Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02473623 2004-07-30
I
WO 00/42947 PCT/USOOJOI53I
TITLE OF THE INVENTION
COVERED ENDOPRUSTHESIS AND DELIVERY SYSTEM
BACKGROUND OF THE INVENTION
, 1. Field of the Invention
The present invention relates to endoprostheses such as stents and
stmt-grafts, and particularly to endoprostheses that are suitable for use in
transjugular intrahepatic portosystemic shunt (TIPS) applications.
2. Description of Related Art
It is known that diseased or damaged liver tissue may increase the
resistance .to hepatic perfusion resulting in excessive and often dangerous
fluid
pressure increases in the portal vascular cireulatian. This condition can lead
to
gastrointestinal variceal hemorrhage and pathological conditions such as
ascites.
In order to decompress the portal circulation, a transjugular intrahepatic
portosystemic shunt (TIPS) may be created through the liver tissue by
connecting the portal vein to the inferior vena cava via the hepatic vein.
This
procedure first forms a fairly large puncture (for instance, using a 16 or 18
gauge needle) directty through the liver to allow direct flow between the
portal
vein and the hepatic vein: Next the puncture is Lined viittr a stent (for
example,
an 8-12 mm stent) to form a shunt. The TIPS pro~;edure has proven to be safe
and effective at decompressing the portal system and in controlling acute
variceal hemorrhage. A summary of TIPS procedures using unlined Palmaz
balloon-expandable stents (available from Cordis Carp., Miami Lakes, FL) is
provided in Zemel, et al., °Technicai Advances in 'Transjugutar
intrahepatic
Portosystemic Shunts, 12 RadioGraohics fi15-fi22 X1992).
Unfortunately, for many patients convenfit~rial TIPS procedures may
. provide fairly short-lived improvements. Stenosis or occlusion may occur in
up
to half of ail patients within 6 months following TIPS creation. Blockage of
the
shunt normally occurs due to pseudointimal formation or intimal hyperplastic
response.
CA 02473623 2004-07-30
i
W O 00142947 PCT/US00/01531
2 _
In the article by Nishimine; et al., "improved Transjuguiar Intrahepatic
Portosystemic Shunt Potency with PTFE- Covered Stent-Grafts: Eacperimental
Results in Swine," '196 Radiolaov 341-347 (198b), the authors hypothesize that
shunt potency might be improved if flowing blood could be separated from
leaking bile, the exposed surface of liver parenchyma, and the injured hepatic
vein. in that article the authors describe a device that comprises a thin-
walled
polytetrafluoroethylene (PTFE) graft (available from W. L. Gore ~ Associates,
fnc., Flagstaff, AZ) having single-body Gianturco-Rosch 2-stents (available
from Cook Inc., Bloomington, !N) mounted on each end for anchorage. Once
mounted in place, the authors then deployed one or two WALLSTENT scents
(available from Schneider Inc., Minneapolis, MN) within the PTFE graft to
provide mid-shunt radio! support. The authors reported that a PTFE-covered
stent-graft provided improved TIPS potency verses uncovered scents in a
porcine model. However, the authors also indicated that accurate stem-graft
1 ~~ placement was impoc-tant in order to maintain potency, since occlusion
continued to occur when a scent-graft was misplaced in the shunf or if a
portion
of the intrahepatic tract between the portal vein and the hepatic vein was
otherwise left unlined.
Nishimine et at. also reported that the two-step deployment of their
scent-grafts (that is, deploying a graft with anchoring stems first and then
separately, deploying WALLSTENT stems within the graft as a second
procedure) was "cumbersome and technically challenging." Nevertheless, as
the authors explained, this two-part procedure was necessary in order to allow
placement of .the scent and graft combination through a small 10-French (F)
(3.3 mm) sheath.
Accordingly, a fined or otherwise covered stmt=graft device would
appear to be ~rseful in helping to maintain potency in TIPS procedures.
However, a number of problems are presented to someone attempting to
provide an improved TtPS stent-graft.
First, as Nishimine et al. reported; small profile delivery of a stent-graft
device is difficult to accompiish in a single step. Generally percutaneous
delivery of a scent or stem-graft requires the device to be delivered at no
more
than 93-F (4.3 mm). While Nishimine et al. achieved delivery of a stent and
graft through a 10-F sheath, this was accomplished through a cumbersome and
3b challenging two-step procedure in which the separate devices were combined
in-situ. Other authors have reported delivering stent and graft combined
CA 02473623 2004-07-30
wo oo~az9a~ . . PCTIUS0010~53.1
3
structures in one-step,, but have reduired larger introductory profiles to do
so,
on the order of about 14-F to 16-F (4.7-5.3 mm): Haskal, et al, "PTFE
Encapsulated Endovascular Stent-Graft for Transjugular lntrahepatic Shunts:
Experimental Evaluation," Radiology: 205 (i997); Behesti, et a1. "Technical
Considerations in Covering and Deploying a Wallstent Endvprvsthesis for the
Salvage of a Failing Transjugular intrahepatic Portosystemic Shunt," ,lVIR: 9
(198$).
Second, also as Nishimine et al. reported, accurate device sizing and
placement may be critical in order to successfully avoid a biological response
resulting in stenosis or occlusion. Premature device occlusion or shunt
malfunction may be caused by a variety of conditions, including: failing to
maintain proper flaw into and out of the shunt device; failing to position the
device to completely cover the intrahepatic tract between the portal and
fiepatic
veins; or extending the device too far in the portal circulation or inferior
versa
cave.
Third, it appears important that the graft portion of the device inhibits
seepage of bile and other thrombogenic or mitogenic substances into the blood
system. Maintaining bile-exclusion while also seeking to use very thin graft
materials in order to achieve small device delivery profiles presents a
critical
design challenge.
Fourth, the stent-graft also needs to have suffrcient structural strength
to resist distortion during use. Nishimine et al. report that protrusion of
tissue
through an uncovered stem structure may result in up to 40% of pseudointimal
thickness. The present inventors believe that this tissue encroachment may be
one of the reasons that uncovered stmt structures do not perfiorrn very welt.
While any cover should limit the extent of tissue protruding through the 5tent
structure, if is believed desirable to provide a cover with sufficient
strength that
it will resist distortion from the inward force of tissue protrusion. Thus,
the
choice of cover material, as well as its method of attachment to the
underlying
stent, present design challenges. The stent component also needs sufficient
radial strength to prevent the stent-graft from significantly narrowing or
collapsing under pressure from scar or fibrotic proliferation. Again, wifh
respect
to both the stent and the graft components, the amount of radial strength
provided must be balanced against the desire for' thin graft membranes and
minimal stent size needed to create an overall device with small profile.
Also, a
CA 02473623 2004-07-30
W O .00!42947 PCTIUS00/0I 531
4
desired attribute in the TIPS procedure is that the endoprosthesis is flexible
in
order to accommodate the tortuous intrahepatic pathway.
SUMMARY OF THE INVENTION
The present invention is an improved endoprosthetic device specifically
designed to line an intrahbpatic shunt formed within liver tissue in order to
maintain its effectiveness. The present invention addresses the
endoprosthesis itself, as well. as delivery and deployment apparatus,
including
associated catheters, capture and restraining means, and other related
apparatus. The device of the present invention comprises an endoprosthesis
designed to be deployed in the transjugular intrahepatic partosystemic shunt
(TIPS) procedure. The endoprasthesis of the present invention comprises a
two-part construction having a first, covered, segment that is designed to be
positioned within the intrahepatic tract and hepatic vein end a second,
uncovered, segment that is designed to reside in the portal vein upstream of
the shunt. The endoprosthesis of the present invention provides a unique ste-
nt
and graft combination that combines a self expanding stent with high radial
strength with a thin-walled graft material that is resistant to permeation by
bile
and other fluids.
The endoprosthesis of the present invention has numerous desirable
features. First, the covered segment employs a thin bile exclusionary graft
material that protects the shunt from bile infiltration while maintaining
small wall
thicknesses. Second, the device has a unique one-piece construction that
allows it to be easily and accurately placed within the intrahepatic tract.
Third,
the present invention provides a unique mufti-stage deployment procedure that
again aids in the accurate placement of the endaprosthesis. Fourth, the device
is highly flexible, aiding in its positioning and deployment. Fifth, the
device is
highly compactable, again aiding its deployment and minimizing device profile,
Sixth, the device employs a unique self expanding stem pattern that is
believed
to be easier to manufacture, easier to deploy, and safer to use than
alternative
scent patterns. These and other benefits of the present invention will be
appreciated from review of the following description.
CA 02473623 2004-07-30
WO 00/42947 ~ . PCT/US00/01531.
