meant to limit the scope of the present invention, unless specifically
indicated. Because of the
mixed nomenclature currently in use by those of ordinary skill in the art,
reference to a specific weight or
percentage of a bisphosphonate compound in the present invention is on an acid
active weight basis,
unless indicated otherwise herein. Bisphosphonate doses calculated on the
basis of their salt, derivative
or hydrate forms are included within the dosage ranges of the present
invention on the basis of their
bisphosphonic acid active weights. Additionally, the doses of ali hydrate
forms of alendronate are
calculated on the basis of the alendronic acid active weight. For instance,
the doses of the monohydrate,
trihydrate, hemihydrate and all other hydrate forms of alendronate and its
salts, are calculated on the basis
of their alendronic acid active weights. As another example, the phrase "about
70 mg of a bone
resorption inhibiting bisphosphonate selected from the group consisting of
alendronate, pharmaceutically
acceptable salts, derivatives and hydrates thereof, and mixtures thereof, on
an alendronic acid active
weight basis" means that the amount of the bisphosphonate compound selected is
calculated based on 70
mg of alendronic acid.
As used throughout this specification and claims, the terms "bisphosphonic
acid" and
"alendronic acid" include the related bisphosphonic acid forms,
pharmaceutically acceptable salt forms
and equilibrium mixtures of these. The terms include crystalline, hydrated
crystalline, and amorphous
forms of alendronic acid and pharmaceutically acceptable salts thereof. The
term "alendronic acid"
specifically includes, but is not limited to, anhydrous alendronate
monosodium, alendronate monosodium
hemihydrate, alendronate monosodium monohydrate, alendronate monosodium
trihydrate, anhydrous
alendronate dipotassium, and alendronate dipotassium pentahydrate. Alendronate
monosodium
monohydrate and other crystalline forms of alendronate sodium are disclosed in
U.S. Patent No.
6,281,381. Potassium salts of alendronic acid, and hydrates thereof, are
disclosed in International Patent
Publication WO 99/20635.
While it is conventional to dose and calculate the dosages of bisphosphonates
on the
basis of bisphosphonic acid active weight, bisphosphonate dosages can be
calculated and administered
based on other salt or hydrate forms. For example, dosages of the
bisphosphonate risedronate are
calculated based on the weight of the anhydrous risedronate sodium salt.
According to the Physician's
Desk Reference (55'" Edition, page 2664, (2001)), for example, each tablet of
risedronate contains the
equivalent of 5 mg or 30 mg of anhydrous risedronate sodium, in the form of
the hemi-pentahydrate with
small amounts of monohydrate.
The term "cholecalciferol granules" as used herein refers to the granules that
contain
cholecalciferol and may also contain pre-vitamin D3, isomers of vitamin D3,
transesterfied vitamin D3 or
its isomers andlor additional excipients.
- 14-
CA 02467715 2004-05-19
The terms "continuous schedule" or "continuous dosing schedule," as used
herein, mean
that the dosing regimen is repeated until the desired therapeutic effect is
achieved. The continuous
schedule or continuous dosing schedule is distinguished from cyclical or
intermittent administration.
The term "Dry Vitamin D3 100 granules," as used herein, refers to Dry Vitamin
D3 100,
Gelatin Coated, Pharmaceutical Grade granules which are sold commercially by
BASF.
The term "generalized bone loss," as used herein, means bone loss at multiple
skeletal
sites or throughout the skeletal system. The term "localized bone loss," means
bone loss at one or more
specific, defined skeletal sites.
The terms "human in need of treatment," "human in need of prevention," "human
in need
thereof," and "human at risk thereof," as used herein, refer to a human in
need of treatment for a disease
condition, in need of prevention, mitigation, inhibition or reduction of a
disease condition, or at risk of
developing a disease condition, as determined by a clinician or researcher.
The term "1U," as used herein, means International Units. It is customary to
use
International Units (IU) when stating the potency and dosage of vitamin D. One
International Unit (IU)
is defined as the specific biologic activity of 0.025 pg of the crystalline
international standard or pure
vitamin D. Stated in another way, one microgram of vitamin D is approximately
40 International Units.
The terms "mammal in need of treatment," "mammal in need of prevention,"
"mammal
in need thereof," and "mammal at risk thereof," as used herein, refer to a
mammal in need of treatment
for a disease condition, in need of prevention, mitigation, inhibition or
reduction of a disease condition,
or at risk of developing a disease condition, as determined by a clinician or
researcher.
"Once-weekly dosing," as used herein, means that a unit dosage, for example a
unit
dosage of bisphosphonate and a vitamin D compound, is administered once a
week, i.e., once during a
seven-day period, preferably on the same day of each week. In the once-weekly
dosing regimen, the unit
dosage is generally administered about every seven days. A non-limiting
example of a once-weekly
dosing regimen would entail the administration of a unit dosage of the
bisphosphonate and a vitamin D
compound every Sunday. It is customarily recommended that a unit dosage for
once-weekly
administration is not administered on consecutive days, but the once-weekly
dosing regimen can include
a dosing regimen in which unit dosages are administered on two consecutive
days falling within two
different weekly periods.
"Osteophyte" as used herein refers to newly formed bony structures located at
the joint
margins, and their occurrence is strongly associated with the late stage of
osteoarthritis progression. The
current hypothesis is that osteophytes originate from activated periosteum
leading to new cartilaginous
outgrowths that eventually turns into bone by the process of endochondral bone
formation.
"Pharmaceutically acceptable" as used herein with reference to salts, esters,
hydrates and
derivates of a bisphosphonate (such as alendronate) means that the salts,
derivatives or hydrates of the
-15-
CA 02467715 2004-05-19
bisphosphonate has the same general pharmacological properties as the free
acid form from which they
are derived and are acceptable from a toxicity viewpoint.
The term "pharmaceutically effective amount," as used herein, means that
amount of a
compound, for example a bisphosphonate compound or a vitamin D compound, that
will elicit a desired
therapeutic effect or response when administered in accordance with a
treatment regimen. A
pharmaceutically effective amount of bisphosphonate, for example, is an amount
administered according
to a treatment regimen that is sufficient to elicit prevention, reduction,
inhibition or treatment of abnormal
bone resorption, for instance.
The term "pharmaceutical grade," as used herein, means of a sufficient quality
and
potency so as to conform to applicable United States Pharmacopoeia (USP) and
European Pharmacopoeia
(Ph. Eur.) compendial requirements. While at this time there is no USP
monograph for a formulated
vitamin D3 product, an applicable Ph.Eur. monograph has been published. A
"pharmaceutical grade"
cholecalciferol, for example, generally is of a superior grade than the
vitamin D commonly used in
nutritional supplements.
The terms "preventing, inhibiting, reducing or treating," as used herein,
include
addressing abnormal bone resorption (and the resultant physiological
conditions - e.g., osteoporosis)
through the direct or indirect alteration of osteoclast formation or activity,
and encompass prevention,
inhibition, reduction or treatment of bone loss, especially the inhibition of
removal of existing bone either
from the mineral phase and/or the organic matrix phase, through direct or
indirect alteration of osteoclast
formation or activity. These terms also mean addressing other disease states
or conditions in such fashion
as to promote relief from the disease or condition.
The term "until the desired therapeutic effect is achieved," as used herein,
means that a
composition, for example, a bisphosphonate and cholecalciferol composition, is
administered according
to a chosen dosage schedule, or a course of therapy is followed, up to the
time that the clinical or medical
effect sought for the disease or condition is observed by the clinician or
researcher. For methods of
treatment of the present invention, the bisphosphonate compound may be
continuously administered until
the desired change in bone mass or structure is observed. In such instances,
achieving an increase in bone
mass, preventing further reduction in bone mass or replacing abnormal bone
structure with more normal
bone structure, are among the desired objectives. For methods of the present
invention, the
bisphosphonate compound may be continuously administered for as long as
necessary to prevent the
undesired condition. In such instances, maintenance of bone mass density is
often an objective. Non-
limiting examples of administration periods can range from about 2 weeks to
the remaining life span of
the mammal. For humans, administration periods can range from about 2 weeks to
the remaining life
span of the human, preferably from about 2 weeks to about 40 years, more
preferably from about 1 month
- 16-
CA 02467715 2004-05-19
to about 35 years, more preferably from about 6 months to about 30 years, and
most preferably from
about 1 year to about 20 years.
The term "vitamin D," as used herein, means both vitamin DZ and vitamin D3
which have
the chemical structures shown in FIG. 3. The phrase "metabolites of vitamin D"
and "derivatives of
vitamin D," as used herein, mean metabolites and derivatives of vitamin DZ and
vitamin D3. The term
"vitamin D compound," as used herein, means vitamin DZ (ergocalciferol) and
vitamin D3
(cholecalciferol), 7-dehydrochlosterol and pre-vitamin Dz and pre-vitamin D3,
as well as isomers of or
esters of any of 7-dehydrocholesterol, vitamin Dz, vitamin D~, pre-vitamin Dz,
or pre-vitamin D3, or
mixtures thereof. At relevant physiological temperatures, vitamin DZ and
vitamin D3 are at equilibrium
with their respective pre-vitamin isomers, although that equilibrium is
shifted in favor of vitamin DZ and
vitamin D3. In the present invention, the term "vitamin D compound" does not
include metabolites of
vitamin D, such as, for example, 25-hydroxycholecalciferol or calcitriol or
their analogs, nor does the
term include the active hormone calcitriol or its analogs. The terms vitamin
D3 and cholecalciferol, are
used interchangeably herein, unless expressly otherwise indicated.
The present invention provides compositions comprising a bisphosphonate, or
pharmaceutically acceptable salts, derivatives or hydrates of the
bisphosphonate, or mixtures thereof, and
a vitamin D compound. Tn an exemplary embodiment, the bisphosphonate compound
is selected from
alendronate sodium, alendronate sodium monohydrate or alendronate sodium
trihydrate, and the vitamin
D compound is cholecalciferol.
The precise dosage of the bisphosphonate and the vitamin D compound will vary
with the
dosing schedule, the oral potency of the particular bisphosphonate chosen, the
age, size, sex and
condition of the mammal or human, the nature and severity of the disorder to
be treated, and other
relevant medical and physical factors. Thus, a precise pharmaceutically
effective amount cannot be
specified in advance, but can be readily determined by the caregiver or
clinician. Appropriate amounts
can be determined by routine experimentation from animal models and human
clinical studies.
Generally, a pharmaceutically effect amount of bisphosphonate is chosen
according to a continuous
dosing schedule until the desired therapeutic effect is achieved. For humans,
an effective oral dose of
bisphosphonate is typically from about 0.0001 mglkg to about 100 mg/kg body
weight and preferably
about 0.0005 to about 20 mg/kg of body weight for a 75 kg subject.
In embodiments of the present invention, an appropriate amount of the vitamin
D
compound is chosen to provide adequate vitamin D nutrition during the dosing
interval without
interfering with the bisphosphonate's ability to obtain a bone resorption
inhibiting effect. For oral
compositions of the present invention comprising alendronate, pharmaceutically
acceptable salts,
derivatives or hydrates of alendronate, or mixtures thereof, and a vitamin D
compound, an amount of the
vitamin D compound comprises from about 100 IU to about 60,000 IL1. Non-
limiting examples of an oral
-17-
CA 02467715 2004-05-19
amount of the vitamin D compound in embodiments of the present invention
include, but are not limited
to, dosages of 700, 1,400 IU, 2,800 IU, 4,200 IU, 56001U, 7,0001U, 8,400 IU,
14,000 IU, 28,000 IU,
36,000 IU and 60,000 IU of the vitamin D compound.
For oral compositions comprising a vitamin D compound and a pharmaceutically
S effective amount of alendronate, or pharmaceutically acceptable salts,
derivatives or hydrates of the
alendronate, or mixtures thereof, an oral pharmaceutically effective amount of
alendronate typically
comprises from about 0.05 mg to about 1120 mg of the alendronate compound, on
an alendronic acid
weight basis. Non-limiting examples of an oral pharmaceutically effective
amount of alendronate in
embodiments of the present invention include, but are not limited to, dosages
of about 2.5 mg, 5 mg, 8.75
mg, 10 mg, 17.5 mg, 35 mg, 40 mg, 70 mg, 140 mg, 280 mg, 560, and 1120 mg of
alendronate, each on
an alendronic acid weight basis.
A bisphosphonate and vitamin D composition of the present invention is
typically administered in
admixture with suitable pharmaceutical diluents, excipients, or carriers,
suitably selected with respect to a
dosage form for oral administration. Examples of oral dosage forms include
tablets (including
compressed, coated or uncoated), capsules (each of which includes sustained
release or timed release
formulations), hard or soft gelatin capsules, pellets, pills, powders,
granules, elixirs, tinctures, slurries,
effervescent compositions, films, sterile solutions or suspensions, syrups and
emulsions and the like.
Likewise, it may also be administered in intravenous (bolus or infusion),
intraperitoneal, topical (e.g.,
ocular eyedrop), intranasal, inhaled, subcutaneous, intramuscular or
transdermal (e.g., patch) form,
metered aerosol or liquid sprays, drops, ampoules, auto-injector devices or
suppositories all using forms
well known to those of ordinary skill in the pharmaceutical arts. An effective
but non-toxic amount of the
compositions desired can be employed. The compositions are intended for oral,
parenteral, intranasal,
sublingual, or rectal administration, or for administration by inhalation or
insufflation. Formulation of the
compositions according to the invention can conveniently be effected by
methods known from the art, for
example, as described in Remineton's Pharmaceutical Sciences. l7'h ed., 1995.
For example, for oral administration in the form of a tablet, capsule, pellet,
or powder,
the active ingredients can be combined with an oral, non-toxic,
pharmaceutically acceptable inert carrier
such as lactose, starch, sucrose, glucose, methyl cellulose, magnesium
stearate, mannitol, sorbitol,
croscarmellose sodium and the like; for oral administration in liquid form,
e.g., elixirs, syrups, slurries,
emulsions, suspensions, solutions, and effervescent compositions, the oral
drug components can be
combined with any oral, non-toxic, pharmaceutically acceptable inert can-ier
such as ethanol, glycerol,
water and the like. Moreover, when desired or necessary, suitable binders,
fillers, diluents, lubricants,
compression aids, disintegrants, buffers, coatings, and coloring agents can
also be incorporated. Suitable
binders can include but are not limited to starch, gelatin, natural sugars
such as glucose, anhydrous
lactose, free-flow lactose, beta-lactose, and corn sweeteners, natural and
synthetic gums, such as acacia,
-18-
CA 02467715 2004-05-19
guar, tragacanth or sodium alginate, carboxymethyl cellulose, polyethylene
glycol, waxes, and the like.
Lubricants used in these dosage forms can include but are not limited to
sodium oleate, sodium stearate,
magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the
like. Suitable
disintegrants may be one of several modified starches or modified cellulose
polymers, including
crosscarmellose sodium. Diluents, which may be used as compression aids,
include, but are not limited
to, lactose, dicalcium phosphate, cellulose, microcrystalline cellulose, and
the like. Glidants, which
improve the flow characteristics of a powder mixture, may also be utilized in
the present invention.
Examples of glidants include, but are not limited to, colloidal silican
dioxide, talc, and the like. The
compositions used in the present method can also be coupled with soluble
polymers as targetable drug
l0 carriers. Such polymers can include but are not limited to
polyvinylpyrrolidone, pyran copolymer,
polyhydroxylpropyl-methacrylamide, and the like. Additional excipients, such
as those described in U.S.
Patent 5,358,941; U.S. Patent 5,882,656 and PCT Publication WO 95/29679, may
also be utilized.
An embodiment of the present invention, for example, is a 80 mg to 1500 mg
tablet
including about 0.5% to about 90% alendronate sodium by weight, about 1 % to
about 70%
cholecalciferol granule by weight (equivalent to about 0.0005% to about 20%
cholecalciferol by weight),
about 10% to about 80% lactose anhydrous by weight, about 5% to about 50%
microcrystalline cellulose
by weight, about 0.1% to about 5% colloidal silicon dioxide by weight, about
0.5% to about 10%
croscarmellose sodium by weight, and about 0.5% to about 5% magnesium stearate
by weight.
