Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
A SYSTEM AND METHOD FOR AUTOMATIC PROTEIN
SEQUENCING BY MASS SPECTROMETRY
FIELD OF THE INVENTION
The present invention relates generally to a computer implemented method of
determining the amino acid sequence of a protein by automatic interpretation
of mass
spectra of isotopically-labeled C-terminal peptide fragments of the protein.
BACKGROUND
The linear arrangement of amino acids in a protein is elucidated by protein
sequencing. Knowledge of the sequence of a protein is essential to the
techniques of
molecular biology. For example, protein sequence information is a prerequisite
for DNA
cloning and provides information for making oligonucleotide probes and
polymerase chain
reaction (PCR) primers. Furthermore, protein sequencing allows the synthesis
of peptides
to be used in antibody production, enables the identification of proteins of
interest, and
helps characterize recombinant products.
When the sequence of a peptide sample is deduced without any additional
information such as the sequence of a known related peptide, the approach is
known as de
novo sequencing. Despite the progress in genomic DNA sequencing, de novo
sequencing of
proteins and peptides is still required in a biological research environment
since many
experiments are carried out in organisms whose genomes are not sequenced.
The basic method of protein sequencing is Edman degradation, (Ward & Simpson,
"Proteins and Peptides, Isolation for Sequence Analysis of in Molecular
Biology and
Biotechnology, Robert A. Meyers, Ed., VCH Publishers, Inc. (1995), p. 767), a
three-step
chemical process based on N-terminal cleavage. Although laboratory automation
has made
today's practice of the Edman method very efficient, it has several drawbacks,
including
sensitivity to non-protein contaminants (see, e.g., Keen ~ Findlay, "Protein
Sequencing
Techniques" in Molecular Biology and Biotechnology, Robert A. Meyers, Ed., VCH
Publishers, Inc., (1995)).
Chemical sequencing of the C-terminus of a protein can be accomplished by the
thiocyanate method (Schlack & Kumpf, Physiol. Chem., (1926) 154:125-170).
Although
useful for sequencing proteins and peptides that are blocked at the N-
terminus, this method
also has its drawbacks, including the severity of the reaction conditions and
the need to
couple the protein to a solid support (Bailey, J. Chromatog. A, (1995), 705:47-
65).
Ultimately, mass spectrometry (MS) has emerged as an attractive alternative to
chemical methods and has been used to solve sequencing problems that are not
easily
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
handled by conventional techniques of protein chemistry (she, a g , Carr &
Annan,
"Overview of Peptide and Protein Analysis by Mass Spectrometry," in Current
Protocols in
Molecular Biology, Ausubel et al., Eds., John Wiley & Sons, Inc., (1997),
10.21). In mass
spectrometry, the molecular weights of gas-phase ions that are formed from
intact neutral
molecules are determined by separation based on their mass-to-charge (m/z)
ratios.
One effective way of sequencing proteins is the use of mass spectrometry to
determine the molecular weights of peptides in mixtures, such as those
resulting from
proteolytic digestion. The digestion of a protein with a particular enzyme,
e.g., trypsin,
cleaves the protein at specific sites whose locations depend on the amino acid
sequence of
the protein. The result is a collection of peptides that gives rise to a
signature mass
spectrum, often caned a "fingerprint." When mlz values are measured to better
than 0.01%
accuracy, the amino acid composition of a peptide fragment can be reliably
deduced. Thus,
a fingerprint can be utilized to unambiguously identify a protein, or to
verify a translation
product by comparing it to information contained in a 'database of peptide
fingerprints of
known proteins.
Mass spectrometry is not limited to measuring the masses of single species
but,
through the technique of tandem mass spectrometry (MS/MS), can also reveal
structural
information, including peptide sequences. In many mass spectrometry systems,
further
fragmentation of the gas phase ions occurs, either spontaneously, or by
collision with gas
molecules in so-called "collision induced dissociation" (CI17). The
subfragments that are
generated can also be separated from one another by m/z ratio.
A particular advantage of tandem mass spectrometry is that it can provide
amino
acid sequence information for peptides at the picomolar or femtomolar level.
In this
application, tandem mass spectrometry typically uses a first mass analyzer to
select a
particular peptide ion that it permits to undergo fragmentation, for example
by CID, to
produce subfragment ions of the parent peptide or peptide fragment. The
technique also
utilizes a second mass analyzer so that, after initial peptide ionization and
ion selection,
subfragment ions are separated and analyzed. The resulting mass spectra
contain m/z ratios
for the subfragments.
The fragmentation mechanisms undergone by organic molecules, for example
during
CID, have been well-studied. Therefore important structural information can be
revealed by
analyzing the masses of both parent species and their subfragments. In
particular,
molecules tend to preferentially cleave at weak chemical bonds but many
functional groups
remain intact during the fragmentation process. It has been found that the
peptide amide
linkage is particularly susceptible to cleavage under the conditions employed
in MS/MS.
-2-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
Consequently, the tandem mass spectra of peptides conPain beaks corresponding
to
subfragments which differ from one another by single amino acid residues and
can therefore
assist in sequence determination (see, e.g., Hunt et al., (1986), Proc. Natl.
Acad. Sci. USA,
83:6233-6237).
Nevertheless, the problem of analyzing tandem mass spectra remains formidable
for
a number of reasons. First, cleavage at a peptide bond gives rise to a pair of
fragments, one
containing the N-terminus, the other containing the C-terminus. Which of these
fragments
bears the charge after fragmentation is not predictable so that most spectra
contain two
series of fragments: those containing the C-terminus, known as the y-ions
(also called Y"
ions); and those containing the N-terminus, known as the b-ions (also called B-
ions). The
main challenge of de novo sequencing by mass spectrometry is to reliably
recognize the ions
of one series of fragments in an otherwise complicated spectrum.
Second, the fragmentation process is not ideal. Some amide linkages are not
cleaved during Cm so that the differences between some peaks in the MS/MS
spectrum do
not correspond to masses of single amino acid residues but to two or more
residues.
Similarly, some fragmentation occurs within amino acid residues to produce
subfragments
whose masses do not differ from the masses of other subfragments by exact
numbers of
amino acid residues.
Third, the conditions under which peptide samples are ionized often give rise
to
multiply charged ions. Therefore there may be series of peaks in the spectrum
which
correspond to ions of the same fragment bearing different charges. In such
circumstances,
the peaks which correspond to fragments differing by a single amino acid
residue will differ
by a m/z value which is a fraction of the mass of the residue.
Finally, a further problem, which depends upon the resolution of the
instrument
employed, is that it may not be possible to resolve closely spaced peaks that
correspond to
different isotopically substituted forms of the same fragment.
In general, then, the outcome of a typical de novo MS/MS analysis of a
polypeptide
is a spectrum whose interpretation is far from straightforward and which
usually results in
some unidentified members of the peptide sequence.
Hitherto, computational methods for the interpretation of de novo MS/1VIS
peptide
spectra have been only partially successful. Part of the reason is that the
spectra themselves
do not have sufficient sensitivity or resolution to permit thorough analysis.
Another reason
is that the algorithms employed are either too time-consuming to be practical
or not accurate
enough to be useful. For example, in one early approach, measured masses of
peptide
fragments after enzymatic digestion are compared with theoretical peptide
masses from
-3-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
each sequence entry in a database, using the same cleav~.ge'~peci~city. The
comparison
gives a score with which to quantify the goodness of fit (see, e.g., Cottrell,
Pept. Res.,
(1994), 7:115-124; Matsui et al., Electrophoresis, (1997), 18:409-417).
In another approach, theoretical spectra of many possible sequences are
matched
S with the actual spectrum until a good fit is obtained (Eng, J.K., et al.,
"An Approach to
Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a
Protein
Database," Journal of the American Society of Mass Spectrometry, 5:976 - 989,
(1994)). A
drawback with this approach is that there can be a combinatorial explosion
associated with
trying to match many possible sequences of amino acid residues with the set
that gives rise
to a spectrum. In employing approximations to limit the inevitable
combinatorial
explosion, the correct sequence may often be rejected.
In situations where the protein of interest may have a high homology with
proteins
whose sequence is already known, rapid identification of proteins has recently
been
achieved by combining partial sequence data obtained by mass spectrometry with
efficient
l S methods of searching large sequence databases (Neubauer et al., Proc.
Natl. Acad. Sci.
USA, (1997), 94:385-390; Neubauer et al., Nature Genetics, (1998), 20:46-50).
Similarly,
direct analysis of large protein complexes has been accomplished by using
computer
algorithms to correlate acquired peptide fragment mass spectra with predicted
amino acid
sequences in translated genomic databases (Link et al., Nature Biotechnology,
(1999),
17:676-682).
The foregoing techniques will not always impact upon de novo sequencing,
however, because of the dependence upon existing sequence data. In an early
approach to
de novo sequencing by mass spectrometry, mass differences between successive
adjacent
peaks in the spectra were compared with the masses of the amino acids in turn
until a match
was found. A sequence was deduced based on a score associated with the
intensity of the
peaks (Pates, et al., "Computer aided Interpreation of Low Enery MS/MSl Mass
Spectra of
Peptides", in Techniques In Protein Chemistry II, Ed., J. J. Villafranca,
(1991), Academic
Press, Inc., p.477).
In another approach to de novo sequencing, a so-called "spectrum graph" is
derived
from the measured spectrum by assigning a vertex to each peak and constructing
an edge
between pairs of vertices whose masses differ by the mass of an amino acid
residue,
(Dancik, et al., J. Comp. Biol., (1999), 6:327-342). The correct sequence can
be inferred
from the longest path within the graph but only if noise is efficiently
eliminated from the
spectrum. However, this method produces a large number of suggested sequences
with a
scoring probability associated with each, and relies upon carrying out a graph
theoretical
-4-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
technique, the antisymmetric longest path problem, which sale's very poorly
with
increasing peptide length.
The most recent experimental approaches,to protein sequencing by mass
spectrometry have utilized labelling or tagging of peptide sequences. In a
chemical method,
methyl labelling of C-terminus residues by methyl ester formation, for
example,
comparison of spectra for labelled and unlabelled samples, can lead to
sequence data from
characteristic peak spacings in the spectra (Hunt et al., Proc. Natl. Acad.
Sci. USA, (1986),
83:6233-6237). A general drawback of chemical labeling is the chemical
reaction step
involved and the need to obtain spectra for two different samples.
In another labelling method, deuterium.is exchanged for acidic hydrogens along
the
peptide sequence (Sepetov, et al., Rapiel Commun. In Mass Spect., (1993), 7:58-
62).