5. _
DESCRIPTIaN OF THE DRAWINGS
The operation of the present invention should become apparent from
the following description when considered in conjunction with the
accompanying drawings, in which:
Figure 1 is a side elevation view of one embodiment of an
endoprosthesis of the present invention;
Figure 2 is a side elevation view of an endoprosthesis of the present
invention shown mounted in its delivery apparatus;
Figure 3 is a top plan view of an endoprosthesis of the present
invention, the endoprosthesis being normally cylindrical but is represented in
this Figure flat by making a longitudinal cut along the length of the
endoprosthesis and the uncoiling the endoprosthesis along this cut into a flat
sheet. This view shows in decal! the winding pattern of the two segments of
the
76 scent component of the endoprosthesis of the present invention;
Figure 4 is a side eievatian view of an endoprosthesis of the present
invention showing it in its compacted diameter (b) and in its enlarged
deployed
diameter (a);
Figure 5 is a side elevation view of an endoprosthesis of the present
invention shown bent around a rod to demonstrate flexibility via bend radius;
Figure 6a is an end view of an endoprosthesis of the present invention
at its deployed diameter;
Figure 6b is an end view of the endoprosthe.sis of Figure fia, with the
endoprosthesis shown compacted by folding and rolling the endoprosthesis
about a central catheter shaft;
Figure 6c is an end view of the endoprostheais of Figure 6a, with the
endoprosthesis shown compacted by radially compressing the endoprosthesis
about a central catheter shaft;
Figure 6d is an end view of the endoprosthesis of Figure 6a, with the
endoprosthesis shown radiaily compacted by folding into pleats of the
endoprosthesis about a central catheter shaft;
Figure 7 is a side elevation view of an endoprosthesis of the present
invention shown in its deployed diameter and in a compacted, pleated
diameter;
CA 02473623 2004-07-30
WO OOI42947 6 PCTIUSUU/0153-1
Figure 8a is a three-quarter isometric view of one embodiment of a
tapered die used to establish a pleated compacted endoprosthesis of the
present invention;
Figure 8b is a side cross-section view of the tapered die of Figure 8a
along tine 8b-8b;
Figure 9 is a side elevation view of an endoprosthesis of the present
invention having tether lines positioned around the circumference of one end
of
the endoprosthesis in specific locations such as to control the locations
where
the stent forms pleats as it is drawn through a tapered die such as that shown
in Figures 8a and 8b;
Figure 10 is a side elevation view of a compacted endoprosthesis of the
present invention mounted on a delivery catheter and covered with a packaging
sheath;
Figure 11 is a side elevation view of a compacted endoprasthesis of the
present invention shown partially deployed after the packaging sheath has
been removed;
Figure 12 is a side elevation view of the compacted endoprosthesis of
Figure 10 with the packaging sheath and delivery catheter being inserted
through a hemostatic sheath;
Figure 13 is a side elevation view of the endoprosthesis of Figure 12
with the delivery catheter partially advanced into a transjugular sheath with
the
packaging sheath shown partially removed;
Figure 14 is a side elevation view of a compacted endoprosthesis of the
present invention shown advanced into a portal vein;
Figure 15 is a side elevation view of a compacted endoprosthesis of the
present invention shown confined within a hemostatic sheath and located at an
intrahepatic juncture site in a portal vein;
Figure 16 is a side elevation view of a TIP,'S endoprosthesis of the
present invention being partially deployed in the portal vein through
withdrawal
of the hemastatic sheath;
Figure 17 is a side elevation view of the TIPS endoprosthesis of the
present invention being partially withdrawn to properly position the
endoprosthesis within the portal vein, an unconstrained expanded portal stent
segment of the endoprosthesis engaging the intrahepatic juncture to assure
proper placement;
CA 02473623 2004-07-30
WO 00/42947 ~ PCTlUS00101531
Figure 18 is a side elevation view of the TIPS endoprosthesis of the
present invention having been fully deployed within an intrahepatic tract and
the portal vein; .
Figure 19 is a three-quarter isometric view of a portion of an apparatus
used to test the permeability of the cover of the present invention;
Figure 20 is a side elevation view of an endoprosthesis of the present
invention mounted with the fixture elements illustrated in Figure 19;
Figure 21 is a top plan view of a loop device used to measure radial
stiffness of an endoprosthesis of the present invention;
Figure 22 is a three-quarter perspective view of an endoprosthetic
device mounted in the loop device of Figure 19, positioned so as to connect
the
loop device to a tensile tester; and
Figure 23 is a scanning electron micrograph (SEM), enlarged 1000X, of
the surface of an expanded poiytetrafiuoroethylene (PTFE) cover used in the
present invention, having anode and fibril structure and having bent fibrils
incorporated therein.
DETAILED DESCRIPTION OF THE INVENTION
The present invention is an improved impiantable endoiuminai device,
and especially such a device for establishing and maintaining an intrahepatic
portosystemic shunt. As is explained in greater detail below, typically this
procedure is performed endoiuminaity through the jugular vein, connecting the
portal vein to the inferior versa cava by way of hepatic vein. As a result,
the
procedure is commonly referred to as being a "transjuguiar intrahepatic
portosystemic shunt" or abbreviated "TIPS" or "TtPSS." !t should be
appreciated, however, that a shunt through the liver between the Aorta! vein
and the versa cava may be accomplished by other methods and that the
endoprosthesis devices of the present invention are not limited in application
to
transjuguiar insertion. As such, the term "intrahepatic portosystemic shunt"
as
used Herein is intended to include any procedure whereby pressure is relieved
in the portal vein by way of a shunt from the portal to the systemic systems.
As has been explained, typicaliy a shunt is established creating a large
tract {for instance, by puncturing with a 18 or 18 gauge needle and then
3~ ballooning to about 10 mm) directly through the liver to allow connection
between the portal vein and the hepatic vein. Following ballooning, the shunt
is
CA 02473623 2004-07-30
WO 00142947 ' g' PCT/US00/O1'S3:1
generally maintained by lining the puncture with an uncovered scent. The TIPS
procedure has proven to be safe and effective at decompressing the portal
system and in controlling acute variceal hemorfiage. Unfortunately, for many
patients conventional TIPS procedures may provide fairly short-lived
improvements. Stenasis or occlusion may occur in a significant number of
patients. Blockage of the shunt normally occurs due to thrombosis,
pseudointimai formation, or intimal hyperpiastic response. While covered
scents have been employed with some improvement in subsequent occlusive
response, these previous covered stent devices have been diffrcult to properly
position, have had excessively large insertion profiles, andlor are
ineffective at
bile exclusion - bile being linked to increased hyperplastic response.
The present invention addresses all of these defciencies of previous
stent devices by providing an endoprosthesis stent-graft combination that has
a
small introductory profile, is easily positioned and deployed within the
shunt,
and employs a cover that is highly resistant to bile permeation
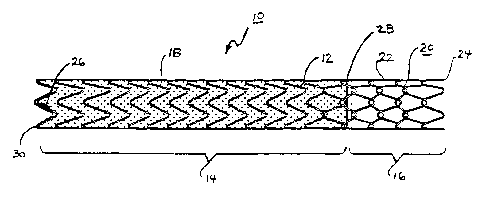
Shown in Figure 1 is one embodiment of an endoprosthesis 10 of the
present invention. The endoprosthesis comprises a two-part stem element 12
having a first segment 14 and a second segment 16. A cover 18 is provided
along the length of the first segment 14, while the second segment 16 is left
uncovered.
The first segment 14 and the cover 18 are adhered together (for
instance by an adhesive and/or by a wrap of an adhered fim) to maintain the
position of the cover 18 on the endoprosthesis 10. The attachment of the cover
to the stent element 12 also prevents the first segment 14 from excessively
longitudinally elongating when longitudinal tension is applied to the
endoprosthesis 10. It is believed preferable that the cover 18 line the
interior
of the scent element l2, as shown, but acceptable results may also be achieved
with the cover 18 placed on the outside of the stent element 12, with the
cover
being placed both inside and outside of the scent element 12, or with the
stent
element 12 being embedded within the cover 18. As such, the term "cover" as
used herein is intended to include any generally continuous material that is
placed inside of andlor placed outside of andlor mounted integrally with the
stent element 12 of the present invention.
As will become clear from the following description, the second segment
16 of the stent is left uncovered to permit its permanent deployment within
the
portal vein and thereby allow perfusion of portal venous branches through the
CA 02473623 2004-07-30
WO 00142947 PCTJUSOO/OIS3.1
9
interstices of 16. In order to prevent the second segment 16 from undergoing
uncontrolled longitudinal elongation, a different stent pattern is employed
along
this uncovered segment. in this embodiment an interlocked (or "chain-finked")
stem pattern 20 is used that prevents the second segment 16 from excessively
longitudinally elongating beyond a predetermined desired length: In the
interlocked stem pattern 20 shown, a single wire 22 is employed that is
wrapped from the cover 18 to a distal end 24 of the endoprosthesis 10 and
then back to the cover 18. This pattern allows the wire 22 to be terminated
within the cover 18 and avoids having a loose end of the wire 22 exposed at
1 D distal end 24 of the endoprosthesis 10. This wrapping pattern is explained
in
greater detail with regard io Figure 3, below.