The weight range of Dry Vitamin D3 100 granules is to ensure 2800 IL1 potency
in each
tablet because the granule contains a potency range of 100,000 IU to 110,000
IU vitamin D3 per gram.
The quantity of lactose anhydrous is adjusted according to the amount of Dry
Vitamin D3 100 granules
added to the tablet in order to maintain a final tablet weight of 325 mg.
Other non-limiting examples of
oral compositions of the present invention comprising a bisphosphonate
compound, such as alendronate,
and a vitamin D compound, are described herein, including in the Examples
below. The Dry Vitamin D3
100 granules contain about 100,000 IL1 vitamin D3 per one gram of granule
weight. Thus, 28 mg of Dry
Vitamin D3 100 granules contains about 28001U of vitamin D3, which is the
equivalent of about 70 pg
vitamin D~.
Bisphosphonate/vitamin D compositions of the present invention, including the
embodiments described herein, may be administered at intervals of once-weekly,
bi-weekly, monthly,
twice monthly, and bi-monthly. For once-weekly dosing with a composition of
the present invention, an
oral pharmaceutically effective amount of alendronate comprises from about
0.05 mg to about 1120 mg
of the alendronate compound, on an alendronic acid active weight basis.
Embodiments of the present
invention providing a weekly oral pharmaceutically effective amount of
alendronate include, but are not
limited to, unit dosages which are useful for preventing osteoporosis
comprising a vitamin D compound
and from about 35 mg to about 70 mg of the alendronate compound; a unit dosage
which is useful for
- 19-
CA 02467715 2004-05-19
treating osteoporosis comprising a vitamin D compound and about 70 mg of the
alendronate compound; a
unit dosage which is useful for treating PageYs disease comprising a vitamin D
compound and about 280
mg of the alendronate compound; and a unit dosage which is useful for treating
metastatic bone disease
comprising a vitamin D compound and about 280 mg of the alendronate compound.
For once-weekly dosing, a pharmaceutically effective amount of a vitamin D
compound
in a bisphosphonate/vitamin D composition of the present invention comprises
from about 100 IU to
about 60,000 IU of vitamin D. Accordingly, in an embodiment of the present
invention, the composition
comprises from about 100 ILT to about 5,600 IU of a vitamin D compound, and a
pharmaceutically
effective amount of alendronate, pharmaceutically acceptable salts,
derivatives or hydrates of
alendronate, or mixtures thereof. In another embodiment, the pharmaceutically
acceptable amount of
alendronate comprises from about 0.05 mg to about 1120 mg, on an alendronic
acid active basis, of
alendronate, pharmaceutically acceptable salts, derivatives or hydrates of
alendronate or mixtures thereof.
For bi-weekly or bi-monthly dosing, a pharmaceutically effective amount of a
vitamin D
compound in a bisphosphonate/vitamin D composition of the present invention
comprises from about 100
IU to about 60,000 IU of vitamin D. In an embodiment of the present invention,
the composition
comprises from about 100 IU to about 8,4001U of a vitamin D compound, and a
pharmaceutically
effective amount of alendronate, pharmaceutically acceptable salts,
derivatives or hydrates of
alendronate, or mixtures thereof. 1n another embodiment, the pharmaceutically
acceptable amount of
alendronate comprises from about 0.05 mg to about 1120 mg, on an alendronic
acid active basis, of
alendronate, pharmaceutically acceptable salts, derivatives or hydrates of
alendronate or mixtures thereof.
For monthly dosing, a pharmaceutically effective amount of a vitamin D
compound in a
bisphosphonate/vitamin D composition of the present invention comprises from
about 100 IU to about
36,000 IU of vitamin D. In an embodiment of the present invention, the
composition comprises from
about 100 ILJ to about 11,2001U of a vitamin D compound, and a
pharmaceutically effective amount of
alendronate, pharmaceutically acceptable salts, derivatives or hydrates of
alendronate, or mixtures
thereof. In another embodiment, the pharmaceutically acceptable amount of
alendronate comprises from
about 0.05 mg to about 1120 mg, on an alendronic acid active basis, of
alendronate, pharmaceutically
acceptable salts, derivatives or hydrates of alendronate or mixtures thereof.
The present invention also encompasses methods for preventing, reducing,
inhibiting and
treating diseases and conditions associated with abnormal bone resorption,
such as osteoporosis. A
person suffering from osteoporosis, i.e., has a bone mineral density (BMD)
which is at least about two or
two and one-half standard deviations below the norm of pre-menopausal women,
would be a candidate
for administration of a composition of the present invention according to a
method of the present
invention. It has been found that vitamin D3, administered in a once-weekly
dose up to seven or more
times than the amounts that would be given on a daily basis, can be
simultaneously co-administered with
-20-
CA 02467715 2004-05-19
a bisphosphonate, such as alendronate, without adversely affecting the
bioavailability of the
bisphosphonate. See, e.g., Example 7. The methods of the present invention do
not have disadvantages
of current methods of treatment which require cumbersome, irregular, or
complicated dosing regimens to
provide adequate vitamin D during bisphosphonate therapy.
As a result, a composition of the present invention, such as a composition
comprising a
bisphosphonate compound, for example alendronate, and a vitamin D compound,
would be effective for
all of the indications for which compositions comprising alendronate or other
bisphosphonates without a
vitamin D compound are effective. The methods and compositions of the present
invention are useful for
reducing or inhibiting bone resorption, and for treating, reducing, inhibiting
or preventing abnormal bone
resorption, and conditions associated therewith. Compositions of the present
invention can thus be used
in humans and other animals to increase bone mass and to prevent, inhibit,
reduce and treat the following
conditions and disease states: bone loss; osteoporosis, including but not
limited to, post-menopausal
osteoporosis, steroid-induced osteoporosis, male osteoporosis, disease-induced
osteoporosis, idiopathic
osteoporosis, and glucocorticoid-induced osteoporosis; osteonecrosis, Paget's
disease; osteoarthritis;
rheumatoid arthritis, other arthritic conditions, abnormally increased bone
turnover; localized bone loss
associated with periprosthetic bone toss or osteolysis; bone fractures;
metastatic bone disease; Gaucher's
disease; avascular necrosis; polyostotic fibrous dysplasia; Charcot's joint;
parasitic disorders;
osteogenesis imperfecta; homocystinuria; lysinuric protein intolerance;
Turner's syndrome;
immobilization; fibrous dysplasia ossificans progressive; fibrogenesis
imperfecta ossium; periodontal
disease; tooth loss; hypercalcemia of malignancy; multiple myeloma;
osteopenia, including but not
limited to, immobilization-induced osteopenia and osteopenia due to bone
metastases; and other bone
diseases and conditions that may be associated with abnormal bone resorption.
The present invention relates to the use of a composition of the instant
invention for the
preparation of a medicament useful in the treatment, reduction, inhibition or
prevention of an arthritic
condition. The present invention also relates to the use of a composition of
the instant invention and an
agent selected from androgen receptor modulator; an inhibitor of osteoclast
proton ATPase; an inhibitor
of HMG-CoA reductase; an osteoblast anabolic agent; calcitonin; Vitamin K2 or
a pharmaceutically
acceptable salts and mixtures thereof, for the preparation of a medicament
useful in the treatment of an
arthritic condition.
In an embodiment of the invention, the arthritic condition is amyloidosis;
ankylosing
spodylitis; bacterial arthritis; basic calcium phosphate crystal deposition
disease; Behcet's disease; bursitis
and tendinitis; CPPD deposition disease; calcific tendonitis; carpal tunnel
syndrome; Ehlers-Danlos
syndrome; enteropathic arthritis; Felty's syndrome; fibromyalgia; gout; fungal
arthritis;
hemoglobinopathy; hemophilic arthropathy; hypertrophic osteoarthropathy;
infectious arthritis;
inflammatory bowel disease; juvenile arthritis; juvenile rheumatoid arthritis;
lupus erythematosus; lyme
-21
CA 02467715 2004-05-19
disease; marfan syndrome; mixed connective tissue disease; multicentric
reticulohistocytosis, myopathies;
myositis; osteoarthritis; osteonecrosis; osteonecrosischondrodystrophy;
polyarteritis; polymyalgia
rheumatica; psoriatic arthritis; Raynaud's phenomenon; reflex sympathetic
dystrophy syndrome; Reiter's
syndrome; relapsing polychondritis; rheumatoid arthritis; rheumatic fever;
sarcoidosis; septic arthritis;
scleroderma; Sjogren's syndrome; spondyloepiphyseal dysplasia; systemic lupus
erythematosus; and viral
arthritis.
An embodiment of the invention is a method of treating, reducing, inhibiting
or
preventing the progression of osteoarthritis in a mammal in need thereof,
comprising administering to the
mammal a therapeutically effective amount of a composition of the instant
invention. It is known in the
literature that osteoarthritis is accompanied with a well-defined changes in
the joints, including erosion of
the articular cartilage surface, peri-articular endochondral
ossification/osteophytosis, and subchondral
bony sclerosis and cyst formation. See Oettmeier R, Abendroth, K,
"Osteoarthritis and bone: osteologic
types of osteoarthritis of the hip," Skeletal Radiol. 1989; 18: 165-74.
Recently, the potential contribution
of subchondral bone sclerosis to the initiation and progression of
osteoarthritis have been suggested.
Stiffened subchondral bone as the joint responding to repetitive impulsive
loading, is less able to
attenuate and distribute forces through the joint, subjecting it to greater
mechanical stress across the
articular cartilage surface. This in turn accelerates cartilage wear and
fibrillate. See Radin, EL and Rose
RM, " Roie of subchondral bone in the initiation and progression of cartilage
damage," Clin. Orthop.
1986; 213: 34-40. Inhibition of excessive subarticular bone resorption by a
composition of the instant
invention could lead to inhibition of subchondral bone turnover, and thus may
have a favorable impact on
osteoarthritis progression.
Another embodiment of the invention is a method of treating, reducing,
inhibiting or
preventing rheumatoid arthritic conditions in a mammal in need thereof,
comprising administering to the
mammal a therapeutically effective amount of a composition of the instant
invention. It is known in the
literature that progressive destruction of the periarticular bone is a major
cause of joint dysfunction and
disability in patients with rheumatoid arthritis. See Goldring SR, "
Pathogenesis of bone erosions in
rheumatoid arthritis" Curr. Opin. Rheumatol. 2002; 14: 406-10. In addition,
generalized bone loss is a
major cause of morbility associated with severe rheumatoid arthritis. The
frequency of hip and spinal
fractures is substantially increased in patients with chronic rheumatoid
arthritis. See Gould A, Sambrook,
P, Devlin J et al, "Osteoclastic activation is the principal mechanism leading
to secondary osteoporosis in
rheumatoid arthritis," J. Rheumatol. 1998; 25: 1282-9. The use of anti-
resorptive agents in the treatment
or prevention of resorption in subarticular bone and of generalized bone loss
represents a rational
approach for pharmacological intervention on the progression of rheumatoid
arthritis. Accordingly, the
compositions of the present invention comprising a bisphosphonate compound and
a vitamin D
-22-
CA 02467715 2004-05-19
compound can be used to treat, reduce, inhibit or prevent the bone loss
associated with rheumatoid
arthritis and other osteoarthritic conditions.
More generally, it is believed that bisphosphonates can be given in the same
formulation
with vitamin D without adversely affecting the bioavailability of the
bisphosphonate. Furthermore, it is
believed that lower doses of vitamin D and higher doses of vitamin D can be
given in the same
formulation with bisphosphonates without affecting their bioavailability. As
an example, a once-weekly
dosage of 2800 IU vitamin D is believed to be effective when administered in
combination with a
bisphosphonate in the compositions of the present invention. It is also known
that vitamin DZ can be used
in place of vitamin D3 with similar results as those found for vitamin D3.
Thus, administration of a
vitamin D compound in the same formulation with a bisphosphonate compound
eliminates the separate
dosing requirements of vitamin D during bisphosphonate treatment and provides
vitamin D nutrition
without adversely affecting the bioavailability and efficacy of the
bisphosphonate.
Patients would benefit from the vitamin D and bisphosphonate combination
because it
provides additional vitamin D nutrition to facilitate normal bone formation
and mineralization and to
I S enhance the efficacy of bisphosphonate treatment. From patient lifestyle
and compliance standpoints, the
methods of the present invention would also be more convenient than daily or
cyclic dosing regimens for
bisphosphonates with additional daily vitamin D administration. As a result of
this invention, patients
may no longer separately need to take vitamin D daily to benefit from
additional vitamin D nutrition
because this invention provides for once-weekly doses of vitamin D. Patients
will not need to keep track
of a complex dosing regimen of separate bisphosphonate and vitamin D
administration. Finally, patients
will be subjected less frequently to the inconvenience of having to take the
bisphosphonate compounds
on an empty stomach and having to fast for at least 30 minutes before or after
dosing. The methods of
the present invention are thus likely to have the advantage of promoting
better patient compliance, and
which in turn can translate into better therapeutic efficacy.
1t is also believed that a bisphosphonate/vitamin D composition would also be
less
irritating to the esophageal, as well as the gastrointestinal system. Since
alendronate could potentially
penetrate into the stratum basale of the stratified squamous epithelium (e.g.,
via its own penetration or via
penetration into a site of local injury caused by abrasive food or other
agent), it could cause an inhibition
of keratinocyte growth, as suggested by its effects on keratinocyte growth in
vitro. See, A. A. Reszka et
al., Mol. Pharmacol., 2001;59(2):193-202. Suppression of growth could slow the
process of epithelial
repair, thus leading to local irritation or ulceration. Autoradiograms of rats
fed radioactive 1,25(OH)Z
vitamin D3 do show evidence of expression of the vitamin D receptor in the
epithelium of the esophagus.
See Stumpf, WE, et al., Histochemistry, 1987;87(1 ):53-8. Levels are below
those seen in the parathyroid
gland.
-23-
CA 02467715 2004-05-19
Therefore, it is believe that coadministration of an active vitamin D3
metabolite (e.g.
1,25(OH)Z cholecalciferol or calcitriol) with alendronate could exacerbate
esophageal irritation through
its differentiative effects on keratinocytes. Both calcitriol and 25-OH-
cholecalciferol have been observed
to effect keratinocytes by inhibiting growth and inducing differentiation. See
K. Matsumoto et al.,
Biochem. Biophys. Acta., 1991;1092(3):311-8. Keratinocyte differentiation is
associated with cell cycle
arrest (growth arrest), and thus the combination of alendronate with an active
vitamin D metabolite could
have synergistic effects on inhibiting growth in the stratum basale. This
could in turn cause a greater
irritant effect. Because oral vitamin D3 (cholecalciferol) requires activation
in the liver and then the
kidney, it is believed that it would not elicit the same local irritant effect
as an active metabolite.
Additionally, it may be possible that normal physiological levels of active
vitamin D3
hormone could assist the body in its attempt to repair sites of local
irritation induced by acute exposure to
alendronate. Because oral vitamin D3 is administered in combination with the
alendronate, and because a
large proportion of the elderly population is vitamin D3 deficient, this may
better enable the body to speed
the healing process along.
It is also believed that a vitamin D/bisphosphonate composition is useful for
the
prevention or treatment of sway. Additionally, it is believed that a vitamin
D/bisphosphonate
composition is useful for reducing falls. It is believed that a vitamin
D/bisphosphonate composition will
increase muscle strength, improve neuromuscular function, reduce body sway and
improve physical
function in elderly people. This would lead to a reduced risk of falls and
thus contribute towards a
reduced risk of bone fractures. Epidemiologic studies demonstrate the high
prevalence of vitamin D
deficiency in elderly in the U.S. See A.N. Exton-Smith et al., Lancet
1966;2:999-1001; RP Heaney et al.,
Osteoporos Int 2000;11:553-5; MJ McKenna, Am J Med 1992;93:69-77; ST Haden et
al., Calcif Tissue
1nt 1999;64:275-9. There is evidence for the effect of vitamin D on extra-
skeletal tissues. See Latham et
al, 2003;51:1219-1226. Additionally, vitamin D receptors have been identified
in muscle tissue and
muscle weakness, limb pain and impaired physical function are well recognized
manifestations of severe
vitamin D deficiency.
