Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
DESCRIPTION
Condensed Heterocyclic Compounds
Technical Field
This invention relates to novel condensed heterocyclic compounds having
pharmacological activity, to a process for their production and to a
pharmaceutical
composition containing the same.
Background Art
Poly (adenosine 5'-diphospho-ribose) polymerise ["poly (ADP-ribose)
polymerise" or "PARP", which is also sometimes called "PARS" for "poly (ADP-
ribose)
synthetase"] is an enzyme located in the nuclei of cells of various organs,
including muscle,
heart and brain cells. PARP plays a physiological role in the repair of strand
breaks in
DNA. Once activated by damaged DNA fragments, PARP catalyzes the attachment of
up
to 100 ADP-ribose units to a variety of nuclear proteins, including histones
and PARP
itself.
Some condensed heterocyclic compound having inhibitory activity of PARP have
been known, for example, in WO95/24379, W098/33802 and W099/11624.
Disclosure of the Invention
This invention relates to novel condensed heterocyclic compound, which have
pharmaceutical activity such as PARP inhibiting activity, to a process for
their production,
to a pharmaceutical composition containing the same and to a use thereof.
One object of this invention is to provide the novel condensed heterocyclic
compound, which have a PARP inhibiting activity.
Another object of this invention is to provide a process for production of the
condensed heterocyclic compound.
A further object of this invention is to provide a pharmaceutical composition
containing the condensed heterocyclic compound as an active ingredient.
Still further object of this invention is to provide a use of the condensed
heterocyclic compound for manufacturing a medicament for treating or
preventing various
diseases, or a method of treating or preventing various diseases by
administering the
condensed heterocyclic compound in an effective amount to inhibit PARP
activity.
Thus, the present invention provides the following.
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
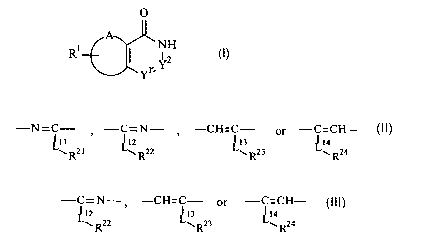
[1] A compound of the formula (I):
O
A~
R1 I 'NH (I)
Yr Y
wherein
Rl is hydrogen, halogen, lower alkyl or lower alkoxy,
A and two adjacent carbon atoms of the six membered ring to be bonded with A
form benzene ring, pyridine ring, or five to seven membered partially
saturated ring optionally containing one or more heteroatom(s) selected from
the group consisting of nitrogen atom, oxygen atom, and sulfur atom,
-Yl=Y2- is -N=C- , -C=N- , -CH=C- or -C=CH-
~11 ~12 ~13 ~14
w R21 w R22 y R23 w R24
[wherein Lll, L12, L13 and L14 is
( 1 ) lower alkylene,
(2) lower alkenylene,
(3) cyclo(lower)alkylene,
(4) cyclo(lower)alkenylene,
(5) diradical of saturated- or unsaturated monocyclic group with one
or more nitrogen atom(s), which is obtained after removal of one
hydrogen atom from said monocyclic group, or
(6) N(R3)-L- (wherein R3 is hydrogen or lower alkyl, and L is
lower alkylene or lower alkenylene), and
R21, R22, R23 and R24 1S
(1) cyclic amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl,
(2) carbocyclic group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
2
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
substituted with lower alkyl, or
(3) amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, nitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl.],
provided that
when A and two adjacent carbon atoms of the six membered ring to be bonded
with A form benzene ring,
then -Yl=Y2- is -C=N-, -CH=C- or -C=CH-
~12 ~13 ~14
wR22 ~R23 wR24
or its prodrug, or their salts.
[2] The compound according to [1], wherein
A~ /
w /\ w
is , , C~ , ,
\N ~
1
x2~
or ~ ,
[wherein X1 and X 2 is N, O or S].
[3] The compound according to [2], wherein
Rl is hydrogen, and
R21, R22, R23 arid R24 is tetrahydropyridyl, piperidyl or piperazinyl, each of
which is
substituted with aryl optionally substituted with halogen.
[4] The compound according to any one of [1], [2] and [3], wherein
L is lower alkylene.
[5] The compound according to any one of [1], [2], [3] and [4], wherein
_Yl-Y2_ is -N=C-
~11
W R21
[6] A pharmaceutically composition comprising a compound of the formula (I):
O
A~
R1 I ~ NH ( I )
Y~:Y
3
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
wherein
R' is halogen, lower alkyl or lower alkoxy,
A and two adj acent carbon atoms of the six membered ring to be bonded with A
form benzene ring, pyridine ring, or five to seven membered partially
saturated ring optionally containing one or more heteroatorn(s) selected from
the group consisting of nitrogen atom, oxygen atom, and sulfur atom,
-yl=ya- is -N=C- ~ -C-N- ~ -CH=C- or -C=CH-
~11 ~12 ~ 13 ~14
wRal ~R22 LwRa3 wR2a
[wherein L11, L12, L13 and L14 is
(1) lower alkylene,
(2) lower alkenylene,
(3) cyclo(lower)alkylene,
l5 (4) cyclo(lower)alkenylene,
(5) diradical of saturated- or unsaturated monocyclic group with one
or more nitrogen atom(s), which is obtained after removal of one
hydrogen atom from said monocyclic group, or
(6) N(R3)-L- (wherein R3 is hydrogen or lower alkyl, and L is
lower alkylene or lower alkenylene), and
Ral, R22, Ra3 and Ra4 is
(1) cyclic amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl,
(2) carbocyclic group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl, or
(3) amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl.],
4
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
provided that
when A and two adjacent carbon atoms of the six membered ring to be bonded
with A form benzene ring,
then -Y1=Y2- is -C=N-, -CH=C- or -C=CH-
~12 ~13 ~14
y R22 y R23 W R24
or its prodrug, or their pharmaceutically acceptable salts, and a
pharmaceutically
acceptable carrier, wherein said compound is present in an amount effective
for
inhibiting PARP activity.
[7] The pharmaceutical composition of [6] for treating or preventing diseases
ascribed by
NMDA- and NO-induced toxicity.
[8] The pharmaceutical composition of [6] for extending the lifespan or
proliferative
capacity of cells or altering gene expression of senescent cells
[9] The pharmaceutical composition of [6] for treating or preventing tissue
damage
resulting from cell damage or death due to necrosis or apoptosis; neural
tissue damage
resulting from ischemia and reperfusion injury, neurological disorders and
neurodegenerative diseases; neurodegenerative diseases; head trauma; stroke;
Alzheimer's disease; Parkinson's disease; epilepsy; Amyotrophic Lateral
Scleosis
(ALS); Huntington's disease; schizophrenia; chronic pain; ischemia and moss
following hypoxia; hypoglycemia; ischemia; trauma; nervous insult; previously
ischemic heart or skeleton muscle tissue; radiosensitizing hypoxic tumor
cells; tumor
cells from recovering from potentially lethal damage of DNA after radiation
therapy;
skin aging; arteriosclerosis; osteoarthritis; osteoporosis; muscular
dystrophy;
degenerative diseases of skeletal muscle involving replicative senescence; age-
related
macular degeneration; immune senescence; AIDS; other immune senescence
diseases; inflammatory bowel disorders (e.g., colitis); arthritis; diabetes;
endotoxic
shock; septic shock; or tumor.
[ 10] A method of inhibiting PARP activity comprising administering a compound
of the
formula:
O
A~
R1 I 'NH (I)
Yr Y
wherein
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Rl is hydrogen, halogen, lower alkyl or lower alkoxy,
A and two adjacent carbon atoms of the six membered ring to be bonded with A
form benzene ring, pyridine ring, or five to seven membered partially
saturated ring optionally containing one or more heteroatom(s) selected from
the group consisting of nitrogen atom, oxygen atom, and sulfur atom,
_yl-y2_ is -N-C- ~ -C=N- ~ -CH-C- or -C=CH-
~11 ~12 ~13 L14
w R21 w R22 y R23 w R24
[wherein Lll, L12, L13 and L14 is
(1) lower alkylene,
(2) lower alkenylene,
(3) cyclo(lower)alkylene,
(4) cyclo(lower)alkenylene,
(5) diradical of saturated- or unsaturated monocyclic group with one
or more nitrogen atom(s), which is obtained after removal of one
hydrogen atom from said monocyclic group, or
(6) N(R3)-L- (wherein R3 is hydrogen or lower alkyl, and L is
lower alkylene or lower alkenylene), and
R21' R22' R23 and R24 15
(1) cyclic amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl,
(2) carbocyclic group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl, or
(3) amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl.],
provided that
when A and two adjacent carbon atoms of the six membered ring to be bonded
6
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
with A form benzene ring,
then -YI=Yz- is -C=N-, -CH=C- or -C=CH
L~z Li3
w Rzz W R23 W Rz4
or its prodrug, or their salts.
[11] A use of a compound of the formula (I):
O
A~
Rl I 'NH (I)
Yl: Y
wherein
Rl is hydrogen, halogen, lower alkyl or lower alkoxy,
A and two adj acent carbon atoms of the six membered ring to be bonded with A
form benzene ring, pyridine ring, or five to seven membered partially
saturated ring optionally containing one or more heteroatom(s) selected from
the group consisting of nitrogen atom, oxygen atom, and sulfur atom,
-Yl=Yz- is -N=C- -C=N- -CH=C- or -C=CH-
> > >
l5 Ln Liz y3 Lea
W R21 W Rz2 W Rz3 W R24
[wherein L11, Liz, L13 and L14 is
( 1 ) lower alkylene,
(2) lower alkenylene,
(3) cyclo(lower)alkylene,
(4) cyclo(lower)alkenylene,
(5) diradical of saturated- or unsaturated monocyclic group with one
or more nitrogen atom(s), which is obtained after removal of one
hydrogen atom from said monocyclic group, or
(6) N(R3)-L- (wherein R3 is hydrogen or lower alkyl, and L is
lower alkylene or lower alkenylene), and
Rzl, Rzz, Rzs and Rz4 is
(1) cyclic amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, nitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)allcoxy and phenyl, and which is optionally
substituted with lower alkyl,
7
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
(2) carbocyclic group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, nitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl, or
(3) amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, nitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
ZO substituted with lower alkyl.],
provided that
when A and two adjacent carbon atoms of the six membered ring to be bonded
with A form benzene ring,
then -Y'=Yl- is -C=N-, -CH=C- or -C=CH-
1~J ~~R22 "WR23 ~~R24
or its prodrug, or their pharmaceutically acceptable salts, for manufacturing
a
medicament for inhibiting PARP activity.
The condensed heterocyclic compound of this invention can be represented by
the
20 following formula (I):
O
A~
R1 I 'NH ( I )
yl=y
wherein
Rl is hydrogen, halogen, lower alkyl or lower alkoxy,
25 A and two adjacent carbon atoms of the six membered ring to be bonded with
A
form benzene ring, pyridine ring, or five to seven membered partially
saturated ring optionally containing one or more heteroatom(s) selected from
the group consisting of nitrogen atom, oxygen atom, and sulfur atom,
_yl=y2_ is -N=C- ~ -C-N- ~ -CH=C- or -C=CH
~11 X12 X13 X14
30 I'WR21 LyRz2 L°wR23 I'wR24
[wherein Lll, L12, L13 and L14 is
8
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
( 1 ) lower alkylene,
(2) lower allcenylene,
(3) cyclo(lower)alkylene,
(4) cyclo(lower)alkenylene,
(5) diradical of saturated- or unsaturated monocyclic
group with one
or more nitrogen
atom(s), which
is obtained after
removal of one
hydrogen atom
from said monocyclic
group, or
(6) N(R3)-L- (wherein R3 is hydrogen or lower alkyl,
and L is
lower alkylene
or lower alkenylene),
and
R21, R2a, Ra3 and R24 is
(1) cyclic amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl,
(2) carbocyclic group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl, or
(3) amino group, which is substituted with phenyl optionally
substituted with one or more suitable substituent(s) selected from the
group consisting of halogen, vitro, lower alkoxy, lower alkyl,
halo(lower)alkyl, halo(lower)alkoxy and phenyl, and which is optionally
substituted with lower alkyl.),
provided that
when A and two adjacent carbon atoms of the six membered ring to be bonded
with A form benzene ring,
then -YI=Ya- is -C=N-, -CH=C- or -C=CH-
yR2z yRa3 "WR24
or its prodrug, or their salts.
The compound (I) or its prodrug, or their salt can be prepared by the
following
processes. In the following formulae, compounds may be prodrugs or their
salts.
9
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Process 1
O
R1 A~ COOR3 NH RI '~'\
~NH
-I- ~ 2~ I
O HaN Lu.R I N%LmRai
(II) (III) (I-a)
or its salt or its salt or its salt
[wherein, R', R21 and A are each as defined above, and R3 is lower alkyl.]
In this process, the compound (I-a) or its salts can be produced by reacting
the
compound (II) or its salt and compound (III) in the presence of base, such as
inorganic
bases, for example, an alkali metal [e.g., sodium or potassium], alkoxide,
hydroxide,
carbonate or bicarbonate thereof, or organic bases such as a trialkylamine
[e.g.,
trimethylamine or triethylamine] or the like.
The reaction is usually carried out in a conventional solvent such as an
alcohol
(e.g., methanol, ethanol or isopropyl alcohol), ether (e.g., tetrahydrofuran,
dioxane,
diethylether), amide (e.g., N, N-dimethylformamide, N, N-dimethylacetamide),
nitrite (e.g.,
acetonitrite), or any other organic solvent which does not adversely affect
the reaction.
The reaction may be usually carried out under cooling to heating since the
reaction
temperature is not critical.
Process 2
O
A~ CONOH2 A
1 Base 1 ~ NH
R ~ ~ as R ( ~ 2s
X3/ Lls'R x3i LIS~R
(IV) (I-b)
or its salts or its salts
[wherein, Rl and A are each as defined above, and X3 is CH or N, Lls has a
same meaning
of Lll or L13, and R25 has a same meaning of R21 or R23.]
In this process, the compound (I-b) can be produced by subjecting the compound
(IV) to cyclization reaction in the presence of base, such as inorganic bases,
for example,
an alkali metal [e.g., sodium or potassium], alkoxide, hydroxide, carbonate or
bicarbonate
thereof, or organic bases such as a triatkylamine [e.g., trimethylamine or
triethylamine] or
the like.
The reaction is usually carried out in a conventional solvent such as water,
an
alcohol (e.g., methanol, ethanol or isopropyl alcohol), ether (e.g.,
tetrahydrofuran, dioxane,
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
diethylether), amide (e.g., N, N-dimethylformamide, N, N-dimethylacetamide),
nitrite (e.g.,
acetonitrile), or any other organic solvent which does not adversely affect
the reaction.
The reaction may be usually carried out under cooling to heating since the
reaction
temperature is not critical.