In order to facilitate placement of the endoprosthesis 10 device, a series
of radiopaque markers 26, 28 are provided along the length of the
endoprosthesis. Marker 26 identifies proximal end 30 of the endoprosthesis 10
and marker 28 identifies the junction between the first segment 14 and the
second segment 16 of the endoprosthesis 10. By employing marker 28 as a
circumferential band that entirely or substantially marks the circumference of
the endoprosthesis 10, this further facilitates correct placement of the
endoprosthesis 10.
The scent element 12 may be formed from a variety of wire materials,
including stainless steel, nickel-titanium alloy (nitinol), tantalum, elgiloy,
various
polymer materials, such as polyethylene terephthalate) (PET) or
palytetrafluoroethylene {PTFE}, or bioresorbable materials, such as
levorotatory polylactic acid (L-PLA) or po(yglycolic; acid {PGA). The
preferred
material comprises a supereiastic material, such as nitinol metal, that will
withstand tight compression in a compacted configuration (diameter) and then
self-expand to a deployed configuration (diameter) once released in place.
Alternatively, the stenf element 12 of the present invention may be
constructed
from a material (e.g., stainless steel) that can be mechanically enlarged in
place, such as through balloon expansion.
For application in a TIPS procedure, the endoprosthesis .10 of the
present invention would typically have dimensions as follows: a length of
about
fi to 12 crn, with a length of about 6 to 10 cm being preferred; a deployed
diameter of about fi to 14 mm, with a diameter of about 8 to 12 mm preferred;
and a wall thickness (comprising both the cover 18 and the stem element 12) of
about 0.1 to 1.0 mm, with about 0.1 to 0.6 mtr~ being preferred. While the
CA 02473623 2004-07-30
WO 00142947 ° ~ O PCTIUS00101531
dimension "diameter" is used herein, it should be understood that this
dimension is intended to define the average cross-sectional dimension of the
device and is not intended to Limit the present invention to devices with
circular
cross-sectional shapes. The device may also have multiple diameters, with the
diameter of section 16, for example, being larger (or smaller) than the
diameter
of section 14. One or both of the sections 14, 1 fi may also be tapered or
otherwise adjusted in dimensions.
The endoprosthesis 10 itself will preferably have a compacted
dimension of less than or equal to 16 French (5.3 rnm), and more preferably a
compacted dimension of less than or equal to 12 F (about 4.0 mm), and even
mare preferably a compacted dimension of less than or equal to 10 F {3.3 mm)
or even 9 F (3.0 mm) or 8 F (2.7 mm) or less. In order to be delivered
percutaneously, the endoprosthesis and its deployment apparatus should have
a diameter of less than about 13 F (4.3 mm), and more preferably less than or
equal to 10 F (3.3 mm): "French" measurements used herein define the size of
a hole through which a device will pass. For example, a device with a
measurement of "10 French" will pass through a 10 French hole (which has a
diameter of 3.3 mm). Again, the device need not have a circular cross-sectiah
in .order to pass through a circular 10 French hole so long as the hole is
large
enough to accommodate the widest crass-section dimension of the device.
The first segment 14 of the endoprosthesis 10 will typically comprise
about 50 to 90 percent of the entire length of the endoprosthesis.
Accordingly,
the first segment 14 will normaNy be about 4 to 8 cm in length and the second
segment 16 will normally be about 1 to 3 cm in length.
The preferred stent element 12 is constructed from a superelastic nitinol
metal with the following properties: having alloy percentage of about 51
nickel, about 49% titanium (for example, SE 508 available from Nitinol Devices
& Components, Fremont, CA, USA); being 40-45'% cold worked; having a
tensile modulus of approximately 35 to 70 x 106 kPa; being electopolished; and
having a wire diameter of about 0.15 to 0.50 mm, and preferably about 0.25 to
0.30 mm.
The cover 18 performs a number of functions in the endoprosthesis 10
of the present invention. First, the cover 18 prevents extrusion of liver
tissue
through the stent element so as to help maintain the full desired diameter of
the
shunt. Second, as has been noted, the cover 18 helps to maintain the
CA 02473623 2004-07-30
WO 00/42947 11 PCTIUSUO/01531
dimensions of the endoprosthesis and prevents unccmtrolled elongation of the
stem element. Third, it is believed to be highly desirable for the cover 18 to
reduce or eliminate bile from permeating into the shuns. Fourth, the cover
allows bending of segment 14 without kinking or compromising the iuminal
surface of the device. Based upon the medical literature, the inventors
believe
that bile permeation into the shunt may contribute to hyperplastic response
and
premature occlusion. It is also desirable that the cover 18 provides all of
these
benefits while contributing minimal additional profile to the endoprosthesis
device.
To this end, it is desirable to provide a cover material that has the
following properties: longitudinal tensile strength wh~sn mounted in the
device
of about 5 to 20 Kgf, and preferably about 10 to 15 Kgf; a permeability as
quantified by a Gurley Number of greater than about 60 seconds per 100.cc or
air per one (1 ) cmz of material; a thickness of about 0.05 to 0.25 mm, with a
thickness of about 0.10 to 0.20 mm preferred; a water entry pressure of about
5 .
to 15 psi (34 to 1.02 kPa) or more, with 7 to 9 psi (48 to 62 kPa) or more
preferred. Test procedures used to establish these values are described in the
Examples below.
As the terms "bile exclusionary," "resistant to bile permeation," "bile
resistant," and similar terms are .used herein, these are intended to
encompass
any cover that has a Gurley Number of greater than about 60 seconds per 100
cc air per a cm2 material. More preferably, the Gtirley Number is greater than
about 70, 80, 90, 100 or more seconds per 100 cc of air per 1 cm2 of material.
The preferred cover 18 material comprises a fluoropolymer and
especially a fluoropolymer comprising expanded polytetrafluoroethylene
(PTFE). For example, a cover may be constructed by forming an ultra-thin
walled expanded PTFE base tube by methods described in British Patents
7,355,373, 1,506,432, published PCTApplication W~ 95105555, and in United
States Patents 3,953,566, 3,962,153, 4,096,227, 4,'187,390, 4,902,423,
5,718,973, 5,735,892, and 5,620,763. This process produces an expanded
PTFE tube having a microstructure of polymeric nodes connected by polymeric
fibrils and defining void spaces therein.
The extruded ultra-thin walled tube preferably has a wall thickness of
about 0.05 to 0.30 mm, with a preferred wall thickness of about 0.05 to 0.2
mm.
CA 02473623 2004-07-30
W0.00142947 ' 12 PCTIUS00101531
The tube preferably has a nominal fibril length of about 5 to 50 micron, with
a
preferred fibril length of about 15 to 35 . Fibril length may be determined
through conventional methods, such as Section 8.2.1.3 of ANSIIAAMI VP20-
1994.
Over the extruded ultra-then walled tube an expanded PTFE film is
heiically wrapped. The film may be manufactured in the following manner:
PTFE fine powder is blended with a hydrocarbon lubricant and paste extruded
through a flat die. The resulting tape is calendered to approximately 50% of
is
original thickness. Next, the tape is then heated to volatilize the
hydrocarbons.
The resulting dried tape is then heated to about 300°C, expanded at a
ratio of
25:1 and then expanded again at a ratio of 8:1, and then sintered at a
temperature of about 370°C to achieve the desired properties.
This produces a low permeability film, with preferred properties of about
0.005 to 0.025 mm in thickness, with a more preferred thickness of about
0.010 to 0.015 mm jas measured by a Mitutoyo Snap Gauge Model Number
2804-10). The frlm preferably has a fibrl length of approximately 50 to 100
micron, with a preferred fibril length of about 70 to 80 microns. The film
further
has a methanol bubble point of about 10 to 40 kPa, and more preferably a
bubble point of about 20 to 30 kPa. Alternatively, a film as described in
United
States Patent 5,476,589 to Bacino, may be employed.
Bubble point may be determined by placing the film in a one inch (2.54
cm) diameter round clamp and completely wetting the sample out with
methanol from above. Pressure is then applied slowly (at the approximate rate
of 40 kPa/min) to the bottom side of the wet out sample until bubbles appear
on
the other side of the sample. The pressure at which the first bubble appears
is
designated as the "bubble point."
Finally, the film has a Frazier permeability of about 0.5 to 3
cm3a;~lseGcm2~e~, @ 125 Pa back pressure, with a preferred Frazier
permeability of about 0.5 to 1.5 cm38;,lsedcm2",~e~, (~ 125 Pa back pressure.