Although this method permits ready differentiation between b-ion and y-ion
series peaks,
the technique is only practical for short peptide sequences (< 10 residues)
and only offers
additional sequence information for those residues with acidic side chains.
Isotopic labelling of the peptide sequence prior to MS/MS analysis with labels
other
than: deuterium has been a desirable technique for some time but requires a
sensitivity which
is hard to obtain with tandem mass spectrometry and usually requires
comparison of spectra
for two samples. An example of a technique in which information can be
obtained from a
single spectrum is described in Gygi et al., "Quantitative analysis of complex
protein
mixtures using isotope-coded affinity tags," Nature Biotech., 17:994-999
(1999). In this
technique, proteins are labeled with a reagent such as iodoacetamide that has
affinity for
sulfhydryl groups. Proteins from one sample are labeled with a normal reagent
and proteins
from another sample are labeled with reagent that has been substituted with 8
deuteriums.
Both samples are combined, and further labeled with a biotin affinity tag
prior to analysis by
mass spectrometry. Peaks from the two samples are separated by 8 mass units.
The
drawback of the method is the need for a cysteine residue on the protein
samples.
Although an application to'$O labelling with a four-sector tandem mass
spectrometer has been reported (Takao et al., Anal. Chem., (1993), 65:2394-
2399) in which
two spectra on a single sample are obtained, the method of analysis is a
simple comparison
of spectra which becomes rapidly impractical for sequences longer than those
reported
(about 10 residues). De novo sequencing of proteins by mass spectrometry has
therefore
been a challenging problem for quite some time.
Recently, however, the sensitivity required for de novo sequencing
isotopically
labeled peptides has been achieved by combining a nanoelectrospray ion source
with a
quadrupole time-of flight tandem mass spectrometer. The approach utilizes an
intrinsic
-5-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
feature of the quadrupole time-of flight device which gtves'rise to a fiigher
sensitivity and
resolution than other types of mass spectrometers (Shevchenko et al., Rapid
Communications in Mass Spectrometry, (1997) 11:1015-1024). Isotopic labeling
of
C-terminal peptide fragments, e.g., by enzymatic digestion of a protein in 1:1
'60/'80 water,
provides a characteristic isotopic distribution for these fragments that can
be readily
identified (Schnolzer et al., Electrophoresis, (1996), 17:945-953). The
principle of the
method is to identify C-terminal fragment ions of a peptide in one spectrum by
their 1:1
'60/'80 isotopic pattern when the peptide has been labeled at its C-terminus
to SO% with'$O
isotopes and to SO% with'60 isotopes before being subjected to a tandem mass
spectrometric analysis. Although two spectra are required, they are both
obtained for the
same sample. The fact that analysis of the difference between the two spectra,
i. e., a
subtraction, is used means that measurements can be made with an enhanced
sensitivity,
leading to identification of a series of peaks from isotopically labelled
subfragments. These
peaks arise from C-terminal ions that differ in mass by one amino acid, a fact
which allows
elucidation of the amino acid sequence.
Nevertheless, the analysis of isotopically labelled spectra remains
complicated for a
number of reasons. The identification of a C-terminal peptide by a
characteristic isotopic
distribution, such as that obtained when digesting a protein in water having a
known
percentage of'80, is made difficult by the natural isotopic abundance of
isotopes such as,
~ ~ inter alia, '3C and 'sN. For example, as a result of these natural
isotopic abundances, two
peaks in a peptide mass spectrum that are separated by 2 Daltons (Da) might
arise from:
peptide subfragment ions having either a'60 atom or a'80 atom at the C-
terminus; peptide
subfragment ions having either a'60 atom at the C-terminus, or one'3C atom and
one'SN
atom, or two '3C atoms, or two 'SN atoms. As the peptides become larger, there
is a greater
chance for incorporation of the less abundant'3C and'SN isotopes, and the
problem of
identifying C-terminal peaks for amino acid sequencing becomes increasingly
difficult.
In summary, existing methods for de novo protein sequencing all have
drawbacks.
Mass spectrometry is a more promising technique for protein sequencing because
it requires
picomolar or even femtomolar amounts of sample and produces highly accurate
spectra.
However, difficulties in spectral interpretation are significant for larger
peptides and
proteins. Accordingly, the present art is in need of an analytical technique
that permits the
sequence of large peptides to be deduced from mass spectra.
Citation of a reference herein shall not be construed as indicating that such
reference
is prior art to the present invention.
-6-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
SUMMARY OF THE INVENTION
The present invention involves the derivation of the amino acid residue
sequence of
a protein or peptide through the automated analysis of differential scanning
mass
spectrometry data. Specifically, the aspect of peptide sequence analysis
addressed by the
present invention is the automated identification of C-terminal, or y ion
peaks, in the mass
spectrometry data. Once y-ion peaks have been identified, peptide sequences
can be
deduced by calculating mass differences between adjacent y-ion peaks and
attributing each
mass difference to a specific amino acid residue. Since a mass spectrum of a
peptide
consists of a large number of peaks, the derivation of the peptide sequence by
human
inspection of a simple difference between a pair of spectra is usually not
straightforward and
rarely fast.
Accordingly, the subject of the present invention is a computer algorithm for
deducing the peptide sequence of a peptide from a pair of MS/MS spectra
obtained on a
partially isotopically labelled sample. The algorithm seeks to compute a
"filtered" spectrum
comprising just the C-terminus set of subfragments (the y-ion series), from
which it is
possible to accurately deduce the amino acid sequence.
v The present invention involves an apparatus for determining the amino acid
residue
sequence of a peptide, comprising: an input device configured to accept mass
spectrometry
data obtained by applying differential scanning mass spectrometry to a sample
of the
~ peptide in which an isotopic label is present in a proportion which is
substantially different
from its natural abundance; a processor configured to execute mathematical
operations on
the mass spectrometry data; and a memory connected to the processor to store:
a first set of
instructions to direct the processor to generate a probability that a peak in
the mass
spectrometry data derives from a y ion subfragment of the peptide wherein the
first set of
instructions are repeatedly executed for each peak in the mass spectrometry
data; a second
set of instructions to direct the processor to produce a filtered mass
spectrum of the peptide,
wherein each peak in the filtered mass spectrum whose intensity is greater
than a threshold
value, is predicted to correspond to a y-ion subfragment of the peptide; and a
third set of
instructions to direct the processor to derive and store in the memory an
amino acid residue
sequence of the peptide from the filtered mass spectrum. In a preferred
embodiment, the
isotopic label is '80, and the proportion is 50%.
According to the technique of differential scanning mass spectrometry, the
mass
spectrometry data comprises a first mass spectrum that has signals from
subfragment ions in
which the isotopic label is both present and absent, and a second mass
spectrum in which
signals from subfragment ions in which the isotopic label is not present are
substantially
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
suppressed. In a preferred embodiment, the probanmty is computed from a
product of a
first scoring value and a second scoring value, wherein the first scoring
value is proportional
to the likelihood that a peak in the first mass spectrum arises from an
isotopic cluster that
comprises a signal from a subfragment ion in which the isotopic label is
absent and also a
signal from a subfragment ion in which the isotopic label is present in the
proportion; and
wherein the second scoring value is proportional to the likelihood that a peak
in the second
mass spectrum arises from an isotopic cluster containing a peak from a
subfragment ion in
which the isotopic label is present in the proportion and in which a peak from
a subfragment
ion in which the isotopic label is absent is effectively suppressed relative
to the first mass
spectrum.
The present invention additionally involves a method for determining the amino
acid
residue sequence of a peptide, the method comprising: accepting mass
spectrometry data
obtained by applying differential scanning mass spectrometry to a sample of
the peptide in
which an isotopic label is present in a proportion which is substantially
different from its
natural abundance; generating a probability that a peak in the mass
spectrometry data
derives from a y-ion subfragment of the peptide wherein the first set of
instructions are
repeatedly executed for each peak in the mass spectrometry data; producing a
filtered mass
spectrum of the peptide, wherein each peak in the filtered mass spectrum~whose
intensity is
greater than a threshold value, is predicted to correspond to a y-ion
subfragment of the
20~ -peptide; and deriving an amino acid residue sequence of the peptide from
the filtered mass
spectrum. According to a preferred embodiment of the present invention, the
method for
determining the amino acid residue sequence of a peptide is executed by a
computer under
the control of a program, the computer including a memory for storing the
program, an
input device configured to accept mass spectrometry data and a processor
configured to
execute mathematical operations on said mass spectrometry data.
BRIEF DESCRIPTION OF THE DRAWINGS
Other advantages of the invention will become apparent upon reading the
following
detailed description and upon reference to the drawings, in which:
Figure 1. A computer system according to the present invention.
Figure 2. A quadrupole time of flight mass spectrometer used in the preferred
embodiment of the invention.
_g_
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
Figure 3. Flow chart of partial isotopic labelling for u~'~' i~ith'a preferrea
embodiment, of the present invention.
Figure 4. Flow chart of a differential scanning method.
Figure 5. Flow chart of an algorithm according to the present invention.
Figure 6. A representative shape of the scoring value Sl" and S2" as a
function of
g" calculated using Equations 3 and 6, respectively.
Figure 7. Spectra showing comparison of unfiltered and filtered peptide
subfragment ion mass spectra.
Figure 8. Representative mass spectrometer for practicing the invention.
~~ DETAILED DESCRIPTION OF THE INVENTION
Introduction
A method of identifying the y-ion peaks of a protein in a tandem mass spectrum
is
described. The term "protein" is used herein in a broad sense which includes,
mutatis
E ynutandis, peptides, polypeptides and oligopeptides, and derivatives
thereof, such as
glycoproteins, lipoproteins, and phosphoproteins, and metalloproteins. The
principal
distinguishing features of such species is that the "protein" comprises one or
more peptide
(-N(-H)C(=O}-) linkages.
The aim of the method is to simplify and automate analysis of the MS/MS
spectra in
such a way that a likely peptide sequence can be proposed. The method is
implemented in a
computer algorithm. It is based on acquiring not just one, but two, fragment-
ion spectra of
peptides from a protein sample which has been enzymatically digested in a
water mixture
comprising known proportions of Ha'$O and HZ'60. The water mixture is such
that the
fractional composition of H2'$O is substantially greater than its natural
abundance and the
conditions are such that the peptide fragments incorporate'8O labels at their
C-termini in
the same proportion as is present in the water mixture. One spectrum is
obtained by
selecting the entire'60f$O isotopic mixture of the peptide for fragmentation
and a second
spectrum is obtained for which only'$O labeled peptide ions are fragmented.