Process 3
O R3 O
H-N'
NH ~R4 R1 A~ I NH R3
~4~Lis~Z (VI) ~4~Lis-NwR4
(V) or its salt (I-c)
or its salt or its salt
[wherein, Rl and A are each as defined above, and X4 is CH or N, Lls has a
same meaning
of Lll or L13, Z is halogen, and
R3
N~R4 is substituted cyclic amino groups or optionally substituted amino
group.]
In this process, the compound (I-c) or its salts can be produced by reacting
the
compound (IV) or its salt and compound (V) in the presence of base, such as
inorganic
bases, for example, an alkali metal [e.g., sodium or potassium], alkoxide,
hydroxide,
carbonate or bicarbonate thereof, or organic bases such as a trialkylamine
[e.g.,
trimethylamine or triethylamine] or the like.
The reaction is usually carried out in a conventional solvent such as an
alcohol
(e.g., methanol, ethanol or isopropyl alcohol), ether (e.g., tetrahydrofuran,
dioxane,
diethylether), amide (e.g., N, N-dimethylformamide, N, N-dimethylacetamide),
nitrite (e.g.,
acetonitrile), or any other organic solvent which does not adversely affect
the reaction.
The reaction may be usually carried out under cooling to heating since the
reaction
temperature is not critical.
11
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Process 4
O R3 O
R NH H N~R4 R H
X5 5
(VI) ~R3
or its salt m
~4
(V) (I-c) R
or its salt or its salt
[wherein, RI and A are each as defined above, and XS is CH or N, L16 has a
same meaning
of Lla or L14, Z is halogen, and
R3
N~R4 is substituted cyclic amino groups or optionally substituted amino
group.]
This reaction can be carried out in the same manner as Process 3.
l5
The compound of the present invention can be purified by any conventional
purification methods employed for purifying organic compounds, such as
recrystallization,
column chromatography, thin-layer chromatography, high-performance liquid
chromatography and the like. The compounds can be identified by conventional
methods
such as NMR spectrography, mass spectrography, IR spectrography, elemental
analysis,
and measurement of melting point.
Some of the starting compounds (II], (III), (IV) and (V) are novel and can be
prepared by the well-known processes or its analogous processes, for example,
the
processes described in the W02000142025 and the processes shown in
Preparations
mentioned below.
Suitable salts of the compounds of the present invention are pharmaceutically
acceptable conventional non-toxic salts and can be an organic acid addition
salt (e.g.
formate, acetate, trifluoroacetate, maleate, tartrate, oxalate,
methanesulfonate,
benzenesulfonate, toluenesulfonate, etc.), an inorganic acid addition salt
(e.g.
hydrochloride, hydrobromide, sulfate, phosphate, etc.), a salt with an amino
acid (e.g.
aspartic acid salt, glutamic acid salt, etc.), or the like.
The "prodrug" means the derivatives of compounds of the present invention
having a chemically or metabolically degradable group, which becomes
pharmaceutically
active after biotransformation.
12
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
The compounds of formula (I) may contain one or more asymmetric centers and
thus they can exist as enantiomers or diastereoisomers. Furthermore certain
compounds
of formula (I) which contain allcenyl groups may exist as cis- or trans-
isomers. In each
instance, the invention includes both mixtures and separate individual
isomers.
The compounds of the formula (I) may also exist in tautomeric forms and the
invention includes both mixtures and separate individual tautomers.
The compound of the formula (I) and its salt can be in a form of a solvate,
which
is included within the scope of the present invention. The solvate preferably
include a
hydrate and an ethanolate.
Also included in the scope of invention are radiolabelled derivatives of
compounds of formula (I) which are suitable for biological studies.
In the above and subsequent description of the present specification, suitable
examples and illustrations of the various definitions, which the present
invention includes
within the scope thereof, are explained in detail as follows.
The term "lower" means a group having 1 to 6 carbon atom(s), unless otherwise
provided.
Suitable "lower alkyl" includes a straight or branched alkyl having 1 to 6, in
particular 1 to 2, carbon atoms. Preferable examples which may be mentioned
are methyl,
ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl and hexyl.
Suitable "lower alkoxy" includes straight or branched alkoxy having 1 to 6, in
particular 1 to 2, carbon atoms. Preferable examples which may be mentioned
are
methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, iso-butoxy, sec-butoxy and
tert-butoxy,
preferably methoxy. Suitable "lower alkylamino" include mono (lower)
alkylamino and di
(lower) alkylamino. Preferable examples which may be mentioned are
methylamino,
dimethylamino, ethylamino, dimethylamino, n-propylamino, isopropylamino,
n-butylamino, iso-butylamirio, sec-butylamino and tert-butylamino, preferably
dimethylamino and diethylamino.
Suitable "lower alkylene" includes a straight or branched alkylene having 1 to
6,
in particular 3, carbon atoms. Preferable examples which may be mentioned are
methylene, ethylene, trimethylene, propylene, methyltrimethylene (1- or 2-
methyltrimethylene) and hexamethylene, preferably trimethylene.
Suitable "lower alkenylene" includes a straight or branched alkenylene having
1
to 6, in particular 3, carbon atoms. Preferable examples which may be
mentioned are
vinylene, propenylene, dimethylpropenylene (e.g., 3,3-dimethylpropenylene,
etc.) and
hexenylene preferably propenylene.
13
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
The term "halogen" means fluoro, chloro, bromo or iodo.
Suitable "halo(lower)allcyl" contains 1 to 4, in particular 1 or 2, carbon
atoms, and
preferably 1 to 9, in particular 1 to 5, identical or different halogen atoms,
preferably
fluorine, chlorine and bromine, in particular fluorine and chlorine. Examples
which may
be mentioned are trifluoromethyl, trichloromethyl, chlorodifluoromethyl,
dichlorofluoromethyl, chloromethyl, bromomethyl, 1-fluoroethyl, 2-fluoroethyl,
2,2-difluoroethyl, 2,2,2-trifluoroethyl, 2,2,2-trichloroethyl and
pentafluoroethyl, preferably
trifluoromethyl.
Suitable "halo(lower)alkoxy" contains 1 to 4, in particular 1 or 2, carbon
atoms,
and preferably 1 to 9, in particular 1 to 5, identical or different halogen
atoms, preferably
fluorine, chlorine and bromine, in particular fluorine and chlorine. Examples
which may
be mentioned are trifluoromethoxy, trichloromethoxy, chlorodifluoromethoxy,
dichlorofluoromethoxy, chloromethoxy, bromomethoxy, 1-fluoroethoxy, 2-
fluoroethoxy,
2,2-difluoroethoxy, 2,2,2-trifluoroethoxy, 2,2,2-trichloroethoxy and
pentafluoroethoxy,
preferably trifluoromethoxy.
The term carbocyclic group intended to mean cyclo(lower)alkyl or
cyclo(lower)alkenyl.
Suitable "cyclo(lower)alkyl" and cyclo(lower)alkyl moiety in the term
"cyclo(lower)alkylene" includes a saturated carbocycle having 3 to 7, in
particular 5 to 6,
carbon atoms. Preferable examples which may be mentioned are cyclopropyl,
cyclobutyl,
cyclopentyl, cyclohexyl and cycloheptyl, preferably cyclopropyl and
cyclohexyl.
Preferable example which may be mentioned as "cyclo(lower)alkylene" are
cyclohexylene (e.g., 1,3- cyclohexylene, 1,4-cyclohexylene, etc.). Suitable
"cyclo(lower)alkenyl" and cyclo(lower)alkenyl moiety in the term
"cyclo(lower)alkenylene" includes a partially saturated carbocycle having 3 to
7, in
particular 5 to 6, carbon atoms. Preferable examples which may be mentioned
are
cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl and cycloheptenyl,
preferably
cyclopentenyl and cyclohexenyl.
Preferable example which may be mentioned as "cyclo(lower)alkylene" are
cyclopentenylene (e.g., 1,3-cyclocyclopent-1-enylene, etc.), cyclohexenylene
(e.g., 1,3-
cyclohex-1-enylene, etc.).
Suitable "heteroaryl" and heteroaryl moiety in the terms
"heteroaryl(lower)alkyl" and
"heteroaromatic acyl" is intended to mean 5- to 7-membered rings having
preferably 1 to 3,
in particular 1 or 2, identical or different heteroatoms. Heteroatoms in the
heteroaryl are
oxygen, sulfur or nitrogen. Examples which may be mentioned are furyl,
thienyl,
pyrazolyl, imidazolyl, triazolyl (e.g., 1,2,3- and 1,2,4-triazolyl, etc.),
isoxazolyl, thiazolyl,
14
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
isothiazolyl, oxadiazolyl (e.g., 1,3,4-, and 1,2,5-oxadiazolyl, etc.),
azepinyl, pyrrolyl,
pyridyl, piperazinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl (e.g.,
1,3,5-, 1,2,4- and
1,2,3-triazinyl, etc.), oxazinyl (e.g., 1,2,4- and 1,2,6-oxazinyl, etc.),
oxepinyl, thiepinyl,
diazepinyl (e.g., 1,2,4-diazepinyl, etc.), preferably thienyl, pyrazolyl,
imidazolyl, thiazolyl,
pyridyl, pyrazinyl.
Suitable "cyclic amino group" are heteroaromatic or aliphatic ring systems
having
one or more nitrogen atoms as the heteroatom, in which the heterocyclic rings
can be
saturated or unsaturated, can be one ring system or several fused ring
systems, and
optionally contain further heteroatoms, such as nitrogen, oxygen and sulfur
and the like.
Cyclic amino groups can furthermore also. denote a spiro ring or a bridged
ring system. The
number of atoms which form cyclic amino groups is not limited, for example in
the case of
a single-ring system, they comprise 3 to 8 atoms, and in the case of a three-
ring system,
they comprise 7 to 11 atoms.
Preferable examples of "cyclic amino group" are described as follows:
(1) examples which may be mentioned of cyclic amino group with saturated
monocyclic
groups with one or more nitrogen atoms) as the heteroatom are azetidinyl (3-
azetidinyl),
pyrrolidinyl (e.g., 1- and 3-pyrrolidinyl, etc.), piperidyl (e.g., piperidine,
4-piperidyl, etc.),
homopiperidino (e.g., hexahydro-1H-azepin-1-yl, etc.), homopiperazinyl (e.g.,
hexahydro-1H-1, 4-diazepin-1-yl, etc.), imidazolidinyl (e.g., 1-
imidazolidinyl, etc.),
piperazinyl (e.g., 1-piperazinyl, etc.), perhydropyrimidinyl (e.g.,
perhydropyrimidin-1-yl,
etc.) or diazacycloheptanyl (e.g., 1,4-diazacycloheptan-1-yl, etc.);
(2) examples which may be mentioned of cyclic amino group with unsaturated
monocyclic groups with one or more nitrogen atoms) as the heteroatom are
pyrrolinyl
(e.g., 2-pyrrolin-1-yl, etc.), pyrrolyl (e.g., 1-pyrrolyl, etc),
tetrahydropyridyl (e.g.,
3,6-dihydro- ((2H)-pyridyl, etc.), pyridyl (e.g., 2-pyridyl, etc.),
tetrahydroazepinyl (e.g.,
2,3,6,7-tetrahydro-1H-azepin-1-yl, 2,3,4,7-tetrahydro-1H-azepin-1-yl, etc.),
imidazolyl
(1-imidazolyl), pyrazolyl, triazolyl, tetrazolyl, tetrazolyl, pyrimidinyl,
pyrazinyl,
pyridazinyl, dihydro-pyridazinyl (e.g., 1,2-dihydro-pyridazin-1-yl, etc.) or
dihydro-pyrimidinyl (e.g., 1,2-dihydro-pyrimidin-1-yl, etc.);
(3) examples which may be mentioned of cyclic amino groups with saturated or
unsaturated monocyclic groups with one to three nitrogen atoms and one to two
sulfur
atoms as heteroatoms are thiazolidinyl (e.g., 3-thiazolidinyl, etc.),
isothiazolinyl (e.g.,
2-isothiazolinyl, etc.) or thiomorpholino;
(4) examples which may be mentioned of cyclic amino groups with saturated or
unsaturated monocyclic groups with one to three nitrogen atoms and one to two
oxygen
atoms as heteroatoms are oxazolyl, isoxazolyl, oxadiazolyl (e.g., 1,2,4-
oxadiazolyl, or
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
1,3,4-oxadiazolyl) or morpholinyl;
(5) examples which may be mentioned of cyclic amino groups with saturated or
unsaturated fused cyclic groups are indolyl (e.g., 1-indolyl, etc.),
dihydrobenzimidazolyl
(e.g., 1,2-dihydrobenzimidazol-1-yl, etc.), perhydropyrrolo[1,2-a]pyrazinyl
(e.g.,
perhydropyrrolo[1,2-a]pyrazin-2-yl, etc.), tetrahydrobenzo[f]isoquinolinyl
(e.g.,
1,4,5,6-tetrahydrobenzo[f]isoquinolin-3(2H)-yl, etc.),
hexahydrobenz[fJisoquinolinyl (e.g.,
cis- and trans-1,4,4a,5,6,1Ob-hexahydrobenz[fJisoquinolin-3(2H)-yl, etc.),
tetrahydropyrido[3,4-b]indolyl (e.g., 1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-
2-yl, etc.)
tetrahydrobenzazepinyl (e.g., 1,2,4,5-tetrahydro-3H-3-benzazepin-3-yl, etc.),
or
dihydroisoquinolinyl (e.g., 3,4-dihydro-2(1H)-isoquinolinyl, etc.);
(6) examples which may be mentioned of cyclic amino groups with spirocyclic
groups
are azaspiro[4,5]decanyl (e.g., 2-azaspiro[4,5]decan-2-yl, etc.),
spiro[1H-indene-1,4'-piperidyl] (e.g., spiro[1H-indene-1,4'-piperidin-1'-yl],
etc.), or
dihydrospiro[1H-indene-1,4'-piperidyl] (e.g.,
2,3-dihydrospiro[1H-indene-1,4'-piperidin-1'-yl], etc.);
(7) examples which may be mentioned of cyclic amino groups bridged
heterocyclic
groups are azabicyclo[2,2,1]heptanyl (e.g., 2-azabicyclo[2,2,1]heptan-7-yl,
etc.), or
diazabicyclo[2.2.1]heptyl (e.g., 2,5-diazabicyclo[2.2.1]kept-2-yl, etc.).
Among the above, preferable "cyclic amino group" included in Rl is
above-mentioned (1) or (2), in which the most preferable one is piperidyl,
tetrahydropyridyl or piperazinyl.
Preferable examples which may be mentioned of "diradical of saturated or
unsaturated monocyclic group with one or more nitrogen atom(s), which is
obtained after
removal of one hydrogen atom from said monocyclic group" are azetidinylene
(e.g., 1,2- or
1,3-azetidinylene), pyrrolidinylene (e.g., 1,2- or 1,3- pyrrolidinylene), or
piperidinylene
(e.g., 1,3- or 1,4-piperidinylene).