Frazier permeability may be determined by placing a sample of material
(referred to as a "membrane") to be tested on a 2~.4 mm diameter circular
clamp. One side of the clamp is sealed such that gaauge pressure can be
supplied to one side of the membrane and flow rates through the membrane
can'be measured. Ambient air (20°C ~ 3 C) is slowly increased on one
side of
the membrane to 125 Pa. The flow of air through the membrane is measured,
CA 02473623 2004-07-30
WO 00142947 ~ 3 PCT/US00/01531
and expressed in units of cubic centimeters of air (cm3~;,) flowing ttirough a
square centimeter of material (cm2,~"e,~,) in a second (s) with a hack
pressure of
125 Pa.
To assemble the material of the cover 18, the extruded ultra thin wall
base tube is mounted over a stainless steel mandrel with approximately the
same outer diameter as the inner diameter of the base tube. The film is slit
to
0.75 inches (19 mm) wide and then helically wrapped in appraxirnately 6 Payers
onto the ultra-thin walled base tube (with an overlap of about 16 mm for each
wrap of the tube). The composite construction is cooked in an air convection
oven for about 15 minutes at about 370°C.
After the composite tube has cooled, "stored length" may be imparted
into the wrapped tube by deforming or bending the 'Fbrils of the expanded
PTFE node and fibril rnicrastructure and setting the material in this
configuration, such as in the following manner. They stored length is imparted
by compressing ("scrunching") the graft along the mandrel longitudinally up
until a point where the graft material begins to buckle. After this
compression,
the construction is heated treated at about 320°C far about 5 minutes.
in order
to achieve greater stretch, the base tube can he over-wrapped with a thin
ePTFE film layer prior to scrunching. This over-wrap resists buckling of the
tube, allowing more longitudinal compression to be imparted. After the
construction is heat treated, the over-wrap is then ennwrapped and removed,
and the composite is carefully removed from the mandrel. Stored length may
also be imparted to the device after the stent has been attached. This can be
accomplished through a similar process except an additional 320°C heat
treatment cycle for approximately 15 minutes is performed after stent
attachment and compression.
After "stored length" has been imparted to the tube (such as through the
above described "scrunching" process), the stent can be slid coaxially over
the
PTFE firm wrapped tube. The stent is preferably constructed of a helical
pattern as described and illustrated herein with no interconnects from row-to-
row, such that the stmt can bend freely. The wrapped tube and stent are then
mounted onto a stainless steel mandrel. An expanded PTFE tube (e.g., a
GORE-TEX~ Vascular Graft, available from W. L. Gore and Associates, Inc.,
Flagstaff, AZ, USA) can be sandwiched between the wrapped tube and the
undersized stainless steel mandrel to function as a cushioning layer. The
apices of the stent are aligned as necessary, and a porous composite film of
CA 02473623 2004-07-30
WO OO1a2947 14 PCTIUSOOI0153.1-
FEP and PTFE are wrapped over the construction in approximately
overlapping layers, with the side of the film containing FEP toward the lumen
of
the graft. Alignment of the stent apices can be accomplished through use of a
filament threaded through apices of adjacent rows of first segment 14.
The cover 18 is preferably attached to the stent element 12 by adhering
the two together through use of a suitable.adhesive, such as fluorinated
ethylene propylene (FEP), polyurethane, cyanoacn~letes, etc. Additionally, the
materials may be bonded together through heat treatment (such as, sintering of
the materials together) or through use of a wrap (for instac~ce a tube, tape,
or
membrane) around the outside of the stent and cover (either continuous or
discontinuous) that is adhered through either a thermoplastic or thermoset
adhesive to the stent and cover. Alternatively, the stem may also be coated
with a thermopolymer or thermoset adhesive and the cover bonded by
reflowing or setting the polymer coating.
The FEP-coated porous expanded PTFE composite film is made by a
process that comprises the steps of:
a) Contacting a porous PTFE film with another layer which is preferably a film
of FEP or alternatively of another thermoplastic pofymer;
b) Heating the composition obtained in step a) to a temperature above the
melting point of the thermoplastic polymer;
c) Stretching the heated composition of step b) while maintaining the
temperature above the melting point of the themloplastic polymer; and
d) Cooling the product of step c).
The FEP adhesive coating on the porous expanded PTFE film may be
either continuous {non-porous) or discontinuous (porous), depending primarily
on the amount and rate of stretching, the temperature during stretching, and
the thickness of the adhesive prior to stretching. The preferred composite
film
has a thickness of about 0.0004" (D.01 mm), a methanol bubble point of about
1.5 psi {10 kPa), a Frazier number of about 21 fi~,;~lrninlftz",~er;~,, @ 0,5"
H20 back
pressure (10.1 crn3~,lseclcrn2",@ 125 Pa back pressure), and an
approximate weight ratio of FEP to PTFE of 18%.
Weight ratio is calculated by using 2.1 g/cc as the density of FEP and
1.5 g/cc as the density of dried tape. The total mass of FEP going into the
expansion process is divided by the total mass of dried PTFE tape going into
the expansion process.
CA 02473623 2004-07-30
WO 00/42947 PC'1'NS00101531
After this wrap, the construction is cooked in an air convection oven at
about 320°C for 15 minutes. After cooking, the con~aruction is cooled
to
ambient temperature and removed from the mandrel.
As has been noted, it is believed to be desirable to exclude bite and
5 other biological liquids from permeating through the cover 18: The above
described cover 9 8 construction produces a material that the inventors
believe
is significantly resistant to transmural fluid (bile) permeation. The ability
of a
material to exclude bile can be approximated by meaasuring the Gurley Number
of the material. The test for this procedure is detailed in the Examples,
below.
i 0 The markers 26, 28 may comprise any suitable radiopaque material that
will appear on a fluoroscope or similar device. Suitable materials may
include:
metal (such as gold, iridium, platinum, or stainless slleel); or filled
polymers
(such as carbon filled expanded polytetrafluoroethylene, or barium filled
FEP).The preferred markers 26, 28 comprise greater than 99.9% gold.
15 Figure 2 illustrates one embodiment of the deployment apparatus 30
that may be used to install an endoprosthesis 10 of the present invention.
This
deployment apparatus 30 comprises: an introducing (or packaging) sleeve 32;
a constraining sleeve 34; a distal shaft 36; a proximal shaft 38; a strain
relief
40; a deployment port 42; a deployment knob 44 mounted within the
deployment port 42 that is connected to a deployment line 46 attached to a
constraining sleeve 34 surrounding the endoprosthesis 10; a side arm adapter
48; a flushing port 50; and a guidewire port 52. A radiopaque marker 54 may
be provided on the distal shaft 36 to aid in the remote positioning of the
endoprosthesis 10. The operation of the deployment apparatus 30 is explained
in detail below with reference to Figures 10 through 18.
The winding pattern of the preferred stent elf:ment 12 of the present
invention is illustrated in Figure 3. As can been appreciated through
examination of this Figure, a single wire 22 may be employed to form both the
first segment 14 and the second segment 16 of the stent element 12. Over the
length of the first segment 14, the wire 22 may be farmed in an undulated or
serpentine pattern 54. Since the cover is used to maintain the relative
positions
of the first undulated pattern 54, each winding 56 of the undulated pattern
around the circumference of the stent element 12 does not need to be attached
to adjacent windings. It should be appreciated that come link, such as a
fiber,
wire extension, etc., may be employed to facilitate alignment of adjacent rows
of windings if desired.
CA 02473623 2004-07-30
WO 00142947 ' PCTIUS00/0153.1
16
Along the length of the uncovered second segment 16, however, some
means should be provided to prevent the second segment 16 from elongating
in an uncontrolled fashion. In the preferred embodiment shown, this is
accomplished by employing the interlocked (or "chain-linked") stent pattern
20.
In this pattern the single wire 22 is wrapped from the first segment 14 to the
distal end 24 and then back to the first segment 14. Along the length of the
second segment 16 the wire 22 is provided with a second undulated pattern 58
along a first pass 60 and a third undulating pattern 62, interlocking with the
second undulating pattern 58 along a second pass 64. By interlocking the
second undulating pattern 58 and the third undulating pattern fit, the stent
pattern 20 permits the second segment 16 to be longitudinally compressed,
thus imparting flexibility; but the scent pattern prevents the second segment
16
from being longitudinally elongated beyond a predetem~ined maximum length.