After acquisition of the'60/'$O and'80 mass spectra, the data are analyzed
using the
computer program product and methods of the present invention in order to
identify the
-9-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
peaks which arise from y-ions. Peaks corresponding to' C-t'~rminal peptide-
subi~agments
can be identified when comparing the two spectra using two criteria. The first
criterion is
their'60/'$O isotopic distribution in the first spectrum which is usually
difficult or
impossible to recognize unambiguously by visual inspection. The second
criterion is the
change in the isotopic distribution of C-terminal subfragment ion peaks when
comparing the
first spectrum with the second. C-terminal ions are identified by having peaks
from
complete'6O/'g0 isotopic distributions in the first spectrum but only peaks
from'80
isotopes in the second spectrum. Non C-terminal ions have the same isotopic
representation
in both spectra since they do not contain the'$O isotope in the proportion
introduced by
enzymatic digestion.
Once all C-terminal fragment ions have been identified, the peptide sequence
can be
deduced by calculating the mass difference between adjacent fragments and from
their order
in the spectrum. The methods and computer program product of the present
invention may
further comprise the calculation of subtracted and filtered mass spectra.
The methods of the present invention may be applied to proteins or peptides of
any
length, provided that machine resolution permits a well-resolved mass spectrum
to be
obtained, in particular as long as the different isotopes can be resolved. The
number of
amino acids which can be read is sequence dependent, so there will be peptides
of say 20
amino acids in length for which only 5 amino acids can be read, whereas there.
may be
20. others that are 25 amino acids in length and for which all 25 residues can
be read: In
general, a tendency is observed that the readable sequence gets shorter when
the peptide
exceeds a certain size. It has been found that peptides up to a size of 3 kDa
(approximately
30 residues) can still be sequenced to a sufficient length ( i.e., it is
possible to read 20 of the
30 amino acids). There is no lower limit for sequencing.
Apparatus
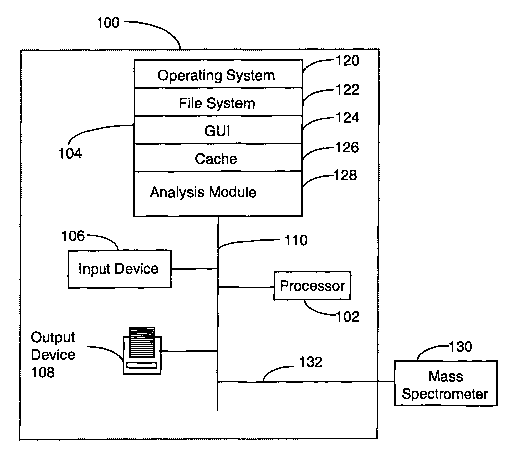
The invention, as shown in figure 1, comprises a system 100 for deducing a
peptide
sequence from mass spectrometry data obtained from mass spectrometer 130.
System 100
comprises a processor 102; a section of memory 104 which will typically
include both high
speed random access memory as well as non-volatile memory (such as one or more
magnetic disk drives); an input device 106, for inputting user-specific
parameters, which
may comprise a keyboard, mouse and/or touch-screen display; an output device
10~ for
printing or displaying the sequence of the protein or peptide, and at least
one bus 110
connecting the processor 102, the memory 104, the input device 106, and the
output device
10~. Though not shown in Figure 1, the system 100 also preferably comprises a
network or
-10-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
other communication interface for communicating with other bbh'ipufers as weld
as other
devices.
The memory preferably stores an operating system 120 for providing basic
system
services, a file system 122, an analysis module 128 configured to analyze mass
spectrometry
data, a cache 126 and optionally a graphical user interface (GIT.~ 124.
Preferably, system 100 acquires mass spectrometry data via data channel 132
from
mass spectrometer 130. In one embodiment of the present invention, the mass
spectrometer
130 is a triple quadrupole mass spectrometer.
The analysis module 128, upon receiving a request to deduce the sequence of
the
peptide or protein from mass spectrometry data, executes instructions which
enable
identification, with substantial probability, which peaks in the mass
spectrometry data
correspond to peptide subfragments in the y-ion series. Once the y-ions are
identified, the
amino acid sequence is determined by calculating the mass differences between
adjacent y
ion peaks. Each mass difference corresponds to the mass of one amino acid
residue. All
amino acids in a peptide chain, except for leucine and isoleucine which have
the same mass
as each other, may be distinguished. The entire protein sequence may be
determined by
concatenating or overlapping separate peptide sequences determined from the
spectra of
different peptide fragments, using principles well known to one skilled in the
art.
This system, when operated in a laboratory environment in conjunction with
mass
spectrometry data can provide an efficient and useful method of deducing the
amino acid
residue sequence of a protein or peptide.
Instrumentation
A mass spectrometer separates ions according to their m/z ratio, the ratio of
their
mass, m to charge, z. In a first stage, a sample is ionized, for example by
electron
bombardment, creating ions that, in a subsequent stage, are accelerated
through an
inhomogeneous electromagnetic field towards a detector. In one embodiment, the
magnetic
field perturbs the trajectories of the ions according to their m/z ratio: an
ion with a small
mass will travel more quickly and be less easily perturbed than a heavier ion;
an ion with a
small charge will be perturbed more than one with a large charge. In practice,
most ions
that are produced carry only a single positive charge, though some ionization
techniques can
readily give rise to multiply charged ions. The production of ions of
different m/z from the
same sample arises for several reasons: the conditions of ionization may cause
the
molecules to dissociate; the ions themselves may subsequently rearrange and
dissociate; and
-11-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
because there are invariably many different isotopic substituerits in the
molecules of a given
sample.
In one embodiment, a triple quadrupole mass spectrometer is used to acquire
peptide
subfragment data. An example of such a machine is an API III from Perkin Elmer
Sciex
(PE-Sciex). In this embodiment, three quadrupoles are used as an ion guide,
the mass filter
and the collision cell. The typical layout of such a mass spectrometer 300 is
shown in
Figure 3, though it is understood that variations on the components of such a
mass
spectrometer are envisaged for practice with the methods of the present
invention.
In tandem mass spectrometry, two stages of mass analysis are used. In the
first
stage, precursor ions are produced from an ionization source 304. In a
preferred
embodiment, electrospray ionization is used to produce the precursor ions. The
precursor
ions are optionally passed through a first quadrupole 306 which acts as an ion
guide. This
ion guide is not usually a mass-selective quadrupole and is usually only
present in triple-
quadrupole machines. Precursor ions pass into a mass filter 310 that selects a
precursor ion
having a particular value of the m/z ratio, or, more generally precursor ions
whose m/z
ratios lie within a narrow range. Currently, it is known that the mass filter
310 which gives
the greatest sensitivity is the quadrupole mass filter. An ion trap can
alternatively be used.
In a preferred embodiment of the present invention, mass filter 310 is a
quadrupole mass
filter. The range of m/z ratios transmitted by the quadrupole mass filter is
known as the
20. transmission window.
In a preferred embodiment, mass spectrometer 300 used to acquire peptide
subfragment data is a quadrupole time of flight ("Q-TOF") mass spectrometer.
An example
of such a machine is the "Q-Tof2" by Micromass, in the United Kingdom.. Such a
machine
employs two quadrupoles. A quadrupole 312 is employed as the mass filter for
precursor
ion selection, and a quadrupole 322 is used in a collision cell 310 where the
precursor ion is
further fragmented into subfragments. A time of flight ("TOF") mass analyzer
340 is used
to examine the subfragment ions. A representative mass spectrometer design for
practicing
the invention is also shown as Figure 8.
Iofzization Techniques:
There are a number of ionization techniques used to produce precursor ions for
mass
spectrometry analysis. These include, but are not limited to, electron
ionization, chemical
ionization, field ionization, field desorption, fast-atom bombardment, plasma
desorption,
laser desorption, and electrospray ionization. The two most commonly-used
ionization
techniques for biomolecule analysis are matrix-assisted laser desorption
ionization
-12-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
("MALDr') and electrospray ionization ("ESI"). The methods of the present
invention are
independent of the ionization technique employed.
In one embodiment of the mass spectrometer utilized with the present
invention,
MALDI is used. MALDI is a specific type of laser desorption in which
biomolecules are
co-crystallized with a large molar excess of a small, ultraviolet radiation-
absorbing organic
acid (matrix). Upon irradiation of the co-crystal with an ultraviolet laser,
matrix molecules
and biomolecules are sent into the gas phase, where protons are transferred
from the matrix
molecules to the biomolecules, thus forming biomolecule precursor ions for
analysis.
MALDI usually gives rise to singly-charged precursor ions which subsequently
undergo
"post-source decay" (PSD) to produce fragment ions. It is not usually
necessary to use
collision-induced dissociation with MALDI, therefore. The MALDI method is
often used in
conjunction with a time of flight mass spectrometer and therefore may be used
with the
methods of the present invention.
In a~preferred embodiment, in use with the present invention, the ionization
source
304 produces precursor ions by ESI, according to which, ions are formed by
spraying a
dilute solution of biomolecules at atmospheric pressure from the tip of a fine
metal .
capillary. The spray creates a fine mist of droplets that become highly
charged in a high
electric field. As the droplets evaporate, the biomolecules pick up one or
more protons
from the solvent to form :ions with single or multiple positive charges. As
the droplets
; shrink, charge repulsion causes the ions to be evaporated from the droplet
surface, which
are then analyzed in the mass spectrometer. In a preferred embodiment of the
mass
spectrometer used with the present invention, ESI is used to generate
precursor ions.
Whereas MALDI can result in extensive fragmentation of the sample and
precursor ions,
ESI results in little to no fragmentation. Furthermore, samples for ESI are in
solution so
that the technique is ideally suited for coupling with purification
techniques, such as HPLC.
lllllass Filters:
In a preferred embodiment of the mass spectrometer in use with the present
invention, a quadrupole mass filter 310 is used to select precursor ions. A
quadrupole mass
filter comprises a quadrupole 312, consisting of two pairs of precisely
parallel metal rods,
with opposite rods being electrically connected. A voltage made up of a direct
current
potential ("DC") and an alternating radiofrequency ("RF") component is applied
to each pair
of rods. Because ions passing through the quadrupole are alternately attracted
to and
repulsed from the rods, they have an oscillating traj ectory, and only those
ions with kinetic
energy in a certain range pass between the rods and out the other side. All
other ions collide
-13-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
with the rods. Since the kinetic energy of any given ion is proportional to
its mass, the
selection of ions is mass-dependent. The ions that pass through the quadrupole
identify the
transmission window of the quadrupole. If the DC and RF amplitudes are varied
together in
such a way that their ratio, DC/RF, remains constant, the center of the
transmission window
can be shifted to other m/z values, and ions with different masses can be
"filtered" through
and analyzed.