A number of prospective, randomized, intervention studies demonstrated the
efficacy of
vitamin D to improve musculoskeltal function and reduce fall risk. Treatment
with vitamin D, and
calcium, has been shown to reduce the incidence of non-vertebral fractures and
to reduce postural sway
and possibly the incidence of falls. See J.K. Dhesi et al., Age and Aging
2002;31:267-271. Additionally,
it has been demonstrated that the number of falls in elderly community-
dwelling patients can be
significantly reduced by treatment with alfacalcidol (1-alfa-hydroxyvitamin
D3), and minimal calcium
intake. See L. Dukas et al, JAGS 2004;52:230-236.
It is also believed that a vitamin D/bisphosphonate composition will enhance
the
absorption of calcium. There have been studies which utilized the active
metabolites of vitamin D to
-24-
CA 02467715 2004-05-19
examine the positive effects on fractional calcium absorption in
postmenopausal women. See M.L.
Holzherr et al., Osteoporosis Int 2000; I 1:43-51; J.C. Gallagher et al., J.
Clin. Endocrinol. Metab., 1980;
51(5):1359-64. Bisphosphonates have also been shown to increase intestinal
calcium absorption in rat
models. See P. Ammann et al., J Bone Miner Res 1993;8(12):1491-8; H. Fleisch
Osteoporos Int
1996;6:166-70; J-P Bonjour Endocrinol Metab 1988;17:E260-E264. However, it is
believed that a
composition of a vitamin D compound and a bisphosphonate would increase
absorption of calcium more
than the additive effect of vitamin D or a bisphosphonate each administered
alone. Additionally, it is
believed that a composition of cholecalciferol and alendronate would greatly
enhance the absorption of
calcium more than a combination of an active form of vitamin D and a different
bisphosphonate. This
increase in calcium absorption would correlate to a reduction in fracture
risk.
The present invention also provides for the use of a composition comprised of
a vitamin
D compound and a bisphosphonate compound comprising a pharmaceutically
effective amount of at least
one bisphosphonate, or a pharmaceutically acceptable salt, derivative or
hydrate of the bisphosphonate, or
mixtures thereof, and one or more active ingredients for the manufacture of a
medicament for the
treatment, reduction, inhibition or prevention, in mammals such as humans, of
the conditions and disease
states identified above.
In further embodiments, the methods and compositions of the present invention
can also
comprise a histamine H2 receptor Mocker (i.e., antagonist) and/or a proton
pump inhibitor, which are
well known therapeutic agents for increasing gastric pH. See, e.g., L.J.
Hixson, et al., Current Trends in
the Pharmacotherapy for Peptic Ulcer Disease, Arch. Intern. Med., vol. 152,
pp. 726-732 (April 1992). It
is found in the present invention that the sequential oral administration of a
histamine H2 receptor blocker
and/or a proton pump inhibitor, followed by a bisphosphonate and vitamin D
composition can help to
minimize adverse gastrointestinal effects. In one embodiment of the present
invention, the histamine H2
receptor blocker and/or proton pump inhibitor is administered from about 30
minutes to about 24 hours
prior, or from about 30 minutes prior to about 12 hours prior, to the
administration of the bisphosphonate
and vitamin D composition. The dosage of the histamine H2 receptor blocker
and/or proton pump
inhibitor will depend upon the particular compound selected and factors
associated with the mammal to
be treated, i.e., size, health, etc. Non-limiting examples of histamine H2
receptor Mockers and/or proton
pump inhibitors include those selected from the group consisting of
cimetidine, famotidine, nizatidine,
ranitidine, omprazole, and lansoprazole.
The present invention further encompasses methods of manufacturing
compositions of
the present invention, including for example pharmaceutical compositions
comprising a bisphosphonate
compound and a vitamin D compound. In an embodiment, a method for preparing an
alendronate-
cholecalciferol formulation, comprises: preparing a powder blend comprising
alendronate; compacting
the powder blend to form an alendronate mixture; milling and blending the
alendronate mixture with
-25-
CA 02467715 2004-05-19
cholecalciferol granules to form a blend; and lubricating and compressing the
blend. In another
embodiment, a method for preparing an alendronate-cholecalciferol solid dosage
form comprises:
blending alendronate, colloidal silicon dioxide, lactose anhydrous,
microcrystalline cellulose, and
croscarmellose sodium to form a pre-blend; blending the pre-blend and
magnesium stearate to form a first
lubricated mixture; roller compacting the first lubricated mixture to form
compacted ribbons; milling the
compacted ribbons to form a lubricated blend; blending the lubricated blend
with cholecalciferol granules
to form a second lubricated mixture; and compressing the second lubricated
mixture into the solid dosage
form.
FIG. 4 depicts a flow-chart of an embodiment of a method of the present
invention for
manufacturing a bisphosphonate/vitamin D compositions of the present
invention. In this embodiment,
the composition is made by a process comprising roller compacting an
alendronate sodium formulation to
form a ribbon, milling of the ribbon produced from the roller compaction step
and then blending with the
extragranular addition of the vitamin D3 formulation. Using, for example, the
active ingredients and
excipients identified in Example 1, this formulation and process results in a
product which satisfies
regulatory requirements for product release and stability of both alendronate
and vitamin D3. As shown
in FIG. 4, in this embodiment, at step 301 a pre-blend of colloidal silicon
dioxide, lactose anhydrous, and
alendronate sodium is prepared. As depicted by step 302, the pre-blend is then
blended with
microcrystalline cellulose and croscarmellose sodium. At step 303, magnesium
stearate is added to form
a lubricated mixture. The lubricated mixture is passed through a roller
compactor and the compacted
ribbons are milled, as indicated at step 304. In the embodiment depicted in
FIG. 4, at step 305 vitamin D3
granules containing about 2800 IU (or the equivalent of about 70 pg) of
vitamin D3 are then added and
blended with the milled granules, with the vitamin D3 granule charge
quantitity adjusted based on both
incoming granule assay and the yield from the roller compaction/milling step.
The resulting mixture is
then compressed at step 306 to form tablets and the compressed tablets are de-
dusted. The resulting
tablets may be packaged in suitable packaging, including for example moisture-
proof and light-tight
blister packs or bottles.
Vitamin D3 (cholecalciferol) and vitamin Dz (ergocalciferol) are water
insoluble,
hydrophobic compounds with a melting point of about 84 °C and about 115
°C, respectively. These
compounds are also highly prone to oxidation and are photolabile, breaking
down into various
degradation products. Vitamin D granule is also prone to segregation. The
stability of vitamin D is thus
affected by the extent and nature of processing as well as the storage
conditions (e.g., exposure to light,
high temperatures, and high relative humidity) of the vitamin D/bisphosphonate
compositions. As a
result, the desire to include vitamin D in the compositions of the present
invention presents a particular
challenge insofar as developing methods of manufacturing and storing vitamin D-
containing
compositions is concerned. Accordingly, there is a need for vitamin
D/bisphosphonate compositions that
-26-
CA 02467715 2004-05-19
have been formulated so as to reduce the degradation of the vitamin D, both
during processing and during
storage. There is also a need for methods of manufacturing such stable
compositions. In addition, there
is a need to develop methods of detecting or measuring the degradation of
vitamin D in vitamin D-
containing compositions, such as those of the present invention. In addition,
because the level of a
particular vitamin D degradant may be very small (on the order of nanograms),
there is a need to develop
methods of measuring or detecting degradation of vitamin D in vitamin-D
containing compositions, such
as those of the present invention, having a limit of quantitation (LOQ)
sufficient to detect the amounts of
vitamin D degradants.
Accordingly, the present invention also provides methods of manufacturing
compositions
comprising a bisphosphonate compound and a vitamin D compound that minimizes
the loss of the
vitamin D compound during manufacture. By controlling the humidity during
manufacture, temperature
and light present during the formulation ofvitamin D or providing the
appropriate finished dosage form
packaging, it is possible to reduce the loss of vitamin D while maintaining
fully its potency. 1n an
embodiment of the instant invention, the temperature during the manufacturing
process is less than or
equal to about 35 °C. In a further embodiment, the temperature is
between about 20 °C to about 30 °C.
1n an embodiment, the relative humidity during the manufacturing process is
less than or equal to about
60% RH. In another embodiment, the relative humidity is between about 20% to
about 40%. In addition,
controls may be placed on the starting moisture levels of not only the vitamin
D components of the
formulation, but also on any excipients that may be present. In a further
embodiment, the relative
humidity is between about 25% to about 35%.
The present invention also encompasses methods of manufacture that comprise an
additional drying step. Thus, as another embodiment, the compositions of the
instant invention may be
manufactured under different conditions (temperature and/or relative humidity)
as described above, and
the moisture content of the manufactured composition may be reduced by drying
the composition. In an
embodiment, the drying may involve drying (with, for example, heat) the
compositions of the present
invention after the solid dosage form has been created. 1n another embodiment,
the drying may involve
film coating of a solid dosage forms (e.g., tablets) of the compositions of
the present invention. In
another embodiment, the drying may also involve packaging the compositions of
the present invention
with appropriate amounts of dessicants or other moieties to reduce moisture
content. In another
embodiment, the drying may involve storing the compositions of the present
invention in storage forms
that reduce moisture and/or light (e.g., aluminum blister packs, moisture-
proof bottles).
In embodiments of the present invention, vitamin D compounds used as a
starting
material may include a free flowing, stabilized granules of vitamin D. In
embodiments, the vitamin D
granules used as a starting material in manufacturing methods of the present
invention are Dry Vitamin
D3 100, Gelatin Coated, Pharmaceutical Grade, sold by BASF. The particles of
vitamin D are dissolved
-27-
CA 02467715 2004-05-19
in medium chain triglycerides in droplets of 1-2 pm embedded in a starch-
coated matrix of gelatin and
sucrose. The dissolved vitamin D can then be stabilized with t-
butylhydroxytoluene (BHT). The vitamin
D granules contain sodium aluminum silicate as a flow aid. One with ordinary
skill in the art would
understand that the amount of vitamin D added to the composition may be need
to be adjusted based the
source of vitamin D and/or the potency of the vitamin D being added. For
example, if Dry Vitamin D3
100 Gelatin Coated, Pharmaceutical Grade granules (BASF) were utilized, one
with ordinary skill would
understand that the granules may have different potencies (e.g., 100,000 IU/g
or 105,000 IU/g or 110,000
IU/g) which would require one to adjust the amount of granules added to the
composition in order to
achieve 2800 IU, or 5600 IU, ofvitamin D in the composition.
The vitamin D contained in an embodiment of the present invention conforms to
the
acceptance criteria of the Ph. Eur, Cholecalciferol Concentrate (Powder Form)
monograph. While at this
time there is no USP monograph for a formulated vitamin D3 product, an
applicable Ph.Eur. monograph
has been published. The inactive ingredients in the compositions of the
vitamin D compounds used in
embodiments of compositions of the present invention (e.g., medium chain
triglycerides, butylated
hydroxytoluene, sucrose, gelatin, modified starch, and sodium aluminum
silicate) are either compendia)
or food grade materials.
1n embodiments of the methods and compositions of the present invention, the
alendronate used as a starting material is compendia) grade alendronate sodium
monohydrate, or
compendia) grade alendronate sodium trihydrate, obtained from Merck & Co,,
Inc.
1n addition, commercially available vitamin D granules can possibly be used in
the
compositions of the present invention, such as those available from Roche,
BASF, or Solvay,
In further embodiments, the present invention provides a kit for conveniently
and
effectively carrying out the methods in accordance with the present invention.
Such kits are especially
suited for the delivery of solid oral forms such as tablets or capsules and in
embodiments include a
number of unit dosages a card having the dosages oriented in the order of
their intended use. An example
of such a kit is a "blister pack." Blister packs are well known in the
packaging industry and are widely
used for packaging pharmaceutical unit dosage forms. If desired, a memory aid
can be provided, for
example in the form of numbers, letters, or other markings or with a calendar
insert, designating the days
in the treatment schedule in which the dosages can be administered.
Alternatively, placebo dosages, or
calcium or dietary supplements, either in a form similar to or distinct from
the bisphosphonate and
vitamin D unit dosages, can be included to provide a kit in which a dosage is
taken every day. In those
embodiments including a histamine H2 receptor andlor proton pump inhibitor,
these agents can be
included as part of the kit.
The present invention also provides a detection method that was developed in
order to
measure the degradation products of the vitamin D3 compounds of the present
invention. Specifically, a
-28-
CA 02467715 2004-05-19
method of measuring the degradation of the pharmaceutical composition may
comprise extracting the
cholecalciferol from the composition into a first solution to form a second
solution, separating a sample
containing cholecalciferol from the second solution, and detecting the amount
of cholecalciferol in the
sample by subjecting the sample to reverse-phase HPLC separation. The
detection method of the present
invention is carried out to detect about 2800 IU to about 5600 IU
cholecalciferol per pharmaceutical
composition. Additionally, the detection method has a limit of quantitation
(LOQ) of cholecalciferol of
less than about 9 ng/mL cholecalciferol.
In an embodiment, the method utilizes a first solution which comprises water,
alcohol,
acetonitrile or mixtures thereof. In a specific embodiment, the first solution
contains about 5% water and
about 95% methanol. An exemplary sample preparation may be extracted of 15
tablets containing 2800
1U of vitamin D each into about 50 mL of about 5% water and about 95%
methanol. Also in an
embodiment, the resulting solution may be stirred for about 10 minutes,
sonicated for about 30 minutes,
and then stirred for an additional 3 hours. In an embodiment, the separating
of the samples may be
carried out by centrifugation, which can be from about 5,000 rpm to about
15,000 rpm. In an
embodiment, the column is a Phenomenex Phenosphere 80~ ODS (1) column (150 x
4.6 mm, 3pm), and
the injection volume is 100 lxL. The samples are eluted down the column and
then detected. In an
embodiment of this method, a 65-minute gradient may be used. A detection
wavelength of about 260 nm
to about 265 nm may also be used. In an embodiment of this method, the
detecting step is accomplished
at a reverse-phase HPLC column temperature of about 25°C. A sample tray
temperature of about 5°C
may be used. In an embodiment, the detecting step comprises reverse-phase HPLC
separation using an
eluant of about 99% acetonitrile and about 1 % of 0.025% phosphoric acid.
In an embodiment, the reverse-phase I-IPLC column that may be used in the
methods of
the present invention include columns that are either only partially endcapped
or not endcapped. The
endcapping process reduces the free silanol groups on the stationary phase,
therefore, it affects the
separation between pre-vitamin D and vitamin D peaks. Attempts to use
endcapped columns were
unsuccessful in providing a peak resolution that was sufficient for the assay
method of the present
invention because any degradate eluting between the two actives would not be
resolved and quantitated.
Indeed, in identifying columns for use with the methods of the present
invention, a vitamin D3 isomer
(0.96RRT) was observed as eluting between two actives found in the
formulation. More method
development using other endcapped columns all showed limited resolution
between pre-vitamin D3 and
vitamin D3 peaks.
Column carbon loading has an impact on the elution of four vitamin D~ ester
adducts,
which are the products of transesterification between either pre-vitamin D3 or
vitamin D3 and the medium
chain triglycerides (Ca and Coo fatty acid esters are present in BASF vitamin
D granules that may be used
in the compositions and methods of the present invention). These esters may
react with the hydroxyl
-29-
CA 02467715 2004-05-19
group of vitamin D3 through a transesterification mechanism to form C8 D3 and
Coo D3 esters. Because of
the long fatty acid chains, these esters are very hydrophobic and interact
with the C~8 stationary phase. A
column with higher carbon loading has more C~8 stationary phase; therefore, it
interacts more strongly
with the esters and retains the esters on the column for a longer period of
time. Accordingly, in an
embodiment of the methods of the present invention, a HPLC column having less
than about 10% carbon
loading may be used. Using a column with lower carbon loading reduced the
interaction between the
stationary phase and the esters, resulting in earlier elution of these peaks.
Results showed that, for
example, using a Platinum EPS Ci8 column with low carbon loading (5%), all
esters were eluted before
minutes when a mobile phase containing 95% acetonitrile/5% water was used.