It has been known that, during major cellular stresses, the activation of PARP
can
rapidly lead to cell damage or death through depletion of energy stores and
PARP
activation play a key role in both NMDA- and NO-induced neurotoxicity (Zhang
et. al.,
Science, 263: 687-89 (1994)). Therefore, the compound possessing PARP
inhibiting
activity, such as the compound (I) of this invention, or pharmaceutically
acceptable salts
are useful in treating and preventing various diseases ascribed by NMDA- and
NO-induced
toxicity. Such diseases include, for example, tissue damage resulting from
cell damage or
death due to necrosis or apoptosis; neural tissue damage resulting from
ischemia and
reperfusion injury, neurological disorders and neurodegenerative diseases;
16
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
neurodegenerative diseases; head trauma; stroke; Alzheimer's disease;
Parkinson's disease;
epilepsy; Amyotrophic lateral Scleosis (ALS); Huntington's disease;
schizophrenia;
chronic pain; ischemia and neuronal loss following hypoxia; hypoglycemia;
ischemia;
trauma; or nervous insult.
It has been demonstrated that PARP inhibitor are useful in deducing infarct
size
(Thiemermann et al, Proc. Natl. Acad. Sci. USA, 94: 679-83 (1997)). Therefore,
the
compound possessing PARP inhibiting activity, such as the compound (I) of this
invention,
or pharmaceutically acceptable salts are useful in treatment and prevention of
previously
ischemic heart or skeleton muscle tissue.
It is also known that PARP is thought to play a role in enhancing DNA repair.
So,
the compound possessing PARP inhibiting activity, such as the compound (I) of
this
invention, or pharmaceutically acceptable salts are effective in treating and
preventing
radiosensitizing hypoxic tumor cells; tumor cells from recovering from
potentially lethal
damage of DNA after radiation therapy.
Further, the compound possessing PARP inhibiting activity, such as the
compound
(I) of this invention, or pharmaceutically acceptable salts are useful in
extending the
life-span and proliferative capacity of cells and altering gene expression of
senescent cells.
They are useful for treating and preventing skin aging; Alzheimer's diseases;
arteriosclerosis; osteoarthritis; osteoporosis; muscular dystrophy;
degenerative diseases of
skeletal muscle involving replicative senescence; age-related macular
degeneration;
immune senescence; AIDS; and other immune senescence diseases.
Still further, the compound possessing PARP inhibiting activity, such as the
compound (I) of this invention, or pharmaceutically acceptable salts are
effective in
treating and preventing inflammatory bowel disorders (e.g., colitis);
arthritis; diabetes;
endotoxic shock; septic shock; or tumor. Also, they are useful in reducing
proliferation of
tumor cells and making synergistic effect when tumor cells are co-treated with
an
alkylating drug.
The compound possessing PARP inhibiting activity, such as the compound (I) of
this invention, or pharmaceutically acceptable salts are effective in treating
and preventing
pituitary apoplexy; conjunctivitis; retinoblastoma; retinopathy; acute retinal
necrosis
syndrome; Sjogren's syndrome.
The compound (I), its prodrug, or their salt can be administered alone or in
the
form of a mixture, preferably, with a pharmaceutical vehicle or carrier.
The active ingredient of this invention can be used in the form of a
pharmaceutical
preparation, for example, in solid, semisolid or liquid form, which contains a
compound (I),
as an active ingredient, in admixture with an organic or inorganic carrier or
excipient
17
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
suitable for external (topical), enteral, intravenous, intramuscular,
parenteral or
intramucous applications. The active ingredient can be formulated, for
example, with the
conventional non-toxic, pharmaceutically acceptable carriers for ointment,
cream, plaster,
tablets, pellets, capsules, suppositories, solution (saline, for example),
emulsion,
suspension (olive oil, for example), aerosols, pills, powders, syrups,
injections, troches,
cataplasms, aromatic waters, lotions, buccal tablets, sublingual tablets,
nasal drops and any
other form suitable for use. The carriers which can be used are water, wax,
glucose,
lactose, gum acacia, gelatin, mannitol, starch paster, magnesium trisilicate,
talc, corn starch,
keratin, paraffin, colloidal silica, potato starch, urea and other carriers
suitable for use in
manufacturing preparations, in solid, semisolid, or liquid form, and in
addition auxiliary,
stabilizing, thickening and coloring agents and perfiunes may be used. The
active
compound is included in a pharmaceutical composition in an effective amount
sufficient to
produce the desired effect upon the process or condition of the diseases.
The active ingredient can be formulated into, for example, preparations for
oral
application, preparations for injection, preparations for external
application, preparations
for inhalation, preparations for application to mucous membranes.
Manunals which may be treated by the present invention include livestock
mammals such as cows, horses, etc., domestic animals such as dogs, cats, rats,
etc. and
humans, preferably humans.
While the dosage of therapeutically effective amount of the compound (I) will
vary depending upon the age and condition of each individual patient, an
average single
dose to a human patient of about 0.01 mg, 0.1 mg, 1 mg, 10 mg, 50 mg, 100 mg,
250 mg,
500 mg, and 1000 mg of the compound (I) may be effective for treating the
above-mentioned diseases. In general, amounts between 0.01 mg/body and about
1,000
mg/body may be administered per day.
In order to illustrate the usefulness of the object compound (I), the
pharmacological test data of the compound (I) are shown in the following.
A. Test Compound
(1) 2-[3-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)propyl]-5,6,7,8-tetrahydro-
4(3H)-quinazolinone
(Compound A: The compound of Example 1 )
(2) 2-[3-(4-Phenyl-3,6-dihydro-1 (2H)-pyridyl)propyl]-
3,5,7,8-tetrahydro-4H-thiopyrano[4,3-d]pyrimidin-4-one
(Compound B: The compound of Example 3-(10))
(3) 4-[4-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)butyl]-1(2H)-phthalazinone
18
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
(Compound C: The compound of Example 7)
(4) 4-[4-(9-Methyl-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)butyl]-
1 (2H)-phthalazinone
(Compound D: The compound of Example 9-(7))
B. PARP inhibitory activity (In vitro assay)
(1) Assay conditions:
The recombinant human PARP (5.3mg protein/ml) were incubated with a test
compound in a 100.1 reaction buffer containing the indicated concentration of
1 mCi/ml
3aP-NAD, SOmM Tris-HCI, 25mM MgCl2, 1mM DTT (dithiothreitol), O.OSmM NAD
(nicotinamido adenine dinucleotide), lmg/ml activated DNA, pH8Ø Incubation
was for
minutes at a room temperature and the reaction was stopped by the addition of
200,1 of
ice-cold 20% trichloroacetic acid followed by rapid filtration through GF/B
filters. The
filters were treated with scintillation fluid and acid-insoluble counts were
measured for
15 quantification of unit activity.
PARP inhibitory activity (%) _
[1-(enzyme activity with test compound)/(enzyme activity with vehicle)] x100
(2) Result
PARP inhibitory activity (ICS~) in test compound.
Test Compound IC50(~,M)
Compound A < 0.5
Compound B < 0.5
Compound C < 0.5
Compound D I ~ o.s
This invention relates to novel Quinazolinone compounds had a potent PARP
inhibitory activity. PARP inhibitors including this invention relates to novel
quinazolinone
compounds were effective in preventing reduction of striatal DA and its
metabolite induced
by MPTP treatment in mice. Therefore, it suggests that these compounds may
have
protective benefit in the treatment of neurodegenerative disease such as
Parkinson's
disease.
Abbreviations used herein have the following meanings:
ABBREVIATION DEFINITION
Me methyl
Et ethyl
19
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
TBu tert-buthyl
Bzl benzyl
Ph phenyl
Ac acetyl
Bz benzoyl
Any patents, patent applications, and publications cited herein are
incorporated by
reference.
Best Mode for Carrying out the Invention
The following Preparation and Examples are given for the purpose of
illustrating
the present invention in detail, but are not to be construed to limit the
scope of the present
invention.
Preparation 1
To a solution of 3,4-difluorobromobenzene (5.81 g) in tetrahydrofuran (50 ml)
was added dropwise n-butyl lithium (19.3 ml) at - 78 °C under nitrogen.
The mixture
was stirred at the temperature for 0.5 hour. To the mixture was added dropwise
a solution
of t-Butyl 4-oxo-1-piperidinecarboxylate (5 g) in tetrahydrofuran (20 ml) at -
78 °C, and
the mixture was stirred for 1 hour, then warmed to 0 °C and stirred for
further 1 hour.
The reaction was quenched with water and extracted with ethyl acetate twice.
The
combined extracts were dried over magnesium sulfate and concentrated. This
crude
t-Butyl 4-(3,4-difluorophenyl)-4-hydroxy-1-piperidinecarboxylate was used for
the next
step without further purification.
Preparation 2
To a solution of t-butyl 4-(3,4-difluorophenyl)-4-hydroxy-1-
piperidinecarboxylate
(8.96 g; net: 7.79 g) in dichloromethane (98 ml) were added in sequence
methanesulfonylchloride (5.77 ml), triethylamine (34.7 ml) and 4-
dimethylaminopyridine
(152 mg). After stirring at room temperature for 2 hours, the mixture was
diluted with
water and extracted with dichloromethane twice. The combined extracts were
dried over
magnesium sulfate and concentrated. A solution of the residue and
triethylamine (34.7
ml) in dichloromethane (98 ml) was stirred at room temperature for 2 days. The
mixture
was diluted with water and the organic layer was separated. The organic
extract was
dried over magnesium sulfate and concentrated. The residue was chromatographed
on
silica gel using 10% ethyl acetate in hexane as an eluent to give t-Butyl
4-(3,4-difluorophenyl)-3,6-dihydro-1 (2H)-pyridinecarboxylate (4.37 g) as an
oil.
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
1H NMR (CDC13, ~): 1.50 (9H, s), 2.40 - 2.60 (2H, m), 3.63 (2H, t, J=5.7 Hz),
3.90 - 4.20 (2H, m), 5.97 (1H, s), 6.80 - 7.40 (4H, m).
Mass (ESI): 318.2 (M+Na)+
Preparation 3
To a solution of t-butyl 4-(3,4-difluorophenyl)-3,6-dihydro-1 (2H)-
pyridinecarboxylate (4.3 g) in ethyl acetate (20 ml) was added dropwise 4N
hydrogen
chloride in ethyl acetate (18.25 ml), and the mixture was stirred at room
temperature
overnight. After evaporation of the mixture, the residue was triturated with
ethyl acetate
and diisopropylether, and the resulting powder was collected, washed with
diisopropylether and dried in vacuo to give 4-(3,4-Difluorophenyl)-1,2,3,6-
tetrahydropyridine hydrochloride (3.25 g).
1H NMR (DMSO-db, &): 2.20 - 4.20 (6H, m), 6.09 (1H, s), 7.00 - 7.80 (3H, m),
9.07 (2H, brs)
Mass (ESI): 196.2 (M+H)+
Preparation 4
To a suspension of L-alanine methyl ester hydrochloride (12.9 g) and
triethylamine (38.6 ml) in dichloromethane (130 ml) was added dropwise
chloroacetylchloride (8.83 ml) at 0 °C. After stirring at 0 °C
for 30 minutes, the mixture
was concentrated and diluted with ethyl acetate (100 ml) and 1N aqueous
hydrochloric
acid (100 ml). The organic layer was separated, washed with water twice, dried
over
magnesium sulfate and concentrated. A solution of the residue in 40% ethyl
acetate in
hexane (200 ml) was treated with silica gel (85 g), and silica gel was removed
by filtration
and washed with 40% ethyl acetate in hexane (200 ml) twice, and the combined
filtrate
was concentrated to give methyl (2S)-2-[(chloroacetyl)amino]propanoate as a
brown oil.
IH NMR (DMSO-d6, 8): 1.30 (3H, d, J=7.3 Hz), 3.64 (3H, s), 4.09 (2H, s),
4.20-4.35 (1H, m), 8.64 (1H, d, J=6.8 Hz)
Mass (ESI): 202.2 (M+Na)+
Preparation 5
A solution of methyl (2S)-2-[(chloroacetyl)amino]propanoate (5 g),
4-chloroaniline (3.55 g) and triethylamine (11.6 ml) in toluene (50 ml) was
stirred at
100 °C overnight. The mixture was diluted with water (100 ml) and
extracted with ethyl
acetate twice. The combined extracts were washed with water and brine, dried
over
magnesium sulfate and concentrated. The residue was chromatographed on silica
gel
21
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
using 50% ethyl acetate in hexane as an eluent to give methyl
(2S)-2-({[(4-chlorophenyl)amino]acetyl}amino)propanoate (3.07 g) as an oil.
'H NMR (DMSO-d6, ~): 1.27 (3H, d, J=7.3 Hz), 3.31 (3H, s), 3.66 (2H, d, J=6.0
Hz), 4.20 - 4.50 (1H, m), 6.12 (1H, t, J=6.0 Hz), 6.54 (2H, d, J=8.8 Hz), 7.1
(2H,
d, J=8.8 Hz), 8.32 (1H, d, J=7.2 Hz).
Mass (ESI): 293.2 (M+Na)+
Preparation 6
A slurry of methyl (2S)-2-({[(4-chlorophenyl)amino]acetyl}amino)propanoate
(3.02 g) and potassium t-butoxide (2.5 g) in toluene was stirred at 80
°C overnight.
After cooling to room temperature, the reaction was quenched with 1N aqueous
hydrochloric acid and extracted with ethyl acetate twice. The combined
extracts were
dried over magnesium sulfate and concentrated. The residue was chromatographed
on
silica gel using 80% ethyl acetate in hexane as an eluent to give
(3S)-1-(4-Chlorophenyl)-3-methyl-2,5-piperazinedione (1.5 g).
1H NMR (DMSO-d6, ~): 1.37 (3H, d, J=7.0 Hz), 4.11 (1H, q, J=7.0 Hz), 4.22 (1H,
d, J=16.6 Hz), 4.32 (1H, d, J=16.6 Hz), 7.30 - 7.60 (4H, m), 8.41 (1H, brs)
Mass (ESI): 261.1 (M+Na)+
Preparation 7
The following compound was prepared in a similar manner to that of Preparation
4.
(1) Ethyl2-((chloroacetyl)amino)-2-methylpropanoate
'H NMR (DMSO-d6, S): 1.14 (3H, t, J=7.1 Hz), 1.36 (6H, s), 3.80 - 4.20 (4H,
m),
8.52 (1H, brs)
Mass (ESI): 230.2 (M+Na)+
Preparation 8
The following compound was prepared in a similar manner to that of Preparation
5.