Production of Subfragment Ions:
In one embodiment of the mass spectrometer used with the present invention,
the
. 10 filtered precursor ions having a particular m/z are sent to a collision
cell 320. In a triple
quadrupole mass spectrometer, the collision cell comprises the third
quadrupole. In a ToF
machine, it typically comprises the second of two quadrupoles. It is
understood that many
machines that are compatible with the present invention utilize collision
cells that comprise
quadrupoles. In machines that utilize ion traps, the ion trap itself is a
collision cell because
ions can be collided with rest gas atoms inside it.
In collision cell 320, the filtered precursor ions collide with uncharged gas
molecules, such as argon or xenon, or dinitrogen, delivered from a source 314.
The kinetic
energy of the precursor ions is partially transformed into vibrational energy,
resulting in the
breaking of the precursor ions'. predominantly weak chemical bonds. Peptide
precursor ions
preferentially fragment at their peptide amide bonds to produce peptide
subfragments. The
resulting subfragment ions are analyzed by the mass analyzer 340.
Mass Analyzers:
In a preferred embodiment, the mass analyzer used is a time of flight ("TOF")
mass
analyzer. In this type of mass analyser, subfragment ions are accelerated
through
accelerating plates 342 and pass into a region that has no external electric
field, known as a
drift tube 344. If all of the subfragment ions entering the drift tube have
the same kinetic
energy, given by /Zmv~ for an ion of mass m and speed v, then since velocity
is inversely
proportional to the square-root of mass, subfragments with larger mass 346
will travel more
slowly than subfragments with smaller mass 348. The heavier subfragment ions
will
therefore reach the detector 350 at the end of the drift tube at a later time
than the lighter
subfragment ions. TOF analyzers are often used in conjunction with MALDI. TOF
analyzers are advantageous in that they have virtually unlimited mass range
and high scan
rates.
-14-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
In a preferred embodiment, the detector 350 is an e~ectxon multiplier, wherein
the
display of the mass spectrum is effectively instantaneous. Detector 350
transmits mass data
to computer system 100, via transmission channel 132.
A limitation of TOF analyzers is that peaks are broadened because not all
members
of the same subfragment ion population have the same kinetic energy. Since the
initial
energy spread is mass dependent, peaks from heavier subfragment ions are
broader. As is
well known to one skilled in the art, the initial kinetic energy distribution
of subfragment
ions entering the drift tube can be decreased by increasing the final
accelerating voltage.
The resolution of the TOF analyzer can also be increased by increasing the
length of the
drift tube, which increases the time difference between arrivals of ions of
different mlz, but
also increases the spread of arrival times of ions having the same mlz. In
another
embodiment of the mass spectrometer used with the present invention, the TOF
analyzer is a
"reflectron" type in which the ions follow a curved path. A reflectron TOF
analyzer slows
the ions down and turns them round before directing them to the detector. When
the ions
turn around the slower ones catch up with the faster ones.
Mass Spectrometry Data
Mass spectrometry data comprises a number of elements, wherein each element
has
an intensity value, I, for a m/z value. The data comprises elements across a
range of m/z
..values. A name of the unit widely used for m/z values is "Thomson" (Th). The
collection
of data comprising intensity values for a range of Thomson is often called a
"mass
spectrum." The m/z values in a mass spectrum are typically separated from one
another by
0.02 Th, but, depending upon resolution, may be separated from one another by
0.01 Th or
0.05 Th.
A "peak" in a mass spectrum is defined by a collection of adjacent elements,
at
which each intensity value is above a threshold intensity value. Mass
spectrometry data
typically also comprises a background intensity, and many low-intensity pieces
of data,
often called noise. The threshold intensity value may be chosen so that noise
is eliminated
from consideration during analysis. Usually a peak intensity is proportional
to its height,
though this approximation may break down for more complex spectra,
particularly for
heavier ions.
Strictly, the overall intensity of a peak is obtained by calculating the area
under the
peak. In one embodiment, the calculation is achieved by a centroiding method.
In
centroiding, for any peak whose width, measured as full-width at half maximum
height
("FWHM"), is at least 0.04 Th, data in a window of width 0.08 Th are merged
into the peak
-15-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
and added up. Centroiding is not generally good enough for the accuracy needed
with the
present invention because separate peaks may be accidentally merged.
Accordingly, in a
preferred embodiment, an integration method is employed for calculating peak
intensities.
This method preferably adds all intensities that are present around a peak
within a window
of about ~ 0.02 Th. Because different subfragment ions within the same
spectrum may have
different charges, a different window should be chosen according to the number
of charges
on the subfragment ion. Accordingly, for a singly charged fragment, the window
is
preferably 0.04 Th; for a doubly charged fragment, the window is preferably
0.02 Th. It is
consistent with the methods of the present invention that other windows may be
chosen
when carrying out peak integration. Indeed it is also possible that different
sized windows
may be chosen over different regions of a mass spectrum.
Most of the chemical elements of which organic molecules are comprised have
more
than one naturally occurring isotope, see Table 1, hereinbelow. Because a mass
spectrum is
made up from signals produced by a large number of ions, the spectrum
comprises a
statistical sampling of all of the naturally occurring isotopes. As molecules
become larger,
the percentage of the population of molecules having one or more atoms of a
heavier isotope
also increases. Consequently, a given subfragment ion does not appear as a
single sharp
peak in the spectrum (except in the case of artificially ensured isotopic
purity). Instead, the
portion of the mass spectrum around the mlz value of a given ion contains a
number of
:.peaks because each of the elements present in the ion has its own
distribution of isotopes in
nature.
Table 1
Isotopic Mass and Abundance Values for Atoms in Proteins
(Taken from, Wapstra & Audi, Nucl. Phys., (1985), A432:1-54)
Element Isotope Mass Natural abundance (%)
Hydrogen 'H 1.007 825 035 99.985
ZH 2.014 1 O l 779 0.01 S
Carbon 'aC 12.000 000 000 98.90
'3C 13.003 354 826 1.10
Nitrogen '4N 14.003 074 002 99.634
isN 15.000108 97 0.366
Oxygen '60 15.994 914 63 99.762
- 16-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
"O 16.999 131 2 0.038
'$O 17.999 160 3 0.200
Sulphur 32S 31.972 070 698 95.02
33S 32.971458 428 0.75
saS 33.96? 866 650 4.21
36S 35.967 080 620 0.02
Therefore, the mass spectrum of a given peptide subfragment will comprise a
n~ber of closely separated peaks, each of which corresponds to a particular
distribution of
isotopes amongst its atoms. If the peptide subfragment attains a single charge
during
ionization, then the closely separated peaks for that subfragment are each
separated by
approximately one m/z unit. The collection of peaks which correspond to
fragments
differing from one another only by isotopic variation is called a cluster.
With the exception
o f laC whose mass is defined to be 12.0000 atomic mass units, no isotope has
an integer
mass. The mass of a peptide molecule with one '3C atom is not exactly the same
as the
mass of the same peptide molecule with one "O atom but no '3C atoms. Therefore
the
peaks within a cluster may be poorly resolved'and may overlap to a great
extent.
The mass of those molecules in which every atom is present as the most
abundant '
: isotope is called the "monoisotopic mass." The monoisotopic mass of a
molecule comprises
a sum of the accurate masses for the most abundant isotopes over all the
atoms. The peak
which corresponds to the monoisotopic mass is typically of lowest mass because
the most
abundant isotope of each element occurring in a protein or peptide has the
lowest mass of all
the isotopes. This peak is not always the most intense, however.
The intensity distribution of the peaks within a cluster is often called an
"envelope"
and its shape is the result of many contributing factors. For very large
molecules, the peak
corresponding to the monoisotopic mass is not necessarily the most intense.
The most
significant contributor to the isotopic peak pattern for biomolecules is '3C.
The occurrences
of the heavy isotopes of oxygen, nitrogen, and sulfur also contribute to the
isotope envelope.
C~.bon has two principal naturally-occurring isotopes: 'ZC, which has a mass
of 12.000000
and a natural abundance of 98.9%; and'3C, which has a mass of 13.003355 and a
natural
abundance of 1.1 %. Irrespective of peptide size, the first peak in the
resolved isotopic
cluster arises from the all'ZC-containing ion. For peptides with masses less
than
approximately 1,800 Daltons (corresponding to peptides containing
approximately 100
c~.bon atoms), this is the most intense peak. However, for peptides with
masses greater
-17-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
than 1,800 Daltons (corresponding to peptides containing more tltan'~b6ut f00
ca9~on
atoms), the first peak in the isotopic cluster will not be the most intense
peak because the all
'ZC-containing ion will no longer be the most abundant, i.e., on average every
molecule in
the sample will contain at least one atom of'3C. In such cases, it may be more
useful to
consider the most intense peak and refer to it as the "average mass."
Manipulation ofData:
A feature of the present invention is the comparison of two mass spectra
obtained for
the same sample, the two spectra differing from one another by the centering
of the
transmission window. Obtaining the difference between the two spectra on the
same sample
by simple subtraction is rarely straightforward and several data processing
operations should
be carried out. One problem is that subtraction may lead to negative peaks due
to phase
mismatches. As an illustration, there may be a peak in both of the two spectra
corresponding to an m/z value of 123. In the first spectrum it may start at
122.85 and end at
123.15; but in the second spectrum the corresponding peak may start at 122.88
and end at
:123.18. In order for subtraction to be effective, precise alignment of the
spectra is required,
a procedure that may be difficult if the phase mismatch is not constant over
the whole range
of the spectrum. In practice, this problem is preferably addressed by "partial
centroiding:"
A bin-width is chosen, ,typically 0.05 Th but which may be as low as 0.02 Th,
according to
the mass resolution of the instrument. The spectra are divided into regular
spacings of this
bin width. If two data points are within the bin-width, their intensities are
added up.
A second complication in the subtraction of spectra is the fact that, under
slightly
different operating conditions (as necessarily arises for the two different
spectra), a pair of
peaks common to both spectra do not necessarily have the same intensity. So,
even when
aligned in phase, the subtraction of one peak from another may not give a
baseline value,
thus giving rise to small positive or negative peaks. Although the spectra can
be scaled so
as to match peak heights to one another, the scaling factor required may vary
over the range
of the spectrum. In a preferred embodiment, the spectrum of the'g0 containing
peptide
subfragment is scaled to overlap with the'60/'80 spectrum. The spectra are
divided into
windows, typically 20 Th wide, though other values, both larger and smaller
are consistent
with the methods of the present invention. In each such window, the highest
peak in the
's0~'sO spectrum is determined with, say, m/z value mP and intensity Il . The
highest peak
in the'g0 spectrum in the range (mP-1, mP+2) is determined, with intensity I2.