Similarly, all four
10 esters were eluted within 26 minutes when another column, Phenosphere ODS
(1) column (7% carbon
loading), was used.
Exemplary chromatographic conditions that may be used in the methods of the
present
invention are listed below:
Flow Rate: 1.2 mL/min
Column Tem erature:25 C
In'ection Volume: 100 L
Mobile Phase. Gradient, A = 0.025% phosphoric
acid, B = 99%Acetonitrile
/1 %A
Run Time: 65 minutes
Column: Phenos here 80 ~, ODS 1 column,
150 x 4.6mm, 3 m
Sam le Tra Tem erature5 C
Detector Wavelength:265 nm
l 5 Gradient Time Table:
T (min)0 16 39 43 57 57.0165
Aqueous51.513 10 0 0 51.551.5
Mixture48.587 90 100 10048.548.5
Using detection methods of the present invention, both pre-vitamin D and
vitamin D
peaks can be quantitated to calculate the total amount of vitamin D in a
sample. Specifically, the methods
of the present invention are sufficiently sensitive and selective, with a
sample of about 2800 IU
cholecalciferol, to distinguish between cholecalciferol, pre-cholecalciferol,
and their isomers, and to
detect one or more cholecalciferol ester adducts, or one or more pre-
cholecalciferol ester adducts.
-30-
CA 02467715 2004-05-19
Three types of potential vitamin D3 degradation products have been observed in
stability
studies for the pharmaceutical compositions of the present invention, which
are described below
(including in Example 6). In the stability studies described below, the tablet
composition that was
studied comprises about 91.4 mg alendronate sodium, about 26.7 mg
cholecalciferol granules, about
131.0 mg microcrystalline cellulose, about 62.4 mg lactose anhydrous, about
9.7 mg croscarmellose
sodium, about 0.8 mg colloidal silicon dioxide, and about 3.l mg magnesium
stearate.
As depicted in FIG. 5, the structure of vitamin D3 includes a conjugated
triene, which
undergoes a variety of thermal and photochemical isomerizations. Five vitamin
D3 isomers have been
identified in exemplary pharmaceutical compositions ofthe present invention
(in this example, using
vitamin D3 (cholecalciferol)): pre-vitamin D3, trans-vitamin D3, and three
additional isomers at 0.78RRT,
0.96RRT and 1.09RRT (which is a measure of retention time of the compound by
high performance
liquid chromatography (HPLC) as described below). Structures for some of these
vitamin D3 isomers are
shown in FIG. 5. Structural conclusions are based on UV, MS, and in some cases
NMR spectroscopy.
Vitamin D and its isomer pre-vitamin D are known to interconvert thermally by
a
sigmatropic 1,7-hydrogen shift. 1n vivo, pre-vitamin D has been shown to be an
immediate precursor to
vitamin D, and both species are found in equilibrium concentrations at
physiological temperatures,
although that equilibrium appears to be largely in favor of vitamin D. Because
both vitamin D and pre-
vitamin D are considered to serve the same physiological function, vitamin D
assays when reported
conventionally comprise the sum of both species. This is consistent with both
USP and Ph.Eur.
monographs for vitamin D3 containing products, for example. Available
stability data indicate that none
of the other isomers will approach the ICH qualification threshold of I .0% by
weight at 24 months, stored
at 25°C/60%RH in appropriate packages.
The most prominent degradation products appear to be vitamin D3 esters formed
by
transesterification reactions of vitamin D3 with the medium chain
triglycerides (MCT) in the vitamin D3
granules used in the compositions of the present invention. Structures for
some of these vitamin D3 ester
adducts are also shown in FIG. 5. The predominant species correspond to n-
octanoate (C8) and n-
decanoate (Cio) esters of vitamin D3. The pre-vitamin D3 ester adducts can be
generated either by
reaction of pre-vitamin D3 with the triglycerides in the vitamin D3 compound
or by thermal conversion
from the vitamin D3 esters. Of the quantifiable degradation products, only the
C8 and C~° vitamin D3
ester adducts appear to increase to any appreciable extent during the
stability study.
Available stability data indicate that these species should not approach the
ICH
qualification threshold of 1.0% weight at 24 months, stored at less than about
30°C and at less than about
30% relative humidity (RH) and they are not expected to give rise to safety
concerns in any event in
embodiments of compositions and methods of the present invention. Studies
further show that after 24
-31 -
CA 02467715 2004-05-19
months when stored at less than about 30°C and less than about 30%RH,
total degradants of the
compositions of the present invention is less than about 5%.
It is also understood that vitamin D can undergo autoxidation through
induction by a free
radical initiator or spontaneously in solid or solution phase to form a
variety of products, some of which
have been identified. Representative characterization ofvitamin D degradation
(and, specifically,
vitamin D3 in this instance) in embodiments of compositions of the present
invention confirms the
autoxidative lability of vitamin D3 in which vitamin D3 was converted to an
oil or amorphous solid and
exposed to temperatures from 20-40°C. Within hours, HPLC analysis
showed extensive destruction of
vitamin D3 and the appearance of many unresolved degradation products
exhibiting very low ultraviolet
(UV) absorption. At longer exposure times, these absorptions continued to
decrease as further reaction
occurred.
A more detailed analysis of the autoxidation of vitamin D3 was carried out
using the free
radical initiator, azo-bis-isobutyronitrile (AIBN). In this experiment, AIBN
was used to initiate the
autoxidation of vitamin D3 in solution. The resulting product profile was
characterized by HPLC using
UV, mass spectrometric (MS) and evaporative light scattering (ELS) detection.
The results showed that:
(a) solution-phase autoxidation also can lead to multiple degradation
products, (b) autoxidation can lead
to gradual destruction of the UV chromophore resulting in an apparent material
loss, while ELS
detection, on the other hand, afforded significantly better mass recovery, and
(c) the mass spectrometric
m/z ratios and in some cases the observed UV/vis spectra confirmed the
oxidative nature of these reaction
products.
A radiolabel study was conducted in an attempt to characterize vitamin D3
degradation in
a granule formulation used in embodiments of compositions of the present
invention comprising about 70
mg alendronate and about 2,800 IU (70 llg) vitamin D3. Tritium labeled vitamin
D3 was utilized as a
means of tracking degraded vitamin D3, independent of changes in UV absorption
characteristics. The
radiolabeled vitamin D3 was incorporated into a formulation that modeled the
vitamin D granules used in
embodiments of compositions of the present invention, and was then analyzed
for stability. The
antioxidant level in the model formulation was at a reduced level from
antioxidant levels considered
desirable for commercial formulations, in order to ensure that degradation
occurred within a reasonable
timeframe. Samples were analyzed after 14 weeks at 40°C/75%RH and
70°C using liquid scintillation
counting (LSC) and reverse-phase high performance liquid chromatography (RP-
HPLC) with
simultaneous UV and online radiodetection. A vitamin D3 loss of approximately
40% was observed for
samples stored at 40°C/75%RH conditions, whereas the low temperature
controls showed good stability.
Results of this analysis are shown in the radiochromatograms for the degraded
samples of FIG. 6, which
show a large region of unresolved degradates, none of which appears to be a
major product. These results
provide further evidence that, when not properly stabilized, vitamin D3
degrades oxidatively into multiple
-32-
CA 02467715 2004-05-19
products with reduced UV absorption and that these products account for loss
of vitamin D3. Based on
these stability analyses, individual autoxidative degradates are not expected
to approach levels that are of
a safety concern in tablet embodiments of compositions of the present
invention.
The present invention also includes methods of measuring the pharmacokinetic
parameters in mammals upon the administration of the compositions of the
present invention. The
pharmacokinetic parameters that may be measured include, for example, total
urinary excretion, urinary
excretion, area under the serum-concentration-versus-time curve (AUC), steady
state maximum plasma
concentration (Cm~), time of Cm~ (T",~), and serum concentration median
apparent half life (t"z) of a
tablet, such as, for example, a tablet comprising about 70 mg alendronate and
about 2,800 IiJ
cholecalciferol. These measurements confirm that embodiments of the
compositions and methods of the
present invention produce pharmaceutically effective levels of alendronate and
cholecalciferol in the
body (the latter as demonstrated by comparison to recommended daily amounts of
a vitamin D compound
in the compositions and methods of the present invention).
In an embodiment, the present invention includes methods of measuring
cholecalciferol
in human serum after administration of a pharmaceutical composition comprising
alendronate and
cholecalciferol, the method comprising: (1 ) administering to a human a
composition comprising
alendronate and cholecalciferol; (2) obtaining from the human a plasma sample;
(3) extracting the
cholecalciferol from the plasma sample to form a first solution; (4) reacting
the cholecalciferol in the first
solution with a dienophile to form one or more diets-alder addition products
of cholecalciferol; (5)
separating the diets-alder addition products of cholecalciferol using high
performance liquid
chromatography (HPLC) separation; and (6) detecting an amount of
cholecalciferol in the sample using
mass spectroscopy. In an embodiment of this method, the dienophile comprises 4-
phenyl-1,2,4-
triazoline-3,5-dione (P-TADO or PTAD). Also, the detecting step may be
conducted in a positive
ionization mode using a heated nebulizer probe, and may further comprise
adding a deuterated internal
standard cholecalciferol to each human plasma sample, and extracting,
reacting, separating, and detecting
the deuterated internal standard cholecalciferol along with the sample
cholecalciferol. This method has a
limit of quantitation (LOQ) of cholecalciferol of less than about 0.5 ng/mL
cholecalciferol when I mL of
plasma is measured. An embodiment of the present invention is a vitamin
D/bisphosphonate composition
wherein a plot of serum concentration of a mammal over 120 hours after
administration of the
composition yields at least one of the following: a least-squares (LS) mean
AUC~o_~ZOn~~ of cholecalciferol
of about 296.4 ng.h/mL, wherein the pharmacokinetic parameters have been
measured without taking into
account baseline cholecalciferol serum concentrations; a least-squares (LS)
mean AUC~o_,iona of about
297.5 ng.h/mL, wherein the pharmacokinetic parameters have been measured by
taking into account
baseline cholecalciferol serum concentrations using a predose 0 hr serum
cholecalciferol concentration as
a covariate; and a least-squares (LS) mean AUC~o_,zo6> of about 143.1 ng.hlmL,
wherein the
-33-
CA 02467715 2004-05-19
pharmacokinetic parameters have been measured by taking into account baseline
cholecalciferol serum
concentrations using a subtraction of estimated baseline cholecalciferol over
the 120 hour period. In
another embodiment, the composition comprises a bisphosphonate and
cholecalciferol wherein a plot of
plasma concentration a mammal over 120 hours after administration of the
composition yields at least
one of the following: a least-squares (LS) mean for steady state maximum
plasma concentration (Cmex) of
over 120 hours of about 5.9 ng/mL, wherein the pharmacokinetic parameters have
been measured without
taking into account baseline cholecalciferol serum concentrations; a least-
squares (LS) mean for steady
state maximum plasma concentration (Cm~) of over 120 hours of about 5.9 ng/mL,
wherein the
pharmacokinetic parameters have been measured by taking into account baseline
cholecalciferol serum
concentrations using a predose 0 hr serum cholecalciferol concentration as a
covariate; and
a least-squares (LS) mean for steady state maximum plasma concentration (Cm~)
of about 4.0 ng/mL,
wherein the pharmacokinetic parameters have been measured by taking into
account baseline
cholecalciferol serum concentrations using a subtraction of estimated baseline
cholecalciferol over the
120 hour period.
The present invention also encompasses a composition wherein a plot of the
plasma
concentration of cholecalciferol of a mammal over 120 hours after
administration of the composition
yields: a steady state maximum plasma concentration (CmaX) of cholecalciferol
at an arithmetic mean time
of occurrence of Cm~ (Tm~) of about 12 hours, and wherein the pharmacokinetic
parameters have been
without taking into account baseline cholecalciferol serum concentrations. In
a further embodiment, the
composition has a plasma concentration median apparent half life (t,n) of the
cholecalciferol of the
composition in mammals that is about 23.8 hours, and the pharmacokinetic
parameters have been
measured by taking into account baseline cholecalciferol serum concentrations
using a subtraction of
estimated baseline cholecalciferol procedure.
In order to determine the pharmacokinetic characteristics of the compositions
of the
present invention, studies of samples from an open-label, randomized, 2-part,
2-period crossover study in
236 healthy non-pregnant women and men age 18 to 65 were conducted. In this
study, described in detail
in Example 7 below, the phannacokinetic parameters (AUCo-izon~~ Cm~~ Tmax~ and
serum concentration
median apparent half life (t"z)) of vitamin D3 administered as a 70-mg
alendronatel2800 IU vitamin D3
combination tablet relative to a 2800 IU vitamin D3 tablet were studied. In
addition, the urinary excretion
of alendronate was studied in the combination tablet in relation to the once-
weekly 70 mg tablet of
FOSAMAX~. In summary, (1) a 70 mg alendronate/2800 IU vitamin D3 combination
tablet according
to the present invention was shown to be bioequivalent to a 70 mg alendronate
tablet with respect to
alendronate bioavailability; (2) the bioavailability of vitamin D3 in the 70
mg alendronate/2800 IU
vitamin D3 combination tablet and in a tablet containing 2800 IU vitamin D3
(without alendronate) was
shown to be similar, and (3) a 70 mg alendronate/2800 IU vitamin D3
combination tablet according to the
-34-
CA 02467715 2004-05-19
present invention was shown to be generally well tolerated. Accordingly, it is
expected, for example, that
once-weekly dosing with a bisphosphonate/vitamin D compound of the present
invention will provide
vitamin D3 blood levels and/or therapeutic effects comparable to the vitamin D
blood levels and/or
therapeutic effects from a recommended daily dose ofvitamin D, such as 400 IU
vitamin D daily, over
the same period as the once-weekly dosing of the bisphosphonate/vitamin D
compound.
These and other embodiments of the present invention are further explained in
the non-
limiting examples that follow.
EXAMPLES
The following examples further describe and demonstrate embodiments within the
scope
of the present invention. The examples are given solely for the purpose of
illustration and are not to be
construed as limitations of the present invention as many variations thereof
are possible without departing
from the spirit and scope of the invention.
EXAMPLE 1
Bisphosnhonate and Vitamin D Tablets
A finished drug product is a combination tablet containing alendronate sodium
(about 70
mg anhydrous free acid equivalent) and vitamin D3 (about 2800 LU. (about 70
fig)), with ingredients
identified in Table 1-1. All of the excipients are compendial and were
selected to achieve maximum
physical and chemical stability.
Table l-1
Tablet Composition
Alendronate
Sodium
70 mg/
Vitamin
D3 2800
LU.
Tablets
in redient m /Tab Wei ht
Alendronate 91.37 28.1
Sodium
Dry Vitamin 26.67 8.2%
D3 100
ranules
Microcrystalline131.0 40.3%
Cellulose
NF
Lactose Anhydrous62.35 19.2%
NF
Croscarmellose9.740 3.0%
-35-
CA 02467715 2004-05-19
Sodium NF
Colloidal 0.8120 0.25%
Silicon
Dioxide NF
Magnesium 3.0870 0,95%
Stearate
NF
_
Total ~ - 325 ~ 100%
The resulting tablets are used in accordance with the methods of the present
invention for preventing,
inhibiting, reducing or treating osteoporosis, for example. Similarly, tablets
comprising other relative
weights of alendronate, on an alendronic acid active basis are prepared
including, but not limited to,
about 2.5 mg, S mg, 8.75 mg, 17.5 mg, 70 mg, 140 mg, 280 mg, 560 mg, or 1120
mg per tablet.
Similarly, tablets comprising other relative weights of vitamin D3 per unit
dosage are prepared including,
but not limited to, about 1,400, 2,800, 5,600, 7,000 IU, 8,400 IU, 14,0001U,
28,000, or 36,000 IL1 per
tablet. Such tablets may be administered at intervals ranging from once-weekly
to bi-monthly.
EXAMPLE 2
Bisphosphonate and Vitamin D Composition
A composition comprising a bisphosphonate and vitamin D may be prepared using
mixing and formulation techniques as described in this specification. A
composition containing about 35
mg of alendronate, on an alendronic acid active basis, and about 5,600 IU of
vitamin D3 may be prepared
using the following relative weights of ingredients.