(1) Ethyl2-({[(4-chlorophenyl)amino]acetyl}amino)-2-methylpropanoate
1H NMR (DMSO-d6, 8): 1.10 (3H, t, J=7.1 Hz), 1.35 (6H, s), 3.61 (2H, d, J=6.0
Hz), 4.00 (2H, q, J=7.1 Hz), 6.54 (2H, d, J=8.8 Hz), 7.09 (2H, d, J=8.8 Hz),
8.17
( 1 H, brs)
Mass (ESI): 321.2 (M+Na)+
22
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Preparation 9
The following compound was prepared in a similar manner to that of Preparation
6.
(1) 1-(4-Chlorophenyl)-3,3-dimethyl-2,5-piperazinedione
'H NMR (DMSO-d6, 8): 1.42 (6H, s), 4.32 (2H, s), 7.20 - 7.70 (4H, m), 8.50
(1H,
brs)
Mass (ESI): 275.1 (M+Na)+
Preparation 10
To a suspension of lithium aluminum hydride (225 mg) in tetrahydrofuran (7.5
ml) was added in portions 1-(4-chlorophenyl)-3,3-dimethyl-2,5-piperazinedione
(0.5 g),
and the mixture was stirred at 50 °C for 3 hours. After cooling to room
temperature, the
reaction was quenched with 1N aqueous sodium hydroxide (0.5 ml). The resulting
precipitates were removed by filtration and washed with ethyl acetate, and
then the
combined filtrate was washed with brine, dried over magnesium sulfate and
concentrated.
A solution of the residue in ethyl acetate was treated with 4N hydrogen
chloride in ethyl
acetate (1 ml), and the mixture was concentrated. The residual oil was
triturated with a
small amount of acetone, and then the resulting powder was collected, washed
with
acetone and dried in vacuo to give 1-(4-Chlorophenyl)-3,3-dimethylpiperazine
hydrochloride (0.22 g).
1H NMR (DMSO-d6, ~): 1.37 (6H, s), 3.00 - 3.40 (6H, m), 7.02 (2H, d, J=9.0
Hz),
7.28 (2H, d, J=9.0 Hz), 9.08 (2H, brs)
Mass (ESI): 225.3 (M+H)+
Preparation 11
The following compound was prepared in a similar manner to that of Preparation
10.
(1) (3S)-1-(4-Chlorophenyl)-3-methylpiperazine hydrochloride
1H NMR (DMSO-d6, 8): 1.29 (3H, d, J=6.5 Hz), 1.80 - 4.30 (7H, m), 6.90 - 7.40
(4H, m)
Mass (ESI): 211.2 (M+H)+
Preparation 12
Amixture of 4-bromochlorobenzene (2 g), 2-amino-2-methyl-1-
(triphenylmethyl)aminopropane (4.83 g), tris(dibenzylideneacetone)dipalladium
(287 mg),
2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (390 mg), sodium t-butoxide (1.4
g) in toluene
23
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
(24 ml) was stirred at 120 °C under nitrogen for 2 hours. After cooling
to room
temperature, the mixture was diluted with diisopropylether and filtered, and
the filtrate was
concentrated. The residue was chromatographed on silica gel using 10°1o
ethyl acetate in
hexane as an eluent to give 2-(4-chlorophenyl)amino-2-methyl-1-
(triphenylmethyl)amino-
propane (2.83 g).
1H NMR (CDC13, ~): 1.30 (6H, s), 1.92 (1H, t, J=6.8 Hz), 2.27 (1H, d, J=6.8
Hz),
3.59 (1H, brs), 6.26 (2H, d, J=8.8 Hz), 6.91 (2H, d, J=8.8 Hz), 7.10 - 7.70
(15H,
m)
Mass (ESI): 463.3 (M+Na)+
Preparation 13
To a solution of 2-(4-chlorophenyl)amino-2-methyl-1-(triphenylmethyl)amino-
propane (2.79 g) in dichloromethane (100 ml) were added in sequence
triethylamine (3.88
ml) and methyl oxalyl chloride (1.16 ml). After stirring at room temperature
for 4 hours.
the mixture was washed with sodium hydrogen carbonate aqueous solution, dried
over
magnesium sulfate and concentrated. The residue was chromatographed on silica
gel
(ethyl acetate/hexane = 1 /9 to 1 / 1 ) to give methyl
f (4-chlorophenyl)-[1,1-dimethyl-2-
((triphenylmethyl)amino)ethyl]amino](oxo)acetate (3.3
g) as an oil.
1H NMR (CDCl3, 8): 1.28 (6H, s), 1.87 (1H, t, J=8.5 Hz), 2.63 (2H, d, J=8.5
Hz),
3.46 (3H, s), 7.10 - 7.70 (19H, m)
Mass (ESI): 549.3 (M+Na)+
Preparation 14
To a solution of methyl
~(4-chlorophenyl)-[l,l-dimethyl-2-
((triphenylmethyl)amino)ethyl]amino](oxo)acetate (3.3
g) in dichloromethane were added in sequence anisole (3.3 ml) and
trifluoroacetic acid (6
ml) at 0 °C. After stirring at this temperature for 2 hours, the
mixture was diluted with
water and extracted with dichloromethane twice. The combined extracts were
dried over
magnesium sulfate and concentrated. A suspension of the residue in 2-propanol
(15 ml)
was stirred at 80 °C in the presence of acetic acid (1 ml) for 2 hours.
The mixture was
cooled to 0 °C, and the resulting precipitates were collected, washed
with 2-propanol and
dried in vacuo (40°C) to give 1-(4-chlorophenyl)-6,6-dimethyl-2,3-
piperazinedione (1.17
g)-
'H NMR (CDC13, 8): 1.34 (6H, s), 3.55 (2H, d, J=3.3 Hz), 7.00 - 7.20 (3H, m),
7.43 (2H, d, J=8.6 Hz)
24
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Mass (ESI): 275.2 (M+Na)+
Preparation 15
To a suspension of 1-(4-chlorophenyl)-6,6-dimethyl-2,3-piperazinedione (0.69
g)
in tetrahydrofuran (25 ml) was added dropwise 2M boran-methyl sulfide complex
in
tetrahydrofuran (6.8 ml) under nitrogen, and the mixture was stirred at room
temperature
overnight. The reaction was quenched with methanol and 12N aqueous
hydrochloric acid
( 1.5 ml) was added. After stirring at 70 °C for 1 hour, the mixture
was cooled to room
temperature, basified with 1N aqueous sodium hydroxide and extracted with
dichloromethane twice. The combined extracts were dried over magnesium sulfate
and
concentrated. The residue was dissolved in dichloromethane, treated with 4N
hydrogen
chloride in ethyl acetate ( 1 ml) and concentrated to give
1-(4-Chlorophenyl)-2,2-dimethylpiperazine hydrochloride (0.46 g) as an
amorphous
powder.
IH NMR (DMSO-d6, ~): 1.09 (6H, s), 2.90 - 3.40 (6H, m), 7.20 (2H, d, J=8.7
Hz),
7.3 8 (2H, d, J=8.7 Hz), 9.3 8 (2H, brs)
Mass (ESI): 225.3 (M+H)+
Preparation 16
Amixture of 4-bromochlorobenzene (1.5 g), cis-2,6-dimethylpiperazine (1.07 g),
trans-dichlorobis(tri-o-tolylphosphine)palladium (II) (185 mg), sodium t-
butoxide (1.09 g)
in toluene (20 ml) was stirred at 100 °C under nitrogen for 3 hours.
After cooling to
room temperature, the reaction was quenched with water and extracted with
dichloromethane twice. The combined extracts were dried over magnesium sulfate
and
concentrated. The residue was chromatographed on silica gel using 5% methanol
in
dichloromethane as an eluent to give 4-(4-Chlorophenyl)-cis-2,6-
dimethylpiperazine (1.46
g) as a solid.
Mass (ESI): 225.3 (M+H)+
Preparation 17
A biphasic solution of (3R,SR)-1-benzyl-3,5-dimethylpiperazine (1.61 g; net:
1.50
g) and di-t-butyldicarbonate (1.61 g) in dichloromethane (20 ml) and 1N
aqueous sodium
hydroxide (20 ml) was stirred at room temperature for 30 minutes. The organic
phase
was separated and the aqueous layer was further extracted with
dichloromethane. The
combined extracts were dried over magnesium sulfate and concentrated in vacuo.
The
residue was dissolved in 20% ethyl acetate in hexane and treated with silica
gel (7.5 g).
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Silica gel was removed by filtration and washed with 20% ethyl acetate in
hexane twice,
and then the combined filtrate was evaporated to afford colerless oil. A
solution of the
residue in methanol was hydrogenated over 10% palladium-on-charcoal (450 mg)
for 3
hours. The catalyst was removed by filtration and the filtrate was
concentrated. The
residue was chromatographed on silica gel (20% ethyl acetate in hexane to 10%
methanol
in dichloromethane), and then the fractions eluted with 10% methanol in
dichloromethane
were combined and concentrated to give t-butyl
(2R,6R)-2,6-dimethyl-1-piperazinecarboxylate (1.32 g) as an oil.
IH NMR (CDCl3, 8): 1.30 (6H, d, J=6.6 Hz), 1.47 (9H, s), 2.71 (2H, dd, J=4.4,
12.6 Hz), 3.15 (2H, dd, J=4.0, 12.6 Hz), 3.70 - 4.00 (2H, m)
Mass (ESI): 237.3 (M+Na)+
Preparation 18
A mixture of t-butyl (2R,6R)-2,6-dimethyl-1-piperazinecarboxylate (1.27 g),
4-bromochlorobenzene (3.4 g), tris(dibenzylideneacetone)dipalladium (0) (271
mg),
2,2'-bis(diphenylphosphino) -1,1'-binaphthyl (369 mg), sodium t-butoxide (2.28
g) in
toluene (26 ml) was stirred at 80 °C under nitrogen overnight. The
mixture was cooled,
diluted with water and extracted with dichloromethane twice. The combined
extracts
were dried over magnesium sulfate and concentrated. The residue was dissolved
in 20%
ethyl acetate in hexane (50 ml) and treated with silica gel (20 g). Silica gel
was removed
by filtration and washed with 20% ethyl acetate in hexane (50 ml) twice, and
then the
combined filtrate was evaporated. To a solution of the residue in
dichloromethane (30
ml) was added dropwise trifluoroacetic acid at 0 °C. After stirring for
1 hour, the mixture
was concentrated, basified with 1N aqueous sodium hydroxide and extracted with
dichloromethane twice. The combined extracts were dried over magnesium sulfate
and
concentrated. The residue was chromatographed on silica gel (30 g) (50% ethyl
acetate in
hexane to 10% methanol in dichloromethane), and the fractions eluted with 10%
methanol
in dichloromethane were combined and concentrated. A solution of the residue
in ethyl
acetate was treated with 4N hydrogen chloride in ethyl acetate (2 ml), and the
resulting
powder was collected, washed with ethyl acetate and dried in vacuo to give
(3R,SR)-1-(4-Chlorophenyl)-3,5-dimethylpiperazine hydrochloride (1.49 g).
1H NMR (DMSO-d6, 8): 1.34 (6H, d, J=6.6 Hz), 3.12 (2H, dd, J=6.4, 13.0 Hz),
3.43 (2H, dd, J=3.3, 13.0 Hz), 6.99 (2H, d, J=9.0 Hz), 7.27 (2H, d, J=9.0 Hz),
9.48
(2H, brs)
Mass(ESI): 225.3 (M+H)+
26
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Preparation 19
The following compound was prepared in a similar manner to that of Preparation
17.
(1) t-Butyl (2S,6S)-2,6-dimethyl-1-piperazinecarboxylate
1H NMR (CDCl3, 8): 1.30 (6H, d, J=6.6 Hz), 1.47 (9H, s), 2.71 (2H, dd, J=4.4,
12.6 Hz), 3.15 (2H, dd, J=4.0, 12.6 Hz), 3.70 - 4.00 (2H, m)
Mass (ESI): 237.3 (M+Na)+
Preparation 20
The following compound was prepared in a similar manner to that of Preparation
18.
(1) (3S,SS)-1-(4-Chlorophenyl)-3,5-dimethylpiperazine hydrochloride
1H NMR (DMSO-d6, 8): 1.34 (6H, d, J=6.6 Hz), 3.12 (2H, dd, J=6.4, 13.0 Hz),
3.43 (2H, dd, J=3.3, 13.0 Hz), 6.99 (2H, d, J=9.0 Hz); 7.27 (2H, d, J=9.0 Hz),
9.48
(2H, brs)
Mass (ESI): 225.3 (M+H)+
Preparation 21
A mixture of 4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (6 g),
4-bromobutyronitrile (3.35 ml) and diisopropylethylamine (16 ml) in
N,N-dimethylformamide (30 ml) was stirred at 80 °C for 3 hours. The
mixture was
diluted with water, extracted with ethyl acetate twice. The combined extracts
were
washed with water three times, dried over magnesium sulfate and concentrated.
The
residue was dissolved in ethyl acetate and treated with silica gel (30 g).
Silica gel was
removed by filtration and washed with ethyl acetate. The combined filtrate was
concentrated to give 4-(4-phenyl-3,6-dihydro-1 (2H)-pyridyl)butanenitrile as
an oil.
1H NMR (CDCl3, 8): 1.75 - 2.10 (2H, m), 2.30 - 2.90 (8H, m), 3.05 - 3.25 (2H,
m),
6.06(lH,s),7.10-7.80(SH,m)
Mass (APCI): 227.40 (M+H)+
Preparation 22
To a suspension of ammonium chloride (2.95 g) in toluene (20 ml) was added
dropwise 2N trimethylaluminium in toluene (27.5 ml) at 0 °C under
nitrogen, and the
mixture was stirred at room temperature for 2 hours. To this aluminum amide
reagent
was added dropwise 4-(4-phenyl-3,6-dihydro-1 (2H)-pyridyl)butanenitrile (2.5
g) in toluene
(10 ml) at room temperature, and this solution was stirred at 80 °C
overnight. The
27
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
reaction mixture was carefully poured into a suspension of silica gel (60 g)
in chloroform
(180 ml). Silica gel was removed by filtration and washed with methanol (200
ml), and
then the combined filtrate was concentrated. The residue was chromatographed
on
aluminum (68 g) using 20% methanol in dichloromethane as an eluent to give
4-(4-phenyl-3,6-dihydro-1(2H)-pyridyl)butanimidamide (2.04 g) as an oil.
IH NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.10 - 2.90 (8H, m), 3.09 (2H, d,
J=2.8 Hz), 6.16 (1H, s), 7.10 - 7.70 (SH, m), 8.69 (3H, brs)
Mass (APCI): 244.33 (M+H)+
Preparation 23
The following compounds were prepared in a similar manner to that of
Preparation 21.