The 20 Th
wide window of the'$O spectrum is scaled with the factor Il/I2. Finally, once
the scaled
spectrum has been subtracted from the unsealed spectrum, noise filtering is
applied to the
-18-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
resulting spectrum: any peak whose width is below some threshold, Say 0.05
'1'h,1s
eliminated.
Partial Isotopic Labeling of Peptides
In a preferred embodiment, the computer program product and methods of the
present invention are for use in conjunction with partial isotopic labeling of
peptide
fragments of a protein and the differential scanning mass spectrometry
technique. Partial
isotopic labeling of the C-termini of peptide fragments can be accomplished by
methods
known to those of skill in the art. A preferred embodiment for use with the
present
invention is shown in Figure 3. Peptides are labeled by enzymatic digestion of
a protein 200
using, inter alia, trypsin, chymotrypsin, or papain, preferably trypsin, in
bulk solvent water,
a known proportion of which is '$Q-labeled water, i.e., H2'$O, step 202.
The known proportion of labelled water is substantially different from the
proportion
of the label found naturally. Preferably, substantially different means
present in an amount
that renders contribution from the natural abundance of the isotope
insignificant when
carrying out mass spectrometry measurements and means present in an amount
that
. facilitates automated analysis of a mass spectrum so that signals from
peptides that have
incorporated label from the labeled water are readily distinguished. In one
embodiment of
the present invention, the protein is digested in the presence of 30% by
volume 'g0=labeled
.water, preferably in the presence of 33% by volume '$O-labeled water, more
preferably in
the presence of 40% by volume '$O-labeled water, or most preferably SO% by
volume'$O-
labeled water. In general, any known proportion between about 30% and about
75% by
volume of'$O-labeled water is suitable for carrying out the methods of the
present
invention. Proportions by volume of about 30% to about 75%'$O-labeled water
are
substantially different from the natural abundance of'g0-labeled water.
Those of skill in the art will recognize that enzymatic digestion of a protein
in, e.g.,
SO% HZ'$O and 50% HZ'60 results in the generation of many peptide fragments.
After
digestion, the peptide fragments are purified and separated, step 204, by,
e.g., gel
electrophoresis or HPLC. The peptide fragments 206 that are produced
are.analysed by
mass spectrometry. Accordingly, hereinafter the term peptide will also include
the term
peptide fragment, as understood to be a peptide that has been produced by
fragmentation of
some longer peptide.
When the enzyme digests the protein, it cleaves a peptide amide bond leaving
at
least one peptide fragment with a free amino group (N-terminus) and a
corresponding
peptide fragment with a trailing carbonyl group (C-terminus). A water molecule
from the
-19-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
bulk solvent water adds to the C-terminus group to produce a~carboxylie acid
gro~rp. Due to
the presence of a known proportion of i80-labeled water, a known proportion of
the cleaved
peptide fragments will have'80 at the C-terminus. The proportion of cleaved
peptide
fragments with'$O at the C-terminus is preferably substantially the same as
the proportion
of'$O-labeled water by volume in the bulk solvent water.
In a preferred embodiment, the known proportion of'$O-labeled water is 50% by
volume. For a particular peptide, digested in bulls solvent water, SO% of
which by volume
is 180-labeled water, for every peptide fragment molecule of mass m with a'6O
atom
incorporated at the C-terminus, there will be approximately one peptide
fragment molecule
with mass m+2 because an'g0 atom is incorporated at the C-terminus. Therefore,
the
peptide fragment and each subfragment of the peptide fragment that includes
the C-terminus
will have the characteristic 1:1 '60 l'80 isotopic distribution that should be
distinguishable
in a mass spectrum as two peaks of similar intensity separated by two mass
units. At lower
resolution, such a pair of peaks may appear to be a single split peak. Thus, y-
ions in a M/S
of such a sample should appear as split peaks or pairs of similar intensity
peaks.
Unfortunately, more often than not, it is not possible to discern visually the
1:1 '6Of$O .
isotopic pattern in a mass spectrum of a peptide fragment. Other ions can
mimic the pair of
peaks or split peak distribution and overlapping subfragment ions with similar
masses can
distort it. For example, as already mentioned, in sufficiently long peptide
sequences, the
,20 peak at m+1 due to molecules with one'3C substituent is at least as big as
the peak at m
corresponding to those molecules with no '3C substituent. If it were
straightforward to
identify y-ion peaks in this way then sequencing of the peptide by inspection
of a mass
spectrum of the isotopic mixture would be feasible.
Although the preferred embodiments of the invention are described herein using
peptides labeled at the C-terminus with'60 and'gO using tryptic digestion in
H2'6O and
Ii~'$O, those of skill in the art will recognize that the present invention
can be used in
conjunction with peptide fragments partially labeled with other isotopes, for
example "O,
and using alternative peptide labeling techniques. It will be understood that
the amounts of
label that should be incorporated may differ for other isotopic labels from
those that are
preferred for'g0 but that one of skill in the art will be able to determine
the amount of label,
different from the natural abundance, that should be utilized in order to
practice the methods
of the present invention.
Furthermore, in principle, there is an analogous labeling scheme for b-ions.
In such
an embodiment, the labeling is preferably on the N-terminus. Isotopic labeling
at the N-
terminus is not as straightforward as isotopic labeling at the C-terminus,
which is readily
-20-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
accomplished at the same time as enzymatic digestion. An'~'N baset~I~beling
scheme i's not,
ideal because there are very few practical reactions which could introduce
such an isotopic
label into the peptide. Thus, labeling at the N-terminus is preferably
accomplished
artificially, for example, by acetylation. An acetylation reaction introduces
a CH3-C(=O)-
group at the N-terminus (see, for example, Pfeifer, T., Rucknagel, P.,
Kuellertz, G., and
Schierhorn, A., "A strategy for rapid and efficient sequencing of Lys-C
peptides by
matrix-assisted laser desorption/ionisation time-of flight mass spectrometry
post-source
decay," Rapid Commun. Mass Spectrum., 13(5):362-9 (1999)). Carrying out the
acetylation
reaction with a mixture of reagents, one ordinary, the other containing a
heavier isotope
could introduce a mixture of isotopes at the N-terminus. Since the acetylation
reaction is an
additional reaction to be performed in such a scheme, isotopic labeling cannot
ordinarily be
accomplished with the same efficiency as with C-terminal labeling during
enzymatic
digestion which is usually required to generate the peptide fragments for
sequencing.
Additionally, the isotopically labeled component is preferably CH3-C(=180)- or
13CH3-13
C(=O), (both of which give a mass shift of 2 Da) which are more expensive than
HZ 1s0,
which can be readily purchased.
The methods of the present invention are not limited to sequence determination
of
peptide fragments obtained by enzymatic digestion of a protein. The sequence
of any
peptide that has been subjected to partial isotopic labelling may be
determined by the
method of the present invention.
Differential Seanning ltlass Spectrometry
In differential scanning mass spectrometry, outlined in Figure 4, two MS/MS
spectra
are obtained for a given peptide fragment 400. A first spectrum, denoted SP1,
is obtained,
step 402, for the mixture of 160 and 180 containing peptide and their
respective
subfragments. A second spectrum, denoted SP2, is obtained, step 406, for just
the 180
containing peptide and its subfragments. Thus, in SP2, signals for the 160
containing
peptide and its subfragment ions are substantially suppressed. In a preferred
embodiment,
these two spectra are collected on the same peptide sample. The first and
second spectra
may be obtained in any order but are separated by a step of re-centering the
transmission
window, step 404. Computational analysis, step 408, of the two spectra can
produce a
substantially clean spectrum for the C-terminus series of subfragments of the
160 containing
peptide. Peaks arising from non C-terminal subfragments always have their
normal isotopic
distribution (irrespective of 1g0 labelling through enzymatic digestion) and
therefore should
-21-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
not remain when the two spectra are subtracted from one ano~her~ Tie peptide
sequence
410 can be obtained from the analysis.
A peptide sample usually contains many different species, for example the
different
peptide fragments which result from enzymatic digestion, or the different
isotopically
substituted forms of a particular peptide or peptide fragment. In a preferred
embodiment,
the different isotopically substituted forms of a particular peptide or
peptide fragment
constitute the sample that is introduced into the mass spectrometer. The
selection of a
precursor ion or precursor ions by appropriate adjustment of the transmission
window
therefore permits analysis of a particular species or a restricted subset of
all of the species.
The performance of precursor ion selection from a quadrupole mass filter
entails a
compromise between resolution and sensitivity. The resolution is determined by
the width
of the transmission window. Although the highest resolution is obtained from
the narrowest
window, the highest resolution also requires the highest sensitivity.
Therefore, operating the
quadrupole mass filter so that it selects a single isotope results in
insufficient transmission
of precursor ions to permit accurate analysis. That is, at the highest
resolution possible, not
enough sample is transmitted to give a useful spectrum at the sensitivity
levels employed.
The transmission window is not uniform, however. That is, ions whose m/z
ratios lie within
the transmission window are not transmitted with equal intensities. The way in
which the
intensity varies across the transmission window of a mass filter is called the
transmission
: curve.
Differential scanning mass spectrometry is based in part on Applicants'
surprising
discovery that, because of the shape of the transmission curve of a quadrupole
mass filter,
the transmission window can be chosen in such a way that ions may effectively
be excluded
without a concomitant loss of sensitivity. Without being bound by any theory,
the shape of
the transmission curve of a quadrupole mass filter is not symmetric around the
selected m/z,
but has a sharply rising flank (towaxd the lower m/z) and an extended, longer
tail (toward
the higher m/z). Because of this characteristic, when the center of a
transmission window of
a constant width is re-centered, i.e., is moved from one m/z to a slightly
higher m/z, so that
ions with the lower m/z are not transmitted. Thus, by moving the window to
higher values,
the lighter'60 isotopes fall out of the transmission window and the'$O
isotopes fall within
it. The transmission window behaves as if it has a sharp cut-off 'edge' at the
lower end of
its m/z range.
The quadrupole mass filter is such that a transmission window corresponding
to,
e.g., 3 Da, can be chosen. If it is centered at an mlz value corresponding to
the mono-
isotopic mass, it transmits both the'60- and 1g0-containing ions of a
particular peptide,
-22-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
giving a first spectrum, SP1. The transmission window is th~'n r~
c~~red'aroun"d a second '
position, at a m/z value corresponding to one mass unit higher, without
changing its width
and thereby without reducing the signal-to-noise ratio, in order to obtain a
second spectrum,
SP2. In its second position, the transmission window effectively prevents
transmission of
the 160-containing ion without affecting transmission of the l80-containing
ion. Therefore,
transmission of the peptide containing the lower molecular weight oxygen
isotope at its C-
terminus is essentially completely suppressed in the second spectrum, SP2. The
second
position of the transmission window permits transmission of ions whose masses
are two
mass units higher than the monoisotopic mass. Although such species include
normal
isotopic variants of the 160-containing species (e.g., those ions containing
two 13C atoms),
their contribution is out-weighed by the contribution from the peptide ions
which have
picked up an unnatural proportion of 180 through enzymatic digestion. In an
alternate
embodiment, the transmission window can be centered at the second position
prior to the
first position.