In redient Per Tablet
Alendronate Monosodium Trih 45.68 m
drate
D Vitamin D3 100 ranules 56 m
Anh drous Lactose, NF 71.32 m
Microc stalline Cellulose, 80.0 m
NF
Ma nesium Stearate, NF 1.0 m
Groscarmellose Sodium, NF 2.0 mg
* Granule contains approximately 100,000 IU per one gram; therefore 56 mg of
the granule is
equivalent to about 5600 IU.
-36-
CA 02467715 2004-05-19
The resulting dosage forms are used in accordance with the methods of the
present invention for
preventing, inhibiting, reducing or treating osteoporosis, for example.
Similarly, dosage forms
comprising other relative weights of alendronate, on an alendronic acid active
basis are prepared
including, but not limited to, about 2.5 mg, 5 mg, 8.75 mg, 17.5 mg, 70 mg,
140 mg, 280 mg, 560 mg, or
1120 mg per tablet. Similarly, dosage forms comprising other relative weights
of vitamin D3 per unit
dosage are prepared including, but not limited to, about 1,400, 2,800, 5,600,
7,000 1U, 8,4001U, 14,000
IU, 28,000, or 36,000 IU per dosage form. Such dosage forms may be
administered at intervals ranging
from once-weekly to bi-monthly. These dosage forms may be, for example,
tablets or capsules
EXAMPLE 3
Alendronate and Vitamin D Tablets
Tablets containing about 70 mg of alendronate, on an alendronic acid active
basis, and
2800 IU of vitamin D3, are prepared using methods disclosed herein, using the
following relative weights
I S of ingredients:
Table 3-1
Composition (per tablet):
Alendronate sodium 91.37 mgt
Silicon Dioxide, Colloidal, 0.81 mg
CAB-O-S1L P
Dry Vitamin D3 100 granules 26.67 mg*
$
Cellulose Microcrystalline 131 mg
NF Avicel PH-102
Lactose NF Anhydrous 63.35 mg
Croscarmellose Sodium Compendia) 9.74 mg
Magnesium Stearate NF (Non-Bovine) 3.09 mg
'Equivalent to 70,0 mg free
acid
Dry V itamin D3 100 granules
also contained medium chain
triglycerides, gelatin,
sucrose, butylated hydroxytoluene,
starch and sodium aluminum
silicate.
*26.67 grams of the Dry Vitaminns
D3 100 granules contai 105,000
IU/g
of
vitamin
D3.
The resulting tablets are used in accordance with the methods of the present
invention for preventing,
inhibiting, reducing or treating osteoporosis, for example. Similarly, tablets
comprising other relative
weights of alendronate, on an alendronic acid active basis are prepared
including, but not limited to,
about 2.5 mg, 5 mg, 8.75 mg, 17.5 mg, 70 mg, 140 mg, 280 mg, 560 mg, or 1120
mg per tablet.
-37-
CA 02467715 2004-05-19
Similarly, tablets comprising other relative weights of vitamin D3 per unit
dosage are prepared including,
but not limited to, about 1,400, 2,800, 5,600, 7,000 IU, 8,400 IU, 14,000 IU,
28,000, or 36,000 IU per
tablet. Such tablets may be administered at intervals ranging from once-weekly
to bi-monthly.
EXAMPLE 4
Effect of Vitamin D, (Powder Forml on Alendronate Absoprtion
To examine the interaction of vitamin D3 (cholecalciferol) in powder form on
alendronate when administered in a single dosage, a two-period, crossover,
study in 14 healthy,
nonpregnant women and men, aged 18 to 85 was conducted. Subjects received one
alendronate 70-mg
tablet in each period. A single dose of vitamin D3 5600 IU was coadministered
with the alendronate
tablet in one of the two periods, based on a computer-generated patient
allocation schedule. When
vitamin D3 was administered with alendronate, the vitamin D3 powder was
reconstituted in 60 mL of
plain tap water and administered to the subject with the alendronate tablet
(Treatment A). The vitamin D3
bottle was rinsed and filled with 60 mL of plain tap water 3 times, each then
administered to the subject.
Therefore, a total volume of 240 mL of plain tap water was administered with
the vitamin D3. When
alendronate was administered alone, a 240-mL volume of plain tap water was
administered with the dose
(Treatment B). At least a l4-day washout separated each period. The treatment
schematic and allocation
are in Table 4-1.
Table 4-1
Treatment Schematic and Allocation
Grou Period Period
1 2
1 (N=7) A B
ANs 0002, 0004, 0005, 0008,
0009, 0011, 0013
2 (N=7) B A
ANs 0001, 0003, 0006, 0007,
0010, 0012, 0014
Treatment A = 70-mg alendronate
tablet with vitamin D, 5600
IU.
Treatment B = 70 m alendronate.
Subjects were sequestered in the study unit the evening prior to each
treatment.
Following an overnight fast (except water), subjects were administered the
respective treatment. Subjects
continued to fast following drug administration until a defined meal was
administered 2 hours postdose.
Clinical supply information is in Table 4-2. The composition and analytical
results for
the alendronate tablet and vitamin D3 used in this study are listed in Tables
4-3 and 4-4
-38-
CA 02467715 2004-05-19
Table 4-2
C'.linical SunnliPc
Dru PotencDosa a Form
Alendronate?0 Tablet
mg
Vitamin 5600 Granules
D3 IU
Table 4-3
Alendronate Tablet Formulation Characteristics
Composition (per tablet):
Alendronate sodium 91.37 mgt
Lactose NF Anhydrous 1 I 3.38 mg
Cellulose Microcrystalline 140.00 mg
NF Avicel 102
Magnesium Stearate Impalpable 1.75 mg
Powder NF
Croscarmellose Sodium NF 3.50 m
T a A
Equivalent to 70.0 mg free
acid.
Table 4-4
Vitamin D3 Granular Powder
Formulation Characteristics
i0
Composition (per bottle):
D vitamin D3 T a 100 CWS/HP51.96 m t
t E uivalent to 5600 (U.
Ah doses were administered following an overnight fast (except for water).
Subjects
were administered a single alendronate 70-mg tablet with 240 mL of plain tap
water. When subjects were
administered the vitamin D3 dose of 5600 IU with alendronate, the subjects
were instructed to resuspend
and co-administer the alendronate 70-mg tablet with the vitamin D3 dose
(supplied in granulated form
and reconstituted in water at the study site). The total volume of liquid
administered with each
alendronate dose was 240 mL. Subjects remained in the fasted state for an
additional 2 hours following
study drug administration and subsequently served a defined meal. Subjects
remained upright for the 2
hours between drug administration and the defined meal. Each dose period was
separated by an interval
of at least 14 days.
Urine specimens for alendronate assay were collected for pharmacokinetic
analyses over
the following intervals: -2 to 0 hour predose, 0 to 8 hours postdose, 8 to 24
hours postdose, and 24 to 36
hours postdose. The urine collection obtained over the 2-hour period just
prior to study drug
-39-
CA 02467715 2004-05-19
administration provided a baseline alendronate determination. All urine
specimens were collected in
preweighed polypropylene containers. For the 0- to 8-, 8- to 24-, and 24- to
36-hour postdose urine
collections, 12.5 grams of boric acid were added to the containers as a
preservative at the beginning of the
timed interval. At the end of each timed collection interval, the entire urine
collection was weighed, the
specific gravity measured, and the net volume determined. The urine specimen
was acidified in situ.
Five mL of 6.0 N Hydrochloride Acid (HCI) were added per 200 mL of urine to
bring the urine specimen
pH to <_2Ø Following acidification, the urine specimen was agitated and a
sample was aliquoted into a
polypropylene container to be stored frozen (-20°C) until high-
performance liquid chromatography
(HPLC) assay was completed. The total urine volume for each period, including
the volume of the boric
acid and HCI, was used to determine total urinary excretion of alendronate for
a given interval.
The analytical method for the determination of alendronate in human urine
involved 3
distinct operations: (1) isolation of the analyte and an internal standard
(pamidronate) from urine, (2)
formation of strongly fluorescent derivatives, and (3) HPLC separation and
fluorescence detection of the
resulting derivatives. Alendronate and the internal standard were co-
precipitated from urine with
naturally present phosphates by the addition of calcium chloride and sodium
hydroxide. The pellet,
isolated by centrifugation, was reconstituted in 1 M hydrochloric acid and
applied to an anion-exchange
diethylamine (DEA) cartridge in acetate-buffered solution at pH 4. Alendronate
was eluted from the
DEA cartridge by a solution of 0.20 M sodium citrate and 0.20 M sodium
phosphate dibasic (adjusted to
pH 9). Alendronate was derived with 2,3-naphthalenedicarboxyaldehyde in the
presence of N-acetyl-D-
penicillamine at room temperature. The derivative was then applied to a non-
silica-based polymeric
column composed of the copolymer of styrene and divinyl benzene. The mobile
phase was initially
composed of 85% 0.025 M sodium citrate, 0.025 M sodium phosphate dibasic (pH
6.95), and 15%
acetonitrile at a flow rate of 1 mL/min. Later-eluting endogenous components
of urine were removed by
increasing acetonitrile to 50%. The assay was validated at between 5 ng/mL and
125 nglmL, in human
urine, with coefficients of variation below 10%. A 5-mL urine sample was
required to obtain the 1-
ng/mL limit of detection.
The total urinary excretion of alendronate for a given interval (-2 to 0, 0 to
8, 8 to 24, 24
to 36 hours) was determined by multiplying the concentration of alendronate in
the analyzed aliquot by
the total urine volume (including boric acid and hydrochloric acid) for the
interval.
A comparison of the total urinary excretion for the 70-mg alendronate tablet
plus 5600
1U vitamin D3 and the 70-mg alendronate tablet alone was performed using an
analysis of variance
(ANOVA) model suitable for a 2-period, crossover design. The ANOVA model
contained factors for
sequence, subject (sequence), period, and treatment. Total urinary excretion
was log-transformed.
Results from the Shapiro-Wilk test for normality, along with plots of
residuals from the model, did not
suggest any departure from the assumptions of the ANOVA model. To estimate the
relative
-40-
CA 02467715 2004-05-19
bioavailability of the 70-mg alendronate tablet plus vitamin D3 versus the 70-
mg alendronate tablet alone,
a 95% CI was computed, based upon the t-distribution, for the GMR for total
urinary excretion.
Additionally, the posterior probability that the true GMR is above the
clinically important bound of 0.50
was also calculated.
One subject was dropped from the above analysis since this particular subject
had urinary
alendronate concentrations for all 3 collection intervals (0 to 8, 8 to 24,
and 24 to 36 hours) below limit of
quantification for both treatments, Due to a slight imbalance in the ordering
of the treatment sequences,
the least-squares means for the total urinary excretion are reported. The data
from that subject was
excluded from analysis.
The least-square means were obtained by back-transformation from the ANOVA
model.
A11 p-values were rounded to 3 decimal places prior to reporting. Results for
which p<_0.050 is reported
are considered statistically significant.
Table 4-5 displays the total urinary excretion of alendronate as a 70-mg
alendronate
tablet plus vitamin D3 and the 70-mg alendronate tablet for each subject.
Summary statistics along with
the GMR, with its corresponding 95% CI, for total urinary excretion of
alendronate are in Table 4-6.
The least-squares geometric mean for total urinary excretion was 183.61 for
the 70-mg
alendronate tablet plus 5600 IU vitamin D3 and 157.97 wg for the 70-mg
alendronate tablet alone. The
GMR and its corresponding 95% C1 for 70-mg alendronate + vitamin D3 relative
to alendronate alone
were 1.16 (0.74, 1.83). The posterior probability that the GMR might be above
the clinically important
bound of 0.50 was 0.999.
Table 4-5
Individual Total Urinary Excretion (ltg) of Alendronate Over 36 Hours
Following Single-Dose Administration of 70-mg Alendronate Tablet
Plus 5600 IU Vitamin D3 and 70 mg Alendronate Administered Alone
70 mg Alendronate
Plus
5600 IU Vitamin 70 m Alendronate
D3
194.22 68.02
311.32 111.11
367.40 290.62
291.47 310.48
181.42 . 113.40
127.41 68,64
494.92 169.86
<LOQt <LOQt
21.75 61.29
185.46 257.75
1 O 1.51 68.73
-41 -
CA 02467715 2004-05-19
97.86 259.69
248.71 341.90
464.51 519.58
Arithmetic 237.54 203.16
Mean
Standard Deviation143.63 140.51
~ <LOQ = Below
limit of quantitation
of l ng/mL.
Table 4-6
Summary Statistics and Geometric Mean Ratio With Corresponding 95% CI
of Total Urinary Excretion (Itg) of Alendronate Over 36 Hours Alendronate
Following Single-Dose Administration of 70 mg Plus Vitamin D3
and 70 mg Alendronate Alone
TreatmentN LS MedianMin Max SD GMR~ 95% Ch
Mean for
GMR
Alendronatel3 183.61194.2221.75494.92260.401.16 (0.74,
1.83)
vitamin
D3
Alendronate13 157.97169.8661.29519.58177.07
Root Mean
Squared
Error
(RMSE)
in log
scale
from
ANOVA
Model
= 0.522
(In wg).
t LS mean=Least-Squares
mean
(back-transformed
from
the log
scale).
$ SD=Back-transformed
Between-Subject
Standard
Deviation.
GMR=Lesst-Squares
mean
ratio
(alendronate
+ vitamin
D3/alendronate).
CI=Confidence
Interval.
EXAMPLE 5
Effect of Vitamin D3 (Contained in an Alendronate/Vitamin D Tabletlon
Alendronate Absomtion
To examine the potential for an interaction between alendronate and orally-
administered
vitamin D3, fourteen healthy adult subjects (6 men, 8 women, ages 33 - 61 yr.)
were administered single
70-mg tablets of alendronate, without vitamin D3, and together with a powdered
dose of vitamin D3,
(5600 IU) suspended in 240 mL of water. This study was of an open, randomized,
crossover two-way
design. The purpose of the study was to obtain a preliminary estimate of the
relative bioavailability of
alendronate following a 70-mg tablet administered with vitamin D3, relative to
alendronate administered
without vitamin D3.
Alendronate was administered orally as a 70-mg tablet in each of the two
periods. In one
period, the tablet was administered with vitamin D3 powder reconstituted in
plain tap water and in the
alternate period the tablet was taken alone with plain tap water. Urine was
collected for two hours
preceding and 36 hours following each dose of alendronate for analytical
determination of excreted
-42-
CA 02467715 2004-05-19
alendronate. Relative bioavailability was estimated based on total urinary
recovery of alendronate over
the 36 hours post-dose.
Urinary recovery of alendronate following the dose of 70-mg alendronate
without
vitamin D3 was 202 ltg with a 90% CI of ( 126 ltg, 279 ltg), recoveries
following the 70-mg dose
administered together with vitamin D3, averaged 238 Itg with a 90% CI of (159
pg, 316 pg). The
geometric mean ratio (90% CI) was estimated at 1.18 (0.80, 1.?4). This
investigation shows that oral
administration of vitamin D3 together with an oral dose of alendronate has
minimal to no effect on the
bioavailability of alendronate.
l0 EXAMPLE 6
Stabilitv Stud of Vitamin D, and Alendronate
The stability of a composition ofthe invention in the form of a combination
tablet
containing alendronate sodium (70 mg anhydrous free acid equivalent) and
vitamin D3 (2800 LU./70 p.g)
has been studied. Table 6-1 contains a the tablet composition of an embodiment
of an
alendronate/vitamin D combination tablet. All of the excipients are compendia)
grade and were selected
to achieve maximum physical and chemical stability.