(1) 4-[4-(3,4-Difluorophenyl)-3,6-dihydro-1(2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, b): 1.60 - 2.00 (2H, m), 2.20 - 2.80 (8H, m), 3.07 (2H, d,
J=2.6 Hz), 6.04 (1H, s), 7.00 - 7.80 (3H, m)
Mass (ESI): 263.3 (M+H)+
(2) 4-[4-(4-Chlorophenyl)-2,2-dimethyl-1-piperazinyl]butanenitrile
1H NMR (DMSO-db, 8): 1.08 (6H, s), 1.50 - 1.80 (2H, m), 2.20 - 2.70 (6H, m),
2.87 (2H, s), 3.00 - 3.20 (2H, m), 6.91 (2H, d, J=9.1 Hz), 7.20 (2H, d, J=9.1
Hz)
Mass (ESI): 292.3 (M+H)+
(3) 4-[(2S)-4-(4-Chlorophenyl)-2-methyl-1-piperazinyl]butanenitrile
IH NMR (DMSO-d6, &): 1.05 (3H, d, J=5.6 Hz), 1.60 - 1.90 (2H, m), 2.00 - 3.60
(11H, m), 6.93 (2H, d, J=9.1 Hz), 7.21 (2H, d, J=9.1 Hz)
Mass (ESI): 278.2 (M+H)~
(4) 4-[4-(4-Chlorophenyl)-3,3-dimethyl-1-piperazinyl]butanenitrile
IH NMR (DMSO-d6, 8): 0.98 (6H, s), 1.60 - 1.90 (2H, m), 2.20 - 3.20 (lOH, m),
7.10 (2H, d, J=8.8 Hz), 7.29 (2H, d, J=8.8 Hz)
Mass (ESI): 292.4 (M+H)+
(5) 4-[(2R,6S)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]butanenitrile
1H NMR (CDC13, ~): 1.16 (6H, s), 1.60 - 3.60 (12H, m), 6.82 (2H, d, J=9.0 Hz),
7.19 (2H, d, J=9.0 Hz)
Mass (ESI): 292.4 (M+H)+
(6) 4-[(2R,6R)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]butanenitrile
1H NMR (DMSO-d6, b): 1.01 (6H, d, J=6.1 Hz), 1.50 - 1.80 (2H, m), 2.20 - 3.30
(lOH, m), 6.91 (2H, d, J=9.0 Hz), 7.20 (2H, d, J=9.0 Hz)
Mass (ESI): 292.2 (M+H)+
28
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
(7) 4-[(2S,6S)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]butanenitrile
1H NMR (DMSO-db, 8): 1.01 (6H, d, J=6.1 Hz), 1.50 - 1.80 (2H, m), 2.20 - 3.30
(lOH, m), 6.91 (2H, d, J=9.0 Hz), 7.20 (2H, d, J=9.0 Hz)
Mass (ESI): 292.2 (M+H)+
(8) 4-[4-(4-Fluorophenyl)-3,6-dihydro-1(2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, 8): 1.60 - 2.00 (2H, m), 2.20 - 2.80 (8H, m), 3.06 (2H, d,
J=3.0 Hz), 6.12 ( 1 H, t, J=3.0 Hz), 7.00 - 7.70 (4H, m)
Mass (ESI): 245.4 (M+H)+
(9) 4-[4-(4-chlorophenyl)-3,6-dihydro-1(2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, b): 1.60 - 1.90 (2H, m), 2.30 - 3.20 (lOH, m), 6.19 (1H, t,
J=3.5 Hz), 7.30 - 7.70 (4H, m)
Mass (APCI): 261.07 (M+H)+
(10) 4-[4-(4-Methylphenyl)-3,6-dihydro-1(2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, 8): 1.60 - 1.90 (2H, m), 2.28 (3H, s), 2.30 - 2.80 (8H, m),
l5 3.07 (2H, d, J=2.7 Hz), 6.09 (1H, s, J=2.7 Hz), 7.13 (2H, d, J=8.0 Hz),
7.31 (2H, d,
J=8.0 Hz)
Mass (APCI): 241.33 (M+H)+
(11) 4-[4-(4-T'rifluoromethylphenyl)-3,6-dihydro-1(2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, b): 1.70 - 2.00 (2H, m), 2.30 - 3.20 (lOH, m), 6.33 (1H, s),
7.50 - 7.70 (4H, m)
Mass (APCI): 295.00 (M+H)+
(12) 4-[4-(4-Methoxyphenyl)-3,6-dihydro-1(2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, b): 1.70 - 2.00 (2H, m), 2.30 - 2.80 (8H, m), 3.74 (3H, s),
6.03 (1H, s), 6.89 (2H, d, J=8.8 Hz), 7.36 (2H, d, J=8.8 Hz)
Mass (APCI): 257.27 (M+H)+
(13) 4-[4-(4-Chlorophenyl)-1-piperazinyl]butanenitrile
1H NMR (DMSO-d6, ~): 1.70 - 1.90 (2H, m), 2.30 - 2.80 (8H, m), 3.12 (4H, t,
J=5.0 Hz), 6.94 (2H, d, J=9.1 Hz), 7.22 (2H, d, J=9.1 Hz)
Mass (APCI): 264.47 (M+H)+
(14) 4-[4-(4-Fluorophenyl)-1-piperazinyl]butanenitrile
1H NMR (DMSO-d6, ~): 1.60 - 2.00 (2H, m), 2.30 - 2.80 (8H, m), 3.07 (4H, t,
J=5.0 Hz), 6.80 - 7.20 (4H, m)
Mass (ESI): 248.3 (M+H)+
(15) 4-[4-(4-Nitrophenyl)-1-piperazinyl]butanenitrile
iH NMR (DMSO-d6, ~): 1.70 - 1.90 (2H, m), 2.20 - 2.80 (8H, m), 3.45 (4H, t,
J=5.0 Hz), 7.03 (2H, d, J=9.4 Hz), 8.05 (2H, d, J=9.4 Hz)
29
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Mass (ESI): 275.3 (M+H)+
Preparation 24
The following compounds were prepared in a similar manner to that of
Preparation 22.
(1) 4-[4-(3,4-Difluorophenyl)-3,6-dihydro-1(2H)-pyridyl]butanimidamide
1H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.20 - 3.30 (lOH, m), 6.05 (1H, s),
7.00 - 7.70 (3H, m)
Mass (ESI): 280.4 (M+H)+
(2) 4-[4-(4-Chlorophenyl)-2,2-dimethyl-1-piperazinyl]butanimidamide
1H NMR (DMSO-d6, b): 1.03 (6H, s), 1.50 - 1.90 (2H, m), 2.20 - 3.30 (IOH, m),
6.92 (2H, d, J=9.0 Hz), 7.21 (2H, d, J=9.0 Hz), 8.45 (3H, brs)
Mass (ESI): 309.3 (M+H)+
(3) 4-[(2S)-4-(4-Chlorophenyl)-2-methyl-1-piperazinyl]butanimidamide
IH NMR (DMSO-d6, ~): 1.04 (3H, d, J=5.5 Hz), 1.60 - 2.00 (2H, m), 2.00 - 3.70
(11H, m), 6.93 (2H, d, J=9.0 Hz), 7.22 (2H, d, J=9.0 Hz), 8.68 (3H, brs)
Mass (ESI): 295.4 (M+H)+
(4) 4-[4-(4-Chlorophenyl)-3,3-dimethyl-1-piperazinyl]butanimidamide
'H NMR (DMSO-d6, 8): 0.99 (6H, s), 1.60 - 1.90 (2H, m), 2.10 - 3.20 (lOH, m),
7.10 (2H, d, J=8.8 Hz), 7.30 (2H, d, J=8.8 Hz), 9.03 (3H, brs)
Mass (ESI): 309.3 (M+H)+
(5) 4-[(2R,6S)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]butanimidamide
1H NMR (DMSO-d6, b): 1.06 (6H, d, J=6.2 Hz), 1.50 - 1.90 (2H, m), 2.10 - 3.90
(lOH, m), 6.92 (2H, d, J=9.1 Hz), 7.21 (2H, d, J=9.1 Hz)
Mass (ESI): 309.3 (M+H)+
(6) 4-[(2R,6R)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]butanimidamide
1H NMR (DMSO-d6, 8): 1.01 (6H, d, J=6.1 Hz), 1.50 -1.90 (2H, m), 2.20 - 3.30
(lOH, m), 6.92 (2H, d, J=9.0 Hz), 7.21 (2H, d, J=9.0 Hz), 8.79 (3H, brs)
Mass (ESI): 309.3 (M+H)+
(7) 4-[(2S,6S)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]butanimidamide
1H NMR (DMSO-d6, S): 1.01 (6H, d, J=6.1 Hz), 1.50 - 1.90 (2H, m), 2.20 - 3.30
(lOH, m), 6.92 (2H, d, J=9.0 Hz), 7.21 (2H, d, J=9.0 Hz), 8.79 (3H, brs)
Mass (ESI): 309.3 (M+H)+
(8) 4-[4-(4-Fluorophenyl)-3,6-dihydro-1 (2H)-pyridyl]butanimidamide
1H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.30 - 2.80 (8H, m), 3.08 (2H, d,
J=2.9 Hz), 6.13 ( 1 H, s), 7.10 - 7.60 (4H, m)
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Mass (ESI): 262.4 (M+H)+
(9) 4-[4-(4-Chlorophenyl)-3,6-dihydro-1 (2H)-pyridyl]butanimidamide
'H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.20 - 2.80 (8H, m), 3.09 (2H, d,
J=2.8 Hz), 6.21 (1H, s), 7.20 - 7.60 (4H, m)
Mass (APCI): 278.07 (M+H)+
(10) 4-[4-(4-Methylphenyl)-3,6-dihydro-1(2H)-pyridyl]butanimidamide
1H NMR (DMSO-d6, ~): 1.70 - 2.00 (2H, m), 2.28 (3H, s), 2.30 - 2.70 (8H, m),
3.08 (2H, d, J=2.7 Hz), 6.11 (1H, s), 7.14 (2H, d, J=8.2 Hz), 7.32 (2H, d,
J=8.2
Hz)
Mass (APCI): 258.33 (M+H)+
(11) 4-[4-(4-(Trifluoromethyl)phenyl)-3,6-dihydro-1(2H)-pyridyl]butanimidamide
1H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.20 - 3.80 (lOH, m), 6.35 (1H, s),
7.50 - 7.90 (4H, m), 8.53 (3H, brs)
Mass (ESI): 312.3 (M+H)~
(12) 4-[4-(4-Methoxyphenyl)-3,6-dihydro-1(2H)-pyridyl]butanimidamide
1H NMR (DMSO-d6, S): 1.70 - 2.00 (2H, m), 2.30 - 2.80 (8H, m), 3.06 (2H, d,
J=3.0 Hz), 3.74 (3H, s), 6.04 (1H, s), 6.90 (2H, d, J=8.8 Hz), 7.36 (2H, d,
J=8.8
Hz)
Mass (APCI): 274.27 (M+H)+
(13) 4-[4-(4-Chlorophenyl)-1-piperazinyl]butanimidamide
1H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.20 - 2.70 (8H, m), 2.90 - 3.30
(4H,
m), 6.94 (2H, d, J=9.1 Hz), 7.23 (2H, d, J=9.1 Hz), 8.97 (3H, brs)
Mass (APCI): 281.20 (M+H)+
(14) 4-[4-(4-Fluorophenyl)-1-piperazinyl]butanimidamide
1H NMR (DMSO-dg, 8): 1.70 - 2.00 (2H, m), 2.20 - 2.80 (8H, m), 3.05 (4H, t,
J=5.0 Hz), 6.80 - 7.20 (4H, m), 8.80 (3H, brs)
Mass (ESI): 265.4 (M+H)+
(15) 4-[4-(4-Nitrophenyl)-1-piperazinyl]butanimidamide
1H NMR (DMSO-d6, 8): 1.70 - 4.00 (14H, m), 7.02 (2H, d, J=9.4 Hz), 8.06 (2H,
d,
J=9.4 Hz)
Mass (ESI): 292.4 (M+H)+
Preparation 25
To a solution of 4-(4-phenyl-3,6-dihydro-1 (2H)-pyridyl)butanenitrile (0.75 g)
in
toluene was added dropwise 1N diisobutylaluminium hydride in hexane (6.63 ml)
at
-78 °C, and the mixture was warmed up to 0 °C. The reaction was
quenched with 1N
31
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
aqueous hydrochloric acid, basified with saturated aqueous sodium hydrogen
carbonate.
The mixture was filtered through celite and the filter cake was washed with
dichloromethane, then the combined filtrate was dried over magnesium sulfate
and
concentrated. The residue was chromatographed on silica gel (80% ethyl acetate
in
hexane to 10% methanol in dichloromethane) to give
4-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)butanal (0.4 g) as an oil.
1H NMR (CDC13, S): 1.90 - 2.30 (2H, m), 2.40 - 2.60 (2H, m), 2.70 - 2.85 (2H,
m),
2.85 - 3.10 (2H, m), 3.50 - 3.70 (2H, m), 6.04 (1H, m), 7.10 - 7.60 (SH, m)
Mass (APCI): 230.27 (M+H)+
Preparation 26
A slurry of 4-benzyloxybutanal, (3-oxo-1,3-dihydro-2-benzofuran-1-yl)-
(triphenyl)phosphonium bromide (560 mg) and triethylamine (7.39 ml) in
tetrahydrofuran
(50 ml) was stirred at room temperature overnight. The resulting precipitates
were
removed by filtration and washed with ethyl acetate, and then the combined
filtrate was
concentrated. The residue was chromatographed on silica gel using toluene as
an eluent
to give an oil, which was dissolved in ethanol and refluxed in the presence of
hydrazine
monohydrate (1.4 g) for 1 hour. The mixture was concentrated, then
dichloromethane and
water were added and the organic layer was separated. The aqueous layer was
further
extracted with dichloromethane, and then the combined extracts were dried over
magnesium sulfate and concentrated. The residue was triturated with
dichloromethane
and diisopropylether, and then the resulting powder was collected, washed with
diisopropylether and dried in vacuo to give 4-[4-(Benzyloxy)butyl]-1(2H)-
phthalazinone
(2.78 g).
IH NMR (DMSO-d6, 8): 1.50 - 2.00 (4H, m), 2.94 (2H, t, J=7.2 Hz), 3.49 (2H, t,
J=6.1 Hz), 4.45 (2H, s), 7.10 - 7.50 (SH, m), 7.70 - 8.20 (3H, m), 8.26 (1H,
dd,
J=1.9, 7.1 Hz), 12.45 (1H, brs)
Mass (ESI): 309.3 (M+H)+
_Preparation 27
To slurry of 4-[4-(benzyloxy)butyl]-1 (2H)-phthalazinone in dichloromethane (5
ml) was added dropwise 1M boron tribromide in dichloromethane (0.97 ml), and
the
mixture was stirred at room temperature for 2 hours. The reaction was quenched
with
water and extracted with dichloromethane twice. The combined extracts were
dried over
magnesium sulfate and concentrated. The residue was triturated with
diisopropylether,
and the resulting powder was collected, washed with diisopropylether and dried
in vacuo to
32
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
give 4-(4-Bromobutyl)-1(2H)-phthalazinone.