As shown in Fig. 2, the selected precursor ions are subsequently passed into a
collision cell 320 wherein the precursor ions are fragmented into
"subfragments."
Subfragments are also identified herein as "peptide subfragments," or
"subfraginent ions."
In the second stage of mass analysis, subfragment ions that are produced from
a precursor
ion are passed into a mass analyzer 340 and thereafter to a detector 350.
In order to accurately assign masses from m/z values in the spectrum, it is
usually
preferable to calibrate the mass analyzer. As is well known to one skilled in
the art,
calibration can take the form of recording a spectrum for a sample whose mass
is known
accurately.
A transmission window of 3 Da is not so narrow that unacceptable loss of
sensitivity
occurs in the resulting spectrum. Therefore, a given fragment of a C-terminal
peptide
digested in 50% H2180 and SO% Ha160, whose 160-containing form has mass m,
will give
rise to two peaks of approximately equal intensity in the first spectrum, and
only one peak in
the second spectrum. The two peaks in the first spectrum SP1 correspond to
fragments with
masses at m and m+2 whereas the single peak in the second spectrum SP2
corresponds to
fragment ions with masses m+2.
Mass resolution is often expressed as the ratio m/Om, where m and m+0m are the
masses of two adjacent peaks of approximately equal intensity to be resolved
in the mass
spectrum. The differential scanning technique requires the mass analyzer 340
and detector
350 to be able to resolve signals for subfragment ions whose molecular masses
differ by at
most about one or two Daltons. Specifically, the peak arising from a peptide
subfragment
- 23 -
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
with mass m, having a 160 atom at the C-terminus, and the pEak ansmg~TO~n the
same
peptide subfragment having mass m+2 because of a 180 atom at the C-terminus,
and both of
which having the same charge, must be resolvable in the spectrum. The larger
the peptide,
the larger the mass of the subfragment ions. Therefore, the resolution of the
analyzer must
be greater for larger peptides, if the m and m+2 peaks are to be resolvable.
Precursor ions created by electrospray ionization often have multiple charges
and
consequently their m/z values are fractions of their masses. Although a doubly
charged
subfragment ion will appear at m/z values one half of its mass, it will be
necessary to
resolve peaks for subfragment ions of mass m and m+2 which are separated by a
single m/z
unit.
Those of skill in the art will recognize that the resolution of the instrument
used to
collect data will influence accurate identification of C-terminal ions.
Although the methods
of the present invention can be practiced on low-resolution machines, such as
triple
quadrupoles, they are preferably carned out on high resolution machines.
If all of the C-terminal peptide subfragment ions can be identified by the
characteristic appearance of the m and m+2 doublet in the'60/'$O spectrum and
by
corresponding suppression of the 160 peak in the l80 spectrum, then the
sequence 216 of the
peptide or protein can, in principle, be "read" from the spectrum by looking
at the m/z
differences between successive peaks in the C-terminal series. All amino acids
except for
leucine and isoleucine, which have the same mass as one another, are
distinguishable from
each other by their characteristic masses and hence m/z values.
In practice, however, peptide sequencing using differential scanning mass
spectrometry is more difficult than simple comparison of spectra.
Identification of the peaks
arising from C-terminal peptide subfragment ions usually cannot be
accomplished by visual
inspection, particularly for longer peptides. The computer program product and
methods of
the invention alleviate this difficulty and allow for fast and accurate
interpretation of mass
spectra acquired using the differential scanning technique, resulting in fast
and accurate
determination of the previously-unknown amino acid sequence of a protein.
Algorithm For Identification of C terminal Peptide Subfragment Ions
The main problem addressed by the algorithms of the present invention is the
identification of y-ions in the mass spectrum of a peptide. The overall
principle is to
compute a filtered spectrum, SS, for the peptide, see Figure 5. The filtered
spectrum is
effectively a simulated spectrum which contains a peak at a m/z value of mP,
if mP
corresponds to a y-ion of a 160 containing peptide. The height of a peak in
the filtered
-24-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
spectrum is analogous to an intensity in a measured spectrum but is cxleulated
by a
cumulative multiplication of factors, each of which indicates the likelihood
that the peak
corresponds to a y-ion. An advantage of a filtered spectrum is that it is also
visually
pleasing and easy to interpret.
The steps that precede production of a filtered spectrum SS are as follows,
with
reference to Figure 5. The charge on the peptide that gives rise to the
spectrum is preferably
ascertained. The starting points are the 160/'x0 mass spectrum SP1 500 and the
180
spectrum SP2 502, from which, the charges on the subfragment ions are deduced,
step 504.
Subsequently, the peak for each subfragment in the'60/ls0 mass spectrum is
analyzed to see
1f whether it corresponds to an 's0-labeled ion, step 506, and a scoring value
S1 508 for each
peak is deduced. Peaks in the's0 mass spectrum are also analyzed to see
whether they
represent'60 containing peptide subfragments whose presence is suppressed in
the's0 mass
spectrum relative to the'60/'s0 mass spectrum, to produce a scoring value S2,
512. It is to
be understood that steps 506 and 510 may be reversed in order without
departing from the
15 scope of the present invention. Finally, scoring values S 1 and S2 are
combined to produce a
filtered spectrum SS, 514. Each of the foregoing steps is now.described in
greater detail.
The algorithm utilizes data for the'60/'s0 spectrum, SP1, and the's0 only
spectrum,
SP2. The principal task of the algorithm is to produce a scoring dataset, SD,
in which every
peak in the '60/180 spectrum is assigned a probability value that it is a y-
ion of a'60
20 .containing peptide subfragment. The filtered spectrum, SS, is then
computed, for every
value mP, according to equation (1):
SS(mP) = SD(mP)*SP1(mP) (1)
25 The final result of the algorithm is to produce a filtered spectrum which
contains computed
m/z values for'60 y ions, with all other ions screened out. It is to be
understood that the
methods of the present invention are equally applicable to calculations of
filtered spectra
that correspond to just parts, or ranges, of the measured spectra. It is not
to be construed
that the methods of the present invention are limited to calculations of
filtered spectra,
30 scoring dataset or scoring values that encompass the entirety of measured
spectra for either
of the positions of the transmission window.
In conjunction with differential scanning mass spectrometry, the
identification of y-
ions using the computer program product and methods of the present invention
is facilitated
by recognizing two essential features of y-ions in the spectra. First, y-ions
have a'60/'s0
35 isotopic distribution in the'6~/'s0 spectrum, SP1. Second, the'60 peaks of
y-ions are
- 25 -
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
suppressed in the'80 spectrum, SP2. These two features are used toi'~afcul~te
an'oveiall
scoring value, for each peak in the'60/'80 spectrum,. which forms part of the
scoring
dataset, SD.
The first step is to deduce a mass value, m, of the fragment which gives rise
to the
peak at position mP. Methods for accomplishing this can be found in:
Uttenweiler-Joseph,
S., Neubauer,G., Christoforidis, S., Zerial, M., and Wilm, M., "Automated de
novo
sequencing of proteins using the differential scanning technique," Proteomics,
1(5):668-682,
(2001), incorporated herein by reference. According to the type of ionization
employed the
subfragment ion giving rise to the peak at mP may be multiply charged. The
electrospray
ionization method typically give rise to multiply charged ions. Methods of
deducing the
number of charges are well-known toahose skilled in the art. The most
straightforward way
of identifying multiply charged ions is to examine the spacing of peaks
associated with
adjacent isotypes. For example, if such peaks are 0.5 m/z units apart, the
ions are double
charged. If the peaks are 0.33 or 0.25 m/z units apart, the ions are triply or
quadruply
charged respectively. More sophisticated methods of interpreting mass spectra
of multiply
. charged ions include those described in U.S. Patent 5,072,11 S, to Zhou,
incorporated herein
by reference.
The overall scoring value, SD(mP), for a peak at mP, which measures the
overall
probability that the peak is the first peak of a doublet arising from a
partially-labeled peptide
:.subfragment, is computed from a product of two factors, equation 2:
SD(mP) = S 1 (mP)*S2(mP) (2)
Sl(rnP) is a first scoring value that is a probability calculated by comparing
the
distribution and intensities of peaks in the envelope around the peak at mP in
the'64/'$O
spectrum, with the expected distribution and intensities of peaks for a
peptide of the same
mass using natural isotopic abundances. Therefore S1(mP) indicates how likely
the peak at
mP arises from a fragment with the'60l'80 ratio resulting from enzymatic
digestion in 50%
HZ'80 and 50% HZ'60, or, in an alternative embodiment, in a water mixture
containing some
other proportion of HZ'$O.
S2(mP) is a second scoring value that is a probability calculated by comparing
the
intensity of the peak at mP in the'60f$O spectrum SP1 with the intensity of
the peak at mP
in the'$O spectrum SP2 and evaluating the degree of suppression of this peak
in the second
spectrum. Therefore S2(mP) indicates how likely the peak at mP corresponds to
the'60
3~ containing y-ion of a peptide.
-26-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
Calculation of a first scoring value SI based on expected andobserv~d
~sott~pic
distributions.
The first step in the method of the present invention calculates a first
probability,
known as a first scoring value, S 1, that a particular peak at position mP
arises from the first
isotope of a '60/180 isotopic cluster in spectrum SP1. For a peptide whose
monoisotopic
mass is mo, the observed isotope envelope comprises contributions from ions
whose masses
are approximately mo+1, mo+2, mo+3, etc. If the monoisotopic species gives
rise to a peak
at mP with intensity Io the envelope will comprise successive peaks, denoted
(mP+1) with
intensity h, (mP+2) with intensity Ia, (mP+3) with intensity I3, and so forth.
If the ions
contributing to the cluster have a single charge, the successive peaks in the
envelope are
separated by approximately one m/z unit. The highest mass that is usually to
be considered
depends on the value of mo, since larger peptides are expected to incorporate
a greater
number of heavy isotopes, and will therefore have more significant peaks in
the isotope
envelope. The observed peak intensities of the isotope envelope, Io, h, Iz and
so forth, are
usually governed by the natural isotopic abundance. The natural isotopic
distribution of
carbon, nitrogen, oxygen and sulfur (Table 1) has been a factor that has
complicated the
interpretation of peptide mass spectra, but in the present invention it can be
used to some
advantage. Because natural abundances are known it is straightforward to
identify when
they are perturbed, for example, by the artificial'6~f$O ratio arising from
enzymatic
digestion in a mixture of 50% Hz'80 and SO% Hz160, and to quantify the extent
to which
they are perturbed.