Table 6-1
Tablet Composition
Alendronate
Sodium
70 mg/
Vitamin
D3 2800
LU. Tablets
In redient m Tab Wei ht
Alendronate Sodium91.37 28.1%
D Vitamin D3 100 26.67* 8.2%
ranules
Microc stalline 131.0 40.3%
Cellulose NF
Lactose Anh drous 62.35 19.2%
NF
Croscarmellose 9.740 3.0%
Sodium NF
Colloidal Silicon 0.8120 0.25%
Dioxide NF
Magnesium Stearate2.275 0.7%
NF
Intra ranular
Magnesium Stearate0.8120 0.25%
NF _
Extra ranular
Total 325 100%
*26.67 grams of the Dry Vitamin D3 100 granules contains 105,000 IU/g of
vitamin D;
-43-
CA 02467715 2004-05-19
The alendronate assay and dissolution methods may employ reversed-phase HPLC
with
pre-column 9-fluorenylmethyl chloroformate (FMOC) derivatization, similar to
methods already reported
for FOSAMAX~ tablets. The vitamin D3 assay and degradates method may also be a
reversed-phase,
gradient HPLC method (RP-HPLC) capable of resolving and quantitating vitamin
D3 and multiple
potential degradation products of vitamin D3. The vitamin D3 content
uniformity and dissolution assays
also employ reversed-phase HPLC. The dissolution method may use a surfactant
medium (1% SDS) due
to the poor aqueous solubility of vitamin Dj Due to the low vitamin D3 potency
(70 fig) of the
combination tablet, one may use three tablets to 500 mL of medium to obtain a
suitable signal.
Fifty-two weeks of assay and degradate data are provided below for a batch of
the
combination tablets stored at 30°C/65%RH and 40°C/75%RH (See
Tables 6-2 and 6-3). These data
demonstrate the acceptable stability of an embodiment of a compositions of the
present invention,
although the data generated do indicate that there is slight degradation of
vitamin D~. Greater
degradation is found at higher temperatures in the aluminum blisters and the
HDPE bottles without
desiccant.
Table 6-2:
Summary of Vitamin D3 Stability Assay Results:
Alendronate Sodium 70 meNitamin D, 2800 LU. Combination Tablets
Vitamin Vitamin
Storage WeekstD3 D3
Condition (% (%
Label Label
Claim) Claim)
75 Foil
ml to
HDPE Foil
bottle Aluminum
w/ Blister
foil
induction
seal,
1
desiccant
4
tablets
er
bottle
Lot Lot Lot Lot Lot Lot
001 002 003 001 002 003
Initial 0 98.6 97.3 99.2 98.6 97.3 99.2
25C/60% 13 NT 97.3 99.5 99.2 97.7 NT
RH
26 99.9 98.0 NT 100.8 NT 99.6
39 99.0 NT 98.1 NT 93.8 97.1
44 99.1 97.1 99.6 97.9 95.4 99.1
52 98.2 96.4 99.3 98.8 94.9 99.3
30C/65% 13 100.1 96.8 NT 99.3 NT 99.1
RH
26 99.7 NT 99.5 NT 96.7 99.8
39 NT 94.4 97.1 97.6 94.3 NT
44 97.3 95.7 97.6 97.9 96.1 98.0
52 97.5 95.4 97.5 97.7 94.1 97.1
40C175% 13 99.3 96.0 99.0 97.1 96.1 97.8
RH
26 97.1 94.5 96.9 96.8 94.7 97.6
The theoretical
timepoint
in weeks
is indicated.
NT=Not
tested.
-44-
CA 02467715 2004-05-19
Table 6-3;
Summary of Vitamin D3 Degradation Stability Results: Alendronate Sodium 70
mg/Vitamin D3 2800 LU.
Combination Tablets, Foil to Foil Aluminum Blister
Degradate Storage Weeks% Label
Condition Claim
vvt%
relative
to Vitamin
D
Lot 001 Lot 002 Lot 003
0.74RRT Initial 0 0.4 0.3 0.3
(traps-vitamin25C/60% 13 0.2 0.3 NT
D3) RH
26 0.2 NT 0.2
39 NT 0.3 0.2
44 0.2 0.3 0.2
52 0.2 0.3 0.2
30C/65% 13 0.2 NT 0.3
RH
26 NT 0.3 0.2
39 0.2 0.3 NT
44 0.2 0.3 0.2
52 0.2 0.3 0.2
40C/75% 13 0.2 0.3 0.2
RH
26 0.1 0.2 0.1
0.78RRT Initial 0 0.0 0.0' 0.0
(vitamin D3 25C/60% 13 0.0 0.1 NT
isomer) RH
26 0.1 NT 0.1
39 NT 0.1 0.1
44 0.1 0.1 0.1
52 0.1 0.1 0.1
30C/65% 13 0.1 NT 0.1
RH
26 NT 0.1 0.1
39 0.2 0.2 NT
44 0.2 0.2 0.2
52 0.2 0.2 0.2
40C/75% 13 0.2 0.2 0.2
RH
2b 0.3 0.3 0.3
0.96RRT Initial 0 0.2 0.3 0.2
(vitamin D3 25C/60% 13 0.2 0.2 NT
isomer) RH
26 0.2 NT 0.1
39 NT 0.2 0.1
44 0.1 0.2 0.1
52 0.1 0.2 0.1
30C/65% 13 0.2 NT 0.2
RH
26 NT 0.2 0.1
39 0.1 0.2 NT
44 0.1 0.2 0.1
52 0.1 0.2 0.1
40C/75% 13 0.1 0.2 0.1
RH
26 0.0 0.0 0.0
1.09RRT Initial 0 NR NR NR
(vitamin D3 25C/60% 13 0.0 0.0 NT
degradate) RH
26 O.Ot NT 0.0;
39 NT 0.0$ 0.0$
44 O.Ox 0.0# O.Ot
-45-
CA 02467715 2004-05-19
Degradate Storage Weeks% Label
Condition Claim
wt% relative
to Vitamin
D
Lot 001 Lot 002 Lot 003
52 0.0$ 0.0 0.0
30C/65% 13 0.0 NT 0.0
RH
26 NT 0.0 0.0
39 0.0' 0.0 NT
44 0.0 0.1 0.0
52 0.1 0.1 0.1
40C/75% 13 0.0$ O.Ot O.Ot
RH
26 0.2 0.2 0.1
1.39RRT Initial 0 0.1 0.1 0.1
(C8 vitamin 25C/60% 13 0.2 0.1 NT
D3 ester) RH
26 0.2 NT 0.2
39 NT 0.2 0.2
44 0.3 0.2 0.3
52 0.3 0.3 0.3
30C/65% 13 0.2 NT 0.2
RH
26 NT 0.2 0.3
39 0.4 0.3 NT
44 0.4 0.4 0.4
52 0.5 0.4 0.5
40C/75% 13 0.3 0.3 0.3
RH
26 0.6 0.5 0.6
1.52RRT Initial 0 0.0 0.0 0.0
(C 10 vitamin 25Cl60% 13 0.0 0.0 NT
D3 ester) RH
26 0.1 NT 0.1
39 NT 0.1 0.1
44 0.2 0.1 0.2
52 0.2 0.2 0.2
30C/65% 13 0.0 NT 0.1
RH
26 NT 0.1 0.2
39 0,2 0.2 NT
44 0.2 0.2 0.2
52 0.3 0.2 0.3
40C/75% 13 0.2 0.2 0.2
RH
26 0.4 0.3 0.4
Total degradatesInitial 0 0.7 0.7 0.6
25C/60% 13 0.6 0.8 NT
RH
26 0.8 NT 0.8
39 NT 1.0 0.9
44 0.9 1.0 0.9
52 1.0 1.1 1.0
30C/65% 13 0.7 NT 0.8
RH
26 NT 1.0 0.9
39 1.0 1.1 NT
44 1.1 i.3 1.1
52 1.3 1.4 1.4
40C/75% 13 0.9 1.1 0.9
RH
26 1.6 1.6 1.5
-46-
CA 02467715 2004-05-19
Degradate ~ Storage ~ Weekst l % Label Claim
Condition TI (wt% relative to Vitamin D)
Lot 001 T Lot 002 Lot 003
timepoint in weeks is indicated.
0.0 represents results <0.1 % or Not Detected.
'=Not tested.
Summary of Vitamin D3 Degradation Stability Results: Alendronate Sodium 70
mg/Vitamin D3 28001.U.
Combination Tablets, 75cc HDPE Bottle, 4 Tablets per Bottle, and One 1-gram
Desiccant
Degradate Storage Weeks'% Label
Condition Claim
wt% relative
to Vitamin
D
Lot 001 Lot 002 Lot 003
0.74RRT Initial 0 0.4 0.3 0.3
(trans-vitamin 25C/60% 13 NT 0.4 0.3
D3) RH
26 0.2 0.3 NT
39 0.2 NT 0.2
44 0.2 0.3 0.2
52 0.2 0.3 0.2
30C/65% 13 0.2 0.3 NT
RH
26 0.2 NT 0.2
39 NT 0.3 0.2
44 0.2 0.3 0.2
52 0.2 0.3 0.2
40C/75% 13 0.2 0.3 0.2
RH
26 0.2 0.2 0.2
0.78RRT Initial 0 0.1 0.0 0.0
(vitamin D3 25C160% 13 NT 0.0 0.0
isomer) RH
26 0.1 0.1 NT
39 0.1 NT 0.1
44 0.1 0.1 0.1
52 0.1 0.1 0.1
30C/65% 13 0.1 0.1 NT
RH
26 0.1 NT 0.1
39 NT 0.1 0.1
44 0.2 0.1 0.1
52 0.2 0.2 0.2
40C/75% 13 0.1 0.1 0.1
RH
26 0.2 0.2 0.2
0.96RRT Initial 0 0.2 0.3 0.2
(vitamin D3 25C160% 13 NT 0.2 0.2
isomer) RH
26 0.1 0.2 NT
39 0.1 NT 0.1
44 0.1 0.2 0.1
52 0.1 0.2 0.1
30C/65% 13 0.2 U.2 NT
RH
26 0.1 NT 0.1
39 NT 0.2 0.1
44 0.1 0.2 -
-47-
CA 02467715 2004-05-19
Degradate StorageWeeks % Label
Condition Claim
wt% relative
to Vitamin
D
Lot 001 Lot Lot 003
002
52 0.1 0.2 0.1
40C/75%13 0.2 0.2 0.1
RII
26 0.0 0.1 0.0~
1.09RRT Initial0 NR NR NR
(vitamin D3 25C/60%13 NT NR NR
degradate) RH
26 O.Ot O.Ot 0.0$
39 O.Ot NT O.Ot
44 0.1 O.Ot O.Ot
52 0.1 O.Ox 0.1
30C/65%13 0.0 0.0~ NT
RH
26 0.1 NT 0.0
39 NT 0.1 0.1
44 0.2 0.1 0.1
52 0.1 0.1 0.2
40C/75%13 0.1 0.1 NR
RH
26 0.2 0.1 0.1
1.39RRT Initial0 0.1 0.1 0.1
(C8 vitamin 25C/60%13 NT 0.1 0.1
D3 ester) RH
26 0.2 0.2 NT
39 0.2 NT 0.2
44 0.2 0.2 0.2
52 0.2 0.2 0.3
30C/65%13 0.1 0.1 NT
RH
26 0.2 NT 0.2
39 NT 0.3 0.3
43 0.3 0.3 0.3
52 0.4 0.3 0.4
40C/75%13 0.2 0.2 0.2
RI-1
26 0.5 0.4 0.5
1.52RRT Initial0 0.0 0.0' 0.0'
(C10 vitamin 25C/60%13 NT 0.0 0.0'
D3 ester) RH
26 0.0 0.0 NT
39 0.1 NT 0.1
44 0.1 0.1 0.1
52 0.1 0.1 0.1
30C/65%13 0.0 0.0 NT
RH
26 0.1 NT 0.1
39 NT 0.1 0.2
44 0.2 0.2 0.2
52 0.2 0.2 0.2
40C/75%13 0.1 0.1 0.1
RI-I
26 0.3 0.2 0.3
Total degradatesInitial0 0.7 0.7 0.6
25C/60%l3 NT 0.7 0.6
RH
26 0.6 0.8 NT
39 0.8 NT 0.8
.
44 ~.0 0.9 0.8
L
-48-
CA 02467715 2004-05-19
Degradate Storage Weeks% Label
Condition Claim
wt% relative
to Vitamin
D
Lot 001 Lot 002 Lot 003
52 1.0 1.0 1.0
30C/65% 13 0.7 0.8 NT
RH
26 0.9 NT 0.9
39 NT 1.1 1,0
44 1.2 1.2 1.1
52 1.2 1.3 1.2
40C175% 13 1.0 1.1 0.9
RH
26 1.2 1.3 1.2
T The theoretical
timepoint in
weeks is indicated.
$ 0.0 represents
results <0.1
% or Not Detected.
NT=Nat tested.
NR=Not re rted.
EXAMPLE 7
Pharmacokinetics of Vitamin Da and Alendronate
An open-label, randomized, two-part, two-period, crossover study was conducted
with
236 healthy non pregnant women and men aged 18 to 65. The study was conducted
in two parts (Parts I
and II) with each consisting of a two-period, crossover design. Each subject
participated in one part of
the study only (i.e., each subject participated only in Part I or only in Part
II). Subjects entered into the
study sequentially within each part of the study, with a washout period of at
least 12 days between
treatment periods within each part of the study. Part I included Treatments A
and B, and Part II included
Treatments A and C. The Treatments consisted of the following: Treatment A -
single dose of a 70 mg
alendronate/2800 IU vitamin D3 combination tablet according to Table 7-3
below; Treatment B - single
dose of a 70 mg alendronate tablet according to Table 7-2 below; Treatment C -
a single dose of a 2800
IU vitamin D3 tablet (containing placebo excipients to replace alendronate)
according to Table 7-4 below.
In Part I, urine was collected starting 2 hours prior to, and over the 36
hours following,
dose administration in each period for determination of total urinary
excretion of alendronate. In Part II,
blood samples were collected for serum vitamin D3 determination in each period
at -24, -18, -12, and -6
hours predose, at 0 hour (just prior to drug administration), and at selected
times over the 120 hours
following dose administration.
Part I of the study evaluated the bioequivalence of alendronate in the 70 mg
alendronate/2800 IU vitamin D3 combination tablet according to Table 7-3
below, and a 70-mg
alendronate tablet according to Table 7-2 below. Part II of the study
evaluated serum pharmacokinetics
(AUCo_,zon« Cm~ of vitamin D3 obtained following administration of the 70 mg
alendronate/2800 IU
vitamin D3 combination tablet and the 2800 IU vitamin D3 tablet. The 2800 IU
vitamin D3 tablet
-49-
CA 02467715 2004-05-19
contained 28001U vitamin D3 and the inactive excipients in the
alendronatelvitamin D3 combination
tablet.
The primary pharmacokinetic parameter in Part I was total urinary excretion of
alendronate from 0 to 36 hours following oral-dose administration.
Determination of total urinary
excretion of alendronate was consistent with previous studies characterizing
the oral bioavailability of
alendronate through urinary excretion, since plasma concentrations following
oral administration are low
and difficult to detect.
The primary pharmacokinetic parameters in Part II were AUCo_,zonr and Cm~ of
vitamin
D3. Blood was collected for determination of serum vitamin D3 concentration at
the following time
points in each period: -24, -18, -12, -6 hours pre-dose, 0 hour (immediately
prior to drug administration),
and 2, 3, 5, 7, 16, 24, 36, 48, 72, 96, and 120 hours post-dose.
Serum vitamin D3 concentrations for 24 hours prior to study drug
administration were
collected to provide an indication of the behavior of endogenous levels of
vitamin D3 over 24 hours in a
controlled environment. Since vitamin D3 is synthesized in the skin via
exposure to ultraviolet light,
subjects were housed in the study unit and not exposed to direct sunlight
during the duration of the
pharmacokinetic-sampling periods (e.g., for 144 hours, from 24 hours pre-dose
until 120 hours post-
dose). Subjects were required to wear sunblock (SPF 45) and limit sun exposure
throughout the entire
study including the washout period. Subjects were also restricted from eating
foods known to be high in
vitamin D3 (e.g., salmon, herring, mackerel, cod, tuna fish, swordfish
oysters, and sardines) as well as
foods known to be supplemented with vitamin D3 (e.g., certain cereals,
fortified milk and some yogurts).
Each subject in Part I received a single oral dose of 70 mg alendronate/2800
IU vitamin
D3 combination tablet and a single oral dose of 70 mg alendronate in a
randomized, crossover fashion.