It should be noted that the interlocked stent pattern 20 also imparts columnar
support when the device is in a radially compressed configuration and less so
when it is deployed.
it is even more desirable for the interlocked stent pattern 20 along
uncovered second segment 16 to provide some degree of resistance to
longitudinal compression. This helps to prevent the second segment 16 from
20-- longitudinally collapsing upon itself once deployed in vivo. It has been
determined that additional longitudinal stiffness may be provided to the
second
segment 16 by using a modified "cross-over stent pattern 63 along one side of
the second segment 16. As is shown, the cross-over pattern 63 links between
every third row of the interlocked stmt pattern 20. un other words, instead of
the winding pattern linking the wire 22 with an adjacent row of the
interlocked
pattern, the wire 22 "crosses over two wires and interlocks with a wire in a
row
two removed from the starting row. Any desired cxoss-over pattern may be
incorporated without departing from the present inw:ntion, including one that
skips one, two, three, four or more rows. The amount of longitudinal stiffness
may be modified using this technique by skipping more rows to impart greater
stiffness or skipping fewer rows to impart lesser stiffness.
Longitudinal stiffness may also be provided or supplemented by
structures that will assist in holding the second segrnent 16 in an extended
position. Stiffness may also be imparted by selectively binding portions of
the
interlocked pattern to each other using threads or wires or similar structures
CA 02473623 2004-07-30
WO 00/42947 ' t ~ PCTIUSOOI01531
(for example, attaching interlocked junction 65 together with a thread that
allows no; or only limited, actuation between pattern 58 and pattern 62).
There are a number of benefits achieved with the uncovered second
segment 16 of the present invention. First, the stent patterns described above
provide a clinically desirable amount of radial stiffness to the second
segment
16. "Radial Stiffness" or "compressive stiffness" signifies the resistance of
the
endoprosthesis of the present invention to undergo circumferential
compression. The testing parameters for radial stiffness are set forth in the
Examples, below. Briefly, this value is the amount of force necessary to
compress the device radialfy at a rate of 12.7 mm device circumference/rnin to
75% of its original diameter with a 1 cm wide loop v~rhile the force is
recorded.
After completing the test, the slope of .the load vs. clispiacement curve is
calculated. This calculation is performed by recordiing the circumferential
displacement and corresponding force at 5% and 20% diametric compression
(i.e., the outer diameter of the stent is 95% and 80°,~° of its
original diameter
loaded with a 0.05 Kgf load, respectively). Slope is then calculated by
dividing
the difference in these two forces by the differences in the con-esponding
displacements. The result, the slope from 5% to 2C)% diametrical compression
of the force verses circumferential displacement, is then multiplied by pi
(If) to
obtain the slope of the load vs. diameter curve, expressed in Kgflmm device
diameter.
It is preferred that the device of the present invention will have a radial
stiffness of at least 0.1 Kgf/mm for the covered segment and at least 0.12
Kgflmm for the uncovered segment. More preferat>ly, the device should have a
stiffness of at feast 0.1, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, or
0.2
Kgf/mm or more for the covered segment, and 0.13, 0.14, 0.15, 0.16, 0.17,
0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, or 0.25 Kgflrnm or more for the
uncovered segment. The radial stiffness of the uncovered second segment 16
helps assure that the inlet to the shunt remains open in use. Additionally,
since
formation of the shunt is sometimes difficult to establish with accuracy, the
radial stiffness of the second segment can assist in widening the portal vein
or
any collateral vein that might have been entered by accident.
Second, the open structure of the second s~:gment allows for continued
flow through the portal vein while simultaneously relieving excess pressure by
way of the shunt. This is believed to be important far maintaining flow to the
liver while also relieving excess pressures.
CA 02473623 2004-07-30
WO 00!42947 l 8 PCTIUS00/01531
As has been noted, it is believed desirable that the endoprosthesis 10 of
the present invention is highly resistant to any longitudinal elongation
beyond
its normal deployed length (that is, the longitudinal length of the device
that it
b assumes upon deployment without tension applied to it). lNith respect to the
uncovered .second segment 16, it is desirable that the second segment 16 will
resist any attempt to elongate it beyond about 2~% of its normal deployed
length. °Nom~al deployed length" comprises the length the second
segment 96
naturally assumes when deployed on a table top, extended by hand to its fully
extended length, and then released without being subjected to further
elongation forces. More preferably, the second segment 16 resists elongation
beyond about 20%, and even more preferably beyond about 15%, and still
more preferably beyond 10%, of its normal deployed length. By the term
"resist any attempt to elongate" beyond a given length, it is meant that the
7 5 second segment 16 has a given maximum longitudinal length that will not be
exceeded under normaP conditions encountered once implanted. This can be
tested in vitro by attaching the device to a 2 kg weight and allow the weight
to
hang under normal gravity for one (1) minute. The second segment 16 should
not elongate more than 20% of its normal deployed length under these
conditions.
The interlocked stmt pattern 20, whereby the wire is doubled back on
itself to form an interlocking pattern, provides a number of benefits. First,
this
winding pattern allows a single wire to be used in the scent element 12,
avoiding possible manufacturing problems iriherent in joining multiple wire
2b components together. Second, this winding pattern allows the wire 22 to be
terminated within the cover 18 and avoids having a loose end 67 of the wire 22
exposed at distal end 24 of the endoprosthesis 10. Pt should be appreciated
that this minimizes the risk that the loose wire end I57 might snag or cause
damage to the vascular system and eliminates the need for a separate
manufacturing step to terminate the wire at the distal end 24 of the stent
element 12. Third, the second segment 16 of the s~tent is very flexible,
allowing
it to be easily bent over a 90° angle with a 2 cm outer circumference.
Fourth,
the second segment 16 of the endoprosthesis tends to rebound when it is
compressed under a force and then the force is removed.
3~ As has been noted, it is believed important to provide an
endoprosthesis for TIPS applications that includes both a stent element and a
CA 02473623 2004-07-30
wo eo~a29a~ ' I9 ' pcrrtrsooiom3~
graft element in a single integral package that has a small compacted
configuration (diameter) and fully operational deployed configuratioci
(diameter). To this end, the present inventors have sought to create an
endoprosthesis device that undergoes substantial growth between the
compacted and deployed states. Figure 4 illustrates, one embodiment of a
TIPS endoprosthesis 10 of the present invention showing it enlarging from a
compacted diameter (b) to in an enlarged deployed diameter (a). It is
desirable
for the ratio of ~ to comprise at least about 2, and more desirable for the
ratio
to comprise about 3 to 5 or more. The .present invention employs a variety of
unique materials and structures to produce highly compacted devices that can
achieve these kinds of ratios.
Another useful attribute of the. endoprosthesis device of the present
invention is that it is constructed from materials that rertder the device
highly
flexible, both in its compacted and in its deployed configurations. 1n this
regard, constructing the cover 18 from a stretchable material, such as the
expanded PTFE material previously described, incorporates "stored length" of
material into the device that allows the device to be readily bent without
kinking. Figure 5.shows a TIPS endoprosthesis 10 of the present inventipn
bent around a rod 66 having a diameter °'d." It is desirable that the
first
covered segment 14 of endoprosthesis 10 of the present invention be capable
of being bent around a 25 mm diameter rod, without kinking, and more
preferably around a 20 mm diameter rod without kinking.
As the term °stored length° is used to describe the cover
18, it is
intended to encompass the material comprising the cover either being elastic
in
nature andlor having suff cient excess material incorporated within the cover
to
allow an outer circumference 68 of the cover 18 to °stretch" without
excessively
buckling an inner circumference 70 of the cover 18. With the preferred cover
material of the present invention, expanded PTFE, stored length may be
achieved by compressing the node and fibril structure of the expanded PTFE in
the nom~at deployed configuration of the device - allowing the outer
circumference 68 to separate the node and fibril structure upon actuation
around a rod while the inner circumference 70 remains compressed or
undergoes further compression prior to buckling. Stored length can also be
achieved by employing elastic materials such as poPyurethane; silicone., ar
other biocompatable elastamers or porous materials, such as .porous PET.
CA 02473623 2004-07-30
WO 00/42947 - 20 PCT/US00/01531
Another suitable material is to construct the cover 18 from a stretchable
material, such as an appropriately proportioned stetchable expanded PTFE
disclosed in United States Patent 4,877,661 to House, et al.
To determine the presence of "stored Length" in a cover material of the
present Invention, a number of tests can be perforn~ed. First, the cover and
device can be tested together to determine if the cover material possesses a
recovery response whereby the cover will longitudinally elongate with the
device when tensioned and then recover to, or close to, its original length
upon
i 0 release of tension. This can be tested by determining the "norms! deployed
length" of covered first segment 14 and then applying moderate longitudinal
tension to the device to determine the maximum length the first segment will
assume while recovering to near 'its original normal deployed length upon
release of tension. To have "stored length" within the meaning of the present
invention, the maximum length of the covered segment shoutd be at least 5%
more than the normal deployed length of the covered segment. More
preferably; the maximum length of the covered segment should be 'l0, 15, or
2Q % or more of the normal deployed length of the covered segment.