The theoretical appearance of a peptide subfragment's isotope envelope may be
accurately modeled by solving a polynomial expression that calculates the
abundance-
weighted sum of the isotopes of each element to the molecular ion cluster.
(Yergey (1983) J.
Mass Spectrom. Ion Process 52:337-349). Examples of formulae which are
employed by
the present algorithm include, but are not limited to:
if (NlpeP >300)
~~ _ -0.015468 + 0.00056164 NI~,~,
h=h+b,
endif
if (Mpep > 1000)
$z = 0.020233 - 0.000039644 lVlpep + 0.00000017749 Mpepz
Iz=Iz+8z
-27-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
endif
if (M~~, > 1800)
83 = -0.0033252 - 0.000052477 MPs, -0.000000049304 Nh,~Z +
0.000000000045306 Nh,~,3
I3=I3+S3
endif
if (Nlp~, >2400)
8a = -0.01038 + 0.00012296 Mp~p - 0.00000014875 lVlpepa +
0.000000000053833 Nlp~,3
Ia = Ia +(Sa
endif
if (1VI~~, > 3100)
~s = - 0.0025452 + 0.0000122914 Mpg - 0.0000000116655 MP~,Z. +
0.000000000010044 Mp~3 + 6.2631 X 10-'s Mp~,a
Is=Is+8s
endif
if (Mpg, > 3500)
86 = -0.53925 + 0.00055053 M~,~, - 0.00000012987 Mpep2
=I6+(~6
endif
In these formulae, a fragment of mass Mpep has a fragment of mass MpeP + n in
its
isotopic cluster. The intensity, In, of fragment lVlpep + n in the envelope
can be calculated by
addition of the term 8". The initial values for all the In's are 0 with the
exception of Io which
is never calculated because it is set to 1. to is the intensity of the
monoisotopic species of
the molecule. I" is the intensity of the n'th isotope after the first (the
n+1' th isotope
altogether).
In order to illustrate an application of these formula, consider the 3rd
isotope above
the initial one. As discussed hereinabove, there may be more than one
contribution to every
isotopic mass. There is a contribution from the'60 containing peptide that has
isotopic
substituents from other elements, so the contribution is calculated taking 83.
But there is
-28-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
also a contribution from the '$O-containing peptide. Since the IBiJ-p~pti~e:is
2 Da header,
the same mass is also the first isotope after the initial one for the 1$O-
labeled peptide. Hence,
for the contribution of the'80 labeled peptide to the I3 peak, the
contribution must be
calculated taking bl. In this case, then, I3 is incremented by both 81 and 83.
These exemplary formulae depend only upon fragment mass, not element type or
chemical composition, nor charge. Accordingly, before applying such formulae,
the charge
on the fragment must be deduced, so that its mass can be found. The formulae
are
approximations derived from average abundances amongst currently known peptide
sequences. The most up to date compilations of peptide sequences that are
suitable for
deriving these formulae include, for example, a "non-redundant" database,
updated~on a
regular basis by the European Bioinformatics Institute (EBI). See for example
http://www.ebi.ac.uk/ also available at fta~//~ embl-heidelberg
delpub/databases/nrdb.
Another similar database compiled by NCB1, can be found at
htt~:l/www.ncbi.nhn.nih.,gLovl.
Note that, the heavier the peptide subfragment ion, the more peaks in the
envelope
have significant intensity. It can be seen that the envelope of a peptide
subfragment whose
monoisotopic mass is 1100, say, will have a fragment at mass 1101, whose
intensity is
calculated by the first formula and a fragment at mass 1.102 whose intensity
is calculated by
the second formula. For a peptide subfragment of mass 1100, contributions at
1103 and
greater are negligible.
In the differential scanning technique, the isotopic envelope of y-ions is
perturbed by
the characteristic isotopic distribution from partial labeling with'80. For
example, if the
peak at mP is the first peak in a y-ion'60/'$O isotope cluster, then the
observed intensities of
the peaks (mP+2), (mP+3) and so forth will be different from those of a
subfragment whose
oxygen content is that naturally occurring. The characteristic doublet of
the'60/'80 isotopic
cluster and the isotope envelope of the '80-containing ion will be
superimposed onto the
isotope envelope of the '60-containing ion. In contrast, the isotopic envelope
of a non-y-ion
will simply follow the expected naturally occurring form. As a result,
although it is
extremely difficult to visually assign peaks in a peptide mass spectrum
arising from y-ions,
the fact that their isotopic envelopes differ considerably from the envelopes
expected for the
naturally occurring distribution of isotopes can be exploited computationally.
The theoretically expected peak intensities, denoted h*, IZ*, I3*, and so
forth, based
on natural abundance of isotopes, for a'60-containing y-ion can be calculated
using a
polynomial expression of the type shown above. The observed and calculated
intensities are
normalized to Io and lo* respectively to permit quantitative comparison. The
scoring value
-29-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
S1 is a function of the difference between the observed and
c~.lcuiatedantei~sities.for each
peak in the isotopic envelope, as shown in equation 3:
Sln(mp) _ ~ (0.001+ e-~~n) (3)
wherein:
~ n = In - In (f)
The absolute value of the difference in intensities, O", is calculated from
I", the observed
intensity for peak (mP+n) and I"*, the intensity calculated for a peak (mP+n)
assuming that
~e peak at mP arises from a'60-containing y-ion. S1"(mP) in equation (3) is
the
contribution to the scoring value S 1 of the peak at mP from the intensity of
the peak at
(mP+n). The fundamental constant, a (~- 2.71828...), is the base of the
natural logarithm.
There are two parameters in equation (3) which have the following effects: ~,
is a
"strength", i.e., a weight given to the scoring value, adjustable according to
how significant
this criterion is to be; 6 is a "sharpness" parameter affecting how quickly
Sl" drops to zero
. with increasing g,. The form of equation (3) is shown in Figure 6(a) wherein
~, =5 and a =
0.25. O values are on the x-axis, S 1 values on the y-axis.
The sharpness parameter, a, determines how fast the scoring values drop to
0.001 *~..
It is preferably not fixed, but calculated from the data itself. The purpose
of the scoring
action is to multiply peaks which have an'g0 isotope with the scoring strength
~, and
peaks which do not have this isotope with a very small value, 0.001 *~,,
according to a
preferred form of equation (3). Since most of the peaks will not have an'g0
isotope (only
the C-terminal fragments have it), the average peak should be multiplied with
a very small
value close to 0.001 *~,. Thus, a is preferably chosen such that the average
peak is
multiplied with about 0.003*~,. This means mathematically:
6 - - ln(0.002) (S)
2
(~ avg )
with Da~g the average of all values determined from this spectrum.
In a preferred embodiment of the present invention, ~, is fixed at the value
10Ø
Values of a and ~, are sensitive to the machine employed and the quality of
the data. It is
within the capability of one skilled in the art to choose values of ~, and a
different from
-30-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
those given here in order to produce better results, according to the sample
and the machine
employed.
The exponential term in equation (3) ensures that large differences (~ in
observed
and calculated intensities, when squared, result in small S1" values. Note
that, because both
spectra are normalized to Io = Io* = l, Slo(mP) =1.001 ~, (~~,). In an
alternative
embodiment, the relative contribution of the two terms in equation (3) can be
adjusted by
separately altering the values of their coefficients, while ensuing that their
sum remains
close to 1Ø
Still other forms for equation (3) are possible without deviating from the
principles
of the present invention.
It is to be noted that actual isotopic abundances of the fragments in the
sample
cannot be measured perfectly either. Two major reasons for this are: that a
given peak
comprises signals from only a small number of ions, so statistically the full
abundance of all
isotopic substituents may not be realized; and that a given peak often
develops with signals
from other ions, giving rise to distortions that cannot be predicted.
For every cluster in the spectrum, a contribution Sln(mP) is calculated for
peaks
(mP+n), according to the mass of the peptide subfragment ion, giving rise to
the isotopic:
envelope. For an unlabelled fragment:. 300 < mo < 1000, just (mP+1) is
considered; for 1000
< ~ < 1800, peaks (mP+1) and (mP+2) are considered. When considering a
potentially 'g~
. labelled fragment, isotopes with mass mo + 2 are also considered, so for 300
< NiJ, < 1000
this includes peaks (mP+1), (mP+2) and (mP+3). It is to be understood that the
methods of
the present invention can also be practiced by computing S 1 over a subset of
clusters in the
spectrum.
Thus, the first scoring value, S 1 (mP), is the measure of similarity between
observed
intensities of peaks in the isotopic envelope around a given peak at mP and
the intensities
calculated for these peaks assuming that the peak at mP is the first isotope
in a'bp/180
isotopic cluster in SP 1. The scoring value takes into account not only the
degree of y-ion
labeling, but also the natural abundance of isotopes, which have traditionally
complicated
the mass spectra of large peptides. A small difference between observed and
calculated
intensities is reflected in a high scoring value, which indicates a high
probability that the
peak at mP is due to a'60-containing y-ion, i.e., the monoisotopic species.
In a final step, S 1-values are preferably normalized to l, by dividing
through the
entire S 1 function by its maximum value after it has been calculated for
every peak mP.
This step effectively converts the scoring values into probabilities.
-31-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
Calculation of a second scoring value based oh the degree o~'su~pYession of a
peak in the
second spectrum.
The second procedure in the method of the present invention is to compute a
second
probability, known as a second scoring value, S2, that a particular peak at mP
arises from the
first isotope of a'60f$O isotopic cluster whose'60 isotopes axe suppressed in
spectrum
SP2. This calculation is achieved by comparing the two spectra, SP1 and SP2,
thereby
determining the amount of suppression of the peak in SP2.
As described hereinabove, the transmission window of a quadrupole mass filter
can
be re-centered to a higher m/z value without being narrowed, so that
transmission of a
lighter isotope is effectively precluded. Use of a constant transmission
window width
ensures constant sensitivity. In this way, the two different spectra, SP1 from
an isotopic
mixture of a particular peptide, and SP2 from only the heavx isotope-
containing peptide,
have similar signal-to-noise ratios.
A peak at mP and intensity Ifl in spectrum SP 1, collected when the quadrupole
transmission window embraces the signals for both the'60 and the'g0 containing
fragments, gives rise to additional peaks denoted (mP+n) each having intensity
II;, due to the
. natural distribution of isotopes and the mixture of substituted fragments.