Subjects in Part II received a single oral dose of 70 mg alendronate/28001U
vitamin D3 combination
tablet and a single oral dose of a 2800 IU vitamin D3 tablet in a randomized
crossover fashion. Doses
were administered with 240 mL of plain tap water following an overnight fast
(except water), beginning
at 2100 hours the evening prior to dosing. Subjects were instructed not to lie
down and to remain upright
(at least at a 45° angle, sitting or standing) between drug
administration and the defined meal. Subjects
fasted until the standard meal, which was administered at 2 hours post-dose.
The procedures for
administration of the alendronate/vitamin D3 combination tablet were the same
as those for alendronate.
The compositions administered in the study are as set forth in the tables 7-1
through 7-4
below:
-50-
CA 02467715 2004-05-19
Table 7-1
Clinical Supplies
Drug Potency Dosage
Form
Alendronate sodium/vitamin70 mg/28001U Tablet
D3
Alendronate sodium 70 mg Tablet
Vitamin D3 28001U Tablet
Table 7-2
70-mg Alendronate Tablet Formulation
tion (uer tab
Alendronate sodium 91.37
mgt
Microcrystalline 140.0
Cellulose NF mg
Lactose Anhydrous 113.4
NF mg
Croscarmellose Sodium 3.50
NF mg
Magnesium Stearate 1.75
NF mg
Content Assay (Alendronic Acid):
Mean 70.42 mg
Ran a 66.5 - 73.5 m
TManufactured by the Merck Manufacturing Division as commercial Alendronate
Sodium 70 mg Tablet product.
~E uivalent to 70.0 m anh drous free acid.
-51-
CA 02467715 2004-05-19
Table 7-3
70-mg Alendronate/2800 IU Vitamin D3 Combination Tablet Formulation
Com osition er tablet
Alendronate Sodium 91.37 mgt
Dry Vitamin D3 100 granules 26.67 mg$
Lactose Anhydrous NF 62.32 mg
Microcrystalline Cellulose 131.00 mg
NF
Colloidal Silicon Dioxide 0.812 mg
NF
Croscarmellose Sodium 9.740 mg
NF
Magnesium Stearate NF 2.275 mg
(Intragranular)
Magnesium Stearate NF 0.812 mg
(Extragranular)
Content Assay (Alendronic
Acid):
Mean 70.5 mg
RSD 0.89
Range 69.7 - 71.6 mg
Content Assay (Vitamin
D3):
Mean 2742 IU
RSD 1.59%
Ran a 2643 - 2822 IU
' Equivalent to 70.0 mg
anhydrous free acid
$ Vitamin D3 Dry Pharm
Grade granules also contained
medium chain triglycerides,
gelatin, sucrose, butylatedrch
hydroxytoluene, sta and
sodium
aluminum
silicate.
Adjusted based on assay.
Quantity specified assumes
and assay of 105,000
LU./g.
Adjusted based on quantity00
of Vitamin D3 100,0 LU./g
added
in
order
to
achieve
final tar et tablet wei
ht of 325 m .
-52-
CA 02467715 2004-05-19
Table 7-4
2800 IU Vitamin D3 Tablet Formulation
Com osition er tablet
Dry Vitamin D3 100 granules 26.72 mgt
Microcrystalline Cellulose 97.68 mg
NF
Lactose Anhydrous NF 46.61 mg
Croscarmellose Sodium 5.34 mg
NF
Colloidal Silicon Dioxide 0.445 mg
NF
Magnesium Stearate NF 1.336 mg
Content Assay (Vitamin
D3):
Mean 2789 IU
RSD 2.7%
Range 2660 - 29571U
'Dry Vitamin D3 100 granules
also contained medium
chain triglycerides,
gelatin,
sucrose, butylated hydroxytoluene,sodium
starch and aluminum
silicate
Adjusted
based on assay. Quantity
specified assumes an
assay of 105,000 LU./g.
Tablet
wei ht varied based on
uanti of vitamin D3
100,000 LU./ added.
The area under the serum concentration-versus-time curve from 0 to 120 hours
postdose
(AUCo_,zon~) was calculated using the unadjusted concentrations of vitamin D3
(Ct) by the trapezoidal
method to the last sample collection. Samples with concentrations lower than
the assay's limit of
quantitation (LOQ) were assigned a value of zero for calculation purposes.
Maximum observed
concentrations (Cm~) and time of Cm~ (Tm~) were obtained by inspection of the
measured concentrations
of vitamin D3 in serum and the actual recorded times of sample collection.
Concentration profiles of
vitamin D3 in serum were also measured in three different ways, two of which
account for baseline
vitamin D3 serum concentrations in the manner discussed in below. AUCo_~zon~,
Cm~ and Tm~ were
calculated in the same manner.
The bioavailability of the 70 mg alendronate tablet/2800 IU vitamin D3
combination
tablet relative to the 70 mg alendronate alone tablet was estimated using the
GMR for the total urinary
excretion of alendronate from the alendronate/vitamin D3 combination tablet
versus the 70-mg
alendronate-alone tablet. The relative bioavailability of the 70 mg
alendronate/2800 IU vitamin D3
combination tablet with respect to 2800-IU vitamin D3 tablet alone was
estimated using the GMR
(alendronate plus vitamin D3/vitamin D3 alone) for AUCo_,zon~ and Cm~.
-53-
CA 02467715 2004-05-19
The vitamin D3 single-dose pharmacokinetics following the administration of 70
mg
alendronate/2800 iL1 vitamin D3 combination tablet and 28001U vitamin D3
tablet were compared using
three different approaches. In the first approach, the vitamin D3
pharmacokinetics for endogenous
vitamin D3 serum concentrations were compared following the administration of
the two treatments.
Using this approach, the vitamin D3 single-dose pharmacokinetics (AUCo_,zo6
and Cm~)
for endogenous vitamin D3 serum concentrations following the administration of
?0 mg alendronate/2800
ILT vitamin D3 combination tablet and 2800 IU vitamin D3 tablet were compared
using an ANOVA model
appropriate for a 2-period, crossover design. Appropriate transformations were
used on the
pharmacokinetic parameters (i.e., log-transformation for AUCo.,zon.> Cm~,
ranks for Tm~, and inverse for
apparent t~). Back-transformed summary statistics and inferential results were
reported. The
assumptions of the ANOVA model were tested far normality. The normality
assumption was generally
satisfied for AUCo_,zon1 and Cm~,
To estimate the bioavailability of vitamin D3 in the 70 mg alendronate/2800 IU
vitamin D3 combination tablet relative to that of the 2800 ILJ vitamin D~
tablet, a 90% CI, based upon the
I S t-distribution, was calculated for the GMR (70 mg alendronate/2800 IU
vitamin D3 combination
tablet/2800 IU vitamin D3 tablet) of AUCo.,zon~ and Cm~ and then compared to
the pre-specified
bioequivalence bounds of (0.80, 1.25). Summary statistics and between-
treatment comparisons were also
provided for the TmaX of vitamin D3.
As another way to measure the pharmacokinetic parameters, consideration of pre-
dose
vitamin D3 in the plasma specifically led to the development of a model for
the observed changes in
vitamin D3 concentrations during the experimental period. This model allowed
for the subtraction of the
contributions from the baseline vitamin D3 serum concentrations and enable the
estimation of
pharmacokinetic parameters arising exclusively from the oral administration of
this compound. The
model rested on the following assumptions: (1) background concentrations
change in an approximately
linear fashion as a function of time (C, = C; + C," ~ t, where C; and C," are
the intercept and slope of a
straight line and t is the number of hours relative to dose administration)
when no exogenous vitamin D3
is administered; (2) the phannacokinetic behaviors of endogenous and ingested
vitamin D3 are
independent of each other, that is, the body handles the endogenously
available vitamin D3 in the same
manner, whether additional doses are ingested or not (treated and untreated in
the context of this study),
and ingested vitamin D3 is also handled similarly in the presence of varying
amounts of this compound in
the body previous to dose administration; (3) return of the concentration
profile to baseline following a
dose takes place in a pharmacokinetic manner similar to that observed when
exogenous compounds (most
drug products) are administered, (i.e., the terminal phase of return to
baseline will be log linear).
Based on these assumptions, a function describing the sum of a baseline of the
form C, _
C; + Cm ~ t and a two-compartment model (See equation below) was fitted to
individual C, vs. t profiles (-
-54-
CA 02467715 2004-05-19
24 to 120 h post-dose) by a least-squares minimization method using a
Generalized Reduced Gradient
nonlinear optimization method implemented in Microsoft EXCEL (Solver Routine).
The fit was
constrained to yield a terminal phase of approach-to-baseline approximating
log linear behavior, The
best fit coefficients for each profile were then used to interpolate the
values of the baseline in the range of
0 to 120 hr post-dose and the interpolated baseline subtracted from each
profile.
C~ =Ci+C,nt Efl2 ka(~ hox),+B2 knr(r-~mg)_~AfB~2 k~,(~-hag)
where
t = time relative to dose administration
C~ = Predicted concentration of vitamin D3 in serum
C; = Predicted value of baseline at t=0
Cm = Slope of predicted baseline
And A, B, kd, ke, and ka are parameters of a two-compartment model with first
order absorption and t,a8 is
the individual delay in absorption following oral administration of vitamin
D3.
Pharmacokinetic parameters measured using this method (AUCo_ixon« Cm~~ Tmax)
were
calculated in the same manner as described using the first measurement method.
Summary statistics for
total urinary excretion of alendronate over 36 hours are presented in Table 7-
5 below. After, a single
dose, the LS means for total urinary excretion of alendronate were 197.5 and
191.9 ltg for the 70 mg
alendronate/2800 IU vitamin D3 combination tablet and 70-mg alendronate-alone
tablet, respectively.
The GMR and corresponding 90% CI for the total urinary excretion of
alendronate (70 mg
alendronate/2800 IU vitamin D3 combination tablet versus the 70 mg alendronate-
alone tablet) was 1,03
(0,91, 1.17). The 90% CI fell within the pre-specified bioequivalence bounds
of (0.80, 1.25).
-55-
CA 02467715 2004-05-19
Table 7-5
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Alendronate (~tg) Total
Urinary Excretion Following Single Dose Administration of 70 mg
Alendronate/2800 IU Vitamin D3
Combination Tablet or 70 mg Alendronate-Alone Tablet
LSt 90%
TreatmentN Mean MedianMin Max SD= GMR CI~~
for
GMR
Alendronate207 197.5 209.811.3 3617.7329.1 1.03 (0.91,
+ Vit
D3 1.17)
Alendronate207 191.9 204.40.1 1629.6522.2
Root Mean
Squared
Error
(RMSE)
in log
scale
from
ANOVA
Model
= 0.778.
t LS=Least-Squares
(Back-transformed
from
the log
scale).
SD=Between-Subject
Standard
Deviation
back-transformed
from
log scale.
GMR=Geometric
Mean
Ratio
(LS Mean
of alendronate
+ vit
D3/LS
Mean
of alendronate).
~~ Cl=Confidence
Interval.
With respect to the plasma measurements, the LS means for vitamin D3
AUCo_~zon~ (not
considering endogenous vitamin D3 serum concentrations) were 296.4 and 337.9
ng~h/mL for the 70 mg
alendronate/28001U vitamin D3 combination tablet and 2800 IU vitamin D3
tablet, respectively (Table 7-
6). The AUCo.izon, GMR (alendronate plus vitamin D3 combination tablet/vitamin
D3 tablet) was 0.88,
with a 90% C1 of (0.81, 0.95).
-56-
CA 02467715 2004-05-19
Table 7-6
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Vitamin D3 AUC°.,zoh~
(ng~hr/mL) Not Considering Endogenous Vitamin D3 Serum Concentrations
Following Single-Dose
Administration of 70 mg Alendronate Plus 2800 IU Vitamin D3 Combination Tablet
or 2800 IU Vitamin
D3 Tablet Alone
Between-
Treatment N LS MeanMedianMin Max SubjectGMR~ 90%
t SD$ ChI
for
GMR
Alendronate/28 296.4 257.5 85.01648.8375.5 0.88 (0.81,
Vitamin 0.95)
D3
Vitamin 28 337.9 309.6 111.91485.9344.2
D3
Alone
Root Mean
Squared
Error
(RMSE)=
0.168
(from
the ANOVA
model).
t Least-square
Means
back-transformed
from the
log scale.
$ SD=Standard
Deviation
back-transformed
from log
scale.
GMR=Geometric
Mean Ratio
(LS mean
of vit
D3+alendronate/LS
mean of
vit D3).
II CI=Confidence
Interval.
The LS means for vitamin D3 CmaX, not considering endogenous vitamin D, serum
concentrations, were 5.9 and 6.6 ng/mL for 70 mg alendronate/2800 IU vitamin
D3 combination tablet
and 2800 IU vitamin D3 tablet, respectively (Table 7-7). The GMR for Cm~
(alendronate plus vitamin D3
combination tablet/vitamin D3 tablet) was 0.89, with a 90% CI of (0.84, 0.95).
The 90% CI for AUCo_,~o,,~
and Cmax GMR not considering for vitamin D3 serum concentrations, fell within
the pre-specified
bioequivalence bounds of (0.80, 1.25).
-57-
CA 02467715 2004-05-19
Table 7-7
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Vitamin D3 Cm~
(ng/mL) Not Considering Endogenous Vitamin D3 Serum Concentrations Following
Single-Dose
Administration of 70 mg Alendronate Plus 2800 IU Vitamin D3 Combination Tablet
or 2800 IU Vitamin
D3 Alone Tablet
Between
TreatmentN LS MeanMedianMin Max -SubjectGMR4 90%
t SD* CI~~
for
GMR
Alendronate/28 5.9 5.3 2.5 17.4 3.3 0.89 (0.84,
Vitamin 0.95)
D3
Vit D3 28 6.6 6.2 3.S 18.1 3.1
Alone
Root Mean
Squared
Error
(RMSE)=
0.138
(from
the ANOVA
model).
t Least-
Square
Means
back-transformed
from
the log
scale.
SD=Standard
Deviation
back-transformed
from
log scale.
GMR=Geometric
Mean
Ratio
(LS mean
of vit
D3+alendronate/LS
mean
of vit
D,).
CI=Confidence
Interval.
Table 7-8 displays the results of statistical analysis for the vitamin D3 Tm~,
obtained from
serum profiles not considering endogenous vitamin D3 serum concentrations. The
median T,"~ for
vitamin D3 with and without alendronate was 12.0 and 9.0 hours, respectively.
No significant between-
treatment difference was observed (p-value>0.200).
1S
Table 7-8
Summary Statistics for Vitamin D3 T,"ax (Hours) Obtained from Serum Profiles
Not Considering
Endogenous Vitamin D3 Serum Concentrations Following Single-Dose
Administration of 70 mg
Alendronate Plus 2800 IU Vitamin D3 Combination Tablet or 2800 IU Vitamin D3
Tablet Alone
Between-
Treatment N MedianMin Max Sub'ect -Value
SD t ~
Alendronate/Vitamin28 12.0 7.0 16.0 2.6 0.978
D3
Vitamin D3 28 9.0 7.0 16.0 2.3
Alone
SD = Standard
Deviation.
t -Value was
com uted usin
rank anal
sis.
-58-
CA 02467715 2004-05-19
The LS means for vitamin D3 AUC0.,zon~ were 297.5 and 336.7 ng-h/mL for 70 mg
alendronate/2800 IU vitamin D3 combination tablet and 2800 IU vitamin D3
tablet, respectively (Table 7-
9). The AUCo_,zon, GMR (alendronate plus vitamin D3 combination tablet/vitamin
D3 tablet) was 0.88,
with a 90% CI of (0.82, 0.95).
Table 7-9
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Vitamin D3 AUCo_,zon.
(ng-h/mL), with Predose Vitamin D3 Concentration at Time=0 as a Covariate,
Following Single-Dose
Administration of 70 mg Alendronate plus 2800 IU Vitamin D3 Combination Tablet
or 2800 IU Vitamin
D3 Alone Tablet
Between
TreatmentN LS MedianMin Max -SubjectGMR~ 90% CII~
Meant SDS for
GMR
Alendronate/28 297.5 257.5 85.0 1648.8376.8 0.88 (0.82,
0.95)
Vitamin
D3
Vitamin 28 336.7 309.6 111.91485.9343.0
D3
Alone '
Root Mean
Squared
Error
(RMSE)=
0.154
(from
the ANOVA
model).
t Least-
Square
Means
back-transformed
from
the log
scale.