With respect to a cover constructed from expanded PTFE material,
stored length can also-be determined by microscopic analysis of the node and
fibril structure of the material. As has been noted, an expanded PTFE material
can have stored length imparted to it by "scrunching" the material and then
heat treating it to heat set the fibrils into a bent configuration. An
expanded
PTFE material having bent fibrils is shown in the aEt~l of Figure 23. When
26 formed in this manner, the fibrils will straighten (thus lengthening the
cover)
when longitudinally tensioned and then returned to their bent configuration
when released from tension (alth.ough the fibrils will "creep" into a straight
configuration if tension is maintained over an extended period of time or the
cover is exposed to high heat in its tensioned con~fguration). For an expanded
PTFE material, the presence of."stored length" can be determined by
examining the material untensioned to determined if the fibril struct:Jre is
similar
to that shown in Figure 23. A test for this propert,~ is described in United
States
Patent 4,877,661 to House et aI_ Again, the maximum length of the covered
segment with an expanded PTFE cover material should be at least 5 % mare
36 than the normal deployed length of the
CA 02473623 2004-07-30
WO 00/42947 2~ PCT/USOOIU1531
covered segment, and more preferably 10, 15, or 20'% or more of the normal
deployed Length of the covered segment.
The term abent around a rod without kinking" is intended to encompass
the endoprosthesis 10 being actuated to place its ends at a 90°. angle
with
respect to each other, as is shown in Figure 5, with no significant folding or
buckling forming in the cover 18 at the point of bending around the rod 6fi.
"Significant folding or buckling". is present.when if there is a
circumferentially
directed crease along the inner circumference 70 that separates the creased
portion of the endoprosthesis more than 1 mm from iihe rod. A Similar test is
described in detail in Section 8.9 of ANSI/AAMI VI? 20-1994.
The compacting of the endoprosthesis 70 of the present invention may
be accomplished through a variety of methods, as is illustrated in Figures 6a
through 6d. Figure 6a is an end view of a TIPS andoprosthesis of the present
invention at its deployed diameter 72. Each of Figures 6b, 6c, and 6d
illustrates how the endoprosthesis 10 may be compacted around a catheter 74
to achieve a compacted diameter 76. Figure 6b is an end view of the TIPS
endoprosthesis of Figure 8a, with the endoprosthesis 10 shown compacted by
folding and rolling the endoprosthesis 10. Figure fir is an end view of the
TIPS
endoprosthesis of Figure 6a, with the endoprosthests 10 shown compacted by
radialiy compressing the endoprosthesis 10. Figure 6d is an end view of the
TIPS endoprosthesis of Figure 6a, with the endoprosthesis 7 0 shown
compacted by folding into pleats 78:
While each of these compacting techniques !~rorks well for many
applications, it has been found that the covered first segment 14 of the stmt
element '!2 of the present invention can catch on itself or the constraining
sleeve 34 during deployment if the covered stent segment is not carefully
handled. irt this regard, as is exptained in greater detail below, it is
believed
that compacting the TIPS endoprosthesis 10 of the present invention with
multiple folds in the form of pleats 78, as shown in Figure 6d, provides the
best
results.
Figure 7 is a side elevation view of an undulated stent 80, shown in a
deployed diameter 82 and in a compacted, pleated diameter 84: As can be
seen, in the deployed diameter 80, the undulated stent pattern has exposed
CA 02473623 2004-07-30
W~ 00142947 2~ PCT/US00101537
rearward facing apices 8fi and forward facing apices 88. V1lith apices facing
in
both forward and rearward directions, the inventors have found that there is a
likelihood that the apices will catch and tangle a frbrous constraining
sleeve.
However, if the stent is compacted by pleating, one direction of apices can be
8 formed within pleat 78 so that it is protected within the folded device
below the
level of the outer circumferential surface of the constrained device. This
construction vastly reduces the risk of entanglement. In the compacted stent
84, it can be seen that only the forward facing apices 88 remain exposed on
the
outer circumference of the compacted device, white all of the rearward facing
apices 86 are tucked within pleats 78 of the compacted device.
it should be noted that the pleated folding methods described herein
can be used to direct apices into a wide variety of folded patterns. As such,
the
terms "forward" and "rearward" facing apices are used only for convenience to
describe sets of apices that face in one direction or an opposite direction,
without regard to the actual direction the device may ultimately be deployed.
It has been found that the endoprosthesis of the present invention can
be compacted to its maximum degree, with .a high compaction efficiency, and
with minimal risk of apex entanglement if a pleated folding method is
employed.
One method of pleating ari endoprosthesis is shown in Figures 8a; 8b, and 9.
Figures 8a and Sb illustrated a fluted tapered die 92 suitable for use in
pleating a 5tent or stmt-graft device. The.tapered die 92 has a large opening
94 at one end and a small opening 96 at its opposite end. A number of raised
flutes 98 are provided within the tapered die separated by grooves 100, each
raised flute 98 corresponding to one desired pleat ~to be farmed in ttie
28 endoprosthesis. The raised flutes 98 and/or the grooves 100 may be formed
by molding or machining the shapes into the die 92. Alternatively, as is
illustrated, the flutes 98 may be formed by attaching one or more evenly
spaced bands 105 per filute around the tapered die 92, such as by using
polyester or nylon thread.
Compacting and pleating may be accomplished by attaching a series of
tether lines 104 to an endoprosthesis 7 0, as shown in Figure 9. The tether
lines 104 may be formed from thin wires, polymer inbers, or other suitable
materials. As is shown, the tether lines 104 are attached along the apices
that
are intended to remain exposed (in this case, forward facing apices 88). The
38 tether lines 104 are centrally joined together at point t 08.
CA 02473623 2004-07-30
WO 00142947 PCT/USOOI01531
23 _
To compact the device, the joined tether lines 104 are then drawn
through the tapered die 92, exiting at small opening 96. Each of the tether
lines
104 is drawn through a groove 100. In this manner, the tethered apices 106
will be folded outwardly and each of the untethered apices will be folded
inwardly by the raised flutes 98 as the device is radialfy crushed. Once
exiting
the small opening 96, the compacted device can then be captured within a
constraining sleeve or capture tube (not shown) for subsequent packaging.
The constraining sleeve or capture tube may also contain means, such as
fongitudina! slots or grooves, to assist in maintaining the pleated compacted
configuration of the endoprosthesis. Alternatively, it is also possible to
pleat the
device using a smooth tapered die with folding ridges (for example,
longitudinally applied polyester lines} attached to tine exterior of the
endoprosthesis prior to compression. The folding ridges may then be removed
following compression.
Packaging of the device of the present invention can be accomplished
through a variety of methods. As should be appreciated from the following
description of the preferred deployment method of the device of the present
invention, it is preferred that .the endoprosthe5is 10 of the present
invention be
packaged to permit multi-stage deployment. One such method to accomplish
this packaging is illustrated in Figures 10 and 11. Figure 10 shows a device
of
the present invention as it is delivered for use. The endoprosthesis (not
shown}
is mounted on a delivery catheter 110 between two ~ofives" 112, 114. Olives
are solid bumps of increased diameter on the catheter sf~aft (for instance,
formed from a polymer, glass, or metal material) that inhibit the compacted
26 endoprosthesis from sliding on the catheter shaft. Olive 114 at the distal
shaft
36 may contain a radiopaque marker 64 to aid in ~iasitioning of the device. An
introducing sleeve 32 is attached over the endoprosthesis. The introducing
sleeve 32 is preferably made of polyethylene or similar material with its
inner
diameter being approximately equal to the outer dimensions of the compacted
endoprosthesis. One or more markers or mechanical stops may be provided
on the introducing sleeve 32 to aid in initial positioning and deploying. A
deployment line 46 can be seen exiting the introducing sleeve 32 and passing
into the delivery catheter 110 (as was previously explained in reference to
Figure 2, the deployment line 46 exits the delivery catheter at the deployment
port 42).
CA 02473623 2004-07-30
WO 00142947 PCTIUS0010153I
24 _
When the intrflducing sleeve 32 is removed, as is shown in Figure 11,
the second segment 16 of the endoprosthesis 10 will expand to close to its
fully
deployed diameter. The remainder of the endoprosthesis 10, however, is
contained within a constraining sleeve 34. In operation, the constrained
device
will pass from the introducing sleeve 32 into a cathei:er tube (of
approximately
equal inner diameter) extending past the ultimate deployment site. Deployment
of the second segment 1 G will occur when the devicE: is separated from the
catheter tube at the deployment site.
The preferred constraining sleeve comprises a plurality of interwoven
threads, such as that disclosed in United States patent No. 6,224,627 to
Armstrong et al. The constraining sleeve 34 can be separately deployed by
actuating deployment line 46.