'H NMR (DMSO-d6, 8): 1.70 - 2.10 (4H, m), 2.96 (2H, t, J=7.3 Hz), 3.61 (2H, t,
J=6.4 Hz), 7.70 - 8.10 (3H, m), 8.27 ( 1 H, d, J=8.2 Hz), 12.47 ( 1 H, brs)
Mass (ESI): 305.0 (M+Na)+
The following compounds were prepared in a similar manner to that of
Preparation 26.
Preparation 28
(1) 4-[5-(Benzyloxy)pentyl]-1(2H)-phthalazinone
1H NMR (DMSO-db, 8): 1.40 - 2.00 (6H, m), 2.80 - 3.70 (4H, m), 4.32 (2H, s),
7.20 - 7.50 (SH, m), 7.70 - 8.10 (3H, m), 8.27 (1H, d, J=7.4 Hz), 12.44 (1H,
brs)
Mass (ESI): 345.3 (M+Na)+
Preparation 29
( 1 ) 4-(5-Bromopentyl)-1 (2H)-phthalazinone
1H NMR (DMSO-d6, ~): 1.30 - 2.00 (6H, m), 2.93 (2H, t, J=7.5 Hz), 3.54 (2H, t,
J=6.7 Hz), 7.70 - 8.20 (3H, m), 8.27 ( 1 H, d, J=7.3 Hz), 12.45 ( 1 H, brs)
Mass (ESI): 317.1 (M+Na)+
~0 Preparation 30
50% Pd/C catalyst (50% wet, 400mg) was added to a solution of
4-(4-biphenylyl)-1,2,3,6-tetrahydropyridine (470mg) in a mixture of
tetrahydrofixran
(lOml), methanol (20m1) and acetic acid (lOml). The mixture was stirred under
hydrogen
at atmospheric pressure until gas absorption ceased. After filtration through
celite and
removal of solvent, the residue was dissolved in a mixture of ethyl acetate
and aqueous
sodium hydrogen carbonate. The aqueous phase was separated and the organic
phase was
washed with brine and dried over magnesium sulfate. Evaporation of the solvent
afforded
4-(4-biphenylyl)piperidine (432mg).
Mass: 238.1 (M+H)+
Preparation 31
To a solution of 4-(4-fluorophenyl)-3,6-dihydro-1(2H)-pyridine (1 g) and ethyl
4-oxopentanoate (0.961 ml) in toluene was added a catalytic amount of p-
toluenesulfonic
acid (54 mg), and the mixture was stirred under reflux to remove librated
water
azeotropically. After stirring for 3 hours, the mixture was cooled and diluted
with
dichloroethane. To the mixture were added~sodium tri(acetoxy)borohydride (3.59
g) and
33
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
acetic acid (0.97 ml) in sequence, and the mixture was stirred at room
temperature for 1
hour. The mixture was diluted with water, neutralized and extracted with
dichloromethane three times. The combined extracts were dried over magnesium
sulfate
and concentrated. The residue was chromatographed on silica gel using ethyl
acetate as
an eluent to give Ethyl 4-[4-(4-fluorophenyl)-3,6-dihydro-1(2H)-pyridyl]-
4-mehtylbutanoate (0.72 g)
'H NMR (DMSO-d6, ~ ) : 0.80 - 4.30(19H, m), 6.13(1H, m), 6.80 - 7.60(4H, m).
Mass(ESI): 306.3 (M+H)+
Preparation 32
A mixture of 4-[4-(trifluoromethyl)phenyl]piperidine hydrochloride (1.18 g),
4-bromobutyronitrile (0.662 ml) and triethylamine (1.86 ml) in N,N-
dimethylformamide
(20 ml) was stirred at 80 °C overnight. The mixture was diluted with
water, extracted
with ethyl acetate twice. The combined extracts were washed with water three
times,
dried over magnesium sulfate and concentrated. The residue was dissolved in
ethyl
acetate and treated with silica gel (10 g). Silica gel was removed by
filtration and washed
with ethyl acetate . The combined filtrate was concentrated to give
4-[4-[4-(trifluoromethyl)phenyl]piperidino]butanenitrile as an oil.
IH NMR (DMSO-d6, ~ ) : 1.40 - 3.20(15H, m), 7.48(2H, d, J=8.2 Hz), 7.65(2H,
d).
Mass(ESI): 297.2 (1VI+H)+
The following compound was obtained according to a similar manner to that of
Preparation 32 .
Preparation 33
4-[4-[4-(Trifluoromethoxy)phenyl]-3,6-dihydro-1 (2H)-pyridyl]butanenitrile
1H NMR (DMSO-d6, ~ ) : 1.60 - 3.30(12H, m), 6.20(1H, m), 7.00 - 7.80(4H, m).
Mass(ESI): 311.2 (M+H)+
Preparation 34
Under a nitrogen atmosphere, 4-bromobutanenitrile (402mg) and triethylamine
(0.76m1) was added successively to a suspension of 4-(4-biphenylyl)piperidine
(430mg) in
N,N-dimethylformamide (Sml) at room temperature. The mixture was stirred for
15
hours at 80°C and, cooled to room temperature. The mixture was poured
into a mixture of
water and chloroform and the aqueous layer was separated. The organic layer
was washed
with brine and dried over magnesium sulfate. The solvent was evaporated and
the residue
34
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
was purified by column chromatography on silica gel eluting with
dichloromethane-acetone to afford 4-[4-(4-biphenylyl)piperidino]butanenitrile
(411mg).
Mass: 305.2 (M+H)+
The following compounds Preparations 35 and 36] were obtained according to a
similar manner to that of Preparation 34.
Preparation 35
4-[4-(3,4-dichlorophenyl)-1-piperazinyl]butanenitrile
'H NMR (CDC13, 8 ) : 1.85(2H, m), 2.4-2.7(8H, m), 3.1-3.2(4H, m), 6.72(1H, dd,
J=9.0,3.0 Hz), 6.95(1H, d, J=3.0 Hz), 7.28(1H, d, J=9.0 Hz).
Mass: 320.0, 322.1 (M+Na)'~
Preparation 36
4-[4-(4-biphenylyl)-1,2,3,6-tetrahydropyridyl]butanenitrile
Mass: 303.2 (M+H)+
Preparation 37
To a suspension of ammonium chloride (1.09 g) in toluene (20 ml) was added
dropwise 2N trimethylaluminium in toluene (10.2 ml) at 0 °C under
nitrogen, and the
mixture was stirred at room temperature for 2 hours. To this aluminum amide
reagent
was added dropwise 4-[4-[4-(trifluoromethyl)phenyl]piperidino]butanenitrile
(1.21 g) in
toluene (20 ml) at room temperature, and this solution was stirred at 80
°C overnight.
The reaction mixture was carefully poured into a suspension of silica gel (15
g) in
chloroform (40 ml). Silica gel was removed by filtration and washed with
methanol (50
ml) twice, and the combined filtrate was concentrated. The residue was
chromatographed
on alumina (30 g) (methanol/dichloromethane = 1/4) to give
4-[4-[4-(trifluoromethyl)phenyl]piperidino]butanamidine (1.40 g) as an oil.
1H NMR (DMSO-d6, ~ ) : 1.30 - 3.80(15H, m), 7.49(2H, d, J=7.9 Hz), 7.70(2H, d,
J=7.9
Hz), 8.75(3H, brs).
Mass(ESI): 314.4 (M+H)+
The following compounds [Preparation 38 to 42] were obtained according to a
similar manner to that of Preparation 37.
Preparation 38
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
4-[4-(4-Fluorophenyl)-3,6-dihydro-1 (2H)-pyridyl]-4-methylbutanamidine
'H NMR (DMSO-d6, ~ ) : 0.80 - 4.40(14H, m), 6.15(1H, m), 6.90 - 7.70(4H, m),
8.80(3H,
brs).
Mass(ESI): 276.2 (M+H)+
Preparation 39
4-[4-[4-(Trifluoromethoxy)phenyl]-3,6-dihydro-1 (2H)-pyridyl]butanamidine
'H NMR (DMSO-d6, ~ ) : 1.50 - 4.00(12H, m), 6.22(1H, m), 7.32(2H, d, J=8.2
Hz),
7.56(2H, d, J=8.2 Hz).
Mass(ESI): 328.3 (M+H)+
Preparation 40
To a solution of 4-(4-fluorophenyl)-3,6-dihydro-1(2H)-pyridine (1 g) and ethyl
4-oxopentanoate (0.961 ml) in toluene was added a catalytic amount of p-
toluenesulfonic
acid (54 mg), and the mixture was stirred under reflux to remove librated
water
azeotropically. After stirring for 3 hours, the mixture was cooled and diluted
with
dichloroethane. To the mixture were added sodium tri(acetoxy)borohydride (3.59
g) and
acetic acid (0.97 ml) in sequence, and the mixture was stirred at room
temperature for 1
hour. The mixture was diluted with water, neutralized and extracted with
dichloromethane three times. The combined extracts were dried over magnesium
sulfate
and concentrated. The residue was chromatographed on silica gel using ethyl
acetate as
an eluent to give Ethyl 4-[4-(4-fluorophenyl)-3,6-dihydro-1 (2H)-
pyridyl]pentanoate (0.72
g)
1H NMR (DMSO-d6, ~ ) : 0.80 - 4.30(19H, m), 6.13(1H, m), 6.80 - 7.60(4H, m).
Mass(ESI): 306.3 (M+H)+
Preparation 41
4-[4-(3,4-dichlorophenyl)-1-piperazinyl]butanamidine
IH NMR (DMSO-d6, 8 ) : 1.6-1.9(2H, m), 2.2-2.6(8H, m), 3.1-3.3(4H, m),
6.94(1H, dd,
J=9.0, 2.5 Hz), 7.15 ( 1 H, d, J=2. 5 Hz), 7.3 9( 1 H, d, J=9.0 Hz).
Mass : 316.2 (M+H)+
Preparation 42
4-[4-(4-biphenylyl)-1,2,3,6-tetrahydropyridyl]butanamidine
Mass : 320.1 (M+H)+
36
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Example 1
A suspension of 4-(4-phenyl-3,6-dihydro-1 (2H)-pyridyl)butanimidamide ( 107
mg), cyclohexanone-2-carboxylic acid ethyl ester (50 mg), potassium carbonate
(568 mg)
in ethanol (5 ml) was stirred at 80 °C overnight. The mixture was
diluted with water and
extracted with dichloromethane twice. The combined extracts were dried over
magnesium sulfate and concentrated. The residue was purified by preparative
thin layer
chromatography on silica gel using 10% methanol in dichloromethane as an
eluent to give
2-[3-(4-Phenyl-3,6-dihydro-1 (2H)-pyridyl)propyl]-5,6,7,8-tetrahydro-
4(3H)-quinazolinone (58 mg) as a colerless powder.
IH NMR (CDC13, 8): 1.40 - 2.20 (6H, m), 2.30 - 3.00 (1 OH, m), 3.10 - 3.40
(2H,
m), 6.10 (1H, s), 7.10 - 7.60 (SH, m)
Mass (APCI): 350.20 (M+H)+
Example 2
The following compounds were prepaxed in a similar manner to that of Example
1.
(1) 2-{3-[4-(4-Chlorophenyl)-2,2-dimethyl-1-piperazinyl]propyl}5,6,7,8-
tetrahydro-
4(3H)-quinazolinone
1H NMR (DMSO-d6, 8): 1.02 (6H, s), 1.40 - 1.90 (6H, m), 2.10 - 2.70 (1 OH, m),
2.83 (2H, s), 2.90 - 3.20 (2H, m), 6.89 (2H, d, J=9.0 Hz), 7.20 (2H, d, J=9.0
Hz),
12.28 (1H, brs)
Mass (ESI): 415.4 (M+H)+
(2) 2-{3-[(2S)-4-(4-Chlorophenyl)-2-methyl-1-piperazinyl]propyl}-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, ~): 1.02 (3H, d, J=5.3 Hz), 1.40 - 3.60 (21H, m), 6.91 (2H,
d,
J=9.1 Hz), 7.20 (2H, d, J=9.1 Hz), 12.18 (1H, brs)
Mass (ESI): 401.2 (M+H)+
(3) 2-{3-[4-(4-Chlorophenyl)-3,3-dimethyl-1-piperazinyl]propyl}-5,6,7,8-
tetrahydro-
4(3H)-quinazolinone
1H NMR (DMSO-d6, 8): 0.96 (6H, s), 1.50 - 2.00 (2H, m), 2.00 - 3.20 (14H, m),
7.07 (2H, d, J=8.7 Hz), 7.29 (2H, d, J=8.7 Hz), 12.13 ( 1 H, brs)
Mass (ESI): 415.4 (M+H)+
(4) 2-{3-[(2R,6S)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]propyl}-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, ~): 1.06 (6H, d, J=6.0 Hz), 1.40 - 1.90 (6H, m), 2.10 - 3.80
37
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
( 12H, m), 6.92 (2H, d, J=9.0 Hz), 7.20 (2H, d, J=9.0 Hz), 12.18 ( 1 H, brs)
Mass (ESI): 415.4 (M+H)+
(5) 2-{3-[(2R,6R)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]propyl}-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, b): 0.99 (6H, d, J=6.1 Hz), 1.40 - 2.00 (6H, m), 2.10 - 3.30
( 14H, m), 6.90 (2H, d, J=8.9 Hz), 7.20 (2H, d, J=8.9 Hz), 12.18 ( 1 H, brs)
Mass (ESI): 415.4 (M+H)+
(6) 2-{3-[(2S,6S)-4-(4-Chlorophenyl)-2,6-dimethyl-1-piperazinyl]propyl}-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
~ 1H NMR (DMSO-d6, S): 0.99 (6H, d, J=6.1 Hz), 1.40 - 2.00 (6H, m), 2.10 -
3.30
(14H, m), 6.90 (2H, d, J=8.9 Hz), 7.20 (2H, d, J=8.9 Hz), 12.18 (1H, brs)
Mass (ESI): 415.4 (M+H)+
(7) 2-{3-[4-(4-Fluorophenyl)-3,6-dihydro-1(2H)-pyridyl]propyl)-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, ~): 1.40 - 2.00 (6H, m), 2.10 - 2.70 (12H, m), 3.04 (2H, d,
J=2:6 Hz), 6.09 ( 1 H, s), 7.00 - 7.60 (4H, m), 12.11 ( 1 H, brs)
Mass (APCI): 368.20 (M+H)+
(8) 2-{3-[4-(4-Chlorophenyl)-3,6-dihydro-1 (2H)-pyridyl]propyl{-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, 8): 1.40 - 2.00 (6H, m), 2.20 - 2.80 (12H, m), 3.04 (2H, d,
J=3 .0 Hz), 6.17 ( 1 H, s), 7.20 - 7.60 (4H, m), 12.11 ( 1 H, brs)
Mass (ESI): 384.3 (M+H)+
(9) 2-{3-[4-(4-Methylphenyl)-3,6-dihydro-1(2H)-pyridyl]propyl]-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, 8): 1.40 - 2.00 (6H, m), 2.10 - 2.80 (15H, m), 3.04 (2H, m),
6.07 ( 1 H, s), 7.12 (2H, d, J=8.0 Hz), 7.3 0 (2H, d, J=8.0 Hz), 12.09 ( 1 H,
brs)
Mass (ESI): 364.4 (M+H)+
(10) 2-{3-[4-(4-(Trifluoromethyl)phenyl)-3,6-dihydro-1(2H)-pyridyl]propyl)-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, 8): 1.45 - 1.75 (4H, m), 1.80 - 2.00 (2H, m), 2.10 - 2.80
(12H, m), 3.08 (2H, d, J=1.4 Hz), 6.31 (1H, s), 7.50 - 7.80 (4H, m)
Mass (ESI): 418.3 (M+H)+
(11) 2-{3-[4-(4-Methoxyphenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}-
5, 6,7, 8-tetrahydro-4(3 H)-quinazolinone
IH NMR (DMSO-d6, 8): 1.40 - 2.00 (6H, m), 2.10 - 3.20 (12H, m), 3.74 (3H, s),
6.00 (1H, s), 6.88 (2H, d, J=8.8 Hz), 7.34 (2H, d, J=8.8 Hz), 12.08 (1H, brs)
38
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Mass (APCI): 380.20 (M+H)+
(12) 2-{3-[4-(4-Chlorophenyl)-1-piperazinyl]propyl}-5,6,7,8-tetrahydro-
4(3H)-quinazolinone
IH NMR (DMSO-d6, ~): 1.40 - 2.00 (6H, m), 2.00 - 3.70 (16H, m), 6.91 (2H, d,
J=9.0 Hz), 7.21 (2H, d, J=9.0 Hz), 12.17 ( 1 H, brs)
Mass (APCI): 387.07 (M+H)+
(13) 2-{3-[4-(4-Fluorophenyl)-1-piperazinyl]propyl}-5,6,7,8-tetrahydro-
4(3H)-quinazolinone
1H NMR (DMSO-d6, b): 1.40 - 2.00 (6H, m), 2.10 - 3.20 (16H, m), 6.80 - 7.20
(4H, m), 12.16 ( 1 H, brs)
Mass (APCI): 371.07 (M+H)+
Example 3
The following compounds were prepared in a similar manner to that of Example
1.