Similarly, in the
second spectrum, collected when the quadrupole transmission window is centered
around
the.fragment containing the'80 isotope at the C-terminus, the peak at mP
has.intensity
denoted by Ko, the peak (mP+n) in the same envelope has intensity denoted by
K".
First, the intensities of the peaks in SP1 arising from the peak at mP are
normalized
to la, and the intensities of the peaks in SP2 arising from the peak at mP are
normalized to
Kfl. If the peak at mP arises from the first isotope in the'60fg0 isotopic
cluster, I~«l0
because this peak is suppressed in the econd spectrum. By making I~=1, the
intensities of
the other peaks, Kl, K2, etc., are set to arkificially high values.
For the calculation of the second scoring value, it is desirable to average
abundant
isotopes of a fragment ion which could be'g0 labeled. For a peptide fragment
whose mo is
less than 1,400 Da, the mP and (mP+2) peaks are considered: if unlabeled, only
the first
isotope, at mo, is abundant; if labeled, both the isotopes mo and mo+2 are
abundant. For mo
that is greater than 1,400 Da, the mP, (mP+1), (mP+2), and (mP+3) peaks are
considered: if
unlabeled, only the first isotope is abundant; if labeled with'$O, all of the
isotopes mo to
mo+4 are abundant. This is because, as mentioned above, for heavier peptide
ions, the
contribution of subfragments containing multiple isotopic substituents
increases. The
choice of 1,400 Da is not fixed and other values in the region of about 1,400
Da can be
chosen without departing from the spirit of the present invention.
-32-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
For each spectrum, the average relative isotopic intensity is calculated by
taking the
average of the intensities of all of the peaks considered. For a particular
ion having mass
mo, the averages are I(ave) and K(ave) for spectra SP1 and SP2, respectively.
Thus, for mo <
1,400 Da, I(ave) _ (lo + I~)/2; for mo > 1,400 Da, I(ave) _ (J.~ + Ii + Ia +
I3)/4.
If the peak at mP is the first isotope in the ~601~80 isotopic cluster, which
is
suppressed in the second spectrum, then K(ave)>I(ave). If the peak at mP is
the first isotope
of a non-y-ion, then K(ave) ~-= I(ave).
An expression for the second scoring value, S2, that measures the degree of
suppression of the peak at mP in the second spectrum is shown in equation 6:
S2n(YYIp)= ~,2(1- 2 624n) (6)
wherein the parameter, ~,2, is a scoring weight given to S2(mP) and 0" is the
difference in
peg intensities between the two spectra, i.e., the peak suppression. In a
preferred
embodiment, the scoring weight parameter is given a value of 5. Figure 6(b)
gives an
example for ~ =5 and a2 =0.25. In Fig. 6(b), O values are on the x-axis, and
S2 values are
on the y-axis.
If the suppression is negative, i.e., if the intensity of a.given isotope in
the second
spectrum is greater than in the first spectrum, then 0 is set to 0
(corresponding to S2 = 0,
i.e., no suppression). As with the formulae for Sl, 62 is a sharpness
parameter since it
determines how fast the scoring values drop to 0 if there is no suppression (
i.e., 0" is small).
The scoring function should have the value ~ for peaks which were'80 labelled.
Such peaks are suppressed in the second spectrum. Since most of the peaks are
not labeled,
~e average peak is not'$Q labelled. For an average peak the scoring value
should be very
low, say 0.002 *~ in a preferred embodiment. Furthermore, a2 is preferably
chosen such
that the average peak is multiplied with a small factor such as 0.002*7~,~.
This means
mathematically that 6 may be expressed as a function of the form:
_ -~(1- ~2)
2 (~ n )~
Wlth L~a~g being the average of all values determined from this spectrum and
(3a being a
parameter that is preferably chosen to be 0.002. Of course, many other
mathematical forms
for a are consistent with the methods of the present invention.
- 33 -
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
If I(ave)>K(ave), then S2(mP) is given a value of 0. This indicates that there
was no
suppression of the peak at mP in the second spectrum, and therefore, it is not
the first peak in
an'60/'s0 isotopic cluster.
A high value of S2(mP) indicates a high probability that the peak arises from
a y ion.
In order to convert S2 values to probabilities, after calculation of all S2
values, the
scoring values are divided through by its maximum value.
Finally, a filtered spectrum SS may be calculated using equation 8, obtained
by
substituting equation (2) into equation (1), to calculate an intensity for a
peak at each value
of mP:
SS(mP)= SP1(mP)* S1(mP)*S2(mP) (8)
The procedure described is preferably repeated for every peak in both spectra,
or according
to choice for as many peaks as are of interest.
As demonstrated hereinabove, the scoring functions for every peak depend on
peak
specific parameters (such as suppression and deviation from the expected
isotopic
distribution for an '$O-labelled peak) and on parameters which can only be
calculated if all
suppressions and all deviations are known (i.e., giving rise to the averaged
values Da~J.
Therefore, in a preferred embodiment, the calculation of the filtered spectrum
starts by
ensuring that all deviations and suppressions are calculated for every peak.
Subsequently,
the scoring values for all peaks are calculated and then the spectrum is
multiplied with the
two scoring functions. It is therefore preferred that no peak cluster is
skipped. All
calculations are done for all peaks always evaluating every peak for its
characteristic
whether this one could be the first of an'60f$O cluster. Even if one peak
could be the first
isotope it would be premature to skip all the peaks belonging to this cluster
since it can not
be determined, a priori that the first peak already evaluated is really the
first one before the
others had not been evaluated for the same purpose. For example, it is
possible that the
second or third peak are much more appropriate first peaks of an'60/'$O
cluster.
Determinatioh ofAmiho Acid Sequences
Having identified a series of y-ions in the mass spectrum of the peptide, it
is possible
to deduce the sequence of the peptide by considering mass differences between
adjacent y-
ion peaks in the spectrum.
The method of differential scanning in conjunction with the algorithm of the
present
invention permits identification of peaks in the mass spectrum which
correspond to the
series of y-ion subfragments. Each ion in this series contains the C-terminus
of the peptide.
-34-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
In ideal conditions of collision induced dissociation, in which the peptide
amide bonds are
preferentially cleaved, each y-ion corresponds to a peptide subfragment
containing an exact
number of amino acid residues. Accordingly, if every peptide amide bond is
cleaved in the
collision chamber, each y ion in the series differs from the nearest y-ion in
mass by the mass
S of an amino acid residue. Because all amino acid residues have unique
masses, except for
leucine and isoleucine whose masses are the same, by calculating the mass
difference
between adjacent y ion peaks, it is possible to identify the exact amino acid
residue which
has been cleaved from the heavier fragment in order to produce the lighter
fragment or to
show that it must be either leucine or isoleucine.
10' In one embodiment of the present invention, once a mass difference has
been
computed for a pair. of adjacent y-ion peaks, the mass difference is compared
with the mass
of each of the 20 naturally occurring amino acid residues in turn until a
match is found. If
the mass difference is the same as that of one of the amino acid residues, the
corresponding
position in the peptide sequence is assigned to that amino acid residue. This
procedure is
15 repeated for each adjacent pair of y ion peaks in the mass spectrum. In a
preferred
embodiment of the present invention, if a mass difference between two adjacent
y-ion peaks
does not correspond to the mass of one of the 20 naturally occurnng amino
acids, the mass
difference is compared to the sums of the masses of all pairs of amino acid
residues to
search for a match. If a match is found with a pair of amino acid masses, the
two amino
20 . acid residues are placed in the sequence. The peptide amide bond between
this pair of
amino acids has not cleaved easily enough in the collision chamber to generate
a separate
subfragment containing each of the pair of residues. In this case, it is not
possible to infer
the order in which the pair of amino acids occurs unless other information,
for example for
other overlapping fragments, is available.
25 In a preferred embodiment of the present invention, the procedure of
matching mass
differences between adjacent y ion peaks is repeated for each distinct peptide
or peptide
fragment produced by enzymatic digestion of the protein. The sequence of each
peptide or
peptide fragment is deduced and the sequence of the protein inferred by
joining or
overlapping the sequences of each fragment, according to methods well known to
one
30 skilled in the art (See, for example, Mann, M., "A shortcut to interesting
human
genes:peptide sequence tags, expressed-sequence tags and computers," Trends in
Biological
Science, (1996), 21:494-495).
REFERENCES CITED
-35-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
All references cited herein are incorporated herein by re~ferL~iic.'e irt
their'entirety and
for all purposes to the same extent as if each individual publication or
patent or patent
application was specifically and individually indicated to be incorporated by
reference in its
entirety for all purposes.
Although the foregoing invention has been described in some detail by way of
illustration and example for purposes of clarity of understanding, it will be
readily apparent
to those of ordinary skill in the art in light of the teachings of this
invention that certain
changes and modifications may be made thereto without departing from the
spirit or scope
of the appended claims.
Many modifications and variations of this invention can be made without
departing
from its spirit and scope, as will be apparent to those skilled in the art.
The specific
embodiments described herein are offered by way of example only, and the
invention is to
be limited only by the terms of the appended claims, along with the full scope
of equivalents
to which such claims are entitled.
Alternate Embodiments
The present invention can be implemented as a computer program product that
includes a
computer program mechanism embedded in a computer readable storage medium. For
instance, the computer program product could contain a number of separate
program
modules that may be stored on a CD-ROM, magnetic disk storage product, or any
other
computer readable data or program storage product. The software modules in the
computer
program product may also be distributed electronically, via the Internet or
otherwise, by
transmission of a computer data signal (in which the software modules are
embedded) on a
carrier wave.
While the present invention has been described with reference to a few
specific
embodiments, the description is illustrative of the invention and is not to be
construed as
limiting the invention. Various modifications may occur to those skilled in
the art without
departing from the true spirit and scope of the invention as defined by the
appended claims.
-36-
SUBSTITUTE SHEET (RULE 26)
CA 02468689 2004-05-27
WO 03/046577 PCT/EPO1/14041
1
SEQUENCE LISTING
<110> EMBLEM
<120> MASS SPECTROMETRY METHOD
<130> 9882-010-228
<160> 1
<170> PatentIn version 3.1
<210> 1
<211> 26
<212> PRT
<213> Unknown
<220>
<223> Description of unknown sequence: Unknown peptide
<400> 1
Leu Phe Val Arg Pro Phe Pro Leu Asp Val Gln G1u Ser Glu Leu Asn
1 5 10 15
Glu Ile Phe Gly Pro Phe Gly Pro Phe Lys
20 25
SUBSTITUTE SHEET (RULE 26)