While the mufti-stage deployment apparatus illustrated in Figures 10
and 11 comprises the preferred embodiment of deployment of the present
invention, it should be appreciated that.different embodiments may achieve
similar results. for instance, the second segment 18 may be contaiiled in its
own constraining apparatus, such as the fibers used in constraining sleeve 34
or other constraining devices or other materials. Conversely, the constraining
sleeve 34 may be formed from a tubular material (dice the. introducing sleeve
32). Additionally, it may be benefccial under certain circumstances to provide
more than a two-stage deployment, such as be providing multiple constraining
sleeves that permit multiple stage deployment of the: first segment 1.4 of the
endoprosthesis 10.
The operation of the endoprosthesis and deployment apparatus of the
present invention carp be appreciated through the fc~ilowing description
illustrated by Figures 12 through 18.
Figure 12 illustrates a compacted endoprosthesis of the present
invention, mounted within introducing sleeve 32, being inserted through a
hemostatic valve 116 into catheter tube 117. The catheter tube 117 has
previously been advanced into the portal vein. A guide wire 118 may be
provided to assist in device placement.
As is shown in Figure 13, the endoprosthesis l0,is advanced out of the
3b introducing sleeve 32 and into catheter tube 117. The delivery catheter is
then
CA 02473623 2004-07-30
WO OU14Z947 PCT/USOU101531
advanced to cause the second segment 16 to become constrained within
catheter tube 117.
As is illustrated in Figures 14 and 15, the endoprosthesis 10 is then
advanced through the catheter tube 117 through the inferior vena cava 119a,
5 the hepatic vein 119b, the intrahepatic tract (shunt) 120 farmed in the
giver
122, and well into the portal vein 124. Radiopaque tip 54 can be aligned with
the end of the catheter tube 117. The radiapaque rnarker 28 can be positioned
distal to the intrahepatic juncture site 126.
In Figure 16 the catheter tube 117 is withdrawn proximally, which
10 permits the second segment 16 to fully expand within the portal vein 124.
As is
shown in Figure 17, the delivery catheter 110 is then withdrawn through the
catheter tube 117 until the unconstrained second segment 16 engages the
intrahepatic juncture 126. Alignment can be confirrned fiuoroscopically by
correct orientation of marker band 28 with or without confirmatpry injections
of
15 radiopaque contrast media.
As is illustrated in Figure 18, once the device is properly aligned, the
constraining sleeve 34 is removed by actuating deployment line 46, allowing
the first segment 14 of the endoprosthesis 10 to fullfy enlarge in place in a
tip-
to-hub direction. If desired; further touch-up of the endoprosthesis 10 can be
20 performed by subsequent balloon dilation of the endoprosthesis 10.
It should be appreciated that the above deployment procedure aligns
the covered portion of the eridoprosthesis within the intrahepatic tract
(shunt)
120. Further, the uncovered second segment 16 permits blood flow both to
enter the shunt 120 and to continue through the portal vein 124. The result is
25 that excess pressure can be relieved from the Aorta( system (through the
shunt) without completely eliminating na.rmal blood flow through portal vein
124.
Without intending to limit the present invention to the specifics
described hereinafter, the following exa~mpfes illustrate how the present
invention may be made.
Example 1
A pin jig is manufactured by turning 304 stainless steel rad stock to an 8
mm diameter. Holes are then drilled in the mandrel (#67 bit) to create the
pattern to wind the stem depicted in Figure 3, with 5 forward facing apices
(10
CA 02473623 2004-07-30
WO 00142947 ~ ' 26~ 1PCT/US00/0153I
holes) per revolution. Beyond each end of the hole pattern on the mandrel,
holes are driNed and tapped to fit 10-32 screws. 1 /32 inch (0:79 mrri)
diameter
x 4 mm length steel pins .are then place into these holes, such that 2 mm of
the
pins are imbedded in the mandrel.
Nitinoi wire of a 0.010 (0.26 mm) nominal diameter (acquired from
Nitinol Devices and Components, Frerriont, CA) is then wound by hand around
this pin jig in the pattern shoyiin in Figure 3 and the ends of the wire
affixed in a
tight configuration to the mandrel by 10-32 screws. The wire and the mandrel
is then set in a convection .oven set a 450°C for 15 minutes, followed
by a rapid
quench in ambient temperature water. After the -mandrel is allowed to cool,
the
wire stent is removed from the mandrel by removing the pins and sliding the
stmt from the mandrel.
Next, as a manufacturing aid 'to assist in alignment of apicfa relative to
each other, a small diameter expanded PTFE filament (for example, CV-8.0
16 suture available from W. L. Gore ~ Associates, Inc., Flagstaff, AZ, USA) is
tied
to the end of the wire on the proximal end of the device 30. The filament is
spirally wound toward the distal end of the device 24. The filament is wound
through apices of adjacent. rows of the stent structure. A simihr winding
alignment procedure is described in U.S. Patent Nos. 6,042,605; 6,361,637 and
6,520,986 to Martin et al. The filament is wound along the length of the first
segment 14, where it may then be fixed to the terminal end of the stmt wire
67.
This winding procedure may be started at either end 24 or 30 of the stmt with
similar results.
Next, a thin-walled expanded PTFE tubular extrusion (wall thickness
2S 0.004 inch (0.1 mm)), as previously described is then circumferentially
wrapped
with five to seven layers of an expanded PTFE film. The expanded PTFE film
has a thickness ofØ0007" (0.02 mm), a density of 0.7 glcc, and a methanol
bubble point of 3 psi ( 21 KPa).
This construction is heat treated for 1.~ minutes at 370°C and
cooled.
The wrapped tube is then removed from the mandrel
Another thin-walled expanded PTFE tube is placed into the lumen of the
wrapped tube. These two tubes are then fitted over an 8 mm mandrel. The
wrapped expanded PTFE tube is then over-wrapped with another layer of the
expanded PTFE film.
38 The construction is then compressed longitudinally by hand to
approximately a third of its original length. The construction and mandrel are
CA 02473623 2004-07-30
WO OOf42947 27 PCTNSUO/Q1531
then heat treated at 320°C for 10 minutes, removed from the oven, and
allowed
to .cool. The outer over-wrap is removed. The construction is then removed
from the mandrel and the graft placed under moderate tension to restore the
graft to approximately 2J3rds of its original length.
After trimming one end of the construction squarely, the stent is slid on
the outside of the PTFE construction and the end of the interlocked section of
the stem (see Figure 1 ) aligned v~ith the squared. end such that one row of .
interlocked apices is covered. The 8 mm mandrel is then inserted into the
PTFElnitinoi construction. The stent apices are then manually adjusted to
~ achieve even spacing of the apices, with the forward facing apices from each
row aligned longitudinally. A PTFEIFEP film, as previously described, is then
over-wrapped with the FEP side down on.the exterior surFace of the section of
the stent with non-interlocked windings. This PTFEIFEP film comprises an
expanded layer of PTFE film with a discontinuous layer of FEP. The f(m has a
thickness of 0.0005 inch (0.01 mm) and a methanol bubble point of 1.2 psi (8
KPa). This wrap is a continuous helical circumferential wrap with overlapping
edges to produce approximately 3-5 layers.
After completing the wrap, the wrap is spot attached using a soldering
iron with a tip temperature of sufficient teat to melt tthe FEP. A sacrificial
compression wrap is then applied by heficaffy wrapping with moderate tension
an expanded PTFE flm circumferentialfy around the exterior of the
construction. The construction i5 then heat treated at 320°C for five
minutes
and allowed to air cool. After the construction has cooled, tie over-wrap is
removed. The construction is again longitudinally compressed approximately
50 % of its original length, making sure that the apices remained aligned. The
construction is over-wrapped with a thin expanded PTFE film that serves as a
compression layer and teat treated at 320°C for 10 minutes. After a
short air
cool, while the tube is still warm, moderate longitudinal tension is applied
to the
device by hand to pull the entire device out to approximately its original
length.
The excess expanded PTFE tube is then trimmed filush with tie stent end.
Threads are attached to the covered end of the device by piercing
through the cover material at each end apex of the device with a needle.
Thread is fed through these holes to form loops, and about a 30.cm from the
device they are tied together wilt even slack. The knotted end of the threads
is
fed sequentially though a PTFE tapered die into a PTFE capture tube. The
tapered die is constructed with an approximate 15° included angle taper
and
CA 02473623 2004-07-30
WO 00/42947 ' 2$ ' PCT/USbb/UI~3I
had five nylon fines (outer diameter = 0.018" (0,46 mm)) positioned
approximately equal distance (i.e., 72°} around the inner circumference
of the
taper, as is shown in Figure 8a. The devices and tapered die are then sprayed
with a refrigerant (MicroFreeze, MicroCare Corporation, Bristol CT) to reduce
the temperature of the device below the nitinol's martinsitic temperature. The
device is immediately drawn through the tapered die into a PTFE capture tube.