(1) 2-{3-[4-(3,4-Difluorophenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}-
3,5,7,8-tetrahydro-4H-thiopyrano[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, ~): 1.70 - 2.00 (2H, m), 2.30 - 3.00 (12H, m), 3.07 (2H, d,
J=2.9 Hz), 3 .40 (2H, s), 6.02 ( 1 H, s), 7.00 - 7.60 (3 H, m), 12.3 5 ( 1 H,
brs)
Mass (ESI): 404.2 (M+H)+
(2) 2-{3-[4-(4-Fluorophenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}
3,5,7,8-tetrahydro-4H- thiopyrano[4,3-d]pyrimidin-4-one
IH NMR (DMSO-d6, b): 1.70 - 2.00 (2H, m), 2.20 - 2.80 (4H, m), 3.04 (2H, d,
J=2.8 Hz), 6.09 (1H, s), 7.00 - 7.60 (4H, m), 12.34 (1H, brs)
Mass (APCI): 386.00 (M+H)+
(3) 2-{3-[4-(4-Chlorophenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}-
3,5,7,8-tetrahydro-4H- thiopyrano[4,3-d]pyrimidin-4-one
IH NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.20 - 3.20 (16H, m), 6.17 (1H, s),
7.20 - 7.60 (4H, m), 12.3 5 ( 1 H, brs)
Mass (APCI): 401.93 (M+H)+
(4) 2-{3-[4-(4-Methylphenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}
3,5,7,8-tetrahydro-4H- thiopyrano[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.27 (3H, s), 2.30 - 2.90 (12H, m),
3 .03 (2H, d, J=2.8 Hz), 3 .3 8 (2H, s), 6.07 ( 1 H, s), 7.12 (2H, d, J=8.2
Hz), 7.3 0
3 5 (2H, d, J=8.2 Hz), 12.3 5 ( 1 H, brs)
Mass (APCI): 382.13 (M+H)+
39
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
(5) 2-{3-[4-(4-(Trifluoromethyl)phenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}-
3,5,7,8-tetrahydro-4H- thiopyrano[4,3-d]pyrimidin-4-one
IH NMR (DMSO-d6, S): 1.70 - 2.00 (2H, m), 2.30 - 3.00 (14H, m), 3.08 (2H, d,
J=2.5 Hz), 6.31 (1H, s), 7.40 - 7.80 (4H, m)
Mass (APCI): 434.33 (M-H)-
(6) 2-{3-[4-(4-Methoxyphenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}-
3,5,7,8-tetrahydro-4H- thiopyrano[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, d): 1.70 - 2.00 (2H, m), 2.20 - 3.20 (14H, m), 3.38 (2H, s),
3.74 (3H, s), 6.00 (1H, s), 6.88 (2H, d, J=8.8 Hz), 7.34 (2H, d, J=8.8 Hz),
12.36
(1 H, brs)
Mass (ESI): 398.3 (M+H)+
(7) 2-{3-[(4-Chlorophenyl)-1-piperazinyl]propyl}-3,5,7,8-tetrahydro-
4H-thiopyrano[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, S): 1.70 - 2.00 (2H, m), 2.20 - 3.20 (16H, m), 3.40 (2H, s),
6.92 (2H, d, J=9.1 Hz), 7.21 (2H, d, J=9.1 Hz), 12.36 ( 1 H, brs)
Mass (APCI): 405.3 (M+H)+
(8) 2-{3-[(4-Fluorophenyl)-1-piperazinyl]propyl}-3,5,7,8-tetrahydro-
4H-thiopyrano [4,3-d]pyrimidin-4-one
1H NMR (DMSO-db, 8): 1.70 - 2.00 (2H, m), 2.20 - 2.90 (12H, m), 3.01 (4H, t,
J=4.6 Hz), 3.40 (2H, s), 6.80 - 7.20 (4H, m), 12.43 (1H, brs)
Mass (APCI): 389.2 (M+H)+
(9) 2-{3-[(4-Nitrophenyl)-1-piperazinyl]propyl}-3,5,7,8-tetrahydro-
4H-thiopyrano[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, S): 1.70 - 2.00 (2H, m), 2.20 - 3.00 (16H, m), 3.40 (2H, s),
7.02 (2H, d, J=9.4 Hz), 8.05 (2H, d, J=9.4 Hz), 12.41 (1H, brs)
Mass (ESI): 416.2 (M+H)+
(10) 2-[3-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)propyl]-3,5,7,8-tetrahydro-
4H-thiopyrano [4,3-d]pyrimidin-4-one
H NMR (DMSO-d6, 8): 1.70 - 2.00 (2H, m), 2.20 - 3.90 ( 16H, m), 6.12 ( 1 H,
s),
7.10 - 7.60 (SH, m), 12.38 (1H, brs)
Mass (APCI): 368.07 (M+H)+
(11) 2-[3-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)propyl]-5,6,7,8-
tetrahydropyrido[4,3-d]pyrimidin-4(3H)-one
1H NMR (DMSO-d6, b): 1.70 - 2.00 (2H, m), 2.20 - 3.80 (16H, m), 6.15 (1H, s),
7.00 - 7.60(SH, m)
Mass (ESI): 351.3 (M+H)+
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
(12) 2-[3-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)propyl]-3,5,7,8-tetrahydro-
4H-pyrano [4,3-d]pyrimidin-4-one
IH NMR (DMSO-db, 8): 1.70 - 2.00 (2H, m), 2.20 - 2.80 (lOH, m), 3.07 (2H, s),
3.80 (2H, t, J=5.5 Hz), 4.29 (2H, s), 6.12 (1H, s), 7.10 - 7.70 (SH, m)
Mass (APCI): 352.2 (M+H)+
Example 4
The following compounds were prepared in a similar manner to that of Example
_l.
(1) 2-[3-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)propyl]-3,5,6,7-tetrahydro-
4H-cyclopenta[d]pyrimidin-4-one
1H NMR (CDC13, ~): 1.70 - 2.30 (4H, m), 2.40 - 3.40 (14H, m), 6.06 (1H, s),
7.00
- 7.60 (SH, m)
Mass (APCI): 336.20 (M+H)+
(2) 2-[3-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)propyl]-3,5,6,7,8,9-hexahydro-
4H-cyclohepta[d]pyrimidin-4-one
'H NMR (CDCl3, 8): 1.00 - 2.40 (8H, m), 2.40 - 3.40 (14H, m), 6.07 (1H, s),
7.00
- 7.60(SH, m)
Mass (APCI): 364.20 (M+H)+
(3) 2- f 3-[4-(4-Fluorophenyl)-3,6-dihydro-1(2H)-pyridyl]propyl}-7,8-dihydro-
3H-thiopyrano [3,2-d]pyrimidin-4(6H)-one
IH NMR (DMSO-d6, 8): 1.70 - 2.20 (2H, m), 2.30 - 3.20 (14H, m), 6.10 (1H, s),
7.00 - 7.60 (4H, m), 12.3 7 ( 1 H, brs)
Mass (ESI): 386.2 (M+H)+
Example 5
A mixture of 4-[4-(4-fluorophenyl)-3,6-dihydro-1 (2H)-pyridyl]butanimidamide
(90 mg) and 2H-pyrido-[2,3-d][1,3]oxazine-2,4(1H)-dione (79 mg) in pyridine (5
ml) was
stirred at 120 °C overnight. The mixture was concentrated and
coevaporated with
toluene twice. The residue was purified by preparative thin layer
chromatography using
10% methanol in dichloromethane as an eluent to give 2- f 3-[4-(4-
Fluorophenyl)-
3,6-dihydro-1(2H)-pyridyl]propyl}-pyrido[2,3-d]pyrimidin-4(3H)-one, which was
converted to the corresponding hydrochloride salt (40 mg) by treatment of 4N
hydrogen
chloride in ethyl acetate .
'H NMR (DMSO-d6, 8): 2.00 - 5.30 (12H, m), 6.18 (1H, s), 7.00 - 7.80 (SH, m),
8.5 5 ( 1 H, dd, J=2.0, 8.0 Hz), 8.93 ( 1 H, dd, J=2.0, 4.7 Hz)
41
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Mass (ESI): 365.5 (M+H)+
Example 6
To a solution of 2-[3-(4-phenyl-3,6-dihydro-1 (2H)-pyridyl)propyl]-
5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4(3H)-one (28 mg) in dichloromethane
(5 ml)
and methanol (1 ml) were added 37% aqueous formaldehyde (0.063 ml) and sodium
triacetoxyborohydride (51 mg) in sequence, then the mixture was stirred at
room
temperature for 2 hours. The reaction was quenched with silica gel (0.2 g) and
concentrated. The residue was chromatographed on silica gel (20% methanol in
dichloromethane) to give 6-Methyl-2-[3-(4-phenyl-3,6-dihydro-1 (2H)-
pyridyl)propyl]-
5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4(3H)-one the objective compound as a
brown
powder.
1H NMR (DMSO-d6, b): 1.70 - 3.20 (21H, m), 6.11 (1H, s), 7.00 - 7.50 (SH, m),
12.26 (1H, brs)
Mass (ESI): 365.4 (M+H)+
Example 7
A suspension of 4-(4-phenyl-3,6-dihydro-1(2H)-pyridyl)butanal (0.18 g),
(3-oxo-1,3-dihydro-2-benzofuran-1-yl)(triphenyl)phosphonium bromide (560 mg)
and
triethylamine (0.328 ml) in tetrahydrofuran (20 ml) was stirred at room
temperature for 3
hours. The reaction was quenched with water and extracted with ethyl acetate
twice.
The combined extracts were dried over magnesium sulfate and concentrated. The
residue
was chromatographed on silica gel using ethyl acetate as an eluent to give
oil, which was
dissolved in ethanol and refluxed in the presence of hydrazine monohydrate (77
mg) for 1
hour. The mixture was concentrated, then dichloromethane and water was added
and the
organic layer was separated. The aqueous layer was further extracted with
dichloromethane, then the combined extracts were dried over magnesium sulfate
and
concentrated. The residue was chromatographed on silica gel (ethyl acetate to
5%
methanol in dichloromethane), and then the fractions eluted with 5% methanol
in
dichloromethane were combined and concentrated. The residue was triturated
with a
mixture of ethyl acetate and diisopropyl ether to give
4-[4-(4-Phenyl-3,6-dihydro-1 (2H)-pyridyl)butyl]-1 (2H)-phthalazinone (46 mg)
as a pale
yellow powder.
'H NMR (DMSO-d6, 8): 1.10 - 1.90 (4H, m), 2.30 - 3.00 (8H, m), 3.07 (2H, d,
3 5 J=2.8 Hz), 6.15 ( 1 H, s), 7.10 - 8.40 (9H, m), 12.45 ( 1 H, brs)
Mass (APCI): 360.07 (M+H)+
42
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Example 8
A mixture of 4-(4-Bromobutyl)-1 (2H)-phthalazinone ( 100 mg),
4-fluorophenyl-1,2,5,6-tetrahydropyridine hydrochloride (91 mg) and
triethylamine (0.149
ml) in N,N-dimethylformamide (5 ml) was stirred at room temperature overnight.
The
mixture was diluted with water and extracted with ethyl acetate twice. The
combined
extracts were washed with water three times, dried over magnesium sulfate and
concentrated. The residue was purified by preparative thin layer
chromatography (10%
methanol in dichloromethane) to give
4-{4-[4-(4-Fluorophenyl)-3,6-dihydro-1(2H)-pyridyl]butyl}-1(2H)-phthalazinone
(70 mg)
as a colorless powder.
1H NMR (DMSO-d6, ~): 1.40 - 2.00 (4H, m), 2.30 - 3.30 (lOH, m), 6.12 (1H, s),
7.00 - 7.60 (SH, m), 7.70 - 8.00 (2H, m), 8.04 (1H, d, J=7.6 Hz), 8.26 (1H, d,
J=7.6 Hz), 12.44 ( 1 H, brs)
Mass (ESI): 378.3 (M+H)+
Example 9
The following compounds were prepared in a similar manner to that of Example
8.