$ SD=Standard
Deviation
back-transformed
from
log scale.
GMR=Geometric
Mean
Ratio
(LS mean
of vit
D3+alendronate/LS
mean
of vit
Dj).
II CI=Confidence
Interval.
The LS means C,"~ of vitamin D3, were 5.9 and 6.6 ng/mL for 70 mg
alendronate/2800
IU vitamin D3 combination tablet and 2800-IU vitamin D3 tablet, respectively,
as shown in Table 7-10
below. The C",~ GMR (70 mg alendronate/2800 Ii1 vitamin D3 combination
tablet/2800 IU vitamin D3
tablet) was 0.90, with a 90% CI of (0.85, 0.95). The 90% CI for AUC0.,zon~ and
Cm~ GMR with pre-dose
vitamin D3 concentration at time=0 as a covariate, fell within the pre-
specified bioequivalence bounds of
(0.80, 1.25).
- 59 -
CA 02467715 2004-05-19
Table 7-10
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Vitamin D3 Cm~
(ng/mL) With Predose Vitamin D3 Concentration at Time=0 as a Covariate,
Following Single-Dose
Administration of 70 mg Alendronate plus 28001L1 Vitamin D3 Combination Tablet
or 2800 IU Vitamin
D3 Alone Tablet
Between
Treatment N LS Median Min Max -SubjectGMR 90%
Mean SD$ ChI
t for
GMR
Alendronate/28 5.9 5.3 2.5 17.4 3.3 0.90 (0.85,
Vitamin 0.95)
D3
Vitamin 28 6.6 6.2 3.5 18.1 3.1
D3
Alone
Root Mean
Squared
Error
(RMSE)=
0.115
(from
the ANOVA
model).
t Least-
Square
Means
back-transformed
from the
log scale.
# SD=Standard
Deviation
back-transformed
from log
scale.
GMR=Geometric
Mean Ratio
(LS mean
of vit
D3+alendronate/LS
mean of
vit D3).
n Cl~onfidence
Interval.
The results of the data analysis of the AUCo.izon« of vitamin D3, measured
using model-
based vitamin D3 baseline concentrations, are summarized in Table 7-11. The LS
means for vitamin D3
AUCo_,zonr measured using model-based vitamin D3 baseline concentrations were
143.1 and 169.1
ng~h/mL for the 70 mg alendronate/2800 IU vitamin D3 combination tablet and
the 2800 IU vitamin D3
tablet, respectively. The AUC0.izon~ GMR (alendronate plus vitamin D3
combination tablet/vitamin D3
tablet) was 0.85, with a 90% CI of (0.76, 0.94). The lower limit of 90% CI
fell just below the pre-
specified lower bound of 0.80.
-60-
CA 02467715 2004-05-19
Table 7-11
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Vitamin D3 AUCo.,ZOn~
(ng~hr/mL), Measured Using Model-Based Vitamin D3 Baseline Concentrations,
Following Single-Dose
Administration of 70 mg Alendronate/2800 IU Vitamin D3 Combination Tablet
Between
TreatmentN LS MedianMin Max -SubjectGMR 90% CIII
Mean SD= for GMR
t
Alendronate/28 143.1 153.5 61.1236.147.7 0.85 (0.76,
Vitamin 0.94)
D3
Vit D3 28 169.1 175.0 107.2251.437.3
Alone
Root Mean
Squared
Error
(RMSE)=
0.224
(from
the ANOVA
model).
t Least-
Square
Means
back-transformed
from
the log
scale.
$ SD=Standard
Deviation
back-transformed
from
log scale.
g GMR=Geometric
Mean
Ratio
(LS mean
of vit
D3+alendronate/LS
mean
of vit
D3).
II CI=Confidence
Interval.
The LS means Cm~ of vitamin D3, measured using model-based vitamin D3 baseline
concentrations, were 4.0 and 4.6 ng/mL for the 70 mg alendronate/2800 IU
vitamin D3 combination tablet
and 2800 IU vitamin D3 tablet, respectively, as shown in Table 7-12. The C",~
GMR (70 mg
alendronate/2800 IU vitamin D3 combination tablet/2800 IU vitamin D3 tablet)
was 0.88, with a 90% CI
of (0.83, 0.93). The 90% CI for Cm~ GMR measured using model-based vitamin D3
baseline
concentrations fell within the pre-specified bioequivalence bounds of (0.80,
1.25).
-61-
CA 02467715 2004-05-19
Table 7-12
Summary Statistics and GMR With Corresponding 90% Confidence Intervals for
Vitamin D3 Cm~
(ng/mL) Measured Using Model-Based Vitamin D3 Baseline Concentrations
Following Single-Dose
Administration of 70 mg Alendronate plus 2800 IU Vitamin D3 Combination Tablet
or 2800 IU Vitamin
D3 Tablet Alone
Between
Treatment N LS Median Min Max -SubjectGMR4 90%
Mean SD$ CI~I
t for
GMR
Alendronate/28 4.0 4.0 1.9 6.0 1.1 0.88 (0.83,
Vitamin 0.93)
D3
Vitamin 28 4.6 4.6 3.0 7.2 0.9
D3
Alone
Root Mean
Squared
Error
(RMSE)=
0.115
(from
the ANOVA
model).
t Least-
Square
Means
back-transformed
from the
log scale.
$ SD=Standard
Deviation
back-transformed
from log
scale
GMR=Geometric
Mean Ratio
(LS mean
of vit
D3+alendronate/LS
mean.
of vit
D3).
~~ CI=Confidence
Interval.
Table 7-13 displays the results of statistical analysis for vitamin D3 Tmax
obtained from
profiles measured using model-based baseline vitamin D3 concentrations. The
median T,"ax for vitamin D3
with or without alendronate was 12.0 and 9.0 hours, respectively. No
significant between-treatment
difference was observed (p-value>0.200).
20
Table 7-13
Summary Statistics for Vitamin D3 Tmax (Hours) Obtained From Profiles Measured
Using Model-Based
Baseline Vitamin D3 Concentrations Following Single-Dose Administration of 70
mg Alendronate Plus
2800 IU Vitamin D3 Combination Tablet or 2800 IU Vitamin D3 Tablet Alone
Between-
Treatment N MedianMin Max Sub'ect -Value
SD t $
Alendronate/Vitami28 12.0 7.0 16.02.6 0.978
n D3
Vitamin D3 28 9.0 7.0 16.02.3
Alone
SD = Standard
Deviation.
-Value was
com uted
usin rank
anal sis.
-62-
CA 02467715 2004-05-19
The results of the data analysis of the t, of vitamin D3, measured using model-
based
vitamin D3 background concentrations, are summarized in Table 7-14. The
harmonic mean apparent t.,,
for vitamin D3 with and without alendronate was 24.0 and 25.5 hours,
respectively. No significant
between-treatment difference was observed (p-value>0.200).
Table 7-14
Summary Statistics for Apparent t,~ (hours) For Vitamin D3 Measured Using
Model-Based Vitamin D3
Baseline Concentrations Following Single-Dose Administration of 70 mg
Alendronate/2800 IU Vitamin
D3 Combination Tablet or 2800 IU Vitamin D3 Alone Tablet
TreatmentN HarmonicJackknife Min MedianMax p-Value$
Mean Between-Subject
SDt
Vit D3+ 28 23.8 11.9 11.524.0 100.8 0.833
Alendronate
Vit D3 28 23.2 18.5 5.3 25.5 188.0
t SD
= Standard
Deviation
t p-Value
was
computed
using
inverse
transformation
EXAMPLE 8
Degradation Detection Method
A method has been developed for the composite assay of vitamin D3 in
combination
alendronate/vitamin D3 tablets (70 mg alendronate/2800 1U vitamin D3). Vitamin
D3 is extracted from 15
tablets in about 50 mL of 5% water/ 95% methanol diluent. The solution is
stirred for 10 minutes, sonicated
for 30 minutes, and stirred for an additional 3 hours. Samples are centrifuged
and 100 ~L of the supernatant
are injected onto a Phenomenex Phenosphere 80A ODS (1) column (150 x 4.6 mm, 3
Vim) for reversed
phase HPLC analysis. The method is a 65-minute gradient method with a
detection wavelength at 265 nm.
Both pre-vitamin D3 and vitamin D3 peaks are quantitated and summed to
calculate the total amount of
vitamin D3 in the sample. The method was validated for satisfactory
specificity, linearity, recovery,
precision, reproducibility, solution stability, sensitivity, and robustness.
-63-
CA 02467715 2004-05-19
Exemplary chromatographic conditions are listed below:
Flow Rate: 1.2 mL/min
Column Tem erature:25 C
In'ection Volume: 100 L
Mobile Phase: Gradient, A = 0.025% phosphoric
acid, B = 99%Acetonitrile
/1 %A
Run Time: 65 minutes
Column: Phenos here 80 tl, ODS 1 column,
150 x 4.6mm, 3 m
Sam le Tra Tem erature5 C
Detector Wavelength:265 nm
Gradient Time Table:
T (min)0 16 39 43 57 57.0165
Aqueous51.513 10 0 0 51.551.5
Mixture48.587 90 100 10048.548.5
Formulation Composition of Combination Tablets (70 mg Alendronate/28001U
Vitamin D3)
Com osition Unit Wei ht Wei ht
m
Alendronate Sodium91.5 28.2%
Dry Vitamin D3 26.7* 8.2%
100 granules
Avicel PH102 13l 40.3%
Lactose, Anhydrous62.2 19.1
Croscarmellose 9.75 3.00%
Sodium
Colloidal Silica 0.81 0.25%
Intragranular Mg 2.28 0.70%
Stearate
Extra ranular M 0.81 0.25%
Stearate
Tablet Wei ht: 325 100%
*26.7 grams of the Dry Vitamin D, 100 granules contains 105,000 IUIg of
vitamin D3
-64-
CA 02467715 2004-05-19
The excipient peaks and degradates with their typical retention time and
relative retention times (RRT) with
respect to vitamin D3 are presented in Table 8-1. Major degradation pathways
of vitamin D3 are
photoisomerization, thermal isomerization, and transesterification, as shown
in F1G. 5.
Table 8-1
Summar,~of Peak Identification for Combination Tablets
Retention Time RRT Classification
minutes
3.08 0.09 Excipient-related
3.63 0.10 Excipient-related
3.88 0.108 Excipient-related
4.05 0.11 Excipient-related
4.23 0.12 Excipient-related
4.78 0.13 Excipient-related
7.53 0.21 Unknown
11.33 0.32 Excipient-related
17.25 0.48 Unknown
17.47 0.49 Excipient-related
18.05 0.50 Excipient-related
20.64 0.57 Excipient-related
26.70 0.74 Trans-vitamin D3
28.15 0.78 Vitamin D3 isomer
31.33 0.87 Pre-vitamin D3
34.42 0.96 Vitamin D3 isomer
35.94 1.00 Vitamin D3
39.17 1.09 Vitamin D3 Isomer
43.45 1.21 Excipient-related
49.85 1.39 C8-vitamin D3 ester
50.42 1.40 C8-pre-vitamin
D3 ester
54.52 1.52 C 10-vitamin D3
ester
55.65 1.55 C10-pre-vitamin
Di ester
-65-
CA 02467715 2004-05-19
The limit of quantitation (LOQ) was determined by injecting different
concentrations of
vitamin D3 solution and selecting the lowest concentration with a signal-to-
noise ratio above 10. The
LOQ was determined as about 9 nglmL (injection volume 100 pL), which is 0.04%
of the method
concentration with an average signal-to-noise ratio of 11.1 for ten replicate
determinations.
EXAMPLE 9
Once-weekly Dosing Re ig'mens
Alendronate and vitamin D tablets or other solid dosageformulations containing
about 70
mg of alendronate, on an alendronic acid active basis, and about 5,600 IU of
vitamin D may be prepared.
(See Examples I, 2, and 3). The tablets or other solid dosage formulations may
be orally administered to
a subject once-weekly, i.e., preferably about once every seven days (for
example, every Sunday), for a
period of at least one year. This method of administration is expected to be
useful and convenient for
treating or preventing osteoporosis while providing vitamin D nutrition. This
method is also expected to
be useful for improving subject acceptance and compliance, and ensuring that
all subjects taking a
bisphosphonate receive adequate vitamin D nutrition.
Alternatively, alendronate and vitamin D tablets or other solid dosage
formulations
containing about 70 mg of alendronate, on an alendronic acid active basis, and
about 2,800 IU of vitamin
D may be prepared. (See, e.g., Examples 1 and 3). The tablets or other solid
dosage formulations may be
orally administered to a subject once-weekly, i.e., preferably about once
every seven days (for example,
every Sunday), for a period of at least one year. This method of
administration is expected to be useful
and convenient for treating or preventing osteoporosis while providing vitamin
D nutrition. This method
is also expected to be useful for improving subject acceptance and compliance,
and ensuring that all
subjects taking a bisphosphonate receive adequate vitamin D nutrition.
Alternatively, alendronate and vitamin D tablets or other solid dosage
formulations
containing about 35 mg to about 70 mg of alendronate, on an alendronic acid
active basis, and 2,8001U
of vitamin D may be prepared. (See, e.g., Example 3). The tablets or other
solid dosage formulations
may be orally administered to a human subject once-weekly, i.e., preferably
about once every seven days
(for example, every Sunday), for a period of at least one year. This method of
administration is expected
to be useful and convenient for treating or preventing osteoporosis while
providing vitamin D nutrition.
This method is also expected to be useful for improving subject acceptance and
compliance, and ensuring
that all subjects taking a bisphosphonate receive adequate vitamin D
nutrition.
Alendronate and vitamin D tablets or other solid dosage formulations
containing about
280 mg of alendronate, on an alendronic acid active basis, and about 5,600 ILJ
of vitamin D may be
prepared. (See, e.g., Examples 2 and 3). The tablets or other solid dosage
formulations may be orally
-66-
CA 02467715 2004-05-19
administered to a subject once-weekly, i.e., preferably about once every seven
days (for example, every
Sunday), for a period of at least one to six months. This method of
administration is expected to be
useful and convenient for treating Paget's disease while providing vitamin D
nutrition. This method is
also expected to be useful for improving subject acceptance and compliance,
and ensuring that all
S subjects taking a bisphosphonate receive adequate vitamin D nutrition.
Alternatively, alendronate and vitamin D tablets or other solid dosage
formulations
containing about 280 mg of alendronate, on an alendronic acid active basis,
and about 2,800 IU of
vitamin D may be prepared. (See, e.g., Example 3). The tablets or other solid
dosage formulations may
be orally administered to a subject once-weekly, i.e., preferably about once
every seven days (for
example, every Sunday), for a period of at least one to six months. This
method of administration is
expected to be useful and convenient for treating Paget's disease while
providing vitamin D nutrition.
This method is also expected to be useful for improving subject acceptance and
compliance, and ensuring
that all subjects taking a bisphosphonate receive adequate vitamin D
nutrition.
Alendronate and vitamin D tablets or other solid dosage formulations
containing about
280 mg of alendronate, on an alendronic acid active basis, and 5,600 IU or
2,800 IU of vitamin D may be
prepared. (See, e.g., Examples 1, 2, and 3). The tablets or other solid dosage
formulations may be orally
administered to a subject once-weekly, i.e., preferably about once every seven
days (for example, every
Sunday). This method of administration is expected to be useful and convenient
for treating metastatic
bone disease while providing vitamin D nutrition. This method is also expected
to be useful for
improving subject acceptance and compliance, and ensuring that all subjects
taking a bisphosphonate
receive adequate vitamin D nutrition.
It will be apparent to those skilled in the art that various modifications can
be made to
this invention of methods and compositions far inhibiting bone resorption
without departing from the
scope or spirit of the invention or of the claims. It is also intended that
the present invention and
appended claims cover modifications, variations and equivalents of the methods
and compositions for
inhibiting bone resorption of the present invention.
-67-