As the endoprosthesis is drawn through the die, a 0.066 inch (1.4 mm) outer
diameter polyethylene shaft is placed within its lumen. The resulting device
can then be removed from the capture tube.
The deployed device is flexible in both the covered and uncovered
portions. The uncovered portion can be compressed longitudinally, and the
stunt elastically recovers dose to its original length when the digital
pressure is
removed. The permeability of the covered portion of the construction is low
and the hoop strength of the device is substantial. The device. is compressed
1 b ~ such that the apices pointing away from the attached threads are tucked
under
the apices pointed toward the thread. This compression technique results in a
small collapsed profile.
Tests performed on this example include:
Kink Diameter:
This device is bent at room temperature around progressively smaller
diametrical mandrels until a kink formed in the covered graft section. The
smallest diameter at v~rhich that device did not kink was recorded. A "kink"
is
present when if there is a crease along the inner circumference 70 that
separates the creased portion of the endoprosthesis more than 1 mm from the
rod. A similar test is described in detail in Section 8.9 of ANSIIAAMI VP 20-
1994, although the current test is performed without internal pressure.
Transmurat Permeability:
To quantify a level of °bile resistance," the inventors used a
measure of
air resistance described by Tappi Standard T 460 om-88 in order to establish a
"Gurfey Number." The measurement was performed using a t3URLEY~""'
Densometer Mode! 4110 permeability tester available from Teledyne (Troy, NY,
tJSA). Briefly, the tester measures the time that is takes to pass 100 cubic
CA 02473623 2004-07-30
WO 00/42947 , ~~ PCTNSOOI03531
centimeters of ambient temperature air through an orifice with a pressure of
4:9
inches of H20 (1.2 kPa).
The fixture apparatus 130 used to test for the "Gurley Number" is shown
in Figure 19. A frst end 132 of a flexible tube 134 is attached to an adapter
13fi. The adapter .136 is attached to the Densometer via a 25.4 mm diameter
circular plate (not shown) that attaches to the adapter 13fi via orifice 138.
Sealing material 140 (e.g., a 1/8 inch (0.32 mm) thick silicone sheet) is
mounted around the orifice 138 to effectuate a seal with the circular plate.
This
assures that the air measured by the Densometer passes through orifice 138.
A second end 142 of the flexible tube 134, which is in direct communication
with orifice 138, is attached to a circular fitting 144. The circular fitting
144 has
an outer diameter 14fi approximately the same as the inner diameter of the
device 10 to be tested. The circular fitting 144 is attached to the device 9
0, as
is shown in Figure 20, using PTFE pipe thread tape to create an air tight
fitting
around the circumference. A sealing plug fitting 148 is provided to attached
to
the opposite end of the device 10 to completely seal the second segment 16
and seat a portion of the first segment 14, as these elements are illustrated
in
Figure 1.
The resistance to flow through each of the frttings with no sample
attached is measured to 1.5 seconds or less per 100 cc of air, which is
significantly less time than all of the samples measured. Prior to taking a
measurement, the test area is calculated by multiplying the separation of the
fittings to which the sample was attached to the inner circumference of the
sample. After measuring the time for 100 cubic centimeters of air to pass
through the sample, this value is normalized by multiplying by the total area
tested. The test areas generally range from 4 to 1 fi cm2, and multiple test
areas are tested for each sample.
Radial Compression Testing
Compression testing is performed using an lNSTRON tensile testing
machine equipped with a heated test chamber set to 37°C. As is shown in
Figures 21 and 22, a loop compression fixture 150 is fabricated using 0.1 mm
thick polyester film. The film is cut into a 20 mm x 1fi0 mm rectangle. On one
end of the rectangle, 5 mm is trimmed from each side from the end for 100
mm, creating an elongated tab 152. Approximately 5 mm past the trimmings in
CA 02473623 2004-07-30
j
WO OOI42947 PCT/US00103531
30 _
the wide section, a slit 154 (approximately 11 mm x 1 mm) is cut in the film
to fit
the tab 152. The 1-cm wide tab 152 is then looped through the slit 154 to form
a variable diameter loop 156. After forming the loop 156, ends 158, 160 of the
film are placed in opposing tensile testing grips of the tensile tester.
Devices to be tested are heated to 37°C for greater than 30
minutes
immediately prior to the testing. inside the loop 156 is placed a device 1fi2
to
be tested and the opposing jaws separated until a pre-load of x.05 kg on the
variable diameter loop is detected. The device is then compressed at a rate of
12.7 mm device circumferencelmin to 75% of its original diameter while the
force is recorded. After completing the test, the slope of the load vs.
displacement curve is calculated. This calculation is performed by recording
the circumferential displacement and corresponding force at 5% and 20%
diametric compression (i.e., the outer diameter of the stmt is 95% and 80% of
its original diameter loaded with a 0.05 laad, respectively). Slope is then
calculated by dividing the difference in these two forces by the differences
in
the corresponding displacements. The result, the slope from 5% to 20%
diametrical compression of the force verses circumferentiat displacement, is
then multiplied by pi (II) to obtain the slope of the load vs. diameter curve,
expressed in kglmm diameter.
Profile:
Profile is equal to the inner diameter of the PTFE capture tube into
which the device is drawn. Within the lumen of the device constrained in the
PTFE capture tube is a 0.055 inch (1.4 mm) outer diameter polyethylene tube
to represent the catheter that the device is loaded.
Scanning Electron Microscope Photomicrographs! Bent fibrils:
Scanning electron microscope photomicrographs (SEMs) are taken of
the iuminal surface. Bent fibrils are measured in a manner consistent with
that
method taught in United States Patent 5,308,6fi4 to House et al. Briefly,
horizontal Lines are drawn on the SEM photomicrographs (3 lines per photo, 3
photos per sample) (total of 9 measurements taken). Horizontal distances (H)
are measured between nodes along this line. Vertical distances (V) of the bent
fibrils are measured from the horizontal fine. A ratio (VlH) is calculated and
CA 02473623 2004-07-30
.~ W
WO 00!42947 31 PCT/US00101531
recorded. An SEM of a cover of the present invention showing bent fibrils
therein is attached as Figure 23.
Internal Corrugations:
8 The internal corrugations of the graft are measured using gauge pins.
Gauge pin testing is accomplished by first testing the internal diameter with
pins until the fit between 'the pin and internal diameter allows the pin to be
Jilted
when suspending only the device. Next, the device is longitudinally
compressed approximately 10% (to simulate bending) and the pin test is
repeated. The percent of the compressed inner diameter to the non-
compressed inner diameter is then calculated.
8mm Device
Testing is perform as described above, with the following results:
Kink Diameter 24 mm
Radial Stiffness Covered section - 0.137 k Imm
g diameter
Uncovered section - 0.193 kg/mmdm",eter
Permeability 213 sec per 104 cc air per
1 cm2",a,e~,~~
Profile 3.2 mm
Bent Fibrils Average ratio 1//H = 0.131
Internal Corrugation 9fi% of original
Example 2
A device similar to the one in described in Example 1 is made, however,
the inner diameter is 10 mm, the stent pattern has 6 apices per' revolution,
the
wire diameter is 0.019 inches (0_28 mm), and the tapered die has 6 nylon lines
(spacing = 60°). Testing is perform as described above, with the
following
2~ results:
10mm Device
CA 02473623 2004-07-30
W000142947 ' 32' PCTIUS00I01531
Kink Diameter 20 mm
Radial Stiffness Covered section - 0.173 k Imm
g diameter
Uncovered section - 0.258 kglmmd;~,f,eer
Permeability 143 sec per 100 cc air per
l cm2",~ena,
Profile 3.6 mm
Bent Fibrils Average ratio VIH = 0.176
Internal Corrugation 99% of original
Example 3
A device similar to the one in Example 1 is made; however, the inner
diameter is 12 mm, the stent pattern has 6 apices per revolution, the wire
diameter is 0.010 inches (0.25 mm), and the tapered die teas 6 nylon lines
(spacing = 60°). Testing is performed as described above, with the
following
results:
l2mm Device
Kink Diameter 24 mm
Radial Stiffness Covered section - 0.103 k lmm
g diameter
Uncovered section - 0.157 kg/mmd;~",e,e~
Permeability 111 sec per 100 cc air per 1
Cm2~e~al
Profile ~ 3.6 mm
Bent Fibrils Average ratio VIH = 0.234
internal Corrugation98% of original
While particular embodiments of the present invention have been
illustrated and described herein, the present invention should not be limited
to
such illustrations and descriptions. It should be apparent that changes and
modifications may be incorporated and embodied as part of the present
invention within the scope of the following claims.