(1) 4-{4-[4-(4-Chlorophenyl)-3,6-dihydro-1(2H)-pyridyl]butyl}-
1 (2H)-phthalazinone
iH NMR (DMSO-d6, ~): 1.40 - 1.90 (4H, m), 2.30 - 2.80 (8H, m), 2.95 (2H, t,
J=7.3 Hz), 3.06 (2H, d, J=2.5 Hz), 6.20 (1H, s), 7.20 - 7.60 (SH, m), 7.70 -
8.00
(2H, m), 8.04 (1H, dd, J=1.5, 7.6 Hz), 8.26 (1H, dd, J=1.5, 7.6 Hz), 12.45
(1H,
brs)
Mass (ESI): 394.2 (M+H)+
(2) 4-{4-[4-(4-Methylphenyl)-3,6-dihydro-1 (2H)-pyridyl]butyl}-
1 (2H)-phthalazinone
1H NMR (DMSO-db, S): 1.40 - 1.90 (4H, m), 2.28 (3H, s), 2.30 - 3.30 (1 OH, m),
6.10 (1H, s), 7.13 (2H, d, J=8.1 Hz), 7.31 (2H, d, J=8.1 Hz), 7.70 - 8.00 (2H,
m),
8.05 ( 1 H, d, J=7.4 Hz), 8.26 ( 1 H, d, J=7.4 Hz), 12.45 ( 1 H, brs)
Mass (ESI): 374.4 (M+H)+
(3) 4-{4-[4-(4-(Trifluoromethyl)phenyl)-3,6-dihydro-1(2H)-pyridyl]butyl}-
1 (2H)-phthalazinone
1H NMR (DMSO-d6, 8): 1.40 - 2.00 (4H, m), 2.30 - 3.30 (lOH, m), 6.34 (1H, s),
7.60 - 8.00 (6H, m), 8.04 ( 1 H, d, J=7.7 Hz), 8.26 ( 1 H, d, J=7.7 Hz), 12.45
( 1 H,
43
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
brs)
Mass (ESI): 428.3(M+H)+
(4) 4-~4-[4-(4-Chlorophenyl)-1-piperazinyl]butyl}-1(2H)-phthalazinone
IH NMR (DMSO-d6, 8): 1.40 - 1.90 (4H, m), 2.20 - 3.70 (lOH, m), 6.92 (2H, d,
J=9.1 Hz), 7.21 (2H, d, J=9.1 Hz), 7.70 - 8.00 (2H, m), 8.04 ( 1 H, d, J=7.4
Hz),
8.26 ( 1 H, d, J=7.4 Hz), 12.45 ( 1 H, brs)
Mass (ESI): 397.3 (M+H)+
(5) 4-{4-[4-(4-Fluorophenyl)-1-piperazinyl]butyl}-1(2H)-phthalazinone
1H NMR (DMSO-d6, 8): 1.40 - 1.90 (4H, m), 2.20 - 3.30 (12H, m), 6.80 - 7.20
(4H, m), 7.70 - 8.00 (2H, m), 8.04 ( 1 H, dd, J=1.6, 7.6 Hz), 8.26 ( 1 H, dd,
J=1.6,
7.6 Hz), 12.45 ( 1 H, brs)
Mass (ESI): 381.3 (M+H)+
(6) 4-{4-[4-(4 Nitrophenyl)-1-piperazinyl]butyl}-1(2H)-phthalazinone
1H NMR (DMSO-d6, b): 1.40 - 2.00 (4H, m), 2.00 - 3.70 (12H, m), 7.02 (2H, d,
J=9.5 Hz), 7.70 - 8.20 (SH, m), 8.26 (1H, dd, J=1.1, 7.7 Hz), 12.45 (1H, brs)
Mass (ESI): 408.3 (M+H)+
(7) 4-[5-(4-Phenyl-3,6-dihydro-1(2H)-pyridyl)pentyl]-1(2H)-phthalazinone
1H NMR (DMSO-d6, ~): 1.20 - 2.00 (6H, m), 2.10 - 3.20 (lOH, m), 6.14 (1H, s),
7.10 - 7.60 (SH, m), 7.70 - 8.10 (3H, m), 8.26 (1H, d, J=7.4 Hz), 12.44 (1H,
brs)
Mass (ESI): 374.4 (M+H)+
(8) 4-[4-(9-Methyl-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)butyl]-
1 (2H)phthalazinone
1H NMR (DMSO-d6, ~): 1.40 - 2.00 (4H, m), 2.40 - 3.20 (8H, m), 3.59 (3H, s),
3.64 (2H, s), 6.80 - 7.20 (2H, m), 7.20 - 8.40 (4H, m), 12.56 ( 1 H, brs)
Mass (ESI): 387.3 (M+H)+
Example 10
Oxalyl chloride (0.193mL, 2.21mmol) was dissolved in dichloromethane (3 mL)
at -78 °C. A solution of dimethylsulfoxide (0.392 mL, 5.52 mmol) in
dichloromethane
(1mL) was added dropwise to that solution, and the mixture was stirred for 10
minutes at
that temperature. A solution of 4-(4-hydroxybutyl)-1 (2H)-isoquinolinone (60
mg, 0.276
mmol) in a mixed solvent of dichloromethane (1 mL) and dimethylsulfoxide (1
mL) was
added dropwise. The mixture was stirred at -78 °C for l5minutes, and at
-45 °C for
minutes. Triethylamine (0.70 mL) was added dropwise, and the mixture was
stirred at
35 0 °C for 1 hour. The crude product was used for next step without
purification. The
crude 4-(1-oxo-1,2-dihydro-4-isoquinolinyl)butanal (59 mg) was dissolved in
44
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
dichloromethane (1 mL), and 4-phenyl-1,2,3,6-tetrahydropyridine (87.9 mg,
0.552 mmol)
was added. Then sodium triacetoxyborohydride (117 mg, 0.552 mmol) and acetic
acid
(0.032 mL, 0.552 mmol) were added to the mixture, and it was stirred at room
temperature
for 15 hours. Purification over silica gel chromatography gave
4-[4-(4-phenyl-3,6-dihydro-1 (2H)-pyridyl)butyl]-1 (2H)-isoquinolinone (24 mg,
24.2 %) as
product.
IH NMR (200MHz, DMSO-db, 8): 1.59 (4H, br s), 2.4-2.7 (8H, m), 3.06 (2H, d,
J=2.9 Hz), 6.15 (1 H, br s), 6.98 ( 1 H, d, J=3.5 Hz), 7.1-7.6 (6H, m), 7.71
(1 H, t,
J=6.7 Hz), 7.78 ( 1 H, d), 8.22 ( 1 H, d, J=8.0 Hz), 11.09 ( 1 H, br s)
Example 11
A suspension of 4-[4-(4-fluorophenyl)piperidino]butanamidine (97 mg), methyl
4-oxotetrahydrothiopyran-3-carboxylate (96 mg), potassium carbonate (509 mg)
in ethanol
(S ml) was stirred at 80 °C overnight. The W fixture was diluted with
water and extracted
with dichloromethane twice. The combined extracts were dried over magnesium
sulfate
and concentrated. The residue was purified by preparative thin layer
chromatography on
silica gel (methanol/dichloromethane =1/9) to give
2-[3-[4-(4-Fluorophenyl)piperidino] propyl]-3,5,7, 8-tetrahydro-4H-thiino [4,3-
d]pyrimidin-
4-one (55 mg) as a colerless powder.
iH NMR (DMSO-d6, ~ ) : 1.00 - 3.70(21H, m), 6.90 - 7.40(4H, m), 12.64(1H,
brs).
Mass(ESI): 388.3 (M+H)+
The following compounds Example 12 to 27 were obtained according to a
similar manner to that of Example 11.
Example 12
2-[3-[4-(4-Methoxyphenyl)piperidino]propyl]-3,5,7,8-tetrahydro-4H
-thiino[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, ~ ) : 1.00 - 3.60(21H, m), 3.71(3H, s), 6.84(2H, d, J=8.7
Hz),
7.13 (2H, d, J=8.7 Hz), 12.47( 1 H, brs).
Mass(ESI): 400.3 (M+H)+
_Example 13
2-[3-[4-(4-Methylphenyl)piperidino]propyl]-3,5,7,8-tetrahydro-4H-
thiino[4,3-d]pyrimidin-4-one
IH NMR (DMSO-d6, 8 ) : 1.30 - 3.70(24H, m), 6.90 - 7.20(4H, m), 12.61(1H,
brs).
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
Mass(ESI): 384.2 (M+H)+
Example 14
2-{3-[4-(4-Fluorophenyl)piperidino]propyl]-5,6,7,8-tetrahydro-4(3H)-
quinazolinone
1H NMR (DMSO-d6, ~ ): 1.30 - 3.20(23H, m), 7.00 - 7.40(4H, m), 12.38(1H, brs).
Mass(ESI): 370.3 (M+H)+
Example 15
2-[3-[4-(4-Chlorophenyl)piperidino]propyl]-5,6,7,8-tetrahydro-4(3H)-
quinazolinone
1H NMR (DMSO-d6, ~ ) : 1.30 - 3.20(23H, m), 7.10 - 7.60(4H, m), 12.36(1H,
brs).
Mass(ESI): 386.4 (M+H)+
Example 16
2-[3-[4-(4-Methylphenyl)piperidino]propyl]-5,6,7, 8-tetrahydro-4(3H)-
quinazolinone
1H NMR (DMSO-d6, ~ ) : 1.20 - 3.20(26H, m), 7.00 - 7.20(4H, m), 12.34(1H,
brs).
Mass(ESI):366.4 (M+H)+
Example 17
2-[3-[4-(4-Methoxyphenyl)piperidino]propyl]-5,6,7,8-tetrahydro-4(3H)-
quinazolinone
1H NMR (DMSO-d6, ~ ) : 1.20 - 3.20(23H, m), 3.71 (3H, s), 6.83(2H, d, J=8.6
Hz),
7.12(2H, d, J=8.6 Hz), 12.35(1H, brs).
Mass(ESI): 382.3 (M+H)+
Example 18
2-[3-[4-[4-(Trifluoromethyl)phenyl]piperidine]propyl]-3,5,7,8-tetrahydro-4H-
thiino[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, 8 ) : 1.50 - 3.60(21H, m), 7.46(2H, d, J=8.2 Hz), 7.64(2H, d,
J=8.2
Hz), 12.65 ( 1 H, brs).
Mass(ESI): 438.3 (M+H)+
_Example 19
2-[3-[4-[4-(Trifluoromethyl)phenyl]piperidino]propyl]-5,6,7,8-tetrahydro-4(3H)-
46
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
quinazolinone
IH NMR (DMSO-d6, 8 ) : 1.30 - 3.20(23H, m), 7.45(2H, d, J=8.1 Hz), 7.64(2H, d,
J=8.1
Hz), 12.36(1H, brs).
Mass(ESI): 420.3 (M+H)+
Example 20
2-[3-[(2R,6R)-4-Chlorophenyl-2,6-dimethyl-1-piperazinyl]propyl]-3,5,7,8-
tetrahydro-4H-thiino[4,3-d]pyrimidin-4-one
'H NMR (DMSO-d6, ~ ) : 0.99(6H, d, J=6.0 Hz), 1.50 - 3.70(18H, m), 6.90(2H, d,
J=9.0
l0 Hz), 7.20(2H, d, J=9.0 Hz), 12.45(1H, brs).
Mass(ESI): 433.1 (M+H)+
Example 21
2-[3-[4-(4-Fluorophenyl)-3,6-dihydro-1 (2H)-pyridyl]-3-methylpropyl]-3,5,7,8-
tetrahydro-4H-thiino[4,3-d]pyrimidin-4-one hydrochloride
1H NMR (DMSO-d6, 8 ) : 0.80 - 5.20(20H, m), 6.20(1H, m), 7.00 - 7.70(4H, m).
Mass(ESI): 400.1 (M+H)+
Example 22
2-[3-[4-[4-(Trifluoromethoxy)phenyl]-3,6-dihydro-1 (2H)-pyridyl]propyl]-
3,5,7,8-tetrahydro-4H-thiino[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, 8 ) : 1.70 - 3.60(18H, m), 6.18(1H, m), 7.31(2H, d, J=8.1
Hz),
7.53(2H, d, J=8.1 Hz), 12.37(1H, brs).
Mass(ESI): 452.2 (M+H)+
Example 23
2-[3-[4-[4-(Trifluoromethoxy)phenyl]-3,6-dihydro-1 (2H)-pyridyl]propyl]-
5,6,7,8-tetrahydro-4(3H)-quinazolinone
1H NMR (DMSO-d6, ~ ) : 1.40 - 3.20(20H, m), 6.18(1H, m), 7.31(2H, d, J=8.2
Hz),
7.53 (2H, d, J=8.2 Hz), 12.13 ( 1 H, brs).
Mass(ESI): 434.2 (M+H)+
Example 24
2-[3-[4-(4-biphenylyl)piperidino]propyl]-3,5,7,8-tetrahydro-4H-
thiino[4,3-d]pyrimidin-4-one
47
CA 02474434 2004-07-23
WO 03/063874 PCT/JP03/00708
1H NMR (DMSO-d6, ~ ) :1.5-2.2(8H, m), 2.3-2.65(2H, m), 2.65-2.9(4H, m), 2.9-
3.1(2H,
m), 3.2-3.6(SH, m), 7.2-7.7(9H, m).
Mass: 446.4(M+H)+
Example 25
2-[3-[4-(3,4-dichlorophenyl)-1-piperazinyl]propyl]-5,6,7,8-tetrahydro-4(3 H)-
quinazolinone
IH NMR (DMSO-d6, ~ ) : 1.5-1.9(6H, m), 2.2-2.6(12H, m), 3.0-3.2(4H, m),
6.9(1H, dd,
J=9.0, 2.8 Hz), 7.09( 1 H, d, J=2.8 Hz), 7.3 8 ( 1 H, d, J=9.0 Hz), 12.18 ( 1
H, br s).
Mass: 421.1, 423.2 (M+H)+
Example 26
2-[3-[4-(3,4-dichlorophenyl)-1-piperazinyl]propyl]-3,5,7,8-tetrahydro-
4H-thiino[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, ~ ) : 1.7-1.85(2H, m), 2.2-2.6(8H, m), 2.7-2.9(4H, m), 3.0-
3.2(4H,
m), 3.39(2H, s), 6.90(1H, dd, J=9.0, 2.5 Hz), 7.10(1H, d, J=2.5 Hz), 7.37(1H,
d, J=9 Hz),
12.4(1H, br s).
Mass: 441.1, 439.1(M+H)+
Example 27
2-[3-[4-(4-biphenylyl)-1,2,3,6-tetrahydropyridyl]propyl]-3,5,7,8-tetrahydro-4H-
thiino[4,3-d]pyrimidin-4-one
1H NMR (DMSO-d6, ~ ) : 1.7-2.0(2H, m), 2.3-2.9(8H, m), 3.09(2H, s), 3.2-
3.6(6H, m),
6.20(1H, s), 7.3-7.9(9H, m), 12.4(1H, s).
Mass:444.2(M+H)